

Abstract
Arthropod-borne viruses (arboviruses) comprise a significant and ongoing threat to human health, infecting hundreds of millions annually. Three such arboviruses include circumtropical dengue, Zika, and chikungunya viruses, exhibiting continuous emergence primarily via Aedes mosquito vectors. Nicaragua has experienced endemic dengue virus (DENV) transmission involving multiple serotypes since 1985, with chikungunya virus (CHIKV) reported in 2014–2015, followed by Zika virus (ZIKV) first reported in 2016. In order to identify patterns of genetic variation and selection pressures shaping the evolution of co-circulating DENV serotypes in light of the arrival of CHIKV and ZIKV, we employed whole-genome sequencing on an Illumina MiSeq platform of random-amplified total RNA libraries to characterize 42 DENV low-passage isolates, derived from viremic patients in Nicaragua between 2013 and 2016. Our approach also revealed clinically undetected co-infections with CHIKV. Of the three DENV serotypes (1, 2, and 3) co-circulating during our study, we uncovered distinct patterns of evolution using comparative phylogenetic inference. DENV-1 genetic variation was structured into two distinct co-circulating lineages with no evidence of positive selection in the origins of either lineage, suggesting they are equally fit. In contrast, the evolutionary history of DENV-2 was marked by positive selection, and a unique, divergent lineage correlated with high epidemic potential emerged in 2015 to drive an outbreak in 2016. DENV-3 genetic variation remained unstructured into lineages throughout the period of study. Thus, this study reveals insights into evolutionary and epidemiologic trends exhibited during the circulation of multiple arboviruses in Nicaragua.
Keywords: Dengue virus, Chikungunya virus, Nicaragua, Phylogenetics, Evolution, Positive selection
1. Introduction
Arthropod-borne viruses (arboviruses) and their rapid emergence pose a significant threat to public health. Three such emerging arboviruses include dengue virus (DENV), chikungunya virus (CHIKV), and Zika virus (ZIKV). The global disease burden for all three of these arboviruses is significant, with DENV the most common, causing an estimated 390 million infections per year (Bhatt et al., 2013; Liu-Helmersson et al., 2014) and 50 million to 100 million symptomatic cases annually (Stanaway et al., 2016; Cattarino et al., 2020). CHIKV has emerged more recently and gained global momentum after arriving in the Americas in late 2013. By 2015, it had caused over 1.3 million suspected cases in the Caribbean, Latin America, and the United States (http://www.who.int/mediacentre/factsheets/fs327/en/). ZIKV began emerging on a global scale in 2007 in the South Pacific, causing massive epidemics in the Americas starting in 2015 when it was first reported (Kindhauser et al., 2016), although studies suggest it may have arrived as early as 2014 (Faria et al., 2017). DENV and ZIKV are close relatives within the genus Flavivirus, family Flaviviridae (Lindenbach et al., 2007; Simmonds et al., 2017), whereas CHIKV is a member of the genus Alphavirus, family Togaviridae (Chen et al., 2018). In spite of their taxonomic differences, all three viruses are transmitted primarily by the same mosquito species, Aedes aegypti (Linnaeus), the Yellow Fever mosquito, and Aedes albopictus (Skuse), the Asian Tiger mosquito (Smith, 1956; Gubler, 1998). The potential impact of these viruses is significant, estimated for DENV alone as placing 3.9 billion people in 128 countries at risk of infection (Brady et al., 2012).
In addition to overlapping geographically and by major vectors, all three of these viruses cause substantial asymptomatic infections or nonspecific symptoms (e.g., febrile illness, with rash, myalgia, and/or arthralgia) that challenge clinical diagnosis (Paixão et al., 2018). In the case of DENV, severe life-threatening disease (dengue hemorrhagic fever/dengue shock syndrome; DHF/DSS) is rare and develops depending on multiple factors including host immune status and genetics, the amount of virus inoculated by the arthropod vector, and virus genetic makeup (Gubler and Kuno, 1997; Phuong et al., 2004; Deen et al., 2006; WHO, 2009). For example, DENV occurs as four phylogenetically and antigenically distinct serotypes (DENV-1 to − 4; Zanotto et al., 1996; Holmes and Twiddy, 2003; Rico-Hesse, 2003, Gubler et al., 2007) that elicit cross-reactive immune responses and can impact disease severity through antibody-dependant enhancement (Halstead, 1988; Katzelnick et al., 2017; Salje et al., 2018). Furthermore, certain virus genotypes, which continue to evolve and differentiate (Rico-Hesse, 1990; Vasilakis et al., 2007; Klungthong et al., 2008; Araujo et al., 2009), have been associated with disease and transmission phenotypes (e.g., Anderson and Rico-Hesse, 2006; Mota and Rico-Hesse, 2009; Andrade et al., 2016). The close relationship between DENV and ZIKV also drives cross-reactive immune responses in populations where they co-circulate, with epidemiologic and disease implications (Gordon et al., 2019; Rodriguez-Barraquer et al., 2019)
Nicaragua is a tropical country (~11° to 15°N) where Aedes aegypti is well established in close proximity to people. All four serotypes of DENV have been circulating in Nicaragua for decades, though serotype prevalence and dominance fluctuate annually (Kouri et al., 1991; Harris et al., 2000; Hammond et al., 2005; Balmaseda et al., 2006; Balmaseda et al., 2010; OhAinle et al., 2011). Hyperendemicity, where multiple serotypes co-circulate in the same transmission season, is increasingly common (PAHO, 2015; Ramos-Castaneda et al., 2017). One of the first major dengue outbreaks occurred in Managua, Nicaragua, in 1985, resulting in over 17,000 cases, and was caused by co-circulating DENV-1 and −2 (Kouri et al., 1991). Vector control and eradication efforts were initiated in response, and dengue outbreaks remained limited except for periodic transmission of DENV-1, DENV-2, and DENV-4 in the early 1990s. However, in 1994–95, an introduction of DENV-3 caused a major epidemic with disease including DHF/DSS (Centers for Disease Control and Prevention CDC, 1995; Guzman et al., 1996; Harris et al., 1998). Since then, DENV has remained hyperendemic in Nicaragua despite vector control efforts, causing periodic severe outbreaks linked to virus genetic variation interacting with host immune status (OhAinle et al., 2011). CHIKV was first reported in Nicaragua in 2014 (Wang et al., 2016). ZIKV was first reported in Nicaragua in January 2016 (Zambrana et al., 2018) but was probably present as early as March 2015 (Thézé et al., 2018).
In addition to vector eradication efforts, numerous dengue vaccines are in development (Hombach, 2007; Whitehead et al., 2007), as are ZIKV (Gaudinski et al., 2018; Modjarrad et al., 2018) and CHIKV (e.g., Edelman et al., 2000; Wang et al., 2008) vaccines. To inform control efforts, including vaccine design and testing where cross-reactive immune responses are risk factors, virus surveillance for country-specific epidemiological data is needed along with a better understanding of the major patterns of virus evolution. The Nicaraguan Pediatric Dengue Cohort Study (PDCS) and Nicaraguan Dengue Hospital-based Study were designed as long-term cohort and hospital-based prospective surveillance studies, respectively, to provide essential epidemiologic, clinical, and virologic data, including seroprevalence, incidence, host immune status, disease syndrome, and infecting virus (Kuan et al., 2009; OhAinle et al., 2011; Narvaez et al., 2011). We leverage these data to elucidate trends in virus evolution and improve our understanding of the natural history of emerging infectious diseases.
In this study, we characterize DENV molecular evolution in Nicaragua during a period that includes the regional emergence of CHIKV and ZIKV. Based on second-generation sequencing of multiple co-circulating DENV serotype samples from the PDCS and complementary Hospital-based study, we employ phylogenetic inference to elucidate genetic variation, viral relationships and origins, and evolutionary patterns including selection pressures that may be shaping virus diversification over time. The results of our study provide insight into the evolutionary dynamics of DENV associated with epidemic patterns in Nicaragua during a period of arbovirus emergence.
2. Materials and methods
2.1. Study population and design
DENV-positive samples were collected as part of the PDCS and Dengue Hospital-based study established in Nicaragua in 2004 and 2005, respectively (Kuan et al., 2009; Balmaseda et al., 2010); Narvaez et al., 2011). This study included a subset of viruses isolated from 2013 to 2016, each sample with its corresponding record of serotype and year of isolation. The PDCS (2004 to present) follows ~3800 children between the ages of 2–14 years old who receive primary health care from study physicians at the Health Center Sócrates Flores Vivas (HCSFV) (Kuan et al., 2009) and agree to seek medical care at the first sign of fever. Acute- and convalescent-phase samples (days 1–5 and days 14–21 post-onset of symptoms, respectively) are collected from participants who present to the health center with suspected dengue or undifferentiated fever. The Dengue Hospital-based Study (2005 to present) derives samples from patients with suspected dengue presenting to the Infectious Diseases Unit of the National Pediatric Reference Hospital, Hospital Infantil Manuel de Jesús Rivera (HIMJR), in Managua. Blood samples are collected during the acute phase as well as at 14–28 days and 3, 6, 12, and 18 months.
All samples in both studies are tested for DENV, CHIKV, ZIKV virus by multiplex real-time RT-PCR (Waggoner et al., 2016a; Waggoner et al., 2016b) (CHIKV and ZIKV since 2014 and 2015, respectively). Samples from symptomatic DENV infections were analyzed further by serotype-specific RT-PCR for the detection of viral RNAs (Kuan et al., 2009; Waggoner et al., 2013), and a subset was subjected to virus isolation in C6/36 cells (Balmaseda et al., 1999).
2.2. RNA extraction and cDNA synthesis
DENV strains isolated from virus-infected C6/36 cell culture were transported from Nicaragua to UC Berkeley (Harris laboratory), then to the California Academy of Sciences. The number of virus passages in cell culture was kept to one to two passages to minimize selection in vitro. Viral genetic material was extracted from 300 to 400 μl of cell culture supernatant using AgenCourt RNAdvance Cell V2 kit (Beckman Coulter Genomics, USA) according to the manufacturer’s guidelines with slight modifications. Reverse transcription and the first-strand cDNA synthesis were performed with random hexamer primers and Superscript® III First-Strand Synthesis System (Thermofisher, USA). This was followed by the second-strand cDNA synthesis using Klenow exo-enzyme (New England Biolabs, USA).
2.3. Library preparation and next-generation sequencing
To prepare samples for next-generation sequencing, all cDNA samples derived from cell culture supernatant were purified using Zymo Clean & Concentrator kit (Zymo Research, USA). DENV library preparation was performed using Nextera® XT DNA Library Preparation Kit (Illumina, USA) according to the manufacturer’s instructions with modifications. Briefly, 3 nanograms of input cDNA with 4 μl index was used per reaction (rather than the suggested 1 ng input with 5 μl index). Samples were tagged with a molecular ID tag (MID), which allowed for multiplex sequencing using the Nextera® XT Index Kit (Illumina, USA). Libraries were quantified and assessed for quality using Agilent 2100 Bioanalyzer (Agilent Technologies, USA) and Qubit 2.0 Fluorometer (Life Technologies, USA) prior to equimolar pooling of samples. Sequencing was performed on the Illumina MiSeq platform using pairedend, dual-indexed sequencing and multiple MiSeq sequencing reagent kits, including MiSeq reagent Nano Kit v2 (2 × 250 bp), MiSeq reagent kit v2 (2 × 250 bp), and MiSeq reagent kit v3 (2 × 300 bp), at the Center for Comparative Genomics at the California Academy of Sciences.
No-template controls (NTCs) were included from the RNA extraction to library preparation step and were quantified along with the samples. The quantification results demonstrated the absence of nucleic acid material in the NTCs. Thus, they were not included in the sequencing reaction.
2.4. Data analysis
Raw sequence reads were uploaded to CLC Genomics Workbench 7.0.3 (https://www.qiagenbioinformatics.com/). Reads were filtered for quality and adaptors trimmed using default parameters. High quality reads were mapped using default parameters to complete reference genomes for each DENV serotype from GenBank (NC_001477 (DENV-1), NC_001474 (DENV-2), and NC_001475 (DENV-3), and NC_002640 (DENV-4). To identify non-DENV viruses that could be present in the samples, the reference genomes for CHIKV (NC_004162) and ZIKV (NC_012532.1) were also used to map targets for raw reads. Virus consensus sequences were extracted in CLC Genomics Workbench 7.0.3 using default parameters and exported for phylogenetic analyses. We have empirically determined that when there were at least 800 DENV reads in the sample, sufficient coverage could be obtained for the genomes, which resulted in a reliable phylogenetic relationship.
Raw unprocessed sequencing reads are available through the NCBI Short Read Database as part of BioProject TBD. The assembled whole and partial virus genomes are available through the NCBI Nucleotide database, and their accession numbers are TBD (these will be available upon request and prior to publication).
2.5. Phylogenetic analysis
We analyzed virus consensus genomes from Nicaragua in a phylogenetic context that included publicly available complete-genome sequences from Central and South America and the Caribbean along with representatives from the major genotypes. Our analytical protocol started with an initial broad dataset with as many publicly available sequences (e.g., NCBI, nextstrain.org) for a given serotype as possible for comprehensive coverage over time as well as geographic region, with a particular focus on samples from Central and South America, and the Caribbean. We also blasted our sample genomes in NCBI to pull in top hits of our samples’ closest relatives that may have been missed in the first approach. From this large comprehensive phylogeny, we then pruned out redundant sequences, retaining representative sequences of phylogenetic structure, time and geography, so that the phylogenetic tree was more manageable and visually clear, while maintaining the topology and metadata patterns recovered in the initial comprehensive tree. Distinct alignments were produced for each DENV serotype as well as for CHIKV (no ZIKV was recovered from our samples) using MAFFT on XSEDE (7.305), implemented in the CIPRES Science Gateway (Miller et al., 2010). Phylogenetic trees were estimated using two different methods: maximum likelihood (ML) phylogenetic trees were estimated using RAxML-HPC Black-Box (8.2.10) via CIPRES Science Gateway (Miller et al., 2010), and Bayesian tree estimation was conducted using BEAST2 (Bayesian Evolutionary Analysis for Sampling Trees) and the BEAGLE package (Bouckaert et al., 2014; Drummond et al., 2006). The underlying model of evolution for phylogenetic estimation was a General Time Reversible (GTR) + Gamma4 nucleotide substitution model. We also used BEAST2 and the Bayesian Markov chain Monte Carlo approach (Bouckaert et al., 2014; Drummond et al., 2006) to test for consistency within our DENV phylogenetic framework (Supplementary Figs. S1 to S3 show phylogenetic trees of DENV-1 to − 3, respectively, from the BEAST2 analysis).
2.6. Testing for natural selection
Natural selection can be detected statistically as an elevated (positive selection) or suppressed (negative selection) rate of nucleotide substitutions that modify the translated amino acid (delta non-synonymous or ds) relative to the rate of nucleotide substitutions that do not modify the translated amino acid (delta synonymous or dN). The ratio of non-synonymous (dN) to synonymous (dS) mutations (ω ratio) was evaluated using the DataMonkey web-server and the following models: branch site unrestricted statistical test for episodic diversifying selection (BUSTED), mixed-effects model of evolution (MEME), and adaptive branch site random-effects likelihood method for episodic diversifying selection (aBSREL). The three chosen models were used to test for selection across branches (aBSREL), individual sites (MEME), and entire genes (BUSTED). The general time-reversible (GTR) model was used as the nucleotide substitution model. Starting trees were inferred by the neighbor-joining method with significance levels of p < 0.1 or a Bayes Factor of >50. Prior to selection analyses, alignments were screened for recombination events using the HyPhy package of the DataMonkey web-server (www.datamonkey.org) (Delport et al., 2010), and none were found.
3. Results
Dengue virus culture supernatants derived from over 67 serum/plasma samples from the Nicaraguan PDCS and Hospital-based study collected between 2013 and 2016 were sequenced, 42 of which had sufficient genome coverage (at least 800 mapped reads per sample after quality control and trimming) for phylogenetic analysis (Supplementary Table 1). All DENV recovered fell into one of 3 serotypes: DENV-1, − 2, or − 3. No DENV-4 was detected, consistent with clinical reporting. Eight instances of CHIKV co-infections of clinical DENV samples were identified, of which three had adequate genome coverage for phylogenetic analysis (Supplementary Table 1). No instances of ZIKV coinfection were confirmed. Several of the samples that did not meet the coverage criterion showed evidence of contamination by a mosquito densovirus, specifically Culex pipiens pallens densovirus strains 0507JS11 (Genbank Accession No. FJ805445.1) and YN05217 (Genbank Accession No. EF579771.1), presumably acquired in cell culture.
The different DENV serotypes analyzed showed distinct patterns of circulation (Fig. 1) and evolution. DENV-1 is endemic to Nicaragua and has periodically circulated since 2004–2005. This serotype shows peak relative abundances in terms of the number of cases per month as recently as 2013 and 2014, before apparently disappearing from the PDCS and Hospital-based study populations (Fig. 1). Phylogenetic analysis indicates that DENV-1 isolates from Nicaragua fall within the American/African V genotype (Fig. 2). However, within this genotype, Nicaraguan samples diverge into two distinct lineages, both of which appear to have co-circulated since their introduction into the country. Both lineages include samples from as early as the mid-2000s, suggesting they diversified at the same time. Importantly, these lineages continued to co-circulate and both include samples of DENV-1 from 2013 (there is only a single sample from 2014 in our dataset), suggesting that they were equally abundant during this peak year. Our selection analysis does not reveal any statistical support for the role of positive selection in the diversification of these two Nicaraguan lineages. Based on our phylogenetic inference, which incorporated publicly available DENV-1 sequences from other countries, both lineages from Nicaragua have roots in strains from South America that were circulating in the late 1990s (Supplementary Fig. S1).
Fig. 1.
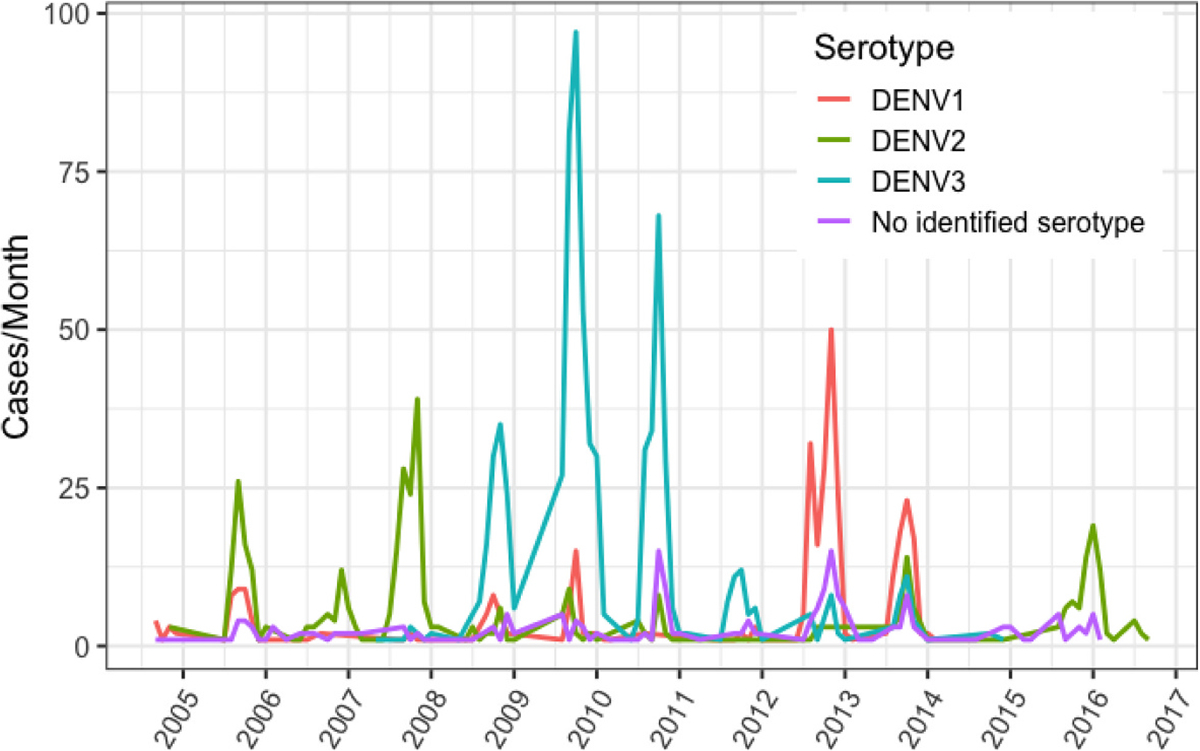
Dengue virus cases per month in Nicaragua from the combined Pediatric Dengue Cohort Study (PDCS) and Hospital-based study, color-coded by serotype, 2005–2016.
Fig. 2.
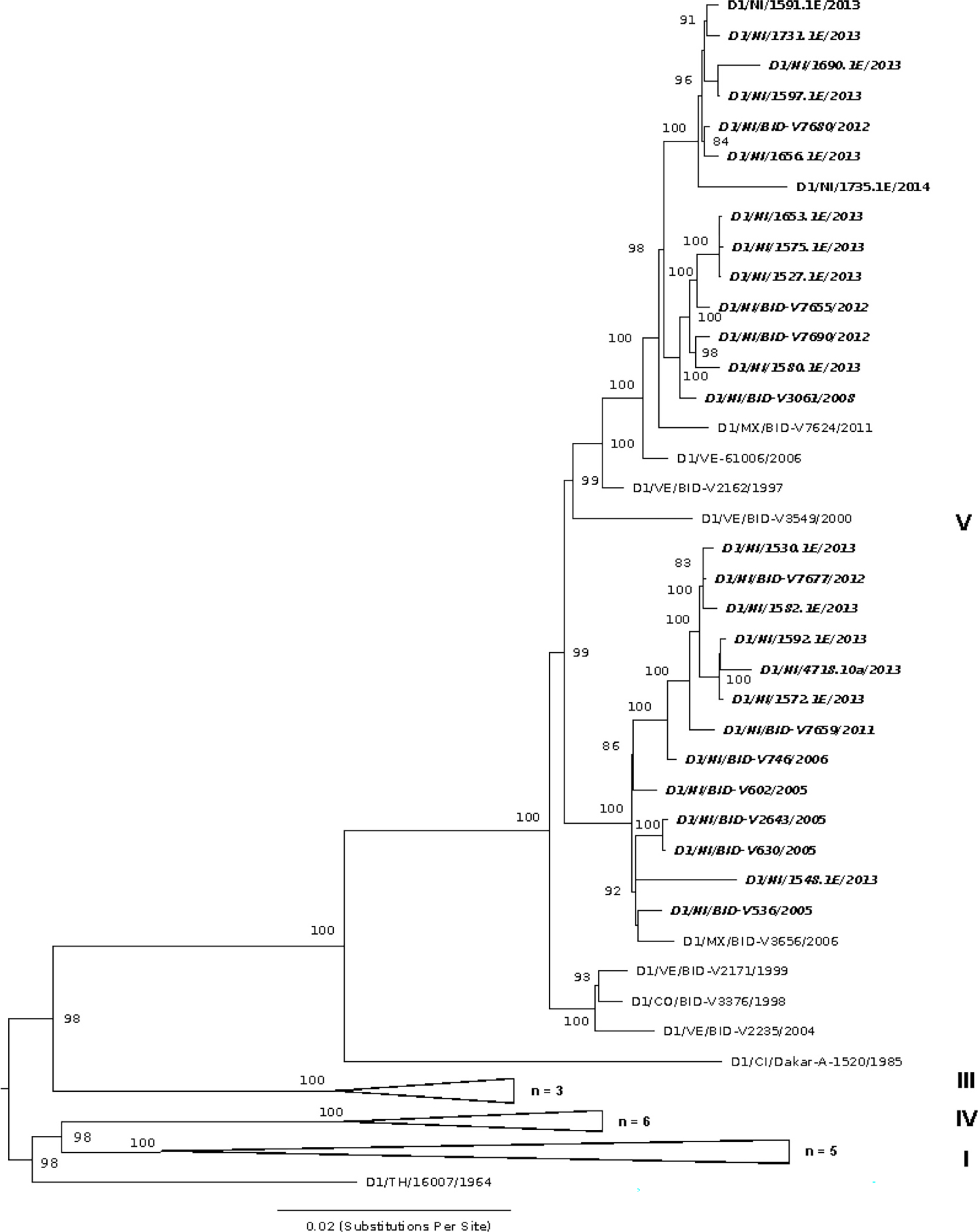
Maximum likelihood (ML) phylogeny of DENV-1 in the region, including 16 genomes from Nicaragua generated in this study, along with 52 previously published sequences. Sequences are labeled by ISO country code, sample identifier, and year of infection. Nicaragua-derived sequences are shown in italics. The tree is mid-point rooted. Relevant major genotypes (of which there are five (I-V) (Holmes and Twiddy, 2003) to six (VI) (Pyke et al., 2016) in total) are labeled on the right. Bootstrap values are shown for each clade based on ML replicates (until convergence) generated in RAxML (Stamatakis et al., 2008).
DENV-2 has been notable for epidemics in Nicaragua associated with severe disease starting in 2005 (Fig. 1), where a combination of virus genotype and host prior immunity proved particularly virulent over multiple transmission seasons (OhAinle et al., 2011). DENV-2 in Nicaragua falls within the Asian/American genotype, which has dominated the Nicaraguan landscape since 1999 (Balmaseda et al., 1999; Hammond et al., 2005; Balmaseda et al., 2010). DENV-2 further diversified into three lineages (NI-I, -IIa and -IIb; OhAinle et al., 2011), one of which, NI-I, was more virulent in people with previously acquired DENV-1 immunity, whereas NI-IIb was more virulent in people with previously acquired DENV-3 immunity (OhAinle et al., 2011). Lineage replacement of NI-I by NI-IIb became evident by 2008, and our data shows that this fit lineage (Quiner et al., 2014) persisted into 2016 (Fig. 3), in spite of dropping to undetectable levels in years prior (Fig. 1). Nicaraguan DENV-2 strains sequenced from 2013 to 2016 all fall within the NI-IIb clade, which consists of 2 main sublineages and includes foreign samples from the region, suggesting geographic exchange with other countries in the region (e.g. Mexico, Guatemala, Honduras). There are several synapomorphies (unique shared derived mutations at the nucleotide level) that distinguish this NI-IIb lineage from prior Nicaraguan DENV-2 NI-I and NI-IIa clades, and tests for selection indicate strong statistical support for episodic positive selection. First, MEME results indicated episodic, diversifying, positive selection at 22 sites (p-value threshold 0.05); one of these sites, AA 1375, has an estimated ω of 51 and is shared by a lineage of 2016 NI samples indicated by an * in Fig. 3. Second, BUSTED results also indicated gene-wide episodic diversifying selection (LRT, p-value = 0.000 ≤ 0.05; unconstrained model ω3 estimated as 5.89 for 0.92% of sites) (aBSREL results did not report evidence of branch-based selection). The 2015–2016 DENV-2 lineage was associated with increasing epidemic activity and severe disease in 2015–2016, continuing until 2019 (unpublished), based on national surveillance clinical and public health records.
Fig. 3.
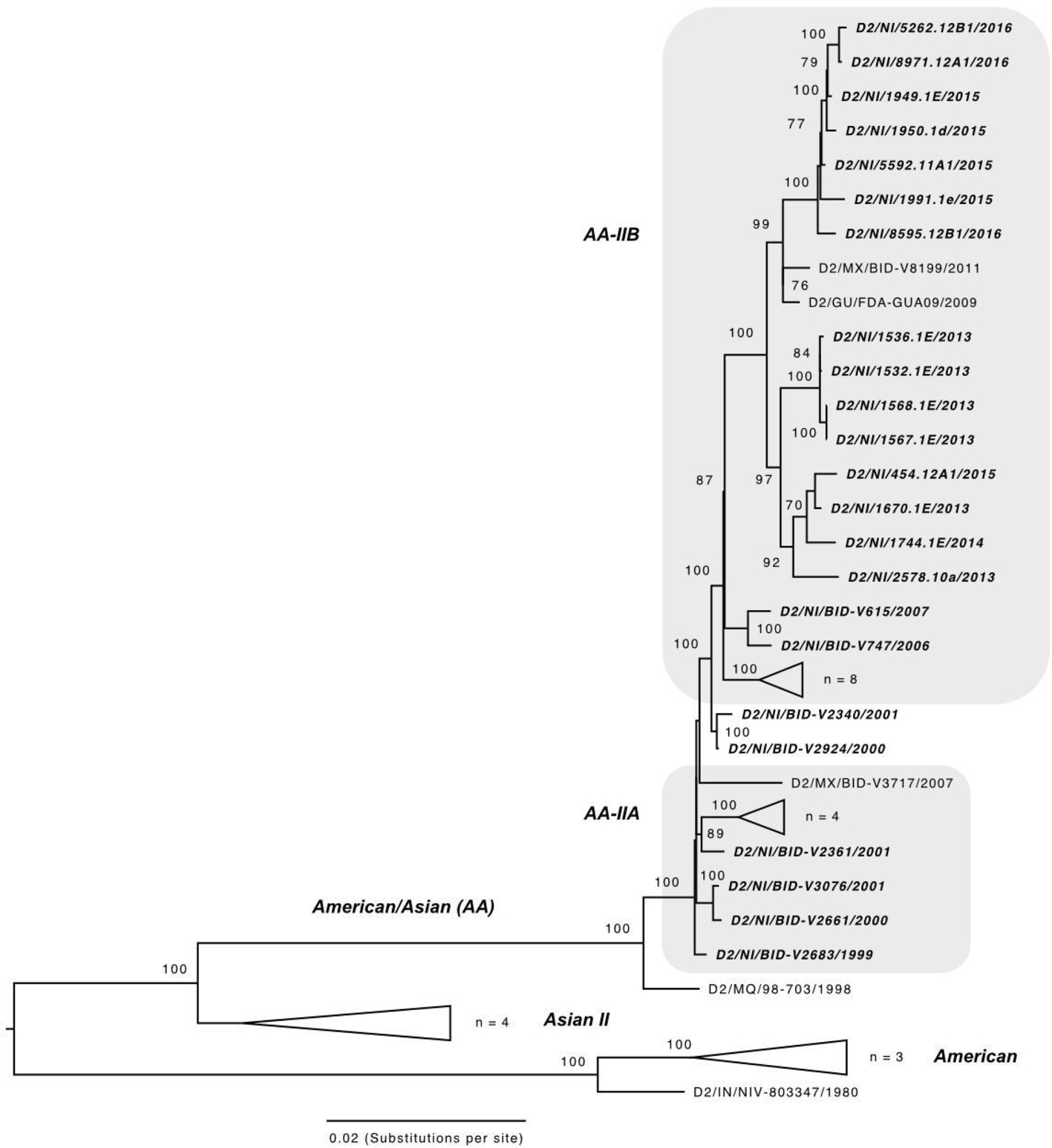
Maximum likelihood (ML) phylogeny of DENV-2 in the region, including 15 genomes from Nicaragua generated in this study, along with 51 previously published sequences. Sequences are labeled by ISO country code, sample identifier, and year of infection. Nicaragua-derived sequences are shown in italics and fall within the American/Asian (AA) genotype. Nicaraguan clades noted by OhAinle et al. (2011) as related to disease severity are labeled AA-IIA and AA-IIB and shaded grey. The tree is mid-point rooted. Other relevant major genotypes (of which there are six, Chen and Vasilakis, 2011, Twiddy et al., 2002) are labeled on the left. Bootstrap values are shown for each clade based on ML replicates (until convergence) generated in RAxML (Stamatakis et al., 2008).
DENV-3 dominated in Nicaragua between 2009 and 2011, then circulated at lower levels until 2014 after which it was no longer detected (Fig. 1). The DENV-3 isolates from Nicaragua fall within genotype III (Indian subcontinent) but have not continued to diversify into structured lineages based on year of collection (Fig. 4).
Fig. 4.
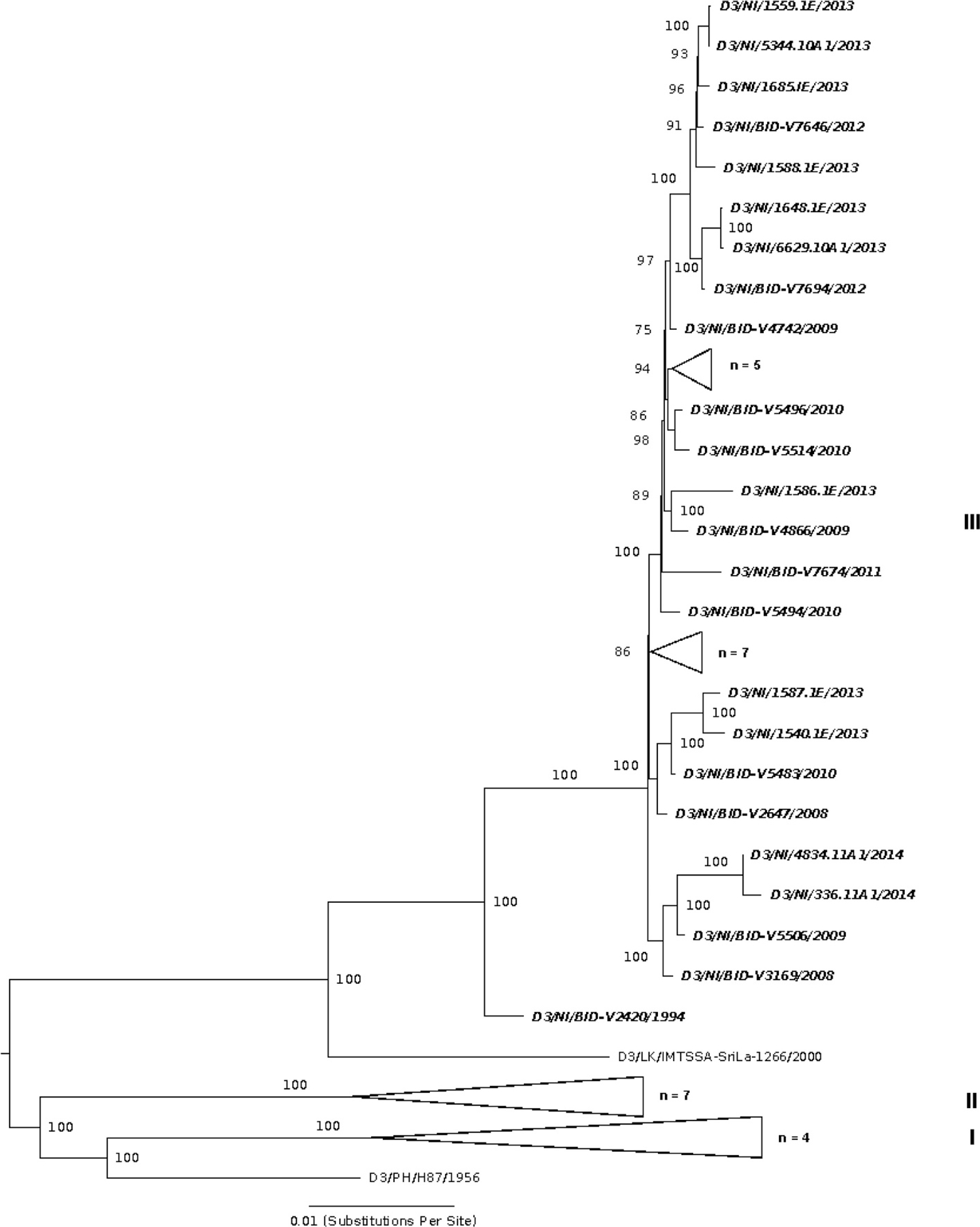
Maximum likelihood (ML) phylogeny of DENV-3 in the region, including 11 genomes from Nicaragua generated in this study, along with 59 previously published sequences. Sequences are labeled by ISO country code, sample identifier, and year of infection. Nicaragua-derived sequences are shown in italics. The tree is mid-point rooted. Relevant major genotypes (of which there are four (I-IV) (Holmes and Twiddy, 2003; Lanciotti et al., 1994; Messer et al., 2003) to five (V) (Diaz et al., 2006) in total) are labeled on the right. Bootstrap values are shown for each clade based on ML replicates (until convergence) generated in RAxML (Stamatakis et al., 2008).
Within our DENV-positive samples (both PDCS and Hospital populations), eight presumptive CHIKV co-infections were detected that traditional screening had missed, three of which yielded consensus genomes with sufficient coverage for further analysis. One of these samples had a high number of reads (over 830,356 total CHIKV reads, or 15.3% of raw reads) while the other two had lower coverage (Supplementary Table 1). All three of these CHIKV co-infections were associated with DENV-2 infections and fall within the CHIKV Asian genotype (Supplementary Fig. 4). Interestingly, they group together and are distinct from other Nicaraguan CHIKV sequences.
4. Discussion
Taking advantage of the ongoing cohort and hospital studies in Nicaragua since 2004 and 2005, respectively, we demonstrate contrasting patterns of evolution among co-circulating DENV serotypes 1, 2, and 3 over recent years, including prior to the introduction and during local emergence of both CHIKV and ZIKV. We also confirmed and characterized three cases of DENV-2 co-infected with CHIKV.
DENV-2 was the only serotype to show evidence of positive selection and was characterized by the emergence of a lineage as early as 2013 associated with high levels of DENV-2 activity in the 2015–2016 epidemic season, also notable for more severe disease. DENV-2 viruses sampled between 2013 and 2016 descended from a lineage (NI-IIB, Fig. 3) shown to be more fit than NI-I (Quiner et al., 2014) and to cause more severe dengue disease in humans with prior DENV-3 immunity (OhAinle et al., 2011).
DENV-1 and −3 were both circulating in 2012 and 2013, although DENV-1 was dominant (Fig. 1), when this DENV-2 lineage first emerged in 2013 from the NI-IIB clade that has been circulating in Nicaragua since 2009 (Anes et al., 2011; OhAinle et al., 2011). DENV-2 disease severity and phenotype associated with this IIB lineage depends not only on prior DENV exposure but also viral genetic sequence (OhAinle et al., 2011). Virus genetic sequence has been noted before to play a role in disease severity and/or correlates of epidemic intensity (Rico-Hesse, 1990; Rico-Hesse et al., 1997; Bennett et al., 2003; Messer et al., 2003; Messer et al., 2002; Rico-Hesse, 2007; OhAinle et al., 2011). Our 2016 samples indicate that NI-IIB has completely replaced earlier lineages, suggesting that these viruses are more fit than earlier strains. In fact, studies in Aedes aegypti mosquitoes and Aedes aegypti cell lines (e.g., Aag2) indicated a fitness advantage of NI-IIB over NI-I (Quiner et al., 2014). DENV-2 has a history of lineage replacement of less virulent autochthonous strains by more virulent variants, such as when the Asian-American genotype II displaced the American genotype in numerous countries in the region (Cologna et al., 2005; Rico-Hesse, 2007). The absence of lineage replacement (in DENV-1) and even the formation of lineages (in DENV-3) in Nicaragua for the other serotypes may suggest that DENV-2 is uniquely under selection specifically due to immunological pressures it experiences in a hyperendemic background.
Both positive selection and importation of new strains can be drivers of lineage turnover in DENV transmission dynamics. Positive selection and adaptive evolution have been reported in DENV circulating in the region (e.g., Bennett et al., 2003, Guzmán et al., 2000; Nagao and Koelle, 2008), although purifying selection is more common (Holmes, 2003; Klungthong et al., 2004; Wittke et al., 2002). In terms of importation, Nicaragua has opportunities to exchange DENV with neighboring Central American, Caribbean, and South American countries where hyperendemicity is common and among which gene flow has been noted previously (Bennett et al., 2006; Foster et al., 2004; Uzcategui et al., 2001; Twiddy et al., 2002). Both DENV-1 and DENV-2 phylogenies (Figs. 2 and 3, respectively) indicate exchange between neighboring countries; for example, DENV-2 from Nicaragua in 2013 appears associated with viruses collected in Guatemala (D2/GU/FDA-GUA09/2009) and Mexico (D2/MX/BID-V8199/2011).
Some limitations exist in our study. Our samples yielded different numbers of virus sequence reads from sample to sample, possibly due to different levels of input into the culture itself (e.g., samples from individuals varying widely in viremia) or variability in sample handling, despite our utmost care in consistently adhering to protocols.
In addition, we included no-template controls (NTCs) in the RNA extraction and library preparation steps. Since our quantification methods prior to sequencing did not detect the presence of nucleic acid material in the NTCs, we did not include them in our sequencing reaction. To help ensure that template sequences were not contaminants, we eliminated samples with less than 800 viral reads from our analyses, a cut-off we empirically determined to provide sufficient coverage for both DENV and CHIKV genomes for phylogenetic inference. In the future, a more direct quantification of DENV reads in NTCs would help inform this cutoff, and the inclusion of sequencing controls (including NTCs) should be adopted.
Our study provides insights into DENV evolutionary dynamics before and during the introduction of two additional arboviruses into Nicaragua, ZIKV, and CHIKV, transmitted by the same vector. Because CHIKV and ZIKV infections can yield similar clinical symptoms to DENV infection in an arboviral hyperendemic region, specific diagnosis and detection pose a challenge. Because of the low specificity and sensitivity in many arboviral tests (e.g. Arrieta et al., 2019), co-infections continue to add to the difficulties in determining the causative agent (i.e., DENV, CHIKV, or ZIKV) in an arboviral hyperendemic region where pathogens yield similar clinical symptoms. Our culture-based RNA sequencing approach is both sensitive and highly discerning in the analysis phase, besides becoming increasingly cost-effective. Furthermore, our approach revealed co-infections of CHIKV that had not been identified in DENV-positive samples using standard multiplex real-time RT-PCR testing.
The Asian genotype of CHIKV first reached the Americas in a series of epidemics on St. Martin Island, French West Indies, in 2013 (Leparc-Goffart et al., 2014). Shortly after its introduction to the Americas, chikungunya cases were first reported in Nicaragua in July of 2014, leading to epidemics in 2014 and 2015 (Balmaseda et al., 2015; Wang et al., 2015; Tan et al., 2018; Gordon et al., 2018). Our results are consistent with this introduction of CHIKV into Nicaragua. Interestingly, the three CHIKV samples derived from DENV co-infections uncovered in this study form a distinct group relative to other similar (in terms of location and time period), publicly available CHIKV samples that are presumably not derived from co-infections (Wang et al., 2015; Tan et al., 2018). That co-infecting CHIKV associated with DENV-2 would be genetically distinct warrants further study (Supplemental Fig. 4).
To the best of our knowledge, this is the first study documenting the evolutionary patterns of multiple DENV serotypes during the introduction and emergence of both CHIKV and ZIKV within Central America. The unique study design of the Nicaragua hospital and cohort studies along with our total-RNA sequencing approach allowed us to characterize contrasting evolutionary patterns among the three DENV serotypes as well to detect co-infecting CHIKV. Importantly, we revealed the ongoing emergence of an adaptive lineage of DENV-2 between 2013 and 2016 associated with disease severity in 2015–2016. Long-term studies such as the Nicaraguan hospital and cohort studies are invaluable for providing an important source of samples for comparative studies of virus evolution and emergence over time, particularly capturing inapparent and co-infecting viruses. Further analysis is underway to determine the long-term co-evolutionary patterns and selection pressures operating in autochthonous transmission arenas involving dengue and other similarly vectored viruses such as ZIKV and CHIKV.
Supplementary Material
Acknowledgement
We thank Saira Saborío Galo for isolation of the viruses analyzed in this study. We would like to thank our study team at the Health Center Sócrates Flores Vivas (HCSFV), the Hospital Infantil Manuel de Jesús Rivera, and the Laboratorio Nacional de Virología at the Centro Nacional de Diagnóstico y Referencia, Ministry of Health, as well as the Sustainable Sciences Institute, in Nicaragua for their dedication and high-quality work. We are deeply grateful to the study participants and their families willing to contribute their time and effort to allow these cohort studies to continue in order to understand dengue virus transmission and evolution.
This research was funded by National Institutes of Health (NIH) Grant P01 AI106695 (to E.H.) and NIH/National Institute of Allergy and Infectious Diseases (NIAID) 1U01AI151788 (to E.H., supporting S.V.E. and P.T.). The PDCS and PDHS were supported by NIH Grants P01 AI106695 (to E.H.), U19 AI11861 (to E.H.), R01 AI099631 (to A.B.), and U54 AI65359 (to A.B. as Project Leader), as well as the FIRST Grant (to E.H.) from the Bill and Melinda Gates Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Ethics statement
The virus isolates obtained for this publication were isolated from participants of prospective cohort studies, which were approved by the Institutional Review Boards (IRBs) at the University of California, Berkeley and the Nicaraguan Ministry of Health. Written consent was obtained from a parent or guardian, or if the guardian was illiterate, the consent form was read aloud in the presence of a witness and the guardian’s thumbprint was obtained in lieu of a signature, as approved by the IRBs. Verbal assent was obtained from all children aged 6 years and older.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.meegid.2020.104680.
Data statement
The assembled whole and partial virus genomes generated in this study have been deposited in GenBank under accession number: MZ008438-MZ008478.
References
- Anderson JR, Rico-Hesse R, 2006. Aedes aegypti vectorial capacity is determined by the infecting genotype of dengue virus. Am. J. Trop. Med. Hyg 75 (5), 886–892. 10.4269/ajtmh.2006.75.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade CC, Young KI, Johnson WL, Villa ME, Buraczyk CA, Messer WB, Hanley KA, 2016. Rise and fall of vector infectivity during sequential strain displacements by mosquito-borne dengue virus. J. Evol. Biol 29 (11), 2205–2218. 10.1111/jeb.12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anes G, Morales-Betoulle ME, Rios M, 2011. Circulation of different lineages of dengue virus type 2 in Central America, their evolutionary time-scale and selection pressure analysis. PLoS One 6 (11), e27459. 10.1371/journal.pone.0027459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo JMG, Nogueira RMR, Schatzmayr HG, Zanotto PMD, Bello G, 2009. Phylogeography and evolutionary history of dengue virus type 3. Infect. Genet. Evol 9, 716–725. [DOI] [PubMed] [Google Scholar]
- Arrieta G, Mattar S, Villero-Wolf Y, et al. , 2019. Evaluation of serological test of Zika in an endemic area of flavivirus in the Colombian Caribbean. Ann. Clin. Microbiol. Antimicrob 18, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmaseda A, Sandoval E, Pérez L, Gutiérrez CM, Harris E, 1999. Application of molecular typing techniques in the 1998 dengue epidemic in Nicaragua. Am. J. Trop. Med. Hyg 61 (6), 893–897. [DOI] [PubMed] [Google Scholar]
- Balmaseda A, Hammond SN, Tellez Y, Imhoff L, Rodriguez Y, 2006. High seroprevalence of antibodies against dengue virus in a prospective study of schoolchildren in Managua, Nicaragua. Tropical Med. Int. Health 11, 935–942. [DOI] [PubMed] [Google Scholar]
- Balmaseda A, Mercado JC, Matute JC, Tellez Y, Saborío S, Hammond SN, Standish K, Nuñez A, Henn MR, Holmes EC, Gordon A, Coloma J, Kuan G, Harris E, 2010. Trends in patterns of dengue transmission in a pediatric cohort study in Nicaragua. J. Infect. Dis 201, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmaseda A, Gordon A, Gresh L, Ojeda S, Saborio S, Tellez Y, Sanchez N, Kuan G, Harris E, 2015. Clinical attack rate of chikungunya in a cohort of Nicaraguan children. Am. J. Trop. Med. Hyg 94 (2), 397–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett S, Holmes E, Chirivella M, Rodriguez D, Beltran M, Vorndam V, Gubler D, McMillan W, 2003. Selection-driven evolution of emergent dengue virus. Mol. Biol. Evol 20 (10), 1650–1658. 10.1093/molbev/msg182. [DOI] [PubMed] [Google Scholar]
- Bennett S, Holmes E, Chirivella M, Rodriguez D, Beltran M, Vorndam V, Gubler D, McMillan W, 2006. Molecular evolution of dengue 2 virus in Puerto Rico: positive selection in the viral envelope accompanies clade reintroduction. Journal of General Virology 87 (4), 885–893. [DOI] [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. , 2013. The global distribution and burden of dengue. Nature 496, 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert RR, Heled J, Kuehnert D, Vaughan TG, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ, 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol 10 (4), e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady Oliver J., Gething Peter W., Bhatt Samir, Messina Jane P., Brownstein John S., Hoen Anne G., Moyes Catherine L., Farlow Andrew W., Scott Thomas W., Hay Simon I., 2012, Aug. Refining the global spatial limits of dengue virus transmission by evidence-based consensus. PLoS Negl. Trop. Dis 6 (8) e1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattarino L, Rodriguez-Barraquer I, Imai N, Cummings DAT, Ferguson NM, 2020. Mapping global variation in dengue transmission intensity. Sci. Transl. Med 12 (528) 10.1126/scitranslmed.aax4144 eaax4144. [DOI] [PubMed] [Google Scholar]
- Chen R, Vasilakis N, 2011. Dengue Quo tu et quo vadis? Viruses 3, 1562–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC), 1995. Dengue type 3 infection-Nicaragua and Panama, October-November 1994. MMWR Morb Mortal Wkly Rep 44 (2), 21–24. [PubMed] [Google Scholar]
- Chen R, Mukhopadhyay S, Merits A, Bolling B, Nasar F, Coffey LL, Powers A, Weaver SC, ICTV Report Consortium, 2018. ICTV virus taxonomy profile: Togaviridae. J. Gen. Virol 99, 761–762. [DOI] [PubMed] [Google Scholar]
- Cologna R, Armstrong PM, Rico-Hesse R, 2005. Selection for virulent dengue viruses occurs in humans and mosquitoes. J. Virol 79 (2), 853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deen JL, Harris E, Wills B, Balmaseda A, Hammond SN, Rocha C, Dung NM, Hung NT, Hien TT, Farrar JJ, 2006. The WHO dengue classification and case definitions: time for a reassessment. Lancet 368, 170–173. [DOI] [PubMed] [Google Scholar]
- Delport W, Poon AF, Frost SD, Kosakovsky Pond SL, 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz FJ, Black WC, Farfan-Ale JA, Lorono-Pinto MA, Olson KE, Beaty BJ, 2006. Dengue virus circulation and evolution in Mexico: a phylogenetic perspective. Arch. Med. Res 37, 760–773. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Ho SYW, Phillips MJ, Rambaut A, 2006. Relaxed Phylogenetics and dating with confidence. PLoS Biol 4 (5), e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman R, Tacket CO, Wasserman SS, Bodison SA, Perry JG, Mangiafico JA, 2000. Phase II safety and immunogenicity study of live chikungunya virus vaccine TSI-GSD-218. Am. J. Trop. Med. Hyg 62, 681–685. [DOI] [PubMed] [Google Scholar]
- Faria NR, Quick J, Claro IM, Thézé J, de Jesus JG, Giovanetti M, Kraemer MUG, Hill SC, Black A, da Costa AC, 2017. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 546, 406–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JE, Bennett SN, Carrington CV, Vaughn H, McMillan WO, 2004. Phylogeography and molecular evolution of dengue 2 in the Caribbean basin, 1981–2000. Virology 324, 48–59. [DOI] [PubMed] [Google Scholar]
- Gaudinski MR, Houser KV, Morabito KM, Hu Z, Yamshchikov G, Rothwell RS, et al. , 2018. Safety, tolerability, and immunogenicity of two Zika virus DNA vaccine candidates in healthy adults: randomised, open-label, phase 1 clinical trials. Lancet 391 (10120), 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Gresh L, Ojeda S, Chowell-Puente G, Gonzalez K, Sanchez N, Saborio S, Mercado JC, Kuan G, Balmaseda A, Harris E, 2018. Differences in transmission and disease severity between two successive waves of chikungunya. Clin. Infect. Dis 67 (11), 1760–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Gresh L, Ojeda S, Katzelnick LC, Sanchez N, Mercado JC, Chowell G, Lopez B, Elizondo D, Coloma J, Burger-Calderon R, Kuan G, Balmaseda A, Harris E, 2019. Prior dengue virus infection and risk of Zika: a pediatric cohort in Nicaragua. PLoS Med 16 (1), e1002726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler DJ, 1998. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev 11, 480–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler DJ, Kuno G, 1997. Dengue and Dengue Hemorrhagic Fever CAB International, London. [Google Scholar]
- Gubler DJ, Kuno G, Markoff L, 2007. Flaviviruses. In: Knipe DM, Howley PM, Griffin D, Lamb M, Roizmen B, Straus SE (Eds.), Fields Virology, 5th edn. Lippincott, Williams & Wilkins, Philadelphia, Pennsylvania, pp. 1153–1252. [Google Scholar]
- Guzman MG, Vasquez S, Martı’nez E, Alvarez M, Rodrı’guez R, et al. , 1996. Dengue en Nicaragua, 1994: Reintroduccion del serotipo 3 en las Americas. Bol. Oficina Sanit. Panam 121, 102–110. [PubMed] [Google Scholar]
- Guzmaá MG, Kouri G, Valdes L, Bravo J, Alvarez M, Vazques S, Delgado I, Halstead SB, 2000. Epidemiologic studies on Dengue in Santiago de Cuba, 1997. Am. J. Epidemiol 152 (9), 793–799. [DOI] [PubMed] [Google Scholar]
- Halstead SB, 1988. Pathogenesis of dengue: challenges to molecular biology. Science 239 (4839), 476–481, 29. [DOI] [PubMed] [Google Scholar]
- Hammond SN, Balmaseda A, Perez L, Tellez Y, Saborío SI, Mercado JC, Videa E, Rodríguez Y, Perez MA, Cuadra R, Solano S, Rocha J, Idiaquez W, Gonzalez A, Harris E, 2005. Differences in dengue severity in infants, children, and adults in a three-year hospital-based study in Nicaragua. Am. J. Trop. Med. Hyg 73, 1063–1070. [PubMed] [Google Scholar]
- Harris E, Roberts TG, Smith L, Selle J, Kramer LD, 1998. Typing of dengue viruses in clinical specimens and mosquitoes by single- tube multiplex reverse transcriptase PCR. J. Clin. Microbiol 36, 2634–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris E, Videa E, Perez L, Sandoval E, Tellez Y, 2000. Clinical, epidemiologic, and virologic features of dengue in the 1998 epidemic in Nicaragua. Am. J. Trop. Med. Hyg 63, 5–11. [DOI] [PubMed] [Google Scholar]
- Holmes EC, 2003. Patterns of intra- and interhost nonsynonymous variation reveal strong purifying selection in dengue virus. J. Virol 77, 11296–11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EC, Twiddy SS, 2003. The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evol 3, 19–28. [DOI] [PubMed] [Google Scholar]
- Hombach J, 2007. Vaccines against dengue: a review of current candidate vaccines at advanced development stages. Rev. Panam. Salud Publica 21 (4), 254–260. [DOI] [PubMed] [Google Scholar]
- Katzelnick LC, Gresh L, Halloran ME, Mercado JC, Kuan G, Gordon A, et al. , 2017. Antibody-dependent enhancement of severe dengue disease in humans. Science 358 (6365), 929–932. 10.1126/science.aan6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindhauser MK, Allen T, Frank V, Santhana RS, Dye C, 2016. Zika: the origin and spread of a mosquito-borne virus. Bull. World Health Organ 94, 675C–686C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klungthong C, Zhang C, Mammen MP Jr., Ubol S, Holmes EC, 2004. The molecular epidemiology of dengue virus serotype 4 in Bangkok, Thailand. Virology 329, 168–179. [DOI] [PubMed] [Google Scholar]
- Klungthong C, Putnak R, Mammen MP, Li T, Zhang C, 2008. Molecular genotyping of dengue viruses by phylogenetic analysis of the sequences of individual genes. J. Virol. Methods 154 (1–2), 175–181. [DOI] [PubMed] [Google Scholar]
- Kouri G, Valdez M, Arguello L, Guzman MG, Valdes L, 1991. Dengue epidemic in Nicaragua, 1985. Rev. Inst. Med. Trop. Sao Paulo 33, 365–371. [PubMed] [Google Scholar]
- Kuan G, Gordon AL, Avilés W, Ortega O, Hammond SN, Elizondo D, Nuñez A, Coloma J, Balmaseda A, Harris E, 2009. The Nicaraguan Pediatric Dengue Cohort Study: study design, methods, use of information technology, and extension to other infectious diseases. Am. J. Epidemiol 170, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciotti RS, Lewis JG, Gubler DJ, Trent DW, 1994. Molecular evolution and epidemiology of dengue-3 viruses. J. Gen. Virol 75, 65–75. [DOI] [PubMed] [Google Scholar]
- Leparc-Goffart I, Nougairede A, Cassadou S, Prat C, De Lamballerie X, 2014. Chikungunya in the Americas. Lancet 383 (9916), 514. [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, Thiel HJ, Rice CM, Martin MA, Roizmen B, 2007. Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, Griffen D, Lamb RA, Straus SE (Eds.), Fields Virology, 5th edn. Lippincott Williams and Wilkins Philadelphia, Pennsylvania, pp. 1101–1113. [Google Scholar]
- Liu-Helmersson J, Stenlund H, Wilder-Smith A, et al. , 2014. Vectorial capacity of Aedes aegypti: effects of temperature and implications for global dengue epidemic potential. PLoS One 9, e89783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer WB, Vitarana UT, Sivananthan K, Elvtigala J, Preethimala LD, Ramesh R, Withana N, Gubler DJ, De Silva AM, 2002. Epidemiology of dengue in Sri Lanka before and after the emergence of epidemic dengue hemorrhagic fever. Am. J. Trop. Med. Hyg 66 (6), 765–773. [DOI] [PubMed] [Google Scholar]
- Messer WB, Gubler DJ, Harris E, Sivananthan K, de Silva AM, 2003. Emergence and global spread of a dengue serotype 3, subtype III virus. Emerg. Infect. Dis 9, 800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T, 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE), 14 Nov. 2010, New Orleans, LA, pp. 1–8. [Google Scholar]
- Modjarrad K, Lin L, George SL, Stephenson KE, Eckels KH, Barrera, La, De, R.A., et al. , 2018. Preliminary aggregate safety and immunogenicity results from three trials of a purified inactivated Zika virus vaccine candidate: phase 1, randomised, double-blind, placebo-controlled clinical trials. Lancet 391 (10120), 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mota J, Rico-Hesse R, 2009. Humanized mice show clinical signs of dengue fever according to infecting virus genotype. J. Virol 83 (17), 8638–8645. 10.1128/JVI.00581-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao Y, Koelle K, 2008. Decreases in dengue transmission may act to increase the incidence of dengue hemorrhagic fever. Proc. Natl. Acad. Sci 105 (6), 2238–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narvaez F, Gutierrez G, Perez MA, Elizondo D, Nuñez A, Balmaseda A, Harris E, 2011. Evaluation of the traditional and revised WHO classifications of dengue disease severity. PLoS Negl. Trop. Dis 5, e1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OhAinle M, Balmaseda A, Macalalad AR, Tellez Y, Zody MC, Saborío S, Henn MR, 2011. Dynamics of dengue disease severity determined by the interplay between viral genetics and serotype-specific immunity. Sci. Transl. Med 3 (114), 114ra128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paixão ES, Teixeira MG, Rodrigues LC, 2018. Zika, chikungunya and dengue: the causes and threats of new and re-emerging arboviral diseases [published correction appears in BMJ Glob. Health 3 (1), 7. e000530corr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan American Health Organization (PAHO), 2015. Data, Maps and Statistics, Region of the Americas (By Country and Year) PAHO. http://www.paho.org/hq/index.php?option=com_topics&view=rdmore&cid=6290&Itemid=40734&lang=en. [Google Scholar]
- Phuong CX, Nahn NT, Kneen R, Thuy PT, Van Thien C, Nga NT, Thuy TT, Solomon T, Stepniewska K, Wills B, 2004. Clinical diagnosis and assessment of severity of confirmed dengue infections in Vietnamese children: is the World Health Organization classification system helpful? Am. J. Trop. Med. Hyg 70, 172–179. [PubMed] [Google Scholar]
- Pyke AT, Moore PR, Taylor CT, Hall-Mendelin S, Cameron JN, Hewitson GR, Pukallus DS, Huang B, Warrilow D, van den Hurk AF, 2016. Highly divergent dengue virus type 1 genotype sets a new distance record. Sci. Rep 6, 22356. 10.1038/srep22356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiner CA, Parameswaran P, Ciota AT, Ehrbar DJ, Dodson BL, Schlesinger S, Kramer LD, Harris E, 2014. Increased replicative fitness of a dengue virus 2 clade in native mosquitoes: potential contribution to a clade replacement event in Nicaragua. J. Virol 88 (22), 13125–13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Castaneda J, Barreto dos Santos F, Martinez-Vega R, de Araujo JMG, Joint G, Sarti E, 2017. Dengue in Latin America: systematic review of molecular epidemiologic trends. PLoS Negl. Trop. Dis 10.1371/journal.pntd.0005224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico-Hesse R, 1990. Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virology 174 (2), 479–493. [DOI] [PubMed] [Google Scholar]
- Rico-Hesse R, 2003. Microevolution and virulence of dengue viruses. Adv. Virus Res 59, 315–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico-Hesse R, 2007. Dengue virus evolution and virulence models. Clin. Infect. Dis 44 (11), 1462–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico-Hesse R, Harrison LM, Salas RA, et al. , 1997. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology 230, 244–251. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Barraquer I, Costa F, Nascimento EJM, Nery N, Castanha PMS, Sacramento GA, et al. , 2019. Impact of preexisting dengue immunity on Zika virus emergence in a dengue endemic region. Science 363 (6427), 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salje H, Cummings DAT, Rodriguez-Barraquer I, Katzelnick LC, Lessler J, Klungthong C, et al. , 2018. Reconstruction of antibody dynamics and infection histories to evaluate dengue risk. Nature 557 (7707), 719–723. 10.1038/s41586-018-0157-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P, Becher B, Bukh J, Gould EA, Meyers G, Monath T, Muerhoff S, Pletnev A, Rico-Hesse R, Smith DB, Stapleton JT, ICTV Report Consortium, 2017. ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol 98, 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CEG, 1956. The history of dengue in tropical Asia and its probable relationship to the mosquito Aedes aegypti. J. Trop. Med. Hyg 11, 171. [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J, 2008. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol 57 (5), 758–771. [DOI] [PubMed] [Google Scholar]
- Stanaway JD, Shepard DS, Undurraga EA, Halasa YA, Coffeng LE, Brady OJ, Hay SI, Bedi N, Bensenor IM, Castañeda-Orjuela CA, Chuang TW, Gibney KB, Memish ZA, Rafay A, Ukwaja KN, Yonemoto N, Murray CJL, 2016. Feb 10. The global burden of dengue: an analysis from the Global Burden of Disease study 2013. Lancet Infect. Dis 10.1016/S1473-3099(16)00026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Pickett BE, Shrivastava S, Gresh L, Balmaseda A, Amedeo P, Hu L, Puri V, Fedorova NB, Halpin RA, LaPointe MP, Cone MR, Heberlein-Larson L, Kramer LD, Ciota AT, Gordon A, Shabman RS, Das SR, Harris E, 2018. Differing epidemiological dynamics of Chikungunya virus in the Americas during the 2014–2015 epidemic. PLoS Negl. Trop. Dis 12 (7) e0006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thézé J, Li T, du Plessis L, Bouquet J, Kraemer MUG, Somasekar S, Yu G, de Cesare M, Balmaseda A, Kuan G, Harris E, Wu CH, Ansari MA, Bowden R, Faria NR, Yagi S, Messenger S, Brooks T, Stone M, Bloch EM, Busch M, Muñoz-Medina JE, González-Bonilla CR, Wolinsky S, López S, Arias CF, Bonsall D, Chiu CY, Pybus OG, 2018. Genomic epidemiology reconstructs the introduction and spread of Zika virus in Central America and Mexico. Cell Host Microbe 23 (6). 10.1016/j.chom.2018.04.017, 855–864.e7. Jun 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twiddy SS, Farrar JJ, Vinh Chau N, Wills B, Gould EA, Gritsun T, Lloyd G, Holmes EC, 2002. Phylogenetic relationships and differential selection pressures among genotypes of dengue-2 virus. Virology 298 (1), 63–72. 10.1006/viro.2002.1447. [DOI] [PubMed] [Google Scholar]
- Uzcategui NY, Camacho D, Comach G, Guello de Uzcategui R, Holmes EC, et al. , 2001. Molecular epidemiology of dengue type 2 virus in Venezuela: evidence for in situ virus evolution and recombination. J. Gen. Virol 82, 2945–2953. [DOI] [PubMed] [Google Scholar]
- Vasilakis N, Holmes EC, Fokam EB, Faye O, Diallo M, Sall AA, Weaver SC, 2007. Evolutionary processes among sylvatic dengue type 2 viruses. J. Virol 81, 9591–9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner JJ, Abeynayake J, Sahoo MK, Gresh L, Tellez Y, et al. , 2013. Single-reaction, multiplex, real-time RT-PCR for the detection, quantitation, and serotyping of dengue viruses. PLoS Negl. Trop. Dis 7 (4), e2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner JJ, Ballesteros G, Gresh L, Mohamed-Hadley A, Tellez Y, Sahoo MK, et al. , 2016. My. Clinical evaluation of a single-reaction real-time RT-PCR for pandengue and chikungunya virus detection. J. Clin. Virol 78, 57–61. 26991052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner JJ, Gresh L, Mohamed-Hadley A, Ballesteros G, Davila MJ, Tellez Y, et al. , 2016b. Single-reaction multiplex reverse transcription PCR for detection of Zika, chikungunya, and dengue viruses. Emerg. Infect. Dis 22 (7), 1295–1297. 27184629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Volkova E, Adams AP, Forrester N, Xiao SY, Frolov I, Weaver SC, 2008. Sep 15. Chimeric alphavirus vaccine candidates for chikungunya. Vaccine 26 (39), 5030–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Saborio S, Gresh L, Parameswaran P, Eswarappa M, Balmaseda A, Harris E, 2015. Chikungunya virus sequences across the first epidemic in Nicaragua, 2014–2015. Am. J. Trop. Med. Hyg 94 (2), 400–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Saborio S, Gresh L, Eswarappa M, Wu D, Fire A, Parameswaran P, Balmaseda A, Harris E, 2016. Chikungunya virus sequences across the first epidemic in Nicaragua, 2014–2015. Am. J. Trop. Med. Hyg 94 (2), 400–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead SS, Blaney JE, Durbin AP, 2007. Prospects for a dengue virus vaccine. Nat. Rev. Microbiol 5 (7), 518–528.10. [DOI] [PubMed] [Google Scholar]
- WHO, 2009. Dengue Hemorrhagic Fever: Diagnosis, Treatment, Prevention, and Control World Health Organization, Geneva, Switzerland. [Google Scholar]
- Wittke V, Robb TE, Thu HM, Nimmannitya S, Kalayanrooj, Vaugh DW, Endy TP, Holmes EC, Aaskov JG, 2002. Extinction and rapid emergence of strains of dengue 3 virus during an interepidemic period. Virology 301, 148–156. [DOI] [PubMed] [Google Scholar]
- Zambrana JV, Bustos Carrillo F, Burger-Calderon R, Collado D, Sanchez N, Ojeda S, Carey Monterrey J, Plazaola M, Lopez B, Arguello S, Elizondo D, Aviles W, Coloma J, Kuan G, Balmaseda A, Gordon A, Harris E, 2018. Sep 11. Seroprevalence, risk factor, and spatial analyses of Zika virus infection after the 2016 epidemic in Managua, Nicaragua. Proc. Natl. Acad. Sci. U. S. A 115 (37), 9294–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotto PMDA, Gould EA, Gao GF, Harvey PH, 1996. Population dynamics of Flaviviruses revealed by molecular phylogenies. Proc. Natl. Acad. Sci. U. S. A 93, 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The assembled whole and partial virus genomes generated in this study have been deposited in GenBank under accession number: MZ008438-MZ008478.