

Abstract
Background
Patients with ALK‐rearranged non‐small cell lung cancer (ALK+ NSCLC) inevitably acquire resistance to ALK inhibitors. Longitudinal monitoring of cell‐free plasma DNA (cfDNA) next‐generation sequencing (NGS) could predict the response and resistance to tyrosine kinase inhibitor (TKI) therapy in ALK+ NSCLC.
Methods
Patients with ALK+ NSCLC determined by standard tissue testing and planned to undergo TKI therapy were prospectively recruited. Plasma was collected at pretreatment, 2 months‐post therapy, and at progression for cfDNA‐NGS analysis, Guardant 360.
Results
Among 92 patients enrolled, circulating tumor DNA (ctDNA) was detected in 69 baseline samples (75%): 43 ALK fusions (62.3%) and two ALK mutations without fusion (2.8%). Two patients showed ALK‐resistance mutations after ceritinib; G1202R, and co‐occurring G1202R and T1151R. Eight patients developed ALK resistance mutations after crizotinib therapy; L1196M (n = 5), G1269A (n = 1), G1202R (n = 1), and co‐occurring F1174L, G1202R, and G1269A (n = 1). Absence of ctDNA at baseline was significantly associated with longer progression‐free survival (PFS; median 36.1 vs. 11.4 months, p = 0.0049) and overall survival (OS; not reached vs. 29.3 months, p = 0.0200). ctDNA clearance at 2 months (n = 29) was associated with significantly longer PFS (25.4 vs. 11.6 months, p = 0.0012) and OS (not reached vs. 26.1 months, p = 0.0307) than those without clearance (n = 22). Patients with co‐occurring TP53 alterations and ALK fusions at baseline (n = 16) showed significantly shorter PFS (7.28 vs. 13.0 months, p = 0.0307) than those without TP53 alterations (n = 25).
Conclusions
cfDNA‐NGS facilitates detection of ALK fusions and resistance mutations, assessment of prognosis, and monitoring dynamic changes of genomic alterations in ALK+ NSCLC treated with ALK‐TKI.
Keywords: anaplastic lymphoma kinase‐rearranged (ALK+) NSCLC, cell‐free DNA, liquid biopsy, next‐generation sequencing
To investigate whether longitudinal monitoring of cfDNA‐NGS could predict the response and resistance of TKI therapy in ALK+ NSCLC, we recruited prospectively 92 patients with ALK+ advanced NSCLC determined by standard tissue testing and planned for TKI therapy. NGS of cfDNA is useful not only for the detection of ALK fusions and resistance mutations, but also for assessing prognosis and monitoring the dynamic changes of genomic alterations in ALK+ NSCLC treated with ALK‐TKI.

1. INTRODUCTION
Rearrangement of anaplastic lymphoma kinase gene (ALK) is oncogenic driver which accounts for 3%–5% of patients with non‐small cell lung cancer (NSCLC). 1 , 2 During the last decade, significant efforts from academia and the pharmaceutical industry have led to the development of numerous ALK tyrosine kinase inhibitors (TKI). Since the first‐generation ALK inhibitor crizotinib 3 was the standard therapy for newly diagnosed ALK positive NSCLC, more potent CNS penetrating second generation ALK inhibitors such as ceritinib, 4 alectinib, 5 brigatinib, 6 and lorlatinib 7 have become the standard treatment as first‐line therapy.
Remarkable therapeutic efficacies were observed with these ALK TKIs, however, essentially many patients inevitably develop acquired resistance within 1 or 2 years. The resistance mechanisms of ALK TKI are categorized into “on‐target” mechanisms such as ALK secondary resistance mutation or amplification, where oncogenic dependencies on ALK kinase persist, and “off‐target” mechanisms such as activation of bypass pathway or lineage changes, where the cancer cells can survive and proliferate regardless of ALK activity. 8 Of note, the incidence and spectrum of secondary mutations in the ALK tyrosine kinase domain is variable among ALK TKIs. L1196M or G1269R is found in 4 ~ 7% of patients resistant to crizotinib, whereas G1202R mutation is observed in 20%–40% of patients resistant to ceritinib, alectinib, or brigatinib. 9 Furthermore, several ALK secondary mutations represent differential sensitivity to the type of ALK TKIs. 9 Therefore, the elucidation of resistance mechanisms is critical to decide subsequent treatment strategies in ALK positive NSCLC who experience progression to ALK TKIs.
Comprehensive next‐generation sequencing (NGS) tests covering all relevant genomic biomarkers by repeat tissue biopsy may be preferred to determine the resistance mechanism. However, tissue NGS tests have challenges regarding tissue availability, quality, and long turn‐around time. Considering intra‐tumoral or inter‐tumoral heterogeneity, small tissue biopsy may not represent the overall genomic status of tumor. To overcome these limitations, NGS of cell‐free plasma DNA (cfDNA‐NGS) has gained substantial attention as an alternative strategy. 10 , 11 , 12 Further, liquid biopsy can make it possible to perform longitudinal monitoring during ALK TKI treatment as noninvasive approach. 13
Technological advances in cfDNA‐NGS has broadened the clinical use in various solid tumors. 14 , 15 , 16 Prior work reported that cfDNA‐NGS test was applicable and useful in ALK positive NSCLC patients, 17 , 18 , 19 , 20 , 21 , 22 but the small number of patients studied still remain a limitation to determine the predictive role of cfDNA‐NGS in the ALK positive NSCLC patients who were being treated with TKI.
This study was initiated to evaluate the role of cfDNA‐NGS not only for the detection of ALK fusions and resistance mutations, but also for assessing prognosis and monitoring the dynamic changes of genomic alterations in ALK positive NSCLC patients treated with ALK TKIs in prospective manner.
2. PATIENTS AND METHODS
2.1. Study population
Patients diagnosed with ALK rearranged advanced or recurrent NSCLC who were treated with ALK TKIs were enrolled. Patients previously treated with cytotoxic chemotherapy or ALK TKI therapy were not excluded from the study. The choice of ALK TKI was made by the physician's discretion. The ALK positive NSCLC was diagnosed by ALK immunohistochemical stain (IHC) using the Ventana anti‐ALK (D5F3), FISH, or NGS. Tumor responses were evaluated every two cycles (8 weeks) according to Response Evaluation Criteria in Solid Tumors (RECIST version 1.1). This study protocol was reviewed and approved by the institutional review boards of Samsung Medical Center (SMC 2013_09‐075, Seoul, Korea). Written informed consent was obtained from each patient. The study was conducted in accordance with the Declaration of Helsinki and the Guidelines for Good Clinical Practice.
2.2. Sample collection and plasma preparation
We prospectively collected plasma before ALK TKI therapy, after 2 months of TKI therapy, and at disease progression. Peripheral blood was collected into a 10‐ml K2‐EDTA vacutainer (Becton Dickinson) and centrifuged at 1500× g for 15 min within 2 h of collection. The supernatants were sequentially centrifuged at 13,000× g for 10 min to remove residual blood compartments. The supernatant was transferred to a 1.5 ml E‐tube and stored at −80°C until use (Figure S1A).
2.3. Cell‐free plasma DNA‐NGS test
Plasma samples were retrospectively analyzed using the Guardant360 test (Guardant Health). Single nucleotide variants (SNVs), insertions, and deletions (indel) were assessed in 74 genes (Figure S1B); rearrangements and copy number alterations were evaluated in selected genes according to the manufacturer. All the detected variants were analyzed by Guardant360 bioinformatic pipeline. After excluding the germline variants that were confirmed by the pipeline, somatic variants were considered as circulating tumor DNA (ctDNA). 23
2.4. Statistical analysis
Statistical analyses were performed using Prism software version 9.0 (GraphPad). The nonparametric Mann–Whitney U‐test was used to compare two groups. Paired values were compared using the nonparametric Wilcoxon matched‐pairs signed‐rank test. For survival analysis, log‐rank (Mantel‐Cox) test was performed. The hazard ratio for group comparison was calculated by Mantel–Haenszel test. Hazard ratios and corresponding 95% confidence intervals were calculated using the Cox proportional hazards model. We consider p values <0.05 as statistically significant. Correlations were analyzed by Spearman's rank correlation coefficient. All tests were the two‐sided test.
3. RESULTS
3.1. Patient characteristics
Ninety‐two patients were enrolled between April 2015 and July 2019. Baseline patients' clinical characteristics are summarized in Table 1. The median age was 55 (range, 21–79), and 70% were female. About two‐thirds of the patients (68.5%) were never smokers. Eighty‐eight patients (95.7%) had adenocarcinomas, three patients (3.3%) had squamous cell carcinomas, and one patient (1.1%) had neuroendocrine carcinoma. Seventy‐five patients (81.5%) had metastatic disease and 17 patients (18.5%) had recurrent disease. Thirty‐one patients (33.7%) had brain metastases at baseline assessment. Eighty‐one patients (88.0%) received ALK TKI as first‐line (crizotinib, n = 59; alectinib, n = 22), nine patients (9.8%) received TKI as second‐line (alectinib, n = 5; crizotinib, n = 2; ceritinib, n = 1; brigatinib, n = 1), and two patients (2.2%) were treated as third‐line (alectinib, n = 1; lorlatinib, n = 1) (Table S1).
TABLE 1.
Patient characteristics
| Total | N = 92 |
|---|---|
| Age, year | |
| Range | 21–79 |
| Median | 55 |
| Gender, n (%) | |
| Female | 62 (67.4) |
| Male | 30 (32.6) |
| Smoking history, n (%) | |
| Current | 18 (19.6) |
| Former | 11 (12.0) |
| Never | 63 (68.5) |
| Tumor histology, n (%) | |
| Adenocarcinoma | 88 (95.7) |
| Squamous cell carcinoma | 3 (3.3) |
| Neuroendocrine carcinoma | 1 (1.1) |
| Stage of the disease, n (%) | |
| Metastatic | 75 (81.5) |
| Recurrent | 17 (18.5) |
| Brain metastases, n (%) | |
| Yes | 31 (33.7) |
| No | 61 (66.3) |
| Prior ALK TKI treatments, n (%) | |
| 0 | 81 (88.0) |
| ≥1 | 11 (12.0) |
| ALK TKI, n (%) | |
| Crizotinib | 61 (67.3) |
| Alectinib | 28 (30.4) |
| Brigatinib | 1 (1.1) |
| Ceritinib | 1 (1.1) |
| Lorlatinib | 1 (1.1) |
Among the enrolled 92 patients, six patients showed early progression and death within 2 months (Figure S2). At the cutoff date of April 20, 2020, the median duration of follow‐up was 19.2 months (range, 0.2 to 60.6). Thirty patients (32.6%) continued to receive ALK TKI without progression and 62 patients (67.4%) demonstrated disease progression.
3.2. Genomic profiles by cfDNA‐NGS in ALK positive NSCLC patients at baseline
Comprehensive genomic profiles were analyzed across 92 ALK positive NSCLC patients using cfDNA‐NGS at baseline (Figure 1). Among 409 different alterations identified from the cfDNA‐NGS at baseline, 205 (50.1%) were annotated as somatic alteration. There was no correlation between the concentration of cfDNA and the age of patients (Spearman correlation, r = −0.03048, p = 0.7730) (Figure S3). Cell‐free circulating tumor DNA (ctDNA), which is annotated as somatic alteration in cfDNA‐NGS, was detected in 69 baseline samples (75%). Among 74 genes covered by Guardant360, ALK alterations (fusion or mutation) were the most common (48.9%) followed by TP53 alterations (mutation or indel) (31.5%). Other frequent somatic mutations include GNAS, AR, CDKN2A, EGFR, KIT, APC, ESR1, MYC, BRCA1, and BRCA2, fusion in ROS1, and copy number gains in ERBB2 and MYC which accounts for less 5%.
FIGURE 1.
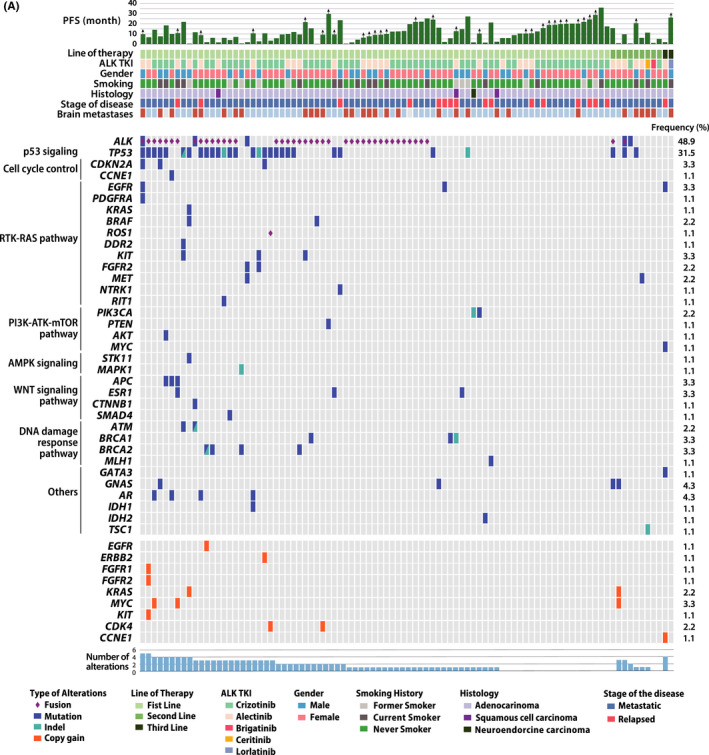
The landscape of somatic alterations in cfDNA‐NGS in ALK positive NSCLC. Heatmap of the genomic landscape of ctDNA and clinicopathologic characteristics of patients with ALK positive NSCLC matched with Swimmer's plot for progression‐free survival
3.3. Detection of ALK alterations by cfDNA‐NGS in ALK positive NSCLC patients at baseline
To evaluate the diagnostic performance of cfDNA‐NGS, we analyzed the detection rate of ALK alterations by cfDNA‐NGS in the ALK rearranged NSCLC patients who had been diagnosed by ALK IHC, FISH, or targeted NGS from tumor tissue.
Among 69 patients with detectable ctDNA at baseline, 40 patients had ALK fusion without ALK mutation, three patients had ALK fusion with ALK mutation, and two patients had ALK mutation without ALK fusion (Figure 2A). There was no significant correlation or pattern between the detection of ALK alteration in cfDNA‐NGS and the type of ALK test at diagnosis (Table S2). Fusions included EML4‐ALK v1 (n = 19), EML4‐ALK v3 (n = 14), CLTC‐ALK (n = 1), TPM3‐ALK (n = 1), GCC2‐ALK/CLIP4‐ALK (n = 1), and other EML4‐ALK fusions (n = 7). A patient with EML4‐ALK v3 and ALK V1045M (SMC‐068), a patient with EML4‐ALK v3 and ALK R1120W (SMC‐092) and a patient with ALK H1228Q mutation (SMC‐017) had no history of prior ALK TKI therapy. On the other hand, one patient (SMC‐051) who demonstrated EML4‐ALK variant 3 fusion with G1202R mutation had received crizotinib for 39.2 months, and the other patient (SMC‐089) with ALK G1202R and T1151R without ALK fusion received ceritinib for 35.2 months before study enrollment.
FIGURE 2.

Longitudinal analysis of cfDNA‐NGS in ALK positive NSCLC patients who were receiving ALK tyrosine kinase inhibitors. Plasma specimens were collected before ALK TKI treatment start, 2 months after ALK TKI treatment, and at progression. Ninety‐two baseline sample, 58 2‐month follow‐up sample, and 35 at‐progression sample were analyzed by cfDNA‐NGS. The result of cfDNA‐NGS analysis was clustered into three groups; ALK alterations, somatic alterations other than ALK, and no somatic alteration. (A–C) Pie charts demonstrate the frequencies of patients by groups and more detailed information of ALK fusions or ALK acquired mutations across serial cfDNA‐NGS (A, before ALK TKI treatment, B, 2‐month follow‐up and C, at progression)
3.4. cfDNA‐NGS during ALK TKI therapies in patients with ALK positive NSCLC
Plasms samples were collected at 2 months of ALK TKI treatment and at the time of progression for longitudinal monitoring. Ten out of 96 patients had progressed within 2 months after enrollment. Among 86 patients, 58 patients were analyzed for cfDNA‐NGS at 2 months after ALK TKI treatment (Figure S2). Twenty‐six patients (44.8%) revealed detectable ctDNA, where 22 patients (84.6%) demonstrated somatic alterations other than ALK, and four patients (15.4%) demonstrated ALK alterations, including EMK4‐ALK variant 3 (n = 1), EML4‐ALK other variants (n = 1), V1045M (n = 1), and G1202R (n = 1) (Figure 2B).
At the data cutoff, 60 patients (65.2%) had progressed on the ALK TKI therapy. Among 35 (58.3%) samples analyzed for cfDNA‐NGS at progression, 25 samples (71.4%) had detectable ctDNA, where 13 samples (37.1%) had ALK alterations, and 12 samples (34.3%) had somatic alterations other than ALK. In patients with detectable ctDNA, 52.0% of the patients (n = 13) had ALK alterations. Three patients had ALK fusions without ALK mutations (EML4‐ALK variant 1 (n = 1), EML4‐ALK variant 3 (n = 1), and TPM3‐ALK (n = 1)), four patients had ALK fusions with ALK mutations (EML4‐ALK variant 1 with L1196M mutation (n = 1), EML4‐ALK variant 3 with G1269A mutation (n = 1), TPM3‐ALK with L1196M mutation (n = 1), and both GCC2‐ALK and CLIP4‐ALK with L1196M mutation (n = 1)), and six patients had ALK mutations without ALK fusions (G1202R (n = 2), L1196M (n = 2), V1045M (n = 1), and co‐occurrence of G1269A, G1202R, and F1174L (n = 1)) (Figure 2C).
3.5. Longitudinal transitions of genomic alterations using cfDNA‐NGS during therapy in ALK positive NSCLC patients
We traced the longitudinal transition of genomic alterations in the cfDNA‐NGS analysis at baseline, 2‐month follow‐up, and progression from the ALK positive NSCLC patients during TKI treatment (Figure S4). Patients were clustered into four categories according to cfDNA‐NGS results; ALK alterations, somatic alterations other than ALK, no somatic alterations, and no cfDNA detected (neither germline nor somatic alteration). Most patients with no somatic alterations at baseline (15 out of 21) continued to show no somatic alterations at 2 months. In contrast, patients with ALK alterations at baseline evolved into variable categories at 2 months; no somatic mutations (n = 13), somatic alterations other than ALK (n = 9), ALK alterations (n = 4), and no ctDNA detected (n = 2). Of note, a substantial proportion of (20 of 29) the patients representing no somatic alterations at 2 months continued ALK TKI treatment without progression. Further, two‐third of patients (20 of 30) receiving ALK TKI without progression had revealed no somatic alterations at 2 months.
3.6. Associations of ctDNA detection at baseline and 2 months after TKI treatment with clinical outcomes in ALK positive NSCLC patients
The absence of detectable ctDNA at baseline (n = 23) was associated with significantly longer progression survival (PFS) (median 36.1 vs. 11.4 months, the Hazard ratio (HR) 0.46, 95% confidence interval (CI) 0.27–0.79, p = 0.0049) and overall survival (OS) (median not reached vs. 29.3 months, HR 0.40, 95% CI 0.18–0.87, p = 0.0200) than those with detectable ctDNA (Figure 3A). Multivariate Cox regression modeling identified the presence of ctDNA at baseline as an independent negative prognostic factor for PFS (HR 2.26, 95% CI 1.12–4.55, p = 0.023) and OS (HR 3.33, 95% CI 1.00–11.02, p = 0.049) (Figure S5). In subgroup analysis of the patients who received crizotinib as first‐line TKI, the absence of detectable ctDNA at baseline (n = 16) was also associated with significantly longer PFS (median 36.1 vs. 10.6 months, HR 0.37, 95% CI 0.20–0.69, p = 0.0019) and OS (median not reached vs 28.3 months, HR 0.40, 95% CI 0.15–0.90, p = 0.0282) than those with detectable ctDNA (Figure S6A). Moreover, patients with higher number of somatic mutations (≥3, median) (median) at baseline (n = 33) showed significantly shorter PFS (7.0 vs. 14.9 months, HR 0.35, 95% CI 0.19–0.65, p = 0.0008) and OS (14.3 vs. 39.5 months, HR 0.33, 95% CI 0.16–0.70, p = 0.0036) than those with the lower number of somatic mutations (<3, median) (Figure 3B).
FIGURE 3.

Survival analysis in the patients with ALK positive NSCLC according to longitudinal analysis of cfDNA‐NGS. Kaplan–Meier curves for progression‐free survival and overall survival of ALK positive NSCLC patients according to the detection of ctDNA at baseline (A), the number of somatic alterations at baseline (B), and detection of ctDNA at 2‐month follow‐up (C)
Given the possible association of ctDNA concentration with tumor burden, 24 , 25 the concentration of ctDNA in plasma was estimated by multiplying the concentration of cfDNA in plasma and the sum of mutation allele fraction that annotated from cfDNA‐NGS analysis algorithm. 26 Among 69 patients with detectable ctDNA, the median of ctDNA concentration was 69.9 pg/ml (range 3.2–35071.2 pg/ml). Patients with higher ctDNA concentration (>69.9, median) at baseline (n = 35) were associated with a trend toward to shorter PFS (9.9 vs. 14.1 months, HR 0.60, 95% CI 0.34–1.08, p = 0.0880) but significantly shorter OS (16.8 vs. 39.5 month, HR 0.47, 95% C.I. 0.21–0.98, p = 0.0437) than those with lower ctDNA concentration (n = 34) (Figure S6B).
Patients with clearance of ctDNA at 2 months (n = 29) had significantly longer PFS (median 25.4 vs. 11.6 months, HR 0.26, 95% CI 0.11–0.61, p = 0.0022) and significantly longer OS (not reached vs. 26.1 months, HR 0.18, 95% C. 0.04–0.92, p = 0.0388) than those without clearance (n = 22) regardless of type of ALK TKIs (Figure 3C).
3.7. Co‐occurrence of TP 53 alterations was associated with poor outcomes in ALK positive NSCLC
TP53 alteration (mutation or indel) (40.0%) was the most frequent co‐occurrent alteration among the patients with ALK‐rearranged NSCLC (Figure 4A). Other frequent somatic mutations, such as BRCA2, APC, and AR were also found.
FIGURE 4.

Survival analysis according to co‐occurrence of TP53 alteration in the patients with ALK positive NSCLC. (A) Frequencies of co‐occurring alteration among ALK positive NSCLC patients (N = 45) (B) Kaplan–Meier curves for progression‐free survival and overall survival of ALK positive NSCLC patients under first‐line ALK TKI according to the co‐occurrence of TP53 mutation with ALK alteration
Patients with co‐occurring TP53 alterations with ALK fusions at baseline (n = 16) showed shorter PFS for first‐line ALK TKI (7.28 vs. 13.0 month, HR 2.69, 95% CI 1.10–6.58, p = 0.0307) than those without TP53 alteration (n = 25). However, there is no difference in OS according to TP53 alterations (26.1 vs. 28.3 month, HR 1.89, 95% CI 0.67–5.38, p = 0.2309) (Figure 4B).
Considering the biological heterogeneity according to the type of TP53 mutation, 27 we mapped the type and location of the TP53 alterations from the 16 patients with co‐occurring TP53 alterations and ALK fusions in baseline cfDNA (Figure S7A). Fifteen patients with missense mutations (62.5%), five patients with nonsense mutations (31.3%), and one patient with a frameshift mutation (6.25%) were identified. In subgroup analysis, type of TP53 mutation was not significant factor to demonstrate the differences of PFS in comparing TP53 wild type to TP53 mutant (Figure S7B–D).
3.8. Resistance mechanisms identified by cfDNA‐NGS at the time of progression in ALK positive NSCLC treated with ALK TKI
Eight patients developed ALK resistance mutations after crizotinib therapy: L1196M (n = 5), G1269A (n = 1), G1202R (n = 1), and co‐occurring F1174L, G1202R, and G1269A (n = 1). Two patients developed ALK resistance mutations after ceritinib: G1202R (n = 1), and co‐occurring G1202R and T1151R (n = 1). Among 35 patients who performed cfDNA‐NGS at progression, patients with detectable ctDNA (n = 25) showed significantly shorter post‐progression survival than those without detectable ctDNA (n = 10; 16.5 vs. undefined months, HR 3.43, CI 1.13–10.37, p = 0.0291) (Figure S8C).
3.9. Activation of the bypass signaling was identified by longitudinal monitoring cfDNA‐NGS in ALK positive NSCLC patients who were resistant to ALK TKI
Matching analysis of genomic alterations by cfDNA‐NGS between baseline and at progression identified not only ALK mutation but also bypass signaling activation. A 53‐year‐old patient (SMC‐036) initially presented lung, pleural, and bone metastases showing robust tumor shrinkage with crizotinib. However, after 4 months of crizotinib, progression occurred. Compared to baseline cfDNA‐NGS results L1196M mutation along other genomic alterations ERBB2 copy number amplification and CCNE1gene amplification were newly identified at progression. Further, the ctDNA percentage of SMAD4 p.L536P mutation (from 8.06% to 17.35%) and TP53 p.Y234* was increased at progression. After progression, patient received alectinib but response was observed only in lung and pleura but progressed in the bone. Finally, patient died shortly of disease progression after chemotherapy. (Figure 5A).
FIGURE 5.
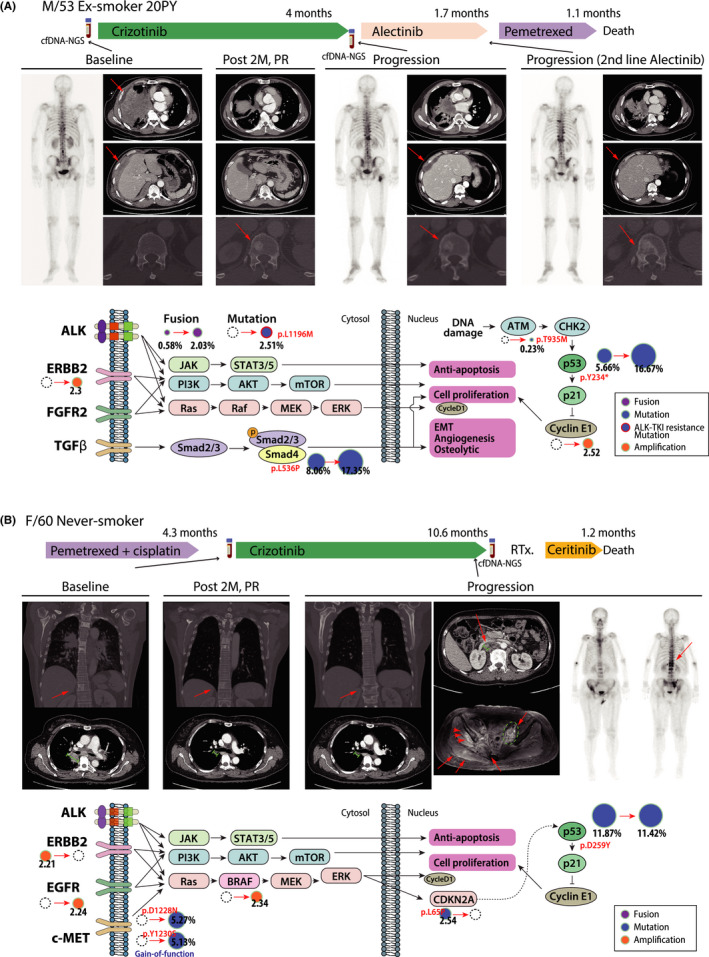
Patients who were annotated bypass signaling activation by cfDNA‐NGS monitoring showed resistance to ALK‐TKI. (A, B) The treatment summary is displayed on the upper panel. The images of whole‐body bone scan and enhance computerized tomography (CT) during treatment are displayed on the middle panel. A diagram of the signaling pathway demonstrates the principal changes between pretreatment and at progression in cfDNA‐NGS and the resistance mechanisms of ALK acquired resistance mutation and bypass signaling activation. The size of the circle represents allele frequency (%) of fusion, indel or mutation, or copy number
Another patient who did not reveal ALK alterations in ctDNA showed bypass pathway activation in the analysis of cfDNA‐NGS at progression, which was suggestive of resistance mechanism against ALK TKI (Figure 5B). A 60‐year‐old woman (SMC‐20) showed a partial response to crizotinib. At progression, no ALK mutation was noted but activation of c‐met pathway including MET Y1230 (or Y1248) (p.Y1230S, NM_000245.2:c.3689A > C, 5.13%) mutation and MET D1228N (or D1246N) (p.D1228N, NM_000245.2:c.3682G > A, 5.27%), which are located on kinase domain of MET along with amplification of BRAF. This patient experienced rapid progression to ceritinib as salvage therapy.
4. DISCUSSION
In this study, we prospectively collected serial plasma samples from 92 patients with ALK rearranged NSCLC who were treated with ALK TKIs and performed comprehensive genomic analysis using cfDNA‐NGS.
We found that ctDNA was detected in 75% of baseline samples and among these 62.3% of ALK fusions and 2.8% of ALK mutations without fusion were identified by cfDNA‐NGS, which is consistent with previous studies. 17 , 19 , 21 Further, cfDNA‐NGS was able to detect not only EML4‐ALK variants but also various fusion partners of ALK fusion, suggesting high performance of NGS with liquid biopsy. 18 , 20 , 21
Our data represent the landscape of genomic alterations in ctDNA from TKI‐naïve ALK positive NSCLC patients. At baseline, cfDNA‐NGS can detect not only ALK gene alterations, but also other coexisting somatic mutations TP53, CDKN2A, EGFR, etc. and copy number variants like ERBB2 and MYC, although the clinical significance of these co‐occurring alterations should be further determined. Most of previous studies with cfDNA were focused to detect ALK resistance mutations in ALK positive NSCLC patients treated with ALK TKI as second‐line therapy. 17 , 18 , 19 The proportion of treatment naïve ALK positive NSCLC patients was less than 50% and small number of patients were evaluated. In contrast, 75 of 92 (81%) patients enrolled in this study were ALK TKI naïve status. Moreover, the plasma samples were prospectively collected, and a large number of patients were longitudinally monitored. Further, with long term follow‐up, we can evaluate the role of cfDNA‐NGS analysis as predictive and prognostic value in ALK positive NSCLC patients treated with TKI.
We also found that the absence of detectable ctDNA at baseline was associated with longer PFS and OS. Further, patients with clearance of ctDNA at 2 months after ALK TKIs showed significantly longer PFS and OS that those without clearance. These results confirmed previous studies, 28 , 29 suggesting that patients with non‐shedder ALK positive NSCLC exhibit less aggressive tumor biology. Besides, patients without detectable ctDNA at baseline were likely to show undetectable ctDNA at 2 months after TKI therapy (16/23) and to demonstrate durable ALK TKI response (12 ongoing therapies among 23 patients). However, due to small number of progression event, the response of ALK‐TKI did not show any significant correlation either the presence of ctDNA at baseline (Table S3) or at 2 months (Table S4). As far as we know, this is the first report that demonstrates the predictive value of cfDNA‐NGS in ALK positive NSCLC based on the PFS and OS data.
At progression, cfDNA‐NGS detected various ALK mutations including L1196M, G1269A, G1202R, F1174L, and T1151R which are known to be resistance to ALK TKIs. Since the spectrum of resistance ALK mutation becomes more complex with sequential use of ALK TKIs or upfront use of second or third generation ALK TKIs, the identification of resistance ALK mutation would be more critical. Tissue biopsy is considered golden standard to elucidate the genomic alterations but the acquisition of adequate tissue at the time of progression is more challenging. Although patients were not treated with other ALK inhibitors according to the ALK mutation status at progression because specimens were retrospectively analyzed in our study, these different ALK mutations might guide treatment decision given the differential sensitivity to the type of ALK TKIs. Given that, cfDNA‐NGS is reasonable approach in clinical practice as alternative measure to detect ALK resistance mutation especially in case of sequential treatment of ALK TKIs.
It has been reported that coexistence of genomic alterations such as TP53 is associated with poor outcome in oncogenic driver in NSCLC. 30 In this study, patients with co‐occurring TP53 alterations and ALK fusions at baseline were associated with shorter PFS and OS than those without TP53 alterations, which is consistent with previous results. 31 In subgroup analysis for TP53 missense mutation and nonsense mutation, while subtype showed trends for shorter PFS compared to TP53 wild type, there were not significant differences in PFS between TP53 mutant subtype and TP53 wild type. Larger number of patients may be necessary for investigating clinical or biological significance according to the type of TP53 mutation in ALK positive NSCLC.
Various agents targeting p53 pathway for cancer therapy are being investigated. 32 Eprenetapopt, mutant p53 reactivator, demonstrated promising efficacy in TP53‐mutant myelodysplastic syndrome or acute myeloid leukemia. 33 In solid tumor, a phase 1b/2 clinical trial of Eprenetapopt in combination with pembrolizumab is ongoing in patients with advanced or metastatic solid cancer (NCT04383938). 34 TP53 mutation was reported as favorable prognostic marker in the patients who treated with immune checkpoint inhibitors (ICIs), and as the association of high tumor mutation burden and activated immune cell infiltration. 35 , 36 As TP53 mutation was associated with higher genetic instability in ALK positive NSCLC, 37 we identified that the number of detected somatic mutations in cfDNA in the patients with TP53 mutations was significantly higher than those with TP53 wild type (Figure S7E). Considering this rationale, ICIs or the combination therapy of TP53 targeting agent with ICIs can be effective in the ALK positive NSCLC patients with co‐occurring TP53 mutations.
To expand PFS and OS in ALK positive NSCLC, it is important to identify and target the patient‐specific resistance mechanism of ALK TKIs accurately. Although second biopsy is vital for profiling the resistance mechanism at progression, on‐treatment biopsy is not always applicable due to the limitation of approach or patient's medical condition. Performing cfDNA‐NGS at progression in ALK positive NSCLC could be prognostic and informative. The patients with no somatic alteration detected cfDNA had better post‐progression survival than those with somatic alterations (Figure S6C), so that the physician could have detected and responded to another progression more rapidly. NGS of cfDNA at progression in ALK positive NSCLC can guide the choice of ALK TKI based on the profile of ALK resistance mutation. From the matching the result of cfDNA‐NGS at progression with subsequent therapies retrospectively, ceritinib was more active for L1196M mutation than alectinib (Figure S8). Further, with comprehensive analysis of genomic alterations using cfDNA‐NGS, activation of other bypass signaling can be identified and compared between baseline and at the time of progression (Figure S8). Representative two cases indicate that the resistance mechanism to ALK TKIs is quite complex and will be variable among patients (Figure 5). For example, in case of SMC‐036, newly identified ERBB2 amplification along with L1196M after crizoinib might explain why patient was initially well responded to alectinib but experienced progressed shortly. Another case (SMC‐020) who developed c‐met mutation without alteration of ALK mutation did not response to ceritinib, can be attributed to the activation of c‐met pathway. In this case, c‐met inhibitor with second generation of ALK TKI might be beneficial. Taken together, real‐time monitoring of genomic alterations during ALK TKI treatment using cfDNA‐NGS as noninvasive method would be valuable to make treatment decision.
There are limitations to this study. The patients in this study were treated with heterogeneous. As the patients had been enrolled for a long period of time (from April 2015 to July 2019), the patients were treated with crizotinib, alectinib, ceritinib, brigatinib, or lorlatinib that were accessible depending on the period. Patient samples were prospectively collected but analysis was conducted in retrospective manner. Hence, patients were not treated with matched targeted therapy according to the resistance mechanism in real‐time. Since most of patients were treated with crizotinib as first‐line, data are limited for resistance to upfront use of second generation ALK TKIs, where most of patients are ongoing without progression. Further analysis will be available in the future. Nevertheless, to the best of our knowledge, our study is one of the largest prospective study to evaluate the role of cfDNA‐NGS in clinical practice.
In conclusion, NGS of cfDNA is useful not only for the detection of ALK fusions and resistance mutations, but also for assessing prognosis and monitoring the dynamic changes of genomic alterations in ALK positive NSCLC treated with ALK‐TKI.
CONFLICT OF INTEREST
S. Olsen is an employee of and holds ownership interest in Guardant Health. M. Lefterova is an employee of Guardant Health. J. Odegaard is an employee of and holds ownership interest in Guardant Health. No potential conflicts of interest were disclosed by the other authors.
AUTHOR CONTRIBUTIONS
Minsuk Kwon, Steve Olsen, and Myung‐Ju Ahn wrote the manuscript. Steve Olsen and Myung‐Ju Ahn initiated the study concept. Minsuk Kwon, Bo mi Ku, Sehhoon Park, Hyun‐Ae Jung, Jong‐Mu Sun, Se‐Hoon Lee, Jong‐Mu SunA., and Myung‐Ju Ahn analyzed the clinical data. Minsuk Kwon, Bo mi Ku, Martina Lefterova, and Justin Odegaard analyzed the genomic data. Hyun‐Ae Jung, Jong‐Mu Sun, Se‐Hoon Lee, Jong‐Mu SunA., Keunchil Park, and Myung‐Ju Ahn supervised the patient enrollment and participated and handled the study participants. All authors approved the final manuscript.
Supporting information
Supplementary Figure 1A
Supplementary Figure 1B.
Supplementary Figure 2.
Supplementary Figure 3.
Supplementary Figure 4.
Supplementary Figure 5.
Supplementary Figure 6.
Supplementary Figure 7.
Supplementary Figure 8.
Supplementary table 2
Supplementary table 1
Supplementary Table 3
Supplementary Table 4
Kwon M, Ku Bm, Olsen S, et al. Longitudinal monitoring by next‐generation sequencing of plasma cell‐free DNA in ALK rearranged NSCLC patients treated with ALK tyrosine kinase inhibitors. Cancer Med. 2022;11:2944‐2956. doi: 10.1002/cam4.4663
Funding information
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (No.NRF‐2017M3A9G5060259).
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Koivunen JP, Mermel C, Zejnullahu K, et al. EML4‐ALK fusion gene and Efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14(13):4275‐4283. doi: 10.1158/1078-0432.ccr-08-0168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Inamura K, Takeuchi K, Togashi Y, Nomura K., Ninomiya H., Okui M., Satoh Y., Okumura S., Nakagawa K., Soda M., Lim Choi Y., Niki T., Mano H., Ishikawa Y. EML4‐ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol Jan 2008;3(1):13–7. doi: 10.1097/JTO.0b013e31815e8b60 [DOI] [PubMed] [Google Scholar]
- 3. Shaw AT, Kim D‐W, Nakagawa K, et al. Crizotinib versus chemotherapy in AdvancedALK‐positive lung cancer. N Engl J Med. 2013;368(25):2385‐2394. doi: 10.1056/nejmoa1214886 [DOI] [PubMed] [Google Scholar]
- 4. Shaw AT, Kim TM, Crinò L, et al. Ceritinib versus chemotherapy in patients with ALK‐rearranged non‐small‐cell lung cancer previously given chemotherapy and crizotinib (ASCEND‐5): a randomised, controlled, open‐label, phase 3 trial. Lancet Oncol. 2017;18(7):874‐886. doi: 10.1016/s1470-2045(17)30339-x [DOI] [PubMed] [Google Scholar]
- 5. Peters S, Camidge DR, Shaw AT, et al. Alectinib versus Crizotinib in untreated ALK‐positive non–small‐cell lung cancer. N Engl J Med. 2017;377(9):829‐838. doi: 10.1056/nejmoa1704795 [DOI] [PubMed] [Google Scholar]
- 6. Camidge DR, Kim HR, Ahn M‐J, et al. Brigatinib versus Crizotinib in ALK‐positive non–small‐cell lung cancer. N Engl J Med. 2018;379(21):2027‐2039. doi: 10.1056/nejmoa1810171 [DOI] [PubMed] [Google Scholar]
- 7. Shaw AT, Bauer TM, De Marinis F, et al. First‐line Lorlatinib or Crizotinib in advanced ALK‐positive lung cancer. N Engl J Med. 2020;383(21):2018‐2029. doi: 10.1056/nejmoa2027187 [DOI] [PubMed] [Google Scholar]
- 8. Lin JJ, Riely GJ, Shaw AT. Targeting ALK: precision medicine takes on drug resistance. Cancer Discov. 2017;7(2):137‐155. doi: 10.1158/2159-8290.Cd-16-1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, Dagogo‐Jack I, Gadgeel S, Schultz K, Singh M, Chin E, Parks M, Lee D, DiCecca RH, Lockerman E, Huynh T, Logan J, Ritterhouse LL, le LP, Muniappan A, Digumarthy S, Channick C, Keyes C, Getz G, Dias‐Santagata D, Heist RS, Lennerz J, Sequist LV, Benes CH, Iafrate AJ, Mino‐Kenudson M, Engelman JA, Shaw AT Molecular mechanisms of resistance to first‐ and second‐generation ALK inhibitors in ALK‐rearranged lung cancer. Cancer Discov Oct 2016;6(10):1118–1133. doi: 10.1158/2159-8290.CD-16-0596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kwapisz D. The first liquid biopsy test approved. Is it a new era of mutation testing for non‐small cell lung cancer? Ann Translat Med. 2017;5(3):46. doi: 10.21037/atm.2017.01.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oxnard GR, Thress KS, Alden RS, et al. Association between plasma genotyping and outcomes of treatment with Osimertinib (AZD9291) in advanced non–small‐cell lung cancer. J Clin Oncol. 2016;34(28):3375‐3382. doi: 10.1200/jco.2016.66.7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu Y‐L, Lee V, Liam C‐K, et al. Clinical utility of a blood‐based EGFR mutation test in patients receiving first‐line erlotinib therapy in the ENSURE, FASTACT‐2, and ASPIRATION studies. Lung Cancer. 2018;126:1‐8. doi: 10.1016/j.lungcan.2018.10.004 [DOI] [PubMed] [Google Scholar]
- 13. Douillard J‐Y, Ostoros G, Cobo M, et al. Gefitinib treatment in EGFR mutated Caucasian NSCLC: circulating‐free tumor DNA as a surrogate for determination of EGFR status. J Thorac Oncol. 2014;9(9):1345‐1353. doi: 10.1097/jto.0000000000000263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34(5):547‐555. doi: 10.1038/nbt.3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ståhlberg A, Krzyzanowski PM, Jackson JB, Egyud M, Stein L, Godfrey TE. Simple, multiplexed, PCR‐based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic Acids Res. 2016;44(11):e105. doi: 10.1093/nar/gkw224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Narayan A, Carriero NJ, Gettinger SN, et al. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error‐suppressed multiplexed deep sequencing. Cancer Res. 2012;72(14):3492‐3498. doi: 10.1158/0008-5472.can-11-4037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dagogo‐Jack I, Brannon AR, Ferris LA, et al. Tracking the evolution of resistance to ALK tyrosine kinase inhibitors through longitudinal analysis of circulating tumor DNA. JCO Precis Oncol. 2018;2:1‐14. doi: 10.1200/po.17.00160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dagogo‐Jack I, Rooney M, Lin JJ, et al. Treatment with next‐generation ALK inhibitors fuels plasma ALK mutation diversity. Clin Cancer Res. 2019;25(22):6662‐6670. doi: 10.1158/1078-0432.ccr-19-1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horn L, Whisenant JG, Wakelee H, et al. Monitoring therapeutic response and resistance: analysis of circulating tumor DNA in patients with ALK+ lung cancer. J Thorac Oncol. 2019;14(11):1901‐1911. doi: 10.1016/j.jtho.2019.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Madsen AT, Winther‐Larsen A, McCulloch T, Meldgaard P, Sorensen BS. Genomic profiling of circulating tumor DNA predicts outcome and demonstrates tumor evolution in ALK‐positive non‐small cell lung cancer patients. Cancer. 2020;12(4):947. doi: 10.3390/cancers12040947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Tian P‐W, Wang W‐Y, et al. Noninvasive genotyping and monitoring of anaplastic lymphoma kinase (ALK) rearranged non‐small cell lung cancer by capture‐based next‐generation sequencing. Oncotarget. 2016;7(40):65208‐65217. doi: 10.18632/oncotarget.11569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCoach CE, Blakely CM, Banks KC, et al. Clinical utility of cell‐free DNA for the detection of ALK fusions and genomic mechanisms of ALK inhibitor resistance in non–small cell lung cancer. Clin Cancer Res. 2018;24(12):2758‐2770. doi: 10.1158/1078-0432.ccr-17-2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Odegaard JI, Vincent JJ, Mortimer S, et al. Validation of a plasma‐based comprehensive cancer genotyping assay utilizing orthogonal tissue‐ and plasma‐based methodologies. Clin Cancer Res. 2018;24(15):3539‐3549. doi: 10.1158/1078-0432.ccr-17-3831 [DOI] [PubMed] [Google Scholar]
- 24. Sozzi G, Conte D, Mariani L, et al. Analysis of circulating tumor DNA in plasma at diagnosis and during follow‐up of lung cancer patients. Cancer Res. 2001;61(12):4675‐4678. [PubMed] [Google Scholar]
- 25. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med. 2014;6(224):224ra24. doi: 10.1126/scitranslmed.3007094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz Jr LA Circulating mutant DNA to assess tumor dynamics. Nat Med 2008/09/01 2008;14(9):985–990. doi: 10.1038/nm.1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goldstein I, Marcel V, Olivier M, Oren M, Rotter V, Hainaut P. Understanding wild‐type and mutant p53 activities in human cancer: new landmarks on the way to targeted therapies. Cancer Gene Ther. 2011;18(1):2‐11. doi: 10.1038/cgt.2010.63 [DOI] [PubMed] [Google Scholar]
- 28. Aggarwal C, Thompson JC, Black TA, et al. Clinical implications of plasma‐based genotyping with the delivery of personalized therapy in metastatic non–small cell lung cancer. JAMA Oncol. 2019;5(2):173‐180. doi: 10.1001/jamaoncol.2018.4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong SQ, Raleigh JM, Callahan J, et al. Circulating tumor DNA analysis and functional imaging provide complementary approaches for comprehensive disease monitoring in metastatic melanoma. JCO Precis Oncol. 2017;1:1‐14. doi: 10.1200/po.16.00009 [DOI] [PubMed] [Google Scholar]
- 30. Canale M, Petracci E, Delmonte A, et al. Impact of TP53 mutations on outcome in EGFR‐mutated patients treated with first‐line tyrosine kinase inhibitors. Clin Cancer Res. 2017;23(9):2195‐2202. doi: 10.1158/1078-0432.ccr-16-0966 [DOI] [PubMed] [Google Scholar]
- 31. Kron A, Alidousty C, Scheffler M, et al. Impact of TP53 mutation status on systemic treatment outcome in ALK‐rearranged non‐small‐cell lung cancer. Ann Oncol. 2018;29(10):2068‐2075. doi: 10.1093/annonc/mdy333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu J, Cao J, Topatana W, et al. Targeting mutant p53 for cancer therapy: direct and indirect strategies. J Hematol Oncol. 2021;14(1):157. doi: 10.1186/s13045-021-01169-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sallman DA, Dezern AE, Garcia‐Manero G, et al. Eprenetapopt (APR‐246) and Azacitidine in TP53‐mutant myelodysplastic syndromes. J Clin Oncol. 2021;39(14):1584‐1594. doi: 10.1200/jco.20.02341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dumbrava EE, Mahipal A, Gao X, et al. Phase 1/2 study of eprenetapopt (APR‐246) in combination with pembrolizumab in patients with solid tumor malignancies. J Clin Oncol. 2021;39((15_suppl)):TPS3161. doi: 10.1200/JCO.2021.39.15_suppl.TPS3161 [DOI] [Google Scholar]
- 35. Lin X, Wang L, Xie X, et al. Prognostic biomarker TP53 mutations for immune checkpoint blockade therapy and its association with tumor microenvironment of lung adenocarcinoma. Front Mol Biosci. 2020;7:602328. doi: 10.3389/fmolb.2020.602328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Long J, Wang A, Bai Y, et al. Development and validation of a TP53‐associated immune prognostic model for hepatocellular carcinoma. EBioMedicine. 2019;42:363‐374. doi: 10.1016/j.ebiom.2019.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alidousty C, Baar T, Martelotto LG, et al. Genetic instability and recurrent MYC amplification in ALK‐ translocated NSCLC: a central role of TP53 mutations. J Pathol. 2018;246(1):67‐76. doi: 10.1002/path.5110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1A
Supplementary Figure 1B.
Supplementary Figure 2.
Supplementary Figure 3.
Supplementary Figure 4.
Supplementary Figure 5.
Supplementary Figure 6.
Supplementary Figure 7.
Supplementary Figure 8.
Supplementary table 2
Supplementary table 1
Supplementary Table 3
Supplementary Table 4
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.