

Abstract
Bacteria of the Burkholderia pseudomallei (BP) group pose a global health threat, causing the infectious diseases melioidosis, a common cause of pneumonia and sepsis, and glanders, a contagious zoonosis. A trait of BP bacteria is a conserved gene cluster coding for the biosynthesis of polyketides (malleicyprols) with a reactive cyclopropanol unit that is critical for virulence. Enzymes building this warhead represent ideal targets for antivirulence strategies but the biochemical basis of cyclopropanol formation is unknown. Here we describe the formation of the malleicyprol warhead. We show that BurG, an unusual NAD+-dependent member of the ketol-acid reductoisomerase family, constructs the strained cyclopropanol ring. Biochemical assays and a suite of eight crystal structures of native and mutated BurG with bound analogues and inhibitors provide snapshots of each step of the complex reaction mechanism, involving a concealed oxidoreduction and a C–S bond cleavage. Our findings illustrate a remarkable case of neofunctionalisation, where a biocatalyst from central metabolism has been evolutionarily repurposed for warhead production in pathogens.
Subject terms: Biosynthesis, Enzymes
Burkholderia pseudomallei is a species of bacteria that poses a global health threat and, more generally, bacteria of the Burkholderia pseudomallei group cause severe diseases that are recalcitrant to treatment with antibiotics. Now, it has been shown how these infamous pathogens repurpose the widespread enzyme BurG to produce a reactive cyclopropanol head group found in the virulence-promoting malleicyprol toxins. Interrupting the synthesis of the cyclopropanol warhead is a potential route for developing antivirulence treatments.
Main
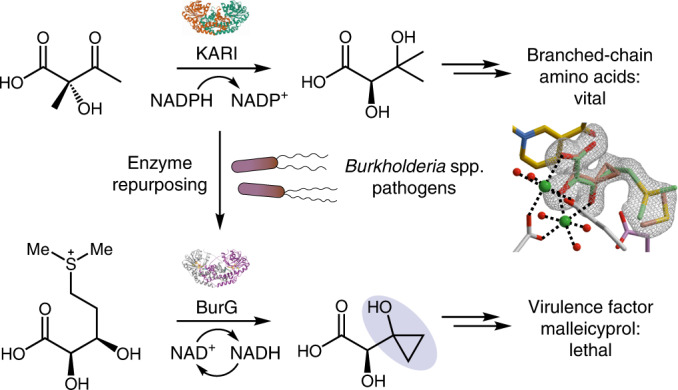
A holistic view on the molecular mechanisms underlying Burkholderia pseudomallei (BP)-mediated diseases is urgently needed for the development of therapies, including vaccination and antivirulence strategies. Beyond typical disease-promoting factors1 such as secretion systems, capsular polysaccharides, a protein toxin2 and siderophores3–5, recent studies indicate that the role of the specialized secondary metabolome of BP bacteria has been greatly underestimated6,7. All members of the BP complex, including the closely related, yet less virulent model organism Burkholderia thailandensis, harbour a highly conserved gene locus (bur/mal, Fig. 1a) coding for an unusual polyketide synthase8,9. According to animal10,11 and cell-based assays using targeted mutants8,12, this gene cluster is critical for virulence. The isolated metabolite (burkholderic acid/malleilactone 1; Fig. 1b) initially assigned as product of the assembly line, however, could not explain the observed phenotypes8,9. Reinvestigation of the metabolome revealed previously overlooked cyclopropanol-substituted congeners named malleicyprols (bis-malleicyprol 2 and malleicyprol 3; Fig. 1b), which show high toxicity in cell assays and in a nematode model13. The effect of malleicyprols is attributed to a cyclopropanol warhead that is known to readily form β-keto radicals upon single-electron oxidation and ring opening (Fig. 1c)14–16. Consequently, the previously detected propanone-substituted metabolite 1 represents the inactive form. Whereas mutational analyses, in vitro assays and isotope-labelling experiments indicated that the reactive cyclopropanol derives from methionine via the zwitterionic gonyol 4 (Fig. 1b and Extended Data Fig. 1)17, the key biosynthetic steps that install the cyclopropanol warhead have remained obscure.
Fig. 1. Malleicyprols, virulence factors encoded by conserved locus in BP group pathogens.

a, Gene cluster coding for malleicyprol biosynthesis. KARI, ketol-acid reductoisomerase b, Structures of malleicyprols 2 and 3 and their degradation product 1, and selected steps in the biosynthesis of the C5 building block. See Extended Data Fig. 1. for detailed biosynthetic pathway. c, Ring-opening of cyclopropanols mediated by single-electron oxidation.
Extended Data Fig. 1. Model of biosynthetic pathway to malleicyprol.

More detailed model of the biosynthetic pathway to malleicyprol.
Results and discussion
The elusive cyclopropanol precursor
Elucidating the biochemical basis of cyclopropanol formation was challenging because no other pathways to cyclopropanol-bearing natural products18 were known and the bur/mal gene clusters did not point to plausible biocatalysts. Hence both the substrate and the timing of the reaction were unclear. Therefore, we first sought to elucidate the steps downstream of gonyol formation by systematically inactivating uncharacterized genes of the bur/mal cluster in the engineered malleicyprol overproducer strain B. thailandensis Pbur9. Monitoring metabolic changes by comparative liquid chromatography–high-resolution mass spectrometry (LC-HRMS) analyses we noted that a mutant lacking burG, in lieu of malleicyprols, produces a new compound (5) with an m/z of 195.0687 (M + H+; Fig. 2a and Supplementary Fig. 1) and a deduced chemical formula of C7H15O4S (Fig. 2b). Its polarity and molecular composition suggested that 5 represents a biosynthetic intermediate that could arise from hydroxylation of gonyol (C7H15O3S). Isolation of 5 for a full structure elucidation proved to be challenging due to its high polarity. Eventually, using an optimized purification protocol, we obtained 5 as its trifluoroacetate (TFA) salt in sufficient amount (1.3 mg l−1) and purity to unequivocally elucidate its structure. NMR data suggested that 5 is a sulfonium-substituted carboxylic acid with two hydroxy substituents at the α and β positions (Fig. 2c). We confirmed the structure of 5 (named gonydiol) and determined the anti-configuration of the hydroxy groups by chemical synthesis (Supplementary Fig. 2) of anti- and syn-configured reference compounds and NMR comparisons (Fig. 2c). We reasoned that 5 could result from hydroxylation of 4 by the gene product of burC, a putative α-ketoglutarate-dependent dioxygenase. To reconstitute the biotransformation, we cloned burC, heterologously produced His6-tagged BurC in Escherichia coli, and synthesized enantiomerically pure R- and S-4 as potential substrates (Extended Data Fig. 2). In the in vitro assays BurC specifically transforms S-gonyol into anti-gonydiol (Fig. 2d). Consequently, the absolute configuration of natural gonydiol, a precursor of the cyclopropanol-bearing building block, is 2R,3R.
Fig. 2. Substrate identification and reconstitution of enzymatic cyclopropanol formation.

a, Metabolomics analysis of B. thailandensis Pbur versus B. thailandensis Pbur BurG::Kan (malleicyprol null mutant) cell extracts (biological triplicates, n = 3). Data filtered for sulfur-containing compounds; see Supplementary Fig. 1 for non-filtered analysis. b, Chemical complementation of the null mutant with 6 restores malleicyprol production (isoforms 2a and 2b) to wild-type level. c, Structure elucidation of isolated (isol.) gonydiol 5, and comparison of key 1H NMR shifts with synthetic (synth.) racemic syn- and anti-isoforms. COSY, correlated spectroscopy; HMBC, heteronuclear multiple bond correlation. d, Formation of 5 catalysed by hydroxylase BurC in assays with addition of R-4 or S-4; top trace: reference (5). e, Detection of DMS as product of BurG reactions by GC-MS. f, Mirror plot of enzymatic assay (top) of BurG incubated with 5 versus denatured BurG (bottom) indicates a compound with the m/z value of 6 as enzymatic product; see Extended Data Fig. 3 for chromatogram. g, BurG-catalysed transformation of 5 to 6. h, Monitoring of enzymatic turnover of isolated 5 and synthetic rac-syn-5 or rac-anti-5 into 6 via HR-LCMS. i, Steady-state kinetics of formation of 6 from rac-anti-5; mean and standard deviation obtained from two independent preparations (n = 2). j, Phylogenetic analysis of BurG, orthologues and canonical KARIs. The scale bar indicates amino acid substitutions per site. k, Alignment of biosynthetic gene cluster encoding for BurG orthologues clustered with the genes associated with biosynthesis of 5.
Extended Data Fig. 2. Absolute configuration of gonydiol.
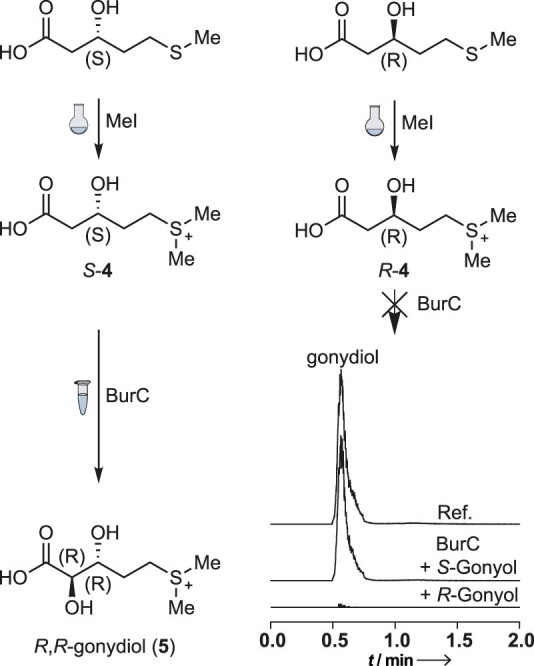
Synthesis of R- and S-gonyol (4) and subsequent enzymatic transformation to gonydiol (5) establishes the absolute configuration of naturally occurring gonyol and gonydiol. HR-LCMS extracted ion monitoring of gonydiol (m/z of 195.0686 in positive-ion mode) after addition of R-4 or S-4 to reaction mixtures containing the hydroxylase BurC.
Discovery of a cyclopropanol synthase
Accumulation of 5 in the burG::Kan mutant strongly suggested that this metabolite is the substrate for BurG downstream of the pathway towards malleicyprol. A sequence similarity search using BLAST predicted that BurG belongs to the family of ketol-acid reductoisomerases (KARIs; EC 1.1.1.86), well-characterized enzymes that play key roles in the biosynthesis of branched-chain amino acids19. To study the function of BurG, we heterologously produced a His6-tagged variant in E. coli and incubated purified His6-BurG with isolated and synthetic substrates. We noted the characteristic smell of dimethylsulfide (DMS) when opening closed reaction tubes of assays containing natural or the synthetic anti-5. In contrast, DMS was not detectable in assays containing either the syn-isomer or heat-denatured BurG. We corroborated these findings by solid-phase microextraction–gas chromatography–mass spectrometry (SPME–GC–MS) gas-phase analysis of the respective assays (Fig. 2e).
Consequently, BurG catalyses a specific C–S bond cleavage reaction to form DMS and a second reaction product, probably composed of the remaining five carbon atoms of 5. By comparison of the averaged mass spectra of total ion currents we identified a compound (6, named trigonic acid) with m/z 131.0349 that is only present in the positive assays with intact BurG (Fig. 2f and Extended Data Fig. 3). The deduced chemical formula of C5H8O4 for the uncharged species of 6 is in agreement with a compound resulting from the elimination of DMS from 5 (C7H15O4S [M + H]+) and indicated an additional ring double-bond equivalent compared to 5. We thus anticipated that BurG catalyses a cyclization reaction, tentatively yielding a cyclopropanol residue as in 6 (Fig. 2g). To prove the proposed structure of 6 we synthesized rac-6 and confirmed its identity with the enzymatically formed product by HR-LCMS (Fig. 2h; see Supplementary Fig. 3 for tandem MS). Moreover, complementation of B. thailandensis Pbur burG::Kan cultures with synthetic rac-6 fully restored malleicyprol production (Fig. 2b). To elucidate the absolute configuration of 6, we synthesized enantiomerically enriched R-6 (e.e. 82%) by asymmetric Sharpless bishydroxylation20,21 and proved its identity with the product of the enzyme assay by chiral HR-LCMS analysis (Extended Data Fig. 4). These results not only demonstrate that the cyclopropanol-substituted molecule 6 in its R-configuration is a true pathway intermediate but also that BurG catalyses an unusual cyclization reaction yielding the malleicyprol warhead.
Extended Data Fig. 3. Identification of trigonic acid (6).
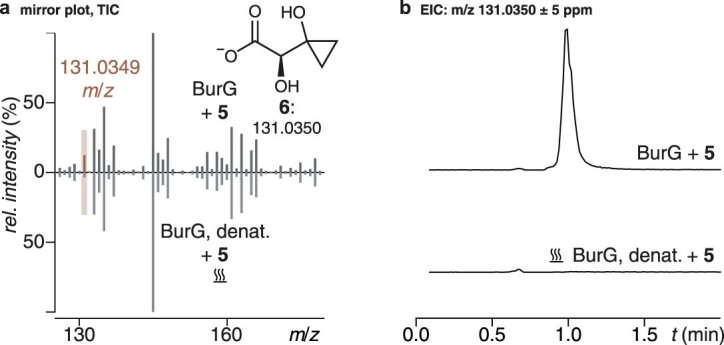
a, Mirror plot of enzymatic assay (top) of BurG incubated with 5 vs. denatured BurG (bottom) indicates a compound with the m/z value of 6 as enzymatic product. b, Extracted ion chromatogram (EIC) in negative ion mode of the enzymatic assay (top) vs. denatured BurG (bottom).
Extended Data Fig. 4. Determination of stereochemistry of trigonic acid (6).

Left panel: stereoselectivity of the asymmetric dihydroxylation (AD)-mix β leads to mainly S-configurated products20; Bn: benzyl protecting group. Consequently AD-mix α forms mainly R-configurated products. Chiral HR-LCMS (bottom left) validates the successful asymmetric synthesis with AD-mix α after hydrolysis of the formed ethyl ester (Et). Right panel: chiral HR-LCMS of asymmetric synthesis products (ref.) vs. enzymatically formed 6 shows the stereochemistry of enzymatically formed 6 to be of R-configuration.
BurG catalyses a transient redox reaction
The formation of a strained cyclopropane ring markedly differs from the biotransformations known for canonical KARIs. Notably, the genomes of BP bacteria harbour additional KARI genes (ilvC) within the operons for branched-chain amino acid biosynthesis. A phylogenetic analysis (Fig. 2j and Supplementary Fig. 4) of BurG-like enzymes and native KARIs revealed a separate clade for tentative cyclopropanol synthases within the KARI family. We identified nine other organisms (Fig. 2k) that harbour burG orthologues along with all genes required for gonydiol biosynthesis.
HHpred analyses22 predicted the typical KARI Rossmann fold domains for these BurG-type enzymes, pointing to the binding of a nucleotide cofactor. Indeed, HR-LCMS analysis of a denatured BurG preparation revealed the presence of the nucleotide cofactor NAD+ (Supplementary Fig. 5). This finding is surprising because KARIs are known to prefer NADPH as cofactor. In this context it is noteworthy that considerable effort has been directed towards engineering KARIs to utilize NADH instead of NADPH23 and to discover rare NADH-preferring KARIs for biotechnological purposes24.
To test whether NAD+ is essential for the BurG-mediated cyclopropanol formation we removed the cofactor from the enzyme preparation by using an extensive dialysis protocol. In the absence of NAD+, BurG is incapable of forming 6, yet the activity can be restored by addition of NAD+. Addition of NADH, however, cannot restore the reaction (Fig. 3a). These results provide evidence that bound NAD+ is essential for the BurG-catalysed reaction.
Fig. 3. Concealed redox cycle catalysed by BurG.

a, Gonydiol 5 transformation assay with NAD+-free BurG with addition of NAD+, NADH or in the absence of cofactor; top: 6 as reference. b, Full view of the 3D structure of BurG co-crystallized with the KARI inhibitor 7 (left). A and B, subunits; N and C, termini. The black rectangle displays the area for the zoom in the substrate binding channel (middle) and active site zoom (right) (PDB 7PCG). GOL, glycerol. c, Steady-state kinetics of NADH-mediated reduction of hydroxypyruvate 8 to glyceric acid 9 through BurG; mean and standard deviation as obtained from two independent protein preparations (n = 2). d, Scheme of the transformation of hydroxamate 10 to hemiaminal 11. e, 3D structure of BurG co-crystallized with 11 after incubation with 10 and NADH (PDB 7PCL). Two magnesium atoms (A and B) are shown as green spheres. f, BurG-mediated oxidation of carba-gonydiol 12 to the enolate 13. g, 3D structure of BurG with 13 obtained after co-crystallization with 12 and NAD+ (PDB 7PCM). h, Single amino acid substitution (E232Q) renders BurG inactive. i, 3D structure of BurG E232Q co-crystallized with 12 (PDB 7PCO).
To gain more insight into the reaction mechanism and the role and position of NAD+ in the enzyme, we performed protein crystallography. Due to the close phylogenetic relation between BurG and the KARIs, we turned to surrogates that had proven successful in previous studies in obtaining ligand-bound three-dimensional (3D) structures such as the KARI inhibitor cyclopropane-1,1-dicarboxylate 7 (ref. 19). We raised crystals of holo-BurG in the absence or presence of 7 as surrogate and solved the structures at a resolution of 1.8 Å and 1.9 Å, respectively. The obtained 3D structure of the complex (Fig. 3b; PDB 7PCG) clearly displays 7 bound between two magnesium ions with NAD+ in close proximity, thus enabling its participation in biocatalysis (Fig. 3b). The amino acid residues Asn48 and Asp51 exclude the negatively charged phosphate group of NADP+ from binding in the active centre of BurG (Extended Data Fig. 5). A co-crystallized glycerol molecule (used as cryoprotectant) maps the entire specificity pocket next to the catalytic centre. The positioning of NAD+ in the active site of BurG indicates that an oxidation event occurs during cyclopropanol formation. Monitoring the BurG assays by HR-LCMS, however, did not show the presence of any oxidized derivatives of 5 or 6 (or any other pathway intermediates). Therefore, BurG is either not redox proficient or a transient redox step occurs during its catalytic cycle. Such a cryptic (or covert) redox reaction would first form NADH by substrate oxidation, followed by NAD+ regeneration through ligand reduction.
Extended Data Fig. 5. Binding mode of NAD+ in the active site of BurG.
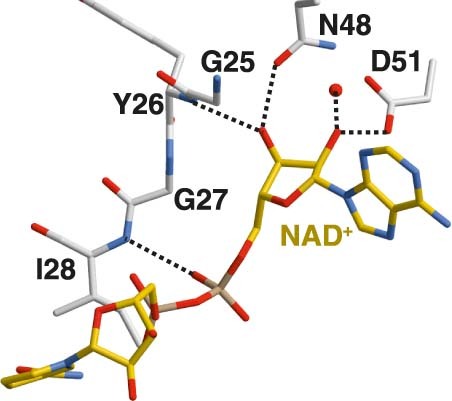
The amino acid residues Asn48 and Asp51 exclude the negatively charged phosphate group of NADP+ (as commonly found in canonical KARIs) from binding in the active centre of BurG.
To investigate the redox proficiency of BurG and its ability to regenerate NAD+ from NADH we devised an absorbance-based assay with the substrate surrogate hydroxypyruvate 8. BurG was incubated with 8 and NADH and the light absorption of the dinucleotide at 340 nm was monitored spectrophotometrically. A clearly observed signal decrease corresponds to the formation of NAD+ and glyceric acid 9, the reduced form of 8 (Fig. 3c). In substrate saturation kinetics we determined a kcat of 16.3 ± 1.1 min−1 (Fig. 3c and Supplementary Fig. 6) for this reaction, which resembles the kcat for ketone reduction by other KARIs19. Further evidence for the redox proficiency of BurG was gleaned from protein crystallization experiments with the previously co-crystallized19 KARI inhibitor N-hydroxy-N-isopropyloxamic acid (IpOHA, 10) in the presence of NADH (Fig. 3d; PDB 7PCL). The 3D structure at 2.05 Å resolution unexpectedly shows that the hydroxamate 10 was reduced to the corresponding hemiaminal 11, as evidenced by the tetrahedral geometry of the surrogate in the 3D structure (Fig. 3e). Notably, the stereochemical course of the reduction is identical with the native product of BurG. Taken together, BurG is a redox-proficient enzyme that promotes cyclopropanol formation via a transient and thus concealed oxidation event. This finding is surprising because characterized enzymes that recycle their NAD+ cofactors are scarce25–27.
To provide further experimental evidence for the proposed transient redox cycle (Extended Data Fig. 6) we captured the enzyme in a state with bound NADH after the oxidation event occurred. Therefore, we synthesized the carba analogue of gonydiol, rac-12, which cannot be converted into a cyclopropanol (Fig. 3f), and performed protein crystallography. Analysis of the obtained 3D structure with a resolution of 2.05 Å of BurG with rac-12 revealed that the surrogate had been converted into the oxidized form 13 that is complexed between the two magnesium atoms and NADH in the active site of the enzyme (Fig. 3g; PDB 7PCM). The planar geometry of the complexed ligand indicates an sp2 hybridization of both the α and β carbons, indicating that 13 is stabilized in the enolic form. The transformation of rac-12 into the trapped enolate 13 provides evidence for an intermediary oxidation during the catalytic cycle of BurG. We obtained similar results in co-crystallization assays with BurG, NAD+ and the substrate surrogate hydroxypyruvate 8. By analogy to 13, the crystal structure depicts a planar geometry of 8 indicating it is bound in its enolized form to the active site of BurG (Extended Data Fig. 7; PDB 7PCI).
Extended Data Fig. 6. Cryptic redox cycle.
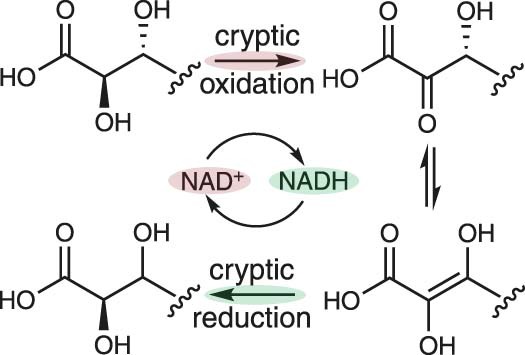
Scheme representing the general concept of a cryptic redox cycle.
Extended Data Fig. 7. Complex structure of BurG co-crystallized with hydroxypyruvate (8) (PDB ID: 7PCI).
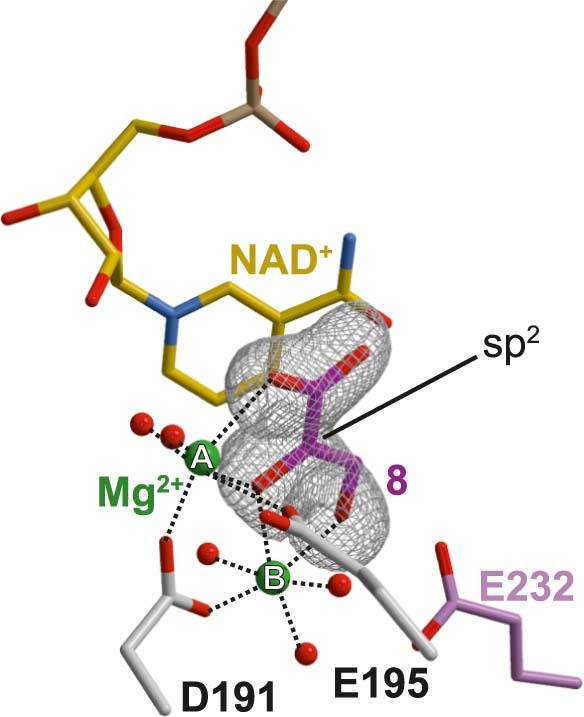
The FO-FC electron density map (contoured to 3 σ) for 8 bound to the Mg-atoms is shown as grey mesh. The ligand has been omitted for phasing. E232 is located on subunit B and marked in pink.
Mechanism of cyclopropanol formation
Our structural analysis revealed that the carboxylate of a glutamic acid side chain (E232) is in close proximity (3 Å) to the β carbons of the substrate surrogates and thus probably facilitates the deprotonation event necessary for enol formation. To probe the function of this putative catalytic residue we generated a point-mutated variant (E232Q) of BurG and assayed its activity with the native substrate gonydiol. As predicted, the mutation hampers the transformation of gonydiol into the cyclopropanol derivative 6 (Fig. 3h). The redox proficiency of BurG E232Q remained intact as indicated by a kcat of 8.2 ± 0.3 min−1 for the NADH-mediated reduction of 8 (Supplementary Fig. 7). Co-incubations of BurG E232Q with rac-12 yielded crystals for which we solved a 3D structure at a resolution of 1.55 Å. In contrast to native BurG, rac-12 was not oxidized by BurG E232Q, as indicated by the tetrahedral geometry of the complexed surrogate (Fig. 3i; PDB 7PCO). The absence of oxidized 13 in BurG E232Q may indicate a role of E232 in shifting the internal redox equilibrium towards the oxidized state by deprotonating the substrate and forming the enolate. By analogy to glycerate oxidation, the oxidized state is probably energetically less favoured.28 The obtained near-atomic resolution structure with BurG E232Q and 12 enabled us to analyse the bound ligand and determine its absolute configuration (2R,3R). The observed chirality of the bound substrate confirmed our stereochemical analysis that gonydiol had the 2R,3R configuration.
Apart from its function as a catalytic base, E232 also plays an active role in substrate recognition and positioning as we gleaned from crystallization assays with gonydiol and BurG. We solved a 3D structure (at 1.6 Å) for which the electron density map shows a mixed population of two subsets of the converted substrate (Fig. 4a and Extended Data Fig. 8; PDB 7PCN). In one subset the active centre is populated by the enzymatic reaction products 6 and DMS. The second subset clearly depicts the backbone of gonydiol, which was oxidized into its enolate 14. Intermediate 14 is firmly held in place by ionic interaction of its sulfonium moiety with E232 and cation–π interaction with Y133 (Fig. 4b). In contrast, the neutral carba analogues 12 and 13 show a higher degree of freedom in their tail regions (Fig. 4c) due to the lack of ionic placement as visible in their crystallographic B factor.
Fig. 4. Parallels and differences between biocatalysis of BurG and canonical KARIs.

a, Structures of BurG with the reaction products 6 and DMS and the trapped enolate intermediate 14 after co-crystallization with gonydiol 5 (PDB 7PCN). Two magnesium atoms (A and B) are shown as green spheres. b, Interaction of active site residues of BurG with 6, 14 and DMS. c, Structural overlay of BurG bound to 12, 13 and 14 indicates a strong interaction of the positively charged sulfonium moiety of 14 with E232 as compared to neutral 12 and 13. d, Tight lock of the oxygen-functionalized compounds 6 and 7 through interaction with Mg2+ ions in the active centre of BurG. Wapo indicates the binding sites of water molecules as found in the apo structure that are replaced by ligand atoms in the depicted structures. e, Structure of unliganded holo-BurG illustrates a hydroxide ion bridged between two Mg2+ centres (PDB 7PCC). f, Comparison of BurG and KARIs displays a similar binding mode of substrates in the Mg2+ cluster. g, Proposed reaction mechanism19 for KARIs with cyclopropane transition state. h, Reaction mechanism proposed for BurG involving a concealed (transient) redox reaction.
Extended Data Fig. 8. Complex structure of BurG co-crystallized with gonydiol (5) (PDB ID: 7PCN).

The 2FO-FC electron density maps (grey meshes, contoured to 1.0 σ) and the FO-FC maps (green meshes, contoured to 3.0 σ) are depicted for the respective ligand. a. 6 and DMS (shadowed in grey) have been omitted for phasing. b. 14 (shadowed in grey) has been omitted for phasing. The FO-FC electron density maps clearly depict the excluded ligands, proving that 14, 6 and DMS are all bound to the active site. The occupancy of each ligand was set to 0.5. E232 is located on subunit B and marked in pink.
The head region (C1–C3) of all co-crystallized ligands was consistently found to be firmly locked in a conserved binding mode that is facilitated by interactions with the two Mg2+ ions of the enzyme (see Fig. 4d,e for unliganded holo structure). In this head region, BurG shows remarkable similarity with regards to substrate structure and binding when compared to canonical KARIs (Fig. 4f). The 3D structure obtained for the holo version of BurG (PDB 7PCC) illustrates that the binding sites of the Mg-bound oxygen functionalities are populated by aqueous residues when no ligand is present (Fig. 4e). In addition to similarities in substrate binding and redox capability, canonical KARIs are thought to contain a cyclopropane transition state19 in their catalytic cascade (Fig. 4g). In the case of BurG, formation of 6 is probably driven by a gain in entropy and removal of the volatile DMS from the chemical equilibrium after cyclopropanol synthesis. Moreover, the enzyme surrounds the cationic sulfonium group of the substrate with a hydrophobic binding pocket lined with Met, Tyr and Val residues, which favour loss of the charge and thus promote catalysis (Fig. 4b).
Based on these results, we devised a plausible reaction mechanism for the BurG-mediated cyclopropanol formation (Fig. 4h). First, by analogy to the initial step in the catalytic cycle of canonical KARIs29, the hydroxide bridged between the magnesium centres (shown in the ligand-free holo structure of BurG; Fig. 4e), would deprotonate the α-hydroxy moiety of 5, thus forming the corresponding alkoxide. Next, the two magnesium ions would complex the alkoxide and position it above the pyridinium moiety of the NAD+ cofactor. In addition, the glutamate residue (E232) would ionically interact with the positively charged sulfonium group to firmly position the substrate in the binding pocket. Subsequent hydride transfer from the C-α of gonydiol to NAD+ (yielding NADH), and deprotonation at the β carbon by E232 would result in an enolate intermediate (14), as mimicked by hydroxypyruvate and the oxidized carba-gonydiol analogue 13. The ideal positioning of C-α relative to the hydride-accepting NAD+-carbon (Extended Data Fig. 9) renders an alternative mechanism involving oxidation at C-β unlikely. Quenching of the negative charge at E232 through proton transfer would disrupt the ionic interaction, destabilize the sulfonium and trigger cyclization. Next, an intramolecular nucleophilic attack at the δ position would result in cyclopropyl ring formation and C–S bond cleavage followed by hydride transfer from NADH to the si face of the C-α atom. The proposed stereochemical course of hydride transfer is in line with the determined stereochemistry of the final product 6 and the orientation found of all surrogates in the active site of the enzyme (Fig. 4a). This key reductive step would regenerate the NAD+ cofactor, leaving the overall redox reaction neutral and retaining the configuration at C2. Our structural analysis indicates that the outlined reaction sequence takes place in a closed conformation of the active site (see Extended Data Fig. 10 for comparison of closed and open/apo conformations). This enzymatic envelope would protect the oxidized and highly reactive intermediates from unwanted side reactions. Hence, final expulsion of the reaction products from the active site would prepare the enzyme for the start of a new catalytic cycle.
Extended Data Fig. 9. Cofactor and ligand orientation in the active site of BurG and KARIs.

The structural comparison between BurG and KARIs illustrates the preferred reaction trajectories of hydride transfer in the distinct enzymes. The interactions of the ligand with the two Mg2+ ions (black dots) indicate that Cα is the preferred carbon atom for catalysis. a, Structure of the catalytically inactive BurG E232Q mutant in complex with surrogate 12. The ligand adopts ideal orientation for hydride transfer to NAD+ (2.3 Å distance, angle between C(NAD+)HCα: 174°, PDB code: 7PCO). b, Structure of BurG co-crystallized with 12 and NAD+. The electron density shows the ligand oxidized to 13 resulting in NADH. Glu232 acts as base and is key to hydride transfer. Distance and angle between the cosubstrate and Cα (2.8 Å, C(NADH)HCα: 123°) are unfavourable for the back reaction. The replacement of sulfur by carbon in surrogate 13 prevents the elimination reaction and keeps the metabolite in its position (PDB code: 7PCM). c, Structure of BurG co-crystallized with 10 and NADH. The electron density shows the Cα-atom of the ligand in sp3 configuration. Therefore, the molecule represents the carbinolamine 11 and the cosubstrate is oxidized to NAD+. Distance and angle between the cosubstrate and Cα allow for a back reaction (2.1 Å, C(NAD+)HCα: 168°). However, formation of the alkoxide is favoured by the interactions with both Mg2+ ions (PDB code: 7PCL). d, Active site architecture of the KARI from Staphylococcus aureus (PDB code: 6AQJ). Crystallization of the enzyme was carried out in presence of 10 and NADPH. It is reported19 that hydride transfer from NADPH to the nitrogen of 10 takes place, hereby forming NADP+ and converting the ligand to 16 via water (W) release. The complex structure lacks defined H-bonds between cosubstrate and ligand.
Extended Data Fig. 10. Structural overlays.

a, Structural overlay of BurG (holo) bound to 11 (PDB code: 7PCL, coloured in pink) with apo BurG (PDB code: 7PCE, coloured in grey). The root mean square deviation (rmsd) between both structures is 1.2 Å over 283 Cα-atoms (353 residues in total). BurG (apo) displays increased flexibility (high temperature factors) and individual secondary structure elements are shifted compared to BurG (holo). As a result, the catalytic centre in apo BurG is accessible to cofactors and ligands (open conformation). In contrast, the active site of BurG (holo) is sequestered in a closed chamber. b, Structural overlay of BurG (holo) (PDB code: 7PCL, coloured in pink) with the ketol-acid reductoisomerase (KARI, holo) from Staphylococcus aureus (PDB code: 6AQJ, coloured in grey). The KARI from S. aureus is bound to 10 while BurG is bound to 11 (the reduced form of 10). Both structures adopt the closed conformation and display a rmsd of 1.4 Å over 288 Cα-atoms (sequence identity 35 %).
Discussion
We reason that the KARI-fold is ideally positioned as enzymatic ancestor from primary metabolism to be repurposed for gonydiol binding, a transient redox reaction, and cyclopropanol construction by elimination of DMS. The fact that canonical KARIs facilitate their reaction sequence to branched carbon backbones through a cyclopropane transition state hints to the evolutionary trajectory (neofunctionalisation) towards the cyclopropanol synthase BurG. Inheritance of the redox capability of KARIs allows BurG to activate its substrate gonydiol as the electrophilic intermediate 14. This activation is vital for intramolecular electrophilic displacement of DMS as leaving group and subsequent cyclopropanol formation. Most characterized biosynthetic pathways towards cyclopropyl30–35 moieties, as in the genotoxin colibactin36,37, for example, also employ electrophilic displacement reactions38. Generation of the enolic nucleophile, however, is commonly achieved via a thioester linkage between enzyme and substrate, and in all studied cases either methylthioadenosine or chloride are employed as leaving groups. The KARI-mediated reaction discovered here may inspire chemical engineers who seek to incorporate highly strained rings in a biocatalytic fashion39. Our work offers the prospects of finding new cyclopropanol-forming enzymes and the corresponding natural products. More importantly, our mechanistic and structural study of the key enzyme responsible for malleicyprol biosynthesis sets the basis for selectively inhibiting the formation of virulence-conferring toxins from pathogenic B. pseudomallei complex bacteria.
Methods
Bacterial strains and general culture conditions
The B. thailandensis wild-type strain E264 (DSM13276) was obtained from DSMZ. Cultures of B. thailandensis E264, B. thailandensis Pbur and B. thailandensis Pbur burG::Kan were either grown in MM99 liquid medium, LB-liquid or LB-agar medium at 30 °C. Cultures of mutant strains were supplemented with either tetracycline (45 μg ml−1, B. thailandensis Pbur) or tetracycline (45 μg ml−1) in addition to kanamycin (150 μg ml−1, B. thailandensis Pbur burG::Kan). Liquid cultures were grown in 300 ml (50–75 ml culture volume) and 1 litre (250 ml culture volume) baffled Erlenmeyer flasks with orbital shaking at 30 °C (B. thailandensis) or 37 °C (E. coli). To obtain biomass for metabolite extraction, the grown cells were pelleted by centrifugation (6,000g, 10 min).
Suicide plasmid preparation for B. thailandensis mutant
A gene fragment (2,349 bp) containing burG was PCR-amplified using primers Isomerase-fw and Isomerase-rv with 5 Prime Extender Polymerase (VWR International). The amplicon was purified using the GFX PCR DNA and Gel Band Purification Kit (GE Healthcare), and the obtained DNA was cloned into the pGEM T-easy vector (Promega). The resulting plasmid (pGEM-Isomerase) was verified by sequencing (GATC biotech) and restricted with the restriction enzyme SmaI and treated with SAP (Shrimp Alkaline Phosphatase, New England Biolabs). pGEM-Kan was restricted by NotI and blunted by Klenow treatment. This blunted kanamycin resistance cassette gene was then cloned into the SmaI-restricted plasmid, generating the Kan-inserted suicide plasmid pGEM-burG::Kan.
Preparation of gene insertion mutant B. thailandensis Pbur burG::Kan
B. thailandensis E264 Pbur was precultured in LB medium supplemented with tetracycline (45 μg ml−1) for 16 h at 30 °C. The cultured cells were inoculated in LB medium with tetracycline (45 μg ml−1) and grown at 30 °C until an OD600 of 0.4–0.6 was reached. Subsequently, the culture broth was centrifuged and the supernatant was removed. Precipitated cells were resuspended in sucrose solution (300 mM) and centrifuged. After repeating this washing step three times, the washed cells were resuspended in 300 mM sucrose and subjected to electroporation (200 kV, Eporator, Eppendorf) with pGEM-burG::Kan (1–10 µg). The transformed cells were then cultured in LB medium (1 ml) for 4 h at 30 °C with shaking and subsequently plated on LB-agar plates supplemented with tetracycline (45 µg ml−1) and kanamycin (150 μg ml−1). After 3 days, positive colonies were observed and confirmed by colony PCR with the primer pair Isomerase fw2 and Isomerase rv2 using Taq Polymerase (New England Biolabs). See Supplementary Fig. 9 for PCR verification.
Preparation of His6-tagged BurC
The gene encoding BurC was amplified by PCR using the primer pair burC4-fw-NdeI and burC-rv-EcoRI. The obtained PCR product was subsequently ligated into the pCR2.1 vector. After sequence analysis (GATC biotech), the gene fragment was restricted with NdeI and EcoRI and then cloned into the NdeI and EcoRI sites of pET28a(+) vector, generating pET28a-burC. To prepare a seed culture, E. coli BL21 (DE3) pET28-burC was grown overnight in LB medium with 50 μg ml−1 of kanamycin. An aliquot (2.5 ml) of this seed culture was used to inoculate 250 ml LB medium supplemented with kanamycin (50 μg ml−1) in a 1 litre baffled Erlenmeyer flask. The culture was grown at 37 °C with orbital shaking until an OD600 of 0.5–0.7 was reached. Protein production was induced by addition of isopropylthiogalactoside (IPTG; 0.5 mM as a final concentration) followed by incubation at 15 °C for 18 h. Cells were harvested by centrifugation (9,000g, 4 °C, 10 min) and frozen at −20 °C until further use. For protein extraction, the cells were resuspended in 50 mM Tris–HCl (pH 8.0), 200 mM NaCl, 10% glycerol, 0.5 mM dithiothreitol, lysozyme (200 μg ml−1), and phenylmethylsulfonyl fluoride (100 μM), followed by incubation at 37 °C for 30 min. Then, the cells were lysed using a sonicator (Bandelin Sonopuls HD2200), and centrifuged (9,200g, 4 °C, 35 min) to obtain a clear lysate. The cleared lysate was loaded onto a Ni-IDA agarose (Biontex) column with manual handling. Equilibration and washing buffers were 50 mM Tris–HCl (pH 7.5), 500 mM NaCl, 10 mM imidazole and 30 mM imidazole, respectively; at least 10 column volumes were used for each step. The elution was carried out using a stepwise increase to 500 mM imidazole in the same buffer. The obtained fractions were analysed for target protein by SDS–polyacrylamide gel electrophoresis (SDS–PAGE; molecular weight of His6-BurC, 39.9 kDa). Protein containing fractions were dialysed against 20 mM sodium phosphate buffer (pH 7.2) and 200 mM NaCl, and concentrated using an Amicon Ultra-30K concentrator (Merck Millipore). See Supplementary Fig. 10 for SDS–PAGE analysis.
Preparation of His6-tagged BurG
The gene encoding BurG was amplified by PCR using the primer pair burG-fw-NheI and burG-rv-HindIII. The obtained amplicon was ligated into the pJET1.2 vector and verified by sequence analysis (GATC biotech). Subsequently, the gene fragment was restricted with NheI and HindIII and then cloned into the NheI- and HindIII-restricted pET28a(+) vector, generating pET28a-burG. To prepare a seed culture, E. coli BL21 (DE3) pET28a-burG was grown overnight in LB medium supplemented with 50 μg ml−1 of kanamycin. An aliquot (2.5 ml) of this seed culture was used to inoculate 250 ml LB medium supplemented with kanamycin (50 μg ml−1) in a 1 litre baffled Erlenmeyer flask. The culture was grown at 37 °C with orbital shaking until an OD600 of 0.5–0.7 was reached. Protein production was induced by addition of IPTG (0.5 mM as final concentration), followed by incubation at 15 °C for 18 h. Cells were harvested by centrifugation (9,000g, 4 °C, 10 min) and frozen at −20 °C until further use. For protein extraction, the cells were resuspended in 50 mM Tris–HCl buffer (pH 8.0) supplemented with 200 mM NaCl, 10% glycerol, 0.5 mM dithiothreitol, lysozyme (1 mg ml−1) and PMSF (100 μM), followed by incubation at 37 °C for 30 min. Then, the cells were lysed using a sonicator (Bandelin Sonopuls HD2200), and centrifuged (9,200g, 4 °C, 35 min) to obtain a clear lysate. The cleared lysate was applied to a Ni-IDA agarose (Biontex) column with manual handling. Equilibration and washing buffers were 50 mM Tris–HCl (pH 7.5), 500 mM NaCl, 10 mM imidazole or 30 mM imidazole, respectively; at least 10 column volumes were used for each step. The elution was carried out using a stepwise increase to 500 mM imidazole in the same buffer. The obtained fractions were analysed for target protein by SDS–PAGE (molecular weight of His6-BurG, 40.6 kDa). Protein containing fractions were dialysed against 50 mM Tris–HCl (pH 7.5), 200 mM NaCl buffer and concentrated using an Amicon Ultra-30K concentrator (Merck Millipore). See Supplementary Fig. 10 for SDS–PAGE analysis.
Generation of pET28a-burG_E232Q
A codon-optimized, point-mutated synthetic burG E232Q gene fragment was obtained from Integrated DNA Technologies. This gene fragment was ligated into a pJET1.2 vector (Thermo Fisher Scientific) and introduced into E. coli XL1 Blue by electroporation. The obtained plasmid pJET-burG_E232Q was verified by sequencing (GeneWiz) followed by restriction with HindIII and NheI. This obtained gene fragment was cloned into an HindIII- and NheI-restricted pET28a vector, yielding pET28-burG_E232Q. (See Supplementary Information for sequence.)
Thermal stability of BurG and BurG E232Q
The thermal stability of BurG and BurG E232Q protein preparations was determined by using a Tycho NT.6 (NanoTemper Technologies) monitoring the fluorescence of each protein preparation at 330 and 350 nm during a thermal ramp. Inflection temperatures (Ti BurG, 60.5 ± 0.1 °C; Ti BurG E232Q, 61.85 ± 0.05 °C) were obtained in technical duplicates at a concentration of 70 µM enzyme supplemented with 2 mM MgSO4 and 4 mM NAD+.
Dialysis of BurG preparations
A preparation of His6-tagged BurG was diluted with denaturing buffer (pH 8.0) to a final concentration of 8 M urea, 100 mM Tris–HCl and 200 mM NaCl. The resulting solution (1 ml) was pipetted into a dialysis bag (Spectra-Por, 3,500 dalton molecular weight cut-off, Spectrum Labs) and the bag was incubated in 3 litres of the same buffer as mentioned above at 4 °C for 24 h. Regular buffer exchange resulted in a final dilution factor of 6.4 × 1017. Next, the denaturing buffer was exchanged for refolding buffer (0.5 M l-arginine, 50 mM Tris–HCl and 100 mM NaCl, pH 8.0) followed by incubation for 12 h at 4 °C with regular buffer exchange to remove residual urea. This was followed by the removal of l-arginine through a further buffer exchange step to reaction buffer (50 mM Tris–HCl and 100 mM NaCl, pH 8.0) and incubation for 5 h at 4 °C with regular buffer exchange. Complete removal of NAD+ was validated by analysis of the refolded protein preparation via LC-HRMS. The resulting NAD+-free solution was subjected to further gonydiol transformation assays.
Substrate saturation kinetics (spectrophotometry)
The reduction activity of BurG (E232Q) was measured with hydroxypyruvate and NADH/NADPH as substrates in varying concentrations in semi-micro disposable ultraviolet cuvettes (Brand) with a final reaction volume of 0.9 ml. Purified tag-free BurG (E232Q) as prepared for protein crystallization (1 µM final concentration) was added to Tris–HCl buffer (100 mM, pH 7.5) containing MgSO4 (10 mM) and NADH/NADPH and the resulting mixture incubated at 30 °C for 5 min in a UV-1800 spectrophotometer (Shimadzu). Subsequently, hydroxypyruvate was added, and the absorption at 340 nm was monitored spectrophotometrically during the incubation at 30 °C. Each substrate concentration was measured in duplicate with enzyme obtained from independent preparations. No decrease in signal was observed when NADPH was used instead of NADH under similar conditions. The molarity of NADH at each time point measured in 1 s intervals was calculated by using a calibration curve obtained on the same instrument. Michaelis–Menten parameters were determined with one substrate at a fixed concentration (2 mM hydroxypyruvate or 0.1 mM NADH) while varying the second substrate at concentrations ranging from 0.020 to 1.5 mM for hydroxypyruvate (0.020–9 mM in the case of BurG E232Q) and from 6.68 to 75 µM for NADH. Notably, substrate inhibition occurred when concentrations higher than 2.25 mM hydroxypyruvate were used with wild-type BurG. Initial velocities were determined from the slope of the NADH degradation divided by the enzyme concentration (v0/[E0]) and plotted against the respective substrate concentration. The obtained curves were fitted to the Michaelis–Menten equation by nonlinear regression using GraphPad Prism v.8.2.1. Kinetic parameters were determined for each enzyme batch separately, averaged and are given with their geometric mean and interval. See Supplementary Figs. 6 and 7 for obtained Michaelis–Menten plots and parameters. Concentrations of used protein solutions were determined on a NanoDrop One (Thermo Fisher Scientific) with a calculated extinction coefficient of 24,870 M−1 cm−1 for BurG in technical triplicates.
Substrate saturation kinetics (LC-HRMS)
The kinetic parameters of the formation of 6 from 5 as catalysed by BurG were determined with the same Orbitrap LC-HRMS system as described below. Enzyme reactions consisted of Tris–HCl buffer (100 mM, pH 7.5), MgSO4 (10 mM), NAD+ (1 mM) and purified tag-free BurG as prepared for protein crystallization (1.5 µM final concentration), and were incubated in 1.6 ml glass vials with a glass insert at 30 °C in the autosampler of the ultra-high-performance liquid chromatography (UHPLC) system. To start the enzymatic reaction, synthetic rac-anti-5 as TFA salt was added in varying concentrations (final concentration, 91.4–1041.7 µM; final reaction volume, 48 µl) and the resulting mixture was promptly and repeatedly (six time points per concentration) injected onto a Nucleoshell Bluebird 2.7, C18 column (50 × 2 mm, Macherey-Nagel) run under isocratic conditions (solvent, H2O + 0.1% HCOOH; run time, 1 min; column oven temperature, 40 °C; post-column cooler, 25 °C; flow rate, 0.45 ml min−1; injection volume, 2 μl) for mass spectrometric analysis. The mass spectrometer was operated in positive selected ion monitoring mode with a scan range of 106.5–156.6 m/z and a resolution of 7,500. TFA (112.98559 m/z) was used as lock mass. The m/z of 6 (131.0350 ± 10 ppm) was monitored via the extracted ion chromatogram, the resulting chromatograms smoothed (gaussian, nine points) and the area under the curve determined via the Genesis Peak Detection algorithm in XCalibur Qual Browser v.4.3.73.11 software (Thermo Fisher Scientific). A calibration curve with 6 injected in varying concentrations (0.0023–0.133 mM) dissolved in a matrix-matched solution (100 mM Tris–HCl buffer pH 7.5, 10 mM MgSO4, 1 mM NAD+) was recorded on the same system in technical duplicates (standard weighed in independently) and used to convert the determined peak areas to concentrations. Initial velocities were determined from the slope of the formation of 6 divided by the enzyme concentration (v0/[E0]) and plotted against the respective substrate concentration. The obtained curves were fitted to the Michaelis–Menten equation by nonlinear regression using GraphPad Prism v.8.2.1. Kinetic parameters were determined for each enzyme batch separately, averaged and are given with their geometric mean and interval. To represent the kinetic parameters for the eutomer ((2R,3R)-5), the averaged KM was divided by two. Concentrations of BurG preparations were determined on a NanoDrop One (Thermo Fisher Scientific) as mentioned above.
Metabolite extraction for comparative LC-HRMS analysis
B. thailandensis Pbur and B. thailandensis Pbur burG::Kan were independently grown in 300 ml baffled Erlenmeyer flasks filled with 100 ml of MM9 medium that had been supplemented with either tetracycline (45 μg ml−1) or tetracycline (45 μg ml−1) plus kanamycin (150 μg ml−1) at 30 °C with orbital shaking for 24 h. Subsequently, the cells were harvested from a 50 ml aliquot by centrifugation (8,000g, for 15 min). The resulting cell pellet was resuspended in 25 ml methanol, sonicated and subsequently incubated for 60 min at room temperature. The centrifugation and extraction steps were repeated a second time and both obtained fractions were combined and concentrated in vacuo to yield crude extracts. These extracts were dissolved in 4 ml of methanol, and 100 µl of the resulting solution was then diluted with 200 µl methanol and filtered through a PTFE syringe filter. Subsequently the extracts were subjected to LC-HRMS analysis for a metabolomics analysis using the software Compound Discoverer 2.1 SP1 (Thermo Fisher Scientific). Both genotypes were analysed using a metabolomics workflow with and without a pattern scoring node to filter for sulfur-containing metabolites. The obtained metabolic profiles were compared using differential analysis.
Chemical complementation of B. thailandensis Pbur burG::Kan
Trigonic acid 6 (400 µM final concentration) was added to MM9 medium (75 ml in a 300 ml baffled Erlenmeyer flask) supplemented with tetracycline (45 μg ml−1) and kanamycin (150 μg ml−1) and inoculated with a preculture of B. thailandensis Pbur burG::Kan to a final OD600 of 0.1. Controls consisted of B. thailandensis Pbur (omitting the kanamycin) and B. thailandensis Pbur burG::Kan without addition of 6 prepared in the same way. All cultures were incubated at 30 °C with orbital shaking for 24 h. The obtained cultures were extracted with EtOAc three times, and the organic phase dried over Na2SO4 and concentrated under reduced pressure. The obtained crude extracts were redissolved in methanol, filtered through a PTFE syringe filter and subjected to LC-HRMS. Similar results were obtained on independent days with independent starting cultures (biological triplicates in total).
In vitro hydroxylation of gonyol
Purified His6-BurC (400 nM final concentration) was added to Tris–HCl buffer (50 mM, pH 7.5) containing ascorbate (0.1 mM), α-ketoglutarate (1 mM) and FeSO4 (0.1 mM). The reaction was started by addition of R- or S-gonyol (4; each as TFA salt; 150 µM final concentration). As a control, purified His6-BurC was heat-inactivated at 80 °C for 25 min and used in the same way. The resulting mixtures were incubated at 30 °C and aliquots taken at various time points (20, 50, 180, 210 and 360 min). Each aliquot was diluted with the same volume of methanol (typically 12.5 µl), filtered through a PTFE syringe filter and directly subjected to LC-HRMS analysis (Extended Data Fig. 2). Similar results were obtained on independent days with independent protein preparations (biological triplicates in total).
In vitro cyclopropanol synthesis
Purified His6-BurG or His6-BurG E232Q (5 µM final concentration) were added to Tris–HCl buffer (100 mM, pH 7.5) containing MgSO4 (2 mM) and NAD+ (1 mM) and the resulting mixture incubated at 30 °C for 5 min. Subsequently anti-, syn- or natural gonydiol (5; each as TFA salt; 1.3 mM final concentration) were added followed by incubation at 30 °C with aliquots taken at various time points (typically after 60 min). Each aliquot was diluted with the same volume of methanol, then filtered through a PTFE syringe filter and directly subjected to LC-HRMS analysis.
Mg2+ can be substituted with Co2+ (in the form of CoSO4) to achieve formation of trigonic acid. However, Mg2+ cannot be substituted with Mn2+ or Zn2+. Sole addition of Mg2+ suffices (when no dialysis under denaturing conditions is performed during enzyme purification) to observe enzymatic transformations and no addition of NAD+ is necessary. However, NAD+ was always detected via LC-HRMS in BurG preparations unless dialysis under denaturing conditions followed by refolding of the protein were performed (see above). Dialysed and thus NAD+-free BurG was used in gonydiol transformation assays as outlined above with and without addition of NAD+ or NADH. Assays performed with tag-free BurG/BurG E232Q as prepared for protein crystallography (Protein crystallography) yielded similar results as observed for His6-tagged BurG. Similar results were obtained on independent days with independent protein preparations (assay repeated more than three times in biological replicates).
Headspace SPME–GC–MS detection of DMS
Purified tag-free BurG as prepared for protein crystallization (5 µM final concentration) was added to Tris–HCl buffer (100 mM, pH 7.5) containing MgSO4 (2 mM) and NAD+ (1 mM). Subsequently synthetic rac-anti-gonydiol 5 (as TFA salt, 2 mM final concentration) was added to give a final volume of 200 µl. The resulting reaction mixture was pipetted into a 1.5 ml Eppendorf tube without lid which was placed inside a 20 ml headspace vial. The vial was sealed and incubated at 30 °C for 80 min. A control reaction was carried out in the same way as detailed above with enzyme preparation that had been denatured at 80 °C for 20 min. For headspace SPME a carbon wide range/polydimethylsiloxane fibre 95 µm thick and 10 mm long (Thermo Fisher Scientific) was used. The fibre was incubated at 260 °C for 4 min prior to headspace extraction. The headspace vial was incubated at 70 °C for 5 min followed by headspace extraction with the fibre for 5 min at 70 °C. For analysis, a TRACE 1310 gas chromatograph (Thermo Fisher Scientific) fitted with a BPX5 capillary column, 30 m × 0.25 mm × 0.25 µm (Trajan) coupled to a TSQ 900 mass spectrometer (Thermo Fisher Scientific) was used. The SSL injector was equipped with a topaz split liner (1 mm × 6.3 mm × 78.5 mm, Restek) heated to 250 °C and used in splitless mode. Helium was used as carrier gas at a flow rate of 0.6 ml min−1. The GC oven was set to a temperature of 30 °C with a hold time for 4 min followed by heating to 200 °C for 3.4 min and a subsequent hold time at 200 °C for 2.6 min. The source and transfer line temperatures of the mass spectrometer were 230 and 250 °C, respectively. The scan range of the mass spectrometer was set to 10–300 m/z. DMS was detected via the extracted ion chromatogram at an m/z of 62, and its identity validated through a database search (NIST 2.4) of the obtained electron ionization spectrum and through its identical retention time when compared with a commercial standard.
Isolation of gonydiol 5
Cells obtained from a 2 litre culture of B. thailandensis Pbur burG::Kan were extracted with methanol (2 × 200 ml) and the solvent subsequently removed under reduced pressure. The obtained crude extract was then redissolved in H2O (300 ml) to form a turbid solution which was pelleted by centrifugation at 6,000g for 45 min to yield a clear yellow solution. This solution was then loaded onto 100 g of the cationic exchange resin Amberlite IR-120 in the H+ state. After washing with 300 ml of H2O, bound cations were eluted with 1 litre of HCl (1 M) and the solvent removed through lyophilization. The concentrated eluate was subsequently redissolved in water and further purified via repeated rounds of preparative HPLC using a Phenomenex Synergi Fusion-RP C18 column (80-4, 250 × 21.2 mm) with H2O + 0.1% TFA as mobile phase at a flow rate of 20 ml min−1. Fractions containing the target molecule (as judged by NMR and LC-HRMS analysis) were pooled, concentrated in vacuo and further purified via repeated rounds of preparative HPLC with a SeQuant ZIC-HILIC column (200-5, 250 × 10 mm, Merck) (solvent A, 98% H2O, 2% CH3CN + 0.1% HCOOH, pH 2.7; solvent B, 10% H2O, 90% CH3CN, 5 mM NH4OAc; isocratic conditions: 71% B; flow rate, 5 ml min−1). Fractions containing the target molecule were pooled, concentrated under reduced pressure and analysed via 1H NMR spectroscopy. We noted a distinct difference in the 1H NMR spectra when fractions that were obtained with TFA in the mobile phase were compared to fractions that were obtained with NH4OAc/HCOOH as additive. While the TFA salt of 5 showed two signals resonating at 4.19 ppm (1H) and 4.01 ppm (1H), respectively, both signals merged to one signal resonating at 3.91 ppm (2H) when no TFA was present. As it seemed reasonable that a separation of these signals would aid structure elucidation, we converted the obtained substance to its TFA salt by repeatedly (3×) redissolving it in 1 ml of 1% TFA and subsequent removal of the solvent in vacuo. Through this we obtained 2.3 mg of the TFA salt of 5 as colourless oil (1.15 mg l−1). [α]D20 = − 3.5 (c = 0.36, MeOH); 1H NMR (600 MHz, CD3OD): see Supplementary Table 1; 13C NMR (151 MHz, CD3OD): see Supplementary Table 1; HRMS: calculated for C7H15O4S ([M + H]+), 195.0686; found, 195.0688.
LC-HRMS
LC-HRMS measurements were carried out with an UltiMate 3000 UHPLC (Thermo Fisher Scientific) coupled to a Thermo Fisher Scientific QExactive HF-X Hybrid Quadrupole-Orbitrap equipped with an electrospray ion source.
For general analysis, bacterial extracts were analysed by using a Kinetex 100-1.7 C18 column (50 × 2.1 mm, Phenomenex) and an elution gradient (solvent A, H2O + 0.1% HCOOH; solvent B, CH3CN; 5% B to 100% B in 4.5 min, 100% B for 2 min, 100% B to 5% B in 0.001 min, 5% B for 1.5 min; flow rate, 0.7 ml min−1, injection volume, 2 μl). For metabolomics analysis, extracts were analysed in positive-ion mode by using a Nucleoshell Bluebird 2.7 C18 column (50 × 2 mm, Macherey-Nagel) and an elution gradient (solvent A, H2O + 0.1% HCOOH; solvent B, CH3CN; 0% B for 1 min, 0% B to 98% B in 6 min, 98% B for 3 min, 98% B to 0% B in 0.01 min, 0% B for 2.99 min; flow rate, 0.4 ml min−1, injection volume, 2 μl).
Enzyme assays were analysed on a Nucleodur HTec 100-2 C18 column (100 × 2 mm, Macherey-Nagel) with an elution gradient (solvent A, H2O + 0.1% HCOOH; solvent B, CH3CN; 5 %B for 0.5 min, from 5% B to 100% B in 6.5 min, 100% B for 3 min, 100% B to 5% B in 0.01 min, 5% B for 2.99 min; flow rate, 0.4 ml min−1, injection volume, 2 μl).
Detection of NAD+ (Supplementary Fig. 5) was carried out by using a Luna Omega PS 100-1.6 C18 column (100 × 2.1 mm, Phenomenex) with an elution gradient (solvent A, H2O + 0.1% HCOOH; solvent B, CH3CN; 0% B for 0.5 min, from 0% B to 99% B in 6.5 min, 99% B for 3 min, 100% B to 0% B in 0.01 min, 5% B for 2.9 min; flow rate, 0.4 ml min−1, injection volume, 2 μl).
Chiral LC-HRMS analysis was carried out by using an Astec CHIROBIOTIC R column (100 × 4.6 mm, Supelco) at a constant temperature of 20 °C (solvent A, 33.3 mM NH4OAc; solvent B, MeOH; isocratic conditions: 75% B; flow rate, 0.5 ml min−1).
NMR
NMR spectra were measured on Bruker Avance II 300 MHz, Bruker Avance III 500 MHz and Bruker Avance III 600 MHz spectrometers (600 MHz with cryo probe) in CD3OD and CD3CN. Spectra were referenced relative to the residual solvent peak (CD3OD: δH = 3.30, δC = 49.0 ppm; CD3CN: δH = 1.94, δC = 1.3; 118.3 ppm).
Phylogenetic analysis
Sequences for KARI family proteins were obtained from the NCBI server (https://www.ncbi.nlm.nih.gov/) by Protein BLAST using the databases of non-redundant protein sequences, UniProtKB/Swiss-Prot and PDB. The respective amino acid sequences were aligned using the MAFFT server (https://mafft.cbrc.jp/alignment/server/)40 with MAFFT v.7 and default settings. An unrooted fast maximum-likelihood-based tree was generated using IQ-TREE (https://www.hiv.lanl.gov/content/sequence/IQTREE/iqtree.html)41 with automatic model selection mode (ModelFinder)42, where the LG+G4 was selected. Phylogenetic bootstrap analysis was performed by ultrafast approximate bootstrap with 1,000 bootstrap replicates43. The obtained tree was displayed using MEGA744. See Supplementary Fig. 4 and Table 6 for detailed information.
Reagents, chemical synthesis and compound characterisation
Unless stated otherwise, all reagents were purchased from Sigma-Aldrich or TCI. The procedures for the preparation of synthetic compounds used in this study, and the spectral data, can be found in the Supplementary Information. For preparation of dimethyldioxirane, see ref. 45.
Statistics and reproducibility
Comparative LC-HRMS analysis: biological triplicates (Fig. 2a and Supplementary Fig. 1).
Chemical complementation of gene inactivation mutant with trigonic acid and gene inactivation mutant analysis: biological triplicates (Fig. 2b).
In vitro hydroxylation of gonyol: technical triplicates (Fig. 2d and Extended Data Fig. 2).
Headspace SPME–GC–MS detection of DMS: performed once (Fig. 2e).
In vitro cyclopropanol synthesis: assay repeated more than three times with independent protein preparations (Fig. 2h).
Chiral LC-HRMS of cyclopropanol formation to determine stereochemistry of natural trigonic acid: technical triplicates (Extended Data Fig. 4).
Determination of enantiomeric excess of synthetic R-trigonic acid: technical triplicates (Extended Data Fig. 4).
NAD+ detection in BurG preparations: biological triplicates (Supplementary Fig. 5).
Substrate saturation kinetics; spectrophotometry (8 as substrate): BurG: biological duplicates (Fig. 3c and Supplementary Fig. 6); BurG E232Q: technical duplicates (Supplementary Fig. 7); LC-MS (5 as substrate): biological duplicates (Fig. 2i).
Protein concentration determination: technical triplicates for each preparation.
In vitro cyclopropanol synthesis with BurG E232Q: biological duplicates (Fig. 3h)
Thermal stability of proteins: technical duplicates.
Protein crystallography
Culture conditions
For protein production to be used in protein crystallization experiments, E. coli transformed with a plasmid containing BurG or BurG E232Q was grown in Fernbach shaking flasks at 37 °C containing 3 litres of lysogeny broth supplemented with kanamycin (50 mg l−1). At an OD600 of 0.6, IPTG was added (final concentration 1 mM), and incubation was continued overnight at 20 °C. Subsequently, the cells were harvested by centrifugation (8,000g, 20 min), washed with 0.9% (w/v) NaCl and stored at −20 °C.
Protein purification for crystallization
Frozen bacterial cell mass (25 g) obtained as detailed above (Culture conditions) was thawed in 50 ml of 100 mM Tris–HCl, pH 7.5, containing 500 mM NaCl and 20 mM imidazole/HCl (buffer A). Subsequently, the cells were disrupted by sonification (Branson Digital Sonifier 250, G. Heinemann) and the resulting suspension centrifuged at 40,000g for 20 min at 4 °C in a Sigma 4K15 centrifuge. The supernatant was loaded onto a 5 ml HisTrap HP column (GE Healthcare), which had been equilibrated with buffer A (flow rate, 5 ml min−1) using an ÄKTA Pure system (GE Healthcare). Unbound or loosely associated proteins were removed by washing with buffer A. BurG protein was eluted by applying a 50 ml linear gradient from buffer A to buffer B (100 mM Tris–HCl, pH 7.5, 500 mM NaCl, 500 mM imidazole). Fractions were combined and 200 U of thrombin from bovine plasma (Serva) was added. The solution was dialysed overnight at 4 °C against 20 mM Tris–HCl, pH 7.5, containing 100 mM NaCl and again applied to the HisTrap HP column equilibrated with buffer A. The percolate was concentrated to 1 ml and the solution was applied to a HiLoad 16/60 Superdex 75 pg column (GE Healthcare; flow rate, 1.5 ml min−1). Single peak fractions were concentrated to 40 mg ml−1 using a 10K Amicon Ultra Centrifugal Filter Device (Millipore), and stored at 4 °C. Analytical size exclusion chromatography was carried out with a Superdex 75 Increase 10/300 GL column (GE Healthcare; flow rate, 1 ml min−1). See Supplementary Fig. 11 for SDS–PAGE analysis.
Figure illustration of structural images
The electron densities represent Fo–Fc maps (contoured to 3σ) with the respective ligands omitted for phasing. The following colour code was used: carbon atoms of subunit A (grey), subunit B (pink), E323Q-mutant (dark pink), NAD+ (gold), NADH (golden rod), 6 (brown) 7 (light blue), 8 (orchid), 11 (olive), 12 (cyan, carbon atom for the substituted sulfur atom in dark green), 13 (dark cyan, carbon atom for the substituted sulfur atom in dark green), 14 (light green), 15 (purple), DMS (brown). Nitrogen, oxygen and sulfur atoms are coloured in blue, red and yellow, respectively. The two magnesium atoms (named A and B) are shown as green spheres, water molecules are presented as red balls. Hydrogen bonds are drawn as black dots. Figures were created with the programs Bobscript46 and PyMOL v.2.3 (Schrödinger).
Protein crystallization
Crystallization experiments of BurG variants (17.5 mg ml−1) were performed using the sitting drop vapour diffusion method at 20 °C. For co-crystallization experiments, the appropriate ligand (100 mM stock solution in H2O) was added to BurG wild-type or mutated BurG to a final concentration of 2 mM. Crystallization droplets had a maximum volume of 0.4 µl with a ratio of either 1:1, 2:1 or 3:1 of protein and reservoir solution. BurG showed a strong crystallization preference for polyethylene glycol. Unfortunately, the majority of crystals had disordered layers. Therefore, suitable candidates with optimal lattice packing had to be identified by diffraction measurements. (See Supplementary Information for distinct crystallization parameters.) Crystals were cryoprotected by a 7:3 mixture of mother liquor and 100% (v/v) glycerol and subsequently vitrified in liquid nitrogen.
Protein structure determination
High-resolution datasets of BurG variants were recorded with synchrotron radiation of λ = 1.0 Å at the beamline X06SA, Swiss Light Source (SLS), Paul Scherrer Institute, Villigen, Switzerland. Reflection intensities were evaluated with the program package XDS and data reductions were carried out with XSCALE47 (Supplementary Table 2). The resolution limits were determined based on the following criteria: I/σ(I) > 2.0, Rmerge < 60% and redundancy >3.0. First, we solved the BurG (holo) structure by Patterson search calculations applying PHASER48 and coordinates of the keto acid reductoisomerase from Staphylococcus aureus (PDB 6AQJ)19. Using COOT49 in combination with REFMAC50, the BurG sequence was built in iterative rounds. The model was completed with the cofactors NAD+ and Mg2+. Water molecules were automatically placed with ARP/wARP solvent51. Restrained and TLS (Translation/Libration/Screw) REFMAC refinements yielded excellent Rwork and Rfree values as well as root-mean-square deviation values of bond lengths and angles. For all other datasets BurG (holo) served as starting model for initial phasing. Model building and structural refinement followed the same procedure (Supplementary Table 2). Interestingly, the high-resolution electron density map at 1.6 Å in PDB 7PCN depicts a mixture of 14 and NADH as well as 6, DMS and NAD+ at the active site. Note that 14 and NADH as well as 6, DMS and NAD+ were incorporated with an occupancy of 0.5 each. In the data set PDB 7PCO (BurG_E232Q mutant), enol-oxaloacetate 15 was co-purified from the E. coli lysate. These findings are in contrast to BurG (wild-type) and show that the E232Q BurG mutant has a high affinity for the metabolite. All crystal structures have been deposited in the RCSB Protein Data Bank. See Supplementary Table 2 for molprobity52 clash scores.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41557-022-01005-z.
Supplementary information
Synthetic procedures, distinct crystallization parameters, nucleotide sequence of synthetic burG E232Q, Supplementary Figs. 1–35, Tables 1–6 and references.
Source Data for the Supplementary Information
Acknowledgements
We thank H. Heinecke and A. Perner for NMR and LC-MS measurements, respectively. We thank A. König for support in the protein crystallization experiments. We thank G. Lackner for provision of material for chiral HPLC and F. Kloss for provision of a Tycho NT.6. We are grateful to J. Franke for helpful discussions. Financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Project-ID 239748522–SFB 1127, and Leibniz Award (to C.H.), DFG Project-ID 325871075–SFB 1309 (to M.G.) and the European Regional Development Fund (ERDF) (MassNat) are gratefully acknowledged. H.K. received fellowships from the Daimler and Benz Foundation and the Fonds der Chemischen Industrie (FCI).
Extended data
Source data
Source Data for Figure 2
Source Data for Figure 3
Source Data for Extended Data Figure 2
Source Data for Extended Data Figure 3
Source Data for Extended Data Figure 4
Author contributions
F.T.: conceptualization, formal analysis, investigation, project administration, visualization, writing—original draft, review and editing. K.I.: conceptualization, formal analysis, investigation. M.I-I.: formal analysis, investigation. H.K.: conceptualization, methodology, writing—review and editing; M.G.: conceptualization, formal analysis, investigation, resources, visualization, funding acquisition, writing—review and editing. C.H.: conceptualization, funding acquisition, resources, supervision, writing—review and editing.
Peer review
Peer review information
Nature Chemistry thanks Satish Nair, Kenichi Yokoyama and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
NMR raw files of natural gonydiol and MS raw files used for comparative metabolomics analysis (Fig. 2a and Supplementary Fig. 1) are deposited on Zenodo. (10.5281/zenodo.6554506) and are available without restrictions. All other data are available in the main text, the Supplementary Information, the respective source data files or via the RCSB PDB (7PCC, 7PCE, 7PCG, 7PCI, 7PCL, 7PCM, 7PCN, 7PCO, 7PCT). Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Michael Groll, Email: michael.groll@tum.de.
Christian Hertweck, Email: christian.hertweck@leibniz-hki.de.
Extended data
is available for this paper at 10.1038/s41557-022-01005-z.
Supplementary information
The online version contains supplementary material available at 10.1038/s41557-022-01005-z.
References
- 1.Galyov EE, Brett PJ, DeShazer D. Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu. Rev. Microbiol. 2010;64:495–517. doi: 10.1146/annurev.micro.112408.134030. [DOI] [PubMed] [Google Scholar]
- 2.Cruz-Migoni A, et al. A Burkholderia pseudomallei toxin inhibits helicase activity of translation factor eIF4A. Science. 2011;334:821–824. doi: 10.1126/science.1211915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franke J, Ishida K, Hertweck C. Evolution of siderophore pathways in human pathogenic bacteria. J. Am. Chem. Soc. 2014;136:5599–5602. doi: 10.1021/ja501597w. [DOI] [PubMed] [Google Scholar]
- 4.Franke J, Ishida K, Hertweck C. Plasticity of the malleobactin pathway and its impact on siderophore action in human pathogenic bacteria. Chem. Eur. J. 2015;21:8010–8014. doi: 10.1002/chem.201500757. [DOI] [PubMed] [Google Scholar]
- 5.Franke J, Ishida K, Ishida-Ito M, Hertweck C. Nitro versus hydroxamate in siderophores of pathogenic bacteria: effect of missing hydroxylamine protection in malleobactin biosynthesis. Angew. Chem. Int. Ed. 2013;52:8271–8275. doi: 10.1002/anie.201303196. [DOI] [PubMed] [Google Scholar]
- 6.Biggins JB, Kang HS, Ternei MA, DeShazer D, Brady SF. The chemical arsenal of Burkholderia pseudomallei is essential for pathogenicity. J. Am. Chem. Soc. 2014;136:9484–9490. doi: 10.1021/ja504617n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klaus JR, et al. Secondary metabolites from the Burkholderia pseudomallei complex: structure, ecology, and evolution. J. Ind. Microbiol. Biotechnol. 2020;47:877–887. doi: 10.1007/s10295-020-02317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biggins JB, Ternei MA, Brady SF. Malleilactone, a polyketide synthase-derived virulence factor encoded by the cryptic secondary metabolome of Burkholderia pseudomallei group pathogens. J. Am. Chem. Soc. 2012;134:13192–13195. doi: 10.1021/ja3052156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franke J, Ishida K, Hertweck C. Genomics-driven discovery of burkholderic acid, a noncanonical, cryptic polyketide from human pathogenic Burkholderia species. Angew. Chem. Int. Ed. 2012;51:11611–11615. doi: 10.1002/anie.201205566. [DOI] [PubMed] [Google Scholar]
- 10.Gutierrez MG, Yoder-Himes DR, Warawa JM. Comprehensive identification of virulence factors required for respiratory melioidosis using Tn-seq mutagenesis. Front. Cell. Infect. Microbiol. 2015;5:78. doi: 10.3389/fcimb.2015.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moule MG, et al. Characterization of new virulence factors involved in the intracellular growth and survival of Burkholderia pseudomallei. Infect. Immun. 2016;84:701–710. doi: 10.1128/IAI.01102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klaus JR, et al. Malleilactone is a Burkholderia pseudomallei virulence factor regulated by antibiotics and quorum sensing. J. Bacteriol. 2018;200:e00008–e00018. doi: 10.1128/JB.00008-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trottmann F, et al. Cyclopropanol warhead in malleicyprol confers virulence of human- and animal-pathogenic Burkholderia species. Angew. Chem. Int. Ed. 2019;58:14129–14133. doi: 10.1002/anie.201907324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicolaev A, Orellana A. Transition-metal-catalyzed C–C and C–X bond-forming reactions using cyclopropanols. Synthesis. 2016;48:1741–1768. [Google Scholar]
- 15.Rosa D, Nikolaev A, Nithiy N, Orellana A. Palladium-catalyzed cross-coupling reactions of cyclopropanols. Synlett. 2015;26:441–448. [Google Scholar]
- 16.Shunsuke C, Zhengyan C, Atta Atta EBS, Koichi N. Generation of β-keto radicals from cyclopropanols catalyzed by AgNO3. Chem. Lett. 2006;35:18–19. [Google Scholar]
- 17.Trottmann F, et al. Sulfonium acids loaded onto an unusual thiotemplate assembly line construct the cyclopropanol warhead of a Burkholderia virulence factor. Angew. Chem. Int. Ed. 2020;59:13511–13515. doi: 10.1002/anie.202003958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueoka R, Bortfeld-Miller M, Morinaka BI, Vorholt JA, Piel J. Toblerols: cyclopropanol-containing polyketide modulators of antibiosis in methylobacteria. Angew. Chem. Int. Ed. 2018;57:977–981. doi: 10.1002/anie.201709056. [DOI] [PubMed] [Google Scholar]
- 19.Patel KM, et al. Crystal structures of Staphylococcus aureus ketol-acid reductoisomerase in complex with two transition state analogues that have biocidal activity. Chem. Eur. J. 2017;23:18289–18295. doi: 10.1002/chem.201704481. [DOI] [PubMed] [Google Scholar]
- 20.Brown, S. P. et al. Compounds that inhibit MCL-1 protein. Patent WO2017/147410A1 (2017).
- 21.Kolb HC, VanNieuwenhze MS, Sharpless KB. Catalytic asymmetric dihydroxylation. Chem. Rev. 1994;94:2483–2547. [Google Scholar]
- 22.Gabler F, et al. Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinform. 2020;72:e108. doi: 10.1002/cpbi.108. [DOI] [PubMed] [Google Scholar]
- 23.Brinkmann-Chen S, et al. General approach to reversing ketol-acid reductoisomerase cofactor dependence from NADPH to NADH. Proc. Natl Acad. Sci. USA. 2013;110:10946–10951. doi: 10.1073/pnas.1306073110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brinkmann-Chen S, Cahn JKB, Arnold FH. Uncovering rare NADH-preferring ketol-acid reductoisomerases. Metab. Eng. 2014;26:17–22. doi: 10.1016/j.ymben.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 25.Stein AJ, Geiger JH. The crystal structure and mechanism of 1-l-myo-inositol-1-phosphate synthase. J. Biol. Chem. 2002;277:9484–9491. doi: 10.1074/jbc.M109371200. [DOI] [PubMed] [Google Scholar]
- 26.Thoden JB, Frey PA, Holden HM. Molecular structure of the NADH/UDP-glucose abortive complex of UDP-galactose 4-epimerase from Escherichia coli: implications for the catalytic mechanism. Biochemistry. 1996;35:5137–5144. doi: 10.1021/bi9601114. [DOI] [PubMed] [Google Scholar]
- 27.Yamauchi N, Kakinuma K. Enzymic carbocycle formation in microbial secondary metabolism. The mechanism of the 2-deoxy-scyllo-inosose synthase reaction as a crucial step in the 2-deoxystreptamine biosynthesis in Streptomyces fradiae. J. Org. Chem. 2002;60:5614–5619. [Google Scholar]
- 28.Holzer, H. & Holldorf, A. in Methods of Enzymatic Analysis (ed. Bergmeyer, H.-U.) 260–262 (Academic Press, 1965).
- 29.Tadrowski S, et al. Metal ions play an essential catalytic role in the mechanism of ketol-acid reductoisomerase. Chem. Eur. J. 2016;22:7427–7436. doi: 10.1002/chem.201600620. [DOI] [PubMed] [Google Scholar]
- 30.Gu L, et al. Metamorphic enzyme assembly in polyketide diversification. Nature. 2009;459:731–735. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin WB, Wu S, Jian XH, Yuan H, Tang GL. A radical S-adenosyl-l-methionine enzyme and a methyltransferase catalyze cyclopropane formation in natural product biosynthesis. Nat. Commun. 2018;9:2771. doi: 10.1038/s41467-018-05217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neumann CS, Walsh CT. Biosynthesis of (−)-(1S,2R)-allocoronamic acyl thioester by an Fe(II)-dependent halogenase and a cyclopropane-forming flavoprotein. J. Am. Chem. Soc. 2008;130:14022–14023. doi: 10.1021/ja8064667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramalingam K, Lee KM, Woodard RW, Bleecker AB, Kende H. Stereochemical course of the reaction catalyzed by the pyridoxal phosphate-dependent enzyme 1-aminocyclopropane-1-carboxylate synthase. Proc. Natl Acad. Sci. USA. 1985;82:7820–7824. doi: 10.1073/pnas.82.23.7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT. Cryptic chlorination by a non-haem iron enzyme during cyclopropyl amino acid biosynthesis. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 35.Zha L, et al. Colibactin assembly line enzymes use S-adenosylmethionine to build a cyclopropane ring. Nat. Chem. Biol. 2017;13:1063–1065. doi: 10.1038/nchembio.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vizcaino MI, Crawford JM. The colibactin warhead crosslinks DNA. Nat. Chem. 2015;7:411–417. doi: 10.1038/nchem.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson MR, et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science. 2019;363:eaar7785. doi: 10.1126/science.aar7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thibodeaux CJ, Chang WC, Liu HW. Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis. Chem. Rev. 2012;112:1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coelho PS, Brustad EM, Kannan A, Arnold FH. Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- 40.Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017;20:1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013;30:1188–1195. doi: 10.1093/molbev/mst024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taber DF, DeMatteo PW, Hassan RA. Simplified preparation of dimethyldioxirane (DMDO) Org. Synth. 2013;90:350–357. [Google Scholar]
- 46.Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 47.Kabsch W. XDS. Acta Crystallogr. D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCoy AJ, et al. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 50.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthetic procedures, distinct crystallization parameters, nucleotide sequence of synthetic burG E232Q, Supplementary Figs. 1–35, Tables 1–6 and references.
Source Data for the Supplementary Information
Data Availability Statement
NMR raw files of natural gonydiol and MS raw files used for comparative metabolomics analysis (Fig. 2a and Supplementary Fig. 1) are deposited on Zenodo. (10.5281/zenodo.6554506) and are available without restrictions. All other data are available in the main text, the Supplementary Information, the respective source data files or via the RCSB PDB (7PCC, 7PCE, 7PCG, 7PCI, 7PCL, 7PCM, 7PCN, 7PCO, 7PCT). Source data are provided with this paper.