

Abstract
Background
Lung adenocarcinoma (LUAD) is the major cause of cancer mortality. Traditional prognostic factors have limited importance after including other parameters. Thus, developing a more credible prognostic model combined with genes and clinical parameters is necessary.
Methods
The messenger RNA (mRNA) expression and clinical information from The Cancer Genome Atlas (TCGA)-LUAD datasets and microarray data from three Gene Expression Omnibus (GEO) databases were obtained. We identified differentially-expressed genes (DEGs) between lung tumor and normal tissues through integrated analysis of the three GEO datasets. Univariate and multivariate Cox regression analyses were conducted to select survival-associated DEGs and to establish a prognostic gene signature which was associated with overall survival (OS). The expression of gene proteins was assessed in 180 LUAD tissue microarrays (TMAs) by immunohistochemistry (IHC). We verified its predictive performance with a Kaplan-Meier (KM) curve, receiver operating characteristic (ROC) curve, and Harrell’s concordance index (C-index) and validated it in external GEO databases. Multivariate Cox regression analysis was performed to identify the significant prognostic indicators in LUAD. Furthermore, we established a prognostic nomogram based on TCGA-LUAD dataset.
Results
A three-gene signature was constructed to predict the OS of LUAD patients. The KM analysis, ROC curve, and C-index present a good predictive ability of the gene signature in TCGA dataset [P<0.0001; C-index 0.6375; 95% confidence interval (CI): 0.5632–0.7118; area under the ROC curve (AUC) 0.674] and the external GEO datasets (P=0.05, 0.004, and 0.04, respectively). Univariate and multivariate Cox regression analyses also verified that LUAD patients with low-risk scores had a decreased risk of death compared to those with a high-risk score in TCGA database [hazard ratio (HR) =0.3898; 95% CI: 0.1938–0.7842; P<0.05]. Finally, we constructed a nomogram integrating the gene signature and clinicopathological parameters (P<0.0001; C-index 0.762; 95% CI: 0.714–0.845; AUC 0.8136). Compared with conventional staging, a nomogram can effectively improve prognosis prediction.
Conclusions
The nomogram is closely associated to the OS of LUAD patients. This consequence may be beneficial to individualized treatment and clinical decision-making.
Keywords: Gene Expression Omnibus (GEO), The Cancer Genome Atlas (TCGA), overall survival (OS), lung adenocarcinoma (LUAD)
Introduction
Lung cancer is the most common cause of cancer-related mortality worldwide. In 2021, it was estimated that there are more than 230,000 new cases of lung cancer and 130,000 deaths each year among the newly diagnosed cancers in the United States (1). Non-small cell lung cancer (NSCLC) is the major form of lung cancer, and lung adenocarcinoma (LUAD) is the most common histologic subtype of NSCLC (2,3). The traditional prognosis and initial treatment of NSCLC depend on clinical parameters, tumor stage, histopathology and tumor markers (4). For patients with early-stage diseases, surgical resection is an effective solution. Patients with locally-advanced disease are candidate to radiotherapy associated with systemic therapy, while metastatic disease is treated with systemic treatments. Traditional prognostic factors have limited importance after including other parameters such as Karnofsky-performance status, dose of systemic therapy or radiotherapy, and weight-loss evolved in multivariate analysis (5). Fortunately, our understanding of the molecular pathogenesis of NSCLC has progressed rapidly. For example, targeted therapy is more effective than standard chemotherapy for mutations of epidermal growth factor receptor (EGFR), rearrangements in anaplastic lymphoma kinase (ALK), oncogene c-ROS1 (ROS1), and other molecular alterations.
Additionally, gene chips and public genomic datasets allow researchers to detect and analyze differentially-expressed genes (DEGs) in various cancer types, which may help to identify tumor-associated genes as well as expression signatures with prognostic impact. Determining the gene characteristics of NSCLC tumor tissue may lead us to uncover crucial biological process during LUAD progression or recurrence, which assist in evaluating the prognosis and the possible therapeutic effects (6). An increasing number of literatures have shown that risk models based on multiple types of genes have great potential to predict LUAD prognosis (7-10). Therefore, we sought to establish a credible prognostic model combined with multiple genes via bioinformatics analysis and clinical parameters.
In this study, we integrated three LUAD datasets from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) to identify reliable DEGs in LUAD. Furthermore, we performed univariate Cox regression analysis and multivariate Cox proportional hazards regression analysis to identify survival-related DEGs. We constructed a risk score model using gene expression from TCGA-LUAD dataset. Subsequently, we used survival analyses, such as Kaplan-Meier (KM) analysis and a receiver operating characteristic (ROC) curve, to verify the model’s predictive performance. We also studied the molecular mechanisms underlying the gene prediction models. Finally, we established a nomogram combining the novel gene signature with clinicopathological parameters to predict the OS of LUAD patients. The detailed workflow of this study is provided in Figure 1. We present the following article in accordance with the TRIPOD reporting checklist (available at https://tlcr.amegroups.com/article/view/10.21037/tlcr-22-444/rc).
Figure 1.
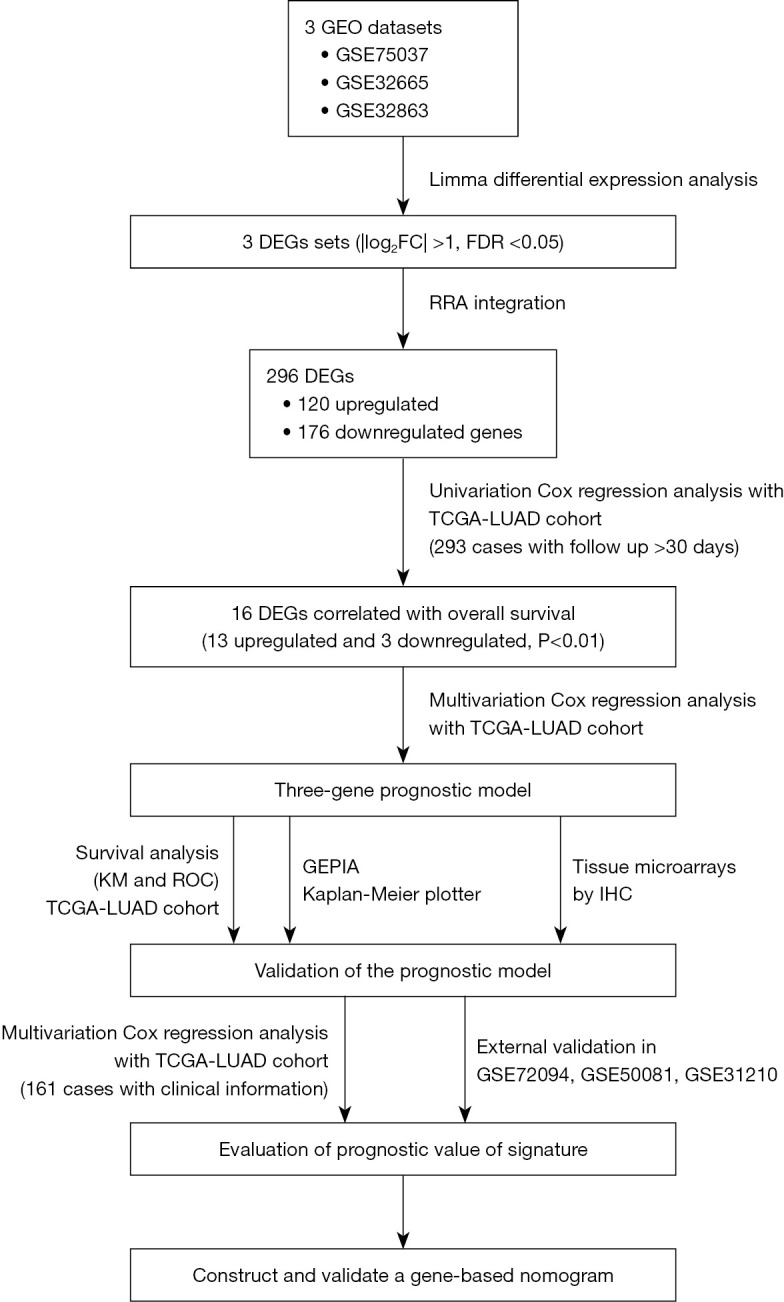
Flowchart displaying the procedure of establishing the gene prognostic model and nomogram of LUAD applied in our study. GEO, Gene Expression Omnibus; DEGs, differentially-expressed genes; FC, fold change; FDR, false discovery rate; TCGA, The Cancer Genome Atlas; LUAD, lung adenocarcinoma; KM, Kaplan-Meier; ROC, receiver operating characteristic; GEPIA, Gene Expression Profiling Interactive Analysis; IHC, immunohistochemistry.
Methods
Gene expression and clinical data
The LUAD messenger RNA (mRNA) expression data and related clinical information were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). We used the keywords “Lung cancer”, “Lung adenocarcinoma”, and “LUAD” for retrieval. Studies based on “Homo sapiens” described as “Expression profiling by array” were included for the next step of screening. “Cell lines” and “xenografts” were excluded from the research. Three gene expression microarray datasets (GSE32863, GSE75037, and GSE32665) were selected and downloaded to screen DEGs. These datasets met the following requirements: (I) contained human lung tissue samples; (II) contained tumor and normal lung tissue samples; and (III) contained at least 100 samples. Moreover, we selected three datasets (GSE72094, GSE50081, and GSE31210) for subsequent validation of the prognostic gene signature, along with the follow-up information. The probes were matched to gene symbols using the manufacturer-provided annotation file. We used the median ranking value to calculate the expression value if multiple probes matched a single gene. The expression data were normalized and log2-transformed for further analysis.
Harmonized RNA sequencing data (HTSeq-counts) and associated clinical information for LUAD were downloaded from TCGA (https://portal.gdc.cancer.gov/; up to November 20, 2020). A total of 551 samples were retrieved. After preliminary screening, 411 samples were selected, including 368 tumor samples and 43 normal tissue samples (available online: https://cdn.amegroups.cn/static/public/tlcr-22-444-1.xlsx). Forty-three cases of normal samples, 74 cases with a follow-up time of ≤30 days, and one sample with metastasis were removed. Thus, 293 cases with relevant tumor tissues and clinical information were ultimately included in the study. The gene expression data of TCGA-LUAD dataset were normalized by variance stabilizing transformation (VST) with R package “Deseq2” (11) for further analysis. The research was conducted in accordance with the Declaration of Helsinki (as revised in 2013).
Analysis of DEGs and integrated microarray dataset analysis
We conducted differential gene expression analysis between tumor and normal tissues based on the GEO dataset via the Limma package in R software (12). Significantly up- and downregulated genes were defined as those with |log2fold change (FC)| >1, P<0.05, and a false discovery rate (FDR) <0.05. We drew a volcano plot to display the FC and P values of DEGs between the two comparison groups. The DEGs identified from three GEO datasets were analyzed synthetically via the robust rank aggregation (RRA) method R package “RobustRankAggreg”. P<0.05 was considered statistically significant.
Survival analysis and establishment of the prognostic gene signature
We normalized the gene expression data of TCGA-LUAD dataset via VST with R package “Deseq2”. We then analyzed the association between expression levels of the DEGs identified from the GEO datasets and OS of LUAD patients in TCGA-LUAD dataset using a univariate Cox regression model. DEGs with P<0.01 were considered significant with OS and included for the multivariable Cox regression analysis. Next, we constructed a prognostic gene signature for LUAD patients based on a linear combination of the multivariable Cox regression coefficients (β) multiplied by the mRNA expression levels of candidate prognostic genes. The risk scoring formula was defined as follows (13):
| [1] |
Patients in TCGA-LUAD were then divided into low- and high-risk subgroups according to the gene signature’s optimal cut-off value, as determined by X-tile software (14). KM survival analysis, the area under the ROC curve (AUC), and the concordance index (C-index) were used to evaluate the ability of the prognostic gene signature via ‘survival’, ‘timeROC’ package in R software. We also compared the prognostic value of gene signature with the previously defined risk models proposed by Zhang et al. (15-17).
Validation of prognostic genes in signature
We applied Gene Expression Profiling Interactive Analysis (GEPIA) (http://gepia.cancer-pku.cn/), an online web server based on TCGA and the Genotype-Tissue Expression (GTEX) databases (18), to display the mRNA expression levels of signature genes in LUAD and non-tumor lung tissues. The KM plotter (https://kmplot.com/analysis/index.php?p=service), an online tool commonly used to assess the effects of genes on survival based on The European Genome-phenome Archive (EGA), TCGA, and GEO (Affymetrix microarrays only) databases, was used to apply survival analysis for the signature genes.
Tissue array specimens and immunohistochemistry (IHC)
Lung cancer tissue arrays (HLugA180Su07) were purchased from Shanghai Outdo Biotech Co., Ltd. (Shanghai, China) for IHC analysis, including 98 LUAD tissues and 82 para-cancerous normal tissues. The primary antibodies used included antibodies against PLEK2 (ab121131, Abcam, UK), COL1A1 (ab34710, Abcam), and GPX3 (ab104448, Abcam). We deparaffinized the tissue arrays and retrieved antigens. Next, we incubated tissues with the primary antibody at 4 ℃ overnight, washed them with phosphate-buffered saline (PBS), and then incubated them with a biotin-conjugated secondary antibody. After washing, we incubated the sections with horseradish peroxidase (HRP) complex and visualized them using diaminobenzidine (DAB) (Maixin Biotech, Fuzhou, China). The IHC score was independently related to staining intensity and the percentage of positive cells. The staining intensity was divided into four levels: 0 (no staining), 1 (weak staining), 2 (moderate staining), and 3 (strong staining). The proportion of stained positive cells was defined as: 1 (0–25% positive cells), 2 (26–50% positive cells), 3 (51–75% positive cells), and 4 (76–100% positive cells). The staining intensity and the percentage scores were multiplied to obtain the total score. All the IHC results were reviewed by pathologist in First Affiliated Hospital, School of Medicine, Zhejiang University.
Evaluation of the prognostic value of the signature
To evaluate the significant prognostic values of the gene signature, we performed univariate and multivariate Cox regression analyses in TCGA-LUAD dataset on the gene signature and corresponding clinicopathological parameters, including age, gender, tumor status, histological subtype, residual tumor status, the American Joint Committee for Cancer tumor node metastasis (AJCC TNM) stage, T stage, N stage, and M stage. Parameters with P<0.05 in the univariate analysis were further included in the multivariate Cox regression analysis. Moreover, we used the GSE72094, GSE50081, and GSE31210 datasets with complete clinical information for external validation in the same way. The risk scores for each patient were calculated using the same formula, and the optimal cut-off value for each dataset was determined by X-tile software. P<0.05 was considered statistically significant.
Construction and validation of the gene prognostic nomogram
We constructed a composite nomogram based on the gene signatures and clinicopathological information identified from the univariate and multivariate Cox regression analyses discussed above to predict the 1-, 2-, and 3-year overall survival (OS) of LUAD patients in TCGA dataset using the “rms” package of R software. Based on the nomogram’s total points, patients were divided into two groups by the optimal cutoffs determined by X-tile. KM survival analysis, the AUC of the ROC curve, and a calibration curve comparing the predicted and observed OS chances were applied to evaluate the prognostic performance of the nomogram. We also compared the nomogram’s predictive ability with that of the AJCC staging using the C-index and AUC.
Statistical analysis
We performed statistical analysis in R v. 4.0.3 (ISBN 3-900051-07-0; https://www.r-project.org/) and GraphPad Prism v. 8.02 for Windows, GraphPad Software, San Diego, CA, USA (https://www.graphpad.com/). We calculated the regression coefficients and hazard ratio (HR) with 95% confidence interval (CI) using univariate and multivariate Cox regression analyses. All statistical tests were two-sided, P<0.05 was considered as statistically significant.
Results
Identification of DEGs obtained from three GEO datasets
According to the flow chart shown in Figure 1, we identified a total of 2,628, 968, and 919 DEGs between tumor and normal tissues via Limma package in R software from the GSE75037, GSE32665, and GSE32863 gene expression profiles, respectively. Details of the GEO datasets in this study are displayed in Table 1. Of them, 1,157, 444, and 316 genes were upregulated, while 1,471, 524 and 603 genes were downregulated in the GSE75037, GSE32665, and GSE32863 datasets, respectively (Figure 2A). A total of 296 DEGs, composed of 120 upregulated and 176 downregulated genes, were identified after integrated analysis using the RRA method (available online: https://cdn.amegroups.cn/static/public/tlcr-22-444-2.xlsx). The top 20 up- and downregulated DEGs are shown in Figure 2B. Hierarchical clustering analysis indicated various DEG expression patterns between tumor and normal tissues (Figure 2C; Figure S1).
Table 1. Details of the GEO datasets included in this study.
| Datasets | Platform | Sample size (tumor/normal) | Application |
|---|---|---|---|
| GSE75037 | Illumina HumanWG-6 v3.0 expression beadchip | 166 (83/83) | Identification of DEG |
| GSE32665 | Illumina human-6 v2.0 expression beadchip | 179 (87/92) | Identification of DEG |
| GSE32863 | Illumina HumanWG-6 v3.0 expression beadchip | 116 (58/58) | Identification of DEG |
| GSE72094 | Rosetta/Merck Human RSTA Custom Affymetrix 2.0 microarray [HuRSTA_2a520709.CDF] | 386 (386/0) | External validation |
| GSE50081 | [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | 130 (130/0) | External validation |
| GSE31210 | [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | 204 (204/0) | External validation |
GEO, Gene Expression Omnibus; DEG, differentially-expressed gene.
Figure 2.

Identification of DEGs in LUAD between tumor and normal lung tissues. (A) Volcano plot for DEGs screened from the GEO profiles (GSE75037, GSE32665, and GSE32863). (B) The heatmap of the top 20 upregulated and downregulated DEGs screened by integrated analysis of the GEO datasets. The upregulated DEGs were shown in red while the downregulated DEGs were shown in blue. The value of each column represented the value of log2FC. (C) Representative heatmap of the DEGs after integrated analysis in GSE75037 indicated that the 296 DEGs can effectively distinguish tumors from non-tumor tissues. DEGs, differentially-expressed genes; LUAD, lung adenocarcinoma; GEO, Gene Expression Omnibus; FC, fold change.
Identification of survival-related DEGs and development of the three-gene prognostic signature
We included 293 patients from TCGA-LUAD dataset with a follow-up period of >30 days in the following survival analyses. The patients’ clinical characteristics are listed in Table 2. Based on the univariate Cox regression model, 16 DEGs were identified as being significantly associated with OS (P<0.01; Table 3). A prognostic gene signature, including PLEK2, COL1A1, and GPX3, was developed by multivariate Cox analysis (Table 3). The downregulated GPX3 with a HR <1 was considered a tumor suppressor, whereas the upregulated COL1A1 and PLEK2 with a HR >1 were regarded as oncogenes. Meanwhile, candidate genes were analyzed using X-tile software to identify the optimal OS cutoff values, and the patients were divided into low- and high-risk groups based on these data. KM survival analysis displayed that three genes were significantly related with patient OS (P<0.05) in TCGA-LUAD dataset (Figure 3A). The risk score was calculated as follows:
Table 2. Clinical characteristics of patients in TCGA-LUAD dataset and the three independent GEO datasets.
| Clinical features | TCGA-LUAD | GSE72094 | GSE50081 | GSE31210 |
|---|---|---|---|---|
| Samples sizes | 293 | 386 | 130 | 204 |
| Age (years), mean ± SD | 64.86±9 | 71.5±1.5 | 61.83±1.745 | 48±10 |
| Follow time (days), mean ± SD | 740.94±627.5 | 1,439±190 | 1,510.55±8.578 | 1,201.5±458.5 |
| Survival status, n | ||||
| Death | 83 | 109 | 54 | 30 |
| Survival | 210 | 277 | 76 | 174 |
| Sex, n | ||||
| Male | 131 | 168 | 67 | 95 |
| Female | 162 | 218 | 63 | 109 |
| Stage, n | ||||
| I | 162 | 246 | 93 | 162 |
| II | 62 | 65 | 37 | 42 |
| III | 47 | 56 | ||
| IV | 15 | 14 | ||
| Unknown | 7 | |||
| Smoking status, n | ||||
| Ever | 291 | 94 | 99 | |
| Never | 30 | 23 | 105 | |
| Unable to determine | 65 | 13 | ||
| Kras status, n | ||||
| Wt | 254 | |||
| Mut | 132 | |||
| Tp53 status, n | ||||
| Wt | 290 | |||
| Mut | 96 | |||
| Stk11 status, n | ||||
| Wt | 323 | |||
| Mut | 63 |
TCGA, The Cancer Genome Atlas; LUAD, lung adenocarcinoma; GEO, Gene Expression Omnibus; SD, standard deviation; Wt, wild-type; Mut, mutated.
Table 3. Identification of the gene expression signature by univariate and multivariate Cox regression analyses.
| No. | Gene | Univariate analysis* | Multivariate analysis** | |||||
|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |||
| 1 | GPX3 | 0.7399 | 0.6117–0.895 | 0.0019 | 0.6238 | 0.4789–0.8125 | 0.0005 | |
| 2 | PTPRM | 1.3738 | 1.1002–1.7154 | 0.0051 | ||||
| 3 | ASPM | 1.2755 | 1.0745–1.514 | 0.0054 | ||||
| 4 | CENPF | 1.2543 | 1.0607–1.4833 | 0.0081 | ||||
| 5 | TK1 | 1.3054 | 1.0733–1.5878 | 0.0076 | ||||
| 6 | PRR36 | 0.7927 | 0.6663–0.9431 | 0.0088 | ||||
| 7 | PLEK2 | 1.3833 | 1.1464–1.6691 | 0.0007 | 1.3876 | 1.0767–1.7883 | 0.0114 | |
| 8 | SLC2A1 | 1.2589 | 1.0766–1.4721 | 0.0039 | ||||
| 9 | FAM83A | 1.2135 | 1.0796–1.3639 | 0.0012 | ||||
| 10 | MIF | 1.4026 | 1.1054–1.7797 | 0.0054 | ||||
| 11 | PRC1 | 1.3344 | 1.0801–1.6486 | 0.0075 | ||||
| 12 | GJB2 | 1.1823 | 1.0597–1.319 | 0.0027 | ||||
| 13 | HMMR | 1.3209 | 1.105–1.579 | 0.0022 | ||||
| 14 | ANLN | 1.3082 | 1.1129–1.5377 | 0.0011 | ||||
| 15 | KCNK5 | 0.7836 | 0.6821–0.9003 | 0.0006 | ||||
| 16 | COL1A1 | 1.1987 | 1.0522–1.3655 | 0.0064 | 1.2173 | 1.02–1.4528 | 0.0293 | |
*, the 16 DEGs were significantly associated with OS (P<0.01) according to univariate Cox regression analysis; **, we then performed multivariate Cox regression analysis on these 16 DEGs to identify the most informative gene set for survival prediction. Finally, the three genes marked in grey in the table were selected for multivariate Cox regression analysis and generation of a prognostic risk model according to their respective regression coefficients. HR, hazard ratio; CI, confidence interval; DEGs, differentially-expressed genes; OS, overall survival.
Figure 3.

Development of the gene signatures and performance evaluation in TCGA dataset. (A) Survival analysis of candidate genes in TCGA-LUAD datasets. (B) Distribution of the risk score, survival status of patients, and the mRNA expression heatmap in TCGA dataset. (C) Survival analysis of the gene signature. Patients from TCGA dataset were divided into two groups according to the optimal cut-off values of the risk scores calculated by X-tile. (D) ROC curves of the 1-, 2-, and 3-year OS of the gene signature. TCGA, The Cancer Genome Atlas; LUAD, lung adenocarcinoma; ROC, receiver operating characteristic; AUC, area under the ROC curve; OS, overall survival; CI, confidence interval; mRNA, messenger RNA.
Risk score = (0.1446053 × expression value of COL1A1) + (−0.2426827 × expression value of GPX3) + (0.2697514 × expression value of PLEK2).
We calculated the optimal cutoff values for the risk scores using X-tile software. Patients were stratified into two groups (cutoff value =2.03) in TCGA-LUAD dataset. The distribution and survival status of LUAD patients were plotted based on the best cutoff value. The heatmap indicated that PLEK2 and COL1A1 tended to have higher expression in high-risk patients, while GPX3 was more highly expressed in low-risk patients (Figure 3B). The KM survival curves revealed significantly lower OS in the high-risk group compared to the low-risk group (P<0.0001) (Figure 3C). The time-dependent ROC curve and C-index were applied to assess the prognostic values of the three genes’ risk score (Figure 3D). The AUCs for the 1-, 2-, and 3-year OS predictions for the risk scores were 0.674, 0.658, and 0.691, respectively. The C-index of the risk score was 0.6375 (95% CI: 0.5632–0.7118). We also compared the ability of the risk score with three previously established gene signatures. The AUCs of the risk scores was close to those of the gene signatures (Figure S2A-S2C), and the risk score’s C-index was also close to those of the gene signatures (0.6375 vs. 0.6497, 0.6327, and 0.6164). Thus, the three-gene signature performed well at predicting the OS of LUAD patients.
Validation of prognostic genes with GEPIA and KM plotter
We applied GEPIA to validate the expression levels of the three genes. The mRNA expression levels of PLEK2 and COL1A1 were significantly increased in LUAD tumor tissue. In contrast, those of GPX3 were significantly decreased compared to normal tissues (Figure 4A). We performed a survival analysis using the KM plotter and demonstrated that the three gene’s expressions were related to LUAD prognosis. Patients with a high expression of PLEK2 or COL1A1, or a low GPX3 expression, had a worse prognosis (P<0.05) (Figure 4B).
Figure 4.

Prognostic genes validation with GEPIA and the KM plotter. (A) COL1A1 and PLEK2 had higher expression levels in the LUAD specimen compared to the normal specimen, while GPX3 had the opposite expression in the GEPIA tumor database. T, tumor tissues (red); N, normal tissues (gray). *, P<0.05. (B) The prognostic information of the three genes in LUAD was demonstrated using the KM plotter. The red curve represents the high expression group, and the black curve represents the low expression group. The representative IHC images and prognostic scores of the genes in LUAD vs. adjacent tissue. (C) Expression of PLEK2, COL1A1, and GPX3 in LUAD tissue and adjacent tissue (DAB/hematoxylin staining; magnification 200×). (D) The expression levels of the three genes were analyzed in 98 LUAD tissues and 80 adjacent tissues. PLEK2 and COL1A1 were more highly expressed in LUAD tissue, while GPX3 exhibited an opposite expression trend (P<0.05). LUAD, lung adenocarcinoma; HR, hazard ratio; IHC, immunohistochemistry; GEPIA, Gene Expression Profiling Interactive Analysis; KM, Kaplan-Meier; DAB, diaminobenzidine.
Tissue array specimens and IHC
To further validate our findings, we examined the protein expression levels of the three genes by IHC in an independent cohort of LUAD patients (HLugA180Su07). Compared with adjacent tissue, COL1A1 and PLEK2 were mainly expressed in the cytoplasm and displayed higher levels in LUAD tissues than adjacent tissues (Figure 4C). The tumor tissues’ IHC scores were significantly higher than those of adjacent tissues (P<0.0001) (Figure 4D). In contrast, the GPX3 protein levels in tumors were lower than those in adjacent tissues’ (P<0.05) (Figure 4C,4D).
Assessment of prognostic factors in TCGA-LUAD
A total of 161 patients from TCGA-LUAD dataset, whose complete clinical information was provided, including age, gender, tumor status, histological subtype, residual tumor status, AJCC TNM stage, T stage, N stage, and M stage, were included in the analysis (Table 4, available online: https://cdn.amegroups.cn/static/public/tlcr-22-444-3.xlsx). We identified the prognostic indicators of OS for lung cancer using univariate and multivariate Cox regression analyses. The univariate analysis indicated that risk score, tumor status, residual tumor, and pathological stages were significantly related with OS of LUAD patients (Table 5). Multivariate analysis shown that risk score was independent risk factor for OS (P<0.05; Table 5). After adjusting for tumor status and clinical stage in the multivariate Cox analysis, LUAD patients with a low-risk score had a lower risk of death compared to those with a high-risk score (HR =0.3898; 95% CI: 0.1938–0.7842; P<0.05; Table 5). Notably, after adjusting for the known risk factors for survival, the gene signature demonstrated a robust performance in predicting the OS of LUAD patients.
Table 4. Baseline characteristics of patients in TCGA-LUAD dataset with complete clinical information.
| Clinical features | Mean ± SD/n |
|---|---|
| Risk score | 2.119±0.05303 |
| Age (years) | 64.56±10.29 |
| Follow time (days) | 554.5±341.5 |
| Sex | |
| Male | 71 |
| Female | 90 |
| Tumor status | |
| Tumor free | 115 |
| With tumor | 46 |
| Histologic diagnosis | |
| LUAD-NOS | 102 |
| LUAD-specified | 59 |
| Residual tumor | |
| R0 | 142 |
| R1 | 8 |
| Rx | 11 |
| Stage | |
| I | 89 |
| II | 37 |
| III | 28 |
| IV | 7 |
| T | |
| T1 | 57 |
| T2 | 84 |
| T3 | 12 |
| T4 | 6 |
| Tx | 2 |
| N | |
| N0 | 101 |
| N+ | 60 |
| M | |
| M0 | 112 |
| M1 | 7 |
| Mx | 42 |
| Subdivision | |
| R | 94 |
| L | 67 |
TCGA, The Cancer Genome Atlas; LUAD, lung adenocarcinoma; NOS, not-otherwise-specified; SD, standard deviation.
Table 5. Univariate and multivariate Cox regression analyses for risk scores on the OS of patients with LUAD.
| Parameters | Univariate analysis | Multivariate analysis | |||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | ||
| Risk group | |||||||
| High risk | 1 | 1 | 1 | 1 | 1 | 1 | |
| Low risk | 0.3780 | 0.2015–0.7091 | 0.00244** | 0.3898 | 0.1938–0.7842 | 0.00825** | |
| Age | 0.994979 | 0.9685–1.022 | 0.7156 | ||||
| Sex | |||||||
| Male | 0.8182 | 0.448–1.494 | 0.514 | ||||
| Female | 1 | 1 | 1 | ||||
| Tumor status | |||||||
| Tumor free | 1 | 1 | 1 | 1 | 1 | 1 | |
| With tumor | 5.0491 | 2.587–9.856 | 2.09e-06*** | 5.35 | 2.559-11.18 | 8.28e-06*** | |
| Histologic diagnosis | |||||||
| LUAD-NOS | 1 | 1 | 1 | ||||
| LUAD-specified | 0.6688 | 0.3488– 1.282 | 0.226 | ||||
| Residual tumor | |||||||
| R0 | 1 | 1 | 1 | 1 | 1 | 1 | |
| R1 | 4.4435 | 1.6802–11.752 | 0.00265** | 0.8514 | 0.2455–2.954 | 0.7999 | |
| Rx | 2.3276 | 0.7012–7.726 | 0.16755 | 1.329 | 0.3082–5.727 | 0.7030 | |
| Stage | |||||||
| I | 1 | 1 | 1 | 1 | 1 | 1 | |
| II | 2.6297 | 1.187–5.827 | 0.01723* | 1.259 | 0.1988–7.977 | 0.8066 | |
| III | 3.3567 | 1.598–7.049 | 0.00138** | 1.6 | 0.1858–13.79 | 0.6686 | |
| IV | 3.9349 | 1.101–14.066 | 0.03506* | 0.5796 | 0.04838–6.945 | 0.6669 | |
| T | |||||||
| T1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| T2 | 1.486 | 0.7244–3.047 | 0.28009 | 1.15 | 0.5201–2.542 | 0.7303 | |
| T3 | 1.523 | 0.3267–7.098 | 0.59231 | 0.6339 | 0.1138–3.532 | 0.6029 | |
| T4 | 4.979 | 1.5392–16.105 | 0.00736** | 4.459 | 0.9936–20.01 | 0.0509 | |
| Tx | 6.889 | 0–∞ | 0.99750 | – | – | – | |
| N | |||||||
| N0 | 0.3460 | 0.1845–0.6488 | 0.000938*** | 0.645 | 0.1033–4.029 | 0.6390 | |
| N+ | 1 | 1 | 1 | 1 | 1 | 1 | |
| M | |||||||
| M0 | 1 | 1 | 1 | ||||
| M1 | 2.04708 | 0.6187–6.773 | 0.241 | ||||
| Mx | 0.91775 | 0.4546–1.853 | 0.811 | ||||
| Subdivision | |||||||
| L | 1 | 1 | 1 | ||||
| R | 1.09535 | 0.6013–1.995 | 0.766 | ||||
*, P<0.05; **, P<0.01; ***, P<0.001. OS, overall survival; LUAD, lung adenocarcinoma; NOS, not-otherwise-specified; HR, hazard ratio; CI, confidence interval.
Validation of the prognostic value of the signature using external GEO datasets
We used three external datasets (GSE50081, GSE72094, and GSE31210) to validate the predictive ability of the prognostic signature (Table 2). Risk scores were calculated using the same formula for each patient, and patients were divided into high- and low-risk groups using the same method. We found that patients with lower risk scores had a better chance of survival than those with higher risk scores (P=0.0016, 0.0033, and 0.0001, respectively; Figure 5A and Figures S3,S4). The heatmap displayed that PLEK2 and COL1A1 tended to have higher expression in high-risk patients, while GPX3 had higher expression in low-risk patients (Figure 5B).
Figure 5.

External validation of the prognostic gene in the GSE31210 dataset. (A) KM survival curves of the gene signature. Patients from the GSE31210 dataset were divided into two groups according to the optimal cut-off values for their risk scores (calculated by X-tile). (B) Distribution of the risk scores, patients’ survival status, and the mRNA expression heatmap in the GSE31210 dataset. (C) ROC curves of the 1-, 2-, and 3-year OS predictions of the gene signature. (D) The prognostic value of the gene signature was evaluated using the multivariate Cox model. *, P<0.05; **, P<0.01. ROC, receiver operating characteristic; AUC, area under the ROC curve; OS, overall survival; CI, confidence interval; KM, Kaplan-Meier; mRNA, messenger RNA.
We then assessed the prognostic power through ROC analysis and C-index. The ROC analysis for GSE31210 and GSE50081 is shown in Figure 5C and Figures S3,S4. The C-index of the risk score in the GSE31210, GSE50081, and GSE72094 datasets were 0.6322 (95% CI: 0.5416–0.7228), 0.6213 (95% CI: 0.5463–0.6962), and 0.5930 (95% CI: 0.535–0.651), respectively. Moreover, after adjusting for covariates, patients with a low-risk score still had a significantly lower risk of death (P=0.05, 0.004, and 0.04, respectively; Figure 5D, Figures S3,S4). Therefore, external validation showed that the prognostic signature performed well at predicting OS in LUAD patients.
Developing and validating a prognostic nomogram
We used these 161 patients from TCGA dataset to build a prognostic nomogram in order to predict the 1-, 2-, and 3-year OS of LUAD patients (Figure 6A). Risk score, tumor status, and pathological stage were selected to establish the nomogram model. The patients were divided into two risk groups according to the total point of the nomogram. The KM plot effectively discerned that those with higher scores had significantly poorer OS than the low-risk group (P<0.0001) (Figure 6B). The AUCs of the 1-, 2-, and 3-year OS of the nomogram were 0.8136, 0.7281, and 0.8324, respectively (Figure 6C). The AUCs of the 1-, 2-, and 3-year OS of the gene signature were 0.7025, 0.6476, and 0.6941, respectively. Additionally, the AUCs of the 1-, 2-, and 3-year OS of the AJCC stage were 0.7394, 0.6668, and 0.6539, respectively (Figure S5). The C-index of the nomogram was 0.762 (95% CI: 0.714–0.845), while that of the AJCC stage was 0.635 (95% CI: 0.547–0.678) and the signature was 0.646 (95% CI: 0.562–0.730), which suggested that the prognostic nomogram may performed best in predicting OS. These data demonstrated that the nomogram had better predictive ability than the AJCC-stage and gene signature for the 1-, 2-, and 3-year OS. The calibration curve for predicting the 1-, 2-, and 3-year OS demonstrated that the nomogram performed well at predicting OS of LUAD patients (Figure 6D). In the third year, when the predicted OS was >80%, the nomogram may underestimate mortality; however, when the predicted OS was <80%, the nomogram may overestimate mortality.
Figure 6.

The performance of the nomogram in predicting the prognosis in TCGA-LUAD dataset. (A) A prognostic nomogram predicting the 1-, 2-, and 3-year OS of LUAD patients. (B) The nomogram’s KM survival curves. Patients from TCGA-LUAD dataset were stratified into two risk groups according to optimal cutoff values of the nomogram (calculated by X-tile software). (C) ROC curves of the 1-, 2-, and 3-year OS predictions of the nomogram. (D) Calibration plot of the 1-, 2-, and 3-year OS of the nomogram. TCGA, The Cancer Genome Atlas; LUAD, lung adenocarcinoma; ROC, receiver operating characteristic; AUC, area under the ROC curve; OS, overall survival; CI, confidence interval; KM, Kaplan-Meier.
Discussion
The 5-year survival rate of lung cancer patients with localized-stage disease is 59%, while those with advanced stage is only 6% (1). LUAD is the most common subtype of lung cancer, and its mechanisms of pathogenesis and metastasis are diverse and heterogeneous. A single clinical parameter has a poor power of prognostic prediction. Outcomes vary between recovery and recurrence, even in patients with similar clinical and pathological features. Despite the recent advances in the biological characterization of LUAD, the molecular mechanisms behind its carcinogenesis remain elusive. Hence, it is necessary to identify a precise and effective prognostic signature to predict the survival of patients with LUAD. In the current study, we constructed a three-gene prognostic model to stratify LUAD patients into different risk groups for OS. These three genes were all significantly associated with the OS of LUAD patients. Among them, PLEK2 and COL1A1 are tumor-promoting genes, while GPX3 is a tumor suppressor gene.
Previous studies have indicated that COL1A1 is an extracellular matrix protein, and its overexpression is vitally related to breast (19), stomach (20), liver (21), and colon (22) tumors. COL1A1 is highly expressed in human breast cancer, and its overexpression promotes breast cancer metastasis (23,24). Additionally, a study has demonstrated that COL1A1 might promote colorectal cancer metastasis by regulating the WNT/planar cell polarity (PCP) pathway (25). Another study has elucidated that COL1A1 was highly expressed in hepatocellular carcinoma (HCC), and its expression was significantly related to HCC disease progression through epithelial-mesenchymal transition (EMT) (21). Studies have also shown that compared to adjacent normal tissues, NSCLC tends to overexpress COL1A1 (26), which is correlated with the expression of hypoxia markers (27). Thus, COL1A1 is a reliable biomarker and recognized therapeutic target for different types of cancer.
Previous study has found that increased PLEK2 expression might be a specific prognostic biomarker for poor progression-free survival (PFS) in LUAD patients. At the single-cell level, its expression is significantly positively correlated with LUAD cell invasion, cell cycle abnormalities, DNA damage, and DNA repair irregularities. Promoter hypomethylation may be a potential mechanism leading to its upregulation (28). Transforming growth factor (TGF)-β-mediated EMT plays a crucial role in tumor invasion and metastasis. A study has reported that PLEK2 was significantly up-regulated in NSCLC cells activating TGF-β1, and was negatively correlated with OS in NSCLC. In terms of mechanism, the PLEK2-SHIP2 axis promotes NSCLC EMT and migration via the TGF-β/PI3K/Akt signaling pathway (29). Additionally, researchers have shown that PLEK2 promotes tumorigenesis and metastasis in other types of cancer (30). Therefore, PLEK2 may be a new prognostic marker.
The protein encoded by GPX3 belongs to the glutathione peroxidase family, which protects cells from oxidative damage by catalyzing glutathione’s organic hydroperoxides and hydrogen peroxide (H2O2) reduction. The role of GPX3 in cancer has not yet been elucidated. Research indicates that GPX3 has a dichotomous effect in different tumor types, both as a tumor suppressor protein and a pro-survival protein. Some studies have demonstrated that the lack of GPX3 expression in tumor tissues is related to poor prognosis and chemotherapy resistance in patients with LUADs and low-grade gliomas. In recent years, the application of GPX3 as a tumor suppressor in a variety of cancers has received extensive attention (31,32). Other studies have indicated that redox signals mediated by GPX3 can inhibit tumors in lung cancer cell lines by inhibiting the Erk-NF-κB-cyclin B1 signaling pathway, leading to cell cycle arrest in the G2/M phase (33-35). However, GPX3 expression is elevated in other tumor tissues, and its high expression is related to poor prognosis in patients with diseases like gastric cancer and lung squamous cell carcinoma (36).
After identifying these three prognostic genes, we constructed and investigated the three-gene prognostic signature for its prognostic value in LUAD patients. Patients in high-risk groups showed significantly poorer prognoses than those in the low-risk group in TCGA-LUAD dataset. As demonstrated by the results, our prognostic signature was superior to or comparable with those reported in three previous studies. The AUC and C-index confirm its predictive value. Additionally, the prognostic model was an independent and significant factor for assessing the patients’ prognoses depending on the univariate and multivariate Cox regression analyses.
We also verified the three-gene signature’s prognostic value in external GEO datasets. To improve the prognosis predictive ability of the three-gene prognostic signature, we integrated the prognostic model and conventional clinicopathological parameters, including AJCC-stage and tumor status, to construct a nomogram that can more accurately predict patient prognosis. As a supplement to AJCC staging, the nomogram presented superior predictive ability in terms of the 1-, 2-, and 3-year OS prediction, according to the survival analysis, C-index, and time- dependent ROC analysis, which is conducive to clinical decision-making and personalized treatment. In our nomogram’s calibration chart, a perfect agreement was observed between the predicted and the observed results. Therefore, our prognostic nomogram based on these three genes can help clinicians predict the survival outcome of LUAD patients and provide a reference for treatment guidance rather than a single routine clinical parameter. Based on TCGA-LUAD data combined with the three GEO datasets and external validation, this study provides solid evidence supporting the prognostic value of the gene signature in LUAD patients. Additionally, there are only three candidate genes in our signature, which will be more convenient and maneuverable in future clinical applications.
However, this study has certain limitations that should be noted. Firstly, our study was retrospective, the sample size was small, and the patients’ information came from TCGA and GEO databases, which are restricted. Thus, it is necessary to verify the gene signature using a sufficient number of LUAD examples. Additionally, the clinical parameters were not adjusted in the three validated GEO datasets because the related information was unavailable from the GEO database. Finally, the specific mechanisms behind these prognostic genes in the pathogenesis and development of LUAD are not fully understood and need to be examined more thoroughly in the future.
Conclusions
In our study, we identified a three-gene model and a prognostic nomogram combined with gene signature and clinicopathological parameters to predict the OS of LUAD. Our prognostic model was closely associated with the prognosis of LUAD, which may facilitate discovering potential therapeutic targets and clinical decision-making.
Supplementary
The article’s supplementary files as
Acknowledgments
We gratefully acknowledge the participation of the study participants and technical support of the Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). We appreciate the great support from Dr. Yoshinobu Ichiki (National Hospital Organization, Japan) and Dr. Francesco Facchinetti (Paris-Saclay University, France) in improving the quality of this paper. We also appreciate the academic support from the AME Lung Cancer Collaborative Group.
Funding: This work was supported by the National Natural Science Foundation of China, China (No. 31700690), the Zhejiang Provincial Natural Science Foundation (No. LQ19H160023), and the Project of Clinical Scientific Research of Zhejiang Medical Association (No. 2018ZYC-A19).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The research was conducted in accordance with the Declaration of Helsinki (as revised in 2013).
Footnotes
Reporting Checklist: The authors have completed the TRIPOD reporting checklist. Available at https://tlcr.amegroups.com/article/view/10.21037/tlcr-22-444/rc
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://tlcr.amegroups.com/article/view/10.21037/tlcr-22-444/coif). FF received personal fees from BMS and Roche for editorial activities, from BeiGene for advisory board. All authors report that this research gets technical support from the Shanghai Outdo Biotech Co., Ltd. (Shanghai, China), and promise that there are no conflicts of interest between the company and the authors. The other authors have no conflicts of interest to declare.
References
- 1.Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics, 2021. CA Cancer J Clin 2021;71:7-33. 10.3322/caac.21654 [DOI] [PubMed] [Google Scholar]
- 2.Inamura K. Clinicopathological Characteristics and Mutations Driving Development of Early Lung Adenocarcinoma: Tumor Initiation and Progression. Int J Mol Sci 2018;19:1259. 10.3390/ijms19041259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543-50. 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buddharaju LNR, Ganti AK. Immunotherapy in lung cancer: the chemotherapy conundrum. Chin Clin Oncol 2020;9:59. 10.21037/cco.2020.01.05 [DOI] [PubMed] [Google Scholar]
- 5.Nieder C, Nestle U, Ukena D, et al. Tumor markers as prognostic factors in non-small-cell bronchial carcinoma. Strahlenther Onkol 1995;171:587-93. [PubMed] [Google Scholar]
- 6.Niemira M, Collin F, Szalkowska A, et al. Molecular Signature of Subtypes of Non-Small-Cell Lung Cancer by Large-Scale Transcriptional Profiling: Identification of Key Modules and Genes by Weighted Gene Co-Expression Network Analysis (WGCNA). Cancers (Basel) 2019;12:37. 10.3390/cancers12010037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu F, Zhan X, Zheng X, et al. A signature of immune-related gene pairs predicts oncologic outcomes and response to immunotherapy in lung adenocarcinoma. Genomics 2020;112:4675-83. 10.1016/j.ygeno.2020.08.014 [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Wu L, Ao H, et al. Prognostic implications of autophagy-associated gene signatures in non-small cell lung cancer. Aging (Albany NY) 2019;11:11440-62. 10.18632/aging.102544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu F, Huang X, Li Y, et al. m6A-related lncRNAs are potential biomarkers for predicting prognoses and immune responses in patients with LUAD. Mol Ther Nucleic Acids 2021;24:780-91. 10.1016/j.omtn.2021.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang C, Zhang Z, Zhang G, et al. Clinical significance and inflammatory landscapes of a novel recurrence-associated immune signature in early-stage lung adenocarcinoma. Cancer Lett 2020;479:31-41. 10.1016/j.canlet.2020.03.016 [DOI] [PubMed] [Google Scholar]
- 11.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao J, Guo C, Ma Z, et al. Identification of a novel gene expression signature associated with overall survival in patients with lung adenocarcinoma: A comprehensive analysis based on TCGA and GEO databases. Lung Cancer 2020;149:90-6. 10.1016/j.lungcan.2020.09.014 [DOI] [PubMed] [Google Scholar]
- 14.Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res 2004;10:7252-9. 10.1158/1078-0432.CCR-04-0713 [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Zhang Z, Yu Z. Identification of a novel glycolysis-related gene signature for predicting metastasis and survival in patients with lung adenocarcinoma. J Transl Med 2019;17:423. 10.1186/s12967-019-02173-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma C, Luo H, Cao J, et al. Identification of a Novel Tumor Microenvironment-Associated Eight-Gene Signature for Prognosis Prediction in Lung Adenocarcinoma. Front Mol Biosci 2020;7:571641. 10.3389/fmolb.2020.571641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z, Qi F, Li F. Establishment of a Gene Signature to Predict Prognosis for Patients with Lung Adenocarcinoma. Int J Mol Sci 2020;21:8479. 10.3390/ijms21228479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 2017;45:W98-W102. 10.1093/nar/gkx247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, Wang L, Yang H, et al. Identification of potential genes related to breast cancer brain metastasis in breast cancer patients. Biosci Rep 2021;41:BSR20211615. 10.1042/BSR20211615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y, Li J, Luo H, et al. Differential Expression of COL1A1, COL1A2, COL6A3, and SULF1 as Prognostic Biomarkers in Gastric Cancer. Int J Gen Med 2021;14:5835-43. 10.2147/IJGM.S321265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma HP, Chang HL, Bamodu OA, et al. Collagen 1A1 (COL1A1) Is a Reliable Biomarker and Putative Therapeutic Target for Hepatocellular Carcinogenesis and Metastasis. Cancers (Basel) 2019;11:786. 10.3390/cancers11060786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 2012;30:1-22. 10.1146/annurev-immunol-100311-102839 [DOI] [PubMed] [Google Scholar]
- 23.Meng C, He Y, Wei Z, et al. MRTF-A mediates the activation of COL1A1 expression stimulated by multiple signaling pathways in human breast cancer cells. Biomed Pharmacother 2018;104:718-28. 10.1016/j.biopha.2018.05.092 [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Shen JX, Wu HT, et al. Collagen 1A1 (COL1A1) promotes metastasis of breast cancer and is a potential therapeutic target. Discov Med 2018;25:211-23. [PubMed] [Google Scholar]
- 25.Zhang Z, Wang Y, Zhang J, et al. COL1A1 promotes metastasis in colorectal cancer by regulating the WNT/PCP pathway. Mol Med Rep 2018;17:5037-42. 10.3892/mmr.2018.8533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oleksiewicz U, Liloglou T, Tasopoulou KM, et al. COL1A1, PRPF40A, and UCP2 correlate with hypoxia markers in non-small cell lung cancer. J Cancer Res Clin Oncol 2017;143:1133-41. 10.1007/s00432-017-2381-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwai M, Tulafu M, Togo S, et al. Cancer-associated fibroblast migration in non-small cell lung cancers is modulated by increased integrin α11 expression. Mol Oncol 2021;15:1507-27. 10.1002/1878-0261.12937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Li T, Hu B, et al. PLEK2 Gene Upregulation Might Independently Predict Shorter Progression-Free Survival in Lung Adenocarcinoma. Technol Cancer Res Treat 2020;19:1533033820957030. 10.1177/1533033820957030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu DM, Deng SH, Zhou J, et al. PLEK2 mediates metastasis and vascular invasion via the ubiquitin-dependent degradation of SHIP2 in non-small cell lung cancer. Int J Cancer 2020;146:2563-75. 10.1002/ijc.32675 [DOI] [PubMed] [Google Scholar]
- 30.Wang F, Zhang C, Cheng H, et al. TGF-β-induced PLEK2 promotes metastasis and chemoresistance in oesophageal squamous cell carcinoma by regulating LCN2. Cell Death Dis 2021;12:901. 10.1038/s41419-021-04155-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nirgude S, Choudhary B. Insights into the role of GPX3, a highly efficient plasma antioxidant, in cancer. Biochem Pharmacol 2021;184:114365. 10.1016/j.bcp.2020.114365 [DOI] [PubMed] [Google Scholar]
- 32.Liu Q, Bai W, Huang F, et al. Downregulation of microRNA-196a inhibits stem cell self-renewal ability and stemness in non-small-cell lung cancer through upregulating GPX3 expression. Int J Biochem Cell Biol 2019;115:105571. 10.1016/j.biocel.2019.105571 [DOI] [PubMed] [Google Scholar]
- 33.Ozeki M, Tamae D, Hou DX, et al. Response of cyclin B1 to ionizing radiation: regulation by NF-kappaB and mitochondrial antioxidant enzyme MnSOD. Anticancer Res 2004;24:2657-63. [PMC free article] [PubMed] [Google Scholar]
- 34.Androic I, Krämer A, Yan R, et al. Targeting cyclin B1 inhibits proliferation and sensitizes breast cancer cells to taxol. BMC Cancer 2008;8:391. 10.1186/1471-2407-8-391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.An BC, Choi YD, Oh IJ, et al. GPx3-mediated redox signaling arrests the cell cycle and acts as a tumor suppressor in lung cancer cell lines. PLoS One 2018;13:e0204170. 10.1371/journal.pone.0204170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang C, Worley BL, Phaëton R, et al. Extracellular Glutathione Peroxidase GPx3 and Its Role in Cancer. Cancers (Basel) 2020;12:2197. 10.3390/cancers12082197 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as