

Abstract
Introduction
Alzheimer disease (AD) and related dementias are characterized by damage caused by neuropathological lesions in the brain. These include AD lesions (plaques and tangles) and non‐AD lesions such as vascular injury or Lewy bodies. We report here an assessment of lesion association to dementia in a large clinic‐based population.
Methods
We identified 5272 individuals with neuropathological data from the National Alzheimer's Coordinating Center. Individual lesions, as well as a neuropathological composite score (NPCS) were tested for association with dementia, and both functional and neurocognitive impairment using regression models.
Results
Most individuals exhibited mixed pathologies, especially AD lesions in combination with non‐AD lesions. All lesion types were associated with one or more clinical outcomes; most even while controlling for AD pathology. The NPCS was also associated with clinical outcomes.
Discussion
These data suggest mixed‐type pathologies are extremely common in a clinic‐based population and may contribute to dementia and cognitive impairment.
Keywords: Alzheimer disease, amyloid angiopathy, cognitive decline, dementia, hippocampal sclerosis, Lewy bodies, neuritic plaques, neurofibrillary, neuropathology, tangles, vascular dementia
1. INTRODUCTION
Alzheimer disease (AD) is the most common cause of dementia, accounting for 60% to 80% of all dementia cases. AD and related dementias (ADRDs) affect >50 million individuals worldwide. 1 Upon autopsy, most AD dementia patients show the hallmark brain lesions of AD: neurofibrillary tangles 2 , 3 , 4 and amyloid plaques, including both diffuse plaques 4 and neuritic plaques. 3 , 5 , 6 Often, patients display other non‐AD neuropathological lesions such as Lewy bodies, 7 , 8 cerebral amyloid angiopathy, 9 , 10 , 11 , 12 , 13 arteriolosclerosis, 11 , 14 , 15 hippocampal sclerosis, 15 , 16 and vascular brain injury. 9 , 12 , 17 These “related dementias” contribute to cognitive impairment and have been reported to increase risk for dementia when comorbid with AD. 18 , 19 , 20 , 21 , 22 , 23
Neuropathological lesions are often considered as distinct disease processes, occurring in isolation. However, there is increasing evidence that dementia of mixed pathologies is more common than previously recognized. Community‐based studies, such as the Honolulu‐Asian Aging Study, 24 90+ study, 18 the Adult Changes in Thought (ACT) study, 25 and the Religious Orders Study (ROS) and Rush Memory and Aging Project (MAP) 22 , 26 have highlighted the continuum of dementia by investigating the effects and commonality of mixed neuropathologies in dementia. These studies show an estimated 50% to 75% of dementia cases have multiple lesion types present at death. 18 , 19 , 21 , 22 , 23 , 25 These studies show some inconsistencies, possibly from the small sample size for many of the studies, or due to differences in study design (Table S1 in the Supporting Information).
One key study showed that in addition to AD lesions, neocortical Lewy bodies, hippocampal sclerosis, low brain weight, and microinfarcts were associated with cognitive impairment in analyses of two population‐based autopsy cohorts. 24 The first population was a predominantly non‐Hispanic white and female group with 334 participants from the 1997 Nun Study 17 ; the second population was a predominantly Japanese American and male group with 774 participants from the 2002 Honolulu‐Asia Aging Study. 27 Here, we tested the findings of these earlier studies using a clinic‐based population from the NIH‐funded Alzheimer Disease Research Centers (ADRCs), together with data from the National Alzheimer's Coordinating Center (NACC). 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 Compared to previous studies, our sample size is larger, and more heterogeneous, composed of both male and female participants (46.30% female), and includes some racial/ethnic diversity (Table S1). In addition, our sample is more geographically representative as it includes participants from the 28 nationwide ADRCs, spanning 17 states. Further, we consider additional neuropathologic lesions that have not been studied in previous works.
RESEARCH IN CONTEXT
Systematic review: The authors began with a literature review. The contribution of lesion comorbidity and mixed pathology has been highlighted in previous studies, but predominantly in community cohorts. We explore similar associations in a larger and more heterogeneous clinic‐based cohort.
Interpretation: Mixed pathologies in AD dementia are common and are associated with more severe functional and neurocognitive impairment in a large clinic‐based cohort of 5272 individuals.
Future directions: With these results, we are interested in (1) investigating the rate of progression and cognitive decline as a function of neuropathologic lesion combinations, (2) integrating genetic information to identify genetic variants associated with neuropathologic lesions, and (3) exploring determinants of AD resilience to cognitive decline, resistance to neuropathology, and other dementias.
2. METHODS
2.1. Study participants
Participants were enrolled and assessed clinically through the nationwide ADRC program. The ADRCs consist of NIH‐funded memory and aging clinics throughout the United States that provide diagnosis and medical services to dementia patients and their families, as well as participate in dementia and aging‐related research. Participants are enrolled and assessed by individuals sites under a consistent protocol and data are then transferred to the NACC. Data for this study were derived from the Uniform Data Set (UDS) of the NACC, together with the NACC neuropathology form (accessed July 2019). 30 As such, this study consists of individuals who presented at memory and aging clinics, who consented to inclusion in research, and later went on to participate in an autopsy program. While the ADRCs are not an intentionally operated for the support of longitudinal cohort studies, participants often visit the clinic multiple times over many years, leading to multiple clinical assessments for most participants.
The UDS consists of participant clinical, medical, and demographic data. The neuropathology form includes detailed information on neuropathological features from brain autopsy reports. 31 In this analysis, we consider all participants with both UDS and neuropathology form (N = 5272). For participants with more than one clinical assessment we only consider the clinical data from their final visit (eg, last assessment prior to death). All participants (or representatives) provided written informed consent; all protocols and assessments were performed with approval by the appropriate institutional internal review boards.
2.2. Neuropathology phenotypes and variable coding
2.2.1. Primary neuropathology phenotypes
Lesion types, variables used, and coding are described in detail in Supplemental Text 1. Briefly, lesion types studied include neurofibrillary tangles, neuritic plaques, diffuse plaques, Lewy bodies, cerebral amyloid angiopathy, arteriolosclerosis, hippocampal sclerosis, and vascular brain injury (as measured by infarcts/lacunes, hemorrhages, microinfarcts, and white matter rarefaction). 3 Specific NACC variables and code mappings used are noted in Supplemental Text 1. Lesions were scored into three categories of increasing severity and numerical value ranging from none or mild (0) to moderate (0.4) to severe (1). 24 Because of differences in form version coding in the UDS, hippocampal sclerosis is represented as a binary variable of absent or present.
2.2.2. Vascular brain injury
For vascular brain injury the relevant variables were coded into binary variables and then summed up (ranging from 0 to 4). Then, we assigned those with a sum of 0 as none/mild (0), 1 as moderate (0.4), and 2 to 4 as severe (1) to better align with the other variable coding).
2.2.3. Neuropathology composite score
A neuropathology composite score (NPCS) was created by summing the 8 lesion scores noted above; thus, ranging from 0 to 8.
2.2.4. Other variables and codings
We also considered brain weight on autopsy (NACC variable NPWBRWT) as in previous studies 24 (Supplemental Text 2). Analyses are described in the Supplementary Material but were excluded here due to high levels of missing data. Individuals were broadly categorized by degree of AD neuropathology according to NIA‐AA Alzheimer's disease neuropathologic change (ADNC), when available (NACC variable NPADNC). When unavailable we used the neurofibrillary tangles, neuritic plaques, and diffuse plaques to categorize individuals using NIA‐REAGAN/CERAD criteria.
Those lacking all AD lesions were “No AD Pathology”; those with any combination of AD lesions were “Any AD Pathology”; those with either a low ADNC or AD pathology not meeting NIA‐REAGAN criteria were further classified as “Low AD Pathology”; those with intermediate/high ADNC or meeting NIA‐REAGAN criteria were classified as “Intermediate or high AD Pathology.”
2.3. Clinical outcomes
Dementia was derived from the NACC variable NACCUDSD, an outcome assessed entirely from clinical data. Neurocognitive impairment was measured by the Mini‐Mental State Examination (MMSE) 35 , 36 (NACCMMSE variable), which ranges from 0 to 30 with lower scores indicating poorer cognitive function. Functional impairment was measured by the Clinical Dementia Rating (CDR®) Dementia Staging Instrument 37 sum of boxes (CDR‐SOB) as measured by the NACC variable CDRSUM, which ranges from 0 to 18, with higher scores indicating poorer functioning.
2.4. Exclusion criteria
Individuals were excluded if they had no UDS form completed, did not have the NACC neuropathology form (must have version 9 or 10), were missing more than three neuropathology variables of interest, and/or were <50 years old by their last clinic visit.
2.5. Assessing missingness
Overall variable‐level missingness is low (see Table 1). We used the MICE package version 3.11 in R to impute the indices of the missing NP variables. 38 The outcome variables (NACCUDSD, NACCMMSE, and CDRSUM) were not included in the prediction matrix of the imputation scheme. Clinical outcome variables were not imputed.
TABLE 1.
Characteristics of the participants
| Demographics | |
|---|---|
| Total number of participants, N | 5272 |
| Sex, % F | 46.30% |
| Hispanic, % | 3.77% |
| African American, % | 3.68% |
| Age at final exam, mean (SD) | 78.52 (±11.14) |
| Clinical characteristics | |
| Number of visits, mean (SD) | 3.28 (±2.31) |
| Years from final exam to death, mean (SD) | 2.65 (±2.07) |
| Individuals with dementia, N (%) | 4106 (78%) |
| MMSE score, mean (SD) | 17.52 (±9.14) |
| CDR‐SOB score, mean (SD) | 8.88 (±6.18) |
| Neuropathology characteristics | None/Mild (%) | Moderate (%) | Severe (%) | Missing (%) |
|---|---|---|---|---|
| Neurofibrillary tangles | 24.8 | 22.3 | 51.5 | 1.4 |
| Neuritic plaques | 36.1 | 18.7 | 45.1 | 0.1 |
| Diffuse plaques | 25.9 | 15.4 | 51.5 | 7.2 |
| Lewy bodies | 65.5 | 3.9 | 26.4 | 4.2 |
| Cerebral amyloid angiopathy | 67.4 | 19.4 | 11.0 | 2.2 |
| Arteriolosclerosis | 50.2 | 28.1 | 12.4 | 9.2 |
| Hippocampal sclerosis | 84.5 | – | 11.7 | 3.8 |
| Vascular brain injury | 41.1 | 32.8 | 19.9 | 6.2 |
| NPCS, mean (SD) | 2.98 (±1.70) | |||
Abbreviations: CDR‐SOB, Clinical Dementia Rating Sum of Boxes; MMSE, Mini‐Mental State Examination; NPCS, Neuropathology Composite Score; SD, standard deviation.
2.6. Atypical neuropathological findings
Cognitive resilience was defined as those with a high burden of neuropathological lesions but who maintained cognitive function. High lesion burden was defined as the upper quartile of the NPCS among the cognitively intact (no dementia, mild cognitive impairment [MCI], or other cognitive impairment per the NACCUDSD variable, with minimal impairment per the CDR‐SOB and, if available, MMSE). Minimal cognitive impairment was defined as an MMSE of 24 or higher per previous reports 39 , 40 and a CDR‐SOB of 4 or less per previous reports. 41 . Neuropathological resistance was defined as those with no or minimal pathological burden (lower quartile of NPCS) despite advanced age (upper quartile of age). Other dementias were defined as dementia (per NACCUDSD) with no or minimal neuropathological lesions (median or lower NPCS among the cognitively intact).
2.7. Statistical analyses
Statistical analyses were conducted in R version 4.0.2. 42 Spearman correlation and the corrgram R package were used to assess correlations. 43 Primary outcome variables were dementia, functional impairment via CDR‐SOB, and cognitive impairment via MMSE.
2.7.1. Analysis of dementia outcome
Logistic regression was used to model the association between dementia and the neuropathologic lesions. For this analysis we only included individuals with a status of either dementia or normal cognition; MCI or other mild impairment were excluded (NACCUDSD). In total, 4106 participants (87%) had dementia and 612 (13%) had normal cognition (Total N = 4718).
2.7.2. Analysis of neurocognitive and functional impairment outcomes
Linear regression was used to model the association between neurocognitive or functional impairment and the neuropathologic lesions using the raw scores of the MMSE and CDR‐SOB as linear outcomes. The MMSE data were available for 4032 individuals and CDR‐SOB for 5272. To replicate analyses performed in White et al., we also considered impairment as an ordinal trait as part of a supplemental analysis. 24
2.7.3. Statistical models
For each clinical outcome two primary models were used: (1) a full model with each lesion as a separate variable, and (2) a model with the NPCS. Covariates for age at clinical exam, sex, a binary variable for African American race/ethnicity, and a binary variable for Hispanic/Latino race/ethnicity were included in all models. Age at exam and age at death were nearly collinear (R 2 = 0.98); therefore, we only included age at exam in the models. A sensitivity analysis of age at death is described in the Supplemental Material. In primary models, the variables were treated as additive terms and therefore the odds ratios (ORs) and betas reflect a one‐unit change in the variable (none/mild vs severe in lesions; 0 to 1 to 2, etc in the NPCS). For the dementia model, pseudo R 2 was estimated using the DescTools package in R. 44 , 45 To test the robustness of these models the supplemental material contains several secondary analyses: ordinal parameterizations of lesions (Table S2), ordinal parameterizations of clinical outcomes (Table S3), no imputed/missing data (Table S4).
3. RESULTS
3.1. Cohort characteristics
The full dataset consisted of 5272 individuals. The majority of participants (72%) had multiple clinic visits ranging from 2 to 13 with a median of 3 assessments. Only the MMSE and CDR‐SOB from the final assessment before death were used in this study. Age at final exam ranged from 50 to 110 years old with a mean of 78.5 (±11.1). Time interval between each follow‐up exam varied. Additionally, time interval between final exam and death varied, ranging from 0 to 13 years with a mean of 2.65 years (±2.08) (Figure S1). Descriptive statistics and exclusion criteria are shown in Table 1 and Figure 1. AD lesions were the most common lesion types, with 4448 individuals (84%) presenting with one or more AD lesions. Vascular lesions were the next most common, with 52.9% of participants positive for vascular brain injury and 40.5% positive for arteriolosclerosis. Only 229 participants (4%) were negative for all lesion types. The average NPCS in the full imputed dataset (N = 5272) was 2.98 (±1.7) (Figure 2). Figure 2A shows the distribution of the total burden of neuropathology as measured by the NPCS. When this burden is split by quartiles (Figure 2B) it was evident that the AD lesions drove most of the distribution, particularly the middle quartiles. The non‐AD lesions were not appreciably different between the 2nd and 3rd quartiles, but, as expected, increased in the 4th quartile. The three AD lesions were highly correlated with each other, and moderately correlated with CAA, a related amyloidosis; vascular brain injury and arteriolosclerosis were also correlated (Figure S2).
FIGURE 1.
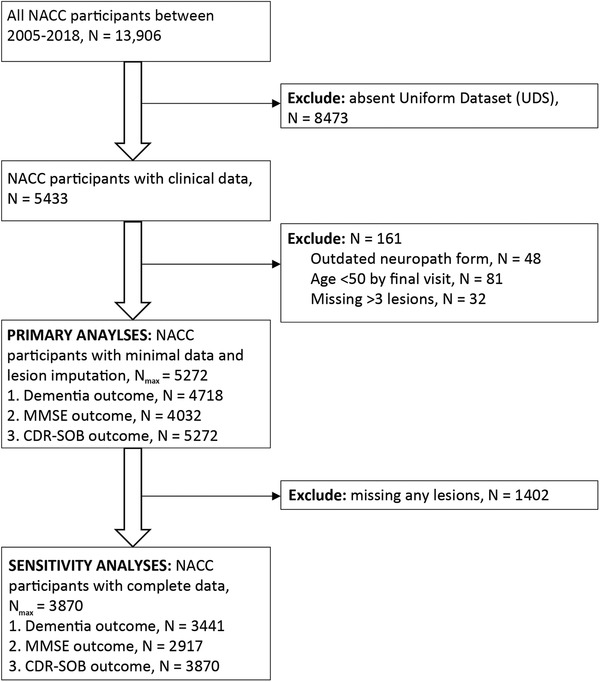
Flow chart of exclusion criteria and analyses. The flowchart shows exclusion criteria and sample sizes in the dataset. The primary dataset allows three or fewer missing lesions to be imputed and analyzed. The clinical outcomes have varying sample sizes as we do not impute the outcome variables. The “complete” data do not allow any missing lesions and were only used in some of the sensitivity analyses. Abbreviations: CDR‐SOB, Clinical Dementia Rating Sum of Boxes; MMSE, Mini‐Mental State Examination; NACC, National Alzheimer's Coordinating Center
FIGURE 2.
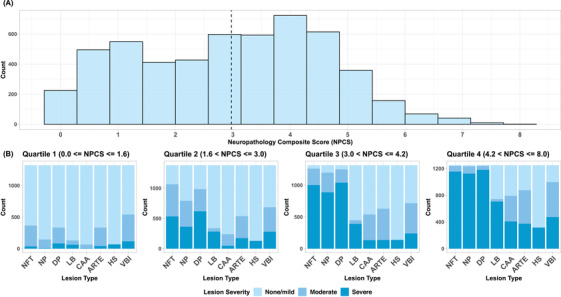
Distributions of neuropathology composite score (NPCS) and lesion severity. (A) Shows the overall distribution of the NPCS (N = 5272). The dashed line notes the mean NPCS of 2.98 (±1.7 SD). This histogram represents the distribution of the NPCS from one imputation with the multivariate imputation by chained equation (MICE) package in R software. Other distributions will vary slightly in number per bin, but will follow the same general shape of this distribution. (B) Shows the distribution of individual lesions, split by quartile of the NPCS. Abbreviations: ARTE, arteriolosclerosis; CAA, cerebral amyloid angiopathy; DP, diffuse plaques; HS, hippocampal sclerosis; LB, Lewy bodies; NFT, neurofibrillary tangles; NP, neuritic plaques; NPCS, neuropathology composite score; VBI, vascular brain injury
Among the cognitively intact (no dementia, CDR‐SOB ≤4, and MMSE ≥24 if available; N = 604) neuropathological lesions were very common (Figure S3). Only 9% of the cognitively intact had no lesions; 33% had one or more moderate lesions (with none severe); and 40% had one or more severe lesions. The median NPCS was 1.4 among this group, indicating that at least half the cognitively intact had neuropathological lesions greater than one severe and one mild/moderate lesion (Figure S3).
Mixed pathologies were very common in the full dataset (Table 2). Of those with any AD pathology (neurofibrillary tangles, neuritic plaques, diffuse plaques; N = 4448), 87% had at least one non‐AD lesion present (N = 3870), and 61% (N = 2728) had one or more severe non‐AD lesions. The rates of mixed pathology did not vary greatly between those deemed “low” AD pathology versus “intermediate/high” AD pathology (Table 2).
TABLE 2.
Categorization of the participants by types of AD pathologic changes and comorbid lesions
| AD pathology | ||||
|---|---|---|---|---|
| No AD pathology | Low | Int/High | ||
| Comorbid non‐AD lesions | None | 229 (28%) | 155 (17%) | 423 (12%) |
| Moderate only | 254 (31%) | 281 (31%) | 861 (24%) | |
| 1 severe | 263 (32%) | 291 (32%) | 1386 (39%) | |
| 2 or more severe | 78 (9%) | 178 (20%) | 873 (25%) | |
| Total | 824 | 905 | 3543 | |
For participants with NACC neuropathology form v10, Low versus Int/High pathology was assessed through NIA‐AA ADNC categorization (NACC variable NPADNC). For individuals with form v9, NIA‐REGAN/CERAD diagnostic criteria were used: “Low” being AD lesions present, but not meeting NIA‐REAGAN criteria; “Int/High” being AD pathology meeting NIA‐REAGAN criteria.
3.2. Association between lesions and dementia
In our analyses of dementia (N = 4718) we observed a significant association between neurofibrillary tangles, neuritic plaques, and dementia (tangles, OR = 11.46, P‐value < 2E‐16; neuritic plaques, OR = 2.33, P‐value = 2.56E‐05) (Table 3); these ORs reflect a one‐unit change in the variable (none/mild vs severe). Among the other non‐AD lesion, hippocampal sclerosis showed the strongest association (OR = 7.37, P‐value = 3.39E‐12), followed by arteriolosclerosis (OR = 2.4, P‐value = 7.59E‐6). Neither diffuse plaques nor vascular brain injury as assessed by the NACC variables collected was associated with dementia in the full model. The NPCS also showed a significant association with dementia (OR = 2.39, P‐value < 2E‐16) when modeled as an additive effect (on the log scale).
TABLE 3.
Association between neuropathology lesions and clinical endpoints
| Dementia (N = 4718) | MMSE (N = 4032) | CDR‐SOB (N = 5272) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | P‐value | β | SE | P‐value | β | SE | P‐value | |
| Age | 0.90 | 0.89‐9.12 | <2E‐16 | 0.23 | 0.01 | <2E‐16 | −0.12 | 0.01 | <2E‐16 |
| Sex | 0.65 | 0.53‐8.05 | 6.56E‐05 | 0.59 | 0.25 | 1.88E‐02 | −0.17 | 0.15 | 2.65E‐01 |
| Hisp | 1.86 | 1.00‐3.47 | 5.18E‐02 | −3.03 | 0.66 | 5.12E‐06 | 2.60 | 0.40 | 6.68E‐11 |
| AA | 0.67 | 0.40‐1.13 | 1.37E‐01 | −2.94 | 0.66 | 9.51E‐06 | 1.07 | 0.40 | 7.94E‐03 |
| NFT | 11.46 | 7.91‐11.66 | <2E‐16 | −6.84 | 0.43 | <2E‐16 | 4.16 | 0.27 | <2E‐16 |
| NP | 2.33 | 1.57‐3.46 | 2.46E‐05 | −2.14 | 0.47 | 4.66E‐06 | 1.54 | 0.29 | 1.23E‐07 |
| DP | 0.88 | 0.64‐1.20 | 4.26E‐01 | 1.27 | 0.42 | 2.40E‐03 | −1.19 | 0.26 | 5.41E‐06 |
| LB | 2.25 | 1.67‐3.03 | 8.92E‐08 | −0.91 | 0.30 | 2.21E‐03 | 0.49 | 0.18 | 6.27E‐03 |
| CAA | 1.80 | 1.14‐2.84 | 1.19E‐02 | −1.97 | 0.42 | 2.62E‐06 | 0.84 | 0.25 | 8.03E‐04 |
| ARTE | 2.40 | 1.64‐3.51 | 7.59E‐06 | −1.66 | 0.42 | 9.60E‐05 | 1.25 | 0.25 | 8.79E‐07 |
| HS | 7.37 | 4.25‐12.78 | 3.39E‐12 | −2.64 | 0.38 | 6.64E‐12 | 2.22 | 0.23 | <2E‐16 |
| VBI | 1.09 | 0.82‐1.45 | 5.72E‐01 | 0.64 | 0.36 | 7.80E‐02 | −0.54 | 0.22 | 1.55E‐02 |
| Age | 0.91 | 0.90‐0.91 | <2E‐16 | 0.24 | 0.01 | <2E‐16 | −0.14 | 0.01 | <2E‐16 |
| Sex | 0.68 | 0.56‐0.85 | 3.45E‐03 | 0.45 | 0.26 | 8.08E‐02 | −0.04 | 0.16 | 8.10E‐01 |
| Hisp | 2.21 | 1.21‐4.05 | 1.01E‐02 | −3.52 | 0.68 | 2.83E‐07 | 2.90 | 0.41 | 1.32E‐12 |
| AA | 0.70 | 0.42‐1.16 | 1.70E‐01 | −2.74 | 0.68 | 6.23E‐05 | 0.98 | 0.41 | 1.82E‐02 |
| NPCS | 2.39 | 2.21‐2.59 | <2E‐16 | −2.01 | 0.08 | <2E‐16 | 1.19 | 0.05 | <2E‐16 |
The upper portion of the table shows results from the full models with lesions parameterized individually, including imputed data. The bottom portion of the table notes the NPCS model. Effect sizes are presented as odds ratios (OR) for the dementia outcome and βs for MMSE and CDR‐SOB outcomes. Bolded items denote P‐value < 0.05. Note that reduced MMSE is indicative of increased impairment, hence the negative effect sizes under the MMSE model.
Abbreviations: AA, African American; ARTE, arteriolosclerosis; CAA, cerebral amyloid angiopathy; CDR‐SOB, Clinical Dementia Rating Sum of Boxes; CI, confidence interval; DP, diffuse plaques; Hisp, Hispanic/Latino; HS, hippocampal sclerosis; LB, Lewy bodies; MMSE, Mini‐Mental State Examination; NFT, neurofibrillary tangles; NP, neuritic plaques; NPCS, neuropathology composite score; SE, standard error; VBI, vascular brain injury.
We assessed model fit using the McKelvey and Zavoina pseudo R 2 estimates. Four models were assessed: a “null” reduced model (age, sex race/ethnicity), an AD model (reduced model plus AD lesions), a full model (reduced model plus all neuropath variables), and an NPCS model (reduced model plus NPCS). The R 2 values were 0.225, 0.495, 0.590, and 0.518, respectively, (see Supplemental Text 3). To replicate White et al., we also analyzed the NPCS as a categorical variable (quartiled, with 0 NPCS as a referent group). 24 This model showed a stronger association with dementia among individuals with upper quartiles of NPCS relative to those with lower (Q4 vs referent: OR = 50.43, P‐value = < 2E‐16.; Q2 vs referent: OR = 6.18, P‐value = 1.55E‐14; see Table S2 for details).
3.3. Association between lesions and neurocognitive impairment
In our assessment of MMSE (N = 4032), neurofibrillary tangles showed the strongest association (β = ‐6.84, P‐value < 2E‐16; Table 3). Vascular brain injury was not associated with MMSE. Similar results were seen in the ordinal logistic regression models (see Table S3). In the full model, all 8 lesions and covariates capture 29% of the variability in the MMSE outcome (R 2 = 0.286). Meanwhile, the reduced model (covariates only) explained only 9.8% of the variability of MMSE (R 2 = 0.098) and the AD model explained 26.5% (R 2 = 0.265). This means that the remaining five non‐AD lesions explain an additional 2% of the total MMSE variability after accounting for AD, age, sex, and race/ethnicity. The NPCS was significantly associated with MMSE (β = ‐2.01, P‐value < 2E‐16), and the model explained 23.5% of the variation in MMSE (R 2 = 0.235). Finally, to replicate White et al., we analyzed the NPCS as an ordinal factor split by quartiles. 24 As in the ordinal analysis for dementia, the effect sizes for upper quartiles of NPCS had stronger associations with MMSE than lower quartiles (Table S2).
3.4. Association between lesions and functional impairment
Analyses of CDR‐SOB (N = 5272) showed similar results as MMSE analyses. Neurofibrillary tangles showed the strongest effect, followed by hippocampal sclerosis, neuritic plaques, and arteriolosclerosis (Table 3), with diffuse plaques and vascular brain injury effect sizes in the opposite direction from the other lesions. Similar results were seen in the ordinal logistic regression models (see Table S3). In the full model, 24% of the variability in the CDR‐SOB is explained (R 2 = 0.237) while the “null” reduced model explained only 8.1% of the variability (R 2 = 0.081) and the AD model (3 AD lesions + covariates) explained 21.4% of the variability (R 2 = 0.214). Finally, the NPCS was significantly associated with CDR‐SOB (β = 1.19, P‐value ≤ 2E‐16); the NPCS model with covariates explained 19% of the variability in CDR‐SOB (R 2 = 0.188). As in the ordinal analyses for dementia and MMSE, the effect sizes for upper quartiles of NPCS had stronger associations with CDR‐SOB than lower quartiles (Table S2).
3.5. Atypical clinical and neuropathological findings
There were 604 cognitively intact individuals (Figure S3). Rates of cognitive resilience (high burden of pathology while maintaining cognitive function) were moderate: 154 participants (25% of all cognitively intact subjects). Few individuals met criteria for neuropathological resistance (advanced age with minimal pathology, NPCS ≤1.4). Only 158 individuals (11% of the 1416 individuals over age 86 at last visit) remained cognitively intact with minimal pathology. Other dementias (dementia with minimal neuropathology) occurred in 749 participants (18% of 4106 with dementia, with NPCS ≤1.4). Many of these had clinical diagnoses for other dementias (eg, frontotemporal lobar degeneration, progressive supranuclear palsy, etc); however, in 29% of these individuals, AD was still clinically diagnosed as the primary disease etiology (NACC variable NACCETPR) (Table S5).
3.6. Time interval sensitivity analysis
One potential limitation to our approach is that the interval between the last clinical assessment and death may impact the effect size estimates in the statistical modeling. Specifically, longer intervals between clinical assessment and death may increase heterogeneity and decrease the magnitude of associations. To test this we performed a sensitivity analysis on interval between last clinical assessment and death (see Supplemental Text 4 and Figure S1). Briefly, we did note a reduction in the magnitude of effect sizes when using the full dataset, relative to those restricted to shorter intervals (see Tables S6 to S9). However, overall the effect sizes were largely robust against longer intervals.
4. DISCUSSION
We present here a large study of the neuropathology in the aging brain (N = 5272) of a clinic‐based population. This study confirms the high rate of AD lesions and related dementia lesions in combination. Most individuals showed some combination of AD lesions (84%) and very few individuals completely lacked the assessed pathologic changes (4%). The percentage of individuals with AD lesions is higher than seen in community‐based studies, such as ROS (60.6) and MAP (59%), 22 but very similar to what has been seen in other considerably smaller (N = 392) clinic‐based cohorts like the clinical core of ROS (83.7%). 22 Of the individuals with AD pathologic changes, 70% had at least one severe lesion from a related dementia. The rate of mixed pathologies is higher than most rates previously reported in other cohorts, likely a reflection of the more heterogeneous clinical populations. Compared to community‐based cohorts, individuals in NACC had more Lewy Bodies, more hippocampal sclerosis, and more mixed pathology. This study also found a high rate of lesions among the cognitively intact, consistent with findings from previous studies. 18 , 22 , 24 , 26 It is not clear if this is due to cognitive resilience (either from cognitive reserve or compensation) or “censoring” due to death.
We observed a strong association between increasing severity of individual lesions and dementia, neurocognitive impairment (MMSE), and functional impairment (CDR‐SOB). The largest effect sizes were seen in neurofibrillary tangles, neuritic plaques, and hippocampal sclerosis. The effect sizes of neurofibrillary tangles and neuritic plaques align well with previous findings in community‐based studies. 18 , 24 , 26 Hippocampal sclerosis is often understudied in combination with the primary AD lesions. In the few community‐based studies that do include hippocampal sclerosis, similar effect sizes were seen. 18 , 24 , 26 Its relatively large contribution to dementia severity indicates it should regularly be included in neuropathologic studies of dementia. The findings were also consistent across a variety of modeling and analysis approaches (see Supplemental Material), lending support to the robustness of the findings.
Diffuse plaques were not associated with the dementia outcome in the full model, likely because of the high correlation between the AD lesions (see Figure S2). A reduced model including diffuse plaques (but lacking tangles and neuritic plaques) did show association with dementia (P‐value ≤2E‐16). Vascular brain injury was also not significantly associated with dementia in the full model, though the effect size was positive. This may be due to inconsistencies in how vascular phenotypes were assessed or coded across ADRCs or could represent a truly weaker effect on dementia outcome (and thus weaker statistical power). A reduced model that included vascular brain injury did show a nominal association with dementia (OR = 1.37, P‐value = 0.01), suggesting some of the signal from vascular brain injury may be captured by the correlation with the arteriolosclerosis phenotype (R 2 = 0.38; see Figure S2).
In the models of MMSE and CDR‐SOB diffuse plaques and vascular brain injury showed effects in the opposite direction than the other lesions types (ie, they look “protective”). This change in direction, however, was not seen in reduced models (diffuse plaques alone, or vascular brain injury alone). Sensitivity analyses suggest that the direction change is due to the correlations among lesions (diffuse correlated with other AD lesions; vascular brain injury correlated with arteriolosclerosis; see Tables S10 and S11) rather than a real “protective” effect. Correlated independent variables in a multivariate regression can have very different main effects relative to simple linear regression of a single variable. In particular, if the variable coding does not reflect the true underlying association (eg, linear coding vs a multiplicative or threshold‐based effect, or unaccounted interactions) the second variable may pick up residual signal in an unanticipated direction. In this case, the diffuse plaque variable is likely picking up residual effects left over from the other AD lesions, and vascular brain injury is picking up residual effects from arteriolosclerosis. The implication is that, in terms of clinical impact and risk, the AD lesions should not be modeled in isolation. Similarly, the vascular phenotype should not be modeled in isolation either. Future studies should explore other, more granular ways of assessing these lesions, their interactions, and modeling their associations with clinical outcomes.
For the MMSE and CDR‐SOB models, we observed increases in the total variability explained when adding the 5 non‐AD lesions to an AD lesions only model. The dementia models showed similar increase in goodness of fit when adding the related‐dementia lesions (see Supplemental Text 3). This confirms that, while AD lesions dominate the models, there is still utility in assessing and modeling other brain lesions. This study also confirms the cumulative effect of multiple lesion types. In particular, upper quartiles of NPCS showed much stronger associations with MMSE and CDR‐SOB than lower quartiles (Table S2). This effect was seen across all three clinical outcomes (dementia, neurocognitive decline, and functional impairment). As such, we recommend, whenever possible, the assessment of multiple neuropathological lesions and types in autopsy programs, even beyond the primary outcomes (eg, assessing Lewy bodies and vascular endpoints in a primarily AD/dementia cohort).
The ascertainment scheme of this study does limit the generalizability. The dataset is weighted towards those with AD or memory impairment and thus is not representative of the general population. Additionally, while the study does include individuals from underrepresented populations (notably African American and Hispanic), the rate of participation in autopsy programs from these populations tends to be low. We also note that more detailed and specific neuropsychiatric and functional measures may improve analyses. For example, the MMSE has been updated to the Montreal Cognitive Assessment (MoCA) in more recent versions of NACC protocols, 46 and more advanced psychometric approaches are available for assessing impairment in specific cognitive domains (eg, executive, memory predominant, visuospatial, etc). 47 , 48 , 49
Taken together, these results confirm the utility of neuropathological collections and show mixed‐type pathologies may be an important contributor to the likelihood and severity of dementia in clinic‐based populations. Further approaches are required to understand the role of these lesions in rates and progression of disease, influence on impairment in specific neuropsychiatric domains, the role of mitigating factors such as education, and the contribution of genetic factors such as APOE genotype or other genetic risk factors.
CONFLICT OF INTEREST
MC reports no conflicts of interest.
ERM, MAPV, and GWB report grant funding through the NIH.
GS reports grant funding through the NIH and an honorarium from the BrightFocus Foundation for grant review.
WKS reports grant funding through the NIH and an honorarium from the University of Chicago for speaking at a seminar.
WK reports grant funding through the NIH, honoraria for serving on external advisory committees (Boston U ADRC, USC ADRC, UCI ADR, Mt. Sinai ADRC), honoraria for grant review (NIH: NIA/CSR), and travel support from the Alzheimer Association for participation in a symposium.
TM reports grant funding through the NIH and Farmer Family Foundation, royalty payments from UpToDate, honoraria and travel support for invited lectures, and has patents pending on novel chemical matter for treatment or imaging of neurodegenerative diseases.
DG is an unpaid member of the American Society for Human Genetics career development committee.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
This work was supported in part by the NIH National Institutes on Aging (grant number R01 AG062695). The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA‐funded ADRCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P30 AG062428‐01 (PI James Leverenz, MD) P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P30 AG062421‐01 (PI Bradley Hyman, MD, PhD), P30 AG062422‐01 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Thomas Wisniewski, MD), P30 AG013854 (PI Robert Vassar, PhD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P30 AG062429‐01 (PI James Brewer, MD, PhD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG053760 (PI Henry Paulson, MD, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG049638 (PI Suzanne Craft, PhD), P50 AG005136 (PI Thomas Grabowski, MD), P30 AG062715‐01 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), P50 AG047270 (PI Stephen Strittmatter, MD, PhD).
Godrich D, Martin ER, Schellenberg G, et al. Neuropathological lesions and their contribution to dementia and cognitive impairment in a heterogeneous clinical population. Alzheimer's Dement. 2022;18:2403–2412. 10.1002/alz.12516
REFERENCES
- 1. Fleming R, Zeisel J, Bennett K. 2020. World Alzheimer Report 2020: Design Dignity Dementia: Dementia‐related Design and the Built Environment. Vol 1. London, England: Alzheimer's Disease International; 2020. [Google Scholar]
- 2. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wolf DS, Gearing M, Snowdon DA, Mori H, Markesbery WR, Mirra SS. Progression of regional neuropathology in Alzheimer disease and normal elderly: findings from the Nun Study. Alzheimer Dis Assoc Disord. 1999;13(4):226‐231. [DOI] [PubMed] [Google Scholar]
- 5. Mirra SS, Hart MN, Terry RD. Making the diagnosis of Alzheimer's disease: a primer for practicing pathologists. Arch Pathol Lab Med. 1993;117:132‐144. [PubMed] [Google Scholar]
- 6. Hyman BT, Trojanowski JQ. Editorial on consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56(10):1095‐1097. [DOI] [PubMed] [Google Scholar]
- 7. Schneider JA, Arvanitakis Z, Yu L, Boyle PA, Leurgans SE, Bennett DA. Cognitive impairment, decline and fluctuations in older community‐dwelling subjects with Lewy bodies. Brain. 2012;135:3005‐3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89:88‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol. 2011;69:320‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boyle PA, Yu L, Nag S, et al. Cerebral amyloid angiopathy and cognitive outcomes in community‐based older persons. Neurology. 2015;85:1930‐1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arvanitakis Z, Capuano AW, Leurgans SE, Buchman AS, Bennett DA, Schneider JA. The relationship of cerebral vessel pathology to brain microinfarcts. Brain Pathol. 2017;27:77‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kryscio RJ, Abner EL, Nelson PT, et al. The effect of vascular neuropathology on late‐life cognition: results from the SMART project. J Prev Alzheimer's Dis. 2016;3:85‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olichney JM, Hansen LA, Hofstetter CR, Lee JH, Katzman R, Thal LJ. Association between severe cerebral amyloid angiopathy and cerebrovascular lesions in Alzheimer disease is not a spurious one attributable to apolipoprotein E4. Arch Neurol. 2000;57:869‐874. [DOI] [PubMed] [Google Scholar]
- 14. Ighodaro ET, Abner EL, Fardo DW, et al. Risk factors and global cognitive status related to brain arteriolosclerosis in elderly individuals. J Cereb Blood Flow Metab. 2017;37:201‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crystal H, Schneider J, Bennett D, Leurgans S, Levine S. Associations of cerebrovascular and Alzheimer's disease pathology with brain atrophy. Curr Alzheimer Res. 2014;11(4):309‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP‐43 pathology in aging and Alzheimer disease. Ann Neurol. 2015;77:942‐952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Snowdon DA. Aging and Alzheimer's disease: lessons from the Nun Study. Gerontologist. 1997;37(2):150‐156. [DOI] [PubMed] [Google Scholar]
- 18. Kawas CH, Kim RC, Sonnen JA, Bullain SS, Trieu T, Corrada MM. Multiple pathologies are common and related to dementia in the oldest‐old: the 90 + Study. Neurology. 2015;85(6):535‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017;134(2):171‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jellinger KA, Attems J. Challenges of multimorbidity of the aging brain: a critical update. J Neural Transm. 2015;122(4):505‐521. [DOI] [PubMed] [Google Scholar]
- 21. Brenowitz WD, Hubbard RA, Keene CD, et al. Mixed neuropathologies and estimated rates of clinical progression in a large autopsy sample. Alzheimer's Dement. 2017;13(6):654‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimer's Dis. 2009;18(3):691‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rahimi J, Kovacs GG. Prevalence of mixed pathologies in the aging brain. Alzheimer's Res Ther. 2014;6(9):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. White LR, Edland SD, Hemmy LS, et al. Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu‐Asia Aging Studies. Neurology. 2016;86(11):1000‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Montine TJ, Sonnen JA, Montine KS, Crane PK, Larson EB. Adult Changes in Thought study: dementia is an individually varying convergent syndrome with prevalent clinically silent diseases that may be modified by some commonly used therapeutics. Curr Alzheimer Res. 2012;9(6):718‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious Orders Study and Rush Memory and Aging Project. J Alzheimer's Dis. 2018;64(s1):S161‐S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. White L. Brain lesions at autopsy in older Japanese‐American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu‐Asia aging study. J Alzheimer's Dis. 2009;18(3):713‐725. [DOI] [PubMed] [Google Scholar]
- 28. Beekly DL, Ramos EM, van Belle G, et al. The National Alzheimer's Coordinating Center (NACC) database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18:270‐277. [PubMed] [Google Scholar]
- 29. Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord. 2007;21(3):249‐258. [DOI] [PubMed] [Google Scholar]
- 30. Besser L, Kukull W, Knopman DS, et al. Version 3 of the National Alzheimer's Coordinating Center's Uniform Data Set. Alzheimer Dis Assoc Disord. 2018;32(4):351‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Besser LM, Kukull WA, Teylan MA, et al. The revised National Alzheimer's Coordinating Center's neuropathology form‐available data and new analyses. J Neuropathol Exp Neurol. 2018;77:717‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weintraub S, Salmon D, Mercaldo N, et al. The Alzheimer's Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord. 2009;23(2):91‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weintraub S, Besser L, Dodge HH, et al. Version 3 of the Alzheimer Disease Centers’ Neuropsychological Test Battery in the Uniform Data Set (UDS). Alzheimer Dis Assoc Disord. 2018;32(1):10‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morris JC, Weintraub S, Chui HC, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord. 2006;20(4):210‐216. [DOI] [PubMed] [Google Scholar]
- 35. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189‐198. [DOI] [PubMed] [Google Scholar]
- 36. Kukull WA, Larson EB, Teri L, Bowen J, McCormick W, Pfanschmidt ML. The Mini‐Mental State Examination score and the clinical diagnosis of dementia. J Clin Epidemiol. 1994;47:1061‐1067. [DOI] [PubMed] [Google Scholar]
- 37. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412‐2414. [DOI] [PubMed] [Google Scholar]
- 38. Van Buuren S, Groothuis‐Oudshoorn K. mice: Multivariate Imputation by Chained Equations in R. Journal of Statistical Software. 2011;45(3):1‐67. [Google Scholar]
- 39. Tombaugh TN, McDowell I, Kristjansson B, Hubley AM. Mini‐Mental State Examination (MMSE) and the Modified MMSE (3MS): a psychometric comparison and normative data. Psychol Assess. 1996;8:48‐59. [Google Scholar]
- 40. Crum RM, Anthony JC, Bassett SS, Folstein MF. Population‐based norms for the Mini‐Mental State Examination by age and educational level. JAMA. 1993;269:2386‐2391. [PubMed] [Google Scholar]
- 41. O'Bryant SE, Waring SC, Cullum CM, et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer's research consortium study. Arch Neurol. 2008;65:1091‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. R Core Team . R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2021. https://www.R‐project.org/ [Google Scholar]
- 43. Wright K corrgram: Plot a Correlogram. R package version 1.13. 2018. https://CRAN.R‐project.org/package=corrgram
- 44. Harel O. The estimation of R 2 and adjusted R 2 in incomplete data sets using multiple imputation. J Appl Stat. 2009;36(10):1109‐1118. [Google Scholar]
- 45. Signorell A, Aho K & Alfons A et al. DescTools: Tools for Descriptive Statistics. R package version 0.99.40. 2021. https://cran.r‐project.org/package=DescTools
- 46. Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695‐699. [DOI] [PubMed] [Google Scholar]
- 47. Shen J, Crane P, Gao S. A latent variable model approach for assembling and scoring screening tests for dementia. J Alzheimer's Dis. 2003;5:399‐407. [DOI] [PubMed] [Google Scholar]
- 48. Mukherjee S, Trittschuh E, Gibbons LE, Mackin RS, Saykin A, Crane PK. Dysexecutive and amnesic AD subtypes defined by single indicator and modern psychometric approaches: relationships with SNPs in ADNI. Brain Imaging Behav. 2012;6(4):649‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Choi S, Mukherjee S, Gibbons LE, et al. Development and validation of language and visuospatial composite scores in ADNI. Alzheimer's Dement Transl Res Clin Interv. 2020;6(1):e12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information