

Abstract
Breast cancer (BC) affects over 250,000 women in the US each year. Drug-resistant cancer cells are responsible for most breast cancer fatalities. Scientists are developing novel chemotherapeutic drugs and targeted therapy combinations to overcome cancer cell resistance. Combining drugs can reduce the chances of a tumor developing resistance to treatment. Clinical research has shown that combination chemotherapy enhances or improves survival, depending on the patient’s response to treatment. Combination therapy is a highly successful supplemental cancer treatment. This review sheds light on intrinsic resistance to BC drugs and the importance of combination therapy for BC treatment. In addition to recurrence and metastasis of BC, the article discussed biomarkers for BC.
Keywords: Breast cancer, intrinsic/acquired resistance, breast cancer recurrence, breast cancer metastasis, breast cancer biomarkers, combination therapies
Introduction
The number of women diagnosed with BC in the US is estimated to be over 250,000 annually [1]. Better treatment choices have helped to reduce BC mortality in developed countries [2]. The advanced stage of BC diagnosis is likely a result of a failure in current BC prevention methods [3], which may have also contributed to an increase in BC incidence [1]. BC is diagnosed in one in eight women aged 85 or older in high-income countries. Preventing breast cancer may be the most cost-effective and socially beneficial strategy [4]. Different subtypes of BC are categorized by their histological appearance and the presence of hormone receptors and growth factors, like ER, PR, and ERBB2. There is a correlation between ER-positive breast cancer and cancer mortality [5-7]. BC is caused by a combination of inherited and non-hereditary risk factors. Various breast cancer-related single nucleotide polymorphisms are frequent in addition to BRCA1 and BRCA2 mutations [8].
Chemoresistance is a key issue in breast cancer treatment. Understanding their chemoresistance mechanisms is challenging. Additional effective treatments are urgently needed to address the drug resistance and improve current therapeutic regimens. The problem can be addressed with innovative agents and carriers of drugs and a combination of treatments. Other emerging medicines, like gene therapy and immune-based therapies, are being evaluated for their ability to overcome drug resistance [9,10]. This review will discuss the intrinsic drug resistance to breast cancer therapy, breast cancer recurrence, metastasis, predictive biomarkers for breast cancer, and combination therapy to combat breast cancer.
Breast cancer metastasis
The metastatic process ends after a series of sequential steps have been completed [11]. If tumor cells have already invaded the surrounding host tissue, they can spread to lymphatic or blood vessels and other organs and tissues [12]. The cancer cells distribute throughout the body via blood and lymphatic veins. Angiogenesis and proliferation occur when tumor cells are in cell cycle arrest, preventing tumor cells from entering the target organ’s parenchyma [13]. Both apoptosis and immunosuppression must be avoided [14]. If these phases successfully spread cancer, there may be “metastases of metastases” [15]. In its early stages, breast cancer can damage the auxiliary lymph nodes, lungs, bones, brain, liver, and peritoneum. BC that has spread to the bones is a regular occurrence. The metastatic spread of breast cancer tumors to the bones is approximately 67 percent. Breast tumors with luminal B (79%) and luminal A (70%) are more likely to metastasize to the bones, whereas HER2+ and TNBC (basal-like) have 60% and 40% chances, respectively. Breast cancer metastases are often found in the liver, auxiliary lymph nodes, and lungs. In about 37% of instances, advanced BC will spread to the liver and lungs, and 30-50% will spread to the auxiliary lymph nodes. TNBC with HER2+ liver metastases is less common than luminal breast cancer. TNBC has a 35% likelihood of metastasizing to the liver, while HER2+ has a 45% chance. TNBC has a higher risk of angiogenesis and infection of auxiliary lymph nodes than other subtypes. Luminal A and B had a reduced likelihood of metastasis in the lungs (25-30%) than TNBC and HER2+ (45-35%). Metastases to the liver, lungs, or brain can substantially reduce a patient’s survival time. Metastases to other organs affect 12.6 percent of cancer patients. TNBC and HER2+ breast tumors reported more metastases in this metastatic zone (25-30%) than Luminal A and Luminal B (5-15%). Less common metastatic locations include the mammary internal chain lymph nodes (10-40%), contralateral breast (6%), and supraclavicular lymph nodes (1-4%) [16]. Figure 1 displays a slew of distant breast cancer metastases.
Figure 1.
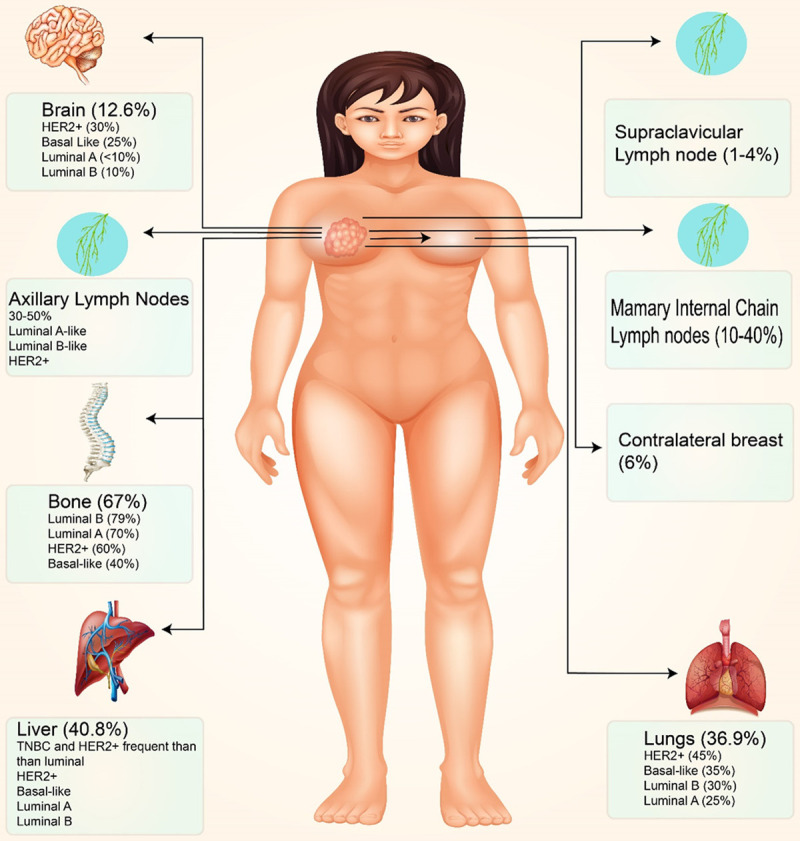
This figure depicts the common locations of breast cancer metastases. Breast cancer can spread to bones, axillary lymph nodes, the liver, and the lungs. A tiny fraction of breast tumors can spread to the inner mammary gland, accounting for 10% to 40% of cases. Breast cancer can spread to different body regions based on the molecular subtype. When comparing luminal tumors to TNBC, lymphatic dissemination in TNBC is less common than in luminal malignancies.
The invasion of malignant cells is the first step in the metastasis process. Before tumor cells invade, their attachment to the extracellular matrix must be altered. The cadherin family, which increases cell-to-cell adhesion, has been linked to BC metastasis [17]. E-cadherin, a cell-to-cell adhesion protein, is required to spread metastatic BC cells [18]. E-cadherin downregulation has been linked to the progression and metastasis of BC, which has a poor prognosis [19,20]. Breast cancer of the lobular type is associated with defective E-cadherin, resulting in its dysfunction [21]. Integrins play a role in tumor cell adhesion to the ECM (34). The ECM is degraded during the invasion process. This enables invasion of tissue boundaries. ECM degradation is aided by metalloproteinases (MMPs) and the urokinase plasminogen activator system (UPA) [22,23]. The UPA levels of BC patients are a reliable predictor of the development of distant metastases [24]. Patients with a good prognosis are also included in this group [25]. According to Huang et al., small interfering RNA (siRNA) suppression of UPA reduced invasion and MMP9 production. MMPs promote ECM proteolysis in breast cancer cell lines that invade the ECM’s invadopodia front [26,27]. Tumor cells must have spread from their original location to other body parts to be considered invasive. Tumor cells can move both individually and in groups [28]. Tumor cells with high differentiation prefer ‘organized migration’ in breast lobular carcinoma [29]. Intercellular adhesion proteins may have structural and functional defects in poorly differentiated cancers that cause cell movement to shift from coordinated to single-cell migration [28]. Intercellular linkages must be established for tumor cells to migrate in groups. Following invasion and intravasation, emboli are discovered in blood and lymphatic arteries [30,31].
Consider the “seed and soil” approach to understanding how cancer spreads. Tumor cells multiply when they contact suitable “soil” [32]. The tumor microenvironment is thought to influence metastasis [33]. The microenvironment in which metastatic tumor cells survive influences their proliferative ability. A favorable microenvironment is required for tumor growth and progression [34]. The microenvironment, which includes fibroblasts, immune cells, endothelial cells, and the ECM, regulates tumor growth [35].
Furthermore, tumor cells create pre-metastatic niches by producing chemicals that enrich the soil before metastasis [36]. The primary tumor sent signals that induced MMP9 synthesis in lung endothelial cells and macrophages, resulting in tumor invasion into the lungs before metastasis [37]. In the pre-metastatic lymph nodes of patients with early-stage BC, there is a cluster of VEGFR-1-positive hematopoietic progenitor cells [36]. Unlike other cancers, breast cancer prefers bone and lung metastases over other organs (such as the liver and brain) [38]. Metastasizing BC cells prefer the bone marrow and lungs as evidence of tissue tropism [39,40]. Chemokines have been found to allow cancer cells to migrate to specific organs.
CXCL12, the chemokine ligand 12 found in breast cancer tissue, is not present in the brain, skin, or skeletal muscle, unlike the CXCR4 receptor and its ligand CXCL12 [41]. Metastatic breast cancer is more likely to spread to CXCL12-producing organs [42]. According to Muller et al., cancer cells can migrate to common sites of metastasis via a connection between CXCR4 and CXCL12 [41]. Tumor vascular growth is also required for metastasis. Angiogenesis is essential for metastasis initiation and progression [43]. It is a distinguishing feature of cancer and a necessary adaptation to the microenvironment for malignancies [44]. As a result of carcinogenesis, pro-angiogenic and anti-angiogenic components become more evenly distributed [45,46]. The ‘angiogenic switch’ process is influenced by some factors, including genetic changes and mechanical stress [47,48]. Tumor vasculature differs from normal vasculature in form, function, and gene expression. Tumor hypoxia is caused by abnormal blood vessels’ insufficient oxygen delivery to the tumor [49]. Tumor cells produce more pro-angiogenic chemicals, leading to abnormal vasculature. As a result, the recursive pattern is repeated indefinitely. Invasive and metastatic mechanisms are activated to avoid the hypoxic environment produced by this cycle [50].
Breast cancer recurrence
BC recurrence accounts for up to half of all fatalities [51]. Experts in breast cancer recurrence have been working worldwide to identify the causes. Other breast cancer subtypes, such as TNBC and all three estrogen, progesterone, and HER2/ErbB2 receptor-negative breast tumors, have also been studied. According to one study, the recurrence rates of breast cancer subtypes differ [52,53]. In the first five years after diagnosis, recurrence rates for ER-negative breast cancers are higher than those for ER-positive BC. A decade after diagnosis, ER-positive breast tumor recurrence rates gradually increase for the next decade before leveling off at 15 years for both subtypes of BC. BC in situ is more likely to recur in women with low estrogen, progesterone, and testosterone levels than in women with high estrogen, progesterone, and testosterone levels [54].
Lymph node metastases are common in late-stage BC and are associated with poor disease-free survival rates [55]. In addition, discovering genetic markers for lymph node metastases may help detect them early and reduce the risk of recurrence [56].
Surgical intervention is an option for patients with advanced BC [57]. The type of surgery depends on the stage of the cancer. BC that is detected early is usually treated with radiation therapy and breast-conserving surgery [58,59]. In most cases, additional radiation treatment is not required following a mastectomy [60]. Age, ductal carcinoma in situ, and lymph node involvement after mastectomy are risk factors for breast conservation therapy. Patients who choose a surgical approach based on clinical and histologic factors will need more personalized treatments and follow-ups [61]. Whether or not a patient had a mastectomy for BC, the chances of recurrence are the same. According to a meta-analysis, radiation therapy lowers the likelihood of recurrence [62]. Cancer patients with advanced BC who receive radiation therapy have been shown to have a 50% reduction in recurrence and death rates. This study discovered several additional variables that may influence radiation efficacy. The surgery required is affected by age, cancer grade, estrogen receptor status, and tamoxifen use.
According to a study, recurrence rates after mastectomy and BCS vary by molecular subtype. The 12,592 participants in the study had 57 percent breast conservation and 43 percent mastectomy. TNBC and HER2-positive cancers had lower recurrence rates after breast-conserving therapy. Postmastectomy recurrence, ER, and PR-positive patients performed better than TNBC and HER2. Patients with HER2-overexpression had a higher risk of recurrence after breast-conserving surgery than TNBC patients. The risk of recurrence in HER2-positive mastectomy patients was not higher than in TNBC patients. Thus, focusing on molecular subgroups is critical in treating breast cancer patients [63]. Some cancers begin recurring before being identified and mistakenly diagnosed as a single tumor. True local recurrences may never spread to other parts of the body. BC was suggested as a viable option for these studies due to the available clinical data. Oncogenes and wound healing (WOWH) theories may also be linked to cancer recurrence [64]. Precancerous lesions, cancer, oncogenesis, wound healing, and cancer recurrence is interconnected. In response to physical, chemical, or biological “wounds”, these oncogenes produce cytokines that cause stem cells to recruit and tissues to remodel (such as chronic inflammation, aging, or reactive oxygen species). All of this leads to the development of malignant tumors.
Intrinsic/acquired drug resistance and mechanisms of resistance to breast cancer therapy
Tumors that do not respond to treatment initially are considered intrinsically resistant, while those that have relapsed after responding initially are considered acquired resistant. Here we will shed light on the Intrinsic/acquired resistance and mechanisms involved in drug resistance.
Intrinsic/acquired drug resistance
Despite advances in BC treatment [65], chemotherapy helps most people with breast cancer but rarely cures metastatic cancer. Drug resistance is either innate or acquired. Innate and acquired resistance differ based on the classification of clinical resistance. Intrinsic resistance occurs when a patient does not improve after a single treatment attempt, whereas acquired resistance occurs after multiple therapy failures [66]. Anti-cancer cells have many built-in mechanisms for surviving chemotherapy treatment. The efflux pumps cause a reduction in drug concentration in cells [67] and a follow-up [68]. Enzymes such as cytochrome p450 and glutathione transferases that break down biochemically might degrade therapeutic substances [69,70]. Drug concentrations may be reduced as a result of poor vascularisation [71,72], ECM interactions, and compounds secreted into the tumor environment [73]. A drug’s intrinsic resistance mechanisms play a key role in establishing the initial response to the drug and affecting subsequent outcomes that can lead to acquired resistance (Figure 2A).
Figure 2.
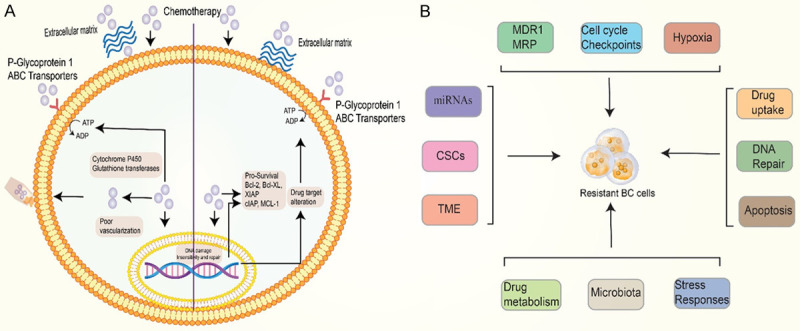
A. Illustrates the Extrinsic and intrinsic factors such as extracellular matrix proteins, P-gp pump and ABC transporter efflux, and cytochrome P450 and glutathione transferases that are involved in the pathogenesis of cancer. B. Resistance to anticancer drugs can be caused by several mechanisms. As shown in the diagram above, cancer cells can survive and relapse in multiple ways. The mechanism includes MDR1 and MRP genes, miRNAs, CSCs, TME, Hypoxia, Cell cycle checkpoints, Drug uptake, DNA repair, Apoptosis, Drug metabolism, Stress response, and microbiota important mechanisms.
Cancer cells’ resistance causes most breast cancer-related fatalities to commonly prescribed treatments. Scientists are working to better understand the genesis of cancer cell resistance and develop novel chemotherapeutic medications or combinations of targeted therapies to improve survival rates. Breast cancer targeted therapy necessitates the expression of specific proteins, such as ER and HER2. Clinical evidence suggests that when there is no ER, endocrine therapy is less likely to be successful [74,75]. Preclinical research suggests that overexpression of HER2 is essential for trastuzumab response [76]. Targeted therapy depletes the ER and HER2 receptors, which is associated with a worse prognosis [77,78]. Targeted therapy may not directly affect ER and HER2 expression. The selection of cancer cells under therapeutic stress is a classic example of clonal selection.
Taxanes and anthracycline are the most potent anti-cancer drugs. Anthracycline causes cell death by binding to topoisomerase II, resulting in DNA breakage [79]. Taxanes attach to α-tubulin and stabilize microtubules, causing mitotic arrest and cell death [80]. Topo-II and α-tubulin are involved in the regulation of target proteins. Topo-II expression and location in the nucleus have influenced anthracycline sensitivity in preclinical mice [81]. Some cell lines’ responses have been linked to Topo-II levels [82]. Topo-II expression fluctuates in tumor tissues, and protein levels do not predict prognosis [83,84]. Topo-II had an AUC of 0.8641 for breast cancer detection, accurately detecting it in 72.22 percent of patients. Compared to those with high or low Topo II and ERBB2 expression, those with low expression showed a considerably higher survival rate [85]. The response to anthracycline in tumors with high topo-II expression has been contradictory in clinical trials, with some studies indicating a better response and others demonstrating no link [81].
Taxanes influence mitosis and intracellular mobility via interfering with microtubule formation. Several isotypes of α-tubulin have been discovered based on variations in the C-terminus [86]. In breast cancer cell lines, tubulin polymerization occurs more slowly, and the stabilizing effect of taxanes is less efficient [87]. Some clinical studies have revealed that patients with high III-tubulin concentrations may not respond well to taxanes [88]; others have identified no such link [89]. In taxanes-resistant breast cancer cells, Let-7c-5p and miR-335 are two significant regulatory variables. CXCL9, CCR7, and SOCS1 are the principal targets of two miRNAs [90].
Anthracycline and taxanes must have high intracellular concentrations to be effective. ATP-binding cassette transporters are drug efflux pumps. The role of ABC transporters (ABCA-ABCG) in human chemotherapeutic resistance is unknown [91,92]. The luminal and biliary canal membranes of the renal tubules and the biliary canalicular cells of the liver aid in detoxification. The luminal membrane of the tubules contains drug efflux pumps [91]. ABCB1 (also known as efflux-related drug resistance in breast cancer) is a multidrug-resistant breast cancer gene that produces P-glycoprotein protein. This protein functions as a mediator. Higher levels of P-gp expression are typically reported in cancer cells that have survived chemotherapy treatment in vitro [93]. P-transmembrane gp sections attach to neutral or positively charged hydrophobic substances, causing the protein to confirm and release the bound drug into the cytoplasm [67]. There is a relationship between taxanes and P-broad gp substrate specificity. A meta-analysis of 31 single-institutional trials demonstrated P-gp expression in almost 40% of breast cancers [94]. Chemotherapy can target a clone of cells with overexpression of P-gp, either directly or by selective selection. However, because P-gp expression varies considerably between studies, using this biomarker to guide treatment selection in solid tumors is problematic. Currently, there is no evidence to support the use of this biomarker to select treatment options for a specific patient’s illness. The MRP1 can transport taxanes, anthracycline, and other hydrophobic substrates [67]. Preclinical research suggests MRP1 is implicated in breast cancer resistance, although early clinical trials have shown that MRP1 does not efficiently identify patients for treatment. Transporter proteins, such as pulmonary resistance-related proteins, are required to deliver drugs into cells [67]. Anthracycline cytoplasmic redistribution could be an effective mechanism for anthracycline resistance.
Mechanisms of breast cancer resistance
Because some breast cancer patients may relapse [95], we need to better understand the resistance mechanisms. The following are examples of breast cancer resistance mechanisms: drugs are degraded by enzyme systems, hormone receptors are influenced, and the cell membrane is involved in drug absorption, transportation, and efflux. In a xenograft model, trastuzumab modulates pro and antiangiogenic effects and normalizes and suppresses human HER2-overexpressing breast cancer vasculature. According to available data, trastuzumab affects cancer cells that are not dependent on VEGF production when injected intravenously [96]. Invivo, a decrease in micro-vessels was observed [97]. Compared to controls, tumor-bearing mice treated with trastuzumab had lower pro-angiogenic factors and higher antiangiogenic factors. The most important mechanisms of trastuzumab resistance in BC are illustrated in Figure 3.
Figure 3.
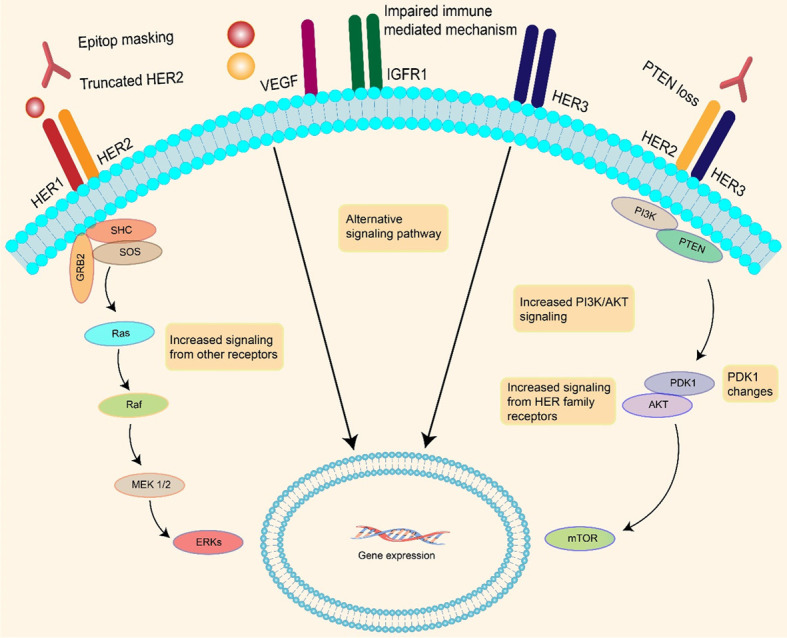
Both HER2 truncation and epitope masking reduce the amount of trastuzumab that can bind to HER2 in breast cancer. PI3K/Akt activity is also increased, and the Phosphodiesterase 1 (PDK1) gene has been changed. These pathways are being triggered at a considerably higher pace than they were previously. The immune system is also impaired.
Twist influences the invasion and metastasis of tumors as an epithelial-mesenchymal transition regulator. Many genes, including E-cadherin, use the E-box transcription initiation sequence. They allow non-invasive cells to invade if the function or expression of E-cadherin is reduced or eliminated. Twist, for instance, can be used to identify the E-box on the promoter of E-cadherin and reduce transcription. As a result, cancer cells in the breast will spread more easily. With the use of Twist, cancer cells can become more resistant to chemotherapy [98]. It has been demonstrated that twist-1 upregulation is linked to chemotherapy resistance [99]. It has also been demonstrated that breast cancers with twist overexpression have a reduced ability to utilize the ER protein [100]. Breast cancer mediates resistance to cancer therapy via different mechanisms (Figure 2B).
MDR1 and MRP gene in conferring resistance to breast cancer therapy
In cancer, the MDR1 gene plays an important role in determining resistance to therapy. P-gp, GST-, and P53 are MDR1-encoded proteins linked to drug resistance [101]. Some potential molecular pathways were identified using aryl pyrimidine androstane derivatives that inhibit MDR1 in adenocarcinoma cells. There is evidence that the acetylation state of aryl pyrimidine influences their MDR1 inhibitory activity. We may discover new MDR1 inhibitors in the future that can reduce drug resistance and retrain cancer cells to respond to treatment [102]. ATP hydrolysis-dependent effluent transporters, such as MDR, are found in cancer cells [103]; MRP, ABCC subfamily, and breast cancer resistant protein are examples of ABC transporters [104]. In the presence of glycoprotein P-170 and P-150 overexpression, glutathione and DNA repair enzymes are increased, and DNA topoisomerase is reduced, promoting drug efflux.
Tumor cells may become resistant to treatment if MRP affects intracellular drug excretion and redistribution. MRP-mediated resistance is less potent than P-gp-mediated resistance [105,106]. The BCRP transporter, unlike P-gp and MRP, is a half-size ABC superfamily member. There are many structural and functional similarities between the two. BCRP methotrexate, mitoxantrone, and camptothecin derivatives [107] have a much lower efflux capacity than P-gp and MRP1 [108].
Cryo-vaulted proteins contain LRP, an ABC transporter protein. In addition to preventing nuclear-targeted drugs from entering the nucleus, LRPs can transport anti-tumor medications into vesicles exocytosing from the cell membrane, reducing intracellular drug concentrations.
Role of miRNAs in mediating resistance to breast cancer treatment
According to a new study, miRNAs may regulate the chemosensitivity of BC cells. In the MCF-7 cell line, miR-451 and miR-326 were found to downregulate MDR-1 and MRP-1 expression, resulting in increased doxorubicin sensitivity [109,110]. A decrease in ERα expression in BC cells can lead to resistance to endocrine therapy [111]. Let-7b/Let-7i, miR-221/222, miR-342-3p, and miR-873 inhibited tamoxifen-resistant breast cancer cells in vitro [112,113]. Furthermore, miRNA influences ERα post-translational modification, affecting endocrine resistance [113]. MDR reversal may be facilitated by miRNA-129-inhibition of ABCB1 expression [114,115]. miR-200c increases chemosensitivity by suppressing TUBB3, ZEB1, and ZEB2 in cancer cells [115]. Paclitaxel resistance can be aided by inhibiting the BAK1 [116]. MiR-218, which targets the BRCA1 gene, regulates cisplatin chemosensitivity in BC [116]. Treatment with curcumin had superior ORR and performance to paclitaxel and placebo after 12 weeks. Intravenous curcumin was well-tolerated and had little effect on patient outcomes [117]. Taxanes’ resistance to their effects is regulated by Let-7c-5p and miR-335-5p. Two miRNAs can also direct their activity to specific downstream cells. Taxanes have no effect on the Let-7c-5p/CCR7/SOCS1 axis or the miR-335-5p/CXCL9 axis. Furthermore, the axes miR-335-5p/CXCL9 and let-7c-5p/CCR7/SOCS1/CXCL9 may influence immune cell chemo-resistance [90].
Cancer stem cells and resistance to breast cancer therapy
Chemoresistance in BC is primarily caused by cancer stem cells (CSCs) [118]. ABC transporters over-expressed in CSCs include P-gp, ABCG2, ABCC1, and ABCB5. CSCs can develop drug resistance by increasing anti-apoptotic levels and repairing DNA [119]. ALDH1 overexpression has also been linked to CSC medication resistance [40]. Notch, Hedgehog [120], and Wnt/β-catenin, all involved in stem cell self-renewal and maintenance, may also be involved in CSC drug resistance. Less Let-7 microRNA is expressed in BC stem cells. The let-7 microRNA can assist BC stem cells in specializing and becoming more chemosensitive [121].
Cancer cells come in various morphologies and genotypes, resulting in a wide range of malignancies. There is intramural as well as patient-specific heterogeneity in cancer. Heterogeneity is aided by random gene expression and chromosomal rearrangement. It is widely assumed that cancer heterogeneity is caused by intrinsic causes [122,123]. Additional tumor cell contacts and the paracrine signal are possible [124,125]. Understanding tumor heterogeneity may result in more effective treatments and decrease drug resistance in breast cancer patients.
Hypoxia-mediated drug resistance to breast cancer therapy
Tumors use glycolysis as their sole source of energy during hypoxia. Hypoxia-induced increases in metabolic enzyme production may render chemotherapy ineffective [126]. Researchers discovered that hypoxia reduced Topo II expression in cancer cells, making them more resistant to chemotherapy [127]. Hypoxia-induced MRP expression influences tumor resistance as well. If the glycolysis process is blocked in anoxic circumstances, drug-resistant cancer cells may become sensitive to chemotherapeutic treatments again [128]. Under hypoxic conditions, HIF-1 forms a heterodimer in both animals and humans. Hyperoxic environments can harm HIF-1, which is sensitive to oxygen concentration. As a result, HIF-1 modulates HIF-1 activity. MDR1 refers to the P-coding gp’s gene MDR1 in the context of hypoxia. MDR1 has hypoxia response elements (HREs) that bind to and influence P-gp expression [129]. HIF-1 and HIF-2 bind to the HRE on the BCRP gene promoter to initiate BCRP expression and, as a result, produce BCRPl overexpression [130,131].
Cancer cells that can move to other body parts are more likely to resist treatment [132,133]. The proton pump (H+)-vacuolar-ATPase (V-ATPase) can change tissue pH. The V-ATPase transfers H+ into the acidic vacuole lumen and/or into the external environment. This proton pump was discovered to be more common in cancer patients [134,135].
When breast cancer cells connect and communicate with stromal cells, they may activate paracrine and neighboring signaling pathways. This could contribute to chemotherapy resistance mediated by stromal cells [136]. HS5 stromal cell signaling reduced the amount of MCF7 breast cancer cells [137].
Tumor microenvironment and resistance to breast cancer therapy
The tumor microenvironment comprises interstitial tissues, extracellular matrix, and stromal cells [138]. The biological elements of the microenvironment include TAM, mesenchymal cells, endothelial cells, and CAF [139]. The cathepsin-dependent function of macrophages can shield cancer cells from the harmful effects of chemotherapy [140]. TAMs and their products regulate cancer cell behavior [141,142]. TAMs can be targeted in conjunction with chemotherapy [143]. When exposed to TM, normal mesenchymal cells are less susceptible to drugs than tumor-resident mesenchymal cells [144]. Research shows mesenchymal stem cells (MSCs) are particularly resistant to paclitaxel [145]. MSCs are also anti-tumors in breast cancer, which is noteworthy [146]. A study revealed an increase in B lymphocytes in draining lymph nodes based on a mouse model of spontaneously metastasized breast cancer lymph nodes. Pathogenic IgG was generated, which activated the HSPA4-binding protein ITGB5 and downstream Src/NF-kB pathways in tumor cells, increasing CXCR4/SDF1-axis-mediated metastasis. There was an increase in serum anti-HSPA4 IgG levels in breast cancer patients with high HSPA4 levels and a poor prognosis in patients with high HSPA4 levels [147].
Drug uptake and resistance to cancer therapy
Developing feasible targeted therapies for breast cancer is difficult due to inherent and/or acquired resistance to chemotherapy. Many signaling pathways and mutations or changes in protein expression have been linked to breast cancer resistance [148]. Evidence suggests that P-gp/MRP1/BCRP, and other members of the drug efflux ABC transporter family, are elevated significantly in resistant breast cancer cells [149]. Further research is required to improve dose-response results and reduce the side effects of de novo and acquired resistance.
Cell cycle checkpoints and resistance to breast cancer therapy
Genomic instability enhances tumor heterogeneity and determines treatment resistance in breast cancer [150]. Cancer cells acquire genetic changes that alter signaling pathways and cellular checkpoints during cancerous transformation. These changes typically prevent genomic instability and uncontrolled cell proliferation. Thus, therapeutic cell cycle targeting has long been considered a viable anti-cancer treatment. In cancer therapy, selective cell cycle inhibitors have been unable to be used due to poor target specificity and undesirable toxicities. We have identified a therapeutic window that permits us to preferentially target the cell cycle in cancer cells through advances in our understanding of the vulnerabilities of malignant cells, resulting in the development of drugs currently under clinical trials [151]. Activation of the G1/S transition is inhibited by CDK4/6-dependent Rb1 inactivation, resulting in resistance to CDK4/6 inhibition. Understanding the relative contributions of different acquired resistance mechanisms in larger clinical datasets will be easier if we examine paired tumor or liquid biopsies obtained before and after treatment. Through a better understanding of these mechanisms, combination therapies and strategies will almost certainly be developed for preventing or overcoming CDK4/6 inhibitor resistance. In preclinical studies, breast cancer models have shown promise from CDK4/6 inhibitors combined with AKT pathway/mTOR inhibitors [152].
Drug metabolism and breast cancer resistance
The redox balance pathways are implicated in drug resistance to various anticancer treatments. Therefore, redox metabolism may be the key to improving medication responsiveness and combating multidrug resistance. Pharmacological impacts affect the circulating as well as intratumorally metabolome of a patient. A metabolic approach can be used to determine whether breast cancer treatments are effective and safe. Drug-resistant breast cancer cells demonstrate increased glycolysis no matter what type of chemotherapy they receive. Nevertheless, the regulation of this enhanced activity varies between resistant breast cancers. Therefore, drug-induced metabolic profiles vary [153].
In addition, the Warburg effect has been linked to drug resistance, suggesting that cancer cells with a high glycolytic rate are better able to resist anticancer treatment [154]. Higher glycolytic rates may restrict lactate production and acidification of the extracellular space. Overexpressed glycolytic regulators such as PDK1 and LDHA are targets for drug-resistant cancers. SKBR3 breast cancer cells produced more glucose-related genes than sensitive cells, resulting in a worse patient outcome. In addition to glucose transporters and glycolytic enzymes, oxidation routes have also been found [155]. ErbB2-positive breast cancer cells that are resistant to trastuzumab are more susceptible to LDHA-induced glycolysis.
Furthermore, using 2-DG with oxamate to block glycolysis resensitizes cells resistant to trastuzumab [156]. Oxamate and paclitaxel-induced synergistic apoptosis in paclitaxel-resistant breast cancer cells [157]. Each type of cancer uses a different metabolic pathway. Different drugs and drug combinations produce different efficacy and toxicity results in different patients. Malignancies and patients resistant to therapy must be detected early and treated with correct drugs.
DNA repair and resistance to breast cancer therapy
Most cancer patients fail due to treatment resistance. DNA repair capacity (DRC) of tumor cells is associated with chemoradiotherapy, targeted therapy, and immunotherapy resistance. Cisplatin is one common chemotherapy agent that damages DNA. Cisplatin was initially effective but then resistant in a mouse model of human lung cancer [158]. DNA damage repair genes and DRC were more abundant in tumors resistant to cisplatin. By inhibiting the NER pathway, tumor cells were more susceptible to cisplatin [159]. It has been shown that 53BP1, a DDR protein involved in breast preservation, is linked with local recurrence in patients with TNBC treated by breast-conserving surgery and radiotherapy [160]. Doxorubicin (DOX) resistance could also be prevented by inhibiting DNA repair kinases in breast cancer cells [161]. In CDK4/6 inhibitors of palbociclib-resistant breast cancer cells, abnormal DNA repair activity was identified; however, PARP inhibitors, olaparib, and niraparib therapy dramatically reduced palbociclib-resistant cancer cell viability [162]. The PARP inhibitor olaparib significantly increased CD8+ T-cell recruitment and activation by stimulating the cGAS/STING pathway in BRCA1-deficient triple-negative breast cancer [163].
Immuno-checkpoint inhibitors and DDR inhibitors are combined for TNBC or ovarian cancer patients, including Niraparib and pembrolizumab (NCT02657889). Phase I, multicentre, dose-escalation trials are conducted on AZD1775 (Adavosertib) and MEDI4736 (durvalumab). DNA repair might play an important role in cancer therapy resistance; evaluating DNA repair phenotypes before treatment would be useful [164].
Stress response and breast cancer resistance
Cancer cells live in a dynamic microenvironment that determines their behavior. Therefore, simple models linking cellular stress with cancer progression should be cautiously treated. Crosstalk between cells adds complexity. ROO levels impact oxidative, metabolic, and genotoxic stress. It will be fascinating to determine the level of ROS necessary to produce a particular stress response in the future. We may be able to develop better techniques to target drug-resistant cancer cells if we view drug resistance as a cellular adaptation to severe conditions. Studying cellular responses to immune inhibitors and activators is essential for cancer immunotherapy.
Several regulatory enzymes and transcription factors (AMPK, HIFα, PGC1α) in the metabolic stress response network can be targeted pharmacologically [165]. In response to MYC-induced metabolic stress, glutaminase and lactate dehydrogenase inhibitors reduce tumor growth by inhibiting the flow of nutrients needed for cancer cell proliferation [166,167]. Several therapeutic options have been considered for MYC-induced metabolic stress, including manipulating the response pathways involved. A triple-negative breast cancer model showed efficacy with inhibition of IRE1/XBP1, and a hepatocellular carcinoma model showed efficacy with inhibition of ARK5/AMPK [168]. Among the three primary methods under evaluation, pre-clinical/clinical studies have focused on targeting cell contractility, solid stress, and ECM stiffness [169,170]. As far as treating cancer with the stiffness of the ECM is concerned, it is more effective in solid tumors like breast cancer combined with celecoxib, β-aminopropionitrile, transforming growth factor- and hedgehog signaling inhibitors [171].
A combination of stress-targeted drugs and standard/conventional therapy could develop more effective therapeutic techniques in the future by targeting cellular stress responses. A better understanding of the stress responses of cancer cells is needed to make these drugs effective.
Apoptosis and breast cancer resistance
Planned cell death, or apoptosis, is critical for tissue homeostasis and the removal of abnormal cells [172]. Several therapies may overcome apoptotic resistance. These include downregulation of anti-apoptotic signaling, death-inducing ligands, and caspase gene expression [173]. It has been shown that death receptor ligands such as Fas-L and TRAIL cause apoptosis in breast cancer cells [174]. TRAIL significantly enhances apoptotic cell death and drug sensitivity when administered with chemotherapy drugs. However, chemotherapy alone cannot induce cell death in breast cancer cells [175].
Furthermore, Curcumin reduced primary and metastatic tumor cell proliferation and induced apoptosis, according to a recent study. Metastatic tumor cells, however, exhibited greater resistance to curcumin’s apoptotic action. DR-5 was linked with resistance to apoptosis. DR-5 was significantly upregulated in primary tumor cells following curcumin treatment, compared with metastatic cells [176].
Microbiota and breast cancer resistance
Researchers have identified gut microbiota dysbiosis as a key factor in BC’s development, treatment, and prognosis in recent years. These processes include proliferation and death of host cells, immune system function, chronic inflammation, oncogenic signaling, hormonal changes, and detoxification processes. Colonization of the gut begins during pregnancy and progresses throughout life. There was a difference in the fecal microbiota composition in newly diagnosed postmenopausal BC patients compared to healthy controls. Bacteria that produce β-glucuronidase modulate enterohepatic circulation and estrogen resorption, increasing the risk of hormone-dependent BC. The risk and prognosis of BC are known to be affected by bacterial metabolites, including phytoestrogens, short-chain fatty acids, lithocholic acid, and cadaverine [177]. Inhibition of DNA synthesis occurs with gemcitabine (2’,2’-difluorodeoxycytidine). Gamma proteobacteria, mostly from the Gammaproteobacteria class, can metabolize gemcitabine in its inactive form (2’,2’-difluorodeoxyuridine). This metabolism requires the bacterial enzyme cytidine deaminase, found only in this family of bacteria. Co-administered ciprofloxacin reversed Gemcitabine resistance caused by intratumoral Gamma proteobacteria in a cancer mouse model, indicating that these bacteria are involved in the drug’s failure [178,179]. A recent study provides evidence for a novel role for local microbiome-immune crosstalk in breast cancer and identifies breast microbial profiles linked to various prognostic clinical variables. This study provides a starting point for further research into how microbial-immune interactions affect breast cancer development and progression. There is also a need for further research on how these microbes mediate resistance to cancer therapies.
Predictive biomarkers for breast cancer
Compared to prognostic biomarkers, predictive biomarkers provide better predictions of patient response. Breast cancer was the first to use treatment efficacy predictors. Although ER and PR measurements have been used in clinical practice for over 40 years, the benefit of Trastuzumab has only been demonstrated in the last 15 years. ER, or ER-alpha, is the most important breast cancer biomarker. The estrogen receptor (ER) must measure all newly diagnosed breast cancers, and recurrent/metastatic lesions must be measured by the estrogen receptor (ER). While prognostic information is useful in endocrine therapy, predictive biomarkers are the primary clinical application of ER. Endocrine therapy should be initiated or considered if an ER test results positively. Hormones are unnecessary in the absence of an ER. Cancer patients are given Endocrine Therapy (ER) in the early, middle, and late stages of the disease [180,181]. Because estrogens, particularly estradiol, are required for the growth of some BC, endocrine therapy is used to treat it.
MYC and cyclin D regulate gene regulatory elements associated with estrogens [182]. Because estrogens promote tumor growth by binding to and activating ER, ER levels correlate with antiestrogen efficacy in BC. In patients with advanced BC who tested positive for estrogen receptors, endocrine therapies such as ovariectomy, adrenalectomy, and hypophysectomy resulted in objective regression. Malignancies that lacked an ER receptor had a very poor prognosis [181]. While it is still possible to use ER to identify advanced patients with breast cancer, it is more commonly used to determine which patients require adjuvant treatment such as oophorectomy and tamoxifen or aromatase inhibitors anastrozole or letrozole, or exemestane. In advanced breast cancer patients, endocrine therapies such as tamoxifen predict response and determine which patients require adjuvant tamoxifen treatment. These treatments slow breast cancer progression by targeting the ER protein. These drugs should only be used by patients who have ER-positive cancer. There is no such thing as drug resistance specific to a class [182]. Although ESR1 wild type is a reliable predictor of therapy benefit, ESR1 mutations are linked to endocrine therapy resistance. These mutations are more likely to be discovered if a patient has recurrent/metastatic breast cancer [183,184]. Most ER ligand-binding domain mutations, such as D538G, Y537S, E380Q, Y537C, or Y537N, cause cancer [183,185]. These mutations resulted in a lack of sensitivity to aromatase inhibitors and the inability of tamoxifen and fulvestrant to inhibit estrogen receptors in vitro [183,184]. The Y537S mutation is most likely responsible for ligand-independent activation [185]. The doses of fulvestrant required to inhibit Y537S are higher than for other ER mutants (D538G, Y337C) and wild-type ER (such as Y537S) [185].
Y537S mutation can cause ligin-independent activation. The only other ER mutants, D538G and Y337C, require a lower hormone dose, whereas the wild type requires a much higher dose [186]. Overexpression of HER2 may help tumor growth by deforming cell membranes. A deformed cell loses its attachments to the extracellular matrix and other cells, increasing the possibilities of invasion [187]. Most HER2-positive breast cancers do not spread; only 15-20% do. Cancers that are HER2-positive are more common in ER-negative women than ER-positive cancers. Amplification/overexpression of the HER2 gene is found in 30% of ER-negative tumor samples but only in 12% of ER-positive tumor samples [188]. Breast cancer biomarkers were first proposed, but they are currently only used to predict the efficacy of anti-HER2 therapy after treatment.
In the United States and Europe, trastuzumab, lapatinib, pertuzumab, and trastuzumab emtansine are currently used to treat HER2-positive breast cancer [189]. Based on the evidence, amplification or overexpression of the HER2 gene appears to be required for these treatments. Anti-HER2 therapies are currently only available to those with the HER2 gene mutation. Trastuzumab, an anti-HER2 treatment, is one of the most studied and approved treatments for metastatic breast cancer. Trastuzumab and chemotherapy resulted in pathologically complete remission in more than half of HER2-positive patients, compared to 19-27 percent of those treated with chemotherapy alone [190]. The current evidence suggests that these treatments require HER2 gene amplification or overexpression. Anti-HER2 therapies are now available for patients with HER2 mutations. The most researched and approved treatment for metastatic breast cancer is chemotherapy. In more than half of HER2-positive patients, trastuzumab plus chemotherapy resulted in pathologically complete tumor remission [190]. Due to the benefits of dual anti-HER2 therapy in neoadjuvant and advanced-stage cancer patients, it is important to identify those who will benefit the most. There should be a list of patients who do not require dual anti-HER2 therapy. Patients with these molecular subtypes had a higher chance of achieving a partial response than patients with other molecular subtypes. Trastuzumab-lapatinib achieved pCR in 41 percent of 101 HER2-enriched patients compared to only 5 percent of non-HER2-enriched patients [191].
HER2 levels are directly related to ER, PR, and other hormones. Because HER2 levels are higher in ER-negative tumors, anti-HER2 therapy may be more effective. According to preliminary anti-HER2 therapy studies, patients with HER2-negative tumors had higher partial response rates than those with HER2-positive tumors. Two monoclonal antibodies plus chemotherapy or two monoclonal antibodies alone produced the highest partial response rates in breast cancer patients. Only 63 percent of patients achieved partial responses in triple therapy, while only 20 percent received the treatment. Patients with receptor-negative cancer responded at 29.1 percent, while those with receptor-positive cancer responded at only 5.9 percent. TILs and PIK3CA mutation status should not be used to decide on anti-HER2 therapy [192].
Therapeutic approaches for breast cancer
The risk of BC can be assessed by using risk assessment tools, and individuals identified as high risks may be given risk-reducing medications. Regarding breast cancer treatment, an individual’s menopausal status influences the medication they choose. The type of medication they choose varies with the stage of the disease. It may evolve into invasive cancer in 40 percent of individuals with Stage 0 ductal carcinoma in situ [193]. There are a number of options available for BC treatment.
Targeting HER2 pathway
Women with BC who have HER2 as a therapeutic target are in the majority. There is also overexpression of HER2 in certain subsets of patients with other solid tumors, including NSCLC, colon cancer, and biliary tract cancer. HER2 can be targeted in several ways, as discussed below.
Targeting HER2 via trastuzumab
The HER2 receptor is one of four tyrosine kinase (TK) receptors (HER1-4), that control vital cell activities (Figure 4) [194]. HER contains extracellular, transmembrane, and intracellular TTK domains. When these domains interact, signaling pathways for proliferation, apoptosis, invasion, metastasis, angiogenesis, and cell differentiation are activated. Herceptin is a type of antibody to be approved to treat HER2-positive BC. Herceptin is designed to attach to the HER2 extracellular area and decrease cell proliferation in HER2-positive BC models [195]. Inhibition of cell signaling is not completely clear in terms of molecular mechanisms, but data suggest that it is more effective against homodimers of HER2 than heterodimers [196,197]. In addition to triggering antibody-dependent cytotoxicity, trastuzumab may cause cell apoptosis [198]. Because of the above reasons, preclinical studies indicate a synergistic impact with certain chemotherapy drugs [199,200]. Trastuzumab was first tested on patients with metastatic BC who had been highly pretreated. The response rates were poor, with only a single example of a complete reaction and 10-15 percent of patients reporting a partial response. Researchers observed that untreated metastatic cancers with HER2-overexpression had higher partial and full responses (26 percent) after single-agent treatment [201]. The inhibition of HER2 only worked in oncogene addiction (gene amplification) cancers. Trastuzumab was also well tolerated but can cause cardiomyopathy in some patients, particularly those who have previously taken doxorubicin or uncontrolled hypertension [202,203]. Trastuzumab interacts synergistically or additively with alkylating medicines, platinum medications, topoisomerase inhibitors, anthracyclines, and taxanes [199,200]. In the most important early trial, chemotherapy with doxorubicin and cyclophosphamide versus chemotherapy plus trastuzumab was compared in HER2+ patients with metastatic disease [203]. FDA approved trastuzumab as a cancer therapy after it significantly increased response rate (50 versus 32%), time to progression (7.4 versus 4.6 months), and overall survival (25 versus 20 months). Importantly, when doxorubicin and trastuzumab were given together, the trial demonstrated a high prevalence of cardiac dysfunction. The past decade has seen many trastuzumab trials in patients with HER2+ metastatic cancer, which showed that it is more effective when taken in conjunction with other drugs such as vinorelbine and docetaxel, and gemcitabine [204].
Figure 4.
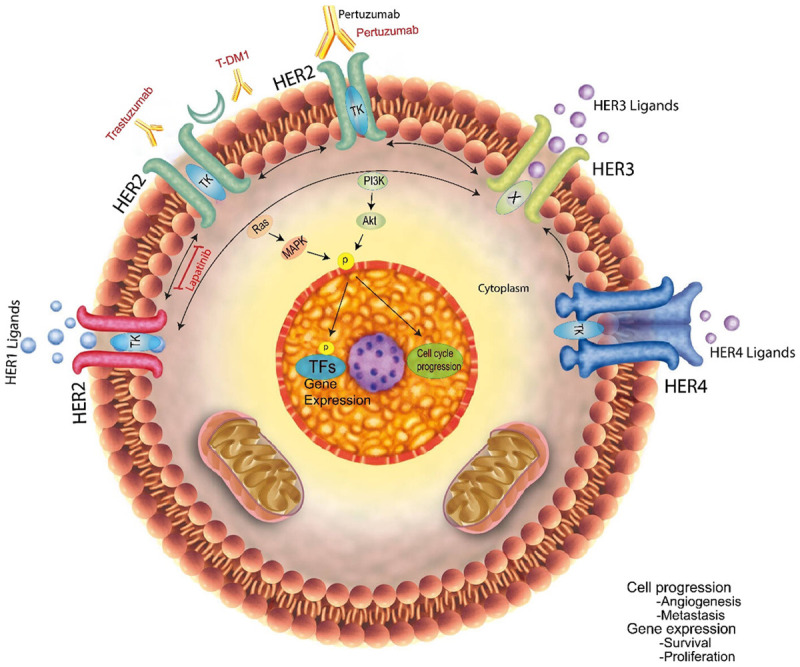
HER2 is a cancer-causing gene with four membrane tyrosine kinase receptors (HER1-HER4), a molecular switch, and more than ten ligands. Ligand binding causes a conformational change in HER1, HER3, and HER4 that facilitates homo- or heterodimerization with another family member. HER2, which is gene-amplified and/or overexpressed in 20% of breast cancers, lacks a ligand and lies in an open conformation in the membrane. Due to its heterodimerization capability and increased expression, HER2 activates itself by homodimerizing with other ligand-bound HER members. In response to dimerization of the HER receptors, their TKs are activated. This initiates a phosphorylation cascade, which triggers downstream signaling pathways, including Ras/p42/44 MAPK and PI3K/AKT. By modifying and phosphorylating transcription factors and other parts of the transcription and cell-cycle machinery, downstream kinases stimulate the transcriptional and cell-cycle machinery. As a result, tumor cells can be stimulated to proliferate, survive, become angiogenic, and metastatic. Because HER3’s TK is inactive (X), the downstream signaling pathway can only be activated by heterodimerization with other HER members. According to the FDA, the HER2-targeted drugs approved for treating HER2+ breast cancer are trastuzumab, pertuzumab, trastuzumab DMT-1, and lapatinib.
As a result of the increased risk of side effects, trastuzumab is usually not given alongside chemotherapy [205,206]. Studies looking at the combination of chemotherapy and trastuzumab have not shown any benefit in OS. The combination of carboplatin to chemotherapy and trastuzumab was studied in the Breast Cancer International Research Group study 007 [205]. Trastuzumab and paclitaxel with carboplatin or without were randomized for 196 patients. In the triple therapy group, response rates were higher. Although OS was not significantly different, doublet chemotherapy was linked with enhanced toxicity, limiting its clinical significance [206]. An indication of congestive heart failure is the most serious contraindication to HER2-targeted treatments [207]. Trastuzumab and pertuzumab were similarly effective as first-line treatments for HER2-positive metastatic BC patients. After removing patients with de novo illness from the trial, taxanes were found not to affect progression-free survival (PFS) [208].
HER2-targeted therapy offered improved responses, longer PFS, longer TTP, and longer OS when combined with chemotherapy in the first-line setting. In this study, HER2-targeted therapy combined with chemotherapy was linked to improved MBC treatment results as a first-line treatment.
Targeting HER2 via lapatinib
A tyrosine kinase inhibitor small molecule called lapatinib targets EGFR1 and human HER2. After oral treatment, lapatinib has a half-life of 24 hours, with peak plasma levels occurring within 4 hours and steady-state levels establishing 6-7 days later [209]. For HER2-positive MBC resistant to anthracyclines, taxanes, or trastuzumab-based regimens, lapatinib and capecitabine are authorized as second-line combination therapy [209]. When used in conjunction with chemotherapy, lapatinib is also effective as first-line therapy, though it may not be as effective as trastuzumab-based therapy [210,211]. Lapatinib has been studied as a first-line treatment in two phase III studies, one of which compared lapatinib to a placebo. Paclitaxel plus lapatinib or placebo were administered to participants who had never received chemotherapy for metastatic illness [210]. The MA.31 trial compared paclitaxel 80 mg/m2 three times a week with lapatinib or trastuzumab as first-line anti-HER2 therapy, followed by anti-HER2 monotherapy [211]. According to the existing evidence, trastuzumab-based regimens are still regarded standard of therapy in this situation.
Targeting HER2 via neratinib
Upon administration, neratinib binds to and inhibits the phosphorylation of EGFR, HER2, HER4, AKT, and MEK in HER2-overexpressing BC cell lines [212]. The orally administered drug, neratinib, inhibits several EGFR family members by interfering with their respective catalytic domains. Four hundred and seventy-nine women with HER2-positive recurring and/or metastatic BC were randomized evenly between neratinib-paclitaxel and trastuzumab-paclitaxel in the NEfERT-T study [213]. Using neratinib-paclitaxel as a first-line treatment for metastatic HER2-positive BC has been found to delay the onset of and reduce the incidence of central nervous system side effects. Diarrhea (30.4 percent) and neutropenia were the most common adverse effects (12.9 percent).
Targeting HER2 via toralizumab
The dimerization of HER2 with HER1, HER3, and HER4 results in the strongest mitogenic stimulus, activating PI3K and MAPK and therefore promoting cell survival and growth [214]. HER2 dimerization with other HER receptors is prevented by toralizumab, a potent inhibitor of double HER2 signaling [215]. Although epratuzumab and trastuzumab bind different epitopes of HER2, the antibodies are more effective when used together than if used alone since they block signals from multiple epitopes [215]. Phase II studies demonstrated that epratuzumab, whether given alone or combined with trastuzumab or other cytotoxic drugs, was generally well tolerated; they also suggested combining epratuzumab and trastuzumab boosted efficacy for early and advanced HER2-positive BC [216]. The CLEOPATRA study showed that epratuzumab, trastuzumab, and docetaxel were more effective than placebo, trastuzumab, and docetaxel [217].
The CLEOPATRA study found that epratuzumab increased PFS in all biomarker subgroups, including ER-negative and ER-positive women [218]. Patients with PIK3CA alpha or low HER2 protein levels had a considerably better prognosis than those with high HER2 protein levels (P 0.05). Patients with malignancies that expressed wild-type PIK3CA had a higher median survival rate than those with mutant Pik3CA in both the control and epratuzumab groups. Patients with MBC who got lapatinib with capecitabine but not T-DM1 had a worse prognosis. New biomarkers have the potential to refine and optimize therapy for specific patient subgroups in the future.
Targeting the hedgehog signaling pathway in breast cancer
There are various strategies for inhibiting the Hh pathway, including antibodies that target Hh ligands, SMO inhibition, and GLI [219,220]. Cyclopamine has limited clinical effectiveness due to its low potency and poor solubility [221]. SMO inhibitors such as sonidegib [222] and taladegib [223,224] are FDA-approved to treat advanced cell carcinoma. Other SMO inhibitors, including saridegib [225], taladegib [223], and glasdegib [226], are also being tested. In basal cell carcinomas, SMO mutations can cause secondary resistance [227]. Due to its unique method of action compared to other SMO antagonists, SMO antagonist itraconazole appears to preserve Hh inhibition even in mutants resistant to drug-resistant SMOs [228].
The GLI1 protein is activated by pathways other than the canonical ones in several cancers, including breast cancer. Direct activation of the GLI1 protein is one mechanism of resistance to SMO inhibitors. GLI might be treated effectively by targeting it with drugs like PI3K inhibitors and other chemotherapy agents [229]. In preclinical studies, GANT61 is the most effective antagonist. However, no clinical data demonstrate the efficacy of GANT58 [219,230]. Vismodegib and GANT61 reduced tumor growth in another xenograft model using TUBO cells [231]. With GANT61, the degree of tumor regression and the duration of tumor remission were significantly longer than with vismodegib, an SMO inhibitor. In 80 percent of mice, GANT61 produced complete tumor regression and remission, and these animals remained tumor-free for 30 weeks [231]. In GLI1 overexpressing breast cancer cell lines, GANT61 significantly reduced cell proliferation, motility, and invasion [232]. This research provides significant preclinical evidence that GANT61 could be used in clinical trials in BC patients.
In xenograft mouse models derived from human patients, SMO inhibitor treatment increased the susceptibility of tumors to docetaxel chemotherapy [233]. The result was the phase I “3 + 3” trial of sonidegib combined with docetaxel for metastatic TNBC. Patients with metastatic TNBC benefited clinically from combination therapy in 3/10 cases (3/10 at recommended doses in phase 2), one of whom had a complete response [234]. No dose-limiting toxicities were observed in the clinical trial with vismodegib and standard neoadjuvant chemotherapy, cyclophosphamide, and epirubicin. Patients are being included in ongoing clinical trials to see the treatment’s influence on the pathological complete response rate in patients with locally advanced TNBC.
Targeting notch signaling pathway in breast cancer
A study using ER+ BC lines identified Notch activation as a mechanism of resistance to endocrine therapy, and the potential use of genetic or pharmacologic treatments to inhibit Notch1 or Notch4 reversed it. The first Notch inhibitor was a GSI, which inhibited the final cleavage of NotchIC and inhibited Notch target gene activation. When combined with tamoxifen, the GSI significantly reduced the growth of ER+ T47D cells and led to extensive cell death in T47D-A18 xenograft tumors [235]. In mice, cells taken from tumors treated with RO4929097 GSI reduced tumor-initiating capacity and ALDH activity 90 days after implantation [236]. Compared to LY-411,575, MRK-003, and Z-Leu-Leu-Nle-CHO GSIs, Z-Leu-Leu-Nle-CHO or MRK003 GSIs led to significant reductions in mammosphere formation, whereas LY-411,575 had a non-significant reduction [237]. In vitro and in vivo, MRK-003 has provoked apoptosis in mammosphere-derived stem-like cells, suggesting a possible method of targeting BCSCs using a GSI [237,238].
Using monoclonal antibodies, one can directly target Notch receptors and ligands. Monoclonal antibodies suppress interactions between Notch receptors and their ligands. Using this strategy, one can selectively target Notch signaling in tumors while avoiding the unintended side effects of GSIs. Based on clinical studies of TNBC, OMP-59R5 has shown stable disease efficacy against Notch2 and Notch3 receptors [239]. MCF-7 and MDA-MB-231 cells responded dramatically to an antibody targeting EGF repeats within Notch1’s ligand-binding domain. The key point is that it specifically targets BCSCs by modulating stem cell gene expression and inducing apoptosis [240].
Researchers have worked to develop strategies to inhibit transcription by targeting Notch receptors. A class of agents called “stapled peptides” can block MAML1 interaction with NotchIC to prevent transcription [241]. Hydrocarbon-stapled peptides generated from MAML1’s dominant-negative variant SAHM1 bind to the Notch1-CSL transcriptional complex in T-cell acute lymphoblastic leukemia and block Notch1 target gene expression [241]. There have been investigations to determine whether not only MAML1 but also CSL can be targeted to inhibit Notch signaling [242]. Inhibiting CSL expression or activity was another directly targeted strategy that decreased breast cancer cell proliferation and growth [243,244]. Targeting the Notch pathway’s downstream signaling components and its transcriptional partners could provide a more focused and potentially more effective strategy for treating Notch-driven breast cancer.
Targeting Wnt signaling in breast cancer
The past few decades have seen hundreds of inhibitors developed. These inhibitors inhibit Porcupine, Fzds, DVLs, TNKS 1/2, and TCF/Catenin and co-activators. Since Porcupine inhibitors such as LGK974, have Wnt-targeting and anticancer activity, they have recently received considerable attention [245-247]. Historically, blocking Wnt signaling by targeting Fzds has been a mainstay strategy. Niclosamide is an FDA-approved antihelminth. It has been tested for its ability to inhibit Fzd1 and may hold the most promise of any Fzd inhibitor [248]. One of the potential drugs for BC and other solid tumors is OMP-18R5, a monoclonal antibody that targets multiple FZDs. Indium 111 and Yttrium 90 are also used as radioactive labels on three antibodies against Fzd10. One of these antibodies is suited for radiotherapy of metastatic synovial sarcoma [249,250]. The FDA approves Lincomycin as an ingredient in poultry feed for killing BCSCs [251]. Further studies have shown that it blocks the phosphorylation of LRP by Wnt [252]. Monomethyl auristatin E (MMAE) inhibits tubulin, thus killing LGR5-positive cancer cells selectively [253]. A small-molecule inhibitor called Sulindac is among the most promising Dvl inhibitors in development. Sulindac is an anticancer drug that is FDA-approved and has been shown to inhibit not only cyclooxygenase 1/2 (COX1/2) but also cyclooxygenase 3 (CYP3A), a metabolic enzyme that is involved in the formation of cancer [254]. The hypothesized mechanism of Sulindac’s actions is that it inhibits the activity of cyclooxygenase 1/2 (COX1/2) as well as Dvl in its PDZ domain [255-257].
Inhibition and degradation of β-Catenin are undeniably effective approaches to inhibiting the canonical Wnt signaling. To date, only two drugs have been identified that are directly targeting β-Catenin [258] and NRX-252262 [259]. These compounds also enhance the destruction of β-Catenin by activating CK1α, which is a small molecule that targets β-Catenin [260-262], GSK3β [263], and Axin [264]. With hexachlorophene, Siah-1-induced degradation of β-Catenin can also be triggered without requiring a destruction complex. Several small-molecule inhibitors are currently being developed to block TNKS1/2’s important role in Axin breakdown mediated by Axin. 2X-121 is the most promising candidate for treating BC and ovarian cancer [265]. The phase I clinical trial for PRI-724, an anti-β-Catenin/CBP drug, is underway for advanced solid tumors. It disrupts the connection between β-Catenin and BCL9 or CBP [266]. TBL1-related protein (TBLR1) and β-Catenin then collaborate to push TLE and HDAC1 out of the way, enhancing Wnt target gene expression [266]. There is a phase I clinical trial for BC2059 [266], a β-Catenin/TBL1 disruptor, to treat desmoid tumors [267]. The findings showed apicularen A and bafilomycin A1 as a potent Wnt signaling inhibitor related to vacuolar H+-adenosine triphosphatase (V-ATPase) [268]. Even though KY02111’s direct targets are not SM04690, it has entered a phase II clinical trial for knee osteoarthritis [269], and SM04690 remains unclear [270].
Conclusion
Cancer cells that are resistant to traditional treatments cause the majority of breast cancer fatalities. Scientists are developing new chemotherapeutic drugs or targeted therapy combinations to improve BC treatment. Endocrine therapy’s effectiveness percentage is reduced when there is no ER expression. Topo-II properly identified 72.22 percent of breast cancer patients. Those with low TopoII and CERBB2 expression survived longer than those with high or low expression. Chemotherapy can directly or indirectly target P-gp-overexpressing cells. Anacyclines, taxanes, and vinca alkaloids are examples of P-broad gp substrates. This biomarker’s used in determining a patient’s treatment options.
EGFR positivity is found in less than 1% of luminal A breast cancers. Lymph node metastases are strongly linked to poor survival rates in late-stage breast cancer, regardless of type. These individuals require more personalized therapy and follow-up measures based on clinical and histological characteristics. The recurrence rate following mastectomy and BCS varies according to the molecular subtype of breast cancer. After breast-conserving surgery, TNBC patients showed lower recurrence rates than HER2+ patients. Some cancers may begin to recur before they are diagnosed, resulting in misdiagnosing a single tumor. Oncogenes may play a role in cancer recurrence. “Metastases of metastases” may arise if cancer spreads through these phases.
The ECM is disrupted during the invasion, facilitating tissue border penetration. MMPs in breast cancer cells stimulate proteolysis on the invasion front of the ECM. Protease-dependent mesenchymal mobility and amoeboid migration are used by tumor cells to disseminate. According to research, bone and lung metastases outperform liver and brain metastases. Cancer cells can migrate to certain organs by using a protein known as a chemokine. Metastasis cannot begin without angiogenesis. Tumor hypoxia occurs when the blood supply to a tumor is insufficient. When Twist recognizes the E-box on E-cadherin promoters, transcription is reduced. Twist has the potential to make cancer cells more resistant to chemotherapy. Twist-overexpressed breast tumors are unable to use ER protein.
MDR1 inhibitors can reduce drug resistance and reprogram cancer cells to react to the therapy. If MRP hinders drug excretion and redistribution, tumor cells may become resistant to treatment. Drug resistance mediated by MRP is weaker than P-global drug resistance.
Nonspecific biodistribution of chemotherapy agents, dose-limiting toxicity, and drug resistance are some of the drawbacks of current cancer treatment methods. As a result, the development of new cancer therapies like liposomal drug delivery systems has accelerated.
Acknowledgements
This study was supported by the Shanxi province basic research program (Grant No. 20210302124230) to Jia Cui.
Disclosure of conflict of interest
None.
Abbreviations
- BC
Breast Cancer
- ER
Estrogen Receptor
- PR
Progesterone Receptor
- ERBB2
Erb-b2 Receptor Tyrosine Kinase 2
- HERE2
Human Epidermal Growth Factor Receptor 2
- Topo-2
Topoisomerase II
- AUC
Area Under Curve
- MRP1
Multidrug Resistance-associated Protein 1
- TNBC
Tiple Negative Breast Cancer
- VEGFR1
Vascular Endothelial Growth Factor Receptor 1
- NER
Nucleotide Excision Repair
- BER
Base Excision Repair
- NHEJ
Non-homologues End Joining
- BAK1
Bcl-2 Antagonist Killer 1
- ABCG2
ATP-Binding Cassette Transporter G-2
- ABCC1
ATP-binding cassette sub-family C-1
- ABCB5
ATP-binding cassette sub-family B-5
- HIF-1
hypoxia-inducible factor-1
- DLMC
Doxorubicin-liposome-microbub-ble-complex
- US
Ultrasound
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Narod SA, Iqbal J, Miller AB. Why have breast cancer mortality rates declined? J Cancer. 2015;5:8–17. [Google Scholar]
- 3.Althuis MD, Dozier JM, Anderson WF, Devesa SS, Brinton LA. Global trends in breast cancer incidence and mortality 1973-1997. Int J Epidemiol. 2005;34:405–412. doi: 10.1093/ije/dyh414. [DOI] [PubMed] [Google Scholar]
- 4.Britt KL, Cuzick J, Phillips KA. Key steps for effective breast cancer prevention. Nat Rev Cancer. 2020;20:417–436. doi: 10.1038/s41568-020-0266-x. [DOI] [PubMed] [Google Scholar]
- 5.Glass AG, Hoover RN. Rising incidence of breast cancer: relationship to stage and receptor status. J Natl Cancer Inst. 1990;82:693–696. doi: 10.1093/jnci/82.8.693. [DOI] [PubMed] [Google Scholar]
- 6.Li CI, Daling JR, Malone KE. Incidence of invasive breast cancer by hormone receptor status from 1992 to 1998. J. Clin. Oncol. 2003;21:28–34. doi: 10.1200/JCO.2003.03.088. [DOI] [PubMed] [Google Scholar]
- 7.Bigaard J, Stahlberg C, Jensen MB, Ewertz M, Kroman N. Breast cancer incidence by estrogen receptor status in Denmark from 1996 to 2007. Breast Cancer Res Treat. 2012;136:559–564. doi: 10.1007/s10549-012-2269-0. [DOI] [PubMed] [Google Scholar]
- 8.Coughlin SS. Epidemiology of breast cancer in women. Breast cancer metastasis and drug resistance. Adv Exp Med Biol. 2019;1152:9–29. doi: 10.1007/978-3-030-20301-6_2. [DOI] [PubMed] [Google Scholar]
- 9.Ji X, Lu Y, Tian H, Meng X, Wei M, Cho WC. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed Pharmacother. 2019;114:108800. doi: 10.1016/j.biopha.2019.108800. [DOI] [PubMed] [Google Scholar]
- 10.Tufail M. Genome editing: an essential technology for cancer treatment. Medicine in Omics. 2022;4:100015. [Google Scholar]
- 11.Majidpoor J, Mortezaee K. Steps in metastasis: an updated review. Med Oncol. 2021;38:1–17. doi: 10.1007/s12032-020-01447-w. [DOI] [PubMed] [Google Scholar]
- 12.Welch DR, Hurst DR. Defining the hallmarks of metastasis. Cancer Res. 2019;79:3011–3027. doi: 10.1158/0008-5472.CAN-19-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown M, Assen FP, Leithner A, Abe J, Schachner H, Asfour G, Bago-Horvath Z, Stein J, Uhrin P, Sixt M. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science. 2018;359:1408–1411. doi: 10.1126/science.aal3662. [DOI] [PubMed] [Google Scholar]
- 14.Fidler IJ, Gersten DM, Hart IR. The biology of cancer invasion and metastasis. Adv Cancer Res. 1978;28:149–250. doi: 10.1016/s0065-230x(08)60648-x. [DOI] [PubMed] [Google Scholar]
- 15.Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, Ruddy K, Tsang J, Cardoso F. Breast cancer. Nat Rev Dis Primers. 2019;5:1–31. doi: 10.1038/s41572-019-0111-2. [DOI] [PubMed] [Google Scholar]
- 17.Meirson T, Gil-Henn H, Samson AO. Invasion and metastasis: the elusive hallmark of cancer. Oncogene. 2020;39:2024–2026. doi: 10.1038/s41388-019-1110-1. [DOI] [PubMed] [Google Scholar]
- 18.Wendt MK, Taylor MA, Schiemann BJ, Schiemann WP. Down-regulation of epithelial cadherin is required to initiate metastatic outgrowth of breast cancer. Mol Biol Cell. 2011;22:2423–2435. doi: 10.1091/mbc.E11-04-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gould Rothberg BE, Bracken MB. E-cadherin immunohistochemical expression as a prognostic factor in infiltrating ductal carcinoma of the breast: a systematic review and meta-analysis. Breast Cancer Res Treat. 2006;100:139–148. doi: 10.1007/s10549-006-9248-2. [DOI] [PubMed] [Google Scholar]
- 20.Kowalski PJ, Rubin MA, Kleer CG. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 2003;5:1–6. doi: 10.1186/bcr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, van Roy F. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 1995;14:6107–6115. doi: 10.1002/j.1460-2075.1995.tb00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Danø K, Behrendt N, Høyer-Hansen G, Johnsen M, Lund LR, Ploug M, Rømer J. Plasminogen activation and cancer. Thromb Haemost. 2005;93:676–681. doi: 10.1160/TH05-01-0054. [DOI] [PubMed] [Google Scholar]
- 23.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 24.Harbeck N, Kates RE, Schmitt M, Gauger K, Kiechle M, Janicke F, Thomassen C, Look MP, Foekens JA. Urokinase-type plasminogen activator and its inhibitor type 1 predict disease outcome and therapy response in primary breast cancer. Clin Breast Cancer. 2004;5:348–352. doi: 10.3816/cbc.2004.n.040. [DOI] [PubMed] [Google Scholar]
- 25.De Cremoux P, Grandin L, Dieras V, Savignoni A, Degeorges A, Salmon R, Bollet MA, Reyal F, Sigal-Zafrani B, Vincent-Salomon A. Urokinase-type plasminogen activator and plasminogen-activator-inhibitor type 1 predict metastases in good prognosis breast cancer patients. Anticancer Res. 2009;29:1475–1482. [PubMed] [Google Scholar]
- 26.Huang HY, Jiang ZF, Li QX, Liu JY, Wang T, Zhang R, Zhao J, Xu YM, Bao W, Zhang Y. Inhibition of human breast cancer cell invasion by siRNA against urokinase-type plasminogen activator. Cancer Invest. 2010;28:689–697. doi: 10.3109/07357901003735642. [DOI] [PubMed] [Google Scholar]
- 27.Kelly T, Yan Y, Osborne RL, Athota AB, Rozypal TL, Colclasure JC, Chu WS. Proteolysis of extracellular matrix by invadopodiafacilitates human breast cancer cell invasion and ismediated by matrix metalloproteinases. Clin Exp Metastasis. 1998;16:501–512. doi: 10.1023/a:1006538200886. [DOI] [PubMed] [Google Scholar]
- 28.McSherry EA, Donatello S, Hopkins AM, McDonnell S. Molecular basis of invasion in breast cancer. Cell Mol Life Sci. 2007;64:3201–3218. doi: 10.1007/s00018-007-7388-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell CD, Waizbard E. Variability of cell size in primary and metastatic human breast carcinoma. Invasion Metastasis. 1986;6:11–20. [PubMed] [Google Scholar]
- 30.Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125I-5-iodo-2’-deoxyuridine. J Natl Cancer Inst. 1970;45:773–782. [PubMed] [Google Scholar]
- 31.Fidler IJ. The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur J Cancer (1965) 1973;9:223–227. doi: 10.1016/s0014-2964(73)80022-2. [DOI] [PubMed] [Google Scholar]
- 32.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 33.Wang S, Li Y, Xing C, Ding C, Zhang H, Chen L, You L, Dai M, Zhao Y. Tumor microenvironment in chemoresistance, metastasis and immunotherapy of pancreatic cancer. Am J Cancer Res. 2020;10:1937. [PMC free article] [PubMed] [Google Scholar]
- 34.Arneth B. Tumor microenvironment. Medicina (Kaunas) 2020;56:15. doi: 10.3390/medicina56010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. 2020;30:R921–R925. doi: 10.1016/j.cub.2020.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Psaila B, Kaplan RN, Port ER, Lyden D. Priming the ‘soil’ for breast cancer metastasis: the pre-metastatic niche. Breast Dis. 2006;26:65–74. doi: 10.3233/bd-2007-26106. [DOI] [PubMed] [Google Scholar]
- 37.Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, Shipley JM, Senior RM, Shibuya M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2:289–300. doi: 10.1016/s1535-6108(02)00153-8. [DOI] [PubMed] [Google Scholar]
- 38.Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, Ponomarev V, Gerald WL, Blasberg R, Massagué J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115:44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massagué J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, Massagué J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 41.Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 42.Bruce J, Carter DC, Fraser J. Patterns of recurrent disease in breast cancer. Lancet. 1970;1:433–435. doi: 10.1016/s0140-6736(70)90829-9. [DOI] [PubMed] [Google Scholar]
- 43.de Castro Junior G, Puglisi F, de Azambuja E, El Saghir NS, Awada A. Angiogenesis and cancer: a cross-talk between basic science and clinical trials (the “do ut des” paradigm) Crit Rev Oncol Hematol. 2006;59:40–50. doi: 10.1016/j.critrevonc.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 45.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 46.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 47.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 48.Fan F, Schimming A, Jaeger D, Podar K. Targeting the tumor microenvironment: focus on angiogenesis. J Oncol. 2012;2012:281261. doi: 10.1155/2012/281261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 50.Leite de Oliveira R, Hamm A, Mazzone M. Growing tumor vessels: more than one way to skin a cat - implications for angiogenesis targeted cancer therapies. Mol Aspects Med. 2011;32:71–87. doi: 10.1016/j.mam.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Wang X, Wang N, Zhong L, Wang S, Zheng Y, Yang B, Zhang J, Lin Y, Wang Z. Prognostic value of depression and anxiety on breast cancer recurrence and mortality: a systematic review and meta-analysis of 282,203 patients. Mol Psychiatry. 2020;25:3186–3197. doi: 10.1038/s41380-020-00865-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat Rev Cancer. 2010;10:871–877. doi: 10.1038/nrc2933. [DOI] [PubMed] [Google Scholar]
- 53.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 54.Wang SY, Shamliyan T, Virnig BA, Kane R. Tumor characteristics as predictors of local recurrence after treatment of ductal carcinoma in situ: a meta-analysis. Breast Cancer Res Treat. 2011;127:1–14. doi: 10.1007/s10549-011-1387-4. [DOI] [PubMed] [Google Scholar]
- 55.Wang G, Zhang S, Wang M, Liu L, Liu Y, Tang L, Bai H, Zhao H. Prognostic significance of occult lymph node metastases in breast cancer: a meta-analysis. BMC Cancer. 2021;21:875. doi: 10.1186/s12885-021-08582-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cavalli LR. Molecular markers of breast axillary lymph node metastasis. Expert Rev Mol Diagn. 2009;9:441–454. doi: 10.1586/erm.09.30. [DOI] [PubMed] [Google Scholar]
- 57.Noguchi M, Inokuchi M, Noguchi M, Morioka E, Ohno Y, Kurita T. Axillary surgery for breast cancer: past, present, and future. Breast Cancer. 2021;28:9–15. doi: 10.1007/s12282-020-01120-0. [DOI] [PubMed] [Google Scholar]
- 58.Riis M. Modern surgical treatment of breast cancer. Ann Med Surg. 2020;56:95–107. doi: 10.1016/j.amsu.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gieni M, Avram R, Dickson L, Farrokhyar F, Lovrics P, Faidi S, Sne N. Local breast cancer recurrence after mastectomy and immediate breast reconstruction for invasive cancer: a meta-analysis. Breast. 2012;21:230–236. doi: 10.1016/j.breast.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 60.van der Leij F, Elkhuizen PH, Bartelink H, van de Vijver MJ. Predictive factors for local recurrence in breast cancer. Semin Radiat Oncol. 2012;22:100–107. doi: 10.1016/j.semradonc.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 61.Houssami N, Macaskill P, Marinovich ML, Dixon JM, Irwig L, Brennan ME, Solin LJ. Meta-analysis of the impact of surgical margins on local recurrence in women with early-stage invasive breast cancer treated with breast-conserving therapy. Eur J Cancer. 2010;46:3219–3232. doi: 10.1016/j.ejca.2010.07.043. [DOI] [PubMed] [Google Scholar]
- 62.Darby S, McGale P, Correa C, Taylor C, Arriagada R, Clarke M, Cutter D, Davies C, Ewertz M, Godwin J, Gray R, Pierce L, Whelan T, Wang Y, Peto R. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet. 2011;378:1707–1716. doi: 10.1016/S0140-6736(11)61629-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lowery AJ, Kell MR, Glynn RW, Kerin MJ, Sweeney KJ. Locoregional recurrence after breast cancer surgery: a systematic review by receptor phenotype. Breast Cancer Res Treat. 2012;133:831–841. doi: 10.1007/s10549-011-1891-6. [DOI] [PubMed] [Google Scholar]
- 64.Meng X, Zhong J, Liu S, Murray M, Gonzalez-Angulo AM. A new hypothesis for the cancer mechanism. Cancer Metastasis Rev. 2012;31:247–268. doi: 10.1007/s10555-011-9342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tagde P, Kulkarni GT, Mishra DK, Kesharwani P. Recent advances in folic acid engineered nanocarriers for treatment of breast cancer. J Drug Deliv Sci Technol. 2020;56:101613. [Google Scholar]
- 66.Goldie JH. Drug resistance and cancer chemotherapy strategy in breast cancer. Breast Cancer Res Treat. 1983;3:129–136. doi: 10.1007/BF01803555. [DOI] [PubMed] [Google Scholar]
- 67.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 68.Gottesman MM, Pastan IH. The role of multidrug resistance efflux pumps in cancer: revisiting a JNCI publication exploring expression of the MDR1 (P-glycoprotein) gene. J Natl Cancer Inst. 2015;107:djv222. doi: 10.1093/jnci/djv222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P 450 pharmacogenetics and cancer. Oncogene. 2006;25:1679–1691. doi: 10.1038/sj.onc.1209377. [DOI] [PubMed] [Google Scholar]
- 70.Sueyoshi T, Negishi M. Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu Rev Pharmacol Toxicol. 2001;41:123–143. doi: 10.1146/annurev.pharmtox.41.1.123. [DOI] [PubMed] [Google Scholar]
- 71.Saggar JK, Yu M, Tan Q, Tannock IF. The tumor microenvironment and strategies to improve drug distribution. Front Oncol. 2013;3:154. doi: 10.3389/fonc.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 73.Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9:665–674. doi: 10.1038/nrc2714. [DOI] [PubMed] [Google Scholar]
- 74.Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 75.Buzdar AU, Hortobagyi G. Update on endocrine therapy for breast cancer. Clin Cancer Res. 1998;4:527–534. [PubMed] [Google Scholar]
- 76.Duffy MJ. Predictive markers in breast and other cancers: a review. Clin Chem. 2005;51:494–503. doi: 10.1373/clinchem.2004.046227. [DOI] [PubMed] [Google Scholar]
- 77.Mittendorf EA, Wu Y, Scaltriti M, Meric-Bernstam F, Hunt KK, Dawood S, Esteva FJ, Buzdar AU, Chen H, Eksambi S. Loss of HER2 amplification following trastuzumab-based neoadjuvant systemic therapy and survival outcomes. Clin Cancer Res. 2009;15:7381–7388. doi: 10.1158/1078-0432.CCR-09-1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liedtke C, Broglio K, Moulder S, Hsu L, Kau SW, Symmans WF, Albarracin C, Meric-Bernstam F, Woodward W, Theriault RL, Kiesel L, Hortobagyi GN, Pusztai L, Gonzalez-Angulo AM. Prognostic impact of discordance between triple-receptor measurements in primary and recurrent breast cancer. Ann Oncol. 2009;20:1953–1958. doi: 10.1093/annonc/mdp263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nitiss J, Beck W. Antitopoisomerase drug action and resistance. Eur J Cancer. 1996;32:958–966. doi: 10.1016/0959-8049(96)00056-1. [DOI] [PubMed] [Google Scholar]
- 80.Miller ML, Ojima I. Chemistry and chemical biology of taxane anticancer agents. Chem Rec. 2001;1:195–211. doi: 10.1002/tcr.1008. [DOI] [PubMed] [Google Scholar]
- 81.Oakman C, Moretti E, Galardi F, Santarpia L, Di Leo A. The role of topoisomerase IIα and HER-2 in predicting sensitivity to anthracyclines in breast cancer patients. Cancer Treat Rev. 2009;35:662–667. doi: 10.1016/j.ctrv.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 82.Houlbrook S, Addison C, Davies S, Carmichael J, Stratford I, Harris A, Hickson I. Relationship between expression of topoisomerase II isoforms and intrinsic sensitivity to topoisomerase II inhibitors in breast cancer cell lines. Br J Cancer. 1995;72:1454–1461. doi: 10.1038/bjc.1995.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Klumper E, Giaccone G, Pieters R, Broekema G, van Ark-Otte J, van Wering ER, Kaspers GJ, Veerman AJ. Topoisomerase II alpha gene expression in childhood acute lymphoblastic leukemia. Leukemia. 1995;9:1653–1660. [PubMed] [Google Scholar]
- 84.Kaufmann SH, Karp JE, Jones RJ, Miller CB, Schneider E, Zwelling LA, Cowan K, Wendel K, Burke PJ. Topoisomerase II levels and drug sensitivity in adult acute myelogenous leukemia. Blood. 1994;83:517–30. [PubMed] [Google Scholar]
- 85.Hua S, Chuanbo F, Zhonglin W, Shuangjiu Z, Xinwen Z. The expression and prognostic significance of Topo-II and c-erbB-2 in breast cancer. Minerva Med. 2020 doi: 10.23736/S0026-4806.20.06637-9. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 86.Kavallaris M, Kuo D, Burkhart CA, Regl DL, Norris MD, Haber M, Horwitz SB. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Investig. 1997;100:1282–1293. doi: 10.1172/JCI119642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dumontet C, Jordan MA, Lee FF. Ixabepilone: targeting βIII-tubulin expression in taxane-resistant malignancies. Mol Cancer Ther. 2009;8:17–25. doi: 10.1158/1535-7163.MCT-08-0986. [DOI] [PubMed] [Google Scholar]
- 88.Hasegawa S, Miyoshi Y, Egawa C, Ishitobi M, Taguchi T, Tamaki Y, Monden M, Noguchi S. Prediction of response to docetaxel by quantitative analysis of class I and III β-tubulin isotype mRNA expression in human breast cancers. Clin Cancer Res. 2003;9:2992–2997. [PubMed] [Google Scholar]
- 89.Bernard-Marty C, Treilleux I, Dumontet C, Cardoso F, Fellous A, Gancberg D, Bissery MC, Paesmans M, Larsimont D, Piccart MJ. Microtubule-associated parameters as predictive markers of docetaxel activity in advanced breast cancer patients: results of a pilot study. Clin Breast Cancer. 2002;3:341–345. doi: 10.3816/CBC.2002.n.037. [DOI] [PubMed] [Google Scholar]
- 90.Chen D, Bao C, Zhao F, Yu H, Zhong G, Xu L, Yan S. Exploring specific miRNA-mRNA axes with relationship to taxanes-resistance in breast cancer. Front Oncol. 2020;10:1397. doi: 10.3389/fonc.2020.01397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 92.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007–1017. [PubMed] [Google Scholar]
- 93.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 94.Trock BJ, Leonessa F, Clarke R. Multidrug resistance in breast cancer: a meta-analysis of MDR1/gp170 expression and its possible functional significance. J Natl Cancer Inst. 1997;89:917–931. doi: 10.1093/jnci/89.13.917. [DOI] [PubMed] [Google Scholar]
- 95.Hong HC, Chuang CH, Huang WC, Weng SL, Chen CH, Chang KH, Liao KW, Huang HD. A panel of eight microRNAs is a good predictive parameter for triple-negative breast cancer relapse. Theranostics. 2020;10:8771. doi: 10.7150/thno.46142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Izumi Y, Xu L, Di Tomaso E, Fukumura D, Jain RK. Herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416:279–280. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- 97.Wen X, Yang G, Mao W, Thornton A, Liu J, Bast R, Le X. HER2 signaling modulates the equilibrium between pro-and antiangiogenic factors via distinct pathways: implications for HER2-targeted antibody therapy. Oncogene. 2006;25:6986–6996. doi: 10.1038/sj.onc.1209685. [DOI] [PubMed] [Google Scholar]
- 98.Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066–9076. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pham CG, Bubici C, Zazzeroni F, Knabb JR, Papa S, Kuntzen C, Franzoso G. Upregulation of Twist-1 by NF-κB blocks cytotoxicity induced by chemotherapeutic drugs. Mol Cell Biol. 2007;27:3920–3935. doi: 10.1128/MCB.01219-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vesuna F, Lisok A, Kimble B, Domek J, Kato Y, van der Groep P, Artemov D, Kowalski J, Carraway H, van Diest P. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-α. Oncogene. 2012;31:3223–3234. doi: 10.1038/onc.2011.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gao L, Yang Y, Song S, Hong H, Zhao X, Li D. The association between genetic variant of MDR1 gene and breast cancer risk factors in Chinese women. Int Immunopharmacol. 2013;17:88–91. doi: 10.1016/j.intimp.2013.05.025. [DOI] [PubMed] [Google Scholar]
- 102.Gopisetty MK, Adamecz DI, Nagy FI, Baji Á, Lathira V, Szabó MR, Gáspár R, Csont T, Frank É, Kiricsi M. Androstano-arylpyrimidines: novel small molecule inhibitors of MDR1 for sensitizing multidrug-resistant breast cancer cells. Eur J Pharm Sci. 2021;156:105587. doi: 10.1016/j.ejps.2020.105587. [DOI] [PubMed] [Google Scholar]
- 103.Perez EA. Impact, mechanisms, and novel chemotherapy strategies for overcoming resistance to anthracyclines and taxanes in metastatic breast cancer. Breast Cancer Res Treat. 2009;114:195–201. doi: 10.1007/s10549-008-0005-6. [DOI] [PubMed] [Google Scholar]
- 104.Kort A, Durmus S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Brain and testis accumulation of regorafenib is restricted by breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) Pharm Res. 2015;32:2205–2216. doi: 10.1007/s11095-014-1609-7. [DOI] [PubMed] [Google Scholar]
- 105.Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst. 2000;92:1295–1302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- 106.Sodani K, Patel A, Kathawala RJ, Chen ZS. Multidrug resistance associated proteins in multidrug resistance. Chin J Cancer Res. 2012;31:58. doi: 10.5732/cjc.011.10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vtorushin S, Khristenko K, Zavyalova M, Perelmuter V, Litviakov N, Denisov E, Dulesova A, Cherdyntseva N. The phenomenon of multi-drug resistance in the treatment of malignant tumors. Exp Oncol. 2014;36:144–56. [PubMed] [Google Scholar]
- 108.Xinjun M, Menggao D, Haibin Z. A two-layer approach to developing self-adaptive multi-agent systems in open environment. IJATS. 2014;6:65–85. [Google Scholar]
- 109.Kovalchuk O, Filkowski J, Meservy J, Ilnytskyy Y, Tryndyak VP, Chekhun VF, Pogribny IP. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther. 2008;7:2152–2159. doi: 10.1158/1535-7163.MCT-08-0021. [DOI] [PubMed] [Google Scholar]
- 110.Liang Z, Wu H, Xia J, Li Y, Zhang Y, Huang K, Wagar N, Yoon Y, Cho HT, Scala S. Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1. Biochem Pharmacol. 2010;79:817–824. doi: 10.1016/j.bcp.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 111.Levin ER. Extranuclear estrogen receptor’s roles in physiology: lessons from mouse models. Am J Physiol Endocrinol Metab. 2014;307:E133–E140. doi: 10.1152/ajpendo.00626.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.He YJ, Wu JZ, Ji MH, Ma T, Qiao EQ, Ma R, Tang JH. miR-342 is associated with estrogen receptor-α expression and response to tamoxifen in breast cancer. Exp Ther Med. 2013;5:813–818. doi: 10.3892/etm.2013.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muluhngwi P, Klinge CM. Roles for miRNAs in endocrine resistance in breast cancer. Endocr Relat Cancer. 2015;22:R279. doi: 10.1530/ERC-15-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Luan Q, Zhang B, Li X, Guo M. MiR-129-5p is downregulated in breast cancer cells partly due to promoter H3K27m3 modification and regulates epithelial-mesenchymal transition and multi-drug resistance. Eur Rev Med Pharmacol Sci. 2016;20:4257–4265. [PubMed] [Google Scholar]
- 115.Cochrane DR, Spoelstra NS, Howe EN, Nordeen SK, Richer JK. MicroRNA-200c mitigates invasiveness and restores sensitivity to microtubule-targeting chemotherapeutic agents. Mol Cancer Ther. 2009;8:1055–1066. doi: 10.1158/1535-7163.MCT-08-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi Y, Xiong W, Li G, Lu J, Fodstad O. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem. 2010;285:21496–21507. doi: 10.1074/jbc.M109.083337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Saghatelyan T, Tananyan A, Janoyan N, Tadevosyan A, Petrosyan H, Hovhannisyan A, Hayrapetyan L, Arustamyan M, Arnhold J, Rotmann AR. Efficacy and safety of curcumin in combination with paclitaxel in patients with advanced, metastatic breast cancer: a comparative, randomized, double-blind, placebo-controlled clinical trial. Phytomedicine. 2020;70:153218. doi: 10.1016/j.phymed.2020.153218. [DOI] [PubMed] [Google Scholar]
- 118.Gooding AJ, Schiemann WP. Epithelial-mesenchymal transition programs and cancer stem cell phenotypes: mediators of breast cancer therapy resistance. Mol Cancer Res. 2020;18:1257–1270. doi: 10.1158/1541-7786.MCR-20-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhou ZY, Wan LL, Yang QJ, Han YL, Li D, Lu J, Guo C. Nilotinib reverses ABCB1/P-glycoprotein-mediated multidrug resistance but increases cardiotoxicity of doxorubicin in a MDR xenograft model. Toxicol Lett. 2016;259:124–132. doi: 10.1016/j.toxlet.2016.07.710. [DOI] [PubMed] [Google Scholar]
- 120.Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, Tamaki Y, Noguchi S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin Cancer Res. 2009;15:4234–4241. doi: 10.1158/1078-0432.CCR-08-1479. [DOI] [PubMed] [Google Scholar]
- 121.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 122.To KK. MicroRNA: a prognostic biomarker and a possible druggable target for circumventing multidrug resistance in cancer chemotherapy. J Biomed Sci. 2013;20:1–19. doi: 10.1186/1423-0127-20-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gatenby RA, Gillies RJ, Brown JS. The evolutionary dynamics of cancer prevention. Nat Rev Cancer. 2010;10:526–527. doi: 10.1038/nrc2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 2014;343:72–76. doi: 10.1126/science.1241328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li ZW, Dalton WS. Tumor microenvironment and drug resistance in hematologic malignancies. Blood Rev. 2006;20:333–342. doi: 10.1016/j.blre.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 126.Shannon AM, Bouchier-Hayes DJ, Condron CM, Toomey D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat Rev. 2003;29:297–307. doi: 10.1016/s0305-7372(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 127.Tsuruo T, Naito M, Tomida A, Fujita N, Mashima T, Sakamoto H, Haga N. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci. 2003;94:15–21. doi: 10.1111/j.1349-7006.2003.tb01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, Keating MJ, Huang P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613–621. [PubMed] [Google Scholar]
- 129.Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387–3394. [PubMed] [Google Scholar]
- 130.Vannini I, Zoli W, Tesei A, Rosetti M, Sansone P, Storci G, Passardi A, Massa I, Ricci M, Gusolfino D. Role of p53 codon 72 arginine allele in cell survival in vitro and in the clinical outcome of patients with advanced breast cancer. Tumor Biol. 2008;29:145–151. doi: 10.1159/000143400. [DOI] [PubMed] [Google Scholar]
- 131.Martin CM, Ferdous A, Gallardo T, Humphries C, Sadek H, Caprioli A, Garcia JA, Szweda LI, Garry MG, Garry DJ. Hypoxia-inducible factor-2α transactivates Abcg2 and promotes cytoprotection in cardiac side population cells. Circ Res. 2008;102:1075–1081. doi: 10.1161/CIRCRESAHA.107.161729. [DOI] [PubMed] [Google Scholar]
- 132.Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73:1524–1535. doi: 10.1158/0008-5472.CAN-12-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Daniel C, Bell C, Burton C, Harguindey S, Reshkin SJ, Rauch C. The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochim Biophys Acta Mol Basis Dis. 2013;1832:606–617. doi: 10.1016/j.bbadis.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 134.Sun-Wada GH, Wada Y. Vacuolar-type proton pump ATPases: acidification and pathological relationships. Histol Histopathol. 2013;28:805–15. doi: 10.14670/HH-28.805. [DOI] [PubMed] [Google Scholar]
- 135.Fogarty F, O’keeffe J, Zhadanov A, Papkovsky D, Ayllon V, O’connor R. HRG-1 enhances cancer cell invasive potential and couples glucose metabolism to cytosolic/extracellular pH gradient regulation by the vacuolar-H+ ATPase. Oncogene. 2014;33:4653–4663. doi: 10.1038/onc.2013.403. [DOI] [PubMed] [Google Scholar]
- 136.Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint Victor C, Wiemann BZ, Ishwaran H. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159:499–513. doi: 10.1016/j.cell.2014.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Huang J, Woods P, Normolle D, Goff J, Benos P, Stehle C, Steinman R. Downregulation of estrogen receptor and modulation of growth of breast cancer cell lines mediated by paracrine stromal cell signals. Breast Cancer Res Treat. 2017;161:229–243. doi: 10.1007/s10549-016-4052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Faramand R, Jain M, Staedtke V, Kotani H, Bai R, Reid K, Lee SB, Spitler K, Wang X, Cao B. Tumor microenvironment composition and severe cytokine release syndrome (CRS) influence toxicity in patients with large B-cell lymphoma treated with axicabtagene ciloleucel. Clin Cancer Res. 2020;26:4823–4831. doi: 10.1158/1078-0432.CCR-20-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ansell SM, Vonderheide RH. Cellular composition of the tumor microenvironment. Am Soc Clin Oncol Educ Book. 2013;33:e91–e97. doi: 10.14694/EdBook_AM.2013.33.e91. [DOI] [PubMed] [Google Scholar]
- 140.De Palma M, Lewis CE. Macrophages limit chemotherapy. Nature. 2011;472:303–304. doi: 10.1038/472303a. [DOI] [PubMed] [Google Scholar]
- 141.Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-McGuinn KM, Zabor EC, Brogi E, Joyce JA. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25:2465–2479. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Patel SA, Meyer JR, Greco SJ, Corcoran KE, Bryan M, Rameshwar P. Mesenchymal stem cells protect breast cancer cells through regulatory T cells: role of mesenchymal stem cell-derived TGF-β. J Immunol. 2010;184:5885–5894. doi: 10.4049/jimmunol.0903143. [DOI] [PubMed] [Google Scholar]
- 145.Bonomi A, Silini A, Vertua E, Signoroni PB, Coccè V, Cavicchini L, Sisto F, Alessandri G, Pessina A, Parolini O. Human amniotic mesenchymal stromal cells (hAMSCs) as potential vehicles for drug delivery in cancer therapy: an in vitro study. Stem Cell Res Ther. 2015;6:1–10. doi: 10.1186/s13287-015-0140-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sun B, Roh KH, Park JR, Lee SR, Park SB, Jung JW, Kang SK, Lee YS, Kang KS. Therapeutic potential of mesenchymal stromal cells in a mouse breast cancer metastasis model. Cytotherapy. 2009;11:289–298. doi: 10.1080/14653240902807026. [DOI] [PubMed] [Google Scholar]
- 147.Gu Y, Liu Y, Fu L, Zhai L, Zhu J, Han Y, Jiang Y, Zhang Y, Zhang P, Jiang Z, Zhang X, Cao X. Tumor-educated B cells selectively promote breast cancer lymph node metastasis by HSPA4-targeting IgG. Nat Med. 2019;25:312–322. doi: 10.1038/s41591-018-0309-y. [DOI] [PubMed] [Google Scholar]
- 148.Sridharan S, Howard CM, Tilley AMC, Subramaniyan B, Tiwari AK, Ruch RJ, Raman D. Novel and alternative targets against breast cancer stemness to combat chemoresistance. Front Oncol. 2019;9:1003. doi: 10.3389/fonc.2019.01003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fletcher JI, Williams RT, Henderson MJ, Norris MD, Haber M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist Updat. 2016;26:1–9. doi: 10.1016/j.drup.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 150.Bower JJ, Vance LD, Psioda M, Smith-Roe SL, Simpson DA, Ibrahim JG, Hoadley KA, Perou CM, Kaufmann WK. Patterns of cell cycle checkpoint deregulation associated with intrinsic molecular subtypes of human breast cancer cells. NPJ Breast Cancer. 2017;3:9. doi: 10.1038/s41523-017-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Thu KL, Soria-Bretones I, Mak TW, Cescon DW. Targeting the cell cycle in breast cancer: towards the next phase. Cell Cycle. 2018;17:1871–1885. doi: 10.1080/15384101.2018.1502567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M, Rodriguez O, Grueso J, Bellet M, Cortés J, Elliott R, Pancholi S, Baselga J, Dowsett M, Martin LA, Turner NC, Serra V. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76:2301–2313. doi: 10.1158/0008-5472.CAN-15-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Costa B, Vale N. Drug metabolism for the identification of clinical biomarkers in breast cancer. Int J Mol Sci. 2022;23:3181. doi: 10.3390/ijms23063181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Icard P, Shulman S, Farhat D, Steyaert JM, Alifano M, Lincet H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist Updat. 2018;38:1–11. doi: 10.1016/j.drup.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 155.Komurov K, Tseng JT, Muller M, Seviour EG, Moss TJ, Yang L, Nagrath D, Ram PT. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol Syst Biol. 2012;8:596. doi: 10.1038/msb.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhao Y, Liu H, Liu Z, Ding Y, LeDoux SP, Wilson GL, Voellmy R, Lin Y, Lin W, Nahta R, Liu B, Fodstad O, Chen J, Wu Y, Price JE, Tan M. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011;71:4585–4597. doi: 10.1158/0008-5472.CAN-11-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB, Ledoux SP, Tan M. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer. 2010;9:33. doi: 10.1186/1476-4598-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Oliver TG, Mercer KL, Sayles LC, Burke JR, Mendus D, Lovejoy KS, Cheng MH, Subramanian A, Mu D, Powers S, Crowley D, Bronson RT, Whittaker CA, Bhutkar A, Lippard SJ, Golub T, Thomale J, Jacks T, Sweet-Cordero EA. Chronic cisplatin treatment promotes enhanced damage repair and tumor progression in a mouse model of lung cancer. Genes Dev. 2010;24:837–852. doi: 10.1101/gad.1897010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang LE, Yin M, Dong Q, Stewart DJ, Merriman KW, Amos CI, Spitz MR, Wei Q. DNA repair capacity in peripheral lymphocytes predicts survival of patients with non-small-cell lung cancer treated with first-line platinum-based chemotherapy. J. Clin. Oncol. 2011;29:4121–4128. doi: 10.1200/JCO.2010.34.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Neboori HJ, Haffty BG, Wu H, Yang Q, Aly A, Goyal S, Schiff D, Moran MS, Golhar R, Chen C, Moore D, Ganesan S. Low p53 binding protein 1 (53BP1) expression is associated with increased local recurrence in breast cancer patients treated with breast-conserving surgery and radiotherapy. Int J Radiat Oncol Biol Phys. 2012;83:e677–683. doi: 10.1016/j.ijrobp.2012.01.089. [DOI] [PubMed] [Google Scholar]
- 161.Stefanski CD, Keffler K, McClintock S, Milac L, Prosperi JR. APC loss affects DNA damage repair causing doxorubicin resistance in breast cancer cells. Neoplasia. 2019;21:1143–1150. doi: 10.1016/j.neo.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kettner NM, Vijayaraghavan S, Durak MG, Bui T, Kohansal M, Ha MJ, Liu B, Rao X, Wang J, Yi M, Carey JPW, Chen X, Eckols TK, Raghavendra AS, Ibrahim NK, Karuturi MS, Watowich SS, Sahin A, Tweardy DJ, Hunt KK, Tripathy D, Keyomarsi K. Combined inhibition of STAT3 and DNA repair in palbociclib-resistant ER-positive breast cancer. Clin Cancer Res. 2019;25:3996–4013. doi: 10.1158/1078-0432.CCR-18-3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, Cheney EM, Bouwman P, Jonkers J, Rottenberg S, Guerriero JL, Wulf GM, Shapiro GI. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 2019;9:722–737. doi: 10.1158/2159-8290.CD-18-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li LY, Guan YD, Chen XS, Yang JM, Cheng Y. DNA repair pathways in cancer therapy and resistance. Front Pharmacol. 2020;11:629266. doi: 10.3389/fphar.2020.629266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zelenka J, Koncošová M, Ruml T. Targeting of stress response pathways in the prevention and treatment of cancer. Biotechnol Adv. 2018;36:583–602. doi: 10.1016/j.biotechadv.2018.01.007. [DOI] [PubMed] [Google Scholar]
- 166.Fiume L, Manerba M, Vettraino M, Di Stefano G. Inhibition of lactate dehydrogenase activity as an approach to cancer therapy. Future Med Chem. 2014;6:429–445. doi: 10.4155/fmc.13.206. [DOI] [PubMed] [Google Scholar]
- 167.Zhao N, Cao J, Xu L, Tang Q, Dobrolecki LE, Lv X, Talukdar M, Lu Y, Wang X, Hu DZ, Shi Q, Xiang Y, Wang Y, Liu X, Bu W, Jiang Y, Li M, Gong Y, Sun Z, Ying H, Yuan B, Lin X, Feng XH, Hartig SM, Li F, Shen H, Chen Y, Han L, Zeng Q, Patterson JB, Kaipparettu BA, Putluri N, Sicheri F, Rosen JM, Lewis MT, Chen X. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J Clin Invest. 2018;128:1283–1299. doi: 10.1172/JCI95873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liu L, Ulbrich J, Müller J, Wüstefeld T, Aeberhard L, Kress TR, Muthalagu N, Rycak L, Rudalska R, Moll R, Kempa S, Zender L, Eilers M, Murphy DJ. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483:608–612. doi: 10.1038/nature10927. [DOI] [PubMed] [Google Scholar]
- 169.Zhou J, Liu Q, Zhang F, Wang Y, Zhang S, Cheng H, Yan L, Li L, Chen F, Xie X. Overexpression of OsPIL15, a phytochrome-interacting factor-like protein gene, represses etiolated seedling growth in rice. J Integr Plant Biol. 2014;56:373–387. doi: 10.1111/jipb.12137. [DOI] [PubMed] [Google Scholar]
- 170.Xu YG, Liu R, Sui N, Shi W, Wang L, Tian C, Song J. Changes in endogenous hormones and seed-coat phenolics during seed storage of two Suaeda salsa populations. Aust J Bot. 2016;64:325–332. [Google Scholar]
- 171.Esbona K, Inman D, Saha S, Jeffery J, Schedin P, Wilke L, Keely P. COX-2 modulates mammary tumor progression in response to collagen density. Breast Cancer Res. 2016;18:35. doi: 10.1186/s13058-016-0695-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Koren E, Fuchs Y. Modes of regulated cell death in cancer. Cancer Discov. 2021;11:245–265. doi: 10.1158/2159-8290.CD-20-0789. [DOI] [PubMed] [Google Scholar]
- 173.Yang L. Breast Cancer and Molecular Medicine. Springer; 2006. Mechanisms of apoptosis resistance in breast cancer; pp. 841–858. [Google Scholar]
- 174.Lin T, Huang X, Gu J, Zhang L, Roth JA, Xiong M, Curley SA, Yu Y, Hunt KK, Fang B. Long-term tumor-free survival from treatment with the GFP-TRAIL fusion gene expressed from the hTERT promoter in breast cancer cells. Oncogene. 2002;21:8020–8028. doi: 10.1038/sj.onc.1205926. [DOI] [PubMed] [Google Scholar]
- 175.Singh TR, Shankar S, Chen X, Asim M, Srivastava RK. Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer Res. 2003;63:5390–5400. [PubMed] [Google Scholar]
- 176.Kamalabadi-Farahani M, H Najafabadi MR, Jabbarpour Z. Apoptotic resistance of metastatic tumor cells in triple negative breast cancer: roles of death receptor-5. Asian Pac J Cancer Prev. 2019;20:1743. doi: 10.31557/APJCP.2019.20.6.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Alpuim Costa D, Nobre JG, Batista MV, Ribeiro C, Calle C, Cortes A, Marhold M, Negreiros I, Borralho P, Brito M. Human microbiota and breast cancer-is there any relevant link? A literature review and new horizons toward personalised medicine. Front Microbiol. 2021;12:357. doi: 10.3389/fmicb.2021.584332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, Thaiss CA, Reuben A, Livny J, Avraham R, Frederick DT, Ligorio M, Chatman K, Johnston SE, Mosher CM, Brandis A, Fuks G, Gurbatri C, Gopalakrishnan V, Kim M, Hurd MW, Katz M, Fleming J, Maitra A, Smith DA, Skalak M, Bu J, Michaud M, Trauger SA, Barshack I, Golan T, Sandbank J, Flaherty KT, Mandinova A, Garrett WS, Thayer SP, Ferrone CR, Huttenhower C, Bhatia SN, Gevers D, Wargo JA, Golub TR, Straussman R. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156–1160. doi: 10.1126/science.aah5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Panebianco C, Andriulli A, Pazienza V. Pharmacomicrobiomics: exploiting the drug-microbiota interactions in anticancer therapies. Microbiome. 2018;6:92. doi: 10.1186/s40168-018-0483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Colleoni M, Montagna E. Neoadjuvant therapy for ER-positive breast cancers. Ann Oncol. 2012;23:x243–x248. doi: 10.1093/annonc/mds305. [DOI] [PubMed] [Google Scholar]
- 181.McGuire W. Estrogen receptors in human breast cancer: an overview. Estrogen Receptor in Human Breast Cancer. J Clin Invest. 1975 [Google Scholar]
- 182.Carroll J. EJE PRIZE 2016: mechanisms of oestrogen receptor (ER) gene regulation in breast cancer. Eur J Endocrinol. 2016;175:R41–R49. doi: 10.1530/EJE-16-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. 2015;12:573–583. doi: 10.1038/nrclinonc.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, Smith A, Wilson J, Morrow C, Wong WL. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 2017;7:277–287. doi: 10.1158/2159-8290.CD-15-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rimawi MF, Schiff R, Osborne CK. Targeting HER2 for the treatment of breast cancer. Annu Rev Med. 2015;66:111–28. doi: 10.1146/annurev-med-042513-015127. [DOI] [PubMed] [Google Scholar]
- 187.Chung I, Reichelt M, Shao L, Akita RW, Koeppen H, Rangell L, Schaefer G, Mellman I, Sliwkowski MX. High cell-surface density of HER2 deforms cell membranes. Nat Commun. 2016;7:1–11. doi: 10.1038/ncomms12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Anderson WF, Rosenberg PS, Katki HA. Tracking and evaluating molecular tumor markers with cancer registry data: HER2 and breast cancer. J Natl Cancer Inst. 2014;106:dju093. doi: 10.1093/jnci/dju093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Martin M, López-Tarruella S. Emerging therapeutic options for HER2-positive breast cancer. Am Soc Clin Oncol Educ Book. 2016;36:e64–e70. doi: 10.1200/EDBK_159167. [DOI] [PubMed] [Google Scholar]
- 190.Dent S, Oyan B, Honig A, Mano M, Howell S. HER2-targeted therapy in breast cancer: a systematic review of neoadjuvant trials. Cancer Treat Rev. 2013;39:622–631. doi: 10.1016/j.ctrv.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 191.Llombart-Cussac A, Cortés J, Paré L, Galván P, Bermejo B, Martínez N, Vidal M, Pernas S, López R, Muñoz M. HER2-enriched subtype as a predictor of pathological complete response following trastuzumab and lapatinib without chemotherapy in early-stage HER2-positive breast cancer (PAMELA): an open-label, single-group, multicentre, phase 2 trial. Lancet Oncol. 2017;18:545–554. doi: 10.1016/S1470-2045(17)30021-9. [DOI] [PubMed] [Google Scholar]
- 192.Montemurro F, Di Cosimo S, Arpino G. Human epidermal growth factor receptor 2 (HER2)-positive and hormone receptor-positive breast cancer: new insights into molecular interactions and clinical implications. Ann Oncol. 2013;24:2715–2724. doi: 10.1093/annonc/mdt287. [DOI] [PubMed] [Google Scholar]
- 193.Trayes KP, Cokenakes SE. Breast cancer treatment. Am Fam Physician. 2021;104:171–178. [PubMed] [Google Scholar]
- 194.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 195.Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 2000;19:6102–6114. doi: 10.1038/sj.onc.1203973. [DOI] [PubMed] [Google Scholar]
- 196.Ghosh R, Narasanna A, Wang SE, Liu S, Chakrabarty A, Balko JM, González-Angulo AM, Mills GB, Penuel E, Winslow J. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res. 2011;71:1871–1882. doi: 10.1158/0008-5472.CAN-10-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bates M, Sperinde J, Köstler W, Ali S, Leitzel K, Fuchs E, Paquet A, Lie Y, Sherwood T, Horvat R. Identification of a subpopulation of metastatic breast cancer patients with very high HER2 expression levels and possible resistance to trastuzumab. Ann Oncol. 2011;22:2014–2020. doi: 10.1093/annonc/mdq706. [DOI] [PubMed] [Google Scholar]
- 198.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 199.Pegram M, Hsu S, Lewis G, Pietras R, Beryt M, Sliwkowski M, Coombs D, Baly D, Kabbinavar F, Slamon D. Inhibitory effects of combinations of HER-2/neu antibody and chemotherapeutic agents used for treatment of human breast cancers. Oncogene. 1999;18:2241–2251. doi: 10.1038/sj.onc.1202526. [DOI] [PubMed] [Google Scholar]
- 200.Pietras RJ, Pegram MD, Finn RS, Maneval DA, Slamon DJ. Remission of human breast cancer xenografts on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene. 1998;17:2235–2249. doi: 10.1038/sj.onc.1202132. [DOI] [PubMed] [Google Scholar]
- 201.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 202.Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 1999;17:2639–2639. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 203.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 204.Aydiner A. Breast Dis. Springer; 2019. Treatment of HER2-overexpressing metastatic breast cancer; pp. 463–494. [Google Scholar]
- 205.Valero V, Forbes J, Pegram MD, Pienkowski T, Eiermann W, Von Minckwitz G, Roche H, Martin M, Crown J, Mackey JR. Multicenter phase III randomized trial comparing docetaxel and trastuzumab with docetaxel, carboplatin, and trastuzumab as first-line chemotherapy for patients with HER2-gene-amplified metastatic breast cancer (BCIRG 007 study): two highly active therapeutic regimens. J. Clin. Oncol. 2011;29:149–156. doi: 10.1200/JCO.2010.28.6450. [DOI] [PubMed] [Google Scholar]
- 206.Robert N, Leyland-Jones B, Asmar L, Belt R, Ilegbodu D, Loesch D, Raju R, Valentine E, Sayre R, Cobleigh M. Randomized phase III study of trastuzumab, paclitaxel, and carboplatin compared with trastuzumab and paclitaxel in women with HER-2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2006;24:2786–2792. doi: 10.1200/JCO.2005.04.1764. [DOI] [PubMed] [Google Scholar]
- 207.Guarneri V, Lenihan DJ, Valero V, Durand JB, Broglio K, Hess KR, Michaud LB, Gonzalez-Angulo AM, Hortobagyi GN, Esteva FJ. Long-term cardiac tolerability of trastuzumab in metastatic breast cancer: the MD Anderson Cancer Center experience. J. Clin. Oncol. 2006;24:4107–4115. doi: 10.1200/JCO.2005.04.9551. [DOI] [PubMed] [Google Scholar]
- 208.Reinhorn D, Kuchuk I, Shochat T, Nisenbaum B, Sulkes A, Hendler D, Rotem O, Tsoref D, Olitzky O, Goldvaser H, Sarfaty M, Neiman V, Prus J, Gottfried M, Yust-Katz S, Yerushalmi R. Taxane versus vinorelbine in combination with trastuzumab and pertuzumab for first-line treatment of metastatic HER2-positive breast cancer: a retrospective two-center study. Breast Cancer Res Treat. 2021;188:379–387. doi: 10.1007/s10549-021-06198-4. [DOI] [PubMed] [Google Scholar]
- 209.Paul B, Trovato JA, Thompson J. Lapatinib: a dual tyrosine kinase inhibitor for metastatic breast cancer. Am J Health Syst Pharm. 2008;65:1703–1710. doi: 10.2146/ajhp070646. [DOI] [PubMed] [Google Scholar]
- 210.Guan Z, Xu B, DeSilvio ML, Shen Z, Arpornwirat W, Tong Z, Lorvidhaya V, Jiang Z, Yang J, Makhson A. Randomized trial of lapatinib versus placebo added to paclitaxel in the treatment of human epidermal growth factor receptor 2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2013;31:1947–1953. doi: 10.1200/JCO.2011.40.5241. [DOI] [PubMed] [Google Scholar]
- 211.Gelmon KA, Boyle FM, Kaufman B, Huntsman DG, Manikhas A, Di Leo A, Martin M, Schwartzberg LS, Lemieux J, Aparicio S. Lapatinib or trastuzumab plus taxane therapy for human epidermal growth factor receptor 2-positive advanced breast cancer: final results of NCIC CTG MA. 31. J. Clin. Oncol. 2015;33:1574–1583. doi: 10.1200/JCO.2014.56.9590. [DOI] [PubMed] [Google Scholar]
- 212.Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R, Overbeek E. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 213.Awada A, Colomer R, Inoue K, Bondarenko I, Badwe RA, Demetriou G, Lee SC, Mehta AO, Kim SB, Bachelot T. Neratinib plus paclitaxel vs trastuzumab plus paclitaxel in previously untreated metastatic ERBB2-positive breast cancer: the NEfERT-T randomized clinical trial. JAMA Oncol. 2016;2:1557–1564. doi: 10.1001/jamaoncol.2016.0237. [DOI] [PubMed] [Google Scholar]
- 214.Alvarez RH, Valero V, Hortobagyi GN. Emerging targeted therapies for breast cancer. J. Clin. Oncol. 2010;28:3366–3379. doi: 10.1200/JCO.2009.25.4011. [DOI] [PubMed] [Google Scholar]
- 215.Franklin MC, Carey KD, Vajdos FF, Leahy DJ, De Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5:317–328. doi: 10.1016/s1535-6108(04)00083-2. [DOI] [PubMed] [Google Scholar]
- 216.Harbeck N, Beckmann MW, Rody A, Schneeweiss A, Müller V, Fehm T, Marschner N, Gluz O, Schrader I, Heinrich G. HER2 dimerization inhibitor pertuzumab-mode of action and clinical data in breast cancer. Breast Care. 2013;8:49–55. doi: 10.1159/000346837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Swain SM, Baselga J, Kim SB, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A, Heeson S. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372:724–734. doi: 10.1056/NEJMoa1413513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Baselga J, Cortés J, Im SA, Clark E, Ross G, Kiermaier A, Swain SM. Biomarker analyses in CLEOPATRA: a phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J. Clin. Oncol. 2014;32:3753–3761. doi: 10.1200/JCO.2013.54.5384. [DOI] [PubMed] [Google Scholar]
- 219.Gonnissen A, Isebaert S, Haustermans K. Targeting the hedgehog signaling pathway in cancer: beyond smoothened. Oncotarget. 2015;6:13899. doi: 10.18632/oncotarget.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ruch JM, Kim EJ. Hedgehog signaling pathway and cancer therapeutics: progress to date. Drugs. 2013;73:613–623. doi: 10.1007/s40265-013-0045-z. [DOI] [PubMed] [Google Scholar]
- 221.Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171–2179. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Casey D, Demko S, Shord S, Zhao H, Chen H, He K, Putman A, Helms W, Keegan P, Pazdur R. FDA approval summary: sonidegib for locally advanced basal cell carcinoma. Clin Cancer Res. 2017;23:2377–2381. doi: 10.1158/1078-0432.CCR-16-2051. [DOI] [PubMed] [Google Scholar]
- 223.Jin G, Sivaraman A, Lee K. Development of taladegib as a sonic hedgehog signaling pathway inhibitor. Arch Pharm Res. 2017;40:1390–1393. doi: 10.1007/s12272-017-0987-x. [DOI] [PubMed] [Google Scholar]
- 224.Ueno H, Kondo S, Yoshikawa S, Inoue K, Andre V, Tajimi M, Murakami H. A phase I and pharmacokinetic study of taladegib, a Smoothened inhibitor, in Japanese patients with advanced solid tumors. Invest New Drugs. 2018;36:647–656. doi: 10.1007/s10637-017-0544-y. [DOI] [PubMed] [Google Scholar]
- 225.Lee MJ, Hatton BA, Villavicencio EH, Khanna PC, Friedman SD, Ditzler S, Pullar B, Robison K, White KF, Tunkey C. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc Natl Acad Sci U S A. 2012;109:7859–7864. doi: 10.1073/pnas.1114718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Savona MR, Pollyea DA, Stock W, Oehler VG, Schroeder MA, Lancet J, McCloskey J, Kantarjian HM, Ma WW, Shaik MN. Phase Ib study of glasdegib, a hedgehog pathway inhibitor, in combination with standard chemotherapy in patients with AML or high-risk MDS. Clin Cancer Res. 2018;24:2294–2303. doi: 10.1158/1078-0432.CCR-17-2824. [DOI] [PubMed] [Google Scholar]
- 227.Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang ALS. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342–353. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kim J, Aftab B, Tang J, Kim D, Lee A, Rezaee M, Kim J, Chen B, King E, Borodovsky A. Itraconazole and arsenic trioxide inhibit hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23:23–34. doi: 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. Arsenic antagonizes the hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A. 2010;107:13432–13437. doi: 10.1073/pnas.1006822107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Gan GN, Jimeno A. Emerging from their burrow: hedgehog pathway inhibitors for cancer. Expert Opin Investig Drugs. 2016;25:1153–1166. doi: 10.1080/13543784.2016.1216973. [DOI] [PubMed] [Google Scholar]
- 231.Benvenuto M, Masuelli L, De Smaele E, Fantini M, Mattera R, Cucchi D, Bonanno E, Di Stefano E, Frajese GV, Orlandi A. In vitro and in vivo inhibition of breast cancer cell growth by targeting the hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget. 2016;7:9250. doi: 10.18632/oncotarget.7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Riaz SK, Khan JS, Shah STA, Wang F, Ye L, Jiang WG, Malik MFA. Involvement of hedgehog pathway in early onset, aggressive molecular subtypes and metastatic potential of breast cancer. Cell Commun Signal. 2018;16:1–12. doi: 10.1186/s12964-017-0213-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, Skhinas JN, Collot R, Yang J, Harvey K. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9:1–18. doi: 10.1038/s41467-018-05220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ruiz-Borrego M, Jimenez B, Antolín S, García-Saenz JA, Corral J, Jerez Y, Trigo J, Urruticoechea A, Colom H, Gonzalo N. A phase Ib study of sonidegib (LDE225), an oral small molecule inhibitor of smoothened or Hedgehog pathway, in combination with docetaxel in triple negative advanced breast cancer patients: GEICAM/2012-12 (EDALINE) study. Invest New Drugs. 2019;37:98–108. doi: 10.1007/s10637-018-0614-9. [DOI] [PubMed] [Google Scholar]
- 235.Rizzo P, Miao H, D’Souza G, Osipo C, Yun J, Zhao H, Mascarenhas J, Wyatt D, Antico G, Hao L. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008;68:5226–5235. doi: 10.1158/0008-5472.CAN-07-5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Simões BM, O’Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, Alférez DG, Spence K, Santiago-Gómez A, Chemi F. Anti-estrogen resistance in human breast tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity. Cell Rep. 2015;12:1968–1977. doi: 10.1016/j.celrep.2015.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Grudzien P, Lo S, Albain KS, Robinson P, Rajan P, Strack PR, Golde TE, Miele L, Foreman KE. Inhibition of notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation. Anticancer Res. 2010;30:3853–3867. [PubMed] [Google Scholar]
- 238.Kondratyev M, Kreso A, Hallett R, Girgis-Gabardo A, Barcelon M, Ilieva D, Ware C, Majumder P, Hassell J. Gamma-secretase inhibitors target tumor-initiating cells in a mouse model of ERBB2 breast cancer. Oncogene. 2012;31:93–103. doi: 10.1038/onc.2011.212. [DOI] [PubMed] [Google Scholar]
- 239.Takebe N, Nguyen D, Yang SX. Targeting notch signaling pathway in cancer: clinical development advances and challenges. Pharmacol Ther. 2014;141:140–149. doi: 10.1016/j.pharmthera.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sharma A, Paranjape AN, Rangarajan A, Dighe RR. A monoclonal antibody against human notch1 ligand-binding domain depletes subpopulation of putative breast cancer stem-like cells. Mol Cancer Ther. 2012;11:77–86. doi: 10.1158/1535-7163.MCT-11-0508. [DOI] [PubMed] [Google Scholar]
- 241.Moellering RE, Cornejo M, Davis TN, Bianco CD, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Astudillo L, Da Silva TG, Wang Z, Han X, Jin K, VanWye J, Zhu X, Weaver K, Oashi T, Lopes PE. The small molecule IMR-1 inhibits the notch transcriptional activation complex to suppress tumorigenesis. Cancer Res. 2016;76:3593–3603. doi: 10.1158/0008-5472.CAN-16-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Yong T, Sun A, Henry MD, Meyers S, Davis JN. Down regulation of CSL activity inhibits cell proliferation in prostate and breast cancer cells. J Cell Biochem. 2011;112:2340–2351. doi: 10.1002/jcb.23157. [DOI] [PubMed] [Google Scholar]
- 244.Wilson JJ, Kovall RA. Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell. 2006;124:985–996. doi: 10.1016/j.cell.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 245.Jiang S, Zhang M, Zhang Y, Zhou W, Zhu T, Ruan Q, Chen H, Fang J, Zhou F, Sun J. WNT5B governs the phenotype of basal-like breast cancer by activating WNT signaling. Cell Commun Signal. 2019;17:1–19. doi: 10.1186/s12964-019-0419-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, Kasibhatla S, Schuller AG, Li AG, Cheng D. Targeting Wnt-driven cancer through the inhibition of porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110:20224–20229. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Proffitt KD, Madan B, Ke Z, Pendharkar V, Ding L, Lee MA, Hannoush RN, Virshup DM. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013;73:502–507. doi: 10.1158/0008-5472.CAN-12-2258. [DOI] [PubMed] [Google Scholar]
- 248.Chen M, Wang J, Lu J, Bond MC, Ren XR, Lyerly HK, Barak LS, Chen W. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry. 2009;48:10267–10274. doi: 10.1021/bi9009677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Giraudet AL, Cassier PA, Iwao-Fukukawa C, Garin G, Badel JN, Kryza D, Chabaud S, Gilles-Afchain L, Clapisson G, Desuzinges C. A first-in-human study investigating biodistribution, safety and recommended dose of a new radiolabeled MAb targeting FZD10 in metastatic synovial sarcoma patients. BMC Cancer. 2018;18:1–13. doi: 10.1186/s12885-018-4544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Franco Machado J, Silva RD, Melo R, G Correia JD. Less exploited GPCRs in precision medicine: targets for molecular imaging and theranostics. Molecules. 2018;24:49. doi: 10.3390/molecules24010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Lu D, Choi MY, Yu J, Castro JE, Kipps TJ, Carson DA. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc Natl Acad Sci U S A. 2011;108:13253–13257. doi: 10.1073/pnas.1110431108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Gong X, Azhdarinia A, Ghosh SC, Xiong W, An Z, Liu Q, Carmon KS. LGR5-targeted antibody-drug conjugate eradicates gastrointestinal tumors and prevents recurrence. Mol Cancer Ther. 2016;15:1580–1590. doi: 10.1158/1535-7163.MCT-16-0114. [DOI] [PubMed] [Google Scholar]
- 254.Boolbol SK, Dannenberg AJ, Chadburn A, Martucci C, Guo X, Ramonetti JT, Abreu-Goris M, Newmark HL, Lipkin ML, DeCosse JJ. Cyclooxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Res. 1996;56:2556–2560. [PubMed] [Google Scholar]
- 255.Lee HJ, Wang NX, Shi DL, Zheng JJ. Sulindac inhibits canonical Wnt signaling by blocking the PDZ domain of the protein dishevelled. Angew Chem Int Ed Engl. 2009;48:6448–6452. doi: 10.1002/anie.200902981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Piazza GA, Alberts DS, Hixson LJ, Paranka NS, Li H, Finn T, Bogert C, Guillen JM, Brendel K, Gross PH. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997;57:2909–2915. [PubMed] [Google Scholar]
- 257.Chiu CH, McEntee MF, Whelan J. Sulindac causes rapid regression of preexisting tumors in Min/+ mice independent of prostaglandin biosynthesis. Cancer Res. 1997;57:4267–4273. [PubMed] [Google Scholar]
- 258.Hwang SY, Deng X, Byun S, Lee C, Lee SJ, Suh H, Zhang J, Kang Q, Zhang T, Westover KD. Direct targeting of β-catenin by a small molecule stimulates proteasomal degradation and suppresses oncogenic Wnt/β-catenin signaling. Cell Rep. 2016;16:28–36. doi: 10.1016/j.celrep.2016.05.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Simonetta KR, Taygerly J, Boyle K, Basham SE, Padovani C, Lou Y, Cummins TJ, Yung SL, von Soly SK, Kayser F. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat Commun. 2019;10:1–12. doi: 10.1038/s41467-019-09358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, Jernigan KK, Meyers KC, Hang BI, Waterson AG. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat Chem Biol. 2010;6:829–836. doi: 10.1038/nchembio.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Li B, Orton D, Neitzel LR, Astudillo L, Shen C, Long J, Chen X, Kirkbride KC, Doundoulakis T, Guerra ML. Differential abundance of CK1α provides selectivity for pharmacological CK1α activators to target WNT-dependent tumors. Sci Signal. 2017;10:eaak9916. doi: 10.1126/scisignal.aak9916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Jiang S, Zhang M, Sun J, Yang X. Casein kinase 1α: biological mechanisms and theranostic potential. Cell Commun Signal. 2018;16:1–24. doi: 10.1186/s12964-018-0236-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Ewan K, Pająk B, Stubbs M, Todd H, Barbeau O, Quevedo C, Botfield H, Young R, Ruddle R, Samuel L. A useful approach to identify novel small-molecule inhibitors of Wnt-dependent transcription. Cancer Res. 2010;70:5963–5973. doi: 10.1158/0008-5472.CAN-10-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Cha PH, Cho YH, Lee SK, Lee J, Jeong WJ, Moon BS, Yun JH, Yang JS, Choi S, Yoon J. Small-molecule binding of the axin RGS domain promotes β-catenin and Ras degradation. Nat Chem Biol. 2016;12:593–600. doi: 10.1038/nchembio.2103. [DOI] [PubMed] [Google Scholar]
- 265.McGonigle S, Chen Z, Wu J, Chang P, Kolber-Simonds D, Ackermann K, Twine NC, Shie JL, Miu JT, Huang KC. E7449: a dual inhibitor of PARP1/2 and tankyrase1/2 inhibits growth of DNA repair deficient tumors and antagonizes Wnt signaling. Oncotarget. 2015;6:41307. doi: 10.18632/oncotarget.5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Osawa Y, Oboki K, Imamura J, Kojika E, Hayashi Y, Hishima T, Saibara T, Shibasaki F, Kohara M, Kimura K. Inhibition of cyclic adenosine monophosphate (cAMP)-response element-binding protein (CREB)-binding protein (CBP)/β-catenin reduces liver fibrosis in mice. EBioMedicine. 2015;2:1751–1758. doi: 10.1016/j.ebiom.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Fiskus W, Sharma S, Saha S, Shah B, Devaraj SG, Sun B, Horrigan S, Leveque C, Zu Y, Iyer S. Pre-clinical efficacy of combined therapy with novel β-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia. 2015;29:1267–1278. doi: 10.1038/leu.2014.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, Boutros M, Niehrs C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science. 2010;327:459–463. doi: 10.1126/science.1179802. [DOI] [PubMed] [Google Scholar]
- 269.Minami I, Yamada K, Otsuji TG, Yamamoto T, Shen Y, Otsuka S, Kadota S, Morone N, Barve M, Asai Y. A small molecule that promotes cardiac differentiation of human pluripotent stem cells under defined, cytokine-and xeno-free conditions. Cell rep. 2012;2:1448–1460. doi: 10.1016/j.celrep.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 270.Deshmukh V, Hu H, Barroga C, Bossard C, Kc S, Dellamary L, Stewart J, Chiu K, Ibanez M, Pedraza M. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr Cartil. 2018;26:18–27. doi: 10.1016/j.joca.2017.08.015. [DOI] [PubMed] [Google Scholar]