

Abstract
Monoclonal antibodies targeting the programmed cell death protein-1/programmed cell death-ligand 1 (PD-1/PD-L1) and cytotoxic T lymphocyte-associated protein-4 (CTLA-4) axes have permanently changed the therapeutic landscape for multiple tumor types previously associated with a dismal prognosis such as melanoma, non-small cell lung cancer, renal cell carcinoma, bladder cancer, head and neck squamous cell carcinoma, MSI-high colorectal carcinoma, Merkel cell carcinoma, and Hodgkin lymphoma. However, only a subset of patients initially benefits from these inhibitors, and increasing clinical experience indicates that in a substantial proportion of initial responders, lethal secondary resistance ultimately develops months or years later. In this paper we evaluated combination therapy with a Phase 1 oncolytic adenovirus called AdAPT-001, armed with a TGF-β “trap” that binds to and neutralizes the immunosuppressive cytokine, TGF-β, and a checkpoint inhibitor, anti-PD-L1, in PD-L1 resistant tumors. The study, which was performed in an immunocompetent syngeneic ADS-12 mouse model, demonstrated that the combination of AdAPT-001 with PD-L1 blockade reversed PD-L1 resistance, potentially representing a future paradigm shift for patients that are primarily or secondarily resistant to checkpoint inhibitors.
Keywords: Checkpoint inhibitor blockade, PD-L1, CTLA-4, oncolytic virotherapy, oncolytic adenovirus, TGF-β, BETA PRIME (NCT04673942)
Introduction
Tumors hijack inhibitory pathways, so-called immune checkpoints, to evade and subvert attack by innate and adaptive immune cells [1-3]. Upregulation of these immunoinhibitory molecules (e.g., CTLA-4, PD-1, IDO, LAG3, TIM3, TIGIT, BLTA, CD-47, and SIRP-alpha) [4], which are associated with suppression of antitumor immunity, is the basis for the plethora of immune checkpoint blockers (ICBs) in various stages of clinical development.
Even as the ICBs ipilimumab, which targets CTLA-4, nivolumab and pembrolizumab, which target PD-1 and atezolizumab, durvalumab, and avelumab, which target PD-L1 have won regulatory approval in multiple malignancies including melanoma, non-small cell lung cancer, renal cell carcinoma, classical Hodgkin lymphoma, and recurrent or metastatic head and neck squamous cell carcinoma [5] on the basis of clinically significant and durable responses, only a minority of patients <40% in these particular tumor types have shown benefit. Moreover, a subset of patients that initially respond to immunotherapy, subsequently acquire resistance [6] and lethally relapse while patients with tumor types such as MSS-colorectal cancer, pancreatic ductal adenocarcinoma and metastatic castration-resistant prostate cancer are generally known to not respond at all [7].
Dual CTLA-4 and PD-1 or PD-L1 blockade, which is intended to release the “brakes” on the immune response, increases response rates at the expense of significantly more grade 3 or 4 immune related adverse events (irAEs) due to overlapping toxicity profiles compared with monotherapies [8]. Therefore, to overcome checkpoint resistance and to broaden the therapeutic scope of ICBs will require the rational identification and optimization of synergistic combinations with improved therapeutic indices.
One minimally toxic potential “antiresistance” combination candidate is oncolytic viruses (OVs). As immune-modulating platforms that are engineered to selectively invade tumor tissue and self-amplify while simultaneously augmenting antitumor immunity through crosspresentation of tumor antigens and induction of cytotoxic T-cells [9], OVs are generally non-pathogenic to normal tissues with complementary and non-overlapping mechanisms of activity to ICBs [10].
AdAPT-001 is an adenovirus derived from human adenovirus 5 with two key modifications. The first modification is a 50 base pair deletion in the E1A promoter that transcriptionally ‘de-targets’ the virus from normal tissue without attenuation of its ability to replicate in and lyse tumor cells. Tumor selectivity is also related to defective IFN signaling in cancer cells, which make them more susceptible to the cytolytic effects of the virus.
The second modification to the virus involves the insertion of a chimeric gene of TGFβ receptor II fused with the Fc portion of human IgG1 to generate a soluble TGFβR-IgG fusion protein that “traps” and neutralizes the activity of the pro-oncogenic cytokine, TGFβ. As one of the strongest physiologic immunosuppressants [11], TGFβ is thought to significantly contribute to metastasis and progression, making it a prime immunotherapeutic target [12].
AdAPT-001 has two mechanisms of action. 1) As a replicating virus, it causes a true infection in treated tumors to convert the normally immunosuppressive tumor microenvironment into a highly active site, turning immunologically cold tumors hot. 2) It blocks TGFβ from driving regulatory T cell and M2 macrophage differentiation, promoting Th1 and M1 phenotypes. To investigate the activity of the TGFβ trap transgene while minimizing effects from viral infection, a non-replicating virus carrying a mouse homologue of the TGFβ trap was used to treat immunocompetent tumor bearing mice, and combined with anti-PD-L1 antibody to test synergy with checkpoint inhibition.
Materials and methods
Cancer cell lines and adenovirus
Cancer cell line
Murine KRAS mutant lung adenocarcinoma cell line, ADS-12, was established in house from a mouse lung cancer carrying a KRAS G12D mutation, which is the only unmodified mouse cancer cell line known to support replication of human adenovirus type 5 at levels comparable to human cells. ADS-12 were grown in RPMI 1640 complete medium and supplemented with 10% fetal calf serum, L-glutamine (2 mmol l-1), penicillin (100 IU ml-1), and streptomycin (50 μg ml-1). All cell lines were maintained at 37°C in a humidified atmosphere at 5% CO2.
Virus production
Mouse-AdAPT-001 (19kmTGFβR-IgG), which carries a disruption in E1A and a TGFβR-IgG fusion using the mouse isoforms of those genes for immunologic compatibility with an immunocompetent mouse, was produced in HEK-293A cells. The virus was purified and concentrated using Fast-Trap Virus purification and concentration kits (Millipore, Billerica, MA, USA). Aliquots were kept frozen at -80°C.
Mice and tumor induction
129S4/SvJae mice were originally purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were bred and maintained at Moores Cancer Center Vivarium of University of San Diego and experiments were approved by the Institutional Animal Care and Use Committee. Mice 6-8 weeks old of either gender were injected subcutaneously with one million syngeneic ADS-12 cells and allowed to form tumors until they reached >50 mm3 in size.
Treatment
When the average tumor size reached >50 mm3, mice were randomized into treatment groups, 10 mice per group. Treatment involved intratumoral injections of either viral storage buffer or mouse-AdAPT-001 at 109 PFU/dose on days 0, 4, and 8, plus intraperitoneal injections of either phosphate buffered saline (PBS) or 200 µg anti-PD-L1 antibody (clone 10F.9G2, BioXcell) diluted in PBS on days 1, 5, 9, and 13. To monitor growth of subcutaneous tumors, tumor diameters were measured by calipers and volume calculated by 0.5 × L × W2 in which L is the longest diameter and W is the perpendicular diameter. Endpoint criteria for the survival studies included tumor volume exceeding 1500 mm3 or tumor ulceration, which led to sacrifice of mice. Treated, tumor-free mice were s.c. rechallenged with one million ADS-12 cells (counted as day 0 for rechallenge data) on the opposite flank from the primary tumor at least 2 months after complete rejection of primary tumors.
Statistical analysis
Values are expressed as mean ± s.e.m. Comparison between two groups was based on unpaired t-test. A value of P<0.05 was considered to be statistically significant.
Results
Safety
All mice tolerated the treatments without obvious signs of toxicity. Mice treated with mouse-AdAPT-001 and/or anti-PD-L1 antibody gained weight at the same rate as control mice (Figure 1).
Figure 1.
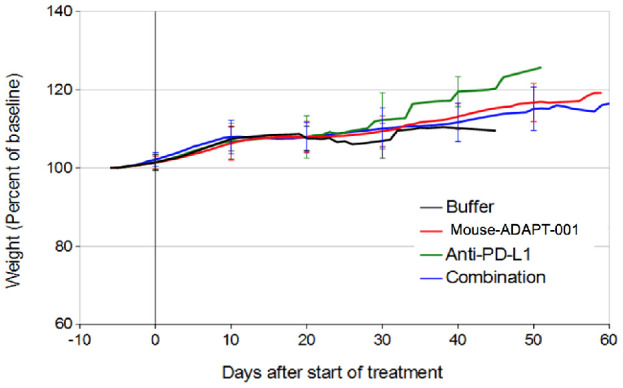
Weight of mice in studies of combination therapy with anti-PD-L1 antibody. 129S4 mice with ADS-12 tumors were treated with intratumoral buffer or mouse-ADAPT-001 on days 0, 4, and 8 and intraperitoneal buffer or anti-PD-L1 antibody on days 1, 5, 9, and 13. Mean ± standard deviation of weight normalized to pre-treatment baseline is plotted for each group. n=10 mice in each group.
Activity
No activity was evident with anti-PD-L1 antibody alone. Treatment with mouse-AdAPT-001 alone in these larger tumors led to complete responses in four of ten mice, and combination therapy led to complete responses in seven of ten mice. Tumor volume was smaller in the combination therapy group compared to mouse-AdAPT-001 alone ten days after starting treatment (P<0.01) (Figure 2). These data further support an immunologic mechanism of action and suggest the presence of synergy between virally mediated TGFβ blockade and systemic immunotherapies, particularly anti-PD-L1 antibody therapy.
Figure 2.
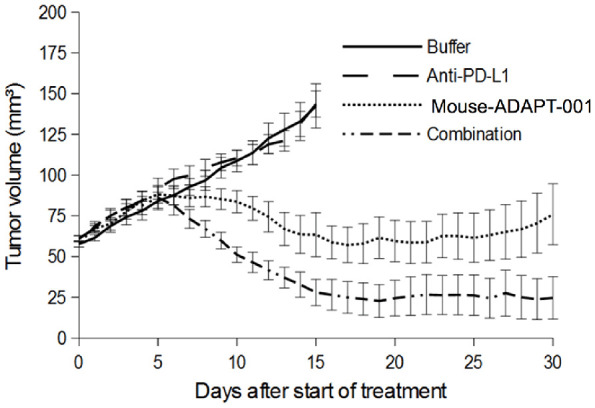
Tumor volumes in studies of combination therapy with anti-PD-L1 antibody. Mean ± SEM tumor volume for each group. n=10 mice per group.
Rechallenge
To determine whether treatment with AdAPT-001 results in immunologic memory, the hallmark of adaptive immunity, the mice that underwent complete tumor regression following AdAPT-001 treatment were rechallenged with an inoculate of ADS-12 cells after primary tumors had not been detected for at least 2 months. No further treatment was given. These treated animals rejected the rechallenged tumors compared with naïve, previously untreated mice in which tumors grew progressively (Figure 3). This result strongly suggests that oncolytic AdAPT-001 therapy generates host antitumor memory T cells that provide long-term protection against tumor relapse.
Figure 3.
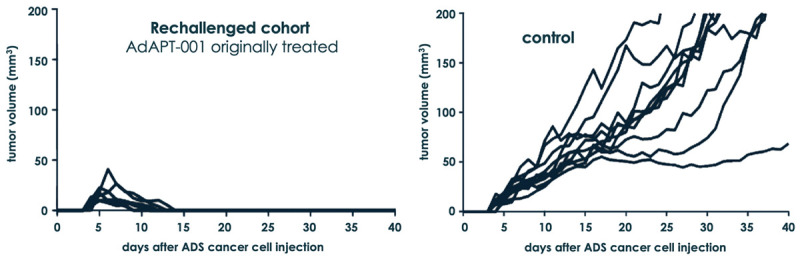
AdAPT-001-treated, tumor-free mice rechallenged with ADS-12 tumor cells on the opposite flank from primary tumor confers long-term protective immunity.
Discussion and conclusion
This is the first published study to demonstrate that AdAPT-001, a first-in-class oncolytic adenovirus armed with a transforming growth factor-β (TGF-β)-neutralizing trap transgene, is safe and synergizes with an anti-PD-L1 checkpoint inhibitor to overcome PD-L1 resistance in an immunocompetent mouse model. The immunocompetency of the mouse model is important to evaluate the impact of an active immune system on overall vector potency and safety; however, murine tumor cells are nonpermissive for human adenoviral infection [13,14]. ADS-12 is, to the best of our knowledge, and as previously described [15], the only immunocompetent syngeneic model in which to test armed oncolytic adenovirus vectors. Alternate animal models such as the cotton rat [16] and the Syrian hamster [17] have previously been used for this purpose but are far from satisfactory or desirable since Ad infection in these species is only semipermissive [18,19].
Tumors have been described as evolving ecosystems in which the cancer cells dynamically interact with their microenvironment, analogous to the biotic associations between different species in a given habitat [20]. A key driver of ecosystem dynamics is the relationship between predator and prey, which in oncology may be represented by the immune system (predator) and the cancer cells (prey). Just as several anti-predation adaptations and strategies occur in nature (e.g., camouflage, venom, body armor etc.), cancer predation defense mechanisms assume several forms including:
• loss of antigenicity through the acquisition of defects or deficiencies in antigen presentation.
• loss of immunogenicity through a lack of immunogenic tumor antigens.
• immunosuppression [21] by which the cancer cells inhibit the production of pro-inflammatory cytokines and dampen cytotoxic T-cell and myeloid responses through overexpression of immune checkpoint molecules.
These strategies, illustrated in Figure 4, collectively result in a state of functional tolerance in which T-cells and myeloid cells not only coexist with (in) the tumor but also support its growth and progression.
Figure 4.
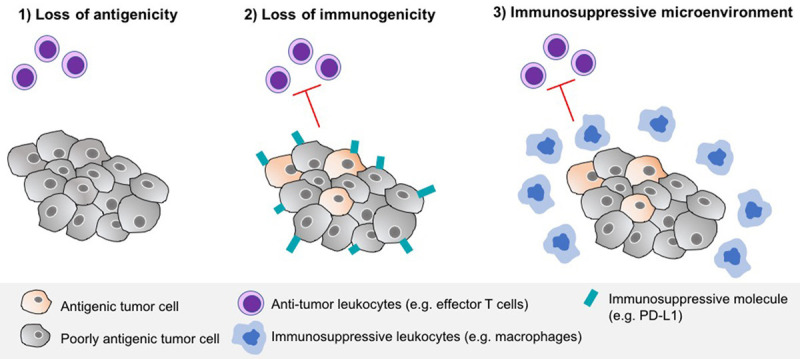
Tumor antipredation strategies against the immune system.
In theory, AdAPT-001, as a lytic virus that not only boosts the adjuvanticity and antigenicity of tumor cells but also neutralizes a key suppressive cytokine, has the potential to address all three of these tolerogenic mechanisms and tilt the balance in favor of the immune system with the induction of long-term protective memory against the tumor to prevent relapse metastases and recurrence. Lysed cancer cells release a plethora of pro-inflammatory cytokines such as IL-6, TNF-α, and interferon that act locally to promote the death of bystander cells [22]. Moreover, since adenoviruses are pathogens, danger signals [23] are also produced in the form of toll-like receptors (TLRs) or damage-associated molecular pattern molecules (DAMPs), hence improving adjuvanticity. In addition, similar to the abscopal or ‘out-of-field’ effect with radiation therapy, capture of released tumor antigens by bystander antigen-presenting cells (APC) following tumor cell lysis is assumed to facilitate cross-priming of an anticancer immune response. On the other hand, as part of a counterregulatory measure to limit excessive inflammation, oncolytic viral infection induces suppressive pathways like PD-L1 [24], which suggests the translational potential for synergistic combinations with immune checkpoint blockers.
As previously discussed, while ICBs have revolutionized the treatment of cancer and their success in a subset of patients with responsive tumor types is unprecedented, the majority either fail to respond initially or develop secondary resistance. The relative refractoriness to ICBs is in part a function of degree and density of lymphocytic infiltration [25] with “cold”, “desert-like” or non-T cell inflamed tumors responding poorly or not at all in contrast to “hot”, or T cell-inflamed tumors that tend to respond well. In fact, it has been suggested 1) that the current ceiling of response rates with ICBs in the range of 20-40% directly correlates with the high occurrence of cold tumors, which constitute the most common immunologic phenotype [26] and 2) that while raising a multipronged immune attack is clearly desirable combinations of checkpoint inhibitors are unlikely to achieve dramatic success rates [27]. Based on the results from this study, one strategy to overcome cancer cell resistance and turn ICB non-responders to responders is with oncolytic viruses (see Figure 5), which may immunologically sensitize (or resensitize) T-cells to checkpoint blockade.
Figure 5.
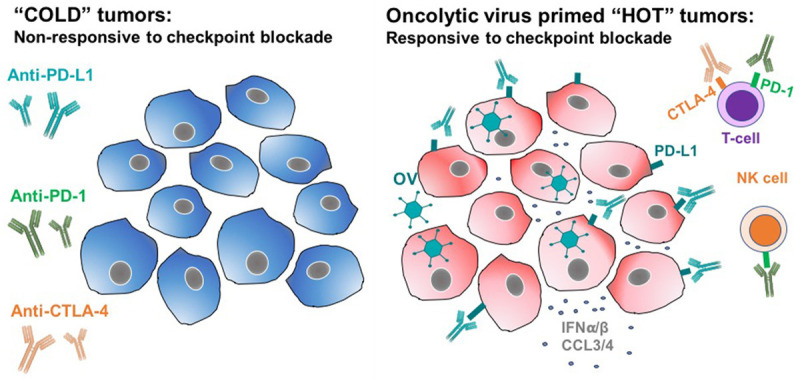
Oncolytic viruses have the potential to convert uninflamed “cold” tumors, so called “immune deserts”, into “hot”/inflamed ones that respond well to checkpoint inhibition.
In summary, this study demonstrates that localized oncolytic infection with AdAPT-001 is safe and abrogates resistance to systemic PD-L1 immunotherapy as well as results in durable protection against syngeneic tumor rechallenge, which strongly supports further evaluation of this combination in upcoming Phase 1 and Phase 2 clinical studies. AdAPT-001 is currently being evaluated in the Phase 1 clinical trial called BETA PRIME with and without a checkpoint inhibitor (NCT04673942).
Disclosure of conflict of interest
None.
References
- 1.Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118:9–16. doi: 10.1038/bjc.2017.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Syn NL, Teng MWL, Mok TSK, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017;18:e731–e741. doi: 10.1016/S1470-2045(17)30607-1. [DOI] [PubMed] [Google Scholar]
- 3.Knaus HA, Kanakry CG, Luznik L, Gojo I. Immunomodulatory drugs: immune checkpoint agents in acute leukemia. Curr Drug Targets. 2017;18:315–331. doi: 10.2174/1389450116666150518095346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stecher C, Battin C, Leitner J, Zettl M, Grabmeier-Pfistershammer K, Höller C, Zlabinger GJ, Steinberger P. PD-1 blockade promotes emerging checkpoint inhibitors in enhancing T cell responses to allogeneic dendritic cells. Front Immunol. 2017;8:572. doi: 10.3389/fimmu.2017.00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maleki Vareki S, Garrigós C, Duran I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit Rev Oncol Hematol. 2017;116:116–124. doi: 10.1016/j.critrevonc.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, Lepage P, Boneca I, Chamaillard M, Kroemer G, Zitvogel L. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44:1255–69. doi: 10.1016/j.immuni.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Gide TN, Wilmott JS, Scolyer RA, Long GV. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin Cancer Res. 2018;24:1260–1270. doi: 10.1158/1078-0432.CCR-17-2267. [DOI] [PubMed] [Google Scholar]
- 8.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larson C, Oronsky B, Scicinski J, Fanger G, Stirn M, Oronsky A, Reid T. Going viral: a review of replication-selective oncolytic adenoviruses. Oncotarget. 2015;6:19976–89. doi: 10.18632/oncotarget.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chesney JA, Puzanov I, Ross MI, Collichio F, Milhem M, Chen L, Kim J, Garbe C, Hauschild A, Andtbacka R. Primary results from a randomized (1:1), open-label phase II study of talimogene laherparepvec (T) and ipilimumab (I) vs I alone in unresected stage IIIB-IV melanoma. J. Clin. Oncol. 2017;35:9509. [Google Scholar]
- 11.Schramm C, Protschka M, Köhler HH, Podlech J, Reddehase MJ, Schirmacher P, Galle PR, Lohse AW, Blessing M. Impairment of TGF-beta signaling in T cells increases susceptibility to experimental autoimmune hepatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2003;284:G525–G535. doi: 10.1152/ajpgi.00286.2002. [DOI] [PubMed] [Google Scholar]
- 12.Pardali K, Moustakas A. Actions of TGFβ as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775:21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A, Hawkins L, Kirn D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6:1134–1139. doi: 10.1038/80474. [DOI] [PubMed] [Google Scholar]
- 14.Silverstein G, Strohl WA. Restricted replication of adenovirus type 2 in mouse Balb/3T3 cells. Arch Virol. 1986;87:241–264. doi: 10.1007/BF01315303. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Hedjran F, Larson C, Perez GL, Reid T. A novel immunocompetent murine model for replicating oncolytic adenoviral therapy. Cancer Gene Ther. 2015;22:17–22. doi: 10.1038/cgt.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prince GA, Porter DD, Jenson AB, Horswoo RL, Chanock RM, Ginsberg HS. Pathogenesis of adenovirus type 5 pneumonia in cotton rats (sigmodon hispidus) J Virol. 1993;67:101–111. doi: 10.1128/jvi.67.1.101-111.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hjorth RN, Bonde GM, Pierzchala WA, Vernon SK, Wiener FP, Levner MH, Lubeck MD, Hung PP. A new hamster model for adenoviral vaccination. Arch Virol. 1988;100:279–283. doi: 10.1007/BF01487691. [DOI] [PubMed] [Google Scholar]
- 18.Ottolini MG, Porter DD, Hemming VG, Hensen SA, Sami IR, Prince GA. Semi-permissive replication and functional aspects of the immune response in a cotton rat model of human parainfluenza virus type 3 infection. J Gen Virol. 1996;77:1739–43. doi: 10.1099/0022-1317-77-8-1739. [DOI] [PubMed] [Google Scholar]
- 19.Thomas MA, Spencer JF, La Regina MC, Dhar D, Tollefson AE, Toth K, Wold W. Syrian hamster as a permissive immunocompetent animal model for the study of oncolytic adenovirus vectors. Cancer Res. 2006;66:1270–6. doi: 10.1158/0008-5472.CAN-05-3497. [DOI] [PubMed] [Google Scholar]
- 20.Nawaz S, Yuan Y. Computational pathology: exploring the spatial dimension of tumor ecology. Cancer Lett. 2016;380:296–303. doi: 10.1016/j.canlet.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 21.Poschke I, Mougiakakos D, Kiessling R. Camouflage and sabotage: tumor escape from the immune system. Cancer Immunol Immunother. 2011;60:1161–71. doi: 10.1007/s00262-011-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers (Basel) 2011;3:3856–3893. doi: 10.3390/cancers3043856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–9. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 24.Bartee E, Li Z. In vivo and in situ programming of tumor immunity by combining oncolytics and PD-1 immune checkpoint blockade. Exp Hematol Oncol. 2017;6:15. doi: 10.1186/s40164-017-0075-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pagès F, Galon J, Fridman WH. The essential role of the in situ immune reaction in human colorectal cancer. J Leukoc Biol. 2008;84:981–987. doi: 10.1189/jlb.1107773. [DOI] [PubMed] [Google Scholar]
- 26.Demaria S, Coleman CN, Formenti SC. Radiotherapy: changing the game in immunotherapy. Trends Cancer. 2016;2:286–294. doi: 10.1016/j.trecan.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cook KW, Durrant LG, Brentville VA. Current strategies to enhance anti-tumour immunity. Biomedicines. 2018;6:37. doi: 10.3390/biomedicines6020037. [DOI] [PMC free article] [PubMed] [Google Scholar]