

Abstract
RNA adopts 3D structures that confer varied functional roles in human biology and dysfunction in disease. Approaches to therapeutically target RNA structures with small molecules are being actively pursued, aided by key advances in the field including the development of computational tools that predict evolutionarily conserved RNA structures, as well as strategies that expand mode of action and facilitate interactions with cellular machinery. Existing RNA-targeted small molecules use a range of mechanisms including directing splicing — by acting as molecular glues with cellular proteins (such as branaplam and the FDA-approved risdiplam), inhibition of translation of undruggable proteins and deactivation of functional structures in noncoding RNAs. Here, we describe strategies to identify, validate and optimize small molecules that target the functional transcriptome, laying out a roadmap to advance these agents into the next decade.
Subject terms: RNA, Drug screening
The potential of therapeutically targeting RNA structures with small molecules is being increasingly recognized. Here, Disney and colleagues review strategies to identify, validate and optimize small-molecule RNA binders. Examples of existing RNA-targeted small molecules, as well as challenges and future directions in the field, are discussed.
Introduction
The first evidence that RNA could be targeted for therapeutic intervention was provided by the discovery of streptomycin in 1944 (ref.1) and the subsequent identification of the bacterial ribosome, a macromolecular RNA–protein complex, as the natural product’s cellular target2. Further insight was provided 20 years later when the first nucleic acid sequence and RNA structure — a tRNA carrying alanine (tRNAAla) — was reported3. The key to understanding the role of tRNA in translation was its 3D fold, required for aminoacylation and hence decoding of mRNA sequence into protein. The discovery of catalytic RNAs4,5 expanded the chemical functions of RNAs far beyond encoding proteins and decoding mRNAs.
The sequence of the human genome further reinforced RNA’s contribution to biology, with far fewer canonical open reading frames (ORFs) observed than expected6. Indeed, organismal complexity is not correlated with the number of ORFs but instead with the number and diversity of noncoding (nc)RNAs6 that function in epigenetic regulation7 and regulate gene expression8, particularly during development. Complementarily, genome-wide association studies (GWAS) have defined biological pathways that are dysregulated in disease, elucidating potential therapeutic targets, both RNA and protein. Such data can be leveraged to enable a bench-to-bedside paradigm in which small molecules bind to and deactivate structured RNAs: a patient’s genome could be sequenced and compared with GWAS to identify the malfunctioning RNA. The targeted therapy would bind a functional structure within the RNA to short-circuit disease pathways.
Promising therapeutic strategies to target RNA include antisense oligonucleotides (ASOs), CRISPR gene editing and small molecules that recognize RNA structures. ASOs and CRISPR editing have been invaluable to the field of chemical biology. However, translating these technologies to the clinic has been challenging owing to difficulties with delivery and significant adverse reactions9,10. Small molecules offer an important alternative with potential for oral bioavailability and blood–brain barrier penetrance, particularly with the wealth of knowledge from medicinal chemistry whereby physicochemical properties can be systematically optimized to improve pharmacokinetics and potency.
Several small molecules that bind RNA structures have been identified that exhibit various modes of action (MOAs), from simple binding to direct cleavage to recruitment of endogenous nucleases (induced proximity). These molecules have been demonstrated to modulate diverse biological processes, such as inhibiting bacterial and viral translation (ribocil and a riboswitch in Escherichia coli11, 2-aminobenzimidazole derivatives and the hepatitis C internal ribosome entry site (IRES)12); inhibiting mRNA translation (synucleozid and α-synuclein13); facilitating alternative pre-mRNA splicing (risdiplam14 and branaplam15 and survival of motor neuron 2 (SMN2)); and inhibiting microRNA (miRNA) biogenesis (a spermine–amidine conjugate and the miR-372 precursor16).
This Review describes foundational methods and strategies to: identify structured, functional RNAs; design and discover lead molecules that bind structured regions; and lead-optimize small molecules, including pharmacophore modelling, structure-based design and targeted degradation. Considerations and future directions for the development of small-molecule RNA binders are highlighted.
Defining RNA structures for small-molecule targeting
Determining RNA structure
An accurate model of RNA structure is key to the design or discovery of small molecules that modulate its function. Computational methods can model RNA structure from sequence, including free energy minimization and phylogenetic comparison. Free energy minimization uses experimentally derived thermodynamic parameters to predict RNA structures17, outputting the minimum free energy structure, assumed to be the functional structure, as well as suboptimal structures. The total free energy of an RNA structure is calculated by summing the free energy of each substructure in the system, such as base pairs, bulges and loops. Dynamic programming algorithms18,19, the basis of programs such as mFold20, RNAfold21 and RNAStructure22, incorporate these experimentally determined thermodynamic parameters, predicting accurately ~70% of the base pairs for RNAs <700 nucleotides long.
Experimental constraints can be integrated into the algorithms to improve the reliability of the prediction22. Unpaired nucleotides can be identified both in vitro and in vivo using chemical modification by dimethyl sulfate (DMS23) or by selective 2′-hydroxyl acylation analysed by primer extension (SHAPE)24–29. Chemical modification either pauses reverse transcriptase (RT), causing truncation of the cDNA (an ‘RT stop’), or induces a mutation, both of which can be read out by high-throughput sequencing. These sites of modification restrain secondary structure predictions in which the extent of modification is correlated with an energetic penalty for a nucleotide to be paired. Transcriptome-wide DMS and SHAPE probing techniques greatly expanded knowledge of the overall RNA structural profiles in cells and enabled investigation of RNA conformational changes under various biological conditions24–26. Although these experimental constraints refine the predicted models, they do not provide information about the 3D fold of the RNA, as provided by X-ray crystallography, NMR spectrometry and cryo-electron microscopy (cryo-EM).
Phylogenetic comparison also provides insight into RNA structure, where genetic differences that preserve secondary structure (covariations: for example, an AU base pair is replaced with GU or GC) can elucidate structure, and conservation suggests selective pressure to retain a functional structure30,31. Covariation-based structural prediction of many RNA structures is highly accurate, as determined by comparison with crystal structures32,33. However, owing to the large dataset required and the complexity of the analysis, automated covariation-based prediction methods remain challenging. Phylogenetic comparison has been coupled with other prediction methods, either in sequence or co-processed, including homology modelling, free energy minimization and chemical modification to improve accuracy34–36, which relies on the quality of alignment and sequence availability.
The folding of RNA is hierarchical, and RNAs can form tertiary structures. Information about RNA 3D structure is especially valuable in uncovering the functional mechanism of RNAs and identifying druggable targets. Although 3D prediction methods are still in their infancy compared with those for proteins, currently available programs, including FARFAR2 (ref.37) (RNA analogue of ROSETTA for protein prediction), MC-Fold/MC-Sym38 and iFoldRNA39, have shown promising results for predicting 3D structures from sequence. Scoring functions that estimate the accuracy of prediction results in which the native structure is unknown have also been developed, such as Rosetta37, RASP40 and ARES41. With the implementation of machine learning techniques, ARES outperforms the other two approaches. Root-mean-square deviation (r.m.s.d.) analysis of predicted structures to known crystal structures demonstrated the power of this approach41.
Biophysical methods such as NMR spectroscopy, X-ray crystallography and cryo-EM have also been extensively used to determine RNA structure42–44. NMR spectroscopy studies also provide information about structural dynamics. Although cryo-EM is typically used to study the structure of molecules of large molecular weight, considerable effort is being exerted to develop approaches that can access smaller RNA structures, for example, as recently reported for a small riboswitch (<40 kDa)45. Computationally assisted cryo-EM has also been introduced to determine the global conformation of RNA molecules, although it does not provide atomic-level resolution46.
Assessing the quality of RNA structural predictions
Many algorithms have been developed that predict RNA structure. However, without a known structure, it is hard to assess the reliability and accuracy of the prediction, which has been addressed by development of various statistical methods.
Partition function calculations have been incorporated into various structure prediction programs, including RNAfold21, Sfold47 and CONTRAfold48. A partition function contains all of the thermodynamic information for a system and quantifies the probability of the predicted structure or substructure therein. Statistics have also been applied to phylogenetic comparisons such as R-scape, which measures the statistical significance of evolutionary covariation, indicative of functionality49. Although R-scape improved the annotation of the consensus structure in 5S rRNA from the Rfam50 database, it did not find significant covariation in the long noncoding (lnc)RNAs HOX antisense intergenic RNA (HOTAIR), steroid receptor RNA activator (SRA) or X-inactive specific transcript (Xist)49, although plausible structures have been predicted by other computational methods51–53. The lack of functional structures in these lncRNAs did not stem from the lack of variance in the phylogenetic tree54, nor does it imply that these lncRNAs do not fold. Instead, they likely form dynamic structures stabilized in the context of other interacting partners, pointing to the importance of knowing the limitations of predictive methods and of experimental validation.
Collectively, these methods demonstrate that predicted structures must be viewed through the lens of statistical power and/or rigour and tempered with in-depth dissection of their biological function. That is, these predictions are hypotheses until further validated.
Defining functional RNA structures in the transcriptome
Functional RNA structures can be found throughout a transcript from the 5′ end to the 3′ end, from untranslated regions to ORFs (Fig. 1). Although data suggest that ORFs are less structured than other regions55–57, highly structured coding sequences have been discovered58,59. Functional structures can be identified computationally (below) or experimentally by using ASOs that sterically block a functional structure60 or by mutational analysis61.
Fig. 1. Human RNAs regulate key biological processes, and their functions are driven by their structures.

a | Examples of structured regions of RNA with important biological functions include IRE (iron-responsive element; translational regulation), splicing modulators (alternative pre-mRNA splicing including those that interact with U1 small nuclear RNA (snRNA)), RNA repeat expansions (aberrant gain-of-function; microsatellite disorders) and Drosha and Dicer processing sites in microRNA (miRNA) precursors. PDB codes: IRE (PDB: 1NBR), intron splicing junctions (PDB: 6VA1, 6HMO), RNA repeat (PDB: 1ZEV). The model of the miRNA structure can be found in ref.108. b | Comparison of RNA and protein as therapeutic targets. Approximately 75% of the human genome is transcribed into RNA, while 1.5% is translated into protein. Common types of drug target and their modes of action are also listed.
Although many RNA structural prediction methods can provide accurate models of RNA secondary structure, they do not predict functionality. Functionality can be predicted by identifying regions of structure that are unusually thermodynamically stable compared with random sequences. The hypothesis is that these stable structures are evolutionarily retained because of selective pressure. ScanFold, a scanning window approach, uses these principles to identify potentially functional structures62, accurately predicting known functional viral structures as well as predicting potential new functional structures in viral and human transcriptomes63,64.
Structural conservation and genetics can also indicate function. For the former, structural motifs that are evolutionarily conserved across species likely have a biological function30,31; for the latter, genetic mutations can cause gain or loss of function. Perhaps the best example of evolutionary conservation of structure and function is rRNA. Indeed, highly conserved structures can be found in bacteria (riboswitches), viruses (IRESs) and humans (IRESs, splicing regulatory elements).
The hunt for targetable, functional RNA structures, particularly in the human transcriptome, has only just begun. Thus far, RNA structures that have been effectively targeted with small molecules (binding produces a downstream biological response), participate in biomolecular interactions with proteins including the ribosome, other RNAs and DNA. Identifying a functional RNA structure only addresses half of the RNA-targeting problem. The other half is discovering or designing chemical matter that selectively binds to the functional structure.
Factors that affect the selectivity of small molecules targeting RNA
Ideally, a small molecule would be completely selective for its RNA target, but in practicality that is likely not required or even achievable. Historically, selective recognition of RNA by small molecules was thought intractable, owing to its perceived lack of structural diversity with only four building blocks, its anionic backbone and the lack of success in high-throughput screening campaigns. Selective recognition is possible as our fundamental understanding of the RNA structures that are targetable and the chemotypes that bind RNA has evolved, alongside advances in RNA structure modelling that provide insight into the functionality of structures.
Binding selectivity and functional selectivity are distinct. Several factors have been identified that drive the cellular (functional) selectivity of ligands that target RNA, including the uniqueness of the structure in the transcriptome, expression of the target compared with off-targets, the relative affinity of the small molecule for on- and off-targets, accessibility of the target site and the functionality of the binding site, where binding to non-functional sites is biologically silent. Regarding target expression, if two or more RNAs have the same binding site, the more highly expressed target will be more occupied by the compound. In the same vein, if two or more RNAs have different structures ligandable by the same compound, the most occupied target will be a composite of relative affinity and expression level. In cases where selectivity is not sufficient, compounds can be designed that target multiple sites within an RNA target simultaneously, thus overcoming the limitations of structural degeneracy (a structure is not unique in the transcriptome).
A few studies have completed transcriptome- and proteome-wide studies that provide insight into the selectivity of RNA-targeting small molecules. These studies have shown that small molecules can exert selective effects across the transcriptome and proteome. Notably, the observed changes and selectivities are similar to those observed for oligonucleotides65. An analogue of the FDA-approved drug risdiplam, which binds to an RNA–protein manifold not solely an RNA, is selective, altering the levels of 12 of 11,174 transcripts and altering the alternative splicing of a subset of mRNAs, including the desired target66. As observed for small molecules that target proteins, the off-targets for RNA-targeting small molecules could be other RNAs, DNA or proteins. Thus far, it appears that the scaffolds that bind to RNA are different from those that bind to proteins (Box 1), as also indicated by the lack of success in identifying selective RNA binders from small-molecule libraries designed for proteins.
Box 1 Commonalities amongst small molecules that bind RNA.
Ideally, small molecules that bind RNA targets would be drug-like and orally bioavailable, as defined by their physicochemical properties. Indeed, analysis of the physicochemical properties of drugs that target proteins have provided guidelines for drug development, such as the Lipinski ‘rule of five’275. RNA, however, is very different from proteins in terms of its chemical composition, its highly electronegative surface potential and its limited buried surface area. It is therefore reasonable to infer that RNA-binding small molecules will have unique properties that do not necessarily fall into traditional rule of five guidelines. Notably, new modalities such as chimeric compounds that target proteins for degradation have many drug discovery efforts lying ‘outside of the rule of five’. Various studies have identified privileged scaffolds and chemotypes that confer affinity for RNA, for example, indole, 2-phenylindole, 2-phenyl benzimidazole, 2-phenylimidazole, methylpyrimidine-2,4-diamine and others67,78,281,282. Further, comparison of the physicochemical properties of RNA-binding molecules with those of FDA-approved drugs available in DrugBank revealed distinct differences109,283,284. In particular, RNA-binding compounds on average have lower octanol–water partition coefficients (LogPs) than protein-binding compounds (ranges from two separate analyses: 0.16 ± 5 versus 2.0 ± 3.5 (ref.109) and 1.02 versus 1.78 (ref.283), respectively), greater topological polar surface areas (156 ± 118 versus 92 ± 144 Å2)109, more hydrogen bond donors (5.2 ± 5 versus 2.0 ± 4) and acceptors (8.6 ± 6 versus 5.0 ± 6)109, and more heteroatom-containing aromatic rings (2.16 versus 1.25)283. An analysis by the Hargrove laboratory suggested that the shape of RNA binders is generally rod-like283. Given the infancy of RNA targeting, these comparisons will likely change as new molecular interactions and activities are catalogued.
A previous analysis investigated the interplay between small-molecule affinity, target complexity and quantitative estimate of drug-likeness (QED)186. QED, a metric of compound quality, evaluates how closely each physicochemical property of a small molecule, measured on different scales, reflects the ideal value and combines them into a single score285. The analysis suggested that the complexity of the RNA target scaled with the affinity for the targets and that drug-like chemical matter emerged when the target RNA structure became more complex186. As there is still much to be understood about the interactions at play that confer high-affinity binding, selectivity and bioactivity, the field will need to continue to revise its view on the properties, including drug-likeness, of RNA-targeted small molecules that are important and further determine whether these properties are target- or target class-dependent. However, a few guideposts have been established. For example, traditional drug-like chemical matter is likely useful for RNAs that adopt complicated binding pockets, that is, those that appear riboswitch-like or protein-like. Notably, complexity should not be confused with uniqueness. Kinase inhibitors bind to an ATP-binding pocket formed by a complicated molecular interaction network. However, they are not unique folds, making the development of selective kinase inhibitors challenging286. Thus, although an RNA may form a complex structure, it does not necessarily indicate a priori that a selective small molecule can be designed or discovered. In other cases, where the RNA is locally structured (a common occurrence for mRNAs, for example), the lack of buried surface area (in comparison with a complex structure) to drive selective interactions could be overcome by binding to multiple structures within an RNA target simultaneously with a single molecule. Although such compounds may be outside of the rule of five, they can be optimized into orally bioavailable medicines.
Strategies to identify small-molecule RNA binders
Target-centric approaches
Various approaches can be used to find small molecules that bind RNA structures in vitro, including fluorescence-based assays, fluorescence resonance energy transfer (FRET)-based approaches67–70 and dynamic combinatorial screening71. The methods described below are target centric, that is, the small molecule’s only choice for binding is a single or a few targets. As with any primary screening assay, secondary analyses are required to identify selective binders. For all fluorescence-based assays described below, care must be taken, as many compounds can interact with the dyes themselves or have emission properties that overlap with the fluorophores.
Affinity mass spectrometry
Affinity selection mass spectrometry (AS-MS) is a label-free method that allows the direct identification of target–ligand complexes by mass spectrometry after separation from unbound ligands by size-exclusion chromatography72,73. Used widely for proteins74, it has only been recently adopted for RNA targets75,76. A variant of AS-MS, named automated ligand identification system (ALIS), uses indirect detection of a target–ligand interaction by dissociating the formed complex before liquid chromatography–mass spectrometry (LC-MS) analysis to identify the bound ligand74–76 (Fig. 2a). In one example of the use of ALIS, synthetic ligands that bound the flavin mononucleotide (FMN) riboswitch were identified75. One challenge associated with AS-MS is the requirement of long small-molecule residence times, such that the complex is still intact after size-exclusion chromatography.
Fig. 2. Methods to identify or design small-molecule RNA binders.

a–c | Methods to identify small molecules that bind RNA. a | Automated ligand identification system (ALIS) is a liquid chromatography–mass spectrometry (LC-MS) method. An RNA target is incubated with a library of small molecules. Unbound ligands are removed by size-exclusion chromatography and then bound ligands are identified by LC-MS. b | Fluorescence-based assays rely on a change in fluorescence upon small-molecule binding to the RNA target. This could be achieved by: displacing a non-selective RNA-binding dye (top); changing the microenvironment of a fluorescent nucleotide analogue (middle); or disrupting donor–acceptor pairs in a fluorescence resonance energy transfer (FRET)-based assay (bottom). c | Microarray-based screening in which a panel of small molecules is pinned to an array surface and incubated with labelled RNA targets, followed by washing and imaging to identify target-binding compounds. d | Compounds functionalized with a cross-linking module (such as diazirine or chlorambucil) and a pull-down tag (such as alkyne or biotin) can be screened against labelled RNA targets by using Chem-CLIP (chemical cross-linking and isolation by pull-down) to identify RNA binders and to map the binding sites. e | Identification of RNA-binding small molecules from a DNA-encoded library (DEL). The library is synthesized on beads, and each building block added during the synthesis is encoded with a DNA tag. The DEL is screened simultaneously for binding to the target of interest and a related RNA to which binding is undesired. The two RNAs are labelled with different fluorophores, and selective binders from the DEL can be identified and isolated by flow cytometry. f,g | Methods to design small molecules that bind RNA. f | Inforna is a lead identification strategy in which the structures present in a cellular RNA are compared with a database of experimentally determined RNA–small molecule interactions. Overlap affords lead targets and lead small molecules. g | Structure-based design of small molecules relies on a model of the structure of the RNA or of the RNA–ligand complex. Both can be used in docking studies while the latter can be used to guide modifications that improve interactions between the RNA and the small molecule. 2-AP, 2-aminopurine; RFU, relative fluorescence units.
Fluorescence-based assays
Another high-throughput assay relies on displacement of a fluorescent dye or compound by a small molecule of interest. Although originally developed to study the binding of DNA structures77, it has also been applied to RNA targets by displacement of the fluorescent dye TO-PRO-1 (refs.78,79) (Fig. 2b), as well as others79–82. An extension of this dye displacement method is an assay with turn-on fluorescence that uses a known fluorescent or fluorescently labelled binding small molecule and an RNA of interest that is labelled on the 5′ or 3′ end with a quencher70.
Another way to assess small-molecule binding can be carried out by replacing an adenine residue with the fluorescent mimic 2-aminopurine (2-AP)83. The fluorescence of 2-AP depends on its microenvironment, which changes upon small-molecule binding (Fig. 2b). This 2-AP assay was originally developed to study the binding of aminoglycosides to bacterial rRNA and the effect of binding on A-site dynamics84, but has been extended to other targets70,85,86. Notably, the position of the 2-AP substitution within the RNA should be carefully chosen to ensure a strong signal. For small RNAs, this can be accomplished by simply substituting each adenine residue. For longer RNAs, such as riboswitches, SHAPE can be used to elucidate where large conformational changes occur upon ligand binding, and hence the optimal position (or postions) for 2-AP substitution85,86.
Many bioactive small molecules have been identified that bind RNAs participating in bimolecular interactions with proteins. To measure inhibition of formation of an RNA–protein interaction or disruption of a pre-formed complex by a small molecule, fluorescence and FRET assays have been developed, particularly for HIV-1 Tat–transactivation response (TAR)87–89 and for RNA repeat expansions–RNA-binding proteins69,90. Labels on the RNA and protein are FRET pairs, and disruption or inhibition of the complex reduces the observed FRET signal (Fig. 2b). Small molecules that bind either the protein or the RNA can reduce FRET, and thus additional investigation is required to confirm that the small molecule binds the RNA as intended.
Microarray-based screening
Small-molecule microarrays (SMMs), created by delivery of minute amounts of compounds to glass slides in a spatial array, were initially used to interrogate protein binding91–94 and later extended to study the binding of aminoglycosides to the rRNA A-site95 and how binding is affected by aminoglycoside modification by resistance enzymes96. SMMs have now been used to screen a wide variety of compounds and RNA targets97–103 (Fig. 2c). One advantage of SMMs is that only a small amount of the small molecule is needed to complete a screen and many thousands of interactions can be profiled at once. Compounds are typically covalently attached to the array. Notably, binding to a surface can be quite different from binding in solution. Small molecules can also be non-covalently attached to agarose-coated microarray surfaces by adsorption104. Although this method can be broadly applied to many compounds104, not all compounds adhere to surfaces.
Fragments
Fragment-based ligand discovery uses libraries of low-molecular-weight compounds to efficiently explore chemical space that might bind the target of interest (Fig. 2d). Although fragments can be screened for binding, such as by NMR spectroscopy (as demonstrated to identify a fragment that binds severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)105), these interactions are often difficult to detect as they are low affinity and have short residence times. To overcome these limitations, small-molecule fragments have been functionalized with photoaffinity groups (fully functionalized fragments; FFFs) that enable capture and identification of bound targets, first applied to proteins106,107 and later to RNA105,108. Low-molecular-weight fragments are ideal for a modular assembly approach in which two fragments are tethered together to bind two structural elements in an RNA target simultaneously, as favourable physicochemical properties can be maintained (discussed below).
DNA-encoded compound libraries
DNA-encoded compound library (DEL) technology is a powerful method to explore chemical space that binds a target biomolecule either in solution or on the solid phase. A DEL is synthesized on beads by split and pool in an iterative process in which one of many building blocks is conjugated and its identity is encoded in a short DNA tag that is ligated to the bead. Compound-functionalized beads are screened for binding typically to a fluorescently labelled target, often in the presence of an off-target that is differentially labelled (Fig. 2e). For RNA targets, a counter-screen can be completed by using an RNA in which the desired binding site has been mutated, akin to mutations for binding analyses (Fig. 2e). For example, a bulged nucleotide could be converted into a base pair or a different bulge, the closing base pairs could be altered and so on. Beads that bind the desired target but not the off-target decoy can be analysed and sorted by flow cytometry. Deep sequencing of the beads identifies the binding compound. Because of the potential of nonspecific binding of the encoding DNA tag, nucleic acids and their binding proteins have been avoided. Thus far, the DEL technology has been applied only twice to nucleic acid targets: RNA109 and highly structured DNA G-quartets110.
Designing small-molecule RNA binders
Rather than screening for chemical matter, RNA binders have been designed using selection-based methods that define the preferred targets of RNA-binding small molecules and structure-based design.
Defining binding landscapes
A selection-based platform, 2D combinatorial screening (2DCS), defines the binding landscape of an RNA-binding small molecule. Small molecules displayed on a microarray select their preferred RNA partners from an RNA library displaying a randomized region in a discrete secondary structure pattern111. The selection experiment is completed under stringent conditions in the presence of a large excess of competitor oligonucleotides that mimic regions common to all members of the library, restricting binding interactions to the randomized region. Fully paired DNA and RNA oligonucleotide competitors or tRNAs are also often used to increase the stringency of the selection. Selected RNAs are analysed by RNA sequencing (RNA-seq)112,113, followed by rigorous statistical analysis of the enrichment of an RNA by the small molecule114. Statistical significance scales with affinity such that the more significant the enrichment of the RNA, the more tightly it binds to the small molecule. This analysis affords a binding landscape, or molecular fingerprint, for each small molecule, where a selective small molecule has few RNAs with statistically significant enrichment and a promiscuous one has many. These binding landscapes inform the ideal target for a small molecule and potential off-targets. Cellular RNA targets can be computationally compared with these molecular fingerprints to inform small-molecule design, as in the lead identification strategy Inforna65 (Fig. 2f). Inforna outputs the targetable sites present in a cellular RNA and the rank order of potential small-molecule binders. This approach has been implemented in both a target-centric and a target-agnostic fashion65.
Structure-based design and docking
First employed for protein targets, structure-based design and docking have enabled the discovery and the optimization of lead compounds for RNA targets115–121 (Fig. 2g). Using well-defined RNA structures, typically elucidated by NMR spectroscopy, small molecules can be designed to fit within binding pockets. Further, NMR studies also enable the prediction of dynamic ensembles of RNA conformations computationally, including short-lived, non-functional species120–125. Small-molecule libraries can then be docked into these ensembles to screen small molecules for selective RNA binding in silico121,126. The significant limitations of these approaches are the quality of the RNA structure, as highly accurate structures are difficult to generate by NMR spectroscopy, as well as the docking programs themselves. Interestingly, an NMR method was recently developed to study conformational biases in HIV-1 TAR and Rev responsive element (RRE) RNAs120. A series of mutations were made to each RNA, and the effect on conformational equilibria was measured by NMR spectroscopy. These spectral studies can estimate the percentage of the RNA folded into a non-functional fold, confirmed by studying the cellular activity of the RNA mutants120.
Phenotypic screening
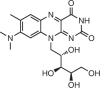
Phenotypic screening is a strategy to identify compounds that affect pathways associated with a specific phenotype and therefore require no knowledge of the MOA or target. These assays design a screen around a biological process, for example, alternative pre-mRNA splicing, inhibition of translation, derepression of protein targets and bacterial growth. Assays completed in mammalian cells typically use generation of either luciferase or a fluorescent protein as a readout. Two small molecules that modulate splicing were discovered from phenotypic screens, the FDA-approved risdiplam66 and branaplam15 (currently in phase II clinical trials) for the treatment of spinal muscular atrophy (SMA)15.
Although phenotypic screens have been executed successfully, challenges remain, largely because the approach is target agnostic. Despite the fact that target validation and mechanistic studies can prove difficult, phenotypic screens have enabled lead optimization, particularly by using structure-based drug design. The reader is referred to ref.127 for a review on the validation of phenotypic screens of RNA targets.
Target validation and selectivity of RNA-targeting small molecules
Direct target engagement is key to defining the MOA of a compound. Below, we describe various target validation methods based on covalent bond formation between the small molecule and the RNA target, cleavage of the RNA target and resistance profiling. Except for resistance profiling, these methods can study target engagement in vitro, in cells or in vivo.
Fortuitously, some methods not only assess engagement of the desired target but also measure selectivity. Here, we note again the difference between binding selectivity and functional selectivity. Binding selectivity, often measured in vitro by introducing point mutations into a model of the binding site of the small molecule, measures relative affinity and hence the extent of target occupancy. Engagement of most structures in a target RNA is biologically silent with no functional consequence. In contrast, functional selectivity measures whether target engagement has a biological effect — the function of the RNA is modulated as assessed by changes in the downstream pathways of the target and by its associated phenotype.
Resistance profiling
Resistance profiling exerts selective pressure to induce mutations in bacterial or viral genomes to confer resistance (Fig. 3a). The genomes of resistant strains are sequenced to identify in which genes mutations have occurred, thus identifying the target of the compound. Such an approach was used to validate the riboswitch target of ribocil (discussed below), roseoflavin and pyrithiamine11,128,129 (Table 1). By comparison with covalent- and cleavage-based target validation methods, mutational resistance profiling does not require synthesis of chemical probes and overall has fewer experimental steps. However, it requires the small molecules to exert sufficient evolutionary pressure to induce mutational resistance, frequently used in cancer biology130.
Fig. 3. Methods of target validation for small molecules that target RNA.

a | Resistance profiling is applicable when the small molecule exerts enough selective pressure to induce mutations that confer resistance. These mutations, identified by sequencing, reveal the targets of the small molecule. b | The target validation method Chem-CLIP (chemical cross-linking and isolation by pull-down) generates a covalent bond between a small-molecule probe and its targets, which are isolated and purified by bead pull-down. Bona fide targets are those enriched in the pulled-down fractions, as compared with the starting cell lysate. Upon co-treating increasing concentrations of the lead compound with Chem-CLIP probe, a dose-dependent restoration of the RNA target in the pulled-down fractions would indicate target engagement of the lead compound. c | ASO-Bind-Map is based on previous studies that show that structured regions of RNA are protected from antisense oligonucleotide (ASO) hybridization and hence ribonuclease (RNase) H degradation135,136. Thus, small molecules that bind to and stabilize the structures of RNA targets can elicit a protective effect against ASO-mediated degradation. d | An RNA degrader can cleave its bound RNA target either directly (bleomycin) or by recruiting endogenous RNases (RIBOTAC). Upon co-treating increasing concentrations of the lead compound with degrader probe, a dose-dependent ablation of degradation would indicate target engagement of the lead compound.
Table 1.
Examples of small molecules that bind to RNAs implicated in infectious disease
| Compound/institution | Associated disease | RNA target | Mode of action | Stage of development | Activity | Refs. |
|---|---|---|---|---|---|---|
|
Compound 1/National Cancer Institute |
Bacterial Infection | Bacterial PreQ1 riboswitch | Modulates riboswitch activity through transcriptional termination | Early discovery | Kd = 490 nM (Bs aptamer domain); Kd = 99 nM (Tt aptamer domain) | 100 |
|
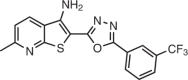
Ribocil/Merck |
Bacterial infection | Bacterial FMN riboswitch | Inhibits translation by binding to the FMN riboswitch | Preclinical | Kd = 16 nM; EC50 (FMN reporter) = 0.2 μM | 11 |
|
Roseoflavin/Yale University |
Bacterial infection | Bacterial FMN riboswitch | Inhibits translation by binding to the FMN riboswitch | Early discovery | Kd = 100 nM | 128 |
|
Pyrithiamine/Yale University |
Bacterial infection | Bacterial FMN riboswitch | Inhibits translation by binding to the FMN riboswitch | Early discovery | Kd = 6 μM (Bacillus subtilis) | 129 |
|
5FDQD/Yale University |
Bacterial infection | Bacterial FMN riboswitch | Inhibits translation by binding to the FMN riboswitch | Early discovery | Kd = 7.5 nM; MIC = 0.5–1 µg ml−1: (antibacterial activity in Clostridium difficile) | 253,254 |
|
Amiloride/Duke University |
Viral infection (HIV-1) | TAR apical loop | Inhibition of the Tat–TAR interaction | Early discovery | CD50 = 4.4 µM (Tat/TAR-displacement FRET) | 160 |
|
Compound 13/University of California, San Diego |
Viral infection (HCV) | HCV IRES IIA domain | Inhibits translation by inducing conformational destabilization of the binding of IRES to ribosome | Early discovery | Kd = 720 nM (29-mer IIA); EC50 = 5.4 μM (HCV-replicon assay) | 158,180 |
|
DMA-155/Duke University, Case Western Reserve University |
Viral infection (SARS-CoV-2) | 5′UTR of SARS-CoV-2 RNA | Inhibits translation by perturbing the interaction between viral RNA and host protein | Early discovery | IC50 = 10 μM (virus titre assay) | 181 |
|
DMA-135/Duke University, Case Western Reserve University |
Viral infection (EV71) | EV71 SLII IRES domain | Inhibits EV71 replication by stabilizing the translation repressive complex SM–SLII–AUF1 | Early discovery | Kd = 500 nM (EV71 SLII RNA); CD50 = 12 µM (EV71 SLII RNA by displacement) | 276 |
|
Compound 19/Yale University |
Fungal infection | Mitochondrial pre-rRNA group II intron | Inhibition of group II intron splicing leading to mitochondria malfunction and fungal growth inhibition | Early discovery |
In vitro splicing inhibition constants (Ki) = 0.36 μM MIC 2–4 μg ml−1 |
154 |
|
Compound 15/National Cancer Institute |
Viral infection (HIV-1) | TAR hairpin | Inhibition of the Tat–TAR interaction | Early discovery | Kd = 49 µM (to 25-2AP TAR); IC50 = 40 µM (Tat/TAR-displacement FRET) | 157 |






CD50, median curative dose; EC50, half maximal effective concentration; EV71, enterovirus 71; FMN, flavin mononucleotide; FRET, fluorescence resonance energy transfer; HCV, hepatitis C virus; IC50, half maximal inhibitory concentration; IRES, internal ribosome entry site; Kd, dissociation constant; MIC, minimum inhibitory concentration; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; TAR, transactivation response; UTR, untranslated region.
Covalent bond formation to measure direct target engagement
Cellular target validation methods for RNA have been recently developed based on covalent bond formation with65,131–133, or cleavage of, the target65. One covalent method, chemical cross-linking and isolation by pull-down (Chem-CLIP; Fig. 3b), developed in 2013 (ref.134), relies on functional modification of small molecules, attaching a cross-linking module (for example, diazirine or chlorambucil) and a purification handle (for example, biotin or an alkyne)65. The RNA-binding module drives target engagement, bringing the cross-linking module into proximity to the RNA such that they react either directly (electrophilic in the case of chlorambucil) or upon irradiation (diazirine). Small molecule–target complexes are captured and purified with beads (streptavidin- or azide-functionalized). Quantitative PCR with reverse transcription (RT–qPCR) or RNA-seq and subsequent statistical analysis are used to analyse the enrichment of RNA targets in the pulled-down fraction, as compared with the starting lysate. Although the exact protocol may vary depending upon the chemistry of cross-linking133 and purification modules, the fundamental idea of Chem-CLIP remains unchanged: transformation of dynamic reversible binding to covalent bonds by cross-linking and amplifying the signal by pull-down enrichment. Chem-CLIP experiments are completed side by side with a probe that lacks the RNA-binding module, controlling for nonspecific reaction of the cross-linking module. Indeed, this approach has validated the RNA targets of small molecules65. This method, however, requires modification of the lead compound, which can be hampered when the synthesis is challenging or if molecular recognition has not been sufficiently defined to inform a site within the small molecule not required for molecular recognition.
Chem-CLIP can also be used to study target binding and functional selectivity, when the pulled-down fractions are analysed by RNA-seq. The enrichment of a transcript indicates the extent of its occupancy by the small molecule, affording the binding selectivity of the small molecule (see ‘Quantifying selectivity’ below). Occupancy of a target RNA is not sufficient for a biological response; the small molecule must bind a functional site. Functional selectivity can be defined by comparing target occupancy with the effect on target expression by RNA-seq analysis upon treatment with the lead compound. Many RNA targets will be occupied but their expression unaffected. Pathway analysis of the RNA-seq data also provides supporting or denying evidence of on-target MOA. Identifying off-targets is key for lead optimization, and fortuitously, Chem-CLIP can identify the exact binding site within a cellular RNA, a method named Chem-CLIP-Map (ref.65).
A variant of Chem-CLIP, competitive-Chem-CLIP (C-Chem-CLIP), defines the targets of the lead compound. Briefly, cells are co-treated with a constant concentration of the Chem-CLIP probe and increasing concentrations of the lead compound. If the two molecules compete for the same binding site, a dose-dependent decrease in target enrichment should be observed as a function of lead compound concentration. C-Chem-CLIP can be used to screen other molecules for binding to the same RNA target to generate a structure–activity relationship (SAR).
Target cleavage to assess occupancy
Complementary cellular target validation strategies have been developed that rely on the competitive cleavage of RNA targets, including ASO-Bind-Map (Fig. 3c) and competition with RNA degraders65 (Fig. 3d).
In ASO-Bind-Map, an ASO gapmer complementary to the target RNA sequence competes with a structure-binding small molecule13. In the absence of small molecule, the ASO induces cleavage by ribonuclease (RNase) H, resulting in reduced abundance of the target RNA. If a small molecule binds to the same region, it impedes hybridization of the ASO and thus diminishes target depletion. This strategy was inspired by studies that used ASOs to probe RNA folding, in which regions of RNA that fold quickly into stable structures are less accessible to ASO hybridization135,136. In contrast to Chem-CLIP, ASO-Bind-Map does not require modification of the lead compound, but does require knowledge of the binding site within the cellular RNA and that the small molecule stabilizes the structure of the RNA sufficiently to impede ASO hybridization. ASO-Bind-Map cannot be used to study selectivity transcriptome-wide, as it would require an ASO to every structure in every target. If an off-target is suspected, ASO-Bind-Map could prove or disprove target engagement.
Methods have also been developed in which the small molecule directly cleaves the RNA target or recruits a nuclease to do so, identifying both on- and off-targets by their depletion upon analysis by RNA-seq, akin to ASOs. To directly cleave RNA targets, a method dubbed small molecule nucleic acid profiling by cleavage applied to RNA (RiboSNAP), uses a lead molecule conjugated to the natural product bleomycin. Bleomycin induces strand scission of both DNA and RNA through a metal-ion and oxidative process137–142. Bleomycin A5 is typically conjugated through its terminal amine, which drives affinity for DNA; alkylation of the amine reduces DNA damage by the conjugate and directs its activity for the RNA target143. As with Chem-CLIP, side-by-side experiments are completed with a probe that lacks the RNA-binding module to control for non-selective reaction of bleomycin. The cellular targets of the small molecule–bleomycin conjugate and the control probe are identified by their depletion in RNA-seq data where bona fide targets are those cleaved only by the conjugate. Likewise, the lead compound can be studied directly in a competition experiment. As with Chem-CLIP, RiboSNAP is a robust method that can measure cellular binding selectivity by analysing the extent to which each transcript is cleaved. Functional selectivity is measured by coupling these cleavage data with the effect of the lead compound on expression levels, where indirect target expression levels are altered by the lead compound, but not depleted by the small molecule–bleomycin conjugate. RiboSNAP can also elucidate the small-molecule binding site within an RNA target.
As an alternative to direct cleavage, RNA targets can be cleaved by small-molecule chimeras that recruit a nuclease, or ribonuclease-targeting chimeras (RIBOTACs)144. A RIBOTAC comprises an RNA-binding module tethered to an RNase L-recruiting small molecule. RNase L, which functions in the antiviral immune response, is normally expressed in minute amounts as an inactive monomer. Its activity is regulated transcriptionally, post-transcriptionally and by an RNase L inhibitor, to tightly control RNase L activity145–147. Upon viral infection, 2′–5′-oligoadenylate (2′-5′A) is synthesized, which dimerizes and activates RNase L, inducing degradation of viral RNA148,149. Previous studies have shown that RIBOTACs recruit and activate RNase L locally and do not elicit an immune response143,150. As with Chem-CLIP and direct cleavage methods, the RNA-binding molecule drives engagement of the target while a nuclease-recruiting module activates RNase L to induce cleavage; bound targets are depleted in RNA-seq analysis. To study targets engaged by the lead compound, a competition experiment can be used in which the levels of bona fide targets are restored as a function of lead compound concentration. For both direct and indirect cleavage methods, pathway analysis can be performed to evaluate on-target and potential off-target effects. An important advantage of cleavage strategies for target validation is that they do not require purification and isolation of cross-linked material; instead targets can be defined by simple RNA-seq analysis.
Quantifying selectivity
Quantifying the effect of a compound across the transcriptome, coupled with defining direct targets and pathway analysis (at the transcriptome and/or proteome level) provides insight into small-molecule selectivity. An important metric to quantify small-molecule selectivity such that small molecules can be compared is the Gini coefficient151,152. The targets of a small molecule are rank ordered by their percentage inhibition, and the cumulative fraction and the total cumulative effect for each are calculated. The former is a function of the number of targets studied and the latter is a weighted inhibition. The two values are plotted against each other, and the Gini coefficient is equal to 1 − 2B where B is the area under the resulting curve153. For a review of Gini coefficients as applied to RNA, the reader is referred to ref.152.
Lead optimization of RNA-targeting small molecules
One advantage of small-molecule therapeutics is that medicinal chemistry efforts can be applied to optimize the initial hit compound, improving both its potency and its physicochemical properties. Below, three broad strategies to lead-optimize RNA-targeting small molecules — including traditional medicinal chemistry as well as structure-guided and modular assembly approaches, which are often integrated synergistically — are discussed.
Traditional medicinal chemistry approaches
Traditional medicinal chemistry optimization typically starts with analogue synthesis or purchase to establish the SAR around the hit compound (Fig. 4a). The bioactivity of an RNA-targeting ligand can be affected by multiple factors, such as binding affinity, cellular permeability and the number of off-targets. SAR is typically defined by a combination of biophysical and cellular assays to evaluate hit analogues. After acquiring sufficient SAR data, pharmacophore modelling and subsequent chemical similarity searching can then diversify and optimize the starting scaffold. Among all scaffolds, those with desirable or optimizable physicochemical properties are prioritized. Traditional medicinal chemistry can be applied to essentially any small-molecule candidate, tempered by the resources required to synthesize or purchase a large number of analogues. Below, we describe how medicinal chemistry approaches were used to optimize risdiplam, a treatment for SMA, and a pyrimido-indole that directs MAPT alternative splicing. The reader is referred to refs.154–159 for other notable examples of lead optimization of RNA-targeted small molecules using medicinal chemistry.
Fig. 4. Strategies for lead optimization of RNA-targeted small molecules.

a | Traditional medicinal chemistry optimization begins with hit expansion and analogue synthesis to generate structure–activity relationships (SARs), which can be used to optimize lead compounds as well as to discover new scaffolds by pharmacophore modelling. b | Structure-guided lead optimization relies on structure modelling of the RNA target to perform virtual screening and to inform compound design based on ligand–RNA interactions. c | Sequence-based lead optimization explores the differences between structures of on-target and off-targets and modifies the lead compound based on structural features unique to the on-target, that is, modular assembly or dimerization. PK, pharmacokinetic; pri, primary.
Structure-guided approaches
Structure-guided approaches for lead optimization rely on sophisticated models of a ligand–RNA complex to define key interactions and identify how positions within the compound’s structure can be modified to improve interactions or eliminate those that are unfavourable (Fig. 4b). Such models are typically experimentally generated by NMR spectroscopy or X-ray crystallography. The dynamic nature of RNA must be considered, as no single static structure can truly represent all possible conformations. To address this limitation, experimental models are often coupled with molecular dynamics simulations to generate an ensemble of structures, where the algorithm simulates RNA conformations on the basis of experimentally determined parameters.
Molecular dynamics-based virtual screening has successfully identified ligands that bind various RNAs, typically using models from NMR spectroscopy or X-ray crystallography. As these structures do not capture the dynamic nature of RNA, and molecular dynamics simulations do not accurately recapitulate RNA electrostatics, docking of small-molecule ligands is not as predictive as it could be. To overcome these challenges, a docking method that uses the dynamic ensemble of RNA conformations from combined NMR and molecular dynamics approaches has been developed, an example of which is provided below126,160. The advantage of structure-based approaches is that they minimize the synthesis efforts by rationally designing analogues, but the limitation is that not all target structures are readily attainable.
Modular assembly approaches
One of the many factors that affect the activity of small molecules that target RNA is the uniqueness of the structural motif throughout the transcriptome. A strategy to optimize small molecules with activity against degenerate functional structures is to exploit adjacent structural elements, that is, a modular assembly approach in which a single small molecule binds to structural elements that are uniquely juxtaposed (Fig. 4c). Indeed, polyvalency — used in various biological processes to enhance avidity and specificity, from cell–cell interactions to gene expression and viral infections — has also been exploited in drug discovery efforts to agonize or antagonize receptors161,162. Such an approach has been taken for RNA targets by modularly assembling two or more RNA-binding modules65,163–166, some with in vivo activity167. One example is described below in which a modular assembly approach broke the degeneracy of two motifs, affording a specific inhibitor of miR-515 biogenesis. Modular assembly has been employed for RNA repeat expansions (Box 2), which display periodic arrays of internal loops.
Box 2 Modular assembly strategy for targeting RNA repeat expansions.
RNA repeat expansions are perhaps ideal targets for a modular assembly strategy. Known to cause more than 40 neurodegenerative and neuromuscular diseases287,288, expanded repeats often adopt a hairpin structure with a periodic array of internal loops. Toxicity is initiated when an RNA repeat expansion reaches a length that corresponds to a shift in structure59,289. Selective recognition of RNA repeat expansions is possible by exploiting the structural differences between the toxic repeats and short non-pathogenic repeats — differences that have been both experimentally and computationally validated59,278,289–291.
Selectivity can also be derived from the effective concentration of the toxic structure. Most expansions have hundreds to thousands of repeats. The effective concentration of the disease-causing structural element is therefore much greater than the concentration of the RNA in which it is embedded and in many potential off-targets. Small molecules are therefore more likely to encounter and engage the toxic repeat and affect its biology selectively. Further, repeats may be particularly sensitive to small-molecule intervention despite their size. For RNA gain-of-function pathology, freeing just a fraction of sequestered protein can improve disease-associated defects292.
Many laboratories have developed dimers that target two internal loops formed by repeat expansions, including those that cause myotonic dystrophy type 1 (DM1)163,293 and type 2 (DM2)294, c9orf72-amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD)295 and so on, although some have mixed MOAs as they also inhibit transcription. In one interesting example with an RNA-centric MOA, a dimer that targets r(CUG)exp was functionalized with azide and alkyne handles293. Upon binding to adjacent sites in the RNA target in patient-derived cells, a proximity-based click reaction afforded higher order oligomers that were formed in cells, that is, an on-site probe synthesis approach that increased potency from the nanomolar range to the picomolar range293. Further, this strategy enabled imaging of the repeats in live, patient-derived cells293. Such an approach was later extended to a tetrazine ligation reaction296.
Modularly assembled small molecules that bind r(CUG) repeat expansions have also been conjugated to bleomycin A5 (Cugamycin; Table 3) to cleave the RNA target selectively in myotubes derived from patients with DM1 and in a mouse model278. Importantly, Cugamycin alleviated DM1-associated defects without inducing DNA damage or lung fibrosis, known side effects of bleomycin137,297. (Please see ‘Target cleavage to assess occupancy’ section for more details about small molecule–bleomycin conjugates.) In vivo, Cugamycin rescued 97% of DM1-associated splicing defects, expression of the muscle-specific chloride ion channel and myotonia, with no significant off-targets. Additionally, small molecule–bleomycin conjugates have been developed for other targets including oncogenic pri-miR-96 (refs.167,298) and the miR-17/92 cluster299 as well as r(CCUG)exp in intron 1 of CNBP, which causes DM2 (ref.300).
Examples of small-molecule RNA binders
Several examples in which small-molecule RNA binders perturb downstream biology in a precise and predicted manner are highlighted below. Notably, the small molecules directly engage a functional site, and simple binding is sufficient to exert a biological effect (Box 1 and Tables 1–3).
Table 3.
Examples of small molecules that bind to RNAs implicated in neuromuscular and neurodegenerative diseases
| Compound/institution | Associated disease | RNA target | Mode of action | Stage of development | Activity | Refs. |
|---|---|---|---|---|---|---|
|
Cugamycin/Scripps Research Institute |
Myotonic dystrophy type 1 | r(CUG)exp in DMPK mRNA | Displaces MBNL1 from binding to r(CUG)exp | Early discovery | Kd = 365 nM (for r(CUG)12); EC25 = 840 nM (rescued splicing defects) | 278 |
|
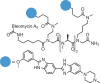
Synucleozid/Scripps Research Institute |
Parkinson disease | Iron-responsive element in α-synuclein mRNA | Inhibits ribosomal assembly and polysome loading onto α-synuclein mRNA | Early discovery | Kd = 1.5 μM; IC50 (neurons) = 0.5 μM | 13 |
|
Mitoxantrone/University of Washington, Seattle |
Alzheimer disease; frontotemporal dementia with parkinsonism-17 | Exon 10–intron junction in MAPT pre-mRNA | Inhibits binding of splicing machinery at the exon 10–intron junction; also inhibits a fungal group II intron277 | Early discovery | Kd <1 μM; EC50 = 700 nM (competition of binding with 25 nM pyrene–neomycin conjugate) | 225,279 |
|
Compound 9/Scripps Research Institute |
Alzheimer disease; frontotemporal dementia with parkinsonism-17 | Exon 10–intron junction in MAPT pre-mRNA | Inhibits binding of splicing machinery at the exon 10–intron junction | Early discovery | Kd = 4.9 μM; IC50 (transfected HeLa cells) = 10 μM; decreased 4R/3R ratio by ~50% in primary neurons at 40 μM | 70,228 |
|
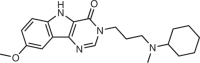
Risdiplam/Roche and PTC Therapeutics |
SMA | SMN2 pre-mRNA exon 7–intron junction | Promotes exon inclusion by stabilizing the binding of the splicing machinery | FDA approved | Kd = 15 nM; EC1.5x (SMA patient-derived fibroblasts) = 7 nM | 14 |
|
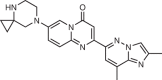
Branaplam |
SMA; HD | SMN2 pre-mRNA exon 7–intron junction | Promotes exon inclusion by stabilizing the binding of the splicing machinery | Phase I/II clinical trial (for SMA: NCT02268552; for HD: NCT05111249) | EC50 (SMN ELISA in SMNΔ7 mouse myoblasts) = 20 nM | 15 |



EC50, half maximal effective concentration; ELISA, enzyme-linked immunosorbent assay; HD, Huntington disease; IC50, half maximal inhibitory concentration; Kd, dissociation constant; SMA, spinal muscular atrophy; SMN, survival of motor neuron.
Targeting functional RNA structures
Many RNA classes have functional sites, for example, Drosha and Dicer processing sites within miRNA precursors, IRESs in viral RNAs and some human mRNAs, bacterial riboswitches, splicing enhancers and silencers in pre-mRNAs, and regulatory structures in 5′ and 3′ untranslated regions (UTRs). Below, we describe the discovery of exemplar small molecules that bind to these functional sites.
Discovery of small molecules that bind viral RNAs
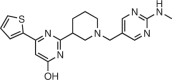
Apart from the bacterial ribosome, noncoding regions have also been investigated as drug targets to treat viral infections such as those caused by HIV160,168 and hepatitis C virus (HCV)12. The first viral regulatory elements targeted by small molecules that interfere with the infection process were HIV TAR and RRE RNAs169–174. Structure-guided approaches have been used to identify new scaffolds for TAR RNA that inhibited Tat-mediated transcriptional activation in a cellular model. One such scaffold, amiloride (Table 1), was later lead-optimized, affording an analogue with >100-fold enhanced affinity compared with the entry compound, and its structural interactions with the RNA target were well characterized126,160.
The HCV mRNA harbours a highly structured and conserved IRES in its 5′UTR, which initiates translation by recruiting and assembling host ribosomes175–177 and can be inhibited by small-molecule binding67,178,179. A mass spectrometry-based screen identified 2-aminobenzimidazole derivatives as both binders and inhibitors of HCV translation (Table 1). The affinity of the lead scaffold was improved ~100-fold after SAR studies158,180.
Another amiloride was discovered that inhibits viral replication of the Betacornoavirus OC43 and SARS-CoV-2 (ref.181). A panel of 23 amilorides was screened for inhibition of the infectivity of OC43, affording three lead molecules. Sequence conservation of the 5′-end of betacoronaviruses suggested a functional role, and this region is predicted to fold into six stem–loop structures (SL1–SL6). Fortuitously, one amiloride bound to SL6, as determined by a dye displacement assay and NMR spectral studies181.
Discovery of an antifungal that targets a group II intron
Group II introns are a class of self-splicing ribozymes found in mitochondria of fungi and yeast but not in mammals182, making them perhaps ideal targets for the development of antifungals. An in vitro screen of ~10,000 compounds for inhibitors of group II intron splicing afforded six hits that served as starting points154. Pharmacophore analysis enabled by SAR studies provided alternative scaffolds with improved physicochemical properties and ultimately a low micromolar inhibitor of group II intron splicing with antifungal activity comparable to that of amphotericin B.
Targeting human miRNAs with small molecules
miRNAs, small noncoding RNAs of 20–25 nucleotides, are key players in post-transcriptional gene regulation, silencing gene expression by association with the Argonaute RNA-induced silencing complex (RISC) and base pairing to the 3′UTR of complementary mRNAs8,183,184. Transcribed by RNA polymerase II as primary transcripts (pri-miRNAs), they are processed stepwise, first by the nuclease Drosha into precursor miRNAs (pre-miRNAs); after export to the cytoplasm, the mature (functional) miRNA is generated by the nuclease Dicer149,185. Fortuitously, their structures can be accurately modelled from sequence, and processing (functional) sites can be deduced from deep sequencing of mature miRNAs. Many studies have shown that Drosha or Dicer processing sites within miRNA precursors are targetable functional structures that can be short-circuited to alleviate disease159,167,186–190.
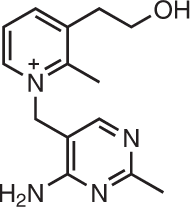
Compounds that bind functional structures in miRNA precursors and selectively inhibit the biogenesis of oncogenic miRNAs have been identified191. In particular, a spermine–amidine conjugate (PA-1) was discovered using an assay that measures enzymatic inhibition of pre-miR-372 processing192 (Fig. 5a and Table 2). In vitro, the molecule, without optimization, bound to the pre-miR-372 Dicer site, inhibited its processing and reduced cancer cell proliferation by derepression of its downstream target large tumour suppressor kinase 2 (LATS2)192. The selectivity of the small molecule was studied miRnome-wide, which revealed that only a small subset of miRNAs were affected. The conjugate also reduced formation of tumour cell spheroids of patient-derived cancer stem cells192. PA-1 was later optimized to afford the pre-miR-372-selective small molecule PA-3 (ref.16) (Table 2). Interestingly, a neomycin-nucleobase-amino acid conjugate that inhibited pre-miR-372 and pre-miR-21 was discovered by the same laboratory; lead optimization by structure-based design afforded a selective inhibitor of pre-miR-21 (ref.193) (Table 2).
Fig. 5. Small-molecule binding to RNA structures elicits biological effects through various modes of action.

a | A small molecule that binds the Dicer site of oncogenic pre-miR-372 inhibits its biogenesis and short-circuits downstream pathways. b | A compound that binds to an iron-responsive element (IRE) in the 5′ untranslated region (UTR) of SNCA mRNA inhibits its translation by mechanically blocking ribosomal assembly and polysome loading onto the mRNA. c | A small molecule that binds to a structural element at the exon 10–intron junction of MAPT pre-mRNA directs alternative splicing towards exon exclusion, reducing the amount of toxic 4R tau produced. d | A small molecule inhibits translation by binding to the flavin mononucleotide (FMN) riboswitch and inducing formation of the sequester loop that hides the start codon. e | Another small molecule, later optimized into an FDA-approved drug for the treatment of spinal muscular atrophy, binds to the exon 7–intron junction in SMN2 pre-mRNA and increases SMN2 protein levels by acting as a molecular glue for the RNA and splicing machinery and promoting exon inclusion. f | A compound binds to polypurine sequences in 5′UTRs of a subset of mRNAs and inhibits translation by acting as a molecular glue for the RNA and eukaryotic initiation factor 4A (eIF4A). IC50, half maximal inhibitory concentration; Kd, dissociation constant; miR, microRNA; MOA, mode of action.
Table 2.
Examples of small molecules that bind to RNAs implicated in cancer
| Compound/institution | Associated disease | RNA target | Mode of action | Stage of development | Activity | Ref. |
|---|---|---|---|---|---|---|
|
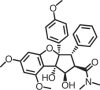
PA-1/Université Côte d’Azur |
Gastric carcinoma | Pre-miR-372 | Inhibits Dicer processing | Early discovery | Kd = 150 nM; IC50 (Dicer cleavage) = 1.06 μM, IC50 20 µM | 192 |
|
PA-3/Université Côte d’Azur |
Gastric carcinoma | Pre-miR-372 | Inhibits Dicer processing | Early discovery | Kd = 110 nM; IC50 (Dicer cleavage) = 0.58 μM | 16 |
|
Neomycin–nucleobase–amino acid conjugate (compound 6b)/Université Côte d’Azur |
Breast cancer | Pre-miR-21 | Inhibits Dicer processing | Early discovery | Kd = 50.7 nM; IC50 (Dicer cleavage) = 1.55 μM | 193 |
|
Targaprimir-515/Scripps Research Institute |
Breast cancer | Pre-miR-515 | Dimeric compound that targets Drosha processing site and adjacent site for enhanced selectivity | Early discovery | Kd = 60 nM; IC50 = 0.2 µM (MCF-7 cells) | 194 |
|
Dovitinib–RIBOTAC/Scripps Research Institute |
Triple-negative breast cancer and Alport syndrome | Pre-miR-21 | Induces degradation of miR-21 via RNase L recruitment | Early discovery | Kd = 3 µM (A bulge 3 × 2 RNA ILL); reduced levels of miR-21 by ~31% at 0.2 µM | 150 |
|
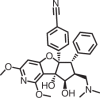
Rocaglamide/UC Berkeley & RIKEN |
Leukaemia | Polypurine sequences in the 5′UTR of a subset of oncogenic mRNAs | Inhibits translation initiation by clamping eIF4A to polypurine RNA sequence in the 5′UTR | Early discovery | Kd = 26 nM (ternary complex between compound, r(AG)10 and eIF4A) | 265 |
|
Zotatifin (eFT226)/eFFECTOR Therapeutics & Inception Therapeutics |
Triple-negative breast cancer and chronic myelogenous leukaemia | Polypurine sequences in the 5′UTR of a subset of oncogenic mRNAs | Inhibits translation initiation by clamping eIF4A to polypurine RNA sequence in the 5′UTR | Phase I–II clinical trial: NCT04092673 | Kd = 21–69 nM (ternary complex between compound, r(AG)3 and eIF4A); IC50 = 10.06 nM (MDA-MB-231 cell proliferation) | 266 |
|
Compound 5/National Cancer Institute |
Breast cancer | MALAT1 | Binds and modulates element for nuclear expression (ENE) triplex | Early discovery | Kd = 2.3 µM; IC50 about 1 µM (RNA level in MMTV-PyMT tumours) | 97 |

EC50, half maximal effective concentration; eIF4A, eukaryotic initiation factor 4A; IC50, half maximal inhibitory concentration; Kd, dissociation constant; miR, microRNA; MMTV-PyMT, mouse mammary tumour virus polyoma middle tumour antigen; RNase, ribonuclease; UTR, untranslated region.
Modular assembly has been used to overcome the degeneracy of functional RNA structures in the transcriptome by targeting two adjacent structures in the RNA target with a single molecule. Such an approach was taken to distinguish between human pri-miR-515 and pri-miR-885 to afford a selective molecule that inhibits only the biogenesis of miR-515 (ref.194) (Fig. 4a and Table 2). The lead molecule, a bis-benzimidazole with steric bulk that ablates DNA binding, prefers a structure harboured in the Drosha processing sites of both pri-miR-885 and pri-miR-515, a 5′UCU/3′AUA internal loop. As expected, the compound inhibited the biogenesis of both miRNAs to a similar extent in cells. To drive the selectivity towards a single target, differences in the secondary structure of the two pri-miRNAs were identified and exploited. A second internal loop that binds the substituted bis-benzimidazole is present adjacent to the Drosha site of miR-515, but not that of miR-885. Therefore, a dimeric compound, dubbed Targaprimir-515, consisting of two copies of the original hit, was designed. By comparison with the monomeric ligand, which showed similar affinity for both RNA targets, Targaprimir-515 had no measurable binding to RNA with a singular binding site and 3,200-fold improvement in affinity to pri-miR-515 (ref.194). The enhancement of selectivity in cells was also evident by miRNA profiling and global proteomics. The self-structure of Targaprimir-515 contributed to the improved in vitro and cellular selectivity194. Collectively, these and other studies65 confirm that bivalent compounds, even those assembled from fragments108, can successfully target RNAs with greater affinity and selectivity than the monomer from which they are derived. Modular assembly has also been applied to target RNA repeat expansions with enhanced potency and specificity (Box 2).
Inhibiting undruggable proteins by targeting the encoding RNA: α-synuclein
Pathogenic proteins are often considered undruggable when they lack a defined structure195,196. One way to drug these intrinsically disordered proteins (IDPs) is to inhibit their translation by targeting the encoding mRNA. Iron-responsive elements (IREs) are small stem–loop structures present in 5′- or 3′UTRs197–199 that bind to iron regulatory proteins (IRPs)200,201. IRP binding to IREs in the 5′UTR prevents ribosome docking and blocks RNA translation whereas binding to the 3′UTR stabilizes the transcript and upregulates translation202. IREs have an important role in cognitive function and hence in neurodegenerative diseases203–207. Small molecules that target IREs and inhibit the translation of these pathogenic proteins have been discovered13,197,208–213.
The aberrant expression and mutation of the IDP α-synuclein, which harbours an IRE in the encoding SNCA mRNA, is linked to Parkinson disease214,215. Targeting the IRE in the SNCA mRNA with small molecules to inhibit its translation is thus a promising strategy216. Synucleozid, a small molecule that binds a bulged adenosine residue within the IRE structure and inhibits SNCA translation, was designed by Inforna13 (Fig. 5b and Table 3). Notably, this bulged adenosine is not present in other IREs. Recognition of the SNCA IRE by synucleozid was dependent upon both the bulged nucleotide and its closing base pairs13; its MOA, by direct target engagement, was reduction of the number of polysomes loaded onto SNCA mRNA. Approximately 90 mRNAs have been identified with IREs or IRE-like elements, which vary in their sequences and structures199; these studies and those mentioned above lay the foundation for regulating translation of other mRNAs with small molecules.
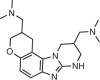
Altering protein isoforms by directing exon exclusion with small molecules: microtubule-associated protein tau
Almost every pre-mRNA goes through a series of processing steps, including alternative splicing (inclusion or exclusion of exons) to produce different protein isoforms217. Unsurprisingly, splice site mutations can alter splicing patterns and cause human diseases218. Studies with oligonucleotides demonstrated that it is indeed possible to rescue splicing defects by binding to and occluding the mutation from the spliceosomal machinery60,219–222, suggesting that small molecules may also be able to direct splicing.
The microtubule-associated protein tau gene (MAPT) produces six tau isoforms by alternative splicing223. Exons 9–12 encode a microtubule-binding domain (MBD), and exon 10 is alternatively spliced to produce isoforms with three (3R) or four (4R) MBDs, with the latter prone to aggregation223. In healthy individuals, the ratio between 3R and 4R is approximately equal; however, a genetic mutation in the splicing regulatory element (SRE) present in exon 10 alters the splicing pattern to produce excess 4R, the cause of frontotemporal dementia with parkinsonism-17 (FTDP-17). One such intronic mutation, dubbed disinhibition dementia parkinsonism amyotrophy complex (DDPAC), converts a GC base pair into a GU wobble pair, destabilizing the structure of the SRE. This destabilization allows U1 small nuclear RNA (snRNA) to more easily bind the SRE and facilitate exon 10 inclusion224. Stabilization of the structure of the SRE with a small molecule might therefore impede U1 snRNA binding and restore a normal splicing pattern. Various small molecules have been identified that bind to the SRE70,225–228 (Fig. 5c and Table 3).
Small molecules with activity in primary neurons were lead-optimized by chemical similarity searching, pharmacophore modelling driven by in vitro and cellular screening and analogue synthesis, structure-based design and docking studies based on the 3D structure of the MAPT SRE bound to several lead molecules, and traditional medicinal chemistry approaches70 (Fig. 4b). This synergistic strategy afforded a drug-like molecule that directed MAPT splicing towards the 3R isoform in primary neurons from a human tau transgenic mouse70.
Targeting RNA structures for degradation
If a functional site has not yet been discovered, various strategies may be applied to modulate RNA function by facilitating its degradation (Fig. 6). Two of these strategies are discussed below; both rely on chemically induced proximity — direct small-molecule cleavage of the target RNA (Box 2) or target degradation achieved by nuclease recruitment (Fig. 6a). Although these examples target known functional structures, the cleavage-based methods eliminate the RNA transcript and thus their MOA does not require binding to a functional site. The advantage of small molecule degraders, like ASOs, is that they are simultaneously target-validation methods. Small molecules can also facilitate degradation of an RNA target by increasing its accessibility to endogenous decay pathways such as the exosome and directing splicing such that the mature mRNA encodes a premature stop codon (Fig. 6b).
Fig. 6. Small-molecule RNA degraders and their mechanisms of action.

a | Degraders can elicit biological functions even if bound to non-functional sites within the RNA target as they cleave the transcript. Simply binding to non-functional sites is in principle biologically silent. Notably, degraders cleave RNA targets sub-stoichiometrically, as the same degrader molecule can cleave more than one RNA transcript by substrate turnover and can be optimized to improve target selectivity, including linker length, substrate preference and cellular localization of the target. b | Mechanisms of small-molecule-facilitated degradation. From left to right: small molecules can bind to intronic RNA repeat expansions that are harboured as retained introns. This causes excision of the intron, which is decayed by the RNA exosome280; RNA-binding compounds can be appended to natural products such as bleomycin that can cause oxidative cleavage of RNA targets selectively; ribonuclease-targeting chimeras induce the proximity of ribonucleases to unnaturally target an RNA for destruction by native quality control pathways; and small molecules can affect pre-mRNA splicing to create a mature mRNA with included exons that contain premature termination codons that trigger decay via nonsense-mediated decay. RNase, ribonuclease.
Nuclease recruitment to cleave RNA targets: ribonuclease-targeting chimeras
Targeted degradation was first demonstrated for proteins, or proteolysis-targeting chimeras (PROTACs)229. PROTACs are chimeric molecules comprising a protein-binding module and an E3 ubiquitin ligase-recognition module, which tags the targeted protein for selective degradation by the proteasome230–234. RIBOTACs have been developed for targeted degradation of RNA144, composed of an RNA-binding module and a RNase-recruiting module that selectively mediates RNA decay (Fig. 6a). RIBOTACs recruit the ubiquitously expressed cellular endoribonuclease RNase L, which functions in the viral immune response (see above)148,149. In its first two iterations, an RNA-binding module that selectively recognizes the Drosha site of pri-miR-96 or the Dicer site of pre-miR-210 was coupled to 2′-5′A4, inducing its selective cleavage in cells144,235.
As 2′-5′A4 reduces drug-likeness, a small-molecule RNase L recruiter was developed143 based on a previously reported small molecule236. A RIBOTAC with this new recruiter was developed to target pre-miR-21 for degradation by recognition of its Dicer processing site143. Notably, the RIBOTAC was more potent than the binder from which it was derived, possessed a prolonged duration of effect and inhibited breast cancer metastasis in a mouse model. Transcriptome-wide studies showed that the RIBOTAC was also more selective than the binder143, as quantified by a Gini coefficient153 and did not elicit an immune response. This enhanced selectivity is a composite of the specificity of the RNA-binding small molecule, the inherent substrate specificity of RNase L and whether the target has an RNase L substrate adjacent to the site at which the RNA-binding small molecule binds, the distance dictated by the linker that tethers the two components of the chimera.
These studies led to the hypothesis that RIBOTACs may allow reprogramming of known drugs for RNA targets. 2DCS selection of the Repurposing, Focused Rescue, and Accelerated Medchem (ReFRAME) library237 indicated that the receptor tyrosine kinase (RTK) inhibitor dovitinib binds the Dicer processing site of pre-miR-21 and therefore might inhibit its cellular processing, albeit at higher concentrations than those required to inhibit RTKs150. Converting the binding molecule dovitinib into a RIBOTAC enhanced its inherent RNA-targeting activity in cells and concomitantly decreased potency against canonical RTK protein targets, shifting selectivity for pre-miR-21 by 2,500-fold (Table 2). Further, the chimera alleviated disease progression in two mouse models caused by miR-21 overexpression: triple-negative breast cancer and Alport syndrome150.
RNA function can indeed be modulated with small-molecule binders or small-molecule degraders, expanding the MOA of RNA-targeting compounds and likely the number of targetable RNAs. Chimeric small molecules that target RNAs set the foundation for new drug discoveries akin to the revolution of PROTACs, enabling inhibition of RNA circuits when the functional site is unknown or absent. It will be exciting to see whether other RNA-modifying enzymes (editing, splicing machinery and so on) can be selectively recruited to RNA targets.
Targeting RNA-associated pathways
Several small molecules that target RNA-associated pathways have been derived from phenotypic screening and are described below.
Ribocil, an antibacterial that targets the FMN riboswitch
Riboswitches are structured noncoding sequences in the 5′ leader of bacterial mRNAs that control gene expression of a downstream ORF61,238–242 (Fig. 5d). Widely distributed across all known phylogenetic groups of bacteria, riboswitches form highly specific binding pockets for small-molecule metabolites, second messengers and inorganic ions243–247. Binding of the small molecule to the receptor (aptamer) domain directs formation of alternative secondary structures in the adjacent regulatory domain (expression platform)244 that modulates transcription or translation of the message248–250. The antibacterial ribocil was identified by a phenotypic screen for inhibitors of the riboflavin biosynthetic pathway in E. coli11 (Fig. 5d and Table 1). Resistance mutations mapped to the FMN riboswitch immediately upstream of the ribB gene11,251, validating the RNA target and elucidating compound MOA. A crystal structure of the riboswitch–ribocil complex revealed that the compound competitively binds to the FMN-binding pocket, using a similar, but not identical, binding mode.
A structure-guided approach to target the FMN riboswitch was also fruitful, yielding the compound 5FDQD (Table 1). Inspection of the crystal structures of the FMN-bound252 and apo-riboswitch253 revealed conformational changes in the RNA that occur upon FMN recognition. An iterative structure-based design strategy was pursued in which, first, structures were analysed for regions that could potentially accommodate chemical changes to FMN; second, a set of structure-guided derivatives was synthesized; third, productive binding was tested using chemical probing and in vitro transcription assays; and fourth, crystal structures of new lead compounds that emerged were determined254,255. The compound that resulted from these efforts, 5FDQD, is an analogue of FMN that binds to its RNA target with activity equipotent to that of the natural effector254,255. The bactericidal activity of this compound is highly selective for Clostridium difficile while having little effect upon diverse other bacteria commonly found in the gut microbiota254. Importantly, in mice, 5FDQD prevented lethal antibiotic-induced C. difficile infection, validating the use of a structure-guided approach to yield potent RNA-targeting therapeutics.
Risdiplam and branaplam, small molecules that target an RNA–protein manifold to direct alternative pre-mRNA splicing
SMA is a genetic disease caused by the deletion or mutation of the survival motor neuron 1 (SMN1) gene, which encodes a catalytic component of a complex responsible for assembly of small nuclear ribonucleoproteins (snRNPs) and hence the spliceosome. Its loss of function in SMA ultimately leads to the degradation of spinal motor neurons, muscle weakness, muscle atrophy and respiratory complications66,256. Fortuitously, humans encode an SMN1 paralogue, SMN2, with the two genes differing by only two nucleotides, one in exon 7 and the other in exon 8. The single-nucleotide polymorphism in exon 7 disrupts a splicing enhancer, resulting in exclusion of SMN2 exon 7 and reduction of the half-life of the encoded protein. If SMN2 exon 7 alternative splicing could be directed towards inclusion, then SMN2 could substitute functionally for loss of SMN1 and hence as a treatment for SMA (Fig. 5e).
A phenotypic screen to discover small molecules that direct splicing of SMN2 such that exon 7 is included identified an orally bioavailable compound, SMN-C3. Subsequent studies showed that SMN-C3 directed endogenous alternative splicing of SMN2 and provided therapeutic benefits in an SMA mouse model, with limited off-target effects. A medicinal chemistry campaign around SMN-C3 generated risdiplam (Evrysdi; Fig. 5e and Table 3), the first small-molecule FDA-approved drug to treat SMA14.
The lead optimization process that afforded risdiplam highlights a traditional medicinal chemistry approach (Fig. 4c). After chemical optimization of SMN-C3 to avoid mutagenicity and to confer favourable pharmacokinetic profiles, the preclinical candidate RG7800 was selected for advancement66,257. As RG7800 caused retinal degeneration14, a medicinal chemistry campaign began around key elements of the chemical structure, particularly with the goal to lower basicity, enhance potency, improve influx into the central nervous system and reduce compound metabolism. A virtual study of a library containing a pyridopyrimidinone central core and a right-hand-side imidazopyridazine fragment was carried out14. Risdiplam emerged from these compounds after a battery of preclinical tests showed high potency for directing SMN2 splicing in vitro and in vivo, reduced basicity, no phototoxicity risk and no formation of active metabolites14.
The challenge was to identify the target of risdiplam and hence its MOA, later determined after a series of studies on a derivative dubbed SMN-C5. NMR spectroscopy revealed that SMN-C5 stabilized an adenosine bulge at the exon 7–intron junction149,258,259, acting as a molecular glue for the ternary complex formed with U1 snRNP, revealed by Chem-CLIP260.
The small molecule branaplam was also identified from a similar phenotypic screen of SMN2 exon 7 alternative splicing, using a mini-gene reporter of a breast cancer 1 (BRCA1) exon 18 mutant that induces exon skipping as a counter-screen15. A series of studies using a related molecule, NVS-SM2, pinpointed that the molecule interacted with 21 nucleotides of the SMN2 5′ splice site, in particular a GA sequence found at the end of exon 7, suggesting involvement of U1 snRNP. NVS-SM2 acts as a molecular glue, as U1 snRNP only bound to SMN2 exon 7 when the small molecule was present15. Branaplam was later discovered to reduce mutant huntingtin (mHTT) protein levels by facilitating pseudo-exon inclusion in the HTT mRNA261. It is currently in clinical trial for the treatment of both SMA and Huntington disease.
The discoveries of risdiplam and branaplam are perhaps the best examples of successful phenotypic drug discovery efforts, as their development did not rely on a specific target but instead on a desired activity.
Rocaglamide, a molecular glue that inhibits translation of polypurine-containing transcripts
Molecular glues that affect the translation of specific mRNAs have also been identified by phenotypic screening, particularly for oncogenic mRNAs that promote proliferation262,263. Notable amongst several interesting compounds is rocaglamide, a member of the flavagline family, a class of bioactive natural products264 (Fig. 5f and Table 2). Rocaglamide has potent anti-tumour activity, specifically inhibiting the translation of a subset of transcripts with polypurine sequences in their 5′UTRs. The compound stabilizes the interaction between eukaryotic initiation factor 4A (eIF4A), an ATP-dependent DEAD-box RNA helicase, and mRNAs that depend on eIF4A binding to unwind them for translation265. Recent studies have afforded rocaglamide analogues with improved potency and physicochemical properties266–268. Thus, translation can be affected not only by small-molecule binding to an mRNA, but also by pharmacological stabilization of transient interactions between transcripts and translation factors, providing specific functional outcomes. Other compounds have been shown to have similar activities, suggesting that medicinal optimization of these compounds could provide small-molecule therapeutics264.
The future of small-molecule targeting of RNA
First viewed as simply an intermediate between DNA and protein, a renaissance in RNA structure and function beginning in the 1980s revealed the complex roles of RNA in homeostasis and disease183,269–273. Although considered a challenging target, modulation of RNA function by structure-binding small molecules is becoming increasingly well established as therapeutically practical. However, tremendous gaps in knowledge remain (Table 4 and Box 3).
Table 4.
Tools and challenges in developing small-molecule therapeutics targeting RNA
| Development step | Tools | Challenges |
|---|---|---|
| Identify druggable RNA targets |
Identify regions in an RNA target that are stably folded by using evolution and prediction restrained by structure mapping experiments Hypothesize a MOA for a binding compound Convert a binder into a degrader if binding alone is not functional |
Validating a predicted RNA structure in cells and identifying factors to prioritize targets that are more easily drugged |
| Identify small molecule binders |
Complete sequence-based design and identify on- and off-targets and routes for optimization Screen compound collections to find binders, considering screening on- and off-targets |
Defining chemotypes in compounds that bind RNA selectively Performing quantitative structure–activity relationships for RNA targets |
| Evaluate bioactivity of small molecules in cells |
Study the effect of RNA target-associated phenotypes and validate by using gain- or loss-of-function Use Chem-CLIP to define cellular on- and off-targets and evaluate the selectivity on the transcriptome and the proteome; correlate Chem-CLIP with -omics studies to define optimization plan |
Correlating binding to function is not straightforward as not all binders will be biologically functional |
| Structure-based lead design and optimization |
Solve structure of complex by NMR spectroscopy and X-ray crystallography, etc. Study pharmacophore model to define features in binding compounds and expand chemical space for target by using chemical similarity searching of pharmacophore and structure-based drug design |
Structure-based drug design and docking for RNA is different than for protein, i.e., electrostatic interactions are a significant driver Force fields need development, as there are limited examples of driving potency and specificity using these approaches |
| Evaluate efficacy and PK in animal models |
Study disease defect in model organisms, including transgenic models In vitro and in vivo PK assays are well established |
Physicochemical properties that potently and specifically affect RNA are unknown; need to be open minded and empirical with compounds, especially those outside rule of five space Studies in model organisms can be challenging especially for noncoding RNAs owing to differences in the human sequence and structure Varied modes of action can complicate PK and pharmacodynamic profiles |
| Preclinical and clinical studies |
Study compound activity in patient tissues, available through many repositories RNA-seq can be incorporated into clinical development pipeline to study efficacy, toxicity, potential off-targets |
Should the pipeline of tests used for oligonucleotide-based medicines be altered? Can this process be streamlined by using RNA-seq and other tools? Differences in sequence and structure in human versus mouse or other species should be considered in efficacy and toxicity studies |
MOA, mode of action; PK, pharmacokinetic(s); RNA-seq, RNA sequencing.
In the protein world, there are target classes that are considered both druggable and undruggable, generally classified by the formation of ordered structures. There is currently not enough knowledge in the RNA world to be able to classify RNA targets; however, most, if not all, bioactive small molecules target stable, functional structures. By identifying regions with unusually stable structures that are evolutionarily conserved, we can gain insight into potential functional structures and expand the druggable transcriptome. Key to the discovery of small molecules that bind these sites is to hypothesize function and hence potential compound MOA. Once the hypothesis is formulated, it must be verified by gain- and loss-of-function studies, particularly the effect on phenotype. Fortuitously, if binding alone is insufficient to modulate RNA function, the binding small molecule can be converted into a degrader. As more data are collected around functional structures, we will be able to tease out factors that contribute to druggablility.
Complementary to identifying high-priority RNA targets is defining physicochemical properties and chemotypes that confer affinity for RNA, which will inform design of RNA-focused small-molecule libraries (Box 1). Such libraries can be used in two ways: first, sequence-based design by defining binding landscapes, which defines both on- and off-targets; and second, screening against defined targets, in which an appropriate counter-screen must be completed to improve the likelihood of selective binding. The resultant datasets refine hypotheses around physicochemical properties and chemotypes that are ideal for RNA targets and hence RNA-focused small-molecule libraries. Notably, these screening collections must maintain chemical diversity to be useful.
Advances in structure-based design and lead optimization for RNA-targeted small molecules are also needed. Force fields that have been developed to enable structure-based design for proteins have not yet been fully customized for RNA, despite recent advances in computational biology. Molecular recognition of RNA by small molecules is driven differently than for protein, specifically the importance of aromatic ring stacking interactions that will require modification of electrostatic parameters. Accurate modelling of electrostatic parameters is key to understanding how small-molecule cores bind to and interact with an RNA target and to guide positioning of functionalities outside the core. Further, small-scale conformational dynamics are important for recognition of RNA targets. In its native state, RNA can have fast exchange between conformations, which are recognized by ligands differently. When the energetic difference between these states is low, there is likely only a modest effect on affinity. When the energy difference between conformations is large, however, the difference in affinity could be dramatic. Such knowledge is key for structure-based design.
Small-molecule binding is often not sufficient for bioactivity. Determining whether binding occurred to the cellular target but produced a biologically silent interaction is now possible with recently developed tools. Correlating target occupancy studies, using methods such as Chem-CLIP, with transcriptome- and proteome-wide studies is a powerful strategy that defines on- and off-targets and when binding elicits a biological response that is not desired. Such data are key to inform lead optimization. Intriguingly, fragment mapping and Chem-CLIP108,133,274, in combination with RNA structure prediction programs such as ScanFold, can provide insight into ligandable cellular RNA structures; biologically silent interactions inform cellular RNAs amenable to targeting by small-molecule-induced degradation.
Translating bioactive small molecules from cells to animals and then to the clinic is a significant hurdle for RNA-targeted small molecules. Of importance will be incorporation of transcriptome-wide analyses in patient tissues that assess efficacy and toxicity into clinical development pipelines, which should also be implemented for oligonucleotide-based modalities. Such studies also inform the range of selectivity that will be acceptable for RNA targets and may be quite different from that of proteins. For in vivo studies, significant differences in the sequence and structure of human and mouse RNAs, particularly noncoding RNAs, can change the activity or selectivity of a compound. As very few RNA-targeted compounds have advanced to animal studies and the clinic, more data will be needed to define pharmacokinetic and pharmacodynamic profiles ideal for preclinical and clinical candidates. As PROTACs and other protein-targeted medicines have changed the view of the rule of five275, we will need to be open to the fact that the physicochemical properties of small molecules that modulate RNA function may be outside the traditional drug-like space.
Box 3 Accelerating development of small molecules targeting RNA.
Key to advancing the RNA-targeting and chemical biology fields is an established pipeline for target validation and MOA studies, a longstanding process for proteins. Compounds with ill-defined mechanism are of less utility in chemical probe and lead medicine development than those that have an established MOA. A pathway for target validation and MOA studies has been previously presented65. The first step in this process is to confirm that the RNA target of interest is indeed driving phenotype, if not previously validated from in vitro or in vivo studies. For miRNAs, forced expression of the miRNA precursor in cells should induce or enhance the observed phenotype, while treatment with an antagomir should alleviate it. Validation for other RNA classes can be completed analogously by forced expression or knockout with ASOs. Small molecules that bind to the target can then be designed or discovered as described above.
Compounds that bind selectively to an RNA target in vitro can be carried forward to cellular studies to investigate whether they have the desired effect on the RNA and its downstream pathways, including phenotype. The effect of the small molecule on the abundance of the target RNA will depend on the target and the MOA of the compound. For example, small molecules that inhibit miRNA biogenesis by simply binding should boost levels of the precursor that it binds, leading to reduction of mature miRNA abundance. Small molecules that degrade the RNA target, whether directly or by recruiting nuclease, should reduce abundance. Further investigation is therefore required when an unexpected effect on RNA abundance is observed to define the compound’s MOA and rule out other effects such as inhibition of transcription. Notably, small molecule binding can alert RNA surveillance and quality control pathways that could induce degradation, and thus an unexpected effect on abundance does not necessarily eliminate an RNA-centric MOA. Instead, cellular target engagement should be confirmed using the methods described above, as should be the case for all RNA-targeting endeavours.
To fully define the effect of an RNA-targeting small molecule on phenotype, gain- and loss-of-function studies should be completed. For the former, forced expression of the RNA target is expected to reduce compound efficacy, as less of the target can be occupied at the same dose. Loss-of-function studies is a multistep process that is MOA-dependent. The first step, regardless of RNA target or MOA, is to express the wild-type RNA and an RNA with point mutations that ablate the binding site of the small molecule in cells with low or no endogenous expression. Here, the small molecule should only alleviate molecular defects and phenotype in wild-type RNA-expressing cells. The second step, applied to all RNA targets, is to confirm that the small molecule has no effect on cells that do not (aberrantly) express the target, typically cells derived from healthy patients. The remaining steps in loss-of-function studies are tailored to compound MOA. For example, bioactive small molecules that modulate miRNA function derepress a downstream protein target that alleviates phenotype. The downstream target can be knocked out chemically or with a small interfering RNA. The small molecule is no longer able to boost abundance of the downstream target and hence alleviate phenotype.
Outlook
As the functions of RNA in both health and disease have expanded and diversified, so has the field of RNA chemical biology, demonstrating that RNA is indeed druggable by small molecules. Further, RNA-targeted small molecules can be designed and lead optimized, the latter using strategies developed for protein targets with important modifications and considerations. As both academia and industry push the frontiers of this field forward, we believe that many more RNA-targeted medicines will reach the clinic in the years to come.
Acknowledgements
This work was funded by the NIH (R35 NS116846, R01 CA249180, UG3 NS116921 and P01 NS099114 to M.D.D. and R01 GM073850 to R.T.B.) and the Department of Defense (W81XWH-19-1-0719 and W81XWH-20-1-0727 to M.D.D).
Glossary
- Antisense oligonucleotides
(ASOs). Single-stranded DNA or RNA oligonucleotides, including combinations thereof that are chemically modified, that are complementary to the sequence of target RNA and can induce RNase H-mediated degradation or sterically block the ribosome.
- Riboswitch
Region of RNA structure located in the 5′ leader of bacterial RNAs that undergoes conformational switching upon small-molecule binding to regulate translation.
- Internal ribosome entry site
(IRES). RNA structural element typically in the 5′ untranslated region that can recruit ribosomes and initiate cap-independent translation.
- MicroRNA
(miRNA). Short noncoding RNA that has important roles in mediating gene expression by guiding Argonaute proteins to their mRNA target via base pairing to the 3′ untranslated region. miRNAs are formed stepwise, first from primary miRNAs to precursor miRNAs by the nuclease Drosha; then from precursor to mature (functional) miRNAs by the nuclease Dicer.
- Pharmacophore
3D arrangement of a molecule or molecular group that confers bioactivity via interactions with the compound’s target.
- Transcriptome
A landscape of all RNAs transcribed from a genome.
- Splicing modulators
Small molecules or proteins that are capable of directing RNA splicing by inducing inclusion or exclusion of exons.
- Functional selectivity
A metric that compares the effect of a compound on the biological activity of the desired target versus its effect on other targets.
- On- and off-targets
An on-target is the biomolecule that a compound is designed to modulate the function of; an off-target is unintendedly modulated by the small molecule.
- RNA sequencing
(RNA-seq). A high-throughput method that analyses gene expression transcriptome-wide.
- Phenotypic screening
A method that screens small molecules on the basis of a desired phenotypic outcome, without knowledge of its actual target.
- Structure–activity relationship
(SAR). Approach that aims to correlate the chemical structure of a compound with its biological activity.
- Ribonuclease-targeting chimeras
(RIBOTACs). Small-molecule chimeras that activate and recruit endogenous ribonucleases to an RNA target to trigger its degradation.
- Gini coefficient
A statistical measurement that quantifies the selectivity of a small molecule, ranging from 0 to 1, where 0 indicates that the compound inhibits every drug target studied equally and 1 indicates a compound selective for one drug target.
- Intrinsically disordered proteins
(IDPs). Proteins that lack well-defined 3D structures and are thus considered undruggable.
- Iron-responsive elements
(IREs). Small stem–loop structures present in 5′ or 3′ untranslated regions (UTRs) that bind to iron regulatory proteins (IRPs). IRP binding to IREs in the 5′UTR prevents ribosome docking and blocks RNA translation whereas binding to the 3′UTR stabilizes the transcript and upregulates translation.
- Proteolysis-targeting chimeras
(PROTACs). Chimeric molecules comprising a protein-binding module and a E3 ubiquitin ligase-recognition module, which tags the targeted protein for selective degradation by the proteasome.
Author contributions
All authors contributed to the writing of the manuscript and preparation of figures.
Competing interests
M.D.D. is a founder of Expansion Therapeutics. R.T.B. serves on the Scientific Advisory Board of Expansion Therapeutics and MeiraGTx. The remaining authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors jointly supervised this work: Robert T. Batey and Matthew D. Disney.
Contributor Information
Robert T. Batey, Email: robert.batey@colorado.edu
Matthew D. Disney, Email: mddisney@me.com
References
- 1.Jones D, Metzger HJ, Schatz A, Waksman SA. Control of Gram-negative bacteria in experimental animals by streptomycin. Science. 1944;100:103–105. doi: 10.1126/science.100.2588.103.b. [DOI] [PubMed] [Google Scholar]
- 2.Waksman SA, Reilly HC, Schatz A. Strain specificity and production of antibiotic substances: V. Strain resistance of bacteria to antibiotic substances, especially to streptomycin. Proc. Natl Acad. Sci. USA. 1945;31:157–164. doi: 10.1073/pnas.31.6.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holley RW, et al. Structure of a ribonucleic acid. Science. 1965;147:1462. doi: 10.1126/science.147.3664.1462. [DOI] [PubMed] [Google Scholar]
- 4.Zaug AJ, Cech TR. The intervening sequence RNA of Tetrahymena is an enzyme. Science. 1986;231:470–475. doi: 10.1126/science.3941911. [DOI] [PubMed] [Google Scholar]
- 5.Stark BC, Kole R, Bowman EJ, Altman S, Altman S. Ribonuclease P: an enzyme with an essential RNA component. Proc. Natl Acad. Sci. USA. 1978;75:3717–3721. doi: 10.1073/pnas.75.8.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai MC, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 9.Bennett CF. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019;70:307–321. doi: 10.1146/annurev-med-041217-010829. [DOI] [PubMed] [Google Scholar]
- 10.Luther DC, Lee YW, Nagaraj H, Scaletti F, Rotello VM. Delivery approaches for CRISPR/Cas9 therapeutics in vivo: advances and challenges. Expert Opin. Drug Deliv. 2018;15:905–913. doi: 10.1080/17425247.2018.1517746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howe JA, et al. Selective small-molecule inhibition of an RNA structural element. Nature. 2015;526:672–677. doi: 10.1038/nature15542. [DOI] [PubMed] [Google Scholar]
- 12.Dibrov SM, et al. Hepatitis C virus translation inhibitors targeting the internal ribosomal entry site. J. Med. Chem. 2014;57:1694–1707. doi: 10.1021/jm401312n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang P, et al. Translation of the intrinsically disordered protein alpha-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl Acad. Sci. USA. 2020;117:1457–1467. doi: 10.1073/pnas.1905057117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ratni H, et al. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA) J. Med. Chem. 2018;61:6501–6517. doi: 10.1021/acs.jmedchem.8b00741. [DOI] [PubMed] [Google Scholar]
- 15.Palacino J, et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 2016;12:304–304. doi: 10.1038/nchembio0416-304c. [DOI] [PubMed] [Google Scholar]
- 16.Becquart C, et al. Exploring heterocycle-spermine conjugates as modulators of oncogenic microRNAs biogenesis. ACS Omega. 2018;3:16500–16508. doi: 10.1021/acsomega.8b02681. [DOI] [Google Scholar]
- 17.Turner DH, Mathews DH. NNDB: the nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res. 2010;38:D280–D282. doi: 10.1093/nar/gkp892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tinoco I, Jr, Uhlenbeck OC, Levine MD. Estimation of secondary structure in ribonucleic acids. Nature. 1971;230:362–367. doi: 10.1038/230362a0. [DOI] [PubMed] [Google Scholar]
- 19.Freier SM, et al. Improved free-energy parameters for predictions of RNA duplex stability. Proc. Natl Acad. Sci. USA. 1986;83:9373–9377. doi: 10.1073/pnas.83.24.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuker M. mFold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lorenz R, et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathews DH, et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl Acad. Sci. USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peattie DA. Direct chemical method for sequencing RNA. Proc. Natl Acad. Sci. USA. 1979;76:1760–1764. doi: 10.1073/pnas.76.4.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bevilacqua PC, Ritchey LE, Su Z, Assmann SM. Genome-wide analysis of RNA secondary structure. Annu. Rev. Genet. 2016;50:235–266. doi: 10.1146/annurev-genet-120215-035034. [DOI] [PubMed] [Google Scholar]
- 25.Sun L, et al. RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol. 2019;26:322–330. doi: 10.1038/s41594-019-0200-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marinus T, Fessler AB, Ogle CA, Incarnato D. A novel SHAPE reagent enables the analysis of RNA structure in living cells with unprecedented accuracy. Nucleic Acids Res. 2021;49:e34. doi: 10.1093/nar/gkaa1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proc. Natl Acad. Sci. USA. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wells SE, Hughes JM, Igel AH, Ares M., Jr Use of dimethyl sulfate to probe RNA structure in vivo. Methods Enzymol. 2000;318:479–493. doi: 10.1016/S0076-6879(00)18071-1. [DOI] [PubMed] [Google Scholar]
- 29.Spitale RC, Flynn RA, Torre EA, Kool ET, Chang HY. RNA structural analysis by evolving SHAPE chemistry. Wiley Interdiscip. Rev. RNA. 2014;5:867–881. doi: 10.1002/wrna.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eddy SR. Computational analysis of conserved RNA secondary structure in transcriptomes and genomes. Annu. Rev. Biophys. 2014;43:433–456. doi: 10.1146/annurev-biophys-051013-022950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gutell RR, Lee JC, Cannone JJ. The accuracy of ribosomal RNA comparative structure models. Curr. Opin. Struct. Biol. 2002;12:301–310. doi: 10.1016/S0959-440X(02)00339-1. [DOI] [PubMed] [Google Scholar]
- 33.Woese CR, Gutell R, Gupta R, Noller HF. Detailed analysis of the higher-order structure of 16S-like ribosomal ribonucleic acids. Microbiol. Rev. 1983;47:621–669. doi: 10.1128/mr.47.4.621-669.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathews DH, Turner DH. Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. J. Mol. Biol. 2002;317:191–203. doi: 10.1006/jmbi.2001.5351. [DOI] [PubMed] [Google Scholar]
- 35.Havgaard JH, Lyngsø RB, Gorodkin J. The FOLDALIGN web server for pairwise structural RNA alignment and mutual motif search. Nucleic Acids Res. 2005;33:W650–W653. doi: 10.1093/nar/gki473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hofacker IL, Fekete M, Stadler PF. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- 37.Watkins AM, Rangan R, Das R. FARFAR2: improved de novo Rosetta prediction of complex global RNA folds. Structure. 2020;28:963–976. doi: 10.1016/j.str.2020.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 39.Sharma S, Ding F, Dokholyan NV. iFoldRNA: three-dimensional RNA structure prediction and folding. Bioinformatics. 2008;24:1951–1952. doi: 10.1093/bioinformatics/btn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Capriotti E, Norambuena T, Marti-Renom MA, Melo F. All-atom knowledge-based potential for RNA structure prediction and assessment. Bioinformatics. 2011;27:1086–1093. doi: 10.1093/bioinformatics/btr093. [DOI] [PubMed] [Google Scholar]
- 41.Townshend RJL, et al. Geometric deep learning of RNA structure. Science. 2021;373:1047–1051. doi: 10.1126/science.abe5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furtig B, Richter C, Wohnert J, Schwalbe H. NMR spectroscopy of RNA. Chembiochem. 2003;4:936–962. doi: 10.1002/cbic.200300700. [DOI] [PubMed] [Google Scholar]
- 43.Lietzke SE, Barnes CL, Kundrot CE. Crystallization and structure determination of RNA. Curr. Opin. Struct. Biol. 1995;5:645–649. doi: 10.1016/0959-440X(95)80057-3. [DOI] [PubMed] [Google Scholar]
- 44.Spahn CM, Penczek PA. Exploring conformational modes of macromolecular assemblies by multiparticle cryo-EM. Curr. Opin. Struct. Biol. 2009;19:623–631. doi: 10.1016/j.sbi.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang K, et al. Cryo-EM structure of a 40 kDa SAM-IV riboswitch RNA at 3.7 Å resolution. Nat. Commun. 2019;10:5511. doi: 10.1038/s41467-019-13494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kappel K, et al. Accelerated cryo-EM-guided determination of three-dimensional RNA-only structures. Nat. Methods. 2020;17:699–707. doi: 10.1038/s41592-020-0878-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding Y, Chan CY, Lawrence CE. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004;32:W135–W141. doi: 10.1093/nar/gkh449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Do CB, Woods DA, Batzoglou S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics. 2006;22:e90–e98. doi: 10.1093/bioinformatics/btl246. [DOI] [PubMed] [Google Scholar]
- 49.Rivas E, Clements J, Eddy SR. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods. 2017;14:45–48. doi: 10.1038/nmeth.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nawrocki EP, et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43:D130–D137. doi: 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fang R, Moss WN, Rutenberg-Schoenberg M, Simon MD. Probing Xist RNA structure in cells using targeted structure-Seq. PLoS Genet. 2015;11:e1005668. doi: 10.1371/journal.pgen.1005668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Somarowthu S, et al. HOTAIR forms an intricate and modular secondary structure. Mol. Cell. 2015;58:353–361. doi: 10.1016/j.molcel.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Novikova IV, Hennelly SP, Sanbonmatsu KY. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res. 2012;40:5034–5051. doi: 10.1093/nar/gks071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rivas E, Clements J, Eddy SR. Estimating the power of sequence covariation for detecting conserved RNA structure. Bioinformatics. 2020;36:3072–3076. doi: 10.1093/bioinformatics/btaa080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505:701–705. doi: 10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ding Y, et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2014;505:696–700. doi: 10.1038/nature12756. [DOI] [PubMed] [Google Scholar]
- 57.Wan Y, et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505:706–709. doi: 10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sobczak K, Krzyzosiak WJ. CAG repeats containing CAA interruptions form branched hairpin structures in spinocerebellar ataxia type 2 transcripts. J. Biol. Chem. 2005;280:3898–3910. doi: 10.1074/jbc.M409984200. [DOI] [PubMed] [Google Scholar]
- 59.Busan S, Weeks KM. Role of context in RNA structure: flanking sequences reconfigure CAG motif folding in huntingtin exon 1 transcripts. Biochemistry. 2013;52:8219–8225. doi: 10.1021/bi401129r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sudarsan N, Barrick JE, Breaker RR. Metabolite-binding RNA domains are present in the genes of eukaryotes. RNA. 2003;9:644–647. doi: 10.1261/rna.5090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andrews RJ, Roche J, Moss WN. ScanFold: an approach for genome-wide discovery of local RNA structural elements-applications to Zika virus and HIV. PeerJ. 2018;6:e6136. doi: 10.7717/peerj.6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moss WN, Priore SF, Turner DH. Identification of potential conserved RNA secondary structure throughout influenza A coding regions. RNA. 2011;17:991–1011. doi: 10.1261/rna.2619511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Leary C, et al. RNA structural analysis of the MYC mRNA reveals conserved motifs that affect gene expression. PLoS ONE. 2019;14:e0213758. doi: 10.1371/journal.pone.0213758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Disney MD. Targeting RNA with small molecules to capture opportunities at the Intersection of chemistry, biology, and medicine. J. Am. Chem. Soc. 2019;141:6776–6790. doi: 10.1021/jacs.8b13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naryshkin NA, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345:688–693. doi: 10.1126/science.1250127. [DOI] [PubMed] [Google Scholar]
- 67.Parsons J, et al. Conformational inhibition of the hepatitis C virus internal ribosome entry site RNA. Nat. Chem. Biol. 2009;5:823–825. doi: 10.1038/nchembio.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carnevali M, Parsons J, Wyles DL, Hermann T. A modular approach to synthetic RNA binders of the hepatitis C virus internal ribosome entry site. Chembiochem. 2010;11:1364–1367. doi: 10.1002/cbic.201000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen CZ, et al. Two high-throughput screening assays for aberrant RNA-protein interactions in myotonic dystrophy type 1. Anal. Bioanal. Chem. 2012;402:1889–1898. doi: 10.1007/s00216-011-5604-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen JL, et al. Design, optimization, and study of small molecules that target tau pre-mRNA and affect splicing. J. Am. Chem. Soc. 2020;142:8706–8727. doi: 10.1021/jacs.0c00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Corbett PT, et al. Dynamic combinatorial chemistry. Chem. Rev. 2006;106:3652–3711. doi: 10.1021/cr020452p. [DOI] [PubMed] [Google Scholar]
- 72.Motoyaji T. Revolution of small molecule drug discovery by affinity selection-mass spectrometry technology. Chem. Pharm. Bull. 2020;68:191–193. doi: 10.1248/cpb.c19-00832. [DOI] [PubMed] [Google Scholar]
- 73.Kiernan UA, Nedelkov D, Niederkofler EE, Tubbs KA, Nelson RW. High-throughput affinity mass spectrometry. Methods Mol. Biol. 2006;328:141–150. doi: 10.1385/1-59745-026-X:141. [DOI] [PubMed] [Google Scholar]
- 74.Annis DA, et al. An affinity selection–mass spectrometry method for the identification of small molecule ligands from self-encoded combinatorial libraries. Int. J. Mass. Spectrom. 2004;238:77–83. [Google Scholar]
- 75.Rizvi NF, et al. Discovery of selective RNA-binding small molecules by affinity-selection mass spectrometry. ACS Chem. Biol. 2018;13:820–831. doi: 10.1021/acschembio.7b01013. [DOI] [PubMed] [Google Scholar]
- 76.Rizvi NF, Nickbarg EB. RNA-ALIS: methodology for screening soluble RNAs as small molecule targets using ALIS affinity-selection mass spectrometry. Methods. 2019;167:28–38. doi: 10.1016/j.ymeth.2019.04.024. [DOI] [PubMed] [Google Scholar]
- 77.Stover JS, Shi J, Jin W, Vogt PK, Boger DL. Discovery of inhibitors of aberrant gene transcription from libraries of DNA binding molecules: inhibition of LEF-1-mediated gene transcription and oncogenic transformation. J. Am. Chem. Soc. 2009;131:3342–3348. doi: 10.1021/ja809083d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tran T, Disney MD. Identifying the preferred RNA motifs and chemotypes that interact by probing millions of combinations. Nat. Commun. 2012;3:1125. doi: 10.1038/ncomms2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Asare-Okai PN, Chow CS. A modified fluorescent intercalator displacement assay for RNA ligand discovery. Anal. Biochem. 2011;408:269–276. doi: 10.1016/j.ab.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang ZF, et al. The hairpin form of r(G4C2)exp in c9ALS/FTD is repeat-associated non-ATG translated and a target for bioactive small molecules. Cell Chem. Biol. 2019;26:179–190. doi: 10.1016/j.chembiol.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang J, Umemoto S, Nakatani K. Fluorescent indicator displacement assay for ligand-RNA interactions. J. Am. Chem. Soc. 2010;132:3660–3661. doi: 10.1021/ja100089u. [DOI] [PubMed] [Google Scholar]
- 82.Wicks SL, Hargrove AE. Fluorescent indicator displacement assays to identify and characterize small molecule interactions with RNA. Methods. 2019;167:3–14. doi: 10.1016/j.ymeth.2019.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jones AC, Neely RK. 2-Aminopurine as a fluorescent probe of DNA conformation and the DNA-enzyme interface. Q. Rev. Biophys. 2015;48:244–279. doi: 10.1017/S0033583514000158. [DOI] [PubMed] [Google Scholar]
- 84.Jean JM, Hall KB. 2-Aminopurine fluorescence quenching and lifetimes: role of base stacking. Proc. Natl Acad. Sci. USA. 2001;98:37–41. doi: 10.1073/pnas.98.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Soulière MF, Haller A, Rieder R, Micura R. A powerful approach for the selection of 2-aminopurine substitution sites to investigate RNA folding. J. Am. Chem. Soc. 2011;133:16161–16167. doi: 10.1021/ja2063583. [DOI] [PubMed] [Google Scholar]
- 86.Soulière MF, Micura R. Use of SHAPE to select 2AP substitution sites for RNA-ligand interactions and dynamics studies. Methods Mol. Biol. 2014;1103:227–239. doi: 10.1007/978-1-62703-730-3_17. [DOI] [PubMed] [Google Scholar]
- 87.Froeyen M, Herdewijn P. RNA as a target for drug design, the example of Tat-TAR interaction. Curr. Top. Med. Chem. 2002;2:1123–1145. doi: 10.2174/1568026023393200. [DOI] [PubMed] [Google Scholar]
- 88.Pascale L, et al. Deciphering structure-activity relationships in a series of Tat/TAR inhibitors. J. Biomol. Struct. Dyn. 2016;34:2327–2338. doi: 10.1080/07391102.2015.1114971. [DOI] [PubMed] [Google Scholar]
- 89.Yang M. Discoveries of Tat-TAR interaction inhibitors for HIV-1. Curr. Drug Targets Infect. Disord. 2005;5:433–444. doi: 10.2174/156800505774912901. [DOI] [PubMed] [Google Scholar]
- 90.Kumar A, et al. Chemical correction of pre-mRNA splicing defects associated with sequestration of muscleblind-like 1 protein by expanded r(CAG)-containing transcripts. ACS Chem. Biol. 2012;7:496–505. doi: 10.1021/cb200413a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.MacBeath G, Koehler AN, Schreiber SL. Printing small molecules as microarrays and detecting protein- ligand interactions en masse. J. Am. Chem. Soc. 1999;121:7967–7968. doi: 10.1021/ja991083q. [DOI] [Google Scholar]
- 92.Fazio F, Bryan MC, Blixt O, Paulson JC, Wong CH. Synthesis of sugar arrays in microtiter plate. J. Am. Chem. Soc. 2002;124:14397–14402. doi: 10.1021/ja020887u. [DOI] [PubMed] [Google Scholar]
- 93.Peng B, Thorsell AG, Karlberg T, Schuler H, Yao SQ. Small molecule microarray based discovery of PARP14 inhibitors. Angew. Chem. Int. Ed. Engl. 2017;56:248–253. doi: 10.1002/anie.201609655. [DOI] [PubMed] [Google Scholar]
- 94.Wang Z, et al. Microarray based screening of peptide nano probes for HER2 positive tumor. Anal. Chem. 2015;87:8367–8372. doi: 10.1021/acs.analchem.5b01588. [DOI] [PubMed] [Google Scholar]
- 95.Disney MD, Seeberger PH. Aminoglycoside microarrays to explore interactions of antibiotics with RNAs and proteins. Chemistry. 2004;10:3308–3314. doi: 10.1002/chem.200306017. [DOI] [PubMed] [Google Scholar]
- 96.Disney MD, Barrett OJ. An aminoglycoside microarray platform for directly monitoring and studying antibiotic resistance. Biochemistry. 2007;46:11223–11230. doi: 10.1021/bi701071h. [DOI] [PubMed] [Google Scholar]
- 97.Abulwerdi FA, et al. Selective small-molecule targeting of a triple helix encoded by the long noncoding RNA, MALAT1. ACS Chem. Biol. 2019;14:223–235. doi: 10.1021/acschembio.8b00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Connelly CM, Abulwerdi FA, Schneekloth JS., Jr. Discovery of RNA binding small molecules using small molecule microarrays. Methods Mol. Biol. 2017;1518:157–175. doi: 10.1007/978-1-4939-6584-7_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Connelly CM, Boer RE, Moon MH, Gareiss P, Schneekloth JS., Jr Discovery of inhibitors of microRNA-21 processing using small molecule microarrays. ACS Chem. Biol. 2017;12:435–443. doi: 10.1021/acschembio.6b00945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Connelly CM, et al. Synthetic ligands for PreQ(1) riboswitches provide structural and mechanistic insights into targeting RNA tertiary structure. Nat. Commun. 2019;10:1501. doi: 10.1038/s41467-019-09493-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sztuba-Solinska J, et al. Identification of biologically active, HIV TAR RNA-binding small molecules using small molecule microarrays. J. Am. Chem. Soc. 2014;136:8402–8410. doi: 10.1021/ja502754f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tran B, et al. Parallel discovery strategies provide a basis for riboswitch ligand design. Cell Chem. Biol. 2020;27:1241–1249.e1244. doi: 10.1016/j.chembiol.2020.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Labuda LP, Pushechnikov A, Disney MD. Small molecule microarrays of RNA-focused peptoids help identify inhibitors of a pathogenic group I intron. ACS Chem. Biol. 2009;4:299–307. doi: 10.1021/cb800313m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Velagapudi SP, et al. Approved anti-cancer drugs target oncogenic non-coding RNAs. Cell Chem. Biol. 2018;25:1086–1094.e1087. doi: 10.1016/j.chembiol.2018.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sreeramulu S, et al. Exploring the druggability of conserved RNA regulatory elements in the SARS-CoV-2 genome. Angew. Chem. Int. Ed. Engl. 2021;60:19191–19200. doi: 10.1002/anie.202103693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Parker CG, et al. Ligand and target discovery by fragment-based screening in human cells. Cell. 2017;168:527–541 e529. doi: 10.1016/j.cell.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang Y, et al. Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat. Chem. 2019;11:1113–1123. doi: 10.1038/s41557-019-0351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Suresh BM, et al. A general fragment-based approach to identify and optimize bioactive ligands targeting RNA. Proc. Natl Acad. Sci. USA. 2020;117:33197–33203. doi: 10.1073/pnas.2012217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Benhamou RI, et al. DNA-encoded library-versus-RNA-encoded library selection enables design of an oncogenic non-coding RNA inhibitor. Proc. Natl Acad. Sci. USA. 2022;119:e2114971119. doi: 10.1073/pnas.2114971119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Litovchick A, et al. Novel nucleic acid binding small molecules discovered using DNA-encoded chemistry. Molecules. 2019;24:2026. doi: 10.3390/molecules24102026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Disney MD, Velagapudi SP, Li Y, Costales MG, Childs-Disney JL. Identifying and validating small molecules interacting with RNA (SMIRNAs) Methods Enzymol. 2019;623:45–66. doi: 10.1016/bs.mie.2019.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Disney MD, et al. Two-dimensional combinatorial screening identifies specific aminoglycoside-RNA internal loop partners. J. Am. Chem. Soc. 2008;130:11185–11194. doi: 10.1021/ja803234t. [DOI] [PubMed] [Google Scholar]
- 113.Costales MG, Childs-Disney JL, Haniff HS, Disney MD. How we think about targeting RNA with small molecules. J. Med. Chem. 2020;63:8880–8900. doi: 10.1021/acs.jmedchem.9b01927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Velagapudi SP, et al. Defining RNA-small molecule affinity landscapes enables design of a small molecule inhibitor of an oncogenic noncoding RNA. ACS Cent. Sci. 2017;3:205–216. doi: 10.1021/acscentsci.7b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem. Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 116.Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257:1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 117.Sledz P, Caflisch A. Protein structure-based drug design: from docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018;48:93–102. doi: 10.1016/j.sbi.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 118.Yadav M, Dhagat S, Eswari JS. Structure based drug design and molecular docking studies of anticancer molecules paclitaxel, etoposide and topotecan using novel ligands. Curr. Drug Discov. Technol. 2020;17:183–190. doi: 10.2174/1570163816666190307102033. [DOI] [PubMed] [Google Scholar]
- 119.Batool M, Ahmad B, Choi S. A structure-based drug discovery paradigm. Int. J. Mol. Sci. 2019;20:2783. doi: 10.3390/ijms20112783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ganser LR, et al. Probing RNA conformational equilibria within the functional cellular context. Cell Rep. 2020;30:2472–2480. doi: 10.1016/j.celrep.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ganser LR, et al. High-performance virtual screening by targeting a high-resolution RNA dynamic ensemble. Nat. Struct. Mol. Biol. 2018;25:425–434. doi: 10.1038/s41594-018-0062-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shi H, et al. Rapid and accurate determination of atomistic RNA dynamic ensemble models using NMR and structure prediction. Nat. Commun. 2020;11:5531. doi: 10.1038/s41467-020-19371-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu B, Shi H, Al-Hashimi HM. Developments in solution-state NMR yield broader and deeper views of the dynamic ensembles of nucleic acids. Curr. Opin. Struct. Biol. 2021;70:16–25. doi: 10.1016/j.sbi.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Q, Sun X, Watt ED, Al-Hashimi HM. Resolving the motional modes that code for RNA adaptation. Science. 2006;311:653–656. doi: 10.1126/science.1119488. [DOI] [PubMed] [Google Scholar]
- 125.Frank AT, Stelzer AC, Al-Hashimi HM, Andricioaei I. Constructing RNA dynamical ensembles by combining MD and motionally decoupled NMR RDCs: new insights into RNA dynamics and adaptive ligand recognition. Nucleic Acids Res. 2009;37:3670–3679. doi: 10.1093/nar/gkp156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stelzer AC, et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011;7:553–559. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bush JA, et al. Systematically studying the effect of small molecules interacting with RNA in cellular and preclinical models. ACS Chem. Biol. 2021;16:1111–1127. doi: 10.1021/acschembio.1c00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Winkler WC, Cohen-Chalamish S, Breaker RR. An mRNA structure that controls gene expression by binding FMN. Proc. Natl Acad. Sci. USA. 2002;99:15908–15913. doi: 10.1073/pnas.212628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sudarsan N, Cohen-Chalamish S, Nakamura S, Emilsson GM, Breaker RR. Thiamine pyrophosphate riboswitches are targets for the antimicrobial compound pyrithiamine. Chem. Biol. 2005;12:1325–1335. doi: 10.1016/j.chembiol.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 130.Lin A, et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019;11:eaaw8412. doi: 10.1126/scitranslmed.aaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Osborn MF, White JD, Haley MM, DeRose VJ. Platinum-RNA modifications following drug treatment in S. cerevisiae identified by click chemistry and enzymatic mapping. ACS Chem. Biol. 2014;9:2404–2411. doi: 10.1021/cb500395z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mortison JD, et al. Tetracyclines modify translation by targeting key human rRNA substructures. Cell Chem. Biol. 2018;25:1506–1518. doi: 10.1016/j.chembiol.2018.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Balaratnam S, et al. A chemical probe based on the PreQ1 metabolite enables transcriptome-wide mapping of binding sites. Nat. Commun. 2021;12:5856. doi: 10.1038/s41467-021-25973-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Guan L, Disney MD. Covalent small-molecule–RNA complex formation enables cellular profiling of small-molecule–RNA interactions. Angew. Chem. Int. Ed. Engl. 2013;52:10010–10013. doi: 10.1002/anie.201301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zarrinkar PP, Wang J, Williamson JR. Slow folding kinetics of RNase P RNA. RNA. 1996;2:564–573. [PMC free article] [PubMed] [Google Scholar]
- 136.Zarrinkar PP, Williamson JR. Kinetic intermediates in RNA folding. Science. 1994;265:918–924. doi: 10.1126/science.8052848. [DOI] [PubMed] [Google Scholar]
- 137.Boger DL, Cai H. Bleomycin: synthetic and mechanistic studies. Angew. Chem. Int. Ed. Engl. 1999;38:448–476. doi: 10.1002/(SICI)1521-3773(19990215)38:4<448::AID-ANIE448>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 138.Ishida R, Takahashi T. Increased DNA chain breakage by combined action of bleomycin and superoxide radical. Biochem. Biophys. Res. Commun. 1975;66:1432–1438. doi: 10.1016/0006-291X(75)90519-7. [DOI] [PubMed] [Google Scholar]
- 139.Burger RM. Cleavage of nucleic acids by bleomycin. Chem. Rev. 1998;98:1153–1170. doi: 10.1021/cr960438a. [DOI] [PubMed] [Google Scholar]
- 140.Magliozzo RS, Peisach J, Ciriolo MR. Transfer-RNA is cleaved by activated bleomycin. Mol. Pharmacol. 1989;35:428–432. [PubMed] [Google Scholar]
- 141.Carter BJ, et al. Site-specific cleavage of RNA by Fe(II).bleomycin. Proc. Natl Acad. Sci. USA. 1990;87:9373–9377. doi: 10.1073/pnas.87.23.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Abraham AT, Lin JJ, Newton DL, Rybak S, Hecht SM. RNA cleavage and inhibition of protein synthesis by bleomycin. Chem. Biol. 2003;10:45–52. doi: 10.1016/S1074-5521(02)00306-X. [DOI] [PubMed] [Google Scholar]
- 143.Costales MG, et al. Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl Acad. Sci. USA. 2020;117:2406–2411. doi: 10.1073/pnas.1914286117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Costales MG, Matsumoto Y, Velagapudi SP, Disney MD. Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc. 2018;140:6741–6744. doi: 10.1021/jacs.8b01233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bisbal C, Martinand C, Silhol M, Lebleu B, Salehzada T. Cloning and characterization of a RNAse L inhibitor. A new component of the interferon-regulated 2-5A pathway. J. Biol. Chem. 1995;270:13308–13317. doi: 10.1074/jbc.270.22.13308. [DOI] [PubMed] [Google Scholar]
- 146.Li XL, et al. RNase-L-dependent destabilization of interferon-induced mRNAs. A role for the 2-5A system in attenuation of the interferon response. J. Biol. Chem. 2000;275:8880–8888. doi: 10.1074/jbc.275.12.8880. [DOI] [PubMed] [Google Scholar]
- 147.Zhou A, Molinaro RJ, Malathi K, Silverman RH. Mapping of the human RNASEL promoter and expression in cancer and normal cells. J. Interferon Cytokine Res. 2005;25:595–603. doi: 10.1089/jir.2005.25.595. [DOI] [PubMed] [Google Scholar]
- 148.Silverman RH. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007;81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Meyer SM, et al. Small molecule recognition of disease-relevant RNA structures. Chem. Soc. Rev. 2020;49:7167–7199. doi: 10.1039/D0CS00560F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang P, et al. Reprogramming of protein-targeted small-molecule medicines to RNA by ribonuclease recruitment. J. Am. Chem. Soc. 2021;143:13044–13055. doi: 10.1021/jacs.1c02248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lorenz MO. Methods of measuring the concentration of wealth. Pub. Am. Stat. Assoc. 1905;9:209–219. [Google Scholar]
- 152.Ursu A, et al. Gini coefficients as a single value metric to define chemical probe selectivity. ACS Chem. Biol. 2020;15:2031–2040. doi: 10.1021/acschembio.0c00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Graczyk PP. Gini coefficient: a new way to express selectivity of kinase inhibitors against a family of kinases. J. Med. Chem. 2007;50:5773–5779. doi: 10.1021/jm070562u. [DOI] [PubMed] [Google Scholar]
- 154.Fedorova O, et al. Small molecules that target group II introns are potent antifungal agents. Nat. Chem. Biol. 2018;14:1073–1078. doi: 10.1038/s41589-018-0142-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Anastasopoulou P, et al. Synthesis of triazole-functionalized 2-DOS analogues and their evaluation as A-site binders. Bioorg. Med. Chem. Lett. 2014;24:1122–1126. doi: 10.1016/j.bmcl.2013.12.125. [DOI] [PubMed] [Google Scholar]
- 156.Iwatani-Yoshihara M, et al. Discovery of allosteric inhibitors targeting the spliceosomal RNA helicase Brr2. J. Med. Chem. 2017;60:5759–5771. doi: 10.1021/acs.jmedchem.7b00461. [DOI] [PubMed] [Google Scholar]
- 157.Abulwerdi FA, et al. Development of small molecules with a noncanonical binding mode to HIV-1 trans activation response (TAR) RNA. J. Med. Chem. 2016;59:11148–11160. doi: 10.1021/acs.jmedchem.6b01450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Seth PP, et al. SAR by MS: discovery of a new class of RNA-binding small molecules for the hepatitis C virus: internal ribosome entry site IIA subdomain. J. Med. Chem. 2005;48:7099–7102. doi: 10.1021/jm050815o. [DOI] [PubMed] [Google Scholar]
- 159.Vo DD, et al. Oncogenic microRNAs biogenesis as a drug target: structure–activity relationship studies on new aminoglycoside conjugates. Chemistry. 2016;22:5350–5362. doi: 10.1002/chem.201505094. [DOI] [PubMed] [Google Scholar]
- 160.Patwardhan NN, et al. Amiloride as a new RNA-binding scaffold with activity against HIV-1 TAR. MedChemComm. 2017;8:1022–1036. doi: 10.1039/C6MD00729E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mammen M, Choi S-K, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 162.Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. Influencing receptor-ligand binding mechanisms with multivalent ligand architecture. J. Am. Chem. Soc. 2002;124:14922–14933. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- 163.Jahromi AH, et al. Developing bivalent ligands to target CUG triplet repeats, the causative agent of myotonic dystrophy type 1. J. Med. Chem. 2013;56:9471–9481. doi: 10.1021/jm400794z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pushechnikov A, et al. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J. Am. Chem. Soc. 2009;131:9767–9779. doi: 10.1021/ja9020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem. Biol. 2012;7:856–862. doi: 10.1021/cb200408a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thadke SA, et al. Design of bivalent nucleic acid ligands for recognition of RNA-repeated expansion associated with Huntington’s disease. Biochemistry. 2018;57:2094–2108. doi: 10.1021/acs.biochem.8b00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Velagapudi SP, et al. Design of a small molecule against an oncogenic noncoding RNA. Proc. Natl Acad. Sci. USA. 2016;113:5898–5903. doi: 10.1073/pnas.1523975113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Le Grice SF. Targeting the HIV RNA genome: high-hanging fruit only needs a longer ladder. Curr. Top. Microbiol. Immunol. 2015;389:147–169. doi: 10.1007/82_2015_434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zapp ML, Stern S, Green MR. Small molecules that selectively block RNA binding of HIV-1 rev protein inhibit Rev function and viral production. Cell. 1993;74:969–978. doi: 10.1016/0092-8674(93)90720-B. [DOI] [PubMed] [Google Scholar]
- 170.Mei H-Y, et al. Inhibition of an HIV-1 Tat-derived peptide binding to TAR RNA by aminoglycoside antibiotics. Bioorg. Med. Chem. Lett. 1995;5:2755–2760. doi: 10.1016/0960-894X(95)00467-8. [DOI] [Google Scholar]
- 171.Wang S, Huber PW, Cui M, Czarnik AW, Mei HY. Binding of neomycin to the TAR element of HIV-1 RNA induces dissociation of Tat protein by an allosteric mechanism. Biochemistry. 1998;37:5549–5557. doi: 10.1021/bi972808a. [DOI] [PubMed] [Google Scholar]
- 172.Ratmeyer L, et al. Inhibition of HIV-1 Rev–RRE interaction by diphenylfuran derivatives. Biochemistry. 1996;35:13689–13696. doi: 10.1021/bi960954v. [DOI] [PubMed] [Google Scholar]
- 173.Park WKC, Auer M, Jaksche H, Wong C-H. Rapid combinatorial synthesis of aminoglycoside antibiotic mimetics: use of a polyethylene glycol-linked amine and a neamine-derived aldehyde in multiple component condensation as a strategy for the discovery of new inhibitors of the HIV RNA Rev responsive element. J. Am. Chem. Soc. 1996;118:10150–10155. doi: 10.1021/ja9612817. [DOI] [Google Scholar]
- 174.Mei HY, et al. Inhibitors of protein-RNA complexation that target the RNA: specific recognition of human immunodeficiency virus type 1 TAR RNA by small organic molecules. Biochemistry. 1998;37:14204–14212. doi: 10.1021/bi981308u. [DOI] [PubMed] [Google Scholar]
- 175.Hellen CU, Pestova TV. Translation of hepatitis C virus RNA. J. Viral Hepat. 1999;6:79–87. doi: 10.1046/j.1365-2893.1999.00150.x. [DOI] [PubMed] [Google Scholar]
- 176.Ji H, Fraser CS, Yu Y, Leary J, Doudna JA. Coordinated assembly of human translation initiation complexes by the hepatitis C virus internal ribosome entry site RNA. Proc. Natl Acad. Sci. USA. 2004;101:16990–16995. doi: 10.1073/pnas.0407402101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Otto GA, Puglisi JD. The pathway of HCV IRES-mediated translation initiation. Cell. 2004;119:369–380. doi: 10.1016/j.cell.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 178.Wang W, et al. Hepatitis C viral IRES inhibition by phenazine and phenazine-like molecules. Bioorg. Med. Chem. Lett. 2000;10:1151–1154. doi: 10.1016/S0960-894X(00)00217-1. [DOI] [PubMed] [Google Scholar]
- 179.Jefferson EA, et al. Biaryl guanidine inhibitors of in vitro HCV-IRES activity. Bioorg. Med. Chem. Lett. 2004;14:5139–5143. doi: 10.1016/j.bmcl.2004.07.066. [DOI] [PubMed] [Google Scholar]
- 180.Dibrov SM, et al. Structure of a hepatitis C virus RNA domain in complex with a translation inhibitor reveals a binding mode reminiscent of riboswitches. Proc. Natl Acad. Sci. USA. 2012;109:5223–5228. doi: 10.1073/pnas.1118699109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zafferani M, et al. Amilorides inhibit SARS-CoV-2 replication in vitro by targeting RNA structures. Sci. Adv. 2021;7:eabl6096. doi: 10.1126/sciadv.abl6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Michel F, Dujon B. Conservation of RNA secondary structures in two intron families including mitochondrial-, chloroplast- and nuclear-encoded members. EMBO J. 1983;2:33–38. doi: 10.1002/j.1460-2075.1983.tb01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 184.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 185.Bushati N, Cohen S. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 186.Warner KD, Hajdin CE, Weeks KM. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018;17:547–558. doi: 10.1038/nrd.2018.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Di Giorgio A, Duca M. Synthetic small-molecule RNA ligands: future prospects as therapeutic agents. MedChemComm. 2019;10:1242–1255. doi: 10.1039/C9MD00195F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Disney MD, Angelbello AJ. Rational design of small molecules targeting oncogenic noncoding RNAs from sequence. Acc. Chem. Res. 2016;49:2698–2704. doi: 10.1021/acs.accounts.6b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pomplun S, Gates ZP, Zhang G, Quartararo AJ, Pentelute BL. Discovery of nucleic acid binding molecules from combinatorial biohybrid nucleobase peptide libraries. J. Am. Chem. Soc. 2020;142:19642–19651. doi: 10.1021/jacs.0c08964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Garner AL, et al. Tetracyclines as inhibitors of pre-microRNA maturation: a disconnection between RNA binding and inhibition. ACS Med. Chem. Lett. 2019;10:816–821. doi: 10.1021/acsmedchemlett.9b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Di Giorgio A, Tran TP, Duca M. Small-molecule approaches toward the targeting of oncogenic miRNAs: roadmap for the discovery of RNA modulators. Future Med. Chem. 2016;8:803–816. doi: 10.4155/fmc-2016-0018. [DOI] [PubMed] [Google Scholar]
- 192.Staedel C, et al. Modulation of oncogenic miRNA biogenesis using functionalized polyamines. Sci. Rep. 2018;8:1667. doi: 10.1038/s41598-018-20053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Maucort C, et al. Design and implementation of synthetic RNA binders for the inhibition of miR-21 biogenesis. ACS Med. Chem. Lett. 2021;12:899–906. doi: 10.1021/acsmedchemlett.0c00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Costales MG, et al. A designed small molecule inhibitor of a non-coding RNA sensitizes HER2 negative cancers to herceptin. J. Am. Chem. Soc. 2019;141:2960–2974. doi: 10.1021/jacs.8b10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer. 2017;17:502–508. doi: 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhou ZD, Tan EK. Iron regulatory protein (IRP)–iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017;12:75. doi: 10.1186/s13024-017-0218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann. N. Y. Acad. Sci. 2004;1012:1–13. doi: 10.1196/annals.1306.001. [DOI] [PubMed] [Google Scholar]
- 199.Volz K. Conservation in the iron responsive element family. Genes. 2021;12:1365. doi: 10.3390/genes12091365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- 201.Maio N, et al. Mechanisms of cellular iron sensing, regulation of erythropoiesis and mitochondrial iron utilization. Semin. Hematol. 2021;58:161–174. doi: 10.1053/j.seminhematol.2021.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Shaw KT, et al. Phenserine regulates translation of beta-amyloid precursor protein mRNA by a putative interleukin-1 responsive element, a target for drug development. Proc. Natl Acad. Sci. USA. 2001;98:7605–7610. doi: 10.1073/pnas.131152998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rogers JT, Cahill CM. Iron-responsive-like elements and neurodegenerative ferroptosis. Learn. Mem. 2020;27:395–413. doi: 10.1101/lm.052282.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lumsden AL, et al. Dysregulation of neuronal iron homeostasis as an alternative unifying effect of mutations causing familial Alzheimer’s disease. Front. Neurosci. 2018;12:533. doi: 10.3389/fnins.2018.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Rogers JT, et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 206.Zhang P, et al. Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free Radic. Biol. Med. 2020;152:227–234. doi: 10.1016/j.freeradbiomed.2020.03.015. [DOI] [PubMed] [Google Scholar]
- 207.Olivares D, Huang X, Branden L, Greig NH, Rogers JT. Physiological and pathological role of alpha-synuclein in Parkinson’s disease through iron mediated oxidative stress; the role of a putative iron-responsive element. Int. J. Mol. Sci. 2009;10:1226–1260. doi: 10.3390/ijms10031226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bandyopadhyay S, et al. Novel 5′ untranslated region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: implications for down syndrome and Alzheimer’s disease. PLoS ONE. 2013;8:e65978. doi: 10.1371/journal.pone.0065978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Utsuki T, et al. Identification of novel small molecule inhibitors of amyloid precursor protein synthesis as a route to lower Alzheimer’s disease amyloid-beta peptide. J. Pharmacol. Exp. Ther. 2006;318:855–862. doi: 10.1124/jpet.106.103309. [DOI] [PubMed] [Google Scholar]
- 210.Canzoneri JC, Oyelere AK. Interaction of anthracyclines with iron responsive element mRNAs. Nucleic Acids Res. 2008;36:6825–6834. doi: 10.1093/nar/gkn774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Venti A, et al. The integrated role of desferrioxamine and phenserine targeted to an iron-responsive element in the APP-mRNA 5’-untranslated region. Ann. N. Y. Acad. Sci. 2004;1035:34–48. doi: 10.1196/annals.1332.003. [DOI] [PubMed] [Google Scholar]
- 212.Tibodeau JD, Fox PM, Ropp PA, Theil EC, Thorp HH. The up-regulation of ferritin expression using a small-molecule ligand to the native mRNA. Proc. Natl Acad. Sci. USA. 2006;103:253–257. doi: 10.1073/pnas.0509744102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Cahill CM, Aleyadeh R, Gao J, Wang C, Rogers JT. Alpha-synuclein in alcohol use disorder, connections with Parkinson’s disease and potential therapeutic role of 5′ untranslated region-directed small molecules. Biomolecules. 2020;10:1465. doi: 10.3390/biom10101465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Stefanis L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012;2:a009399. doi: 10.1101/cshperspect.a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018;109:249–257. doi: 10.1016/j.nbd.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 216.Junn E, et al. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl Acad. Sci. USA. 2009;106:13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ule J, Blencowe BJ. Alternative splicing regulatory networks: functions, mechanisms, and evolution. Mol. Cell. 2019;76:329–345. doi: 10.1016/j.molcel.2019.09.017. [DOI] [PubMed] [Google Scholar]
- 218.Lee Y, Rio DC. Mechanisms and regulation of alternative pre-mRNA splicing. Annu. Rev. Biochem. 2015;84:291–323. doi: 10.1146/annurev-biochem-060614-034316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Bauman J, Jearawiriyapaisarn N, Kole R. Therapeutic potential of splice-switching oligonucleotides. Oligonucleotides. 2009;19:1–13. doi: 10.1089/oli.2008.0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sazani P, Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J. Clin. Invest. 2003;112:481–486. doi: 10.1172/JCI200319547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016;44:6549–6563. doi: 10.1093/nar/gkw533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Scharner J, et al. Hybridization-mediated off-target effects of splice-switching antisense oligonucleotides. Nucleic Acids Res. 2020;48:802–816. doi: 10.1093/nar/gkz1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res. Bull. 2016;126:238–292. doi: 10.1016/j.brainresbull.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 224.Donahue CP, Muratore C, Wu JY, Kosik KS, Wolfe MS. Stabilization of the tau exon 10 stem loop alters pre-mRNA splicing. J. Biol. Chem. 2006;281:23302–23306. doi: 10.1074/jbc.C600143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zheng S, Chen Y, Donahue CP, Wolfe MS, Varani G. Structural basis for stabilization of the tau pre-mRNA splicing regulatory element by novantrone (mitoxantrone) Chem. Biol. 2009;16:557–566. doi: 10.1016/j.chembiol.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lisowiec J, Magner D, Kierzek E, Lenartowicz E, Kierzek R. Structural determinants for alternative splicing regulation of the MAPT pre-mRNA. RNA Biol. 2015;12:330–342. doi: 10.1080/15476286.2015.1017214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Liu Y, et al. Mitoxantrone analogues as ligands for a stem–loop structure of tau pre-mRNA. J. Med. Chem. 2009;52:6523–6526. doi: 10.1021/jm9013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Luo Y, Disney MD. Bottom-up design of small molecules that stimulate exon 10 skipping in mutant MAPT pre-mRNA. Chembiochem. 2014;15:2041–2044. doi: 10.1002/cbic.201402069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sakamoto KM, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zou Y, Ma D, Wang Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019;37:21–30. doi: 10.1002/cbf.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sakamoto KM. Chimeric molecules to target proteins for ubiquitination and degradation. Methods Enzymol. 2005;399:833–847. doi: 10.1016/S0076-6879(05)99054-X. [DOI] [PubMed] [Google Scholar]
- 232.Ottis P, Crews CM. Proteolysis-targeting chimeras: induced protein degradation as a therapeutic strategy. ACS Chem. Biol. 2017;12:892–898. doi: 10.1021/acschembio.6b01068. [DOI] [PubMed] [Google Scholar]
- 233.Crews CM. Inducing protein degradation as a therapeutic strategy. J. Med. Chem. 2018;61:403–404. doi: 10.1021/acs.jmedchem.7b01333. [DOI] [PubMed] [Google Scholar]
- 234.Chamberlain PP, Hamann LG. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019;15:937–944. doi: 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- 235.Costales MG, Suresh B, Vishnu K, Disney MD. Targeted degradation of a hypoxia-associated non-coding RNA enhances the selectivity of a small molecule interacting with RNA. Cell Chem. Biol. 2019;26:1180–1186. doi: 10.1016/j.chembiol.2019.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Thakur CS, et al. Small-molecule activators of RNase L with broad-spectrum antiviral activity. Proc. Natl Acad. Sci. USA. 2007;104:9585–9590. doi: 10.1073/pnas.0700590104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Janes J, et al. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl Acad. Sci. USA. 2018;115:10750–10755. doi: 10.1073/pnas.1810137115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Grundy FJ, Henkin TM. The S box regulon: a new global transcription termination control system for methionine and cysteine biosynthesis genes in Gram-positive bacteria. Mol. Microbiol. 1998;30:737–749. doi: 10.1046/j.1365-2958.1998.01105.x. [DOI] [PubMed] [Google Scholar]
- 239.Gelfand M. A conserved RNA structure element involved in the regulation of bacterial riboflavin synthesis genes. Trends Genet. 1999;15:439–442. doi: 10.1016/S0168-9525(99)01856-9. [DOI] [PubMed] [Google Scholar]
- 240.Mironov AS, et al. Sensing small molecules by nascent RNA. Cell. 2002;111:747–756. doi: 10.1016/S0092-8674(02)01134-0. [DOI] [PubMed] [Google Scholar]
- 241.Nahvi A, et al. Genetic control by a metabolite binding mRNA. Chem. Biol. 2002;9:1043–1049. doi: 10.1016/S1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 242.Winkler W, Nahvi A, Breaker RR. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature. 2002;419:952–956. doi: 10.1038/nature01145. [DOI] [PubMed] [Google Scholar]
- 243.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 244.Polaski JT, Kletzien OA, Drogalis LK, Batey RT. A functional genetic screen reveals sequence preferences within a key tertiary interaction in cobalamin riboswitches required for ligand selectivity. Nucleic Acids Res. 2018;46:9094–9105. doi: 10.1093/nar/gky539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Batey RT, Gilbert SD, Montange RK. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004;432:411–415. doi: 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- 246.Batey RT, Rambo RP, Doudna JA. Tertiary motifs in RNA structure and folding. Angew. Chem. Int. Ed. Engl. 1999;38:2326–2343. doi: 10.1002/(SICI)1521-3773(19990816)38:16<2326::AID-ANIE2326>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 247.Breaker RR. Riboswitches and the RNA world. Cold Spring Harb. Perspect. Biol. 2012;4:a003566. doi: 10.1101/cshperspect.a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Garst AD, Edwards AL, Batey RT. Riboswitches: structures and mechanisms. Cold Spring Harb. Perspect. Biol. 2011;3:a003533. doi: 10.1101/cshperspect.a003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Sherlock ME, Breaker RR. Former orphan riboswitches reveal unexplored areas of bacterial metabolism, signaling, and gene control processes. RNA. 2020;26:675–693. doi: 10.1261/rna.074997.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Wang JX, Breaker RR. Riboswitches that sense S-adenosylmethionine and S-adenosylhomocysteine. Biochem. Cell. Biol. 2008;86:157–168. doi: 10.1139/O08-008. [DOI] [PubMed] [Google Scholar]
- 251.Balibar CJ, et al. Validation and development of an Escherichia coli riboflavin pathway phenotypic screen hit as a small-molecule ligand of the flavin mononucleotide riboswitch. Methods Mol. Biol. 2018;1787:19–40. doi: 10.1007/978-1-4939-7847-2_2. [DOI] [PubMed] [Google Scholar]
- 252.Serganov A, Huang L, Patel DJ. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature. 2009;458:233–237. doi: 10.1038/nature07642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Vicens Q, Mondragón E, Batey RT. Molecular sensing by the aptamer domain of the FMN riboswitch: a general model for ligand binding by conformational selection. Nucleic Acids Res. 2011;39:8586–8598. doi: 10.1093/nar/gkr565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Blount KF, et al. Novel riboswitch-binding flavin analog that protects mice against Clostridium difficile infection without inhibiting cecal flora. Antimicrob. Agents Chemother. 2015;59:5736–5746. doi: 10.1128/AAC.01282-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Vicens Q, et al. Structure–activity relationship of flavin analogues that target the flavin mononucleotide riboswitch. ACS Chem. Biol. 2018;13:2908–2919. doi: 10.1021/acschembio.8b00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Cho S, Dreyfuss G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 2010;24:438–442. doi: 10.1101/gad.1884910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ratni H, et al. Specific correction of alternative survival motor neuron 2 splicing by small molecules: discovery of a potential novel medicine to treat spinal muscular atrophy. J. Med. Chem. 2016;59:6086–6100. doi: 10.1021/acs.jmedchem.6b00459. [DOI] [PubMed] [Google Scholar]
- 258.Campagne S, et al. Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat. Chem. Biol. 2019;15:1191–1198. doi: 10.1038/s41589-019-0384-5. [DOI] [PubMed] [Google Scholar]
- 259.Sivaramakrishnan M, et al. Binding to SMN2 pre-mRNA–protein complex elicits specificity for small molecule splicing modifiers. Nat. Commun. 2017;8:1476. doi: 10.1038/s41467-017-01559-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Wang J, Schultz PG, Johnson KA. Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl Acad. Sci. USA. 2018;115:E4604–E4612. doi: 10.1073/pnas.1800260115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Keller CG, et al. An orally available, brain penetrant, small molecule lowers huntingtin levels by enhancing pseudoexon inclusion. Nat. Commun. 2022;13:1150. doi: 10.1038/s41467-022-28653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Chaidir C, Edrada-Ebel R, Ebel R, Bohnenstengel F, Nugroho BW. Chemistry and biological activity of rocaglamide derivatives and related compounds in Aglaia species (Meliaceae) Curr. Org. Chem. 2001;5:923–938. doi: 10.2174/1385272013375049. [DOI] [Google Scholar]
- 263.Kim S, et al. Silvestrol, a potential anticancer rocaglate derivative from Aglaia foveolata, induces apoptosis in LNCaP cells through the mitochondrial/apoptosome pathway without activation of executioner caspase-3 or -7. Anticancer Res. 2007;27:2175–2183. [PMC free article] [PubMed] [Google Scholar]
- 264.Schulz G, Victoria C, Kirschning A, Steinmann E. Rocaglamide and silvestrol: a long story from anti-tumor to anti-coronavirus compounds. Nat. Prod. Rep. 2021;38:18–23. doi: 10.1039/D0NP00024H. [DOI] [PubMed] [Google Scholar]
- 265.Iwasaki S, Floor SN, Ingolia NT. Rocaglates convert DEAD-box protein eIF4A into a sequence-selective translational repressor. Nature. 2016;534:558–561. doi: 10.1038/nature17978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Ernst JT, et al. Design of development candidate eFT226, a first in class inhibitor of eukaryotic initiation factor 4A RNA helicase. J. Med. Chem. 2020;63:5879–5955. doi: 10.1021/acs.jmedchem.0c00182. [DOI] [PubMed] [Google Scholar]
- 267.Nilewski C, et al. 1-Aminomethyl SAR in a novel series of flavagline-inspired eIF4A inhibitors: effects of amine substitution on cell potency and in vitro PK properties. Bioorg. Med. Chem. Lett. 2021;47:128111. doi: 10.1016/j.bmcl.2021.128111. [DOI] [PubMed] [Google Scholar]
- 268.Nilewski C, et al. Strategic diastereoselective C1 functionalization in the aza-rocaglamide scaffold toward natural product-inspired eIF4A inhibitors. Org. Lett. 2020;22:6257–6261. doi: 10.1021/acs.orglett.0c01944. [DOI] [PubMed] [Google Scholar]
- 269.Kruger K, et al. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 270.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 271.Cech TR, Bass BL. Biological catalysis by RNA. Annu. Rev. Biochem. 1986;55:599–629. doi: 10.1146/annurev.bi.55.070186.003123. [DOI] [PubMed] [Google Scholar]
- 272.Peattie DA, Douthwaite S, Garrett RA, Noller HF. A “bulged” double helix in a RNA–protein contact site. Proc. Natl Acad. Sci. USA. 1981;78:7331–7335. doi: 10.1073/pnas.78.12.7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 274.Velagapudi SP, Li Y, Disney MD. A cross-linking approach to map small molecule–RNA binding sites in cells. Bioorg. Med. Chem. Lett. 2019;29:1532–1536. doi: 10.1016/j.bmcl.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 276.Davila-Calderon J, et al. IRES-targeting small molecule inhibits enterovirus 71 replication via allosteric stabilization of a ternary complex. Nat. Commun. 2020;11:4775. doi: 10.1038/s41467-020-18594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Liu T, Pyle AM. Discovery of highly reactive self-splicing group II introns within the mitochondrial genomes of human pathogenic fungi. Nucleic Acids Res. 2021;49:12422–12432. doi: 10.1093/nar/gkab1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Angelbello AJ, et al. Precise small- molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl Acad. Sci. USA. 2019;116:7799–7804. doi: 10.1073/pnas.1901484116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Donahue CP, Ni J, Rozners E, Glicksman MA, Wolfe MS. Identification of tau stem loop RNA stabilizers. J. Biomol. Screen. 2007;12:789–799. doi: 10.1177/1087057107302676. [DOI] [PubMed] [Google Scholar]
- 280.Angelbello AJ, et al. A small molecule that binds an RNA repeat expansion stimulates its decay via the exosome complex. Cell Chem. Biol. 2021;28:34–45. doi: 10.1016/j.chembiol.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Murchie AI, et al. Structure-based drug design targeting an inactive RNA conformation: exploiting the flexibility of HIV-1 TAR RNA. J. Mol. Biol. 2004;336:625–638. doi: 10.1016/j.jmb.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 282.Foloppe N, et al. A structure-based strategy to identify new molecular scaffolds targeting the bacterial ribosomal A-site. Bioorg. Med. Chem. 2004;12:935–947. doi: 10.1016/j.bmc.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 283.Morgan BS, Forte JE, Culver RN, Zhang Y, Hargrove AE. Discovery of key physicochemical, structural, and spatial properties of RNA-targeted bioactive ligands. Angew. Chem. Int. Ed. Engl. 2017;56:13498–13502. doi: 10.1002/anie.201707641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Childs-Disney JL, et al. A massively parallel selection of small molecule–RNA motif binding partners informs design of an antiviral from sequence. Chem. 2018;4:2384–2404. doi: 10.1016/j.chempr.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat. Chem. 2012;4:90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Bhullar KS, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol. Cancer. 2018;17:48. doi: 10.1186/s12943-018-0804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Paulson H. Repeat expansion diseases. Handb. Clin. Neurol. 2018;147:105–123. doi: 10.1016/B978-0-444-63233-3.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Bernat V, Disney MD. RNA structures as mediators of neurological diseases and as drug targets. Neuron. 2015;87:28–46. doi: 10.1016/j.neuron.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.de Mezer M, Wojciechowska M, Napierala M, Sobczak K, Krzyzosiak WJ. Mutant CAG repeats of huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011;39:3852–3863. doi: 10.1093/nar/gkq1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Sobczak K, de Mezer M, Michlewski G, Krol J, Krzyzosiak WJ. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 2003;31:5469–5482. doi: 10.1093/nar/gkg766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Tian B, et al. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA. 2000;6:79–87. doi: 10.1017/S1355838200991544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Jog SP, et al. RNA splicing is responsive to MBNL1 dose. PLoS ONE. 2012;7:e48825. doi: 10.1371/journal.pone.0048825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Rzuczek SG, et al. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 2017;13:188–193. doi: 10.1038/nchembio.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Wagner-Griffin S, et al. A druglike small molecule that targets r(CCUG) repeats in myotonic dystrophy type 2 facilitates degradation by RNA quality control pathways. J. Med. Chem. 2021;64:8474–8485. doi: 10.1021/acs.jmedchem.1c00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Bush JA, et al. Ribonuclease recruitment using a small molecule reduced c9ALS/FTD r(G4C2) repeat expansion in vitro and in vivo ALS models. Sci. Transl. Med. 2021;13:eabd5991. doi: 10.1126/scitranslmed.abd5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Angelbello AJ, Disney MD. A toxic RNA templates the synthesis of its own fluorogenic inhibitor by using a bio-orthogonal tetrazine ligation in cells and tissues. ACS Chem. Biol. 2020;15:1820–1825. doi: 10.1021/acschembio.0c00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 298.Li Y, Disney MD. Precise small molecule degradation of a noncoding RNA identifies cellular binding sites and modulates an oncogenic phenotype. ACS Chem. Biol. 2018;13:3065–3071. doi: 10.1021/acschembio.8b00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Liu X, et al. Targeted degradation of the oncogenic microRNA 17-92 cluster by structure-targeting ligands. J. Am. Chem. Soc. 2020;142:6970–6982. doi: 10.1021/jacs.9b13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Benhamou RI, et al. Structure-specific cleavage of an RNA repeat expansion with a dimeric small molecule is advantageous over sequence-specific recognition by an oligonucleotide. ACS Chem. Biol. 2020;15:485–493. doi: 10.1021/acschembio.9b00958. [DOI] [PMC free article] [PubMed] [Google Scholar]