

Abstract
Objectives: Joubert syndrome is a spectrum of rare genetic disorders, mainly characterized by a distinctive cerebellar and brain stem malformation called the “molar tooth sign” (MTS), hypotonia, and intellectual disability/developmental delay. Methods: In this study, 4 pediatric cases with developmental delay and oculomotor abnormities were recruited, and submitted to a clinical evaluation and magnetic resonance imaging (MRI) examination. Afterwards, genetic detection with whole exome sequencing (WES) was conducted on the 4 patients. Results: Imaging results demonstrated cerebellar dysplasia in all probands, yet the MTS findings varied in severity. WES detected diagnostic variations in all four probands, which were distributed in four genes, namely CC2D2A, NPHP1, AHI1, and C5orf42. Two variants were novelly identified, which were the CC2D2A: c.2444delC (p.P815fs*2) and the AIH1: exon (15-17) del. In silico analysis supported the pathogenicity of the variations in this study. Conclusions: Our findings expanded the mutation spectrum of Joubert syndrome related disorders, and provided solid evidence to the affected families for further genetic counseling and pregnancy guidance.
Keywords: Joubert syndrome, CC2D2A, NPHP1, AHI1, C5orf42
Introduction
Joubert syndrome (JBTS, MIM #213300), representing a spectrum of rare congenital conditions, was first described by Marie Joubert in 1969 [1] and is characterized mainly by hypotonia, intellectual disability/developmental delay, and a distinctive cerebellar and brain stem malformation called the “molar tooth sign” (MTS) [2]. Other additional features in JBTS patients consist of abnormal respiratory pattern and oculomotor apraxia [3]. In general, the breathing abnormalities improve with age, truncal ataxia develops over time, and acquisition of gross motor milestones is delayed; cognitive abilities are variable, ranging from severe intellectual disability to normal [2,4]. Some JBTS patients may have multisystem organ involvement including retinal dystrophy, ocular colobomas, occipital encephalocele, renal abnormity, hepatic fibrosis, oral hamartomas, polydactyly, and endocrine abnormalities [4,5]. The term “Joubert syndrome and related disorders” (JSRD) was extensively used in recent years because of the strong clinical heterogeneity of these conditions [2].
Estimatedly, the incidence of JBTS ranges between 1/80,000 and 1/100,000 in live births [2]. So far, over 40 genes have been recognized to be responsible for JBTS, in which the vast majority conform to the autosomal recessive inheritance pattern, except for one (the OFD1 gene) which is X-linked [4,6,7]. All of the gene products localize in and around the primary cilium, rendering JBTS a canonical ciliopathy [6]. In about 62%-97% of individuals with clinical manifestations of JSRD, a molecular diagnosis can be established via analyzing these genes, depending on specific detection efficiency in various studies [4,8,9].
These pleiotropic features are typical of a number of disorders of the primary cilium, and make the identification of causative genes challenging given the significant overlap between JBTS and other ciliopathy conditions such as nephronophthisis and Meckel, Bardet-Biedl, and COACH syndromes [3]. Encountering such situations, molecular detection with next generation sequencing has shown robust capacity in the diagnosis of JSRD [7], while clinical and imaging profiling is still of importance to the analysis of subtle differences in phenotypes and their causes [10].
Here in the present study, four young patients with typical clinical and imaging indications of JSRD were recruited in the outpatient department of our center. A comprehensive clinical evaluation and genetic detection using whole exome sequencing (WES) was conducted to explore the causative variations.
Material and methods
This study was approved by the Ethics Committee of Shijiazhuang Obstetrics and Gynecology Hospital (approval No. 20210068), and written informed consent was obtained from all parents of the participants.
Subjects
Between Jun 2016 and Aug 2020, four young children with mental, motor developmental delay of unknown origin were referred to our center with their parents. We carried out a thorough clinical survey including cerebral magnetic resonance imaging (MRI). Subsequently, the peripheral blood samples of all four pedigrees were collected for the following genetic detection.
Genomic DNA extraction
Peripheral blood was collected from the patients and their parents. Genomic DNA was extracted using the QIAamp DNA Blood MiniKit (Qiagen Sciences, USA), and the DNA quality was validated by 1% agarose gels and Qbit DNA Assay Kit in Qubit 2.0 Flurometer (Life Technologies, CA, USA).
Whole-exome sequencing (WES)
WES was carried our as previously described [11]. Briefly, the enrichment of the exonic sequences was conducted by the Sure Select Human Exon Sequence Capture Kit (Agilent, USA). The sequencing libraries were quantified using the Illumina DNA Standards and Primer Premix Kit (Kapa Biosystems, USA), and were massively parallel-sequenced using the Illumina Novaseq6000 platform. After sequencing and filtering out the low-quality readings, the high-quality reads (with quality level Q30>89%) were compared to the human genome reference sequence [hg19]. The GATK software was used to identify suspected pathogenic variants (https://software.broadinstitute.org/gatk/). The CNV variants were called by a routine protocol (https://cnvkit.readthedocs.io/en/stable/pipeline.html). The variations were identified by sequence alignment with the NCBI Reference Sequence using Chromas v2.33. The pathogenicity of the identified variants was then assessed according to the common guidelines issued by the American Association of Medical Genetics and Genomics (ACMG) [12] referring to multiple databases (1000g2015aug_eas, https://www.internationalgenome.org/; ExAC_EAS, http://exac.broadinstitute.org; gnomAD_exome_EAS, http://gnomad.broadinstitute.org/); HGMD®: Human Gene Mutation Database (Professional Version 2019.4) with the Enliven® Variants Annotation Interpretation system (Berry Genomics, China).
Validation by Sanger sequencing and fluorescent quantitative PCR (qPCR)
The suspected diagnostic variant was validated by Sanger sequencing using ABI 3730 Automated Sequencer (Applied Biosystems, USA) according to the manufacturer’s protocol. qPCR was also carried out to verify the deletion variants identified in Case 2 and 3 (detailed methods in Supplementary Material 1).
Analysis of missense variants
The evolutionary conservatism of amino acids (AAs) residue affected by specific missense variants was analyzed using MEGA7 (http://www.megasoftware.net) with default parameters.
Results
Clinical manifestations
Patient 1, a boy first born to his family, was referred to our center due to a delayed milestone at 7 months (hypotonia; could not sit alone; poor head control). The general physical examination result was normal, except for a slight strabismus. Plasma biochemical test, screening for markers of genetic metabolic disease (GMD) and EEG showed no abnormalities. Cerebral MRI indicated that the vermis of cerebellum was smaller, the middle cerebellum fissure was deeper, and the extracerebral space of bilateral frontal part was wider (Figure 1A-C). Follow-up showed that the patient had learned to walk at 15 months, had normal gross motor and language development, had fair fine hand movements, and had the same social and adaptive abilities as children of the same age.
Figure 1.
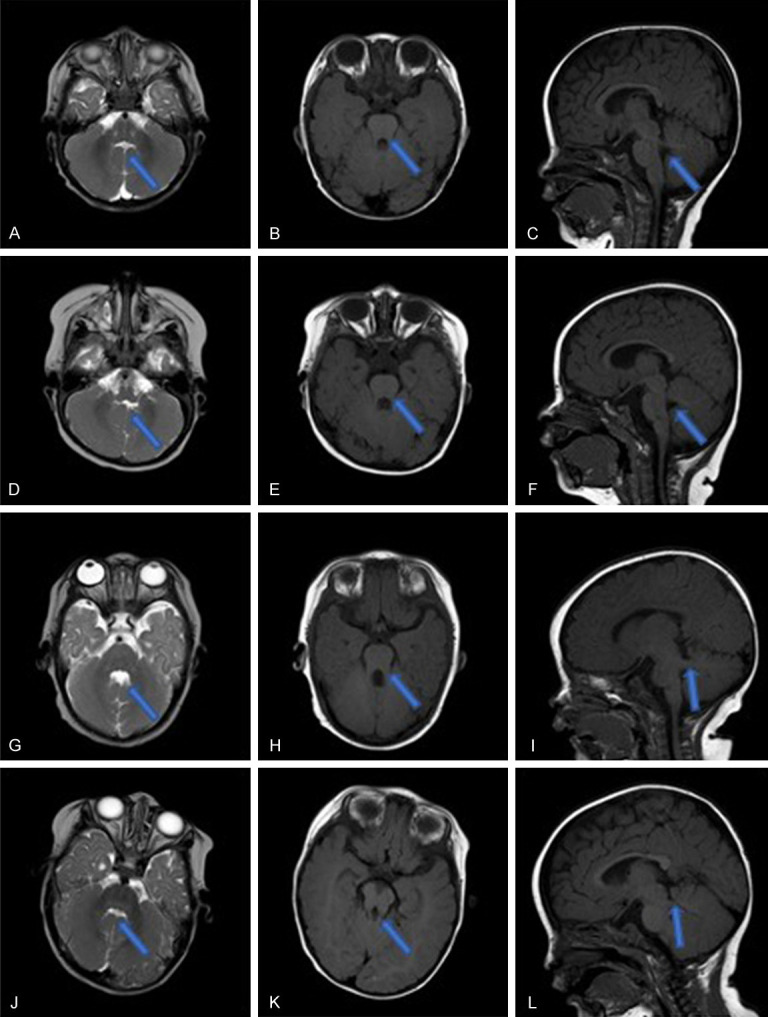
MRI results of the 4 patients. A-C. Patient 1. A. T2WI coronal plane: line-like cerebrospinal fluid between the bilateral cerebellar hemispheres with long T2 signal “midline fissure sign”, the upper part of the fourth ventricle “bat wing”. B. T1WI coronal plane: the midbrain and the enlarged upper cerebellar peduncle form a “grinding sign”. C. T1WI sagittal plane: absence of inferior cerebellar vermis. D-F. Patient 2. D. T2WI coronal plane: line-like cerebrospinal fluid between the bilateral cerebellar hemispheres with long T2 signal “midline fissure sign”, the upper part of the fourth ventricle “bat wing”. E. T1WI coronal plane: the midbrain and the enlarged upper cerebellar peduncle form a “grinding sign”. F. T1WI sagittal plane: absence of inferior cerebellar vermis. G-I. Patient 3. G. T2WI coronal plane: the upper part of the fourth ventricle is “bat wing-shaped”, and the middle part expands into a triangle. H. T1WI coronal plane: the midbrain and the enlarged upper cerebellar peduncle form a “grinding sign”. I. T1WI sagittal plane: the enlarged upper cerebellar peduncle is almost perpendicular to the midbrain. J-L. Patient 4. J. T2WI coronal plane: line-like cerebrospinal fluid between the bilateral cerebellar hemispheres with long T2 signal “midline fissure sign”, the upper part of the fourth ventricle “bat wing”. K. T1WI coronal plane: the midbrain and the enlarged upper cerebellar peduncle form a “grinding sign”. L. T1WI sagittal plane: the enlarged upper cerebellar peduncle is almost perpendicular to the midbrain.
Patient 2, male, the second child in his family, was referred to our outpatient department due to hypotonia and motor retardation at 8 months. The general physical characterization was normal, yet his eyeballs were slightly cohesive. Biochemical, GMD, EEG results were all normal. MRI result generally suggested that the patient had cerebellar dysplasia (Figure 1D-F). Follow-up found that the boy learned to walk at 14 months, his motor and language development was fair, yet the ocular cohesiveness still existed.
Patient 3, male, was referred to our outpatient department due to hypotonia, with poor visual fixation and chasing ability at 2+ months. The general physical characterization was normal, except for the eye movements. Biochemical, GMD, EEG results were all normal. Follow-up found that the patient could only walk 5-6 meters independently, and only speak simple geminate words at 2.5 years old. MRI revealed the cerebellar dysplasia (Figure 1G-I).
Patient 4, male, referred to our outpatient department due to abnormal eye movement at 1 month. The general physical characterization was normal, except that his eyes squinted to one side when he looked at things. Biochemical, GMD, EEG results were all normal. Follow-up to the age of 3 revealed that strabismus was always present, and his motor and language development was slightly behind that of his peers. MRI revealed the cerebellar dysplasia (Figure 1J-L).
Genetic variations
The pedigree diagrams of the enrolled families were depicted in Figure 2 (A for Family 1; D for Family 2; F for Family 3; I for Family 4). Overall, all 4 probands had positive genetic results. Four variations composed by 7 variants were detected, confirming to an autosomal recessive inheritance pattern in all 4 patients. The details of all variants, including the ACMG pathogenic level, are demonstrated in Table 1.
Figure 2.

Genetic variants in the 4 patients. A-C. Patient 1. A. The pedigree diagram; B. Sanger sequencing result for CC2D2A: c.2444delC heterozygous variant; C. Sanger sequencing result for CC2D2A: c.4238G>A heterozygous variant. D, E. Patient 2. D. The pedigree diagram; E. The homozygous deletion of NPHP1 exon (1-20) by the schematic of capture efficiency. F-H. Patient 3. F. The pedigree diagram; G. Sanger sequencing result for AHI1: c.910dupA heterozygous variant; H. The heterozygous deletion of AHI1 exon (15-17) by the schematic of capture efficiency. I-K. Patient 4. I. The pedigree diagram; J. Sanger sequencing result for C5orf42: c.3551G>A heterozygous variant; K. Sanger sequencing result for C5orf42: c.4006C>T heterozygous variant.
Table 1.
Information of the variants identified in this study
| Patient No. | Variant No. | Gene (transcript) | Genomic alteration | Protein alteration | Frequencies in 3 databases* | Revel score* | HGMD* level (PMID*) | ACMG* level (evidence) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | CC2D2A (NM_001080522) | c.2444delC | p.P815fs*2 | 0; 0; 0 | / | / | Likely pathogenic (PVS1+PM2) |
| 2 | c.4238G>A | p.C1413Y | 0.002; 0.004406; 0.004347 | 0.306 | DM (27491411) | Likely pathogenic (PM3_Strong+PM3+BP4) | ||
| 2 | 3 | NPHP1 | del exon (1-20) | null | / | / | DM (multi studies) | Likely pathogenic (PVS1+PM2) |
| 3 | 4 | AHI1 (NM_017651) | c.910dupA | p.T304Nfs*5 | 0; 0; 0 | / | DM (16453322) | Pathogenic (PVS+PS1+PM2) |
| 5 | del exon (15-17) | uncertain | / | / | / | Pathogenic (PVS1+PM2+PM3) | ||
| 4 | 6 | C5orf42 (NM_023073) | c.4006C>T | p.R1336W | 0; 0; 0 | 0.273 | DM (22425360) | Likely pathogenic (PM2+PM3) |
| 7 | c.3551G>A | p.R1184H | 0; 0; 0 | 0.797 | DM (25407461) | Likely pathogenic (PM2+PM3+PM5+PP3) |
1000g2015aug_eas (https://www.internationalgenome.org/); ExAC_EAS (http://exac.broadinstitute.org); gnomAD_exome_EAS (http://gnomad.broadinstitute.org/); Revel: An ensemble method for predicting the pathogenicity of missense variants on the basis of individual tools: MutPred, FATHMM, VEST, PolyPhen, SIFT, PROVEAN, MutationAssessor, MutationTaster, LRT, GERP, SiPhy, phyloP, and phastCons (http://dx.doi.org/10.1016/j.ajhg.2016.08.016); HGMD®: Human Gene Mutation Database (Professional Version 2019.4); PMID: PubMed ID (https://pubmed.ncbi.nlm.nih.gov/); ACMG: The American College of Medical Genetics and Genomics; P: pathogenic; LP: likely pathogenic; VUS: variants of unknown significance.
To be specific, Patient 1 carried a compound heterozygous variation in the CC2D2A gene consisting of 2 variants, c.2444delC (p.P815fs*2) and c.4238G>A (p.C1413Y) (NM_001080522) (Figure 2B, 2C); Patient 2 harbored a homozygous deletion in the NPHP1 gene (exon 1-20) (Figure 2E); Patient 3 had a compound heterozygous variation in AHI1 gene consisting of c.910dupA (p.T304Nfs*5) (NM_017651) and AIH1: exon (15-17) del (Chr6: 135751004-135754394) (Figure 2G, 2H); while Patient 4 had a compound heterozygous variation in the C5orf42 gene consisting of 2 variants, c.4006C>T (p.R1336W) and c.3551G>A (p.R1184H) (NM_023073) (Figure 2J, 2K). Two variants were novelly identified in this study, namely the CC2D2A: c.2444delC (p.P815fs*2) and the AIH1: exon (15-17) del.
Validation with Sanger sequencing and qPCR demonstrated that the variants these patient carried were all inherited from their asymptomatic heterozygous carrier parents, as demonstrated in pedigree diagrams. The detailed results of qPCR validation were included in Supplementary Material 1.
Conservatism analysis of missense variants
In this study, three missense variants were detected, which were CC2D2A: c.4238G>A (p.C1413Y), C5orf42: c.4006C>T (p.R1336W) and C5orf42: c.3551G>A (p.R1184H). We analyzed the AAs they affected. Results indicated that all 3 AAs maintained evolutionary conservatism among species (Figure 3).
Figure 3.
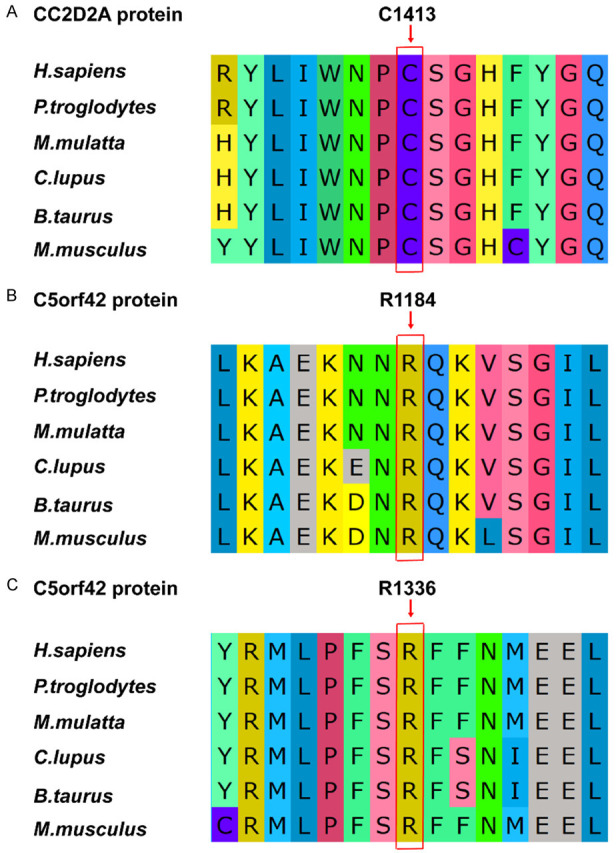
Evolutionary conservatism of 3 amino acids affected by the missense variants in this study. A. The C1413 residue of CC2D2A protein. B. The R1184 residue of C5orf42 protein. C. The R1336 residue of C5orf42 protein.
Discussion
Early clinical differential diagnosis of Joubert syndrome (JBTS) requires profound pediatric experience, as JBTS has overlapping phenotypes with many primary cilium disorders, such as nephronophthisis, Meckel, Bardet-Biedl, and COACH syndromes [13-17]. In this study, 4 patients were all younger than 12 months old with the preliminary and developing indications, which added to the difficulty of diagnosis. Furthermore, the core appearance indication of all 4 patients was oculomotor abnormity. Their MRI results showed typical cerebellar dysplasia, but the MTS varied. This may be because the MTS finding is itself a spectrum and can have milder types [18], such as those caused by mutations in the C5orf42 [19], which is consistent with the situation in our study (Case 4). In other cases, owing to the lack of examination on multiple MRI cuts for subtle findings of MTS and vermis hypoplasia, even experienced neuroradiologists may miss this hallmark [4].
Since the first gene for JSRD, NPHP1, was identified in 2004 [18], over 40 genes have been implicated in its causation accounting for 65%-75% cases [3,4,7]. All of the genes identified so far are localized to or play a role in the function of the sub-cellular structure, the primary cilium, especially at the transition zone of the cilium where it joins the plasma membrane [20]. In our study, 4 patients were positive with diagnostic variations in different genes, reflecting the strong genetic heterogeneity of JBTS. Patient 1 had a compound heterozygous variation in CC2D2A, which encodes a component of a protein complex in the basal body, a ring-like structure that functions in the transition zone at the base of cilia and acts as a barrier to restrict protein diffusion between the plasma and ciliary membranes [21]. In this variation, the c.4238G>A (p.C1413Y) variant was reported to be associated with nephronophthisis-related ciliopathy [22], and indexed as “disease causing” in HGMD. The patient described by Kang et al. did not have extra-renal phenotype [22], which was quite different from Patient 1 in this study. This phenotypic difference was probably owing to the novel variant, c.2444delC (p.P815fs*2), on the other allele. As for Patient 2, the homozygous NPHP1 deletion he carried was considered as the most frequent genetic defect for nephronophthisis, particularly with neurologic involvement [23]. Therefore, attention should be paid to the renal manifestations in cases like Patient 2 in future management. Like CC2D2A, AHI is also a component of the protein complex in the basal body, a ring-like structure that functions in the transition zone at the base of cilia [21]. The variation caused the symptoms of Patient 3 consisted of a sequence variant and an exonic deletion, which were inherited from the parents, respectively. The AHI: c.910dupA (p.T304Nfs*5) variant was reported to cause JBTS [24]; in terms of the micro deletion containing exon 15-17, although it has not been identified before, another ~3.4 kb deletion covering the exon 14-16 of AHI1 was reported to cause JBTS [25], which supports the pathogenicity of the variant in the present study. The 2 variants detected in Patient 4, c.4006C>T (p.R1336W) and c.3551G>A (p.R1184H) in C5orf42 gene, have both been reported, yet with different conditions. The c.4006C>T variant was identified in a French Canadian case of JBTS [26], while the c.3551G>A was associated with a much rarer JBTS subtype, the oral-facial-digital type VI syndrome, characterized by preaxial and mesoaxial polydactyly, hypothalamic hamartoma and other congenital defects along with neurological indications [27]. Using a mouse model, Damerla et al. found that C5orf42, which they called Jbts17, colocalized with Nphp1 in the ciliary transition zone [28]. In C5orf42-knocked-down Xenopus embryos, Toriyama et al. observed ciliopathy-related developmental defects, including failure of neural tube closure, defective Hedgehog signaling, and defective left-right patterning [29]. Moreover, Hong et al. found that human C5orf42 was required for cilium assembly in ciliated cells [30].
Since in all cases, the genes involved in the diagnostic variations fit an autosomal recessive inheritance pattern, those couples still have a 25% risk of having a future pregnancy affected. Therefore, more preventive managements, such as prenatal diagnosis or pre-implantation diagnosis, should be recommended [5,31]. The main limitation of this study was the small sample size included, which made it impossible to trace the association between genotype and phenotype. In addition, necessary functional experiments or biophysical analyses may help to clarify the impact of specific variants.
In conclusion, we identified the causative variations, which were distributed in various genes including CC2D2A, NPHP1, AHI1, and C5orf42, in 4 cases of Joubert syndrome related disorders. Our findings not only extended the spectrum of JSRD mutations, but also highlighted the capability of WES to identify different types of variants.
Acknowledgements
We acknowledge all participants for their support and cooperation in this study. The work was supported by the Key Research and Development Program of Hebei Province (No. 21377720D and No. 182777181).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–825. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- 2.Brancati F, Dallapiccola B, Valente EM. Joubert syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20. doi: 10.1186/1750-1172-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parisi MA. The molecular genetics of Joubert syndrome and related ciliopathies: the challenges of genetic and phenotypic heterogeneity. Transl Sci Rare Dis. 2019;4:25–49. doi: 10.3233/TRD-190041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parisi M, Glass I. Joubert syndrome. 2003 Jul 9 [Updated 2017 Jun 29] In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 1993-2021. [PubMed] [Google Scholar]
- 5.Vilboux T, Doherty DA, Glass IA, Parisi MA, Phelps IG, Cullinane AR, Zein W, Brooks BP, Heller T, Soldatos A, Oden NL, Yildirimli D, Vemulapalli M, Mullikin JC Nisc Comparative Sequencing Program. Malicdan MCV, Gahl WA, Gunay-Aygun M. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med. 2017;19:875–882. doi: 10.1038/gim.2016.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van De Weghe JC, Rusterholz TDS, Latour B, Grout ME, Aldinger KA, Shaheen R, Dempsey JC, Maddirevula S, Cheng YH, Phelps IG, Gesemann M, Goel H, Birk OS, Alanzi T, Rawashdeh R, Khan AO University of Washington Center for Mendelian Genomics. Bamshad MJ, Nickerson DA, Neuhauss SCF, Dobyns WB, Alkuraya FS, Roepman R, Bachmann-Gagescu R, Doherty D. Mutations in ARMC9, which encodes a basal body protein, cause Joubert syndrome in humans and ciliopathy phenotypes in zebrafish. Am J Hum Genet. 2017;101:23–36. doi: 10.1016/j.ajhg.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Abrusco F, Arrigoni F, Serpieri V, Romaniello R, Caputi C, Manti F, Jocic-Jakubi B, Lucarelli E, Panzeri E, Bonaglia MC, Chiapparini L, Pichiecchio A, Pinelli L, Righini A, Leuzzi V, Borgatti R, Valente EM. Get your molar tooth right: Joubert syndrome misdiagnosis unmasked by whole-exome sequencing. Cerebellum. 2021 doi: 10.1007/s12311-021-01350-8. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 8.Bachmann-Gagescu R, Dempsey JC, Phelps IG, O’Roak BJ, Knutzen DM, Rue TC, Ishak GE, Isabella CR, Gorden N, Adkins J, Boyle EA, de Lacy N, O’Day D, Alswaid A, Ramadevi A R, Lingappa L, Lourenço C, Martorell L, Garcia-Cazorla À, Ozyürek H, Haliloğlu G, Tuysuz B, Topçu M University of Washington Center for Mendelian Genomics. Chance P, Parisi MA, Glass IA, Shendure J, Doherty D. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015;52:514–522. doi: 10.1136/jmedgenet-2015-103087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaheen R, Szymanska K, Basu B, Patel N, Ewida N, Faqeih E, Al Hashem A, Derar N, Alsharif H, Aldahmesh MA, Alazami AM, Hashem M, Ibrahim N, Abdulwahab FM, Sonbul R, Alkuraya H, Alnemer M, Al Tala S, Al-Husain M, Morsy H, Seidahmed MZ, Meriki N, Al-Owain M, AlShahwan S, Tabarki B, Salih MA Ciliopathy WorkingGroup. Faquih T, El-Kalioby M, Ueffing M, Boldt K, Logan CV, Parry DA, Al Tassan N, Monies D, Megarbane A, Abouelhoda M, Halees A, Johnson CA, Alkuraya FS. Characterizing the morbid genome of ciliopathies. Genome Biol. 2016;17:242. doi: 10.1186/s13059-016-1099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Surisetti BK, Holla VV, Prasad S, Neeraja K, Kamble N, Yadav R, Pal PK. Clinical and imaging profile of patients with Joubert syndrome. J Mov Disord. 2021;14:231–235. doi: 10.14802/jmd.21066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Hu H, Mu W, Yu M, Chen W, Mi D, Yang K, Guo Q. Case report: exome sequencing identified a novel compound heterozygous variation in PLOD2 causing bruck syndrome type 2. Front Genet. 2021;12:619948. doi: 10.3389/fgene.2021.619948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchison HM, Valente EM. Motile and non-motile cilia in human pathology: from function to phenotypes. J Pathol. 2017;241:294–309. doi: 10.1002/path.4843. [DOI] [PubMed] [Google Scholar]
- 14.Srivastava S, Molinari E, Raman S, Sayer JA. Many genes-one disease? Genetics of nephronophthisis (NPHP) and NPHP-associated disorders. Front Pediatr. 2017;5:287. doi: 10.3389/fped.2017.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartill V, Szymanska K, Sharif SM, Wheway G, Johnson CA. Meckel-gruber syndrome: an update on diagnosis, clinical management, and research advances. Front Pediatr. 2017;5:244. doi: 10.3389/fped.2017.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, Clericuzio C, Demir H, Dorschner M, van Essen AJ, Gahl WA, Gentile M, Gorden NT, Hikida A, Knutzen D, Ozyurek H, Phelps I, Rosenthal P, Verloes A, Weigand H, Chance PF, Dobyns WB, Glass IA. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis) J Med Genet. 2010;47:8–21. doi: 10.1136/jmg.2009.067249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsyth R, Gunay-Aygun M. Bardet-Biedl syndrome overview. 2003 Jul 14 [Updated 2020 Jul 23] In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 1993-2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1363/ [Google Scholar]
- 18.Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DW, McDonald R, Eddy A, Chance PF, Glass IA. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75:82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enokizono M, Aida N, Niwa T, Osaka H, Naruto T, Kurosawa K, Ohba C, Suzuki T, Saitsu H, Goto T, Matsumoto N. Neuroimaging findings in Joubert syndrome with C5orf42 gene mutations: a milder form of molar tooth sign and vermian hypoplasia. J Neurol Sci. 2017;376:7–12. doi: 10.1016/j.jns.2017.02.065. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Gonzalo FR, Reiter JF. Open sesame: how transition fibers and the transition zone control ciliary composition. Cold Spring Harb Perspect Biol. 2017;9:a028134. doi: 10.1101/cshperspect.a028134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2011;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- 22.Kang HG, Lee HK, Ahn YH, Joung JG, Nam J, Kim NK, Ko JM, Cho MH, Shin JI, Kim J, Park HW, Park YS, Ha IS, Chung WY, Lee DY, Kim SY, Park WY, Cheong HI. Targeted exome sequencing resolves allelic and the genetic heterogeneity in the genetic diagnosis of nephronophthisis-related ciliopathy. Exp Mol Med. 2016;48:e251. doi: 10.1038/emm.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konig J, Kranz B, Konig S, Schlingmann KP, Titieni A, Tonshoff B, Habbig S, Pape L, Haffner K, Hansen M, Buscher A, Bald M, Billing H, Schild R, Walden U, Hampel T, Staude H, Riedl M, Gretz N, Lablans M, Bergmann C, Hildebrandt F, Omran H, Konrad M Gesellschaft für Pädiatrische Nephrologie (GPN) Phenotypic spectrum of children with nephronophthisis and related ciliopathies. Clin J Am Soc Nephrol. 2017;12:1974–1983. doi: 10.2215/CJN.01280217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valente EM, Brancati F, Silhavy JL, Castori M, Marsh SE, Barrano G, Bertini E, Boltshauser E, Zaki MS, Abdel-Aleem A, Abdel-Salam GM, Bellacchio E, Battini R, Cruse RP, Dobyns WB, Krishnamoorthy KS, Lagier-Tourenne C, Magee A, Pascual-Castroviejo I, Salpietro CD, Sarco D, Dallapiccola B, Gleeson JG. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol. 2006;59:527–534. doi: 10.1002/ana.20749. [DOI] [PubMed] [Google Scholar]
- 25.Watson CM, Crinnion LA, Berry IR, Harrison SM, Lascelles C, Antanaviciute A, Charlton RS, Dobbie A, Carr IM, Bonthron DT. Enhanced diagnostic yield in Meckel-Gruber and Joubert syndrome through exome sequencing supplemented with split-read mapping. BMC Med Genet. 2016;17:1. doi: 10.1186/s12881-015-0265-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srour M, Schwartzentruber J, Hamdan FF, Ospina LH, Patry L, Labuda D, Massicotte C, Dobrzeniecka S, Capo-Chichi JM, Papillon-Cavanagh S, Samuels ME, Boycott KM, Shevell MI, Laframboise R, Désilets V, Maranda B, Rouleau GA, Majewski J, Michaud JL. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am J Hum Genet. 2012;90:693–700. doi: 10.1016/j.ajhg.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romani M, Mancini F, Micalizzi A, Poretti A, Miccinilli E, Accorsi P, Avola E, Bertini E, Borgatti R, Romaniello R, Ceylaner S, Coppola G, D’Arrigo S, Giordano L, Janecke AR, Lituania M, Ludwig K, Martorell L, Mazza T, Odent S, Pinelli L, Poo P, Santucci M, Signorini S, Simonati A, Spiegel R, Stanzial F, Steinlin M, Tabarki B, Wolf NI, Zibordi F, Boltshauser E, Valente EM. Oral-facial-digital syndrome type VI: is C5orf42 really the major gene? Hum Genet. 2015;134:123–126. doi: 10.1007/s00439-014-1508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damerla RR, Cui C, Gabriel GC, Liu X, Craige B, Gibbs BC, Francis R, Li Y, Chatterjee B, San Agustin JT, Eguether T, Subramanian R, Witman GB, Michaud JL, Pazour GJ, Lo CW. Novel Jbts17 mutant mouse model of Joubert syndrome with cilia transition zone defects and cerebellar and other ciliopathy related anomalies. Hum Mol Genet. 2015;24:3994–4005. doi: 10.1093/hmg/ddv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel AL, Tabler JM, Drew K, Kelly MR, Kim S, Park TJ, Braun DA, Pierquin G, Biver A, Wagner K, Malfroot A, Panigrahi I, Franco B, Al-Lami HA, Yeung Y, Choi YJ, Duffourd Y, Faivre L, Rivière JB, Chen J, Liu KJ, Marcotte EM, Hildebrandt F, Thauvin-Robinet C, Krakow D, Jackson PK, Wallingford JB. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat Genet. 2016;48:648–656. doi: 10.1038/ng.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong H, Joo K, Park SM, Seo J, Kim MH, Shin E, Cheong HI, Lee JH, Kim J. Extraciliary roles of the ciliopathy protein JBTS17 in mitosis and neurogenesis. Ann Neurol. 2019;86:99–115. doi: 10.1002/ana.25491. [DOI] [PubMed] [Google Scholar]
- 31.Haratz KK, Lerman-Sagie T. Prenatal diagnosis of brainstem anomalies. Eur J Paediatr Neurol. 2018;22:1016–1026. doi: 10.1016/j.ejpn.2018.06.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.