

Key Points
Question
What genetic variants or genes are associated with the risk of childhood Hodgkin lymphoma?
Findings
In this genetic association study nested within 3 childhood cancer survivor cohorts, 3 independent single-nucleotide variants (SNVs, formerly single-nucleotide polymorphisms) in the human leukocyte antigen (HLA) locus and 5 loci in non-HLA regions were found to be significantly associated with risk of childhood-onset Hodgkin lymphoma. Two-thirds of previously reported Hodgkin lymphoma–associated non-HLA SNVs were replicated in the analysis.
Meaning
These findings suggest predominantly common and potentially unique genetic etiology between childhood-onset and adulthood-onset Hodgkin lymphoma.
This genetic association study evaluates the association of single-nucleotide variants in the human leukocyte antigen and other loci with childhood-onset Hodgkin lymphoma.
Abstract
Importance
Studies focusing on genetic susceptibility of childhood Hodgkin lymphoma (HL) are limited.
Objectives
To identify genetic variants associated with childhood-onset HL vs adult-onset HL.
Design, Setting, and Participants
This genetic association study was performed with 3 cohorts: the St Jude Lifetime Cohort Study (SJLIFE), initiated in 2007 with ongoing follow-up, and the original and expansion cohorts of the Childhood Cancer Survivor Study (CCSS), initiated in the 1990s with ongoing follow-up. Results of these genome-wide association studies (GWASs) were combined via meta-analysis. Data were analyzed from June 2021 to June 2022.
Main Outcomes and Measures
Childhood HL was the focused outcome. Single-nucleotide variant (SNV, formerly single-nucleotide polymorphism) array genotyping and imputation were conducted for the CCSS original cohort, and whole-genome sequencing was performed for the SJLIFE and CCSS expansion cohort.
Results
A total of 1286 HL cases (mean diagnosis [SD] age, 14.6 [3.9] years), 6193 non-HL childhood cancer cases, and 369 noncancer controls, all of European ancestry, were included in the analysis. Using step-wise conditional logistic regression, the odds ratios (ORs) for each of the 3 independent SNVs identified in the human leukocyte antigen (HLA) locus were 1.80 (95% CI, 1.59-2.03; P = 2.14 × 10−21) for rs28383311, 1.53 (95% CI, 1.37-1.70; P = 2.05 × 10−14) for rs3129198, and 1.51 (95% CI, 1.35-1.69; P = 6.21 × 10−13) for rs3129890. Further HLA imputation revealed 9 alleles and 55 amino acid changes that potentially conferred HL susceptibility. In addition, 5 non-HLA loci were identified: (1) rs1432297 (OR, 1.29; 95% CI, 1.18-1.41; P = 2.5 × 10−8; r2 = 0.55; D′ = 0.75 with previously reported rs1432295, REL); (2) rs2757647 (OR, 1.30; 95% CI, 1.18-1.42; P = 3.5 × 10−8; r2 = 0.59; D′ = 0.83 with previously reported rs6928977, AHI1); (3) rs13279159 (OR, 1.33; 95% CI, 1.20-1.47; P = 1.7 × 10−8; r2 = 0.75; D′ = 1.00 with previously reported rs2019960, PVT1); (4) rs3824662 (OR, 1.52; 95% CI, 1.33-1.73; P = 3.9 × 10−10; r2 = 0.91; D′ = 1.00 with previously reported rs3781093, GATA3); and (5) rs117953624 (OR, 1.98; 95% CI, 1.56-2.51; P = 1.5 × 10−8; minor allele frequency, 0.02), a novel uncommon SNV mapped to PDGFD. Twelve of 18 previously reported genome-wide significant non-HLA SNVs (67%) were replicated with statistically significant results.
Conclusions and Relevance
In this genetic association study, a predominantly common and potentially unique genetic etiology was found between childhood-onset and adulthood-onset HL.
Introduction
Hodgkin lymphoma (HL) originates from B lymphocytes and is characterized by the presence of Reed-Sternberg cells and their variants.1 The remarkably high cure rate of 87% is attributable to advances in therapeutics featuring multiagent chemotherapy with or without radiotherapy.2 HL is most frequently diagnosed among people aged 20 to 34 years (30.7%); children and young adults (<20 years) account for 12.2% of patients with HL.3 Despite its low incidence in children younger than 5 years, HL is the most common cancer among adolescents and young adults aged 15 to 19 years, and it has a favorable 5-year survival, exceeding 95%. However, late effects of chemotherapy and radiotherapy are common after the cure of childhood-onset HL, resulting in accelerated aging4 and excess late mortality.5,6,7
HL is classified into histological subtypes based on cellular composition and architecture; the 2 major subtypes are classical HL (approximately 95% of cases) and nodular lymphocyte predominant HL (approximately 5% of cases).8 Increased familial risk and high concordance between monozygotic twins provide evidence of genetic susceptibility to HL.9 In addition to genetic risk factors, environmental risk factors, such as Epstein-Barr virus (EBV) infection, are also causally related to specific HL subtypes.10
The most significant genetic association for HL is mapped to the human leukocyte antigen (HLA) region. HLA genes serve as the fundamental platform for immune surveillance and responsiveness to internal and external antigens and are implicated in a number of human diseases.11 HLA class I molecules present antigens primarily to cytotoxic T-cells to kill target cells, and HLA class II molecules stimulate antibody production in response to specific antigens. Thus, HLA genes are crucial for the development of autoimmune diseases, infectious diseases,12 and cancers. HL was the first human disease associated with HLA genes,13 and subsequent studies revealed that HLA alleles along with specific single-nucleotide variants (SNVs, formerly single-nucleotide polymorphisms; eg, rs6903608) are significantly associated with HL risk.14,15,16,17 In addition to the strong association between HLA and HL, genetic variants in non-HLA regions have been reported, including rs4459895 (LPP [OMIM 600700])18 and rs1432295 (REL [OMIM 164910]),19 among others.
Although the genetic etiology of HL has been investigated, studies among patients with childhood-onset HL are lacking. Research using cancer survivors to investigate cancer etiology is subject to potential survival bias, but the very favorable survival for pediatric HL makes this a reasonable population for consideration of such investigation. In a recent study, we demonstrated that germline pathogenic variants in BRCA2 are associated with risk of pediatric or adolescent non-HL.20 Here, we followed a similar paradigm to leverage genetic data of HL survivors to identify new genetic variants in HLA and non-HLA regions that are associated with the risk of childhood-onset HL.
Methods
Study Populations
Three large cohorts of survivors of childhood cancer were included in the current genetic association study.21,22,23 The St Jude Lifetime Cohort Study (SJLIFE) was approved by the St Jude institutional review board (IRB), and the Childhood Cancer Survivor Study (CCSS) was approved by the IRB of each participating center. This study is covered by the IRB approval for the 2 cohort studies. The SJLIFE is a retrospective cohort study initiated in 2007 with prospective clinical follow-up of 5-year survivors of childhood cancer treated at the St Jude Children’s Research Hospital. The CCSS is a retrospective cohort study of 5-year survivors of childhood cancer diagnosed and treated at 31 institutions in North America. The CCSS includes an original cohort diagnosed between 1970 and 1986 and an expansion cohort diagnosed between 1987 and 1999. In both studies, self-report or proxy-report questionnaires were used to assess demographic characteristics; clinical information was abstracted from medical records.
Genotype Data
SNV-array genotyping was conducted using the HumanOmni5M-4 version 1 (Illumina) for 5739 childhood cancer survivors in the CCSS original cohort, followed by genome-wide imputation using the Human Reference Consortium Panel as the reference on Michigan imputation server (dbGaP accession: phs001327.v2.p1). Whole-genome sequencing (WGS) with 30 folds was performed on DNA derived from peripheral blood samples from 4853 SJLIFE participants, including 4402 childhood cancer survivors and 451 noncancer controls,24,25 and buccal/saliva samples from 2998 survivors in the CCSS expansion cohort.20 The entire collection of WGS data for the SJLIFE and CCSS expansion cohorts is accessible at the St Jude Cloud.26 All participants providing biological material for DNA analysis provided written informed consent.
Quality Control
Of the 5739 survivors in the CCSS original cohort, 4671 remained after exclusion of 416 individuals of non-European ancestry whose admixture coefficient for 1000 Genomes CEU population was less than 80% and 652 who were also enrolled in SJLIFE.4 The final CCSS original cohort set consisted of 631 HL cases and 4040 non-HL cases (ie, survivors who had any childhood cancer diagnosis except HL) (eFigure 1 in the Supplement). Only variants (n = 8.4 million) with minor allele frequency (MAF) greater than 0.01, Hardy-Weinberg Equilibrium (HWE) P > 1 × 10−6, and having passed other filtering (ie, applying–keep-filtered = PASS setting in vcftools command) were retained for association analysis.
Among the 4402 SJLIFE survivors, 467 were diagnosed with HL. A total of 451 community controls with no history of childhood cancer were included and frequency matched to survivors by age, sex, and race. We subsequently excluded individuals (HL cases and controls) per the following criteria: (1) non-European ancestry with admixture coefficient for the 1000 Genomes CEU population less than 80% (86 case participants; 58 control participants); (2) incorrect or incomplete clinical characteristics (5 control participants); (3) individuals showing cryptic relatedness with another study participant (1 case participant; 19 control participants). Finally, a total of 380 HL cases and 369 controls remained for further analysis (eFigure 1 in the Supplement).
Of the 2998 survivors in the CCSS expansion cohort, 2428 were available for further analyses based on the following exclusions: (1) failure of the coverage and mapping quality control (50 individuals); (2) unexpected duplicates (12 individuals); (3) samples with excessive heterozygosity (97 individuals); and (4) individuals of non-European ancestry with admixture coefficient for 1000 Genomes CEU population less than 80% (411 individuals). Among the 2428 survivors, 275 were diagnosed with HL and considered cases; the 2153 survivors with other diagnoses (non-HL) were considered controls for comparison (eFigure 1 in the Supplement).
Additional quality control was performed to filter low-quality variants derived from WGS data, including the following criteria: (1) minimum genotype quality score of 20; (2) minimum depth of 5; (3) minimum mean depth of 10; (4) HWE P > 1 × 10−6; (5) maximum missing rate of 10% across all samples; (6) MAF greater than 0.01; and (7) exclusion of nonbiallelic loci. A total of 6.9 million SNVs and small insertions and deletions were in common with 8.4 million genotyped or imputed variants from the CCSS original cohort.
Statistical Analysis
Genome-wide association study (GWAS) analyses were performed in each of the 3 cohorts (SJLIFE, CCSS original, and CCSS expansion). A logistic regression model was analyzed for associations between HL cancer status and allelic dose of each variant (0, 1, or 2), with adjustments for sex and the top principal components derived from genotypes. Fixed-effects meta-analysis was used to combine the summary statistics from the 3 GWAS analyses. Heterogeneity between data sets was assessed using I2 as well as P for heterogeneity, calculated from the Cochran Q statistic. Variants with a meta-GWAS P < 5 × 10−8 were deemed genome-wide significant.
To refine associations between HL and HLA regions, step-wise conditional logistic regression was used to identify independent SNVs. First, the top (most significant) SNV from meta-GWAS was added to the list of covariates, and conditional association analysis was performed in each cohort. Then, results of the 3 conditional analyses were combined via meta-analysis to identify the next top SNV (independent secondary signal). This process was repeated until no SNV in the meta-analysis had a P < 5 × 10−8.
PLINK (version 1.90b) was used for association analysis.27 Other statistical analyses were performed with R version 3.6.1 (R Project for Statistical Computing), and 2-sided P ≤ .05 was considered statistically significant. Manhattan plots, regional SNV association results, and quantile-quantile plots were generated with LocusZoom.28 Pairwise linkage disequilibrium (LD) values were obtained by choosing individuals of European ancestry from the 1000 genomes reference panel using LDlink.28
Direct imputing summary association statistics of HLA variants (DISH)19 was used to infer statistical associations between HL risk and HLA alleles, amino acids, and SNVs based on all the variants cataloged in the Type 1 Diabetes Genetics Consortium reference panel comprising 5225 individuals of European ancestry. Biallelic SNVs (n = 28 455) with meta-GWAS summary statistics in the HLA region (Genome Reference Consortium Human Build 38, chr6:28510120-33480577) were fed into the program. Stringent quality control was applied to filter the imputed variants (MAF ≥0.005; imputation score r2 ≥ 0.5) after the DISH imputation.
Results
Meta-analysis of 3 GWAS Data Sets
HL cases were diagnosed between 3.0 and 22.7 years of age (mean [SD] age, 14.6 [3.9] years). The summary statistics from the 3 GWASs were combined with a fixed-effects meta-analysis encompassing 1286 HL cases, 369 noncancer controls, and 6824 other childhood cancer cases, for a total of 6.88 million SNVs. We observed little evidence of genomic inflation by quantile-quantile plot of the meta-GWAS results (λ = 1.021) (eFigure 2 in the Supplement), suggesting little population stratification. The overall landscape of genome-wide associations and significant peaks are illustrated in Figure 1.
Figure 1. Manhattan Plot for Genome-Wide Associations With Hodgkin Lymphoma.
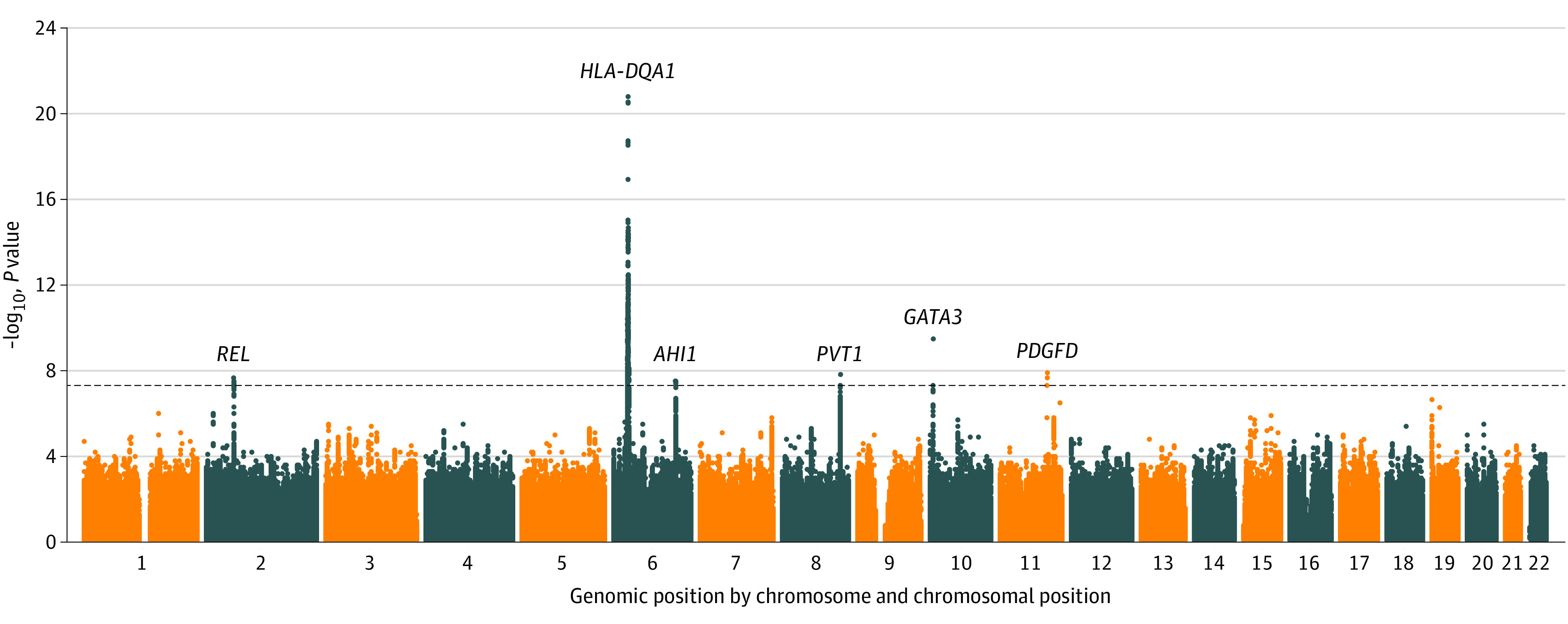
The overall landscape for meta-analysis of 3 genome-wide association studies are shown. The –log10 P values were plotted against the genomic position (x-axis), ie, chromosome and chromosomal position based on Genome Reference Consortium Human Build 38. The black horizontal dotted line represents the genome-wide significance threshold of P = 5 × 10−8. Genome-wide significant peaks were marked at 2p16.1 (REL), 6q21.3 (HLA-DQA1), 6q23.3 (AHI1), 8q24.21 (PVT1), 10p14 (GATA3), and 11q22.3 (PDGFD).
SNVs in the HLA Region
A total of 691 SNVs in the HLA region were significantly associated with HL risk. The strongest association was marked by rs28383311 (OR, 1.80; 95% CI, 1.59-2.03; P = 2.14 × 10−21) (Table 1), in the neighborhood of HLA-DRB1 (OMIM 142857), HLA-DQA1 (OMIM 146880), and HLA-DQB1 (OMIM 604305) genes (Figure 2A), which had a moderate LD (r2 = 0.46, D′ = 1.00) with previously reported SNV rs2858870 near HLA-DRA gene in adolescent and young adult-onset HL.29
Table 1. Association of Independent SNVs in the Human Leukocyte Antigen Locus With Hodgkin Lymphoma Susceptibility.
| SNV | Positionc | Allele | Unconditional logistic regression | Conditional logistic regressiona | Conditional logistic regressionb | ||||
|---|---|---|---|---|---|---|---|---|---|
| Risk | Other | OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | ||
| rs28383311 | 32619234 | A | T | 1.80 (1.59-2.03) | 2.14 × 10−21 | NA | NA | NA | NA |
| rs3129198 | 33103045 | A | G | 1.54 (1.39-1.72) | 2.64 × 10−15 | 1.53 (1.37-1.70) | 2.05 × 10−14 | NA | NA |
| rs3129890 | 32446496 | T | C | 1.23 (1.10-1.37) | 1.71 × 10−4 | 1.48 (1.32-1.65) | 7.04 × 10−12 | 1.51 (1.35-1.69) | 6.21 × 10−13 |
Abbreviaitons: NA, not applicable; OR, odds ratio; SNV, single-nucleotide variant.
Conditional on rs28383311 and rs3129198.
Conditional on rs28383311.
Position according to human reference Genome Reference Consortium Human Build 38.
Figure 2. Regional Plot of Association Results and Recombination Rates for the Hodgkin Lymphoma Risk Loci.

The –log10 P-values (y-axis) are plotted against chromosomal position (x-axis). The black horizontal dotted line represents the genome-wide significance threshold P = 5 × 10−8. The most significant single-nucleotide variant (SNV) in each analysis is marked at the center of the 1-megabase (Mb) region nearby. The color intensity of each SNV represents the extent of linkage disequilibrium (LD) with the lead genetic variant, dark blue (r2 = 0) to red (r2 = 1.0). The light blue line at the bottom of each plot shows genetic recombination rates by using 1000 Genomes Project LD. The bottom tracks show nearby genes mapping to the region of association. Association hits, which have been reported in the EBI genome-wide association study (GWAS) catalog, are also annotated. Diamond-shaped markers indicate the lead SNV in each region, and circles indicate other SNVs in variable extent of LD with the lead SNV. Unconditional logistic regression results were plotted for A, and step-wise conditional logistic regression results were plotted for B and C.
After conditioning on rs28383311, the secondary strongest association peak was marked by rs3129198 (OR, 1.53; 95% CI, 1.37-1.70; conditional P = 2.05 × 10−14) (Table 1), near HLA-DPA1 (OMIM 142880) and HLA-DPB1 (OMIM 142858) genes (Figure 2B). There was no LD (r2 = 0.00, D′ = 0.07) between rs3129198 and rs28383311 (eFigure 3 in the Supplement). However, rs3129198 had a perfect LD (r2 = 1.0, D′ = 1.00) with rs2281389 (HLA-DPB1) in a previous study comprising mostly adulthood-onset HL.17
In the next iteration of step-wise conditional logistic regression, we added rs3129198 and rs28383311 as covariates for adjustments in the model. A tertiary association signal emerged and was marked by the strongest associated SNV rs3129890 (OR = 1.51; 95% CI, 1.35-1.69; P = 6.21 × 10−13) (Table 1) in the neighborhood of HLA-DRA (OMIM 142860) (Figure 2C). The rs3129890 had a very low LD with the primary SNV rs28383311 (r2 = 0.06, D′ = 0.89) or the secondary SNV rs3129198 (r2 = 0.01, D′ = 0.12) (eFigure 3 in the Supplement) and a low LD with rs2395185 (r2 = 0.15, D′ = 1.00) near the HLA-DRA gene, which was reported previously by a study among HL cases diagnosed across a wide age range (16-80 years).30 Notably, rs28383311 and rs3129198 are mapped to HLA class II, whereas rs3129890 is mapped to the juncture between HLA class II and III. Previously reported HL-associated SNVs in the HLA region are provided in the eTable 1 in the Supplement. In addition, 10 of 14 previously reported genome-wide significant HLA SNVs (71%) were replicated with P < .05. Among the replicated SNVs, 8 of 10 (80%) were mapped to HLA class II; rs2248462 (MICB [OMIM 602436]) is mapped to the HLA I, and rs204999 (PRRT1 [OMIM 618297]) is mapped to the HLA III. Most (3 of 4 [75%]) of the remaining insignificant SNVs were mapped to the HLA class I region.
Significant HLA Alleles and Amino Acid Changes
We identified 9 HLA alleles across 3 genes (6 for DQB1 [OMIM 604305], 2 for DPB1 [OMIM 142858], and one for DRB1 [OMIM 142857]) that had P values exceeding genome-wide significance (ie, <5 × 10−8); Seven alleles were positively associated with HL risk, and 2 alleles were negatively associated with it (Table 2). The HLA allele with the strongest association was HLA_DQB1_0603 (P = 9.35 × 10−11; imputed Z = 6.48). An additional set of 55 amino acid changes in HLA genes was significantly associated with HL risk (eTable 2 in the Supplement). Previously reported HLA alleles associated with HL susceptibility are provided in eTable 3 in the Supplement. All 6 HLA alleles in class II genes were significant, including HLA_DQB1_0602 and HLA_DRB1_1501, at the genome-wide significance level, whereas 2 HLA alleles in class I genes were not significant in our study. Regarding the 4 previously reported HL-associated amino acid changes, our analysis replicated the association for AA_DPB1_55_33156577_A (P = 3.6 × 10−3; imputed Z = 2.91) but not for the other 3 amino acid residues in HLA_DPB1 (ie, leucine 35, alanine 69, and valine 84).31
Table 2. Association of HLA Alleles With Hodgkin Lymphoma Susceptibility.
| Marker ID | Positiona | Allele | Imputed Zb | r2predc | Imputed P value | |
|---|---|---|---|---|---|---|
| Effect | Other | |||||
| HLA_DQB1_0603 | 32631061 | Presence | Absence | 6.48 | 0.82 | 9.35 × 10−11 |
| HLA_DQB1_0602 | 32631061 | Presence | Absence | 6.08 | 0.88 | 1.17 × 10−9 |
| HLA_DQB1_06 | 32631061 | Presence | Absence | 5.71 | 0.92 | 1.14 × 10−8 |
| HLA_DQB1_0501 | 32631061 | Presence | Absence | –5.54 | 0.97 | 3.07 × 10−8 |
| HLA_DQB1_05 | 32631061 | Presence | Absence | –5.87 | 0.92 | 4.41 × 10−9 |
| HLA_DQB1_0301 | 32631061 | Presence | Absence | 5.81 | 0.88 | 6.08 × 10−9 |
| HLA_DRB1_1501 | 32552064 | Presence | Absence | 5.84 | 0.89 | 5.28 × 10−9 |
| HLA_DPB1_10 | 33049368 | Presence | Absence | 5.49 | 0.51 | 4.07 × 10−8 |
| HLA_DPB1_1001 | 33049368 | Presence | Absence | 5.49 | 0.51 | 4.07 × 10−8 |
Abbreviation: HLA, human leukocyte antigen.
Postion refers to the genomic coordinates based on Genome Reference Consortium Human Build 37.
Z scores represent association with Hodgkin lymphoma.
The r2pred values represent the assessment of the imputation reliability at each HLA variant.
SNVs in the Non-HLA Region
Our analysis identified the following genome-wide significant SNVs in non-HLA loci (Table 3). First, we identified rs1432297 (OR, 1.29; 95% CI, 1.18-1.41; P = 2.5 × 10−8) at 2p16.1, which is in moderate LD (r2 = 0.52, D′ = 0.73) with previously reported rs1432295,19 mapping to the REL gene. REL encodes c-Rel, which belongs to the Rel family of genes that regulate inflammation and immune response. REL is normally expressed in germinal B-cells and immunoblasts. Moreover, serving as a proto-oncogene, REL also functions in B-lymphocyte survival and proliferation32 and has been identified as a candidate gene for HL risk.33 Second, we identified rs2757647 (OR, 1.30; 95% CI, 1.18-1.42; P = 3.5 × 10−8) at 6q23.3, which is in moderate LD (r2 = 0.59, D′ = 0.83) with previously reported rs6928977,18 mapping to the AHI1 (OMIM 608894) gene. AHI1 is a prerequisite for development of the cerebellum and cortex in humans, and perturbations in AHI1 expression may result in specific subtypes of human leukemia.34 Third, we identified rs13279159 (OR, 1.33; 95% CI, 1.20-1.47; P = 1.7 × 10−8) at 8q24.21, with a high LD (r2 = 0.75, D′ = 1.00) with previously reported rs2019960,19 located at a 82-Kb LD block telomeric to PVT1 (OMIM 165140), which represents a long noncoding RNA associated with acute myeloid leukemia and HL. Fourth, we identified rs3824662 (OR, 1.52; 95% CI, 1.33-1.73; P = 3.9 × 10−10) at 10p14, with a high LD (r2 = 0.91; D′ = 1.00) with previously reported rs3781093,18 mapping to an intronic region of GATA3 (OMIM 131320); GATA3 expression is aberrant in Reed-Sternberg cells and their variants. In addition, a novel uncommon SNV with a large effect size, rs117953624 at 11q22.3 (OR, 1.98; 95% CI, 1.56-2.51; P = 1.5 × 10−8; MAF, 0.02), was identified and mapped to PDGFD (platelet-derived growth factor D; OMIM 609673) (eFigure 4 in the Supplement). Collective evidence supports a role for PDGF in HL pathogenesis,35 and PDGFD is a transforming, angiogenic growth factor.10 In addition, 12 of 18 previously reported genome-wide significant non-HLA SNVs (67%) were replicated with P < .05 (eTable 4 in the Supplement).
Table 3. Association of SNVs in Non–Human Leukocyte Antigen Regions With Hodgkin Lymphoma Susceptibility Based on Meta–Genome-Wide Association Studies.
| SNV | Positiona | Neighboring genes | Allele | OR (95% CI) | P value | |
|---|---|---|---|---|---|---|
| Risk | Other | |||||
| rs1432297 | chr2:60843517 | REL | G | A | 1.29 (1.18-1.41) | 2.50 × 10−08 |
| rs2757647 | chr6:135446416 | AH1 | T | C | 1.30 (1.18-1.42) | 3.52 × 10−08 |
| rs13279159 | chr8:128150801 | PVT1 | G | A | 1.33 (1.20-1.47) | 1.74 × 10−08 |
| rs3824662 | chr10:8062245 | GATA3 | C | A | 1.52 (1.33-1.73) | 3.86 × 10−10 |
| rs117953624 | chr11:104438259 | PDGFD | C | T | 1.98 (1.56-2.51) | 1.45 × 10−08 |
Abbreviations: OR, odds ratio; SNV, single-nucleotide variant.
Position according to human reference Genome Reference Consortium Human Build 38.
Discussion
HL epidemiology is complex, with cases developing across an age spectrum, including children, adolescents, young adults, and older adults. Survival of HL is more favorable among children, adolescents, and young adults compared with older adults. Previous studies have suggested that a difference in HL etiology between children and adolescents vs older adults could explain this difference in survival.36 To investigate the genetic etiology of HL arising during childhood, adolescence, or young adulthood, we conducted a 3-way meta-GWAS analysis by leveraging preexisting data from the SJLIFE and CCSS.
Using step-wise conditional logistic regression methods, we identified 3 independent SNVs (rs28383311, rs3129198, rs3129890) in the HLA locus; most previous studies reported only 1 SNV.17,19,29,30,37,38 The primary and secondary SNVs were in moderate or high LD with previously reported SNVs, but the tertiary SNV had a low correlation with all established HLA SNVs, suggesting both the commonality and difference for genetic factors at the HLA locus underpinning HL risk during childhood and adolescence compared with other ages at onset.
We also identified 5 SNVs at non-HLA loci with genome-wide significance. Four of these SNVs were in moderate or high LD with previously reported SNVs, further validating these findings mapped to genes, such as REL, AHI1, PVT1, and GATA318,19,39 in studies comprising mostly adult-onset HL. Collectively, these findings implicate multiple biological processes, including germinal center dysfunction, disrupted T-cell function, and nuclear factor–κB (NF-κB) activation, in HL pathogenesis40 across age groups. Our identification of rs117361561 (maps to PDGFD), a novel uncommon SNV with a large effect size, is intriguing because PDGFD has a fundamental role in cell growth, differentiation, survival regulation, invasion, and angiogenesis,41 supporting its potential association with cancer progression. Considering that NF-κB may moderate the expression of PDGF-inducible genes,42 the finding of a novel association between rs117361561 and HL risk provides additional evidence to support the role of NF-κB activation in HL pathogenesis, particularly in HL developing in younger people. Future mechanistic studies are needed to demonstrate how these genes function in HL carcinogenesis.
Limitations
This study has limitations. First, we used case-case comparisons in 2 GWAS analyses and a case-control comparison in 1 GWAS analysis. Using other cases as a control set is not ideal and may result in either false-negative or false-positive results. However, we evaluated the heterogeneity across 3 GWASs for reporting genome-wide significant findings to minimize false-positive results. Second, the HL cases included in this study all had 5 or more years of survival; patients who died before 5-year survival were not included. Third, we did not have detailed histologic subtype information, so stratified analysis based on histologic subtype was not carried out. Fourth, EBV infection status was unavailable for all patients with HL in CCSS. Among 380 patients with HL in SJLIFE, 65 (17%) had EBV status, with 15 (23%) EBV positive and 50 (77%) EBV negative. Previous studies have identified low prevalence of EBV-positive HL in adolescent and young adults compared with children younger than 10 years or adults older than 80 years,43 as well as the different etiology between patients with EBV-negative and EBV-positive HL.44 This appears to be consistent with our current results, which showed that none of the 3 HLA SNVs we identified were in the HLA I class implicated in EBV-positive HL.45,46 Fifth, the study was conducted among individuals of European ancestry, so the findings may be not be generalizable to other racial and ethnic groups.
Conclusions
In summary, we refined HL associations in the HLA locus by identifying 3 independent SNVs. In non-HLA regions, we found 1 novel SNV and replicated the majority of previous findings, including 4 SNVs exceeding genome-wide significance. These findings suggest mostly common, as well as potentially unique, genetic factors underpinning HL risk across age groups. Future replication and subsequent functional investigations are warranted to elucidate the genetic architecture and biological mechanisms of HL susceptibility.
eFigure 1. Study Population Flow Diagram
eFigure 2. Quantile-Quantile Plot for the P Values in the –log10 Scale for the Discovery GWAS Meta-analysis
eFigure 3. Pair-wise Linkage Disequilibrium Among 3 Independent SNPs in HLA Region
eTable 1. Meta-GWAS Results for Previously Published HL-Associated SNPs in HLA Region
eTable 2. Significant Amino Acid Changes Associated With HL in HLA Genes
eTable 3. Previously Reported HLA Alleles Associated With HL Susceptibility
eTable 4. Meta-GWAS Results for Previously Published HL-Associated SNPs in Non-HLA Region
eFigure 4. SNV rs117953624 near PDGFD
References
- 1.Küppers R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009;9(1):15-27. doi: 10.1038/nrc2542 [DOI] [PubMed] [Google Scholar]
- 2.Connors JM, Cozen W, Steidl C, et al. Hodgkin lymphoma. Nat Rev Dis Primers. 2020;6(1):61. doi: 10.1038/s41572-020-0189-6 [DOI] [PubMed] [Google Scholar]
- 3.Cancer Stat Facts . Hodgkin lymphoma. Accessed July 7, 2022. https://seer.cancer.gov/statfacts/html/hodg.html
- 4.Qin N, Li Z, Song N, et al. Epigenetic age acceleration and chronic health conditions among adult survivors of childhood cancer. J Natl Cancer Inst. 2021;113(5):597-605. doi: 10.1093/jnci/djaa147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mertens AC, Yasui Y, Neglia JP, et al. Late mortality experience in five-year survivors of childhood and adolescent cancer: the Childhood Cancer Survivor Study. J Clin Oncol. 2001;19(13):3163-3172. doi: 10.1200/JCO.2001.19.13.3163 [DOI] [PubMed] [Google Scholar]
- 6.Castellino SM, Geiger AM, Mertens AC, et al. Morbidity and mortality in long-term survivors of Hodgkin lymphoma: a report from the Childhood Cancer Survivor Study. Blood. 2011;117(6):1806-1816. doi: 10.1182/blood-2010-04-278796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams AM, Mandelblatt JS, Wang M, et al. Accelerated aging and mortality in long-term survivors of childhood cancer: a report from the St. Jude Lifetime Cohort (SJLIFE). J Clin Oncol. 2021;39(15_suppl):10045. doi: 10.1200/JCO.2021.39.15_suppl.10045 [DOI] [Google Scholar]
- 8.Campo E, Harris HL, Jaffe ES, Pileri SA, Thiele J. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. World Health Organization; 2008. [Google Scholar]
- 9.Mack TM, Cozen W, Shibata DK, et al. Concordance for Hodgkin’s disease in identical twins suggesting genetic susceptibility to the young-adult form of the disease. N Engl J Med. 1995;332(7):413-418. doi: 10.1056/NEJM199502163320701 [DOI] [PubMed] [Google Scholar]
- 10.Jarrett RF, Krajewski AS, Angus B, et al. The Scotland and Newcastle epidemiological study of Hodgkin’s disease: impact of histopathological review and EBV status on incidence estimates. J Clin Pathol. 2003;56(11):811-816. doi: 10.1136/jcp.56.11.811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trowsdale J, Knight JC. Major histocompatibility complex genomics and human disease. Annu Rev Genomics Hum Genet. 2013;14:301-323. doi: 10.1146/annurev-genom-091212-153455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulkarni S, Martin MP, Carrington M. The yin and yang of HLA and KIR in human disease. Semin Immunol. 2008;20(6):343-352. doi: 10.1016/j.smim.2008.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amiel J. Study of the leukocyte phenotypes in Hodgkin’s disease. In Teraski PI, ed. Histocompatibility Testing. Munksgaard; 1967:79-81. [Google Scholar]
- 14.Klitz W, Aldrich CL, Fildes N, Horning SJ, Begovich AB. Localization of predisposition to Hodgkin disease in the HLA class II region. Am J Hum Genet. 1994;54(3):497-505. [PMC free article] [PubMed] [Google Scholar]
- 15.Oza AM, Tonks S, Lim J, Fleetwood MA, Lister TA, Bodmer JG. A clinical and epidemiological study of human leukocyte antigen-DPB alleles in Hodgkin’s disease. Cancer Res. 1994;54(19):5101-5105. [PubMed] [Google Scholar]
- 16.Marshall WH, Barnard JM, Buehler SK, Crumley J, Larsen B. HLA in familial Hodgkin’s disease: results and a new hypothesis. Int J Cancer. 1977;19(4):450-455. doi: 10.1002/ijc.2910190403 [DOI] [PubMed] [Google Scholar]
- 17.Moutsianas L, Enciso-Mora V, Ma YP, et al. Multiple Hodgkin lymphoma-associated loci within the HLA region at chromosome 6p21.3. Blood. 2011;118(3):670-674. doi: 10.1182/blood-2011-03-339630 [DOI] [PubMed] [Google Scholar]
- 18.Sud A, Thomsen H, Law PJ, et al. ; PRACTICAL consortium . Genome-wide association study of classical Hodgkin lymphoma identifies key regulators of disease susceptibility. Nat Commun. 2017;8(1):1892. doi: 10.1038/s41467-017-00320-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enciso-Mora V, Broderick P, Ma Y, et al. A genome-wide association study of Hodgkin’s lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3). Nat Genet. 2010;42(12):1126-1130. doi: 10.1038/ng.696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Wilson CL, Armstrong GT, et al. Association of germline BRCA2 mutations with the risk of pediatric or adolescent non-Hodgkin lymphoma. JAMA Oncol. 2019;5(9):1362-1364. doi: 10.1001/jamaoncol.2019.2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howell CR, Bjornard KL, Ness KK, et al. Cohort profile: The St. Jude Lifetime Cohort Study (SJLIFE) for paediatric cancer survivors. Int J Epidemiol. 2021;50(1):39-49. doi: 10.1093/ije/dyaa203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hudson MM, Ehrhardt MJ, Bhakta N, et al. ; Jude Lifetime Cohort . Approach for classification and severity grading of long-term and late-onset health events among childhood cancer survivors in the St. Jude Lifetime Cohort. Cancer Epidemiol Biomarkers Prev. 2017;26(5):666-674. doi: 10.1158/1055-9965.EPI-16-0812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robison LL, Armstrong GT, Boice JD, et al. The Childhood Cancer Survivor Study: a National Cancer Institute-supported resource for outcome and intervention research. J Clin Oncol. 2009;27(14):2308-2318. doi: 10.1200/JCO.2009.22.3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin N, Wang Z, Liu Q, et al. Pathogenic germline mutations in DNA repair genes in combination with cancer treatment exposures and risk of subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol. 2020;38(24):2728-2740. doi: 10.1200/JCO.19.02760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Wilson CL, Easton J, et al. Genetic risk for subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol. 2018;36(20):2078-2087. doi: 10.1200/JCO.2018.77.8589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.St Jude Children’s Research Hospital. St Jude Cloud. Accessed July 8, 2022. https://www.stjude.cloud/
- 27.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559-575. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336-2337. doi: 10.1093/bioinformatics/btq419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cozen W, Li D, Best T, et al. A genome-wide meta-analysis of nodular sclerosing Hodgkin lymphoma identifies risk loci at 6p21.32. Blood. 2012;119(2):469-475. doi: 10.1182/blood-2011-03-343921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Urayama KY, Jarrett RF, Hjalgrim H, et al. Genome-wide association study of classical Hodgkin lymphoma and Epstein-Barr virus status-defined subgroups. J Natl Cancer Inst. 2012;104(3):240-253. doi: 10.1093/jnci/djr516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor GM, Gokhale DA, Crowther D, et al. Further investigation of the role of HLA-DPB1 in adult Hodgkin’s disease (HD) suggests an influence on susceptibility to different HD subtypes. Br J Cancer. 1999;80(9):1405-1411. doi: 10.1038/sj.bjc.6690536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Houldsworth J, Mathew S, Rao PH, et al. REL proto-oncogene is frequently amplified in extranodal diffuse large cell lymphoma. Blood. 1996;87(1):25-29. doi: 10.1182/blood.V87.1.25.25 [DOI] [PubMed] [Google Scholar]
- 33.Barth TF, Martin-Subero JI, Joos S, et al. Gains of 2p involving the REL locus correlate with nuclear c-Rel protein accumulation in neoplastic cells of classical Hodgkin lymphoma. Blood. 2003;101(9):3681-3686. doi: 10.1182/blood-2002-08-2577 [DOI] [PubMed] [Google Scholar]
- 34.Jiang X, Zhao Y, Chan WY, et al. Deregulated expression in Ph+ human leukemias of AHI-1, a gene activated by insertional mutagenesis in mouse models of leukemia. Blood. 2004;103(10):3897-3904. doi: 10.1182/blood-2003-11-4026 [DOI] [PubMed] [Google Scholar]
- 35.Güler N, Yilmaz S, Ayaz S, et al. The platelet-derived growth factor level (PDGF) in Hodgkin’s disease and non-Hodgkin’s lymphoma and its relationship disease activation. Hematology. 2005;10(1):53-57. doi: 10.1080/10245330400020405 [DOI] [PubMed] [Google Scholar]
- 36.Hjalgrim H. On the aetiology of Hodgkin lymphoma. Dan Med J. 2012;59(7):B4485. [PubMed] [Google Scholar]
- 37.Delahaye-Sourdeix M, Urayama KY, Gaborieau V, et al. A novel risk locus at 6p21.3 for Epstein-Barr virus-positive Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev. 2015;24(12):1838-1843. doi: 10.1158/1055-9965.EPI-15-0534 [DOI] [PubMed] [Google Scholar]
- 38.Frampton M, da Silva Filho MI, Broderick P, et al. Variation at 3p24.1 and 6q23.3 influences the risk of Hodgkin’s lymphoma. Nat Commun. 2013;4:2549. doi: 10.1038/ncomms3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cozen W, Timofeeva MN, Li D, et al. A meta-analysis of Hodgkin lymphoma reveals 19p13.3 TCF3 as a novel susceptibility locus. Nat Commun. 2014;5:3856. doi: 10.1038/ncomms4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sud A, Thomsen H, Orlando G, et al. ; PRACTICAL Consortium . Genome-wide association study implicates immune dysfunction in the development of Hodgkin lymphoma. Blood. 2018;132(19):2040-2052. doi: 10.1182/blood-2018-06-855296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Ahmad A, Li Y, et al. Emerging roles of PDGF-D signaling pathway in tumor development and progression. Biochim Biophys Acta. 2010;1806(1):122-130. doi: 10.1016/j.bbcan.2010.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olashaw NE, Kowalik TF, Huang ES, Pledger WJ. Induction of NF-kappa B-like activity by platelet-derived growth factor in mouse fibroblasts. Mol Biol Cell. 1992;3(10):1131-1139. doi: 10.1091/mbc.3.10.1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glaser SL, Lin RJ, Stewart SL, et al. Epstein-Barr virus-associated Hodgkin’s disease: epidemiologic characteristics in international data. Int J Cancer. 1997;70(4):375-382. doi: [DOI] [PubMed] [Google Scholar]
- 44.Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-Barr virus-associated lymphomas. Philos Trans R Soc Lond B Biol Sci. 2017;372(1732):20160271. doi: 10.1098/rstb.2016.0271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diepstra A, Niens M, Vellenga E, et al. Association with HLA class I in Epstein-Barr-virus-positive and with HLA class III in Epstein-Barr-virus-negative Hodgkin’s lymphoma. Lancet. 2005;365(9478):2216-2224. doi: 10.1016/S0140-6736(05)66780-3 [DOI] [PubMed] [Google Scholar]
- 46.Niens M, van den Berg A, Diepstra A, et al. The human leukocyte antigen class I region is associated with EBV-positive Hodgkin’s lymphoma: HLA-A and HLA complex group 9 are putative candidate genes. Cancer Epidemiol Biomarkers Prev. 2006;15(11):2280-2284. doi: 10.1158/1055-9965.EPI-06-0476 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eFigure 1. Study Population Flow Diagram
eFigure 2. Quantile-Quantile Plot for the P Values in the –log10 Scale for the Discovery GWAS Meta-analysis
eFigure 3. Pair-wise Linkage Disequilibrium Among 3 Independent SNPs in HLA Region
eTable 1. Meta-GWAS Results for Previously Published HL-Associated SNPs in HLA Region
eTable 2. Significant Amino Acid Changes Associated With HL in HLA Genes
eTable 3. Previously Reported HLA Alleles Associated With HL Susceptibility
eTable 4. Meta-GWAS Results for Previously Published HL-Associated SNPs in Non-HLA Region
eFigure 4. SNV rs117953624 near PDGFD