

Abstract
High-grade T1 (HGT1) bladder cancer is the highest risk subtype of non–muscle-invasive bladder cancer with unpredictable outcome and poorly understood risk factors. Here, we examined the association of somatic mutation profiles with nonrecurrent disease (GO, good outcome), recurrence (R), or progression (PD) in a cohort of HGT1 patients. Exome sequencing was performed on 62 HGT1 and 15 matched normal tissue samples. Both tumor only (TO) and paired analyses were performed, focusing on 95 genes known to be mutated in bladder cancer. Somatic mutations, copy-number alterations, mutation load, and mutation signatures were studied. Thirty-three GO, 10 R, 18 PD, and 1 unknown outcome patients were analyzed. Tumor mutational burden (TMB) was similar to muscle-invasive disease and was highest in GO, intermediate in PD, and lowest in R patients (P = 0.017). DNA damage response gene mutations were associated with higher TMB (P < 0.0001) and GO (P = 0.003). ERCC2 and BRCA2 mutations were associated with GO. TP53, ATM, ARID1A, AHR, and SMARCB1 mutations were more frequent in PD. Focal copy-number gain in CCNE1 and CDKN2A deletion was enriched in PD or R (P = 0.047; P = 0.06). APOBEC (46%) and COSMIC5 (34%) signatures were most frequent. APOBEC-A and ERCC2 mutant tumors (COSMIC5) were associated with GO (P = 0.047; P = 0.0002). pT1b microstaging was associated with a genomic cluster (P = 0.05) with focal amplifications of E2F3/SOX4, PVRL4, CCNE1, and TP53 mutations. Findings were validated using external public datasets. These findings require confirmation but suggest that management of HGT1 bladder cancer may be improved via molecular characterization to predict outcome.
Significance:
Detailed genetic analyses of HGT1 bladder tumors identify features that correlate with outcome, e.g., high mutational burden, ERCC2 mutations, and high APOBEC-A/ERCC2 mutation signatures were associated with good outcome.
Introduction
With 430,000 new cases diagnosed worldwide annually and 165,000 associated cancer deaths (1), bladder cancer is a major source of morbidity and mortality. At initial diagnosis, approximately 75% of patients have non–muscle-invasive bladder cancer (NMIBC), either confined to the mucosa [Ta and carcinoma in situ (CIS)] or invading the bladder lamina propria but not the muscularis propria (T1). High-grade T1 (HGT1) bladder cancer accounts for 25% to 43% of all NMIBC and has a higher risk of recurrence and progression to invasive disease than other forms of NMIBC (2). Recent data suggest that the risk of recurrence for HGT1 disease is about 40% over 5 years, whereas the risk of progression to muscle-invasive bladder cancer (MIBC) is about 21% at 5 years (2).
HGT1 tumors can present with similar histopathologic characteristics but very different clinical behavior. Consequently, management recommendations range from conservative bacillus Calmette-Guérin (BCG) bladder instillations, to repeat trans-urethral resection of the bladder, to immediate radical cystectomy. Unfortunately, prognostic models and algorithms (EORTC, Cueto; ref. 3) designed for the broader NMIBC populations have not been validated in HGT1. Even though some prognostic factors are incorporated into current treatment guidelines, outcome prediction accuracy for this group of tumors is uncertain (2). Further information on risk factors for recurrence and progression for HGT1 disease based on molecular assessment could enable a personalized treatment approach maximizing the benefit of cystoscopic screening for recurrence, as well as using cystectomy selectively.
We previously performed a meta-analysis of published HGT1 studies addressing the question of which clinicopathologic prognostic factors were predictors of recurrence, progression, and cancer-specific survival. Interestingly, deep lamina propria invasion (microstaging pT1a vs. pT1b) had the greatest negative impact on disease outcome. Lymphovascular invasion, associated CIS, nonuse of BCG, tumor size >3 cm, and older age were also associated with progression and shorter cancer-specific survival (2).
Several attempts have been made to find drivers of progression and recurrence in HGT1 tumors, based on cancer gene mutations, without clear success. Expression signatures using microarrays have also been evaluated, with discordant results across studies (4, 5). Targeted next-generation sequencing has yielded some preliminary results but in limited and retrospective HGT1 patient cohorts (6, 7). A recent metacohort analysis based on publicly available expression data tried to identify potentially lethal profiles of NMIBC, but incomplete clinical annotation prevented definitive conclusions (8). Several authors have concluded that high-risk NMIBC may display MIBC traits (8-10). We have recently reported that HG-NMIBC and MIBC have very similar tumor mutation spectra and overall mutation burden, based on analysis of 472 bladder cancers of various stages (11).
To provide more definitive information on genetic changes associated with progression of HGT1 to MIBC, we analyzed a large set of selected genes in a prospective cohort of HGT1 patients with well-defined clinicopathologic characteristics (12). Adjacent normal tissue was used as a companion specimen when available. The tumor genetic findings were used to assess associations with good outcome (GO, no recurrence), recurrence (R), and progression to MIBC (PD) during 7.4 years of median follow-up. In addition, an exploratory analysis was performed to examine potential genomic differences between pT1a and pT1b microstaging.
Materials and Methods
Patient samples and clinical information
Patients from University Hospital Vall d’Hebron (HVH) were included in a previous protocol registered at clinicaltrials.gov (NCT02113501). An additional cohort of patients managed in the same way at Hospital del Mar was added. Informed consent from the patients was obtained, and studies were conducted in accordance with ethical guidelines as per the Declaration of Helsinki. For the present study, further approval for the genomic analysis was granted by the Dana Farber Cancer Institute, Hospital del Mar, and HVH Ethics Committee.
Patients with extensive concomitant CIS were excluded. Tumors were graded according to the 2004 WHO system (2006) after pathologic assessment of the total specimen.
Three pathologists (I. de Torres, N. Juanpere, and J. Lloreta-Trull) reviewed and selected the cases. An independent review with selection of tumor-rich regions and normal areas for sequencing was done by a fourth pathologist (J. Barletta).
Nucleic acid isolation
Ten μm cores were obtained from 128 formalin-fixed paraffin-embedded (FFPE) tumor samples. DNA was extracted from FFPE tumor areas and “normal” bladder areas using the QIAamp DNA Mini Kit (Qiagen).
DNA sequencing and data processing
Whole-exome sequencing (WES) was performed at the Broad Institute using the pipeline developed by the Cancer Genome Analysis group. Apart from new refinements, all of the methods were used in The Cancer Genome Atlas (TCGA) projects. Briefly, exome capture was performed using the Agilent SureSelect Human All Exon 50 Mb probe set, with 0.2 to 0.5 μg of DNA, and sequencing libraries were prepared by standard methods including addition of indexed sequencing adaptors. Seventy-seven HGT1 tumors and 27 matched normal samples had sufficient DNA for WES. The Picard and Firehose pipelines were used to perform basic alignment of reads using Burrows–Wheeler Aligner (http://bio-bwa.sourceforge.net/bwa.shtml; ref. 13) on the UCSC human reference genome version hg19 yielding BAM files, quality control to make sure tumor and normal are matched; local realignment of reads at site of indels in the reads; identification of somatic single-nucleotide variants (SNV) in the tumor compared to normal (or panel of normals) using the MuTect (14) algorithm (version 1); and identification of somatic insertions and deletions using the Indelocator algorithm. The final output from this analysis is a Mutation Annotation Format (MAF) file that lists all variants detected and additional information, including: the read frequency of the variant allele, the read frequency of the reference allele, category of mutation, genomic coordinates, cDNA coordinates, protein coordinates, and information on whether the gene was found to be mutated in the COSMIC database.
We reviewed all variants in the maf file using IGV 2.4 (15). Variants were removed if there were artifacts due to repetitive sequence or misalignments, known SNPs, had minor allele frequency <5%, were present in intergenic regions or had no effect on coding sequence, or caused a synonymous amino acid change. MAF files generated for this study have been deposited at the European Genome-phenome Archive (EGA), which is hosted by the Center for Genomic Regulation, under accession number EGAS00001004603.
Tumor only analysis
Gene selection
Ninety-five genes were selected for detailed mutation analysis in the TO cohort of samples, based on significantly mutated genes (SMG) identified by TCGA (16) and additional genes selected based on involvement in other urothelial cancer subtypes (Supplementary Table S1A).
Sample selection
The average coverage for each gene for each sample was calculated using the Broad Institute’s internal pipeline Firehose. Samples were removed from further analysis if more than 25 genes (~25% of 96 selected genes) contained an average coverage of less than 30x sequencing depth. Sixty-two of 77 tumor samples met this criterion to be considered for further analysis.
Results
Demographic, clinical, and pathologic data
Seventy-seven clinically annotated samples of HGT1 urothelial carcinoma were obtained from two different centers in Spain (Hospital del Mar and HVH, both in Barcelona). The study cohort began on April 2005 with last assessment performed in February 2014 with a median follow-up of 7.4 years (range, 2–9 years). All patients were treated with BCG following initial diagnosis. Follow-up included cytology every 3 months together with cystoscopy or imaging in alternating manner for the first 2 years and every 6 months until 5 years thereafter (12). Clinical outcome was classified as GO when no recurrence or progression was seen, recurrent disease (R) when recurrent disease with the same stage or less, or progressive disease (PD) when progression to muscle invasion or metastasis was seen after at least 4 years of follow-up. At last assessment, the outcome for this set of patients was GO in 33 patients, R in 10 patients, PD in 18, and 1 with unknown clinical status. High-quality data were obtained from 62 samples by WES, and further analysis was restricted to those samples. Clinicopathologic characteristics of these 62 patients are presented in Table 1.
Table 1.
Clinical, histologic, and genomic variables of 62 HGT1 patients.
| N = 62 | % | |
|---|---|---|
| Gender | ||
| Male/Female | 51/10 | 82.3/16.1 |
| Unknown | 1 | 1.6 |
| Microstaging | ||
| T1a/T1b | 18/13 | 29.0/69.4 |
| Unknown | 1 | 1.6 |
| Pattern | ||
| Papillary | 35 | 56.5 |
| Solid or papillary | 13 | 21.0 |
| Both | 12 | 19.4 |
| Unknown | 2 | 3.2 |
| Multifocality | ||
| Single/Multiple | 26/33 | 41.9/53.2 |
| Unknown | 3 | 4.8 |
| Vascular permeation | ||
| Yes/No | 12/46 | 19.4/74.2 |
| Unknown | 4 | 6.5 |
| Cis | ||
| Yes/No | 17/33 | 27.4/53.2 |
| Unknown/Other | 12 | 19.4 |
| Size | ||
| <3 cm/>3 cm | 20/39 | 32.3/62.9 |
| Unknown | 3 | 4.8 |
| Significantly mutated genes | ||
| TP53 | 25 (20, 4, 1) | 40 |
| KMT2D | 17 (7, 2, 8) | 27 |
| KDM6A | 15 (5, 1, 9) | 24 |
| KMT2C | 13 (11, 0, 2) | 21 |
| CREBBP | 13 (6, 2, 5) | 21 |
| RB1 | 11 (3, 4, 4) | 18 |
| ERBB3 | 10 (9, 1, 0) | 16 |
| ERCC2 | 10 (10, 0, 0) | 16 |
| EP300 | 10 (4, 2, 4) | 16 |
| RHOB | 9 (9, 0, 0) | 14 |
| ERBB2 | 9 (9, 0, 0) | 14 |
| STAG2 | 8 (4, 1, 3) | 13 |
| PIK3CA | 8 (8, 0, 0) | 13 |
| FGFR3 | 8 (8, 0, 0) | 13 |
| ASXL2 | 8 (6, 0, 2) | 13 |
| ARID1A | 7 (0, 1, 6) | 11 |
| BRCA2 | 7 (7, 0, 0) | 11 |
Note: The 17 significantly mutated genes in more than 10% of the samples across 62 tumors by MutSig2CV (q < 0.1) are listed. The total number of mutations is broken down by type (missense, splice site, and nonsense).
Tumor mutational burden analysis
Due to a lack of control tissue or blood DNA samples for the majority of patient samples, tumor-only mutation-calling was performed (see Materials and Methods). After stringent filtering for germline variants using the COSMIC and ExAC databases (17), 20,661 SNVs affecting coding sequence (missense, nonsense, splice site) and 6,816 silent or nonsynonymous mutations were identified. The median mutation rate including both nonsilent and silent variants across 62 TO samples was 8.6 per Mb (median 303 SNVs per sample). Fifteen samples had matched normal tissue, enabling a tumor-normal analysis, which identified 5,956 SNVs with a median of 284 mutations per sample, similar to the median in TO set. To test the performance of the TO analysis, we ignored the matched normal in the 15 TN cases and compared the resulting mutations. The overall sensitivity and precision were 80% and 73% for all nonsilent variants, respectively.
The median nonsilent mutation rate for these 62 samples was 6.5 mutations per Mb, slightly higher than that seen in MIBC in the TCGA analysis where the average and median were 7.7 and 5.5 per Mb, respectively (18). This higher median is likely partially due to the lack of matched normal samples for our TO samples, leading to misidentification of rare germline variants as somatic. However, our previous analysis of a different sample set of patients with bladder cancer at the Dana Farber Cancer Institute (DFCI) also showed a relatively high tumor mutational burden (TMB) in high-grade NMIBC, similar to MIBC but also with the limitations of the lack of normal control (11). Interestingly, there was a significant difference in overall TMB among patients with GO, PD, and R, with median mutation rates of 9.6/Mb, 7.3/Mb, and 5.7/Mb, respectively (Fig. 1A, P = 0.017 by Kruskal–Wallis).
Figure 1.

A, Nonsilent (left) and overall TMB (right) in HGT1-NMIBC. Overall nonsilent TMB good outcome subjects (GO), progressors (PD), and recurrent (R) tumors were 9.6/Mb, 7.3/Mb, and 5.7/Mb, respectively (P = 0.017 by Kruskal–Wallis). B, Outcome association of bladder cancer–related genes (mutated in ≥4 samples). The percentage of samples with nonsilent mutations is shown in the x axis, and the empirical P value (P#) for association with outcome from the random-permutation method (see Materials and Methods), partially correcting heterogeneous mutation burdens among different outcome groups, is shown on the y axis. Black circle, genes with FDR < 0.1; half circle, genes with P value < 0.05. Numbers are the number of mutated samples in the group (GO/R/PD) compared with the total number of samples with mutations in that gene. C, Number of patients with mutations in the 17 SMG grouped by disease outcome (n = 61). ERCC2 is significantly associated with outcome (χ2 test, P < 0.05).
Significantly mutated genes
Seventeen genes were identified as being mutated at a statistically significant rate (SMGs) in this sample set, with mutations identified in more than 10% of the samples (q < 0.1; Fig. 2) by MutSig2CV (19). TP53 mutations were seen in 40% of the samples (25 of 62) and rarely overlapped with MDM2 amplification (2 of 10, P = 0.14 by one-tailed Fisher exact). Notably, alterations in the TP53 pathway (TP53 mutation or MDM2 amplification) were not seen in samples with FGFR3 mutation or amplification (P = 0.03 by one-tailed Fisher exact) or TSC1 mutations or deletions (P = 0.013 by one-tailed Fisher exact), as previously observed (18).
Figure 2.

Landscape of mutations and copy-number alterations in 62 HGT1 bladder cancer samples. Top to bottom: (i) Overall mutation burden, mutations per MB, top row. (ii) Normalized activity of five mutational signatures (two samples with highest COSMIC1 activity actually belong to COSMIC6). (iii) Clinical features, including outcome, squamous histology, micropapillary histology, pT1b, and gender; black, presence of the feature. (iv) Somatic mutations in SMGs with frequency ≥4.8%. (v) Focal CNA for selected genes. (vi) Chromosome arm level CN events.
Alterations in chromatin genes were the most frequent in our cohort. Seven of the 17 SMGs were chromatin modifying or regulatory genes: histone demethylases (KDM6A 24%), histone methyltransferases (KMT2C 21%, KMT2D 27%), histone acetylases (CREBBP 21%, EP300 16%), a member of the SWI/SNF chromatin remodeling complex (ARID1A 11%), and polycomb group genes (ASXL2 13%). These genes are all known to be recurrently and significantly mutated in MIBC (16). In addition, 9% of tumors showed deletions in CREBBP.
Cell-cycle–related genes were the second most frequently altered set of genes, including: CDKN2A deletion (31%), RB1 mutation (18%), and TP53 mutations (40%). DNA damage response (DDR) genes were also mutated at significant frequency (ERCC2 16%, BRCA2 11%, STAG2 13%, and ATM 10%). Finally, several genes of the RAS/RTK/PI3K signaling pathway were found to be frequently mutated (FGFR3 13%, PIK3CA 13%, ERBB2 15%, ERBB3 16%, and RHOB 15%).
Associations between gene mutations and clinical outcome in HGT1
To consider associations between genes with mutations and clinical outcome, we considered the 17 SMGs identified in this cohort, 54 SMGs identified in the TCGA MIBC analysis (16), and 42 genes identified as important in NMIBC in the literature (Supplementary Table S1A), for a total of 95 genes.
The heterogeneous mutation burden across samples is a significant confounding factor in identifying associations of mutated genes with clinical outcome or other covariates. To mitigate this, we used a permutation-based method that maintains the overall number of nonsilent mutations per sample and gene (see Materials and Methods; ref. 20) to identify significant associations. We considered only the 30 genes seen to be mutated in ≥4 samples (single hypermutant phenotype sample due to POLE mutation was excluded) and adjusted empirical P values by FDR (Fig. 1B).
ERCC2 was the only gene for which mutations were strongly associated with GO (9/33 in GO, 0/10 in R, and 1/18 in PD; P# = 0.003, q = 0.1, by random-permutation and P* = 0.022 by one-tailed Fisher exact). Modest enrichment for BRCA2 mutations was also seen in the GO subset (6/33 in GO, 1/10 in R, and 0/18 in PD, P# < 0.05, but corrected q = 0.3).
In contrast, TP53 mutations were modestly enriched in the PD subset (11/33 in GO, 3/10 in R, and 10/18 in PD; Fig. 1C; P# = 0.012 and P* = 0.09, q = 0.23). Several other genes showed modest associations with different clinical outcomes (Fig. 1B), but none were significant after correcting for multiple hypothesis testing, including ATM, ARID1A, AHR, and SMARCB1 in PD; and RHOB and ARID1A in the R subset.
When the same random-permutation method was applied for the association of gene mutations with overall mutation burden (Mann–Whitney P value was used as a statistic for each permutation), ERCC2 mutations were significantly (q = 0.11, P# = 0.003, and P* = 0.00014) associated with TMB, suggesting that the higher mutation burden in GO (vs. PD/R) is due in part to enrichment in ERCC2 mutations. ERBB3, KMT2C, and TP53 mutations were also enriched in high mutation burden samples with P# < 0.05, but were not significant after correcting for multiple hypothesis testing (Supplementary Fig. S1).
Alterations in DDR genes
DDR gene alterations have been described in high-grade NMIBC tumors (but not in low grade) at a rate similar to MIBC and associated with a higher mutational burden (10). To better understand the role of DNA damage and repair processes in HGT1, we assessed 34 genes encompassing major DNA repair pathways (Supplementary Table S1B). We found that 60% of samples (36 of 62) had at least one nonsilent DDR gene mutation (Supplementary Fig. S2) and were more frequent in the GO group (22/33, 65%) compared with R (4/10, 40%) and PD (10/18, 55%; P = 0.003). The presence of DDR gene mutations was also associated with higher mutation burden (Supplementary Fig. S3, P < 0.0001 Mann–Whitney). ERCC2 missense mutations were the most common DDR gene alteration, occurring in 16% (10 of 62) of the cases. Mutations were also seen in BRCA2 in 11%, STAG2 in 13%, and ATM in 10%. Indeed, DDR mutations were no longer associated to GO when removing ERCC2 mutations from the analysis (P = 0.15), suggesting that mutations in ERCC2 were the drivers of this association. Consistent with the association of high TMB with GO, ERCC2-mutated tumors had a significantly higher mutational burden than ERCC2 wild-type (Fig. 1A). When focusing on nucleotide excision repair genes overall (including ERCC2), we found that mutations in this group (after correcting for mutational burden) were predictors of GO.
Mutational signature analysis
Nonnegative matrix factorization (NMF; Bayesian version, SignatureAnalyzer; ref. 21) was used to perform de novo mutation signature analysis of the 62 samples for 27,477 SNVs stratified into 96 base substitution types, and identified five mutation signatures (Fig. 3A). Two distinct APOBEC signatures were identified, APOBEC-A and APOBEC-B (corresponding to COSMIC2 and COSMIC13; named SBS 2 and SBS13 in COSMIC mutational signatures v3), accounting for 30% and 16% of all mutations respectively); a POLE signature (COSMIC10, SBS10 in v3, accounting for 16% of all mutations, seen in a single sample with extremely high TMB, 109 SNVs per Mb); a signature with predominance of C>T_CpG mutations, resembling both COSMIC1 and 6 (SBS 1 and 6), accounting for 5% of all mutations; and a signature resembling COSMIC5 (SBS 5), accounting for 34% (19). APOBEC-mediated mutagenesis (COSMIC2 and 13) was the major source of mutations in this tumor sample set, with overall activity 46%, rising to 55% after removal of the POLE mutations seen in a single sample, and was similar to that seen in the TCGA MIBC cohort (~60%; see Supplementary Materials and Methods).
Figure 3.

A, Mutational signatures of 62 high-grade non–muscle-invasive urothelial tumors identified by a Bayesian nonnegative matrix factorization algorithm (top to bottom). Five distinct mutational signatures were identified that are most similar to COSMIC signatures 1, 2, 5, 10, and 13 (17). Two are due to APOBEC activity (COSMIC2, COSMIC13; APOBEC-A and APOBEC-B, respectively); one is attributable to POLE mutation (COSMIC10); one more uniform mutation signature that resembles COSMIC signature 5; and finally a signature characterized by C>T at CpG that corresponds to the aging signature (COSMIC1). However, two samples in which this signature was predominant (vh122 and vh73, Supplementary Fig. S4A) are MSI high samples, and hence in those two samples, the signature likely reflects the mutagenic effects of defective DNA mismatch repair. B, Association of mutational signatures and SNVs with disease outcome (excluding POLE sample vh102). *, COSMIC1 includes the two samples dominated by COSMIC6 signature. Significant associations (P < 0.05) are indicated by †. Y axis is exponential scale. C, Mutation and focal CN (MutCN) clusters. Normalized strength of association of 46 genetic alterations to the three MutCN clusters is shown. D, Frequency of disease outcome. E, Microstaging by MutCN clusters. Barplots depict the percentage of patients (number of patients in white).
The single, hypermutant sample with TMB 109 SNVs/Mb had more than 95% mutations attributable to the POLE signature and harbored a POLE hotspot dinucleotide mutation, L424V, in addition to N423K in the same read. We also identified two samples with a predominant C>T at CpG mutations, vh122 (≥80%) and vh73 (≥60%), in contrast to less than 20% activity for that signature in the remaining samples (Supplementary Fig. S4A). The mutation spectrum of each of those 2 samples showed a high cosine similarity to each of COSMIC1 (0.93 and 0.92, respectively) and COSMIC6 (0.9 and 0.91, respectively). Because COSMIC6 is known to be associated with defective DNA mismatch repair and commonly found in microsatellite unstable tumors, we searched those samples for mutations in mismatch repair genes. Indeed, vh122 harbored a splice-site mutation in MSH2 (NM_000251.2:c.793–1G>A) annotated as likely pathogenic (in ClinVar; ref. 22), and vh73 had a splice-site mutation in MLH1 (NM_000249.3:c.1989+2T>C).
As previously demonstrated in MIBC (20), the activity of COSMIC5 signature was significantly higher in ERCC2-mutant samples of our cohort (median 280 vs. 87, P = 0.0002 by one-tailed Mann–Whitney, q < 0.1 and P# = 0.002 by random-permutation test). KMT2C mutations were also significantly associated with the COSMIC5 signature after correcting for mutation burden (Supplementary Fig. S1). Furthermore, ERCC2 mutation was associated with overall number of SNVs (P# = 0.004, q = 0.11 by random-permutation test, Supplementary Fig. S1), indicating that the increase of mutation burden in GO patients is partly attributed to the activity of COSMIC5 (median 109 vs. 85, P = 0.22 by Mann–Whitney) in addition to APOBEC mutagenesis. Notably, the activity of COSMIC2 characterized by predominant C>T mutations at TCW (W = A/T) was much higher in GO samples (Fig. 3B, median 51 vs. 16 between GO vs. others, P = 0.047 by Mann–Whitney), although the overall activity of APOBEC mutagenesis (COSMIC2 + COSMIC13) did not reach statistical significance (median 133 vs. 89, P = 0.21 by Mann–Whitney). Although GO samples had somewhat higher overall APOBEC and COSMIC5 activities (Fig. 3B), this was not statistically significant (P = 0.09 and P = 0.08, respectively). C>T_ CpG signature was higher in R, but overall contribution across samples was small. All signature enrichment analyses were performed without the POLE-mutated sample.
In addition, we performed an unsupervised clustering analysis based upon the activity of the five mutational signatures and identified five mutational signature clusters, MSig1–5. Each cluster associated with specific trends in disease outcome; MSig4 was also associated with pT1b microstaging (see Supplementary Results; Supplementary Figs. S4B and S5).
Somatic copy-number alterations
GISTIC analysis supplemented by manual inspection identified focal amplifications (copy ratio > 4) at multiple genes: PVRL4 (44%), YWHAZ (35%), CCND1 (26%), ERBB2 (22%), PPARG (22%), MDM2 (19%), SOX4 (19%), CCNE1 (17%), E2F3 (15%), AHR (13%), FGFR3 (6%), and EGFR (4%). Focal deletions (copy ratio < 0.62) were identified at CDKN2A (31%), TSC1 (19%), CREBBP (9%), and RB1 (6%). PVRL4 amplifications were somewhat enriched in the recurrent tumors (70% in R vs. 40% in PD or GO, P = 0.08 by one-tailed Fisher exact test; Fig. 2). Several other copy-number (CN) changes were associated to a minor degree with outcome: amplifications at 6p22.3, encompassing SOX4 and E2F3, were more frequent in the progressors (33% in PD vs. 13% in GO or R, P = 0.1 by one-tailed Fisher exact test); PPARG amplifications were more frequent in GO (29% in GO vs. 12% in PD or R; P = 0.12 by one-tailed Fisher exact test); CDKN2A deletions (44% in PD or R vs. 18% in GO; P = 0.04 by Fisher exact test) and CCNE1 amplifications (28% in PD or R vs. 7% in GO; P = 0.0.05 by Fisher exact test) were both more frequent in PD or R tumors; and three RB1 deletions were seen only in GO. Chromosomal arm-level somatic copy number alterations (SCNA) involved several chromosomal arms containing genes known to be altered in bladder cancer, including chromosome 9 deletions, chromosome 20 amplification, 8p amplification, and 10q deletions. No particular patterns or enrichments in outcome were observed in arm-level SCNAs (Supplementary Fig. S6, right heatmap).
An unsupervised hierarchical clustering of focal SCNA identified three main clusters (Supplementary Fig. S5, left heatmap). Cluster one was characterized by consistent loss of chromosome 9, similar to the genomic subtype 2 (GS2) seen in Ta (9). Cluster two was characterized by high frequency of CN events with a trend toward enrichment of PD patients. Cluster three is CN quiet with no or a few CNAs, similar to GS1 of Ta (9).
Mutation and CN clusters
The TCGA 2014 study identified three biologically distinct groups in 125 MIBC samples based on mutations in SMGs and focal SCNAs via unsupervised NMF clustering: (i) a “focally amplified” group with enriched focal SCNAs in several genes as well as mutations in KMT2D; (ii) “CDKN2A-deficient/FGFR3-altered” group with enriched papillary histology; and (iii) “TP53/cell-cycle-altered” group with TP53 mutations in nearly all samples as well as frequent RB1 mutations and amplifications in E2F3 and CCNE1 (18). A similar analysis based on gene expression also identified two major genomic circuits in a mixed cohort of NMIBC and MIBC (23): one circuit was characterized by FGFR3 alterations, CCND1 overexpression, 9q, and CDKN2A deletions, and the other was defined by E2F3 amplification, RB1 deletions, and 5p gain. We performed a consensus NMF-based clustering for 46 genetic alterations, comprising mutations in 23 genes (≥5% across samples), focal amplifications and deletions in 13 genes, and 10 recurrent arm-level gain or loss. We consistently identified three clusters based on mutations (Mut) and CN events, which we called MutCN1–3 (Fig. 3C; Materials and Methods).
MutCN1 (n = 19) was characterized by TP53 mutations and focal SCNAs gain in CCNE1, PVRL4, YWHAZ, E2F3-SOX4, and PPARG, mainly corresponding to the “TP53/cell-cycle-altered” group in MIBC. MutCN2 (n = 22) was associated with mutations in RB1 and ERCC2, and several chromatin-modifying genes, CREBBP, KMT2D, KMT2C, and EP300. MutCN3 (n = 20) was associated with mutations and amplification in FGFR3, CDKN2A deletions and amplifications in CCND1 and MDM2, and losses of chromosomes 9, 17, and 20, which is analogous to the “CDKN2A-deficient FGFR3 mutated” group in MIBC.
MutCN clustering was associated with clinical outcome (Fig. 3D, P = 0.04 by Fisher exact test): MutCN1 was associated with progression or recurrence with a much higher proportion, with an overall prevalence of 12/19 (63%) of cases [PD (n = 7, 37%) and R (n = 5, 26%)] compared with GO (n = 7, 37%); MutCN2 had a largely favorable clinical outcome with the highest proportion of GO (n = 16, 73%) relative to PD (n = 6, 27%); MutCN3 was intermediate, comprising an average mix of PD (n = 5,25%), R (n = 5,25%), and GO (n = 10, 50%). MutCN clusters were also significantly associated with microstaging between pT1a versus pT1b (Fig. 3E, P = 0.05 by Fisher exact test), which was also linked with outcome in our original clinical cohort (12). The progression-prone MutCN1 harbored the highest proportion of pT1b (17 of 19; 89%), whereas MutCN2 had fewer pT1b (12 of 22, 55%), and the proportion of pT1b in MutCN3 was intermediate (14 of 20, 70%).
Discussion
HGT1 tumors are clinically challenging because they represent an heterogeneous group of tumors in which histopathologic characteristics have limited ability to predict outcome. The decision to offer conservative local treatment with close surveillance versus radical cystectomy is currently made in the absence of validated prognostic biomarkers. Some clinical and pathologic prognostic factors have been identified in a meta-analysis (2), but lack sufficient power to be used routinely for clinical decision making. Mutation patterns that discriminate between patients with low or high risk of disease progression have not been identified previously (24). In the broader set of NMIBC, several expression signatures have been described with potential ability to predict disease progression to MIBC (4, 5, 25). However, these gene sets show very limited overlap.
In the present study, we performed detailed genetic analysis of HGT1 bladder tumors to identify features that correlate with outcome. One of the main findings in our study is that ERCC2 mutations, the most commonly mutated DDR gene, are strongly associated with GO (P# = 0.003, q = 0.1), defined as lack of recurrence or progression. Interestingly, ERCC2 mutations have also been associated with better outcome in MIBC (23). In addition, we found that high TMB is also linked to good disease outcome, in line with preliminary observations made by Meeks and colleagues in high-risk NMIBC (6).
A comparison of our findings in HGT1 tumors with other genomic datasets highlights and confirms changes between NMIBC and MIBC. Consistent with previous reports (7) and with our recent clinical series at DFCI (11), TMB in HGT1 is higher than in low-grade NMIBC (6) and similar to MIBC (Supplementary Fig. S7). In our cohort, this relatively high mutation burden is mainly due to APOBEC-mediated mutagenesis (COSMIC2 and 13), but also likely due to mutations in ERCC2 that cause its distinctive mutation signature, COSMIC5 (20). We also previously observed a higher prevalence of mutations in DDR genes going from NMIBC to MIBC (20). This higher frequency of DDR mutations can potentially explain acquisition of mutations due to impaired DNA repair, leading to growth advantage and invasive capabilities (26). TP53 and KMT2D mutations are also more frequent in HGT1 than in the overall NMIBC population and similar to MIBC (24, 27, 28). We found a higher frequency of TP53 mutations in progressors compared with patients with GO, conflicting with findings in previous studies (6, 29). Mutation rates for some chromatin modifier genes (e.g., KDM6A) and signaling genes (e.g., FGFR3, PIK3CA, and STAG2) were found lower than in low-grade NMIBC (9, 30) and at similar levels to MIBC (16), confirming our previous findings (11). However, we recognize that the results of these comparisons are tentative, because it is possible that technological, experimental, and/or clinical differences in the different cohorts and analyses may explain some of the differences we have observed.
APOBEC-mediated mutagenesis is the major mutagenic process in this cohort accounting for more mutations than in Ta tumors (9) but similar to TCGA MIBC (16). This progressive increase of APOBEC-mediated mutagenesis compared with low-grade NMIBC might suggest that APOBEC may drive disease progression in NMIBC from low to high grade (27) and subsequent progression to muscle-invasive disease. Interestingly, MSig4, a cluster of tumors based on mutational signatures, identified a subgroup, characterized by very high APOBEC activity with all samples harboring TP53 mutations and high TMB. The likelihood of association of these TP53 mutations to APOBEC compared with TP53 mutations in other samples supports the hypothesis that APOBEC might drive these TP53 mutations (31) through signatures 2 and 13, as recently described (32). In addition, we observed that this cluster was enriched in patients with pT1b (deeper invasion into the lamina propria) and with a trend to poor outcome.
We identified three mutation and CN clusters (MutCN1–3) with similarities to genomic clusters identified in the TCGA study (18). In addition, the clusters that we identified matched subgroups of tumors in other studies of NMIBC (9, 10, 27): (i) the MutCN2 cluster was associated with GO and resembles the GS2 group of Ta tumors (9); (ii) the MutCN3 cluster was also associated with GO, similar to what was observed in gene expression–based class 1 UROMOL tumors (Urobasal in the Lund classification; refs. 10, 27); and (iii) the MutCN1 cluster characterized by cell-cycle alterations and associated with poor outcome is similar to the Genomically Unstable (GU) expression group of the Lund classification (10). The GU group was reported to present higher frequencies of multifocal tumors, concomitant CIS, lymphovascular invasion, and deeper invasion depth compared with Uro cases (33). The GU group has been shown to largely overlap with class 2 UROMOL, characterized by poor prognosis, late cell-cycle genes, and epithelial–mesenchymal transition activation (27). Interestingly, class 2 UROMOL tumors were mostly classified as having high risk of progression by a 12-gene expression prognostic signature (4, 5). Consistent with this, MutCN1 is associated in our series with deeper invasion into the lamina propria, consistent with a more aggressive pattern of genomic alterations in pathologic pT1b microstaging. Of note, this is the first time that pathologic substaging for HGT1 has a defined correlative genomic profile, highlighting the value of this genomic finding in predicting specific pathologic patterns of aggressiveness. It will be of interest to examine whether the MutCN1 cluster features and the 12-gene expression signature directly correlate with each other in HG T1 bladder cancers. Although CDKN2A deletions and CCNE1 amplifications were common events (31% and 25% of patients, respectively), we found a trend, that did not meet significance, associating these CN events with recurrence and progression, consistent with other studies (6, 7).
Despite the homogeneity of this cohort, which comprises exclusively HGT1 tumors with long-term follow-up, our study had several limitations. First, our sample size was moderate, and unfortunately, we were not able to obtain germline DNA for matched tumor-normal analysis in most patients, meaning that we could not confirm that identified variants were somatic and not germline. Nonetheless, analysis of tumor-normal pairs for 15 (24%) samples indicated that overall sensitivity and precision were 80% and 73% for all nonsilent variants called, respectively. This concordance was even higher for the 95 genes important in bladder cancer development that we considered here.
A second major limitation is the lack of analysis of a parallel cohort of low-grade NMIBC and MIBC samples by identical methods. Instead, we compared to prior publications with results of mutation analysis in related sets of bladder cancer. As above, the results of these comparisons are considered tentative, because it is possible that technological, experimental, and/or clinical differences in the different cohorts and analyses may explain some of the observed differences.
A third major limitation is the lack of a second dataset that could be used to validate the predictive associations we have identified. However, we used HGT1 samples from three previous publications to look for support of our findings: 63 samples from Nassar (11), 38 from Pietzak (7), and 78 RNA sequencing samples from Hedegaard (27) cohorts (for more details on the datasets, see Supplementary Materials and Methods). We could confirm the correlations between ERCC2 mutations and TMB [P = 0.057 (11), P = 6.786e-3 (7)], TMB and DDR mutations [P = 8.565e-06 (11), P = 7.553e-4 (7)], and ERCC2 mutations with COSMIC5 [P = 0.002 (11)]. With respect to prognostic associations, ERCC2 mutations were more common in samples from GO patients compared with R/PD [21% vs. 7% (7); 25% vs. 11% (11)]. We could not confirm this finding in the Hedegaard cohort (27) due to low expression of ERCC2 gene across samples, which translated into insufficient coverage to perform variant calling. We confirmed a higher prevalence of DDR mutations in GO patients compared with R/PD [P = 0.01 (11), P = 0.03 (27), P = 0.17 (7)]. TMB was higher in nonrecurring patients [P = 0.08 (7), P = 0.18 (27)] and moderately associated to PFS [P = 0.15 (27)]. Although all of these findings are consistent with the new analysis reported here, they are not statistically significant due to a smaller number of cases being studied, a shorter period of clinical follow-up (24–30 months median follow-up), and to a limited number of events in Nassar series (11).
Thirty-one BCG-treated samples were included in the Nassar dataset (11) and were studied in greater detail. In this subset, there was a stronger association of ERCC2, DDR mutations, and TMB with outcome compared with HGT1 patients who did not receive BCG (Supplementary Table S2), suggesting that high TMB and DDR mutations are prognostic for BCG response. Overall, these external datasets confirmed the association of ERCC2, TMB, and DDR mutations with GO in HGT1 patients. We were not able to validate the relationship of mutational signatures with outcome, likely due to the limited genome coverage of panel sequencing.
Based on our findings, we propose some tentative guidelines to help decide which HGT1 patients can be safely offered conservative bladder-sparing management versus those in whom cystectomy is preferred. (i) Patients with favorable clinicopathologic characteristics and high TMB, or ERCC2 or other DDR gene mutations, may be best considered for a conservative treatment approach with BCG immunotherapy and serial cystoscopy. (ii) Patients with HGT1 tumors harboring TP53 mutations, CDKN2A deletions or focal CCNE1 amplification, or those with MSig4 signature or MutCN1 signature, early cystectomy may be appropriate for prevention of progression to muscle-invasive disease (Fig. 4 and Table 2). As stated, we consider these recommendation to be tentative, which should be validated in independent and larger cohorts.
Figure 4.
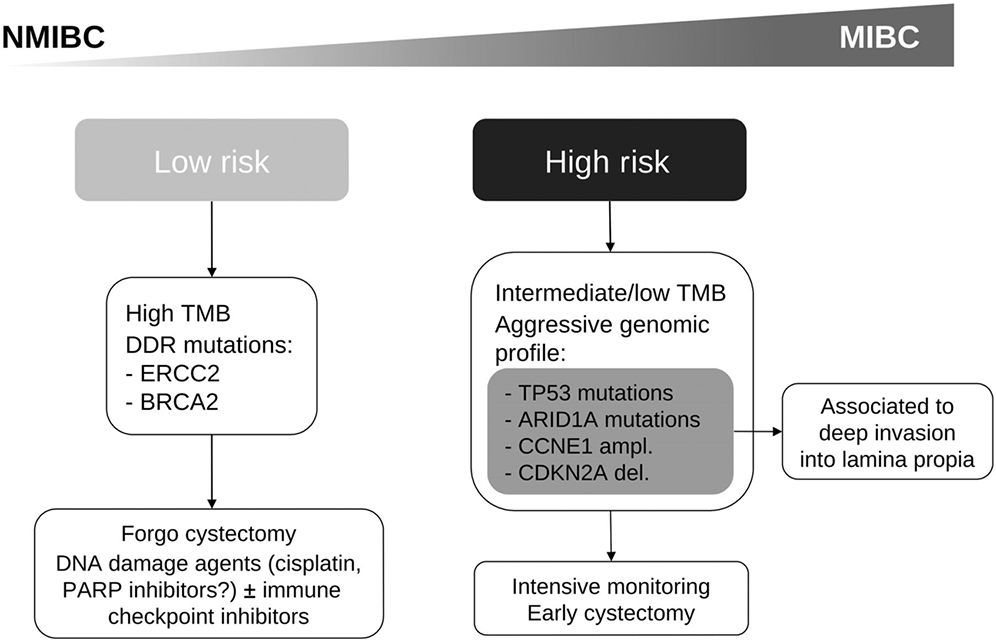
Proposed treatment algorithm based on main genomic characteristics predicting outcome in HGT1 bladder cancer.
Table 2.
Main genomic characteristics predicting outcome in HGT1 bladder cancer.
| Good outcome | Recurrent | Progression | Microstaging pT1b (deeper level of invasion) |
|
|---|---|---|---|---|
| TMB | High | Low | Intermediate | |
| Mutation frequency analysis | ERCC2 (P#< 0.003, q = 0.1) | RHOB and ARID1A (q > 0.1, P# < 0.05) | TP53, ATM, ARID1A, AHR, SMARCB1 (q > 0.1, P# < 0.05) | |
| BRCA2 (P# < 0.05) but q > 0.1 | ||||
| Mutations in DDR genes (P = 0.007) | ||||
| Mutational signature analysis | COSMIC2 (C>T mutations at TCW; P = 0.047) | |||
| COSMIC 5 in ERCC2 mutants (P = 0.0002) | ||||
| MSig clustering | MSig3 (associated with ERCC2 mutations and COSMIC5; 7/7 GO; P = 0.13) | MSig1 (P = 0.04) | MSig4 with high APOBEC activity and TP53 mutations (3/6 PD) | MSig4 (highest APOBEC with TP53 mut; 5/6 pT1b) |
| CN alterations | Focal PVRL4 amplification (P = 0.08) CDKN2A deletion (PD&R, P = 0.04) Focal CCNE1 amplification (PD&R, P = 0.04) |
|||
| MutCN clusters | MutCN2 in 73% of GO (P = 0.04) | MutCN1 cluster-enrich (PD&R, P = 0.04) | MutCN1 clustering has 89% of pT1b samples (P = 0.05; TP53, E2F3.amp, CCNE1.amp, other focal CN events) | |
Note: P#, empirical P value from the random permutation method correcting for heterogeneous mutation burdens among different outcome groups.
The apparent importance of TMB and DDR mutations, especially ERCC2, suggests the possibility that DNA damage agents like PARP inhibitors with or without immune checkpoint inhibitors may be good therapeutic approaches for patients with HGT1 who carry these alterations. As all our patients received BCG, it is possible that high TMB is not a prognostic factor per se, as in MIBC (16), but rather a biomarker of response to intravesical BCG (6). Higher TMB has also been associated with response to immune checkpoint inhibitors in patients with metastatic solid tumors, including bladder (34, 35). However, to formally address this question, it would require analysis of another cohort in which randomization to BCG or not occurred with assessment of outcome.
In summary, high mutational burden, ERCC2 mutations, and high APOBEC-A/ERCC2 (COSMIC2 and COSMIC5) mutation signatures were significant predictors of GO in HGT1. TP53 mutations and CN gain in CCNE1 combined with CDKN2A deletion were associated with disease progression. While awaiting independent validation, our findings suggest consideration of mutational analysis of HGT1 bladder cancers to assess these mutational and CN features, to improve prediction of GO or the risk of recurrence or progression.
Supplementary Material
Acknowledgments
This work has been supported by FIS PI16/00112 and FEDER (Spanish Health Ministry Grant “Fondo de Investigación Sanitaria”) by J. Bellmunt. This study was also possible thanks to a grant from Friends of Dana-Farber (J. Bellmunt), the Retired Professional Fire Fighters Cancer Fund (J. Bellmunt and M.-E. Taplin), and the generous support of Whole Foods Golf Classic for bladder cancer research in memoriam of Christopher Snell (J. Bellmunt and M.-E. Taplin). Also a private donation in the memory of Rich Beaudoin. G. Getz was partially funded by the Paul C. Zamecnick Chair in Oncology at the Massachusetts General Hospital Cancer Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Disclosure of Potential Conflicts of Interest
J. Bellmunt reports grants from FIS PI16/00112 (S) (Spanish Health Ministry Grant & Fondo de Investigación Sanitaria; Spanish National Grant), grants from Friends of Dana-Farber (internal institutional grant), other funding from Retired Professional Fire Fighters Cancer Fund (research support to the institution), and other funding from Whole Foods Golf Classic for bladder cancer research (research support to the institution) during the conduct of the study. A. Rodriguez-Vida reports grants from Takeda, grants and personal fees from MSD, personal fees from Clovis, personal fees from Sanofi, personal fees from BMS, personal fees from Roche, personal fees from Janssen, personal fees from Astellas, and personal fees from Bayer outside the submitted work. K.W. Mouw reports other funding from Pfizer (clinical trial support) outside the submitted work. E.M. Van Allen reports personal fees from Tango Therapeutics, other compensation from Syapse (equity), personal fees and other from Enara Bio (equity), personal fees and other compensation from Manifold Bio (equity), personal fees and other compensation from Monte Rosa (equity), personal fees from Invitae, personal fees from Genome Medical (equity), grants from BMS, and grants from Novartis outside the submitted work. G. Getz reports grants from IBM, grants from Pharmacyclics, and personal fees from Scorpion Therapeutics (founder, consultant, and holds privately held equity) outside the submitted work; in addition, G. Getz has a patent for MuTect issued, a patent for MutSig issued, a patent for POLYSOLVER issued and with royalties paid from Neon Therapeutics, a patent for ABSOLUTE pending, and a patent for Bladder Cancer Classification pending. No potential conflicts of interest were disclosed by the other authors.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol 2017;71:96–108. [DOI] [PubMed] [Google Scholar]
- 2.Martin-Doyle W, Leow JJ, Orsola A, Chang SL, Bellmunt J. Improving selection criteria for early cystectomy in high-grade t1 bladder cancer: a meta-analysis of 15,215 patients. J Clin Oncol 2015;33:643–50. [DOI] [PubMed] [Google Scholar]
- 3.Xylinas E, Kent M, Kluth L, Pycha A, Comploj E, Svatek RS, et al. Accuracy of the EORTC risk tables and of the CUETO scoring model to predict outcomes in non-muscle-invasive urothelial carcinoma of the bladder. Br J Cancer 2013;109:1460–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dyrskjøt L, Reinert T, Novoradovsky A, Zuiverloon TC, Beukers W, Zwarthoff E, et al. Analysis of molecular intra-patient variation and delineation of a prognostic 12-gene signature in non-muscle invasive bladder cancer; technology transfer from microarrays to PCR. Br J Cancer 2012;107:1392–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dyrskjøt L, Reinert T, Algaba F, Christensen E, Nieboer D, Hermann GG, et al. Prognostic impact of a 12-gene progression score in non-muscle-invasive bladder cancer: a prospective multicentre validation study. Eur Urol 2017;72: 461–9. [DOI] [PubMed] [Google Scholar]
- 6.Meeks JJ, Carneiro BA, Pai SG, Oberlin DT, Rademaker A, Fedorchak K, et al. Genomic characterization of high-risk non-muscle invasive bladder cancer. Oncotarget 2016;7:75176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pietzak EJ, Bagrodia A, Cha EK, Drill EN, Iyer G, Isharwal S, et al. Next-generation sequencing of nonmuscle invasive bladder cancer reveals potential biomarkers and rational therapeutic targets. Eur Urol 2017;72:952–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan TZ, Rouanne M, Tan KT, Huang RY, Thiery JP. Molecular subtypes of urothelial bladder cancer: results from a meta-cohort analysis of 2411 tumors. Eur Urol 2019;75:423–32. [DOI] [PubMed] [Google Scholar]
- 9.Hurst CD, Alder O, Platt FM, Droop A, Stead LF, Burns JE, et al. Genomic subtypes of non-invasive bladder cancer with distinct metabolic profile and female gender bias in KDM6A mutation frequency. Cancer Cell 2017;32:701–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sjödahl G, Lauss M, Lövgren K, Chebil G, Gudjonsson S, Veerla S, et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res 2012;18:3377–86. [DOI] [PubMed] [Google Scholar]
- 11.Nassar AH, Umeton R, Kim J, Lundgren K, Harshman LC, Van Allen EM, et al. Mutational analysis of 472 urothelial carcinoma across grades and anatomic sites. Clin Cancer Res 2019;25:2458–70. [DOI] [PubMed] [Google Scholar]
- 12.Orsola A, Werner L, de Torres I, Martin-Doyle W, Raventos CX, Lozano F, et al. Reexamining treatment of high-grade T1 bladder cancer according to depth of lamina propria invasion: a prospective trial of 200 patients. Br J Cancer 2015;112:468–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 2017;171:540–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mouw KW, Cleary JM, Reardon B, Pike J, Braunstein LZ, Kim J, et al. Genomic evolution after chemoradiotherapy in anal squamous cell carcinoma. Clin Cancer Res 2017;23:3214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014;507:315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Tiao G, et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet 2016;48:600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landrum MJ, Kattman BL. ClinVar at five years: delivering on the promise. Hum Mutat 2018;39:1623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Akbani R, Creighton CJ, Lerner SP, Weinstein JN, Getz G, et al. Invasive bladder cancer: genomic insights and therapeutic promise. Clin Cancer Res 2015;21:4514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glaser AP, Fantini D, Shilatifard A, Schaeffer EM, Meeks JJ. The evolving genomic landscape of urothelial carcinoma. Nat Rev Urol 2017;14:215–29. [DOI] [PubMed] [Google Scholar]
- 25.van der Heijden AG, Mengual L, Lozano JJ, Ingelmo-Torres M, Ribal MJ, Fernández PL, et al. A five-gene expression signature to predict progression in T1G3 bladder cancer. Eur J Cancer 2016;64:127–36. [DOI] [PubMed] [Google Scholar]
- 26.Bartek J, Lukas J, Bartkova J. DNA damage response as an anti-cancer barrier: damage threshold and the concept of 'conditional haploinsufficiency’. Cell Cycle 2007;6:2344–7. [DOI] [PubMed] [Google Scholar]
- 27.Hedegaard J, Lamy P, Nordentoft I, Algaba F, Høyer S, Ulhøi BP, et al. Comprehensive transcriptional analysis of early-stage urothelial carcinoma. Cancer Cell 2016;30:27–42. [DOI] [PubMed] [Google Scholar]
- 28.Sanli O, Dobruch J, Knowles MA, Burger M, Alemozaffar M, Nielsen ME, et al. Bladder cancer. Nat Rev Dis Primers 2017;3:17022. [DOI] [PubMed] [Google Scholar]
- 29.Downes MR, Weening B, van Rhijn BW, Have CL, Treurniet KM, van der Kwast TH. Analysis of papillary urothelial carcinomas of the bladder with grade heterogeneity: supportive evidence for an early role of CDKN2A deletions in the FGFR3 pathway. Histopathology 2017;70:281–9. [DOI] [PubMed] [Google Scholar]
- 30.Nordentoft I, Lamy P, Birkenkamp-Demtröder K, Shumansky K, Vang S, Hornshøj H, et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep 2014;7:1649–63. [DOI] [PubMed] [Google Scholar]
- 31.Nikkilä J, Kumar R, Campbell J, Brandsma I, Pemberton HN, Wallberg F, et al. Elevated APOBEC3B expression drives a kataegic-like mutation signature and replication stress-related therapeutic vulnerabilities in p53-defective cells. Br J Cancer 2017;117:113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet 2018;50:1381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patschan O, Sjödahl G, Chebil G, Lövgren K, Lauss M, Gudjonsson S, et al. A molecular pathologic framework for risk stratification of stage T1 urothelial carcinoma. Eur Urol 2015;68:824–32; discussion 35–6. [DOI] [PubMed] [Google Scholar]
- 34.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016;387:1909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019;51:202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.