

Abstract
Subependymal giant-cell astrocytomas (SEGAs) are slow-growing brain tumors that are a hallmark feature seen in 5–10% of patients with Tuberous Sclerosis Complex (TSC). Though histologically benign, they can cause serious neurologic symptoms, leading to death if untreated. SEGAs consistently show biallelic loss of TSC1 or TSC2. Herein, we aimed to define other somatic events beyond TSC1/TSC2 loss and identify potential transcriptional drivers that contribute to SEGA formation. Paired tumor-normal whole-exome sequencing was performed on 21 resected SEGAs from 20 TSC patients. Pathogenic variants in TSC1/TSC2 were identified in 19/21 (90%) SEGAs. Copy neutral loss of heterozygosity (size range: 2.2–46 Mb) was seen in 76% (16/21) of SEGAs (44% chr9q and 56% chr16p). An average of 1.4 other somatic variants (range 0–7) per tumor were identified, unlikely of pathogenic significance. Whole transcriptome RNA-sequencing analyses revealed 190 common differentially expressed genes in SEGA (n = 16, 13 from a prior study) in pairwise comparison to each of: low grade diffuse gliomas (n = 530) and glioblastoma (n = 171) from The Cancer Genome Atlas (TCGA) consortium, ganglioglioma (n = 10), TSC cortical tubers (n = 15), and multiple normal tissues. Among these, homeobox transcription factors (TFs) HMX3, HMX2, VAX1, SIX3; and TFs IRF6 and EOMES were all expressed >12-fold higher in SEGAs (FDR/q-value < 0.05). Immunohistochemistry supported the specificity of IRF6, VAX1, SIX3 for SEGAs in comparison to other tumor entities and normal brain. We conclude that SEGAs have an extremely low somatic mutation rate, suggesting that TSC1/TSC2 loss is sufficient to drive tumor growth. The unique and highly expressed SEGA-specific TFs likely reflect the neuroepithelial cell of origin, and may also contribute to the transcriptional and epigenetic state that enables SEGA growth following two-hit loss of TSC1 or TSC2 and mTORC1 activation.
Introduction
Subependymal giant cell astrocytomas (SEGAs) are slow growing, glioneuronal brain neoplasms that represent 2% of all pediatric brain tumors. SEGAs are seen almost exclusively in patients with tuberous sclerosis complex (TSC, MIM# 191100, 191092). Approximately, 5–10% of patients with TSC develop SEGAs, of which nearly all are diagnosed in childhood [1–4]. TSC is a multisystem neurocutaneous genetic syndrome that affects multiple organ systems, including the central nervous system (CNS) with cortical tubers, white matter heterotopias, subependymal nodules (SENs), and SEGAs [3, 4]. SEGAs have been shown to have either TSC1 or TSC2 biallelic inactivation in about 80% of cases, following the classic Knudson two hit model, leading to complete loss of function of the tuberin-hamartin protein complex and mTORC1 (mechanistic Target Of Rapamycin Complex 1) hyperactivation [1, 5, 6]. Other genetic aberrations, including partial loss of chr22 and BRAF V600E mutation have been reported in a few cases [7, 8].
SEGAs develop from benign, smaller, histologically similar lesions, known as subependymal nodules (SEN), near the foramen of Monro. Although SEGAs are benign histologically, they can cause serious neurological complications, including obstructive hydrocephalus, intractable seizures, and if left untreated can lead to death. Clinical diagnosis is based on neuroimaging of TSC patients where SEGAs are defined as having maximum diameter ≥10mm and/or growth seen on serial scans [2]. There are occasional patients in whom SEGA-like lesions are seen and other features of TSC are not prominent, as well as TSC patients in whom the SEGA location is atypical. Histologically, SEGAs consist of large cells resembling gemistocytic astrocytes that are arranged in fascicles, sheets and nests; the tumor cells show variable expression of glial and neuronal markers, with high levels of cytoplasmic phospho-S6K, phospho-S6, and phospho-Stat3, proteins downstream of mTORC1 [1, 5].
Treatment options for SEGAs include mTORC1 allosteric inhibitors, rapamycin (sirolimus) and everolimus, termed rapalogs, as well as surgical resection. However, resection is challenging due to their deep intracranial location. On the other hand, continuous rapalog treatment is required as tumors can regrow when rapalog therapy is discontinued [2, 9–11].
The evidence that additional genetic events beyond TSC1/TSC2 biallelic inactivation lead to SEGA formation has been limited to date [5, 6]. Previous studies have shown that SEGAs have a distinct gene expression profile compared to periventricular normal brain [7, 12, 13]. However, a comparison to other brain tumors has not been performed previously and several questions remain regarding the pathogenic mechanisms involved in SEGAs.
Herein, we aimed to define the prevalence of other somatic genetic events that might contribute to SEGA formation in a large series of resected SEGA tumors. In addition, we identified differentially expressed genes (DEGs) in SEGAs in comparison to a broad panel of CNS tumors and cortical tubers, in order to provide insight as to their cell of origin, unravel novel aspects of their tumor biology, as well as to identify potential transcriptional driver events for SEGAs.
Materials and methods
Patient recruitment and tumor collection
This study was conducted in compliance with Partners Human Research Committee Institutional Review Board Approval (2011P002651) at the Brigham and Women’s Hospital and in compliance with the Office of Human Research Studies (DF/HCC 10-417) at the Dana Farber Cancer Institute. Signed informed consent or waiver of consent was obtained from patients and/or their guardians, and the sample collection and usage was in accordance with the policies of the institutional review boards at the respective institutions. All samples were de-identified for analysis; we followed the Declaration of Helsinki (World Medical Association Declaration of Helsinki, 2013. https://doi.org/10.1001/jama.2013.281053). The inclusion criteria in the present study were diagnosis of SEGAs, confirmed by histopathological assessment accordingly to the 2016 WHO classification for CNS tumors (Fig. 1a, b) by experienced neuropathologists, and a definite clinical diagnosis of TSC based on current criteria [14–17].
Fig. 1: MRI images and histologic features of SEGAs.
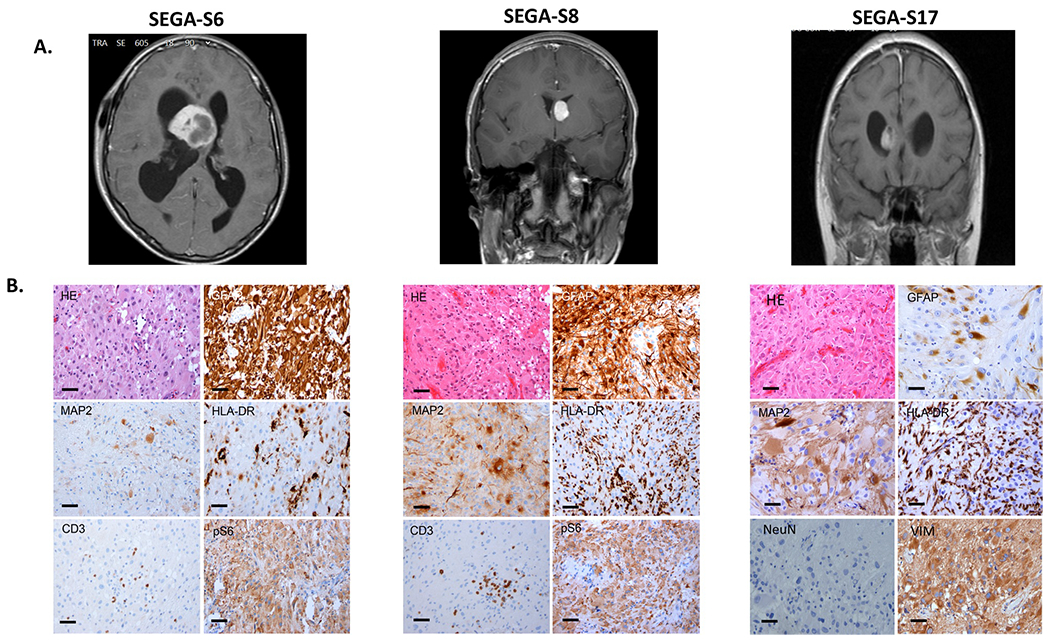
a T1-weighted coronal or sagittal gadolinium-enhanced MRI images showing SEGAs in the right lateral ventricle near the foramen of Monro (SEGA-S6, SEGA-S17), or in a similar location on the left (SEGA-S8) in 3 TSC subjects before surgical resection. b H&E and representative IHC images of the three corresponding SEGA tumors (SEGA-S6, S8, S17). H&E staining shows classical histological SEGA features, with giant cells in a mixed glial background with blood vessels. Variable expression of the glial marker GFAP is seen, with diffuse immunoreactivity in 2 of 3 samples shown here, with less staining for MAP2. HLA-DR staining highlights variable numbers of microglial cells, and CD3 staining shows presence of scant intratumor T lymphocytes; Variable expression of the neuronal marker MAP2 is seen. IHC is negative for the neuronal nuclear marker NeuN, but positive for intermediate filament vimentin (SEGA-S17). Activation of mTORC1 in tumor giant cells is indicated by expression of pS6. Scale bars: 100 μm.
Fresh-frozen surgically resected tumor specimen and/or formalin-fixed paraffin-embedded (FFPE) samples (n = 21) and matching peripheral blood as normal control for comparison were obtained from 20 patients with TSC (male n = 10, female n = 10, age range: 1–47, median = 13.5 years), for exome sequencing analysis. Clinicopathological data were available for all cases (Table 1).
Table 1.
Clinicopathological data for 20 TSC patients with 21 resected SEGA tumors.
| Subject | Age (at surgery)-years | Gender | Tumor location | Tumor size (cm) | Treatment prior to surgery | Treatment | Epilepsy | Age at onset epilepsy | Seizure frequency prior to surgery | Other clinical manifestations |
|---|---|---|---|---|---|---|---|---|---|---|
| SEGA-S1 | 1 | M | Right lateral ventricle | 3 × 3 | Yes | Antiepileptic/sirolimus (non-responder) | Yes (drug-resistance ) | 1 month | Daily | Cortical tubers |
| SEGA-S2 | 2 | M | Left ventricle | 2.5 × 3.5 × 3 | Yes | Antiepileptic | Yes (severe) | 1 month | Daily | Cortical tubers, PKD, cystic tubers, autism, severe ID, renal insufficiency |
| SEGA-S3 | 6 | M | Right ventricle | 6 × 4.4 × 4.5 | Yes | Antiepileptic | Yes | 2 months | 2–3/week | Cortical tubers, cardiac rhabdomyoma, FA, ID, autism, hydrocephalus, vomiting, vision loss |
| SEGA-S4 | 9 | M | Left lateral ventricle | 2 × 1.5 × 0.4 | Yes | Antiepileptic | Yes (with complex partial seizures) | 15 months | Weekly/biweekly subtle seizures | Bicuspid aortic valve, right FR cortical tuber and right TL subcortical tuber |
| SEGA-S5 | 9 | M | Right ventricle (foramen Monro) | 1.2 × 1.2 × 1.4 | Yes | Topical rapamycin 1% ointment for facial lesions | No | N/A | N/A | Cortical tubers, HMM, FA, periungual fibromas, SENS, renal AMLs, above average intellect |
| SEGA-S6 | 14 | M | Intraventricular, cerebrum | 4 | Yes | Antiepileptic | Yes | 4 years | N/A | Cortical tubers, kidney AML, visual loss, normal intellect |
| SEGA-S7 | 14 | M | Two SEGAs, one in right and one in left ventricle | RV: 3.5 × 2 × 2.5, LV: 2.5 × 1.5 × 0.8 | No | N/A | No | N/A | N/A | Cortical tubers, mild FA, normal intellect, acute hydrocephalus |
| SEGA-S8 | 15 | M | Left ventricle | 1.5 | Yes | Antiepileptic | Yes | 10 years | Daily nondisabling (5–6 per day) | Cortical tubers, kidney AML, normal intellect |
| SEGA-S9 | 17 | M | Right ventricle (foramen Monro) | 3.5 × 3 × 2.5 | No | N/A | No | N/A | N/A | History of morning headache and vomiting (raised ICP symptoms related to hydrocephalus), mild ID |
| SEGA-S10 | 23 | M | Ventricle (foramen Monro) | N/A | Yes | Antiepileptic | Yes | 3 years | 2–3 per 6 months | Cortical tubers, kidney AML, normal intellect |
| SEGA-S11 | 1 | F | Right lateral ventricle | 3 × 3 | Yes | Antiepileptic | Yes | 1 month | Daily | Cortical tubers |
| SEGA-S12 | 6 | F | Left lateral ventricle | 6 × 5 × 2 | Yes | Antiepileptic | Yes (drug-resistance ) | 3 days | Daily seizures | Cortical tubers, severe TSC presentation: cortical dysplasia (surgery at age 1), FA, kidney AML, cardiac rhabdomyoma, severe ID, autism, acute hydrocephalus |
| SEGA-S13 | 11 | F | Frontal horn of right lateral ventricle | 2.4 × 1.7 × 2.3 | No | N/A | No | N/A | N/A | Cortical and subcortical tubers, subependymal nodules, FA, shagreen patch, hypomelanotic macules, renal angiomyolipomas, retinal hamartomas, normal development, complaints of blurry vision and some nausea |
| SEGA-S14 | 13 | F | Left lateral ventricle | 4 × 3 | Yes | Antiepileptic | Yes | 17 months | N/A | Cortical tubers |
| SEGA-S15 | 13 | F | Right ventricle | 1 × 2 × 3 | Yes | Antiepileptic | Yes | 9 months | No seizures for >2 years | Cortical tubers, FA, kidney AML |
| SEGA-S16 | 14 | F | Right ventricle | 3.5 × 2.5 × 0.8 | Yes | Antiepileptic | Yes | 12 months | No seizures for >2 years | Cortical tubers, kidney AML, FA, hydrocephalus, vision loss |
| SEGA-S17 | 14 | F | Intraventricular | N/A | No | N/A | No | N/A | N/A | Cortical tubers, kidney AML, normal intellect |
| SEGA-S18 | 22 | F | Left lateral ventricle | 3.5 × 1.5 × 0.6 | Yes | Antiepileptic | Yes (intractable) | N/A | N/A | Cortical tuber, hydrocephalus, fibroadenoma of right breast, hydrocephalus, malignant neoplasm of cerebrum, except lobes and ventricles |
| SEGA-S19 | 47 | F | Right frontal | 1.5 × 0.8 × 0.2 | No | No | No | no | no | FA, SEN, kidney AML, renal cysts, 3 healthy offspring |
| SEGA-S20 | 17 | F | Right lateral ventricle | 1.0 × 0.8 × 0.7 | No | N/A | Yes | 0 | Uncertain | Cortical tubers, ASD, cognitive impairment, behavioral issues, renal AMLs, FA, oral fibromas |
M male, F female, PKD polycystic kidney disease, AML angiomyolipoma, ID intellectual disability, ASD autism spectrum disorder, HMM hypomelanotic macules, FA facial angiofibroma, SEN subependymal nodule, RV right ventricle, LV left ventricle, ICP Increased Intracranial Pressure.
Histopathology studies
Conventional hematoxylin and eosin (H&E) staining was performed. The tumor-cell content was estimated to be at least 80% in all SEGA cases by H&E staining, except for one case (SEGA-S19) in which tumor purity was reduced due to the contamination with non-tumor and inflammatory cells. Immunohistochemistry (IHC) was performed on 5-micron tissue sections as described previously [6, 18]. Antibodies against the following proteins were used for IHC: MAP2 (mouse clone HM2, Sigma 1:100), HLA-DR (mouse clone CR3/43, DAKO, 1:100), CD3 (mouse monoclonal, clone F7.2.38, DAKO; 1:200), pS6 (rabbit polyclonal, Cell Signaling Technology, 1:50), GFAP (rabbit polyclonal, DAKO, 1:4,000), Vimentin (mouse clone V9, DAKO, 1:1,000), NeuN (mouse clone MAB377, Chemicon, 1:2,000); IRF6 (mouse monoclonal, Origene, Cat. No. UM500074, 1:500); SIX3 (rabbit polyclonal, LSBio, Cat. No. LS-B9336–50, 1:4000); VAX1 (mouse monoclonal, Origene, Cat. No. CF811439; 1:100). Information for additional IHC staining is included in Supplementary material.
Exome sequencing methods
Exome hybrid capture, library preparation, massively parallel sequencing (MPS), and bioinformatic analyses were performed at the Broad Institute of MIT and Harvard, following standard methods. Briefly, genomic DNA was sheared in a Covaris sonicator (Covaris, Woburn, MA, USA) to fragments of 200–500 bp, and subject to capture using the MPS Illumina Exome (37.7 Mb of mainly exonic territory; Agilent SureSelect All Exon V2) [19, 20]. Pooled indexed libraries were sequenced on either the HiSeq 2000 or HiSeq 2500 instrument (Illumina platform), using 76 bp paired-end sequencing. The mean coverage for the targeted region was 105x (range: 49–267x) for all tumors and 113x (range: 47–192x) for normal samples. An average of 76% in the targeted region (range: 46–93%) was covered at ≥50x for tumor, whereas it was 92% at ≥50x (range: 80–95%) for normal.
Pre-processing and bioinformatics analysis of MPS data
Demultiplexing/sample deconvolution, base alignment and sequence quality control were performed using Picard tools and the Firehose pipeline at the Broad Institute of MIT and Harvard. Fastq files were generated and unique reads were aligned to the human reference genome GRCh37(hg19) using BWA (v0.7.3a) [http://arxiv.org/abs/1303.3997] and Bowtie 2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml). Variant calling for single nucleotide variants (SNVs) and insertion/deletions (indels) was performed using Haplotype Caller in Genome Analysis Toolkit (GATK, v3.0) Best Practices. Sequencing data were then analyzed using the Cancer Genome Analysis pipeline, as well as custom code in Python, Matlab and Unix to enable the detection of TSC1/TSC2 deleterious sequence variants with low mutant allele frequency (MAF) [5]. Somatic point mutations were called by MuTect (http://www.broadinstitute.org/cancer/cga/mutect/) and short insertions and deletions in sequencing data were identified by SomaticIndelDetector. All variants were annotated using Oncotator (http://www.broadinstitute.org/oncotator). We used ABSOLUTE to estimate tumor purity, tumor cell ploidy, and to determine chromosomal copy-numbers genome-wide [21]. All somatic variant calls were reviewed manually using IGV (https://software.broadinstitute.org/software/igv/), and those reflecting sequencing or other artifacts were excluded. Variant nomenclature was confirmed by Mutalyzer (https://mutalyzer.nl/). SIFT and PROVEAN in silico prediction tools (http://provean.jcvi.org/index.php) were used to assess the functional effects of missense variants. Called variants were validated by either Sanger sequencing (variants with MAF > 10%) or amplicon MPS method (variants with MAF < 10%) for 10 out 21 SEGAs with DNA material available [22, 23].
Whole transcriptome RNA sequencing
Paired end RNA-sequencing (RNA-seq) was performed on 3 SEGA tumors (SEGA-S3, S4, S19 with matching exome data) at the Broad Institute of MIT and Harvard, following standard methods (Illumina platform). Additional RNA-seq raw data were obtained from 13 SEGAs from a previous study [6] and were reanalyzed for uniformity and downstream analyses. After quality assessment and filtering for all SEGA tumors, raw reads were mapped or aligned to the reference genome GRCh37 (hg19) build using the STAR program [24]. VIPER (Visualization Pipeline for RNA-seq analysis) [25] QC analysis for all combined 16 SEGAs revealed high quality sequencing data with a median of ~25 million paired end reads generated for each tumor (range: 21–97M reads). FPKM normalized values for all genes and their isoforms were generated using Cufflinks v2.2.1. Raw data were also converted into RSEM format for comparison to other brain and adult solid tumors from The Cancer Genome Atlas (TCGA) consortium and 10 gangliogliomas [26–29]. We also performed gene fusion analysis to identify any gene rearrangements using FusionInspector (https://github.com/FusionInspector/FusionInspector/), as a previous study reported a single SEGA case with PRRC2B-ALK fusion [30].
Statistical analysis
Statistical analyses were performed using the non-parametric Mann–Whitney U-test in GraphPad Prism software (Graphpad Software Inc., La Jolla, CA). All p-values were corrected applying the Benjamini–Hochberg method. An adjusted p-value/FDR < 0.05 was considered statistically significant. The following convention was used in all figures: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
Results
Clinical and routine diagnostic studies
In this study, we evaluated 21 SEGAs from 20 patients with TSC. Pre-operative brain MRIs demonstrated that these tumors were present on the medial or lateral ventricular wall, which is typical for these lesions (Fig. 1a and Table 1). H&E staining showed classic histologic features for SEGAs, featuring plump cells with abundant glassy eosinophilic cytoplasm and enlarged nuclei with distinct nucleoli (Fig. 1b). IHC showed variable expression of GFAP, MAP2, and HLA-DR; with consistent labeling for pS6, a marker of mTORC1 activation; and variable numbers of CD3+ cells (Fig. 1b).
TSC1/TSC2 mutation analysis of SEGAs
Twenty-one paired SEGA-normal samples were analyzed by exome sequencing. TSC1 and TSC2 germline pathogenic variants were identified in 18 of 20 (90%) patients (6 TSC1 mutations and 12 TSC2 mutations) and were similar to the known pathogenic variant spectrum for these genes (Fig. 2a and Table S1; SEGA-S15 had a large TSC1 deletion, as indicated by focal reduction in read depth for exons 18–23) [4]. Copy neutral loss of heterozygosity (CN-LOH) was seen in the TSC2 region of chr16p in 9 of 12 (82%) tumors from subjects with pathogenic TSC2 variants; size range: 2.2–30.3 Mb (Fig. 2b, Fig. S1 and Table S1). CN-LOH was also seen in chr9q encompassing TSC1 in 6 SEGA tumors from subjects with pathogenic TSC1 variants; size range: 6.7–46 Mb (Fig. 2b, Fig. S1 and Table S1). One tumor from one subject (SEGA-S17), with no germline mutation identified in either TSC1 or TSC2, had CN-LOH in chr16p, suggesting an occult germline mutation in TSC2 (Table S1 and Fig. S1). CN-LOH regions had a variable size on both chromosome 9 and 16, likely reflecting random mitotic recombination events, as seen previously in TSC kidney angiomyolipoma [31].
Fig. 2: Germline and somatic alterations in SEGAs.
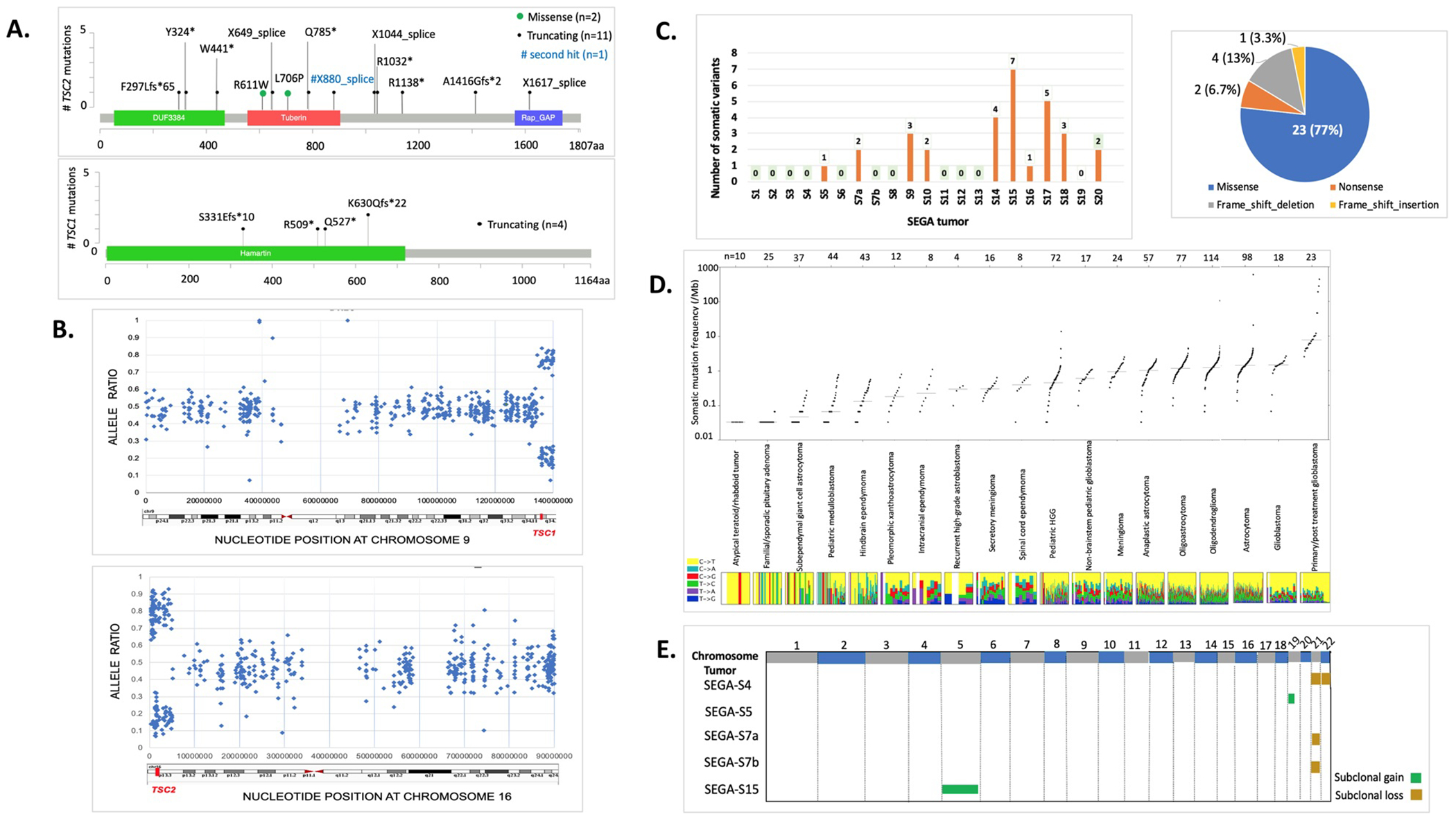
a Position and type of germline and somatic TSC1/TSC2 pathogenic variants in 19 SEGAs. b Examples of Copy Neutral Loss of Heterozygosity (CN-LOH) in two representative SEGAs (SEGA-S9 and S13), encompassing chr9:134,398,493–141,070,719 (6.7 Mb, top) and chr16:304,514–4,942,099 (4.64 Mb, bottom) (GRCh37/hg19). SNP allele frequency (AF) distribution is shown across the entire chromosome with regions of AF skewed from the expected 0.5 (range: 0.4–0.6) in regions surrounding TSC1 (top) or TSC2 (bottom). Each blue dot depicts a single SNV. c Number and type of small somatic variants in 21 SEGA tumors. d Somatic mutation frequency per Mb in SEGAs in comparison to a broad range of pediatric and adult brain tumors. Each dot in the plot represents a different tumor. Y axis is log scale. e Five of 21 (19%) SEGAs harbor subclonal chromosomal copy number gain (green) or loss (brown).
Exome sequence analysis
Exome sequencing revealed 30 somatic point variants/indels in 10 of 21 (45%) tumor-normal matched samples, with a range: 0–7 variants/tumor (overall median 0, average 1.4, Fig. 2c and Table S2). None of these 30 somatic variants occurred in the same gene in different samples and 20 of 30 (67%) were subclonal, determined by ABSOLUTE. Twenty-three of the 30 variants (77%) were missense changes, of which 15 (65%) were classified as likely deleterious/damaging by in silico prediction analysis. None of the genes with somatic alterations were known ‘cancer genes’ [32]. These results suggest that these variants were likely passenger events that do not contribute to SEGA formation. We did not observe any mutations in BRAF in this SEGA cohort (Table S2), similar to our findings in a previous SEGA cohort [5].
We then compared the somatic mutation rate that we observed in SEGAs from our internal cohort (n = 21) including 16 tumors from a previous study [6], with rates that have been reported for a wide variety of other brain tumors (Fig. 2d and Table S3), as well as other TCGA tumors (Fig. S2) [33]. SEGAs in our analysis showed a similar, though slightly lower, mutation burden (SNVs per Mb) to pediatric medulloblastoma and hindbrain ependymoma, and a substantially lower mutation burden than both pediatric and adult low-grade gliomas.
Copy number alteration (CNA) analysis revealed subclonal chromosomal gains and/or losses in 5/21 (24%) tumors at either the arm or whole chromosome level, including chr21 and chr22 loss; and chr5 and chr19p gain (Fig. 2e). These results are similar to those reported previously, in which CNAs were seen in 3/14 (21%) SEGAs for several chromosomes (1, 5, 7, 11, 12, 17, and 19) [6].
Gene expression RNA-seq analyses
We analyzed RNA-seq data for 16 SEGA tumors (see “Materials and methods”) in comparison to the TCGA brain tumor cohort, including low grade gliomas (LGG; n = 530) and TCGA adult glioblastomas (GBM; n = 171), as well as cortical tubers (n = 15), SEN (n = 2), and normal brain samples (n = 11) [6, 29]. This set of 16 SEGA tumors was relatively pure, as reflected by the mutant allele frequency of either TSC1 or TSC2 mutation for each (SEGA-S3, S4, S20 and Martin et al. 2017 [6]).
To discern the potential relationship between SEGA and the large panel of brain tumors studied by TCGA, we performed a de novo clustering analysis for 3060 most variable genes in the combined cohort (n = 745) by consensus Bayesian non-negative matrix factorization, and identified four distinct clusters (Cluster 1–4 in Fig. 3a and Table S4) [34]. Of note, 15 of 16 SEGAs were co-clustered with most GBM samples (95%, 163 out of 171) and 20% of LGG samples (103 out of 530) in Cluster 3. The set of 103 LGG in cluster 3 included 60 astrocytomas, 15 oligoastrocytomas, 16 oligodendrogliomas, and 12 unclassified brain tumors. Note that we recognize that the oligoastrocytoma designation for a subset of gliomas is no longer used; we retain that term here to be consistent with nomenclature used in TCGA project. The single SEGA that was not in cluster 3 was from Martin et al. [6] and was grouped in Cluster 1 with normal brain, suggesting that it was highly contaminated with normal brain.
Fig. 3: Comparison of RNA-Seq expression of SEGAs to other brain tumors and cortical tubers.
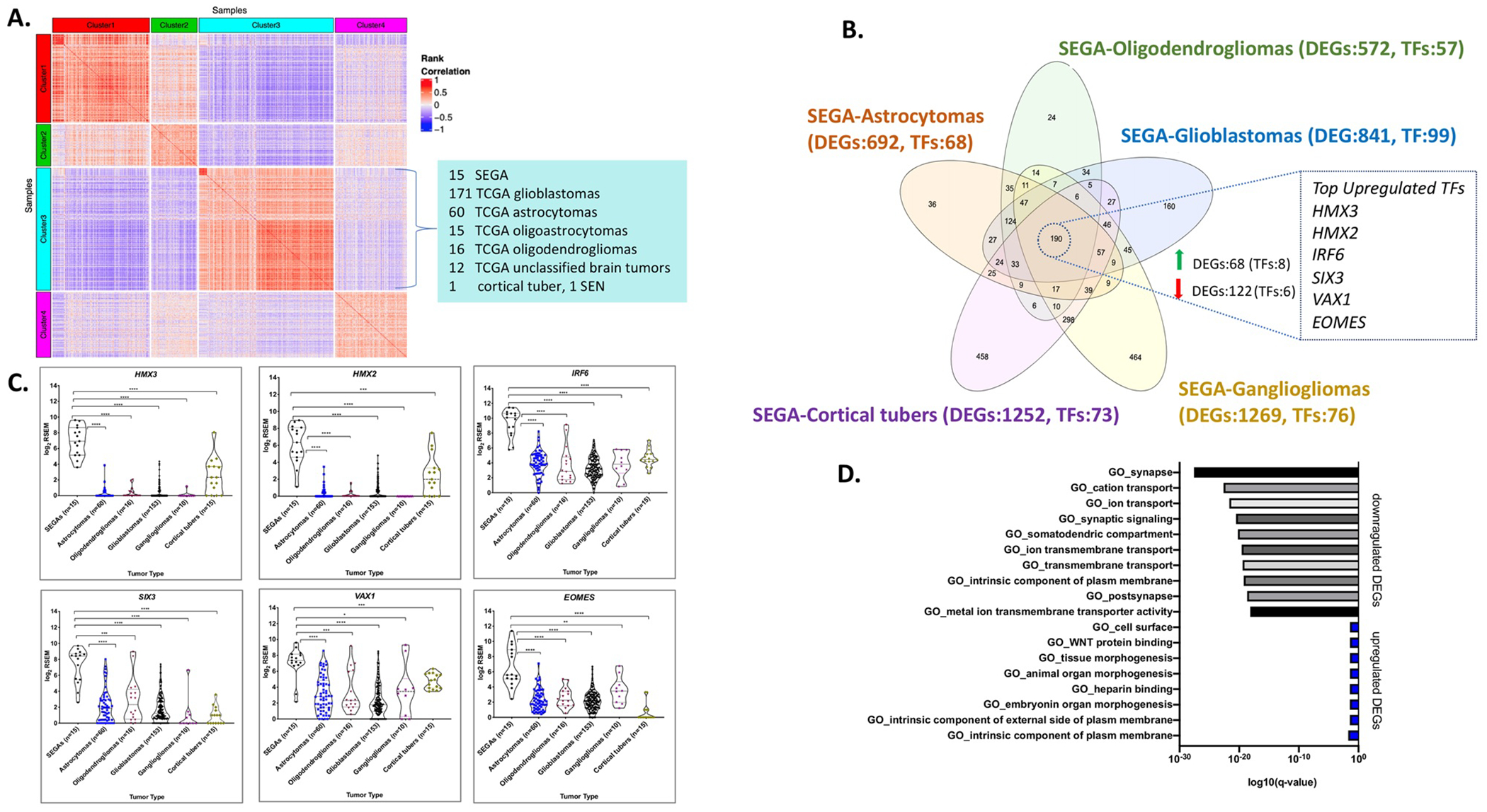
a A heatmap is shown reflecting a de novo clustering analysis using 3060 most variable genes in the combined cohort of: SEGAs (n = 16), TCGA LGG (n = 530), TCGA GBM (n = 171), cortical tubers (n = 15), SEN samples (n = 2), and normal brain samples (n = 11). Pairwise sample by sample Spearman rank correlation was determined and used to generate the heatmap that is shown. Fifteen of 16 (96%) SEGAs are in Cluster 3, form the red square in the upper left corner of Cluster 3, reflecting a high rank correlation among them. Cluster 3 also contains most GBM (n = 163) and a subset of TCGA LGG (n = 103). b Venn diagram of all DEGs (FDR < 0.05) in SEGAs versus each of five other histological subtypes of brain tumors/cortical tubers. Notably, a set of 190 genes were differentially expressed in SEGAs compared to each of the other five sample types. The TFs that are upregulated in SEGAs are shown at right. c Violin dot plots of the top 6 TFs that were differentially and highly expressed in SEGAs compared to other brain tumors and cortical tubers. d GO pathways showing enrichment for the 190 DEGs: top, pathways enriched in genes downregulated in SEGA; bottom, pathways enriched in genes upregulated in SEGA.
To discern genes whose expression was specifically altered in SEGAs, and might contribute to SEGA development, we performed DESeq2 analyses using normalized read counts (RSEM). Pair wise comparisons were made between the SEGAs and each of the histological subtypes of LGG and GBM in Cluster 3 (www.qlucore.com) to identify gene expression differences (Fig. 3a, b) [29, 34, 35]. We identified several hundred genes that were differentially expressed between SEGAs and other tumors at a false discovery rate (FDR)/q-value < 0.05 and p-value < 0.002 (Fig. 3b, Fig. S3a–f and Tables S5–S9).
Analysis of multiple housekeeping genes (e.g., GAPDH, LDHA, SDHA, and ABCF1) across different tumor samples analyzed showed no significant differences in expression, confirming a lack of artifact or bias due to different sample sets, library preparation, and/or sequencing methods (Fig. S4). A set of 190 genes was found to be differentially expressed in SEGAs, all consistently in the same direction (either up or downregulated), in comparison to each of the other sample types (Fig. 3b, Table S10). One hundred fifty one of these 190 common DEGs were also differentially expressed in SEGAs in comparison to normal brain (46 upregulated, 105 downregulated; FDR < 0.05, |log2fold change| ≥ 1) (Table S10).
Since many TFs regulate cell lineage and act as drivers of cancer growth in different cancer types, we chose to focus our analysis on TFs with higher expression in SEGAs compared to these other entities. Fourteen of 190 DEGs were TFs, of which 8 were upregulated (Table 2 and Table S11), including HMX3, HMX2, IRF6, SIX3, EOMES, and VAX1, each with a median fold change ≥ 12 (Fig. 3c and Table 2). We also found that the expression of HMX3, HMX2, SIX3, and VAX1 was much higher in SEGAs than any other TCGA cancer type (2463 tumors of 27 different histological types) and normal tissues {~8500 samples from 30 normal tissue types; the Gene and Tissue Expression (GTEx) project} (Figs. 4, 5) [36]. IRF6 and EOMES were also highly expressed in SEGAs but were also seen at relatively high levels in some other cancer types and normal tissues (Figs. 4, 5). In addition, SIX3 was relatively highly expressed in pituitary gland (Figs. 4, 5). ZBTB20, another TF, was highly expressed in SEGAs compared to all brain tumors and cortical tubers, except for gangliogliomas where it was also highly expressed. ZBTB20 was also much more highly expressed in SEGAs than all normal tissues (Figs. S5, S6).
Table 2.
List of the top 44 upregulated DEGs, common to all pairwise comparisons between SEGAs and other brain tumors/cortical tubers, with a median fold change >= 10 and FDR/q-value < 0.05.
| SEGAs to TCGA astrocytomas | SEGAs to TCGA oligodendrogliomas | SEGAs to TCGA glioblastomas | SEGAs to gangliogliomas | SEGAs to cortical tubers | ||
|---|---|---|---|---|---|---|
| Gene ID | Fold change | Fold change | Fold change | Fold change | Fold change | Median fold change |
| HCRTR2 | 253.2 | 239.6 | 309.5 | 154.7 | 34.9 | 239.6 |
| SFTA3 | 168.1 | 152.6 | 196.7 | 203.5 | 106.3 | 168.1 |
| SPAN8 | 141.8 | 148.4 | 324.7 | 240.7 | 77.1 | 148.4 |
| KCNK12 | 130.3 | 164.3 | 195.4 | 36.0 | 11.7 | 130.3 |
| LHCGR | 111.7 | 85.1 | 102.5 | 14.7 | 60.7 | 85.1 |
| SLN | 81.0 | 125.1 | 46.2 | 1082.1 | 39.3 | 81.0 |
| HMX3 | 79.8 | 70.2 | 76.5 | 88.9 | 17.5 | 76.5 |
| HMX2 | 51.1 | 55.0 | 58.1 | 83.5 | 14.9 | 55.0 |
| SLC14A2 | 39.3 | 35.1 | 55.2 | 53.8 | 86.2 | 53.8 |
| FRZB | 69.6 | 48.1 | 41.1 | 47.0 | 11.7 | 47.0 |
| IRF6 | 37.2 | 44.8 | 71.1 | 50.9 | 28.2 | 44.8 |
| SIX3 | 30.3 | 18.1 | 43.0 | 71.1 | 75.5 | 43.0 |
| TMPRSS2 | 29.3 | 25.2 | 41.9 | 116.8 | 57.3 | 41.9 |
| TRIM63 | 35.4 | 52.2 | 32.2 | 35.8 | 7.1 | 35.4 |
| TRDN | 35.3 | 44.5 | 63.8 | 11.0 | 6.9 | 35.3 |
| GPNMB | 35.3 | 31.2 | 31.5 | 77.0 | 19.9 | 31.5 |
| LOC148145 | 14.9 | 29.7 | 38.0 | 66.4 | 6.0 | 29.7 |
| HGD | 30.3 | 25.9 | 11.1 | 25.2 | 4.3 | 25.2 |
| SLC39A2 | 24.1 | 30.5 | 16.7 | 30.3 | 11.0 | 24.1 |
| FOLR1 | 18.5 | 13.5 | 18.1 | 162.6 | 56.1 | 18.5 |
| SFRP1 | 18.3 | 18.6 | 33.9 | 18.0 | 8.1 | 18.3 |
| ITIH1 | 18.1 | 18.3 | 9.0 | 26.6 | 10.0 | 18.1 |
| TDGF1 | 17.9 | 30.8 | 43.6 | 15.1 | 7.7 | 17.9 |
| LRRN4CL | 8.9 | 22.9 | 7.2 | 39.5 | 16.2 | 16.2 |
| LGR5 | 16.0 | 18.3 | 30.7 | 9.8 | 10.3 | 16.0 |
| EOMES | 15.7 | 13.5 | 16.1 | 6.2 | 46.4 | 15.7 |
| GSTA1 | 16.1 | 18.6 | 13.9 | 15.4 | 8.5 | 15.4 |
| SFRP4 | 8.9 | 11.4 | 16.6 | 15.0 | 31.0 | 15.0 |
| WISP2 | 14.8 | 14.9 | 19.4 | 28.8 | 7.7 | 14.9 |
| GPRC5A | 16.8 | 11.6 | 11.7 | 47.4 | 14.7 | 14.7 |
| HKDC1 | 17.6 | 11.0 | 21.5 | 14.7 | 10.6 | 14.7 |
| GALNT5 | 11.4 | 14.6 | 12.6 | 16.7 | 61.9 | 14.6 |
| SYPL2 | 10.6 | 15.8 | 13.7 | 16.7 | 8.5 | 13.7 |
| TNNC1 | 13.4 | 14.6 | 9.3 | 16.6 | 8.2 | 13.4 |
| KCP | 13.2 | 11.7 | 10.9 | 15.5 | 12.9 | 12.9 |
| ELSPBP1 | 12.8 | 10.9 | 7.6 | 14.2 | 13.2 | 12.8 |
| VAX1 | 14.3 | 11.9 | 33.6 | 12.7 | 5.7 | 12.7 |
| FGFBP2 | 9.7 | 12.6 | 8.4 | 81.9 | 31.4 | 12.6 |
| IP6K3 | 8.7 | 12.5 | 9.6 | 13.8 | 16.4 | 12.5 |
| TMEM200A | 13.9 | 8.8 | 25.6 | 12.5 | 5.1 | 12.5 |
| TTC39A | 11.9 | 25.7 | 12.4 | 10.6 | 8.2 | 11.9 |
| GPR1 | 20.7 | 11.1 | 6.3 | 18.5 | 8.6 | 11.1 |
| F10 | 10.8 | 15.1 | 12.8 | 6.5 | 10.4 | 10.8 |
| HORMAD2 | 10.2 | 10.9 | 10.0 | 5.5 | 4.3 | 10.0 |
Genes highlighted in bold are TFs.
Fig. 4: Box plots for the top six DE (upregulated) TFs in SEGA compared to TCGA tumors (2463 tumors of 27 different histologic types), gangliogliomas and cortical tubers.
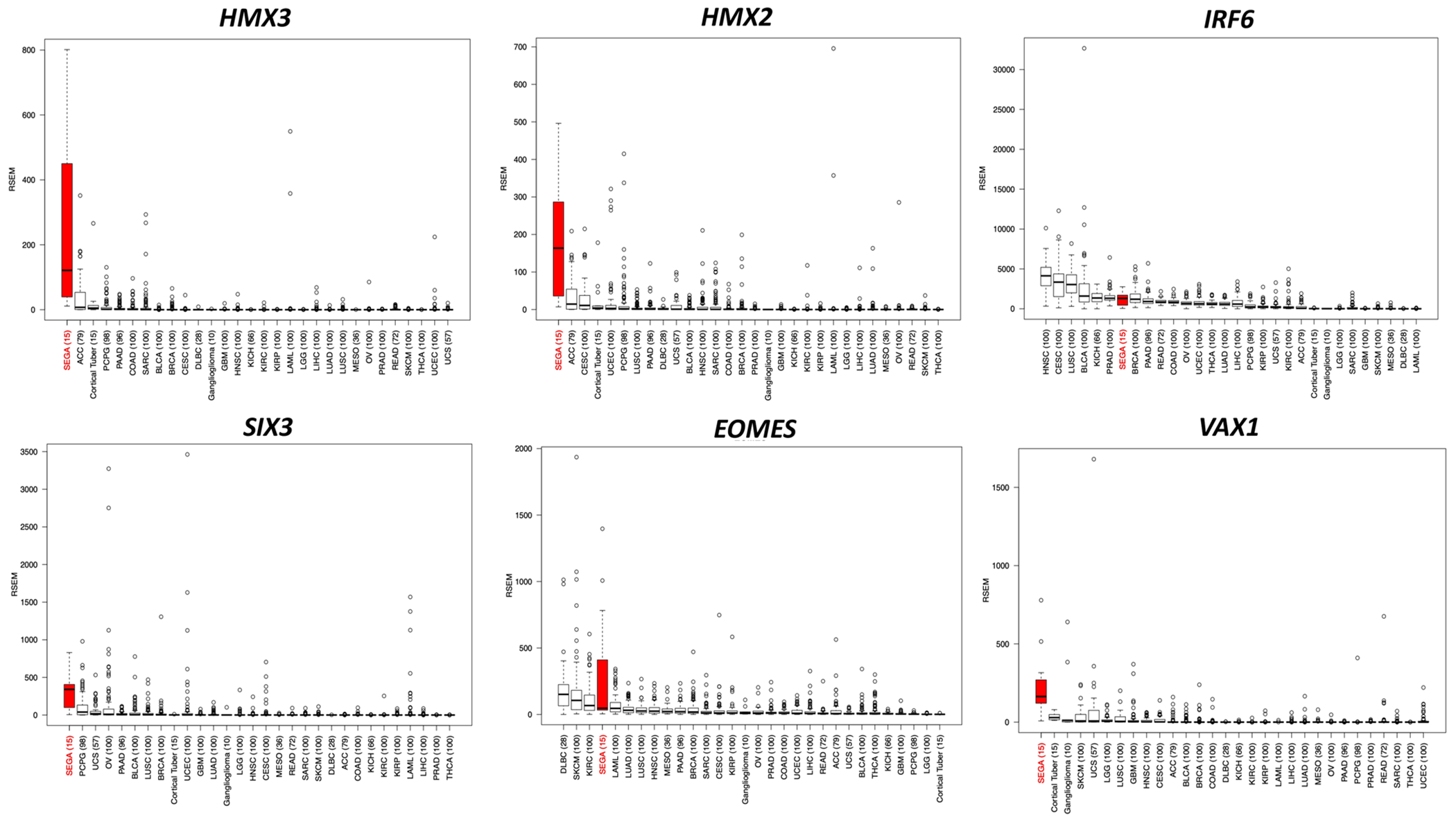
Abbreviations for all TCGA tumor types at: https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations. Gene expression is shown in RSEM values.
Fig. 5: Box plots for the top six DE (upregulated) TFs in SEGA compared to GTEx human normal tissues (~8500 samples from 30 normal tissue types, v6p release).
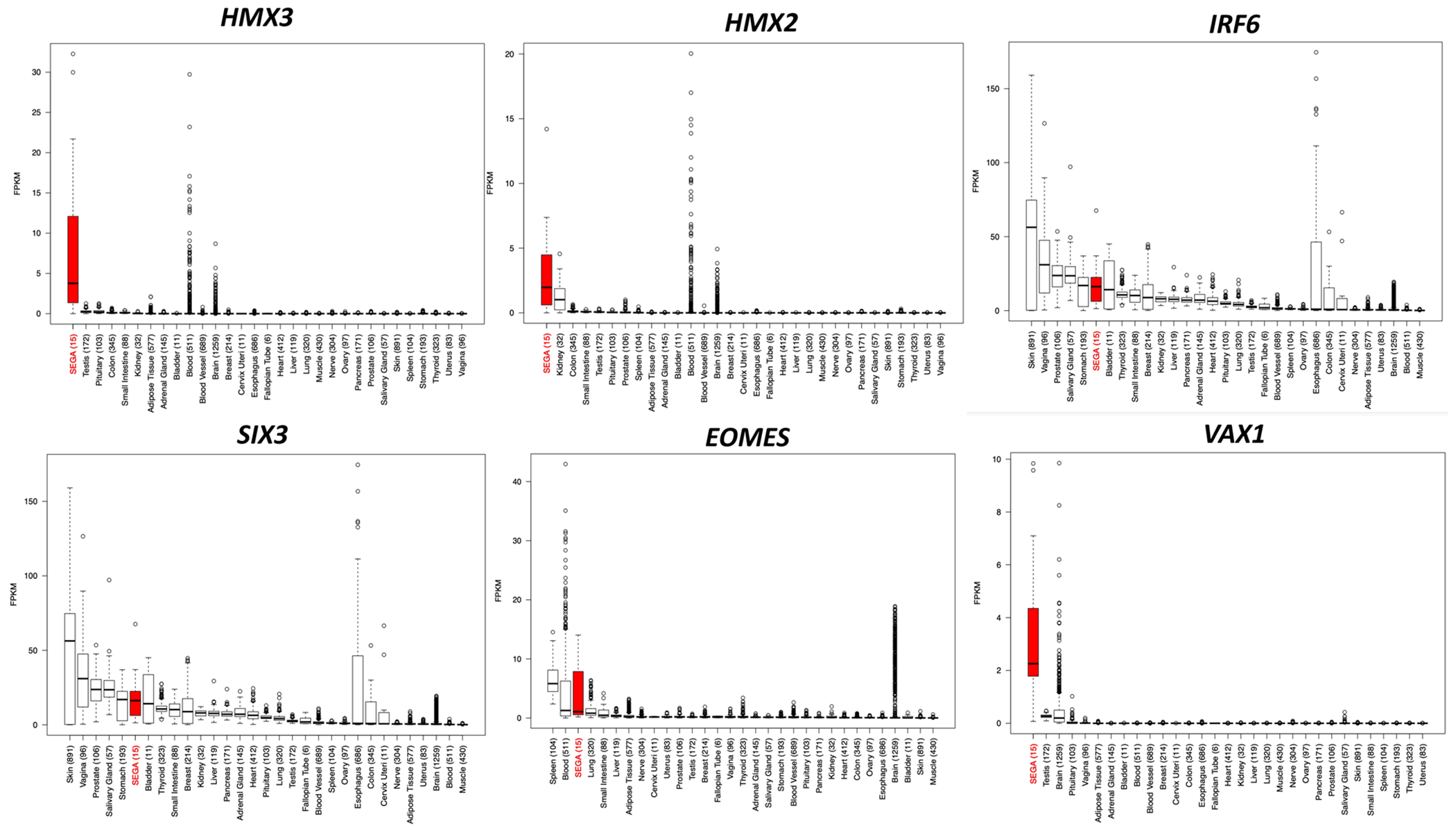
Gene expression is shown in FPKM values.
Considering other differentially expressed genes other than TFs, HCRTR2 was the most highly expressed gene in SEGAs compared to all tumors and normal tissues (Figs. S5, S6). HCRTR2 is a G-protein coupled receptor that binds the hypothalamic neuropeptides orexin A and orexin B and regulates sleep-wakefulness. Of note, a recent study reports that hypothalamic orexin and mTOR activation mediate sleep dysfunction in a mouse model of TSC [37]. GPNMB (Glycoprotein Nmb) was also a top DEG between SEGAs and other pathological entities (Table 2 and Figs. S5, S6). We had previously demonstrated that GPNMB was upregulated in Tsc2 null neuroepithelial cells [38]. No gene fusion events in any of 16 SEGAs analyzed by RNA-Seq were identified.
GSEA pathway enrichment analyses
To examine the biological pathways enriched in SEGAs, we performed Gene Set Enrichment Analysis (GSEA; https://www.gsea-msigdb.org) using all 190 common DEGs from above. Ten GO gene sets were enriched for DEGs that were downregulated in SEGAs (Fig. 3d; top; Table S12); while 8 were enriched for DEGs that were upregulated in SEGAs (Fig. 3d; bottom). The downregulated gene sets were associated with normal brain development, including GO_synapse and GO_synaptic signaling, indicating that even by comparison to these other brain tumors, SEGA have less neuronal differentiation. In contrast, the upregulated gene sets were associated with morphogenesis, cell surface and WNT protein binding.
Immunohistochemistry confirmation
To confirm that the top highly expressed TF in SEGAs, identified by RNA-seq, were also highly expressed at protein level, we performed IHC on SEGAs and multiple other pediatric and adult gliomas (Figs. 6–8) using commercially available antibodies. We stained at least 2–5 sections available per tumor type, including SEGA, cortical tuber, adult glioblastoma, pleomorphic xanthoastrocytoma (PXA), diffuse astrocytoma, oligodendroglioma, subependymoma, as well as normal brain (cortex) (detailed neuropathology evaluation for each stained marker in Table S13). Positivity was defined as moderate-to-strong nuclear or cytoplasmic immunoreactivity in at least 5–25% of cells.
Fig. 6: Representative images of IRF6 immunohistochemistry.
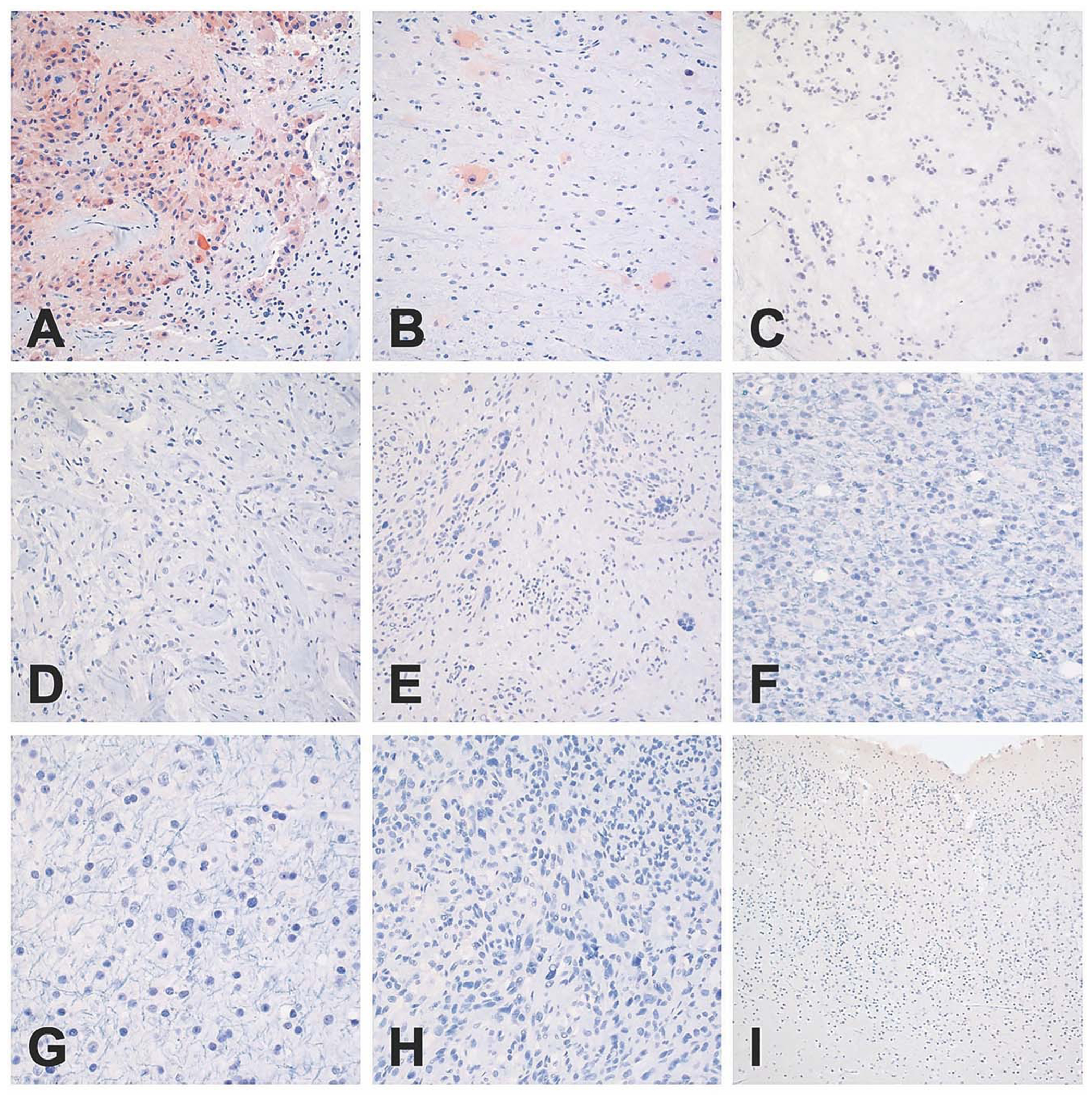
Cytoplasmic staining was observed only in SEGAs (a) and in the balloon neurons of cortical tubers (b). No cytoplasmic immunoreactivity was noted in subependymomas (c), ganglioglomas (d), PXAs (e), oligodendrogliomas (f), astrocytomas (g), glioblastomas (h), or in normal infant cortex (i). Scale bar: 400 μm, PXA: Pleomorphic xanthoastrocytoma.
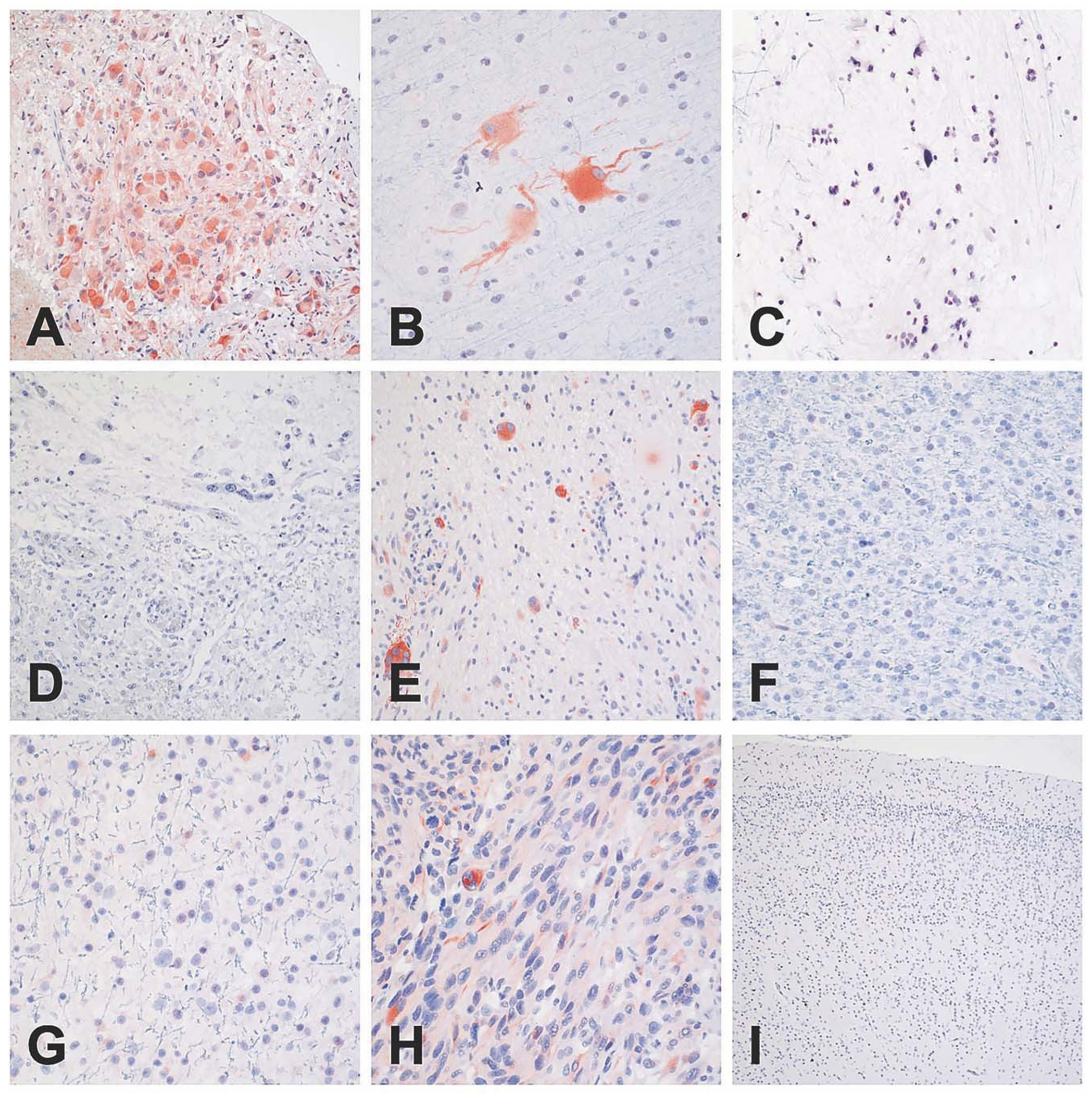
Fig. 8: Representative images of VAX1 immunohistochemistry.
Nuclear staining was observed in all SEGAs (a) and focal staining was seen in abnormal clusters of neurons, consistent with balloon neurons in cortical tubers (b). Occasional examples of subependymoma displayed focal nuclear positivity (c), and a single PXA showed positive staining in large pleomorphic tumor cells (e). No nuclear immunoreactivity was noted in ganglioglomas (d), oligodendrogliomas (f), astrocytomas (g), glioblastomas (h), or in normal infant cortex (i). Scale bar: 400 μm, PXA: Pleomorphic xanthoastrocytoma.
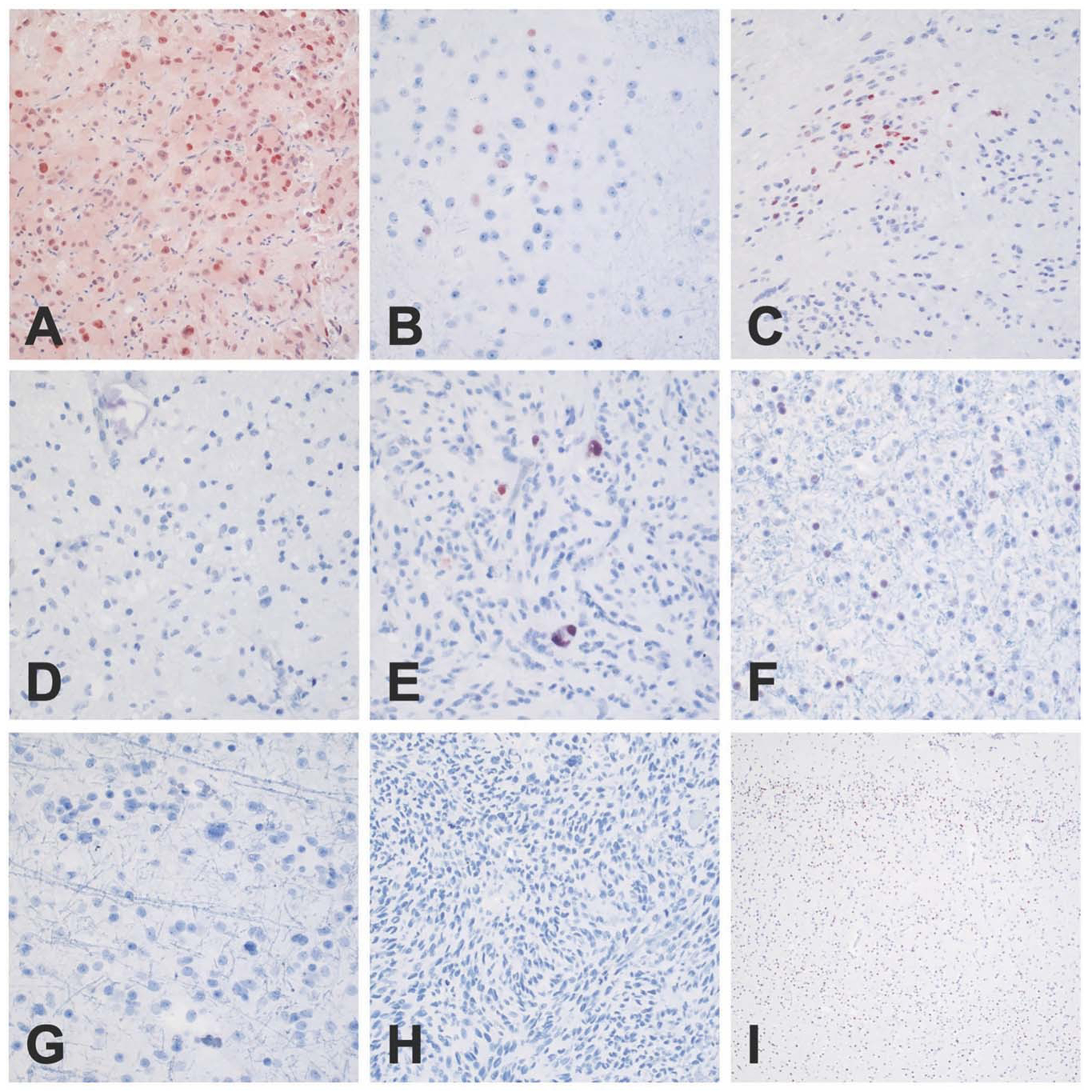
IHC was attempted for each of the top six DE TFs, including HMX3, HMX2, IRF6, SIX3, EOMES, and VAX1, as well as ZBTB20. Antibodies for HMX3 and HMX2 failed to give a reliable signal in SEGA or any other tissue examined. Both IRF6 and SIX3 showed strong cytoplasmic immunoreactivity in nearly all SEGAs examined (3 of 3, 4 of 5, respectively), and no appreciable staining in other tumors types (Figs. 6, 7). IRF6 and SIX3 expression was also seen in the balloon cells of cortical tubers (2 of 3 samples examined). Both VAX1 and ZBTB20 showed strong nuclear immunoreactivity in all SEGAs (3 of 3 and 2 of 2, respectively). VAX1 was highly specific for SEGAs, with little to no staining in other tumor types/lesions (Fig. 8), while ZBTB20 showed moderate to high nuclear staining in all tumors and normal neurons Fig. S7. IHC for EOMES did not show specificity for SEGAs (data not shown).
Fig. 7: Representative images of SIX3 immunohistochemistry.
Strong and diffuse cytoplasmic staining was predominantly limited to SEGAs (a) and in the balloon neurons of cortical tubers (b); however, occasional examples of PXA showed positive staining in large pleomorphic tumor cells (e). Glioblastomas exhibited staining in scattered cells (h). No cytoplasmic immunoreactivity was noted in subependymomas (c), ganglioglomas (d), oligodendrogliomas (f), astrocytomas (g), or in normal infant cortex (i). Scale bar: 400 μm, PXA: Pleomorphic xanthoastrocytoma.
IHC analysis also showed that HCRTR2, the gene with highest differential expression, showed cytoplasmic immunoreactivity in SEGAs and was not seen in other brain tumors/tubers apart from gangliogliomas (Fig. S7). CTSK (cathepsin K), a known marker for kidney angiomyolipoma, another benign tumor seen in the majority of TSC patients, showed very strong cytoplasmic staining in SEGAs (5 of 5, 100%). It was also seen in the balloon cells of cortical tubers (1 of 3, 33%) and in some glioblastoma (1 of 3, 33%) (Fig. S7). GPNMB showed very high expression in SEGA and the balloon cells of cortical tubers but was not seen in normal cortex (Fig. S7).
Unsupervised weighted gene co-expression network analysis (WGCNA)
We also performed unsupervised weighted gene co-expression network analysis (WGCNA), based on pairwise correlations for SEGAs versus TCGA low grade gliomas, glioblastomas, gangliogliomas, and cortical tubers in order to define modules (clusters of co-expressed genes) and intramodular hub genes enriched in SEGAs [39]. WGCNA identified 65 modules of correlating co-expressed genes, that contained 46–1701 genes (median: 102, average: 231). We then determined a correlation score for each module to SEGA tumors, and assigned kME scores to each gene (Tables S14, S15, and Fig. S8). Genes with higher kME scores are considered “hub” genes that may regulate expression within that module. Module ME65, with 47 genes, had the highest correlation score, 0.84, and showed the most consistent difference between SEGAs and the other tumors/cortical tubers. Module ME65 contained 21 of the 190 (11%) common DEGs, including 4 of the 6 TFs showing the largest median expression fold-change; HMX3, HMX2, IRF6, and SIX3. These results suggest that expression of HMX3, HMX2, IRF6, and SIX3 TFs may be co-regulated in a synergistic manner, and that those genes may be master TFs for SEGA, and function as transcriptional drivers of SEGA development. Overall, the 190 DEGs were found in 25 different modules, with module M65, M66, and M85 containing the highest number of co-expressed DEGs (21, 31, 39 genes, respectively).
Discussion
The present study reports multiplatform genomic and expression analyses in a large series of SEGA tumors, with detailed comparison to other brain tumors, other solid tumors, and multiple normal tissues. Exome sequence analysis of 21 matched tumor-normal pairs revealed that the mutational landscape of SEGAs is characterized by consistent biallelic inactivation of either TSC1 or TSC2. In contrast, other somatic mutations appear to be random occurrences, without any duplicate events in this dataset, and very likely do not contribute to SEGA development. The frequency of other somatic mutations (0.80 mutation/Mb) is lower than, but similar to, that seen in TSC-related kidney angiomyolipoma (range 0–12 mutations, 2.1 mutations/Mb; p = 0.016, unpaired Mann–Whitney t-test), as previously reported [31]. CN-LOH was the most common second hit (81%) event in this cohort of SEGAs, similar to our previous reports on both SEGAs and angiomyolipomas [6, 7, 31]. This extremely low somatic mutation rate in SEGAs is similar to a small set of pediatric brain tumors, including atypical teratoid/rhabdoid tumors, pituitary adenoma, and pediatric medulloblastoma [40–44]. Our exome results strongly suggest that biallelic loss of TSC1 or TSC2 is all that is required genetically for SEGA development.
RNA-seq analyses showed that SEGAs have a unique expression profile compared to other brain tumors, having somewhat more similarity to TCGA oligodendrogliomas and cortical tubers, and less to TCGA astrocytomas and gangliogliomas. Seven TFs, HMX3, HMX2, IRF6, SIX3, EOMES, VAX1, and ZBTB20, were highly and relatively uniquely expressed in SEGAs. Little is known about the function of several of these, including potential roles in neurodevelopment. HMX3 and HMX2 are related NKL homeobox transcription factors involved in specification of neuronal cell types and organ development [45, 46]. SIX3 is a sine oculis homeobox TF with a role in eye development, that regulates the proliferation and differentiation of neural progenitor cells through activating transcription of CCND1 and CCND2 [47]. ZBTB20 is also highly expressed uniquely in SEGAs, and is a transcriptional repressor with roles in neurogenesis, glucose homeostasis, and postnatal growth [48]. IRF6 (Interferon regulatory factor 6) plays a role in regulating mammary epithelial cell proliferation, while specifically expressed in SEGAs in comparison to other brain tumors but is expressed in other cancers and normal tissues [49]. These top genes have been reported to be epigenetically regulated bearing histone modifications: H3K27 trimethylation (H3K27me3) mark in brain for HMX2, HMX3, IRF6; H3K4me2 and H3K27me3 in neural precursor cells (NPC) for HMX2, HMX3, and VAX1 [50].
VAX1, SIX3, and IRF6 were all shown to be relatively highly and specifically expressed in SEGAs, suggesting that they may be useful to distinguish SEGAs from histologic mimics, including other brain tumors. However, our efforts to confirm the high and specific expression of these TFs in SEGAs were hampered by the failure of commercially available antibodies against HMX2 and HMX3 in IHC, and failure of attempts at RNA in-situ hybridization for HMX3.
Further studies are warranted to assess the functional importance of each of these TFs in SEGA development. Nonetheless, the key TFs HMX2, HMX3, VAX1, SIX3, and IRF6 may be considered as potential targets for the treatment of SEGAs, independent of, or in combination with mTORC1 inhibitors.
In conclusion, SEGAs have an extremely low somatic mutation burden, apart from TSC1/TSC2, similar to other pediatric brain tumors. Biallelic loss of either TSC1 or TSC2 occurs most commonly due to co-occurrence of a germline small mutation and CN-LOH as a second event, fitting the classic Knudson two hit mechanism. Several TFs, identified by RNA-seq analyses, are highly and relatively uniquely expressed in SEGAs. We consider that these TFs likely reflect the unique developmental state of the neuroepithelial cell in which biallelic loss of TSC1/TSC2 gives rise to SEGAs. They may also be transcriptional drivers of SEGA growth whose expression is required.
Supplementary Material
Acknowledgements
The authors thank all subjects who participated in this study; the clinicians who referred and evaluated the patients and the neurosurgeons who performed the surgical resections; as well as Karthik V. Karnik and Edward R. Kwiatkowski for their work on customized code for MPS analysis, as well as Yana Stackpole for assistance with Qlucore analysis; Stichting Kinderen Kankervrij; Stichting AMC Foundation; Stichting TSC Fonds (EA, AB).
Funding
This work was supported by the Engles Family Fund for Research in TSC and LAM.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Supplementary information The online version of this article (https://doi.org/10.1038/s41379-020-00659-9) contains supplementary material, which is available to authorized users.
Data availability
Access to data which are not available within the article and Supplementary material, can be provided by the authors upon request (direct contact to KG or DJK).
References
- 1.Chan JA, Zhang H, Roberts PS, Jozwiak S, Wieslawa G, Lewin-Kowalik J, et al. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol. 2004;63:1236–42. [DOI] [PubMed] [Google Scholar]
- 2.Kotulska K, Borkowska J, Roszkowski M, Mandera M, Daszkiewicz P, Drabik K, et al. Surgical treatment of subependymal giant cell astrocytoma in tuberous sclerosis complex patients. Pediatr Neurol. 2014;50:307–12. [DOI] [PubMed] [Google Scholar]
- 3.Henske EP, Jóźwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Prim. 2016;2:16035. [DOI] [PubMed] [Google Scholar]
- 4.Salussolia CL, Klonowska K, Kwiatkowski DJ, Sahin M. Genetic etiologies, diagnosis, and treatment of tuberous sclerosis complex. Annu Rev Genomics Hum Genet. 2019;20:217–40. [DOI] [PubMed] [Google Scholar]
- 5.Bongaarts A, Giannikou K, Reinten RJ, Anink JJ, Mills JD, Jansen FE, et al. Subependymal giant cell astrocytomas in Tuberous sclerosis complex have consistent TSC1/TSC2 biallelic inactivation, and no BRAF mutations. Oncotarget. 2017;8:95516–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin KR, Zhou W, Bowman MJ, Shih J, Au KS, Dittenhafer-Reed KE, et al. The genomic landscape of tuberous sclerosis complex. Nat Commun 2017;8:15816. 10.1038/ncomms15816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee D, Cho YH, Kang SY, Yoon N, Sung CO, Suh YL. BRAF V600E mutations are frequent in dysembryoplastic neuroepithelial tumors and subependymal giant cell astrocytomas. J Surg Oncol. 2015;111:359–64. [DOI] [PubMed] [Google Scholar]
- 8.Debiec-Rychter M, Jesionek-Kupnicka D, Zakrzewski K, Liberski PP. Cytogenetic changes in two cases of subependymal giant-cell astrocytoma. Cancer Genet Cytogenet. 1999;109:29–33. [DOI] [PubMed] [Google Scholar]
- 9.Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol. 2014;15:1513–2150. [DOI] [PubMed] [Google Scholar]
- 10.Franz DN, Agricola K, Mays M, Tudor C, Care MM, Holland-Bouley K, et al. Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol. 2015;78:929–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fogarasi A, De Waele L, Bartalini G, Jozwiak S, Laforgia N, Verhelst H, et al. EFFECTS: an expanded access program of everolimus for patients with subependymal giant cell astrocytoma associated with tuberous sclerosis complex. BMC Neurol. 2016;16:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tyburczy ME, Kotulska K, Pokarowski P, Mieczkowski J, Kucharska J, Grajkowska W, et al. Novel proteins regulated by mTOR in subependymal giant cell astrocytomas of patients with tuberous sclerosis complex and new therapeutic implications. Am J Pathol. 2010;176:1878–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bongaarts A, van Scheppingen J, Korotkov A, Mijnsbergen C, Anink JJ, Jansen FE, et al. The coding and non-coding transcriptional landscape of subependymal giant cell astrocytomas. Brain. 2020;143:131–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- 15.Wen PY, Huse JT. 2016 World Health Organization classification of central nervous system tumors. Contin (Minneap Minn). 2017;23:1531–47. [DOI] [PubMed] [Google Scholar]
- 16.Diamandis P, Aldape K. World Health Organization 2016 classification of central nervous system tumors. Neurol Clin. 2018;36:439–47. [DOI] [PubMed] [Google Scholar]
- 17.Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim W, Giannikou K, Dreier JR, Lee S, Tyburczy ME, Silverman EK, et al. A genome-wide association study implicates NR2F2 in lymphangioleiomyomatosis pathogenesis. Eur Respir J. 2019;53(6):1900329. 10.1183/13993003.00329-2019. pii [DOI] [PubMed] [Google Scholar]
- 19.Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher S, Barry A, Abreu J, Minie B, Nolan J, Delorey TM, et al. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 2011;12:R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tyburczy ME, Dies KA, Glass J, Camposano S, Chekaluk Y, Thorner AR, et al. Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet. 2015;11:e1005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giannikou K, Lasseter KD, Grevelink JM, Tyburczy ME, Dies KA, Zhu Z, et al. Low-level mosaicism in tuberous sclerosis complex: prevalence, clinical features, and risk of disease transmission. Genet Med. 2019;21:2639–43. [DOI] [PubMed] [Google Scholar]
- 24.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornwell M, Vangala M, Taing L, Hebert Z, Köster J, Li B, et al. VIPER: visualization pipeline for RNA-seq, a snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinforma. 2018;19:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc, 2012;7:562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016;131:833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ceccarelli M, Barthel FP, Malta TM, Sabetot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016;164:550–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsurubuchi T, Nakano Y, Hirato J, Yoshida A, Muroi A, Sakamoto N, et al. Subependymal giant cell astrocytoma harboring a PRRC2B-ALK fusion: a case report. Pediatr Blood Cancer. 2019;66:e27995. 10.1002/pbc.27995. [DOI] [PubMed] [Google Scholar]
- 31.Giannikou K, Malinowska IA, Pugh TJ, Yan R, Tseng YY, Oh C, et al. Whole exome sequencing identifies TSC1/TSC2 biallelic loss as the primary and sufficient driver event for renal angiomyolipoma development. PLoS Genet. 2016;12:e1006242. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018;174:1034–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive molecular characterization of muscle-invasive bladder. Cancer Cell. 2017;171:540–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang B, Guo D, Han L, Rensing N, Satoh A, Wong M. Hypothalamic orexin and mechanistic target of rapamycin activation mediate sleep dysfunction in a mouse model of Tuberous sclerosis complex. Neurobiol Dis 2020;134:104615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onda H, Crino PB, Zhang H, Murphey RD, Rastelli L, Gould BE, et al. Tsc2 null murine neuroepithelial cells are a model for human tuber giant cells, and show activation of an mTOR pathway. Mol Cell Neurosci. 2002;21:561–74. [DOI] [PubMed] [Google Scholar]
- 39.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bettegowda C, Agrawal N, Jiao Y, Wang Y, Wood LD, Rodriguez FJ, et al. Exomic sequencing of four rare central nervous system tumor types. Oncotarget. 2013;4:572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C, et al. Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro Oncol. 2017;19:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Correa DD, Satagopan J, Martin A, Braun E, Kryza-Lacombe M, Cheung K, et al. Genetic variants and cognitive functions in patients with brain tumors. Neuro Oncol. 2019;21:1297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muskens IS, Zhang C, de Smith AJ, Biegel JA, Walsh KM, Wiemels JL. Germline genetic landscape of pediatric central nervous system tumors. Neuro Oncol. 2019;21:1376–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng Y, Xu Q. Pivotal role of hmx2 and hmx3 in zebrafish inner ear and lateral line development. Dev Biol. 2010;339:507–18. [DOI] [PubMed] [Google Scholar]
- 46.Wang W, Grimmer JF, Van De Water TR, Lufkin T. Hmx2 and Hmx3 homeobox genes direct development of the murine inner ear and hypothalamus and can be functionally replaced by Drosophila Hmx. Dev Cell. 2004;7:439–53. [DOI] [PubMed] [Google Scholar]
- 47.Yu Z, Sun Y, She X, Wang Z, Chen S, Deng Z, et al. SIX3, a tumor suppressor, inhibits astrocytoma tumorigenesis by transcriptional repression of AURKA/B. J Hematol Oncol. 2017;10:115. 10.1186/s13045-017-0483-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie Z, Ma X, Ji W, Zhou G, Lu Y, Xiang Z, et al. Zbtb20 is essential for the specification of CA1 field identity in the developing hippocampus. Proc Natl Acad Sci USA. 2010;10:6510–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo XM, Chen B, Lv JM, Lei Q, Pan YJ, Yang Q. Knockdown of IRF6 attenuates hydrogen dioxide-induced oxidative stress via inhibiting mitochondrial dysfunction in HT22 cells. Cell Mol Neurobiol. 2016;36:1077–86. 10.1007/s10571-015-0301-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kozlenkov A, Li J, Apontes P, Hurd Y, Byne WM, Koonin EV, et al. A unique role for DNA (hydroxy)methylation in epigenetic regulation of human inhibitory neurons. Sci Adv. 2018;4:eaau6190. 10.n26/sciadv.aau6190..26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Access to data which are not available within the article and Supplementary material, can be provided by the authors upon request (direct contact to KG or DJK).