

Abstract

Thermophilic polyester hydrolases (PES-H) have recently enabled biocatalytic recycling of the mass-produced synthetic polyester polyethylene terephthalate (PET), which has found widespread use in the packaging and textile industries. The growing demand for efficient PET hydrolases prompted us to solve high-resolution crystal structures of two metagenome-derived enzymes (PES-H1 and PES-H2) and notably also in complex with various PET substrate analogues. Structural analyses and computational modeling using molecular dynamics simulations provided an understanding of how product inhibition and multiple substrate binding modes influence key mechanistic steps of enzymatic PET hydrolysis. Key residues involved in substrate-binding and those identified previously as mutational hotspots in homologous enzymes were subjected to mutagenesis. At 72 °C, the L92F/Q94Y variant of PES-H1 exhibited 2.3-fold and 3.4-fold improved hydrolytic activity against amorphous PET films and pretreated real-world PET waste, respectively. The R204C/S250C variant of PES-H1 had a 6.4 °C higher melting temperature than the wild-type enzyme but retained similar hydrolytic activity. Under optimal reaction conditions, the L92F/Q94Y variant of PES-H1 hydrolyzed low-crystallinity PET materials 2.2-fold more efficiently than LCC ICCG, which was previously the most active PET hydrolase reported in the literature. This property makes the L92F/Q94Y variant of PES-H1 a good candidate for future applications in industrial plastic recycling processes.
Keywords: polyethylene terephthalate (PET), PET hydrolysis, thermophilic polyester hydrolase, enzyme engineering, crystallization, molecular dynamics, binding modes, kinetics
Introduction
Polyethylene terephthalate (PET), the most abundant synthetic polyester, is widely used in the packaging and textile industries. The global production of PET recently reached 82 million metric tons per year.1 This contributes significantly to the global solid waste stream and environmental plastic pollution. Biocatalytic recycling of PET has emerged as a promising technology that allows the recovery of monomeric building blocks at both laboratory and pilot plant scales.2−5 Although PET hydrolases from a wide range of microorganisms have been identified, only a handful of benchmark enzymes have been extensively engineered for industrial applications.6
As an interfacial biocatalytic reaction, the efficiency of PET degradation is limited by the number of accessible ester bonds at the polymer surface. Amorphous PET polymer chains become more accessible to enzymatic hydrolysis at temperatures approaching the glass transition temperature of PET (Tg, 65–71 °C).6 In aqueous environments, water serves as a plasticizer that lowers the Tg of PET by up to 16 °C.6−8 This effect increases the flexibility of polymer chains at the PET surface layer, allowing degradation at ambient temperatures by mesophilic PET hydrolases, like IsPETase from Ideonella sakaiensis, which have optimal operating temperatures (Topt) of approximately 40 °C.9 Nonetheless, thermophilic and thermostable PET hydrolases have superior degradation activity because of increased substrate accessibility at elevated temperatures.10−12
LCC, a cutinase isolated from a leaf-branch compost metagenome library, is one of the most extensively investigated thermophilic PET hydrolases.13−15 Structure-based protein engineering of LCC has resulted in an F243I/D238C/S283C/Y127G variant (LCC ICCG) that can efficiently depolymerize pretreated postconsumer PET bottles within 10 h at 72 °C. This enables the biocatalytic recycling of PET waste at industrially relevant scales.5 Calcium ions (Ca2+) can boost the thermostability of many bacterial PET hydrolases (changes in Tm (ΔTm) = 10–16 °C and ΔTopt ≥ 10 °C).13,16−18 Engineering of residues in the Ca2+ binding sites has proven to be a useful approach for increasing the Tm of several PET hydrolases by up to 26 °C.5,11,17,19−22 Introduction of a disulfide bridge at this site (D238C/S283C) markedly increased the melting point (Tm) of LCC from 84.7 to 94.5 °C. In a recent study, LCC ICCG was further engineered to obtain an A59K/V63I/N248P variant with a Tm of 98.9 °C.23 Nonetheless, the Topt of this variant for degrading low-crystallinity PET (lcPET, <15% crystallinity) was 74 °C, only 2 °C higher than that for the precursor LCC ICCG.5 At reaction temperatures above 70 °C, water-plasticized lcPET rapidly crystallizes to high-crystallinity PET (hcPET), which is much harder to hydrolyze.6,24,25 The 20% crystallinity threshold for enzymatic hydrolysis can be reached within 3 h at 75 °C.5 This competitive physical aging process of amorphous polymers can therefore considerably reduce the efficiency of enzymatic PET depolymerization, which limits the maximum extent of degradation that can be achieved.25 Therefore, biocatalysts with Topt in the range 72–74 °C, rather than with extremely high Tm values, are desired.6 The mesophilic IsPETase (Tm of 48.7 °C) has also been subjected to protein engineering to improve its thermostability. For example, Cui et al. used a computational strategy to design DuraPETase, a variant with a 31 °C higher Tm and considerably improved PET hydrolysis activity than the wild-type enzyme.26 More recently, a machine-learning-aided approach was used to engineer IsPETase to produce Fast-PETase, a variant with a Tm of 67.1 °C and increased depolymerization efficiency.27
In-depth understanding of the binding mechanisms and noncovalent interactions of insoluble polymeric substrates with biocatalysts can contribute to the design of more efficient PET hydrolases. So far, only two studies have reported structures of crystals soaked with PET analogues. Han et al. reported a structure of the inactive R103G/S131A IsPETase mutant complexed with 1-(2-hydroxyethyl) 4-methyl terephthalate (HEMT).28 Zeng et al. reported the structure of an inactive S165A mutant of LCC ICCG complexed with mono-(2-hydroxyethyl) terephthalate (MHET).23 Consequently, more structures of PET hydrolases with bound substrate analogues are needed. This would enable more precise computational simulations for elucidating the mechanisms of enzymatic depolymerization and predicting more efficient mutants. So far, studies mostly relied on computationally modeled PET substrate conformations.15,29−32 Therefore, we solved crystal structures of two PET hydrolases in complex with PET analogues.
Two highly similar thermophilic PET hydrolases (PES-H1 and PES-H2), derived from a compost metagenome library, were recently disclosed in a patent application.33 PES-H1 demonstrated exceptional hydrolytic activity on amorphous PET films, outperforming wild-type LCC under identical conditions.13,15 Nearly complete depolymerization was achieved after incubation at 70 °C for 24 h.33 We first solved high-resolution structures of both enzymes in their apo form (PDB codes: 7CUV for PES-H1 and 7W69 for PES-H1). While this work was in progress, another crystal structure of PES-H1 (called PHL7 in the publication, PDB code: 7NEI) was solved.34 By soaking with various PET substrate analogues, structures of both PES-H1 and PES-H2 in complex with these ligands were obtained by us. These structures were used for exploring substrate binding modes by molecular dynamics (MD) simulations. Finally, we generated PES-H1 variants with significantly improved activity on both amorphous Goodfellow PET (Gf-PET) films and pretreated postconsumer (‘real-world’) PET waste. These variants outperformed both the wild-type PES-H1 and LCC ICCG.5
Results and Discussion
Structures of PES-H1 and PES-H2
The crystal structures of PES-H1 and PES-H2 were solved at atomic resolutions of 1.45 Å (PDB code: 7CUV) and 1.56 Å (PDB code: 7W69), in the space groups P21 and C2, respectively (Figure S1; Tables S1 and S2). PES-H1 and PES-H2 differ in only four residues (A/E1, L/F209, D/N232, and S/A254), of which only the L/F209 residues are in close vicinity to the catalytic triad (Figure 1). The two hydrolases have similar structures that adopt the canonical α/β-hydrolase fold with a core consisting of nine β-sheets and ten α-helices (Figure S1). Comparison of the apo structure of PES-H1 with the structures of other bacterial PET hydrolases resulted in low root-mean-square deviation (RMSD) values for the protein backbone (Table S3). These highly conserved structures form a distinct subclass of the α/β-hydrolase superfamily.6 PES-H1 has a catalytic triad consisting of S130, H208, and D176 (Figure 1). The nucleophile S130 is located within hydrogen bond distance to be polarized by the base H208, which is in turn stabilized by the acid D176. PES-H1 forms an intramolecular disulfide bridge like structurally similar homologous enzymes (e.g., PDB codes: 5XH3, 3VIS, 4CGI, 4EB0, 4WFJ, and 1JFR). This conserved disulfide bridge (C241/C256) connects two loops between the last β-sheet (Figure S1).
Figure 1.
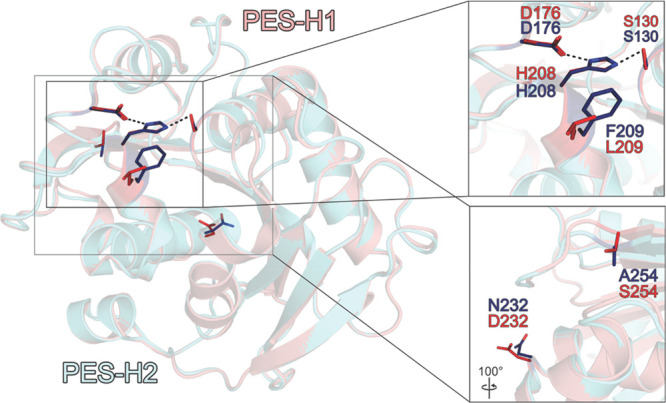
Structural comparison of PES-H1 (salmon, PDB code: 7CUV) and PES-H2 (light blue, PDB code: 7W69). The locations of the variable residues L/F209, D/N232, and S/A254 are indicated. The variable position A/E1 is located at the flexible N-terminal ends of PES-H1 and PES-H2 and are therefore not visible. The S130–H208–D176 catalytic triad is also shown. The amino acid residues are shown as sticks in red for PES-H1 and in dark blue for PES-H2. The inset in the right upper panel demonstrates the close vicinity of the variable position L/F209 to the catalytic triad.
Binding Modes of Substrate Analogues and Degradation Intermediates
Although more than 70 crystal structures of bacterial PET-degrading enzymes are available in the PDB database, the majority of them are apo structures with no substrate bound.6 In order to investigate the mechanism of PET hydrolysis, we solved the structures of PES-H1 in complex with citrate (PDB code: 7E30) and also in complex with the nonhydrolyzable substrate analogue 4-(2-hydroxyethylcarbamoyl) benzoic acid (MHETA) (PDB codes: 7W6C, 7W6O, and 7W6Q; Figure 2B–F), to better understand changes in the protein structure upon binding a PET substrate based on this analogue. We also solved the structures of PES-H2 in complex with polyethylene glycol (PEG) 6000 (PDB code: 7E31) and in complex with bis(2-hydroxyethyl) terephthalate (BHET) (PDB code: 7W66; Figure 2G) (Tables S1 and S2). Based on the structures of PES-H1 (with bound MHETA) and PES-H2 (with bound BHET), six ligand-binding modes were identified. The orientations of the ligands in the substrate binding cavities are distinct but similar to those observed for HEMT bound to IsPETase R103G/S131A28 (PDB code: 5XH3; Figure 2I) and MHET bound to the S165A variant of LCC ICCG23 (PDB code: 7VVE; Figure 2H).
Figure 2.

Comparison of the PET substrate analogue binding modes in the structures of PES-H1, PES-H2, LCC ICCG S165A, and IsPETase R103G/S131A. Single chains (superscripts for chain A or chain B) were extracted from the deposited structures in the PDB database to identify individual binding modes of the soaked ligands within an asymmetric unit. Dashed lines indicate hydrogen bonds (H-bond cut off of 3.5 Å). Interacting residues are shown as sticks and are colored by element: carbon, as for the respective molecule; nitrogen, blue; oxygen, red; sulfur, yellow. Green spheres represent water oxygens. (A) Superimposed structures show the overlapping binding modes of the substrate analogues with the apo structure of PES-H1 (PDB code: 7CUV). (B–F) PES-H1 in complex with MHETA: (B) 7W6CA; (C) 7W6OA; (D) 7W6QA; (E) 7W6OB; and (F) 7W6CB. (G) PES-H2 in complex with BHET (7W66). (H) LCC ICCG S165A in complex with MHET (7VVEA, 7VVEB). (I) IsPETase R103G/S131A in complex with HEMT (5XH3).
While the structures of LCC ICCG S165A (PDB code: 7VVE; Figure 2H) and IsPETase R103G/S131A (PDB code: 5XH3; Figure 2I) exhibited productive ligand conformations with the ester carbonyl carbons interacting with the oxygen of the catalytic serine residue, the PES-H structures revealed multiple noncatalytic intermediate binding modes (IBMs) with the ester bonds (or amide bond in MHETA) positioned too far from S130 for nucleophilic attack. This supports the previous hypothesis that PET hydrolysis may involve dynamic reorientation of polymer chains in the active sites of PET hydrolases.15,35 MHET and BHET have frequently been found to inhibit enzymatic PET depolymerization.36,37 The multiple IBMs suggest an enhanced residence likelihood of these inhibitory degradation intermediates in the substrate-binding groove, where they may prevent productive binding of polyester segments.
The conformational flexibility of predominantly hydrophobic residues surrounding the active site may help to recognize and bind bulky PET substrates during biocatalysis. In the enzyme–substrate complex structures of LCC ICCG S165A (Figure 2H) and IsPETase R103G/S131A (Figure 2I), the aromatic rings of the substrate analogues are T-stacked to W190 and W156, respectively. This effect has been proposed to be very important for PET binding.38 Similar π-stacking interactions with the equivalent W155 were discovered in the PES-H1 structures 7W6C (Figure 2B, F), 7W6OA (Figure 2C), and 7W6QA (Figure 2D). In most IBMs of MHETA ligands (Figure 2B–F), the hydroxyl end of the ethanolamine moiety points away from the catalytic triad. This is partly in agreement with the MHET conformation shown in 7VVE (Figure 2H), which is different from the position of the ethylene glycol (EG) moiety of HEMT bound to IsPETase R103G/S131A (5XH3; Figure 2I). MHETA ligands also interacted with other hydrophobic residues like F62, I178, and L209 in the binding groove of PES-H1. The backbone amide NH groups of M131 and F62 form an oxyanion hole, which is a characteristic shared by many α/β-hydrolases. Terephthalate (TPA) moieties in our structures are found in close vicinity to F62 (equivalent to Y95 in LCC ICCG S165A and Y58 in IsPETase R103G/S131A), rather than close to the catalytic triads as in IsPETase R103G/S131A (Figure 2I) and LCC ICCG S165A (7VVEA; Figure 2H). Furthermore, the hydroxyl end of the ethanolamine moiety forms a hydrogen bond with H184 (Figure 2C,D). The only distinct MHETA binding mode in PES-H1 was observed in 7W6OB (Figure 2E), in which MHETA is bound at the surface of PES-H1, slightly outside the catalytic cavity. This binding mode suggests that substrates can bind at the surface prior to entering deeper into the active site cavity. BHET is more loosely bound to PES-H2 (7W66B; Figure 2G). It is embedded in a groove consisting of S130, H207, W155, I178, H129, F62, and M131. The ester bond between the TPA and EG moieties is closer to S130 but is still outside the catalytic distance, in contrast to the positioning of MHETA in PES-H1. The TPA moiety is close to F62 but far from W155. The second EG moiety is completely exposed to the solvent and has no obvious interactions with any residue.
MD Simulations to Study the Interactions of PES-H1 with a PET Oligomer
To better understand the mechanism of PET hydrolysis, we performed MD simulations of PES-H1 in complex with a modeled oligomer consisting of three repeating PET units (3PET) (Figure S3C) to resemble the binding of a longer polymer segment. PES-H1 remained stable in MD simulations at a time scale of 100 ns as indicated by the steady low backbone RMSD of less than 1 Å compared to the crystal structure (Figure S2). The modeled PES-H1-3PET complex corresponds to a productive state with short distances between the oxyanion hole amides and the central 3PET carbonyl oxygen atom (2.32 and 1.80 Å), and between the carbonyl carbon of an ester in 3PET and the side chain oxygen of the catalytic S130 (2.32 Å). Unfluctuating distances of the 3PET carbonyl carbons to the catalytic S130 verified the productive state of the modeled oligomer during three independent 100 ns MD simulations (Figure S3). Nonetheless, 3PET was slightly flexible in the PES-H1 binding site (Figure S3A). To identify the most favorable 3PET binding modes, we concatenated the trajectories and clustered the 3PET conformations. The four most populated clusters correspond to the four preferred binding modes, each including >5% of all ligand conformations (Figure S4). Cluster 1 represents the 3PET binding mode adopted in about 28% of the conformations (Table S4, Figure S4C). After superimposing the PES-H1 (PDB code: 7CUV) and LCC ICCG S165A structures (PDB code: 7VVE), the central PET unit of cluster 1 and the MHET overlap well with an RMSD of 2.95 Å, while the binding mode of cluster 3 (found in about 8% of all conformations) deviates the most from that of MHET, with an RMSD of 4.01 Å (Figure S4D). This suggests that being adjacent to S130 results in multiple beneficial interactions between PET and the surrounding residues. Evaluating the interaction energies of the four binding poses demonstrated that hydrophobic Lennard-Jones (LJ) interactions between the side chains and the central 3PET unit had a predominant influence on binding. F62, W155, and I178 formed strong LJ interactions (<−10 kJ mol–1), in agreement with their close interactions with soaked MHETA ligands (Figure 2B–D,F,G). H208 and M131 also formed strong LJ interactions (<−7 kJ mol–1) (Figures 1 and 3; Figure S4C). Additionally, T63, A64, and L209 mediate strong LJ interactions (<−7 kJ mol–1) with all three units of 3PET. The side chains of F62 and W155 form stable aromatic interactions with the aromatic rings of 3PET. The orientation of the TPA moieties suggest T-stacking interactions with W155, in agreement with the orientation of MHETA bound to PES-H1 (PDB codes: 7W6CA, 7W6Cb 7W6OA, and 7W6QA) and with earlier studies on other PET hydrolases.12,23 Furthermore, the strong LJ interactions with F62 are in agreement with the close proximity between MHETA and F62. The Coulomb interactions involve fewer residues and are often mediated by backbone atoms of residues, like F62 and M131 from the oxyanion hole, and L92, but can also involve side chains of residues like Q94, H208, N212, and the catalytic S130 (<−7 kJ mol–1). It should be emphasized that the significant contributions by L92 and Q94 are the result of hydrogen bonding with the terminal carboxyl group of the first PET unit, which should only occur when exoscission of polymeric PET substrates takes place. When the total interaction energy is considered, G61, H129, and T157 are also identified as strong PET-binding residues (<−7 kJ mol–1).
Figure 3.

Overview of selected engineering hotspots in PES-H1 for enhancing PET hydrolysis activity and thermostability. (A) A disulfide bridge (R204/S250) is introduced into PES-H1 (PDB code: 7CUV) where a putative Ca2+ binding site (D250/E296) is found in (B) the homologous Cut190. (C) Similarly engineered disulfide bridges in LCC ICCG (C238/C283; PDB codes: 7VVE and 6THT) and (D) TfCut2 (D204/E253; PDB code: 4CG1). (E) The L92F and Q94Y substitutions of PES-H1 were based on the corresponding residues in (F) DuraPETase (F117 and Y119; PDB code: 6KY5). (G–K) Variable amino acid residues were found at the site equivalent to L209 in PES-H1: (G) PES-H1, PDB code 7CUV; (H) PES-H2, PDB code 7W69; (I, J) LCC ICCG S165A, PDB codes 7VVE and 6THT; and (K) IsPETase R103G/S131A, PDB code 5XH3. These residues influence the width of the substrate-binding groove. (L) A number of PES-H1 variants had different PET hydrolyzing activities and thermostabilities. Changes in Tm compared to the wild-type enzyme (ΔTm (°C)) are shown. The percentage relative activity compared to the wild-type enzyme was calculated based on the weight loss of Gf-PET film after 24 h at 72 °C. Red square: wild-type PES-H1. Blue triangles: L209 variants. Orange squares: I178 and F62 variants. Dark blue dots: W155 variants. Light green dot: L92F/Q94Y/R204C/S250C variant. Green dot: R204C/S250C variant with a disulfide bond introduced. Yellow dot: L92F/Q94Y variant.
Engineering PES-H1 for Improved Thermostability and Activity on Amorphous PET Film
Elevated reaction temperatures of up to 75 °C increase polymer chain mobility, making the amorphous fractions of PET more accessible to enzymatic hydrolysis. We investigated enzymatic PET degradation at 72 °C because it is the optimal temperature for the hydrolysis of amorphized PET waste by the reference enzyme LCC ICCG.5 Empirical data have shown that the Tm of a PET hydrolase is usually at least 12 °C higher than its Topt.5,11,13,17 Hence, biocatalysts with a Tm over 85 °C are preferred for efficient PET degradation. When determined in buffers with low ionic strengths (Table S5), PES-H1 had a Tm of 77.1 °C, as determined by nano differential scanning fluorimetry (NanoDSF) and differential scanning calorimetry (DSC). This is in agreement with previously published data.34 In the 1 M potassium phosphate buffer demonstrated by previous studies to be optimal for PET degradation by selected enzymes,33,34,39 the Tm of PES-H1 was increased to 85 °C (Table S5), thereby enabling its application at 72 °C (Figure S5). Because high salt concentrations are not ideal for industrial processes, we investigated the degradation of Gf-PET film by PES-H1 in the presence of lower phosphate buffer concentrations (Figure S6). The results demonstrated a positive correlation between the buffer concentration and the PET degradation performance of wild-type PES-H1 at 72 °C. This is most likely due to the decreased thermostability of PES-H1 at lower buffer concentrations (Table S5).
Many homologous PET hydrolases, including PES-H1 itself, have been found to be stabilized in the presence of Ca2+ ions, as indicated by increased Tm, improved PET-hydrolyzing activity, or both.13,17,18 Ca2+ binding can be mediated by several negatively charged residues. For example, D250 and E296 in the homologous Cut190 from Saccharomonospora viridis were identified by crystallographic investigations to be involved in Ca2+ binding (Figure 3B).40 Ca2+ can be precipitated by buffer ions like phosphate and also by the PET hydrolysis product terephthalate,6,41 making the dependence of PET hydrolase stability on Ca2+ undesirable. This dependence can be significantly mitigated by replacing the Ca2+ binding residues by a disulfide bridge to generate variants that are thermostable in the absence of Ca2+.5,19,11,17,20−22 Accordingly, we generated an R204C/S250C variant of PES-H1 and confirmed disulfide bond formation using Ellman’s reagent (Figure S7). The Tm of this variant was increased by 6.4 °C (Figure 3L, Table S5). Previous studies reported that introducing disulfide bridges at equivalent positions of homologous PET hydrolases resulted in higher increases in Tm (TfCut2, ΔTm = 24.9 °C; IsPETase, ΔTm = 22.3 °C; LCC, ΔTm = 9.8 °C; Cut190, ΔTm = 23.1 °C).5,17,19,21 While more negatively charged amino acids like D250 and E296 in Cut190 (Figure 3B), D238 and S283 in LCC (Figure 3C), and D204 and E253 in TfCut2 (Figure 3D) are frequently observed at these sites, wild-type IsPETase (N204 and S253) and PES-H1 (R204 and S250, Figure 3A) lack these negatively charged residues. This suggests that the generic thermostabilizing effect of an engineered disulfide bridge at this position might be due to a variety of interactions rather than just replacing the Ca2+ binding site. After 24 h at 72 °C, both the wild-type and the R204C/S250C variant of PES-H1 hydrolyzed Gf-PET film (98% depolymerization) almost completely (Figure 4A).
Figure 4.

Characterization of PET-hydrolyzing activity of PES-H1 (wt), the L92F/Q94Y and R204C/S250C variants, and LCC ICCG. (A) Weight loss (in [mg] and [%]) of Gf-PET film determined after enzymatic hydrolysis at 72 °C for 12 and 24 h in 1 M potassium phosphate buffer (pH 8.0). (B–D) Total product release [mM] is shown and used to calculate the depolymerization efficiency [%] with (B) lcPET powder (13%), (C) ball-milled hcPET powder (26%), and (D) grinder-crushed hcPET powder (33%) after 24 h at 72 °C. The total product was defined as the sum of TPA (dark blue), MHET (light blue), and BHET (light gray). Error bars indicate the standard deviation calculated from at least three replicates.
Recently, Nakamura et al. reported an R47C/G89C variant of the homologous hydrolase PET2 and validated the formation of a disulfide bridge by X-ray crystallography.22 Compared to wild-type PET2, the Tm of the R47C/G89C variant of PET2 was increased by 3.1 °C, and its PET hydrolysis activity was 1.3-fold higher. We created a R6C/S49C variant in an attempt to form an equivalent disulfide bridge in PES-H1. This variant was inactive and was therefore not further investigated.
The PES-H1 structure (PDB code: 7CUV, Figure 3E) was compared to the thermostabilized DuraPETase (PDB code: 6KYS, Figure 3F) for identifying additional mutation hotspots. Using Rosetta energy calculations, an L92F/Q94Y variant of PES-H1 was constructed based on the L117F/Q119Y mutations present in DuraPETase. The L92F/Q94Y variant of PES-H1 had a 1.8 °C higher Tm and could almost completely depolymerize Gf-PET film after 24 h at 72 °C (Figures 3L and 4A). Interestingly, the aromatic amino acids that we introduced (L92F/Q94Y) correspond to equivalent residues (F125 and Y127) in LCC ICC.5 Interestingly, LCC ICC was converted to the more active LCC ICCG by removal of the tyrosine residue (Y127G), while introduction of the equivalent tyrosine into PES-H1 (Q94Y) in combination with L92F resulted in increased activity. The L92F/Q94Y variant of PES-H1 outperformed LCC ICCG in the hydrolysis of various PET materials under conditions optimal for PES-H1 (Figure 4A,B). Cui et al. also observed synergistic effects between the equivalent L117F (L92F in PES-H1) and Q119Y (Q94Y in PES-H1) substitutions.26 They argued that these aromatic residues interact with other residues (S214, W185, and Y87) to form an “aromatic tunnel” that facilitates the binding of PET chains to the active site. The hydrophobic nature of the mutations enhances noncovalent interactions with the substrate and improves the overall hydrophobic packing and stability of DuraPETase. The same effect could explain the increased activity and stability of the L92F/Q94Y variant of PES-H1. We introduced the previously discussed disulfide bridge (R204C/S250C) to the L92F/Q94Y variant to generate the L92F/Q94Y/R204C/S250C variant of PES-H1. Although the Tm of this variant was increased by 4.8 °C, it was not further characterized because it hydrolyzed Gf-PET film less efficiently than wild-type PES-H1 (Figure 3L).
Of the four residues that differ between PES-H1 and PES-H2, only L209 is located close to the active site (Figures 1 and 3G). This site has repeatedly been recognized as an engineering hotspot in other homologous PET hydrolases due to its potential interactions with polymeric substrates. Variants with increased specific activity on PET have been created by replacing F at this position with A (TfCut2: F208A), W (LCC WCCG: F243W), or I (LCC ICCG: F243I; Figure 3I,J).5,42 The equivalent S of IsPETase (Figure 3K) has also been replaced with F, but an additional W159H substitution was required for improved PET hydrolysis activity.43 It was recently shown that exchanging L209 in PES-H1 (called PHL7 by Sonnendecker et al.34) to the F found in PES-H2 (Fig 3G,H) reduced PET hydrolysis activity by half. We obtained similar results for the hydrolysis of Gf-PET film by the L209F variant of PES-H1 (Figure 3L). We subjected position L209 to saturation mutagenesis and found that most variants (except L209K, L209R, L209E, L209P, and L209I) resulted in enhanced thermostability. For the L209W, L209F, and L209Y variants, the highest ΔTm in 50 mM phosphate buffer ranged from 3.3 to 3.7 °C (Figure 3L). However, the PET hydrolysis activities of these variants were decreased by more than 15%. Several earlier studies43,44 suggested that the S238F substitution (equivalent to L209F in PES-H1) in the W159H/S238F variant of IsPETase narrowed the active site cleft, potentially enhancing π-stacking between PET and F238. In contrast, Furukawa et al.42 suggested that the corresponding F of TfCut2 restricts access to lcPET, attributing the influence of this residue on catalytic activity to side-chain volume rather than hydrophobicity. Therefore, the role of this important residue is still not fully understood, and it may play different roles in different PET hydrolases.
In addition, we engineered several key residues potentially involved in strong interactions with PET as indicated by structures of PES-H1 and PES-H2 in complex with PET analogues (Figure 2) and MD simulations (Figure S4). These include F62 (Figure 2B–D,G), W155 (Figure 2B–D,F,G), and I178 (Figure 2E). F62 is part of the oxyanion hole. Except for PES-H1, Cut190,18 and Tcur1278,45 other known homologous PET hydrolases have a tyrosine at this position. Introducing this tyrosine into PES-H1 (F62Y) lowered the hydrolytic activity drastically but had no influence on Tm (Figure 3L). The same effects were observed for the F62A variant.
The W155 residue of PES-H1 is equivalent to the W185 residue of IsPETase. The W185 of IsPETase (W130 in Figure 2I due to different residue numbering in the 5XH3 crystal structure) is flexible and can move to open or close access to the active site.43 This wobbling is beneficial for mesophilic PET hydrolases because the PET polymer chain has lower chain mobility at ambient temperatures, and a flexible W185 may facilitate substrate binding.15 This dynamic tryptophan is conserved in many homologous PET hydrolases and is involved in strong interactions with the aromatic rings of PET. We replaced the W155 of PES-H1 to A, F or H, resulting in almost complete loss of activity and considerably reduced stability (Figure 3L). Austin et al. reported similarly reduced activity for the equivalent W185A variant of IsPETase.43 In contrast, the conformation of the corresponding W190 (Figure 2H) in the thermophilic LCC ICCG is fixed by interactions with H218 and F222 (H184 and F188 in PES-H1, Figure 2C,D,F). As a result, LCC ICCG has a more rigid substrate-binding groove, yet it still allows PET binding at elevated temperatures as a result of increased polymer chain mobility.23
The I178 residue is located near the catalytic triad of PES-H1 (Figure 2B–D,F). Previous studies have reported substitutions of this I178 residue in homologous Thermobifida fusca cutinases to smaller residues like S or A, which resulted in PET hydrolyzing activities comparable to that of the wild-type enzyme.46,47 Therefore, we instead substituted I178 to large aromatic residues. However, the I178W, I178F, and I178Y variants lost significant PET hydrolysis activity, and their Tm decreased slightly, suggesting that additional aromatic interactions may hinder productive binding of PET chains. Therefore, we did not further investigate the I178 residue of PES-H1.
Comparing the Hydrolytic Activity of Selected PES-H1 Variants on Different PET Materials
The L92F/Q94Y and R204C/S250C variants of PES-H1 were further investigated using different PET materials. The crystallinity of real-world postconsumer PET waste depends on its thermal history and life cycle.6 Therefore, for biocatalytic recycling at the industrial scale, pretreatment by thermomechanical amorphization and micronization is essential to provide uniformly degradable substrates (in terms of comparable crystallinity, particle size, etc.).5,48 In this study, we used a melt-quenching technique to amorphize postconsumer PET bottles, which were then micronized by various methods. Crystallinity of the PET bottle waste was decreased from 27% to 19% after melt-quenching and increased again to 33% and 26%, respectively, after crushing or ball milling (Table S6). The crystallinity values determined with differently micronized PET powders are in good agreement with two previous studies.48,49 However, the crystallinities of our samples were markedly higher than that reported by Tournier et al., who used other techniques.5 We determined the average molecular masses of selected PET samples by gel permeation chromatography (GPC) and found that values decreased after melt-quenching and micronization (Table S6). Since the particle size can influence the biocatalytic degradability of PET powders, it was important to use similarly sized particles for all assays. Therefore, we sieved all micronized PET samples using a 177 μm mesh to obtain more homogeneous powders.
LCC ICCG and the wild-type PES-H1 were used as reference enzymes. The distinct distribution of charged amino acids at the surfaces of these two enzymes may explain the differences in their buffer requirements (Figure S8). Compared to PES-H1, LCC ICCG does not lose its thermostability and PET degrading activity at a low phosphate buffer concentration (100 mM).5 However, higher phosphate concentrations (up to 1 M) do not reduce the PET hydrolysis activity of wild-type LCC.15,39,50 Since 1 M phosphate buffer is optimal for the activity and stability of PES-H1 while not negatively influencing LCC, we used this buffer for all subsequent PET degradation experiments. As shown in Figure 4A, wild-type PES-H1 and the L92F/Q94Y variant completely depolymerized Gf-PET film in 24 h at 72 °C. Therefore, we also determined the weight loss after 12 h of incubation. The highest weight loss of 37.1 mg (62.4%) was obtained with the L92F/Q94Y variant, and this was 2.3-fold higher than with wild-type PES-H1. The L92F/Q94Y variant also significantly outperformed both the R204C/S250C variant and LCC ICCG in the hydrolysis of Gf-PET film (Figure 4A). Averaged conversion rates of 2.5 mgPET h–1 and 2.3 mgPET h–1 were obtained using wild-type PES-H1 and LCC ICCG, respectively. While the value for PES-H1 is similar to that previously determined by Sonnendecker et al.,34 the value for LCC ICCG is markedly lower than that reported by Tournier et al.5 As explained above, this may in part be due to differences between the PET materials used in different studies. Based on the information provided by the manufacturer, Gf-PET films differ considerably in terms of crystallinity, thickness, molecular mass, and many other polymer parameters, making a direct comparison of data generated in different studies impossible.6
After 24 h at 72 °C, the degradation performance of PES-H1, L92F/Q94Y, R204C/S250C, and LCC ICCG on low-crystallinity (13%) micronized waste PET powder was comparable to that on Gf-PET film. The L92F/Q94Y variant yielded 2.9-fold and 2.2-fold more degradation products than wild-type PES-H1 and LCC ICCG, respectively (Figure 4B). This indicated that, under the applied degradation conditions, the L92F/Q94Y variant was the most active lcPET hydrolase. The R204C/S250C variant was as active as the wild-type PES-H1 in hydrolyzing lcPET materials (Figure 4A,B). However, the R204C/S250C variant was significantly less active than the wild-type against hcPET materials (Figure 4C,D). In contrast, the L92F/Q94Y variant was more active than the wild-type PES-H1 against hcPET powders, forming up to 3.4-fold more degradation products (Figure 4D). For hydrolyzing hcPET powders (Figure 4C,D) generated by different pretreatment methods, LCC ICCG outperformed all PES-H1 variants and yielded up to 4.0-fold more degradation products than the L92F/Q94Y variant (Figure 4C,D). While the crystallinities of the crushed and ball-milled PET powders were clearly higher than the degradable threshold value of approximately 20% suggested by Wei et al.25 and Thomsen et al.,24 LCC ICCG surprisingly released more degradation products from these substrates than from lcPET powder (13% crystallinity) (Figure 4B–D). By comparison with PES-H1 and its variants, the extent of degradation was inversely correlated to the material crystallinity, which is in agreement with previous research on using hydrolases to degrade PET with varying crystallinities.6
In addition to the crystallinity values obtained by DSC, we also determined the number-average (Mn) and weight-average (Mw) molecular masses of the PET powders used for the enzymatic degradation experiments (Table S6). The lower Mn values determined for the hcPET powders derived from ball-milled and grinder-crushed PET bottle waste than that with the grinder-crushed PET film means that the PET bottle powders contained significantly shorter polymers (or even oligomers). When the degradation by PES-H1, its variants, and LCC ICCG (Figure 4B–D) is interpreted in terms of the molecular masses of the PET powders, it appears that Mn had a larger influence on the degradation by LCC ICCG than by PES-H1 and its variants. LCC ICCG seems to hydrolyze shorter polymers more efficiently, while the degradation by PES-H1 and its variants seems to be more influenced by crystallinity. Tournier et al.5 found that LCC ICCG could not efficiently depolymerize the hcPET obtained by their pretreatment methods, but they did not report the molecular masses of their materials. Therefore, a direct comparison of our data to that reported by Tournier et al.5 cannot be made, but our data suggest that not only crystallinity but also average polymer length may influence the enzymatic degradation of PET. This suggests that crystallinity might be only one of several important polymer characteristics that influence enzymatic degradation and that polymer properties should be more extensively characterized in future studies. The superiority of the L92F/Q94Y variant of PES-H1 in degrading lcPET is important since industrially amorphized PET waste has crystallinities below 15%.5 The L92F/Q94Y variant of PES-H1 could therefore be investigated as an alternative to LCC ICCG for PET hydrolysis.
A heterogeneous kinetic model described by the following equation has been widely used for evaluating enzymatic PET hydrolysis.12,45,46,51−53
| 1 |
In this equation, v0 describes the initial reaction rate, kh the hydrolysis rate constant, [S0] the initial substrate concentration, [E0] the enzyme concentration, and KA the adsorption equilibrium constant. We used PET nanoparticles (PET-NP) prepared using previously published protocols51,54 as the substrate to characterize the hydrolysis kinetics catalyzed by PES-H1, its variants, and LCC ICCG. The rates of turbidity change measured at 600 nm as a result of PET-NP degradation were determined in a microplate reader at 70 °C (Figures S9 and S10). The kinetic constants determined according to eq 1 are summarized in Table 1. The initial reaction velocity v0 was defined as the turbidity decrease over a reaction time of 40 min using 0.5 mg mL–1 PET-NP (Figure S9). The L92F/Q94Y variant resulted in the highest hydrolysis rate constant (kh) of 0.065 mL mg–1 min–1, followed by R204C/S250C (0.064 mL mg–1 min–1), wild-type PES-H1 (0.054 mL mg–1 min–1), and LCC ICCG (0.048 mL mg–1 min–1). The adsorption equilibrium constant KA is a parameter to evaluate the enzyme affinity to PET-NP. The R204C/S250C variant had the lowest affinity to PET, while the L92F/Q94Y variant had the highest affinity, which was 1.8-fold higher than that of wild-type PES-H1 and 1.4-fold higher than that of LCC ICCG. The high affinity of the L92F/Q94Y variant to PET nanoparticles demonstrates the importance of the adsorption of enzymes to the surface of PET-NP for efficient hydrolysis. The KA values determined in this study fall in the same range as those determined for many homologous PET hydrolases by other procedures.12,45,46,51,52 In general, the kinetic data, depicted in Table 1 and Table S7 support the observations made using other PET materials (Figure 4A,B). Additionally, we measured the initial hydrolysis rate as a function of the substrate concentration in the range 0–0.5 mg mL–1 using a constant enzyme concentration of 0.7 μM. This enzyme concentration was selected to ensure substrate-saturated conditions (Figure S9). The experimental data fit well to the kinetic model (Figures S9 and S10), demonstrating that under these conditions the model describes the experimental data accurately.
Table 1. Kinetic Parameters for PET-NP Hydrolysis by PES-H1, Its Variants, and LCC ICCG.
| KA [μM–1] | kh [mL mg–1 min–1] | |
|---|---|---|
| PES-H1 wild-type | 4.710 ± 0.087 | 0.054 ± 0.001 |
| L92F/Q94Y | 8.259 ± 0.028 | 0.065 ± 0.002 |
| R204C/S250C | 4.063 ± 0.191 | 0.064 ± 0.001 |
| LCC ICCG | 5.953 ± 0.032 | 0.048 ± 0.001 |
Conclusions
In this study, we solved crystal structures of the thermophilic enzymes PES-H1 and PES-H2 in apo form and notably also in complexes with the PET monomer analogues MHETA and BHET. This enabled the identification of six intermediate binding modes and several key residues involved in binding to PET. MD simulations supported the involvement of these residues in PET binding. Structural analyses and previously identified mutational hotspots were used to select PES-H1 residues for mutagenesis. Several PES-H1 variants with improved thermostability and PET hydrolysis activity were identified. The most active L92F/Q94Y variant outperformed the reference enzyme LCC ICCG in hydrolyzing lcPET materials (<15% crystallinity). Its superior activity over wild-type PES-H1 was also evident with other hcPET materials derived from real-world PET bottle waste. Our detailed structural analyses provide a better understanding of the mechanisms of interfacial biocatalysis, and our engineered variants hold promise for future applications in biocatalytic plastic recycling.
Acknowledgments
A.J. and B.S. gratefully acknowledge the computing time granted through JARA-HPC (project PETaseMD) on the supercomputer JURECA at Forschungszentrum Jülich,55 the hybrid computer cluster purchased from funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) project number INST 208/704-1 FUGG, and the Centre for Information and Media Technology at Heinrich Heine University Düsseldorf. Furthermore, we thank the staff from BL10U2/BL17B/BL17B/BL18U1/BL19U1 beamline of the National Facility for Protein Science in Shanghai (NFPS) at Shanghai Synchrotron Radiation Facility (SSRF) and staff from Beamline 14.1 at BESSY, for assistance during data collection.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.2c02275.
Materials and methods; data collection of crystal structures of PES-H1 and PES-H2 in apo form and in complex with MHETA and BHET; RMSD of selected PET hydrolases; MD simulations; changes in Tm; crystallinities and molecular masses of PET substrates; kinetic parameters of PET-NP hydrolysis; oligonucleotides; structure of PES-H1; stability of PES-H1 in MD simulations; RMSD of 3PET during MD simulations; binding positions of 3PET clusters; degradation of PET at 70 and 72 °C; phosphate dependence; cysteine calibration curve; electrostatic surface display; hydrolysis of PET-NP with varying enzyme concentrations; and hydrolysis of PET-NP with varying substrate concentrations (PDF)
GAFF parameters for each PET unit in 3PET (TXT)
Author Contributions
Conceptualization: R.W., W.L., and U.T.B. Protein crystallographic studies: J.G., Z.L., X.H., C.G.F., G.J.P., M.L., and W.L. Computational simulations: A.J., G.W., J.M., Y.-F.A., D.B., J.D., B.S., and W.L. Protein engineering and characterization: L.P., J.G., and Z.L. Polymer treatment and characterization: Y.C. and W.D. Writing—original draft: L.P., A.J., G.W., C.P.S.B., W.L., and R.W. Writing—review and editing: all authors. Supervision and funding acquisition: W.D., J.D., B.S., W.L., U.T.B., and R.W.
The authors W.L., J.G., Z.L., X.H., and W.D. acknowledge the financial support provided by the National Key Research and Development Program of China (2021YFC2103600 and 2021YFA0910200). The authors U.T.B., R.W., L.P., D.B., J.M., and J.D. gratefully acknowledge the financial support received from the European Union’s Horizon 2020 research and innovation program (MIX-UP, grant 870294; upPE-T, grant 953214; CETOCOEN Excellence, grant 857560) and from the Czech Ministry of Education (CZ.02.1.01/0.0/0.0/16_026/0008451). A.J. is financially supported by the German Federal Environmental Foundation. The authors J.G., X.H., and W.L. also thank the Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-PTJJ-008, TSBICIP-IJCP-003, TSBICIP-KJGG-009-01, and TSBICIP-KJGG-002-06), the Youth Innovation Promotion Association CAS, and the China Scholarship Council for financial support. The authors W.D. and Y.C. acknowledge the financial support provided by the National Natural Science Foundation of China (31961133017).
The authors declare no competing financial interest.
Supplementary Material
References
- Singh A.; Rorrer N. A.; Nicholson S. R.; Erickson E.; DesVeaux J. S.; Avelino A. F. T.; Lamers P.; Bhatt A.; Zhang Y.; Avery G.; Tao L.; Pickford A. R.; Carpenter A. C.; McGeehan J. E.; Beckham G. T. Techno-Economic, Life-Cycle, and Socioeconomic Impact Analysis of Enzymatic Recycling of Poly(Ethylene Terephthalate). Joule 2021, 5 (9), 2479–2503. 10.1016/j.joule.2021.06.015. [DOI] [Google Scholar]
- Wei R.; Tiso T.; Bertling J.; O’Connor K.; Blank L. M.; Bornscheuer U. T. Possibilities and Limitations of Biotechnological Plastic Degradation and Recycling. Nat. Catal. 2020, 3 (11), 867–871. 10.1038/s41929-020-00521-w. [DOI] [Google Scholar]
- Tiso T.; Winter B.; Wei R.; Hee J.; de Witt J.; Wierckx N.; Quicker P.; Bornscheuer U. T.; Bardow A.; Nogales J.; Blank L. M. The Metabolic Potential of Plastics as Biotechnological Carbon Sources – Review and Targets for the Future. Metab. Engin. 2022, 71, 77–98. 10.1016/j.ymben.2021.12.006. [DOI] [PubMed] [Google Scholar]
- Jönsson C.; Wei R.; Biundo A.; Landberg J.; Schwarz Bour L.; Pezzotti F.; Toca A.; Jacques L. M.; Bornscheuer U. T.; Syrén P. Biocatalysis in the Recycling Landscape for Synthetic Polymers and Plastics towards Circular Textiles. ChemSusChem 2021, 14 (19), 4028–4040. 10.1002/cssc.202002666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier V.; Topham C. M.; Gilles A.; David B.; Folgoas C.; Moya-Leclair E.; Kamionka E.; Desrousseaux M.-L.; Texier H.; Gavalda S.; Cot M.; Guémard E.; Dalibey M.; Nomme J.; Cioci G.; Barbe S.; Chateau M.; André I.; Duquesne S.; Marty A. An Engineered PET Depolymerase to Break down and Recycle Plastic Bottles. Nature 2020, 580 (7802), 216–219. 10.1038/s41586-020-2149-4. [DOI] [PubMed] [Google Scholar]
- Wei R.; von Haugwitz G.; Pfaff L.; Mican J.; Badenhorst C. P. S.; Liu W.; Weber G.; Austin H. P.; Bednar D.; Damborsky J.; Bornscheuer U. T. Mechanism-Based Design of Efficient PET Hydrolases. ACS Catal. 2022, 12 (6), 3382–3396. 10.1021/acscatal.1c05856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi R.; Chiavacci P.; Vosa R.; Guerra G. Effect of Moisture on the Crystallization Behavior of PET from the Quenched Amorphous Phase. J. Appl. Polym. Sci. 1991, 43 (6), 1087–1089. 10.1002/app.1991.070430608. [DOI] [Google Scholar]
- Langevin D.; Grenet J.; Saiter J. M. Moisture Sorption in PET Influence on the Thermokinetic Parameters. Eur. Polym. J. 1994, 30 (3), 339–345. 10.1016/0014-3057(94)90297-6. [DOI] [Google Scholar]
- Yoshida S.; Hiraga K.; Takehana T.; Taniguchi I.; Yamaji H.; Maeda Y.; Toyohara K.; Miyamoto K.; Kimura Y.; Oda K. A Bacterium That Degrades and Assimilates Poly(Ethylene Terephthalate). Science 2016, 351 (6278), 1196–1199. 10.1126/science.aad6359. [DOI] [PubMed] [Google Scholar]
- Kawai F. The Current State of Research on PET Hydrolyzing Enzymes Available for Biorecycling. Catalysts 2021, 11 (2), 206. 10.3390/catal11020206. [DOI] [Google Scholar]
- Brott S.; Pfaff L.; Schuricht J.; Schwarz J.; Böttcher D.; Badenhorst C. P. S.; Wei R.; Bornscheuer U. T. Engineering and Evaluation of Thermostable IsPETase Variants for PET Degradation. Eng. Life Sci. 2022, 22 (3–4), 192–203. 10.1002/elsc.202100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronkvist Å. M.; Xie W.; Lu W.; Gross R. A. Cutinase-Catalyzed Hydrolysis of Poly(Ethylene Terephthalate). Macromolecules 2009, 42 (14), 5128–5138. 10.1021/ma9005318. [DOI] [Google Scholar]
- Sulaiman S.; You D.-J.; Kanaya E.; Koga Y.; Kanaya S. Crystal Structure and Thermodynamic and Kinetic Stability of Metagenome-Derived LC-Cutinase. Biochemistry 2014, 53 (11), 1858–1869. 10.1021/bi401561p. [DOI] [PubMed] [Google Scholar]
- Sulaiman S.; Yamato S.; Kanaya E.; Kim J.-J.; Koga Y.; Takano K.; Kanaya S. Isolation of a Novel Cutinase Homolog with Polyethylene Terephthalate-Degrading Activity from Leaf-Branch Compost by Using a Metagenomic Approach. Appl. Environ. Microbiol. 2012, 78 (5), 1556–1562. 10.1128/AEM.06725-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R.; Song C.; Gräsing D.; Schneider T.; Bielytskyi P.; Böttcher D.; Matysik J.; Bornscheuer U. T.; Zimmermann W. Conformational Fitting of a Flexible Oligomeric Substrate Does Not Explain the Enzymatic PET Degradation. Nat. Commun. 2019, 10 (1), 5581. 10.1038/s41467-019-13492-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa T.; Mizushima H.; Ohtsuka J.; Oda M.; Kawai F.; Tanokura M. Structural Basis for the Ca2+-Enhanced Thermostability and Activity of PET-Degrading Cutinase-like Enzyme from Saccharomonospora viridis AHK190. Appl. Microbiol. Biotechnol. 2015, 99 (10), 4297–4307. 10.1007/s00253-014-6272-8. [DOI] [PubMed] [Google Scholar]
- Then J.; Wei R.; Oeser T.; Gerdts A.; Schmidt J.; Barth M.; Zimmermann W. A Disulfide Bridge in the Calcium Binding Site of a Polyester Hydrolase Increases Its Thermal Stability and Activity against Polyethylene Terephthalate. FEBS Open Bio 2016, 6 (5), 425–432. 10.1002/2211-5463.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda M.; Yamagami Y.; Inaba S.; Oida T.; Yamamoto M.; Kitajima S.; Kawai F. Enzymatic Hydrolysis of PET: Functional Roles of Three Ca2+ Ions Bound to a Cutinase-like Enzyme, Cut190*, and Its Engineering for Improved Activity. Appl. Microbiol. Biotechnol. 2018, 102 (23), 10067–10077. 10.1007/s00253-018-9374-x. [DOI] [PubMed] [Google Scholar]
- Zhong-Johnson E. Z. L.; Voigt C. A.; Sinskey A. J. An Absorbance Method for Analysis of Enzymatic Degradation Kinetics of Poly(Ethylene Terephthalate) Films. Sci. Rep. 2021, 11 (1), 928. 10.1038/s41598-020-79031-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Then J.; Wei R.; Oeser T.; Barth M.; Belisário-Ferrari M. R.; Schmidt J.; Zimmermann W. Ca 2+ and Mg 2+ Binding Site Engineering Increases the Degradation of Polyethylene Terephthalate Films by Polyester Hydrolases from Thermobifida fusca. Biotechnol. J. 2015, 10 (4), 592–598. 10.1002/biot.201400620. [DOI] [PubMed] [Google Scholar]
- Emori M.; Numoto N.; Senga A.; Bekker G.; Kamiya N.; Kobayashi Y.; Ito N.; Kawai F.; Oda M. Structural Basis of Mutants of PET -degrading Enzyme from Saccharomonospora viridis AHK190 with High Activity and Thermal Stability. Proteins 2021, 89 (5), 502–511. 10.1002/prot.26034. [DOI] [PubMed] [Google Scholar]
- Nakamura A.; Kobayashi N.; Koga N.; Iino R. Positive Charge Introduction on the Surface of Thermostabilized PET Hydrolase Facilitates PET Binding and Degradation. ACS Catal. 2021, 11 (14), 8550–8564. 10.1021/acscatal.1c01204. [DOI] [Google Scholar]
- Zeng W.; Li X.; Yang Y.; Min J.; Huang J.-W.; Liu W.; Niu D.; Yang X.; Han X.; Zhang L.; Dai L.; Chen C.-C.; Guo R.-T. Substrate-Binding Mode of a Thermophilic PET Hydrolase and Engineering the Enzyme to Enhance the Hydrolytic Efficacy. ACS Catal. 2022, 12 (5), 3033–3040. 10.1021/acscatal.1c05800. [DOI] [Google Scholar]
- Thomsen T. B.; Hunt C. J.; Meyer A. S. Influence of Substrate Crystallinity and Glass Transition Temperature on Enzymatic Degradation of Polyethylene Terephthalate (PET). N. Biotechnol. 2022, 69, 28–35. 10.1016/j.nbt.2022.02.006. [DOI] [PubMed] [Google Scholar]
- Wei R.; Breite D.; Song C.; Gräsing D.; Ploss T.; Hille P.; Schwerdtfeger R.; Matysik J.; Schulze A.; Zimmermann W. Biocatalytic Degradation Efficiency of Postconsumer Polyethylene Terephthalate Packaging Determined by Their Polymer Microstructures. Adv. Sci. 2019, 6 (14), 1900491. 10.1002/advs.201900491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y.; Chen Y.; Liu X.; Dong S.; Tian Y.; Qiao Y.; Mitra R.; Han J.; Li C.; Han X.; Liu W.; Chen Q.; Wei W.; Wang X.; Du W.; Tang S.; Xiang H.; Liu H.; Liang Y.; Houk K. N.; Wu B. Computational Redesign of a PETase for Plastic Biodegradation under Ambient Condition by the GRAPE Strategy. ACS Catal. 2021, 11 (3), 1340–1350. 10.1021/acscatal.0c05126. [DOI] [Google Scholar]
- Lu H.; Diaz D. J.; Czarnecki N. J.; Zhu C.; Kim W.; Shroff R.; Acosta D. J.; Alexander B. R.; Cole H. O.; Zhang Y.; Lynd N. A.; Ellington A. D.; Alper H. S. Machine Learning-Aided Engineering of Hydrolases for PET Depolymerization. Nature 2022, 604 (7907), 662–667. 10.1038/s41586-022-04599-z. [DOI] [PubMed] [Google Scholar]
- Han X.; Liu W.; Huang J.-W.; Ma J.; Zheng Y.; Ko T.-P.; Xu L.; Cheng Y.-S.; Chen C.-C.; Guo R.-T. Structural Insight into Catalytic Mechanism of PET Hydrolase. Nat. Commun. 2017, 8 (1), 2106. 10.1038/s41467-017-02255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneta S.; Arafet K.; Moliner V. QM/MM Study of the Enzymatic Biodegradation Mechanism of Polyethylene Terephthalate. J. Chem. Inf. Model. 2021, 61 (6), 3041–3051. 10.1021/acs.jcim.1c00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S.; Yue Y.; Zheng M.; Li Y.; Zhang Q.; Wang W. Is PETase- and Is MHETase-Catalyzed Cascade Degradation Mechanism toward Polyethylene Terephthalate. ACS Sustain. Chem. Eng. 2021, 9 (29), 9823–9832. 10.1021/acssuschemeng.1c02420. [DOI] [Google Scholar]
- Jerves C.; Neves R. P. P.; Ramos M. J.; da Silva S.; Fernandes P. A. Reaction Mechanism of the PET Degrading Enzyme PETase Studied with DFT/MM Molecular Dynamics Simulations. ACS Catal. 2021, 11 (18), 11626–11638. 10.1021/acscatal.1c03700. [DOI] [Google Scholar]
- Zheng M.; Li Y.; Dong W.; Feng S.; Zhang Q.; Wang W. Computational Biotransformation of Polyethylene Terephthalate by Depolymerase: A QM/MM Approach. J. Hazard. Mater. 2022, 423, 127017. 10.1016/j.jhazmat.2021.127017. [DOI] [PubMed] [Google Scholar]
- Zimmermann W.; Wei R.; Hille P.; Oeser T.; Schmidt J.. New Polypeptides Having a Polyester Degrading Activity and Uses Thereof. EP3517608A1, July 31, 2019.
- Sonnendecker C.; Oeser J.; Richter P. K.; Hille P.; Zhao Z.; Fischer C.; Lippold H.; Blázquez-Sánchez P.; Engelberger F.; Ramírez-Sarmiento C. A.; Oeser T.; Lihanova Y.; Frank R.; Jahnke H.; Billig S.; Abel B.; Sträter N.; Matysik J.; Zimmermann W. Low Carbon Footprint Recycling of Post-Consumer PET Plastic with a Metagenomic Polyester Hydrolase. ChemSusChem 2022, 15 (9), e202101062 10.1002/cssc.202101062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenstein P.; Wei R.; Matysik J.; Song C.. Mechanistic Investigation of Enzymatic Degradation of Polyethylene Terephthalate by Nuclear Magnetic Resonance. In Methods in Enzymology; Elsevier, 2021; Vol. 648, pp 231–252. [DOI] [PubMed] [Google Scholar]
- Barth M.; Honak A.; Oeser T.; Wei R.; Belisário-Ferrari M. R.; Then J.; Schmidt J.; Zimmermann W. A Dual Enzyme System Composed of a Polyester Hydrolase and a Carboxylesterase Enhances the Biocatalytic Degradation of Polyethylene Terephthalate Films. J. Biotechnol. 2016, 11 (8), 1082–1087. 10.1002/biot.201600008. [DOI] [PubMed] [Google Scholar]
- Eugenio E. de Q.; Campisano I. S. P.; de Castro A. M.; Coelho M. A. Z.; Langone M. A. P. Kinetic Modeling of the Post-Consumer Poly(Ethylene Terephthalate) Hydrolysis Catalyzed by Cutinase from Humicola insolens. J. Polym. Environ. 2022, 30, 1627–1637. 10.1007/s10924-021-02301-4. [DOI] [Google Scholar]
- Chen C.; Han X.; Ko T.; Liu W.; Guo R. Structural Studies Reveal the Molecular Mechanism of PETase. FEBS J. 2018, 285 (20), 3717–3723. 10.1111/febs.14612. [DOI] [PubMed] [Google Scholar]
- Schmidt J.; Wei R.; Oeser T.; Belisário-Ferrari M. R.; Barth M.; Then J.; Zimmermann W. Effect of Tris, MOPS, and Phosphate Buffers on the Hydrolysis of Polyethylene Terephthalate Films by Polyester Hydrolases. FEBS Open Bio 2016, 6 (9), 919–927. 10.1002/2211-5463.12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numoto N.; Kamiya N.; Bekker G.-J.; Yamagami Y.; Inaba S.; Ishii K.; Uchiyama S.; Kawai F.; Ito N.; Oda M. Structural Dynamics of the PET-Degrading Cutinase-like Enzyme from Saccharomonospora viridis AHK190 in Substrate-Bound States Elucidates the Ca 2+ -Driven Catalytic Cycle. Biochemistry 2018, 57 (36), 5289–5300. 10.1021/acs.biochem.8b00624. [DOI] [PubMed] [Google Scholar]
- Baker P. J.; Poultney C.; Liu Z.; Gross R.; Montclare J. K. Identification and Comparison of Cutinases for Synthetic Polyester Degradation. Appl. Microbiol. Biotechnol. 2012, 93 (1), 229–240. 10.1007/s00253-011-3402-4. [DOI] [PubMed] [Google Scholar]
- Furukawa M.; Kawakami N.; Tomizawa A.; Miyamoto K. Efficient Degradation of Poly(Ethylene Terephthalate) with Thermobifida fusca Cutinase Exhibiting Improved Catalytic Activity Generated Using Mutagenesis and Additive-Based Approaches. Sci. Rep. 2019, 9 (1), 16038. 10.1038/s41598-019-52379-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin H. P.; Allen M. D.; Donohoe B. S.; Rorrer N. A.; Kearns F. L.; Silveira R. L.; Pollard B. C.; Dominick G.; Duman R.; El Omari K.; Mykhaylyk V.; Wagner A.; Michener W. E.; Amore A.; Skaf M. S.; Crowley M. F.; Thorne A. W.; Johnson C. W.; Woodcock H. L.; McGeehan J. E.; Beckham G. T. Characterization and Engineering of a Plastic-Degrading Aromatic Polyesterase. Proc. Natl. Acad. Sci. U.S.A. 2018, 115 (19), E4350–E4357. 10.1073/pnas.1718804115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson E.; Shakespeare T. J.; Bratti F.; Buss B. L.; Graham R.; Hawkins M. A.; König G.; Michener W. E.; Miscall J.; Ramirez K. J.; Rorrer N. A.; Zahn M.; Pickford A. R.; McGeehan J. E.; Beckham G. T. Comparative Performance of PETase as a Function of Reaction Conditions, Substrate Properties, and Product Accumulation. ChemSusChem 2022, 15 (1), e202101932 10.1002/cssc.202101932. [DOI] [PubMed] [Google Scholar]
- Wei R.; Oeser T.; Then J.; Kühn N.; Barth M.; Schmidt J.; Zimmermann W. Functional Characterization and Structural Modeling of Synthetic Polyester-Degrading Hydrolases from Thermomonospora curvata. AMB Expr. 2014, 4 (1), 44. 10.1186/s13568-014-0044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R.; Oeser T.; Schmidt J.; Meier R.; Barth M.; Then J.; Zimmermann W. Engineered Bacterial Polyester Hydrolases Efficiently Degrade Polyethylene Terephthalate Due to Relieved Product Inhibition: Engineered Polyester Hydrolases. Biotechnol. Bioeng. 2016, 113 (8), 1658–1665. 10.1002/bit.25941. [DOI] [PubMed] [Google Scholar]
- Silva C.; Da S.; Silva N.; Matamá T.; Araújo R.; Martins M.; Chen S.; Chen J.; Wu J.; Casal M.; Cavaco-Paulo A. Engineered Thermobifida fusca Cutinase with Increased Activity on Polyester Substrates. J. Biotechnol. 2011, 6 (10), 1230–1239. 10.1002/biot.201000391. [DOI] [PubMed] [Google Scholar]
- Gamerith C.; Zartl B.; Pellis A.; Guillamot F.; Marty A.; Acero E. H.; Guebitz G. M. Enzymatic Recovery of Polyester Building Blocks from Polymer Blends. Process. Biochem. 2017, 59, 58–64. 10.1016/j.procbio.2017.01.004. [DOI] [Google Scholar]
- Bai C. Structural Changes in Poly(Ethylene Terephthalate) Induced by Mechanical Milling. Polymer 2000, 41 (19), 7147–7157. 10.1016/S0032-3861(00)00048-3. [DOI] [Google Scholar]
- Falkenstein P.; Gräsing D.; Bielytskyi P.; Zimmermann W.; Matysik J.; Wei R.; Song C. UV Pretreatment Impairs the Enzymatic Degradation of Polyethylene Terephthalate. Front. Microbiol. 2020, 11, 689. 10.3389/fmicb.2020.00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel K.; Wei R.; Pfaff L.; Breite D.; Al-Fathi H.; Ortmann C.; Estrela-Lopis I.; Venus T.; Schulze A.; Harms H.; Bornscheuer U. T.; Maskow T. Enzymatic Degradation of Polyethylene Terephthalate Nanoplastics Analyzed in Real Time by Isothermal Titration Calorimetry. Sci. Total Environ. 2021, 773, 145111. 10.1016/j.scitotenv.2021.145111. [DOI] [PubMed] [Google Scholar]
- Wei R.; Oeser T.; Barth M.; Weigl N.; Lübs A.; Schulz-Siegmund M.; Hacker M. C.; Zimmermann W. Turbidimetric Analysis of the Enzymatic Hydrolysis of Polyethylene Terephthalate Nanoparticles. J. Mol. Catal. B: Enzym. 2014, 103, 72–78. 10.1016/j.molcatb.2013.08.010. [DOI] [Google Scholar]
- Scandola M.; Focarete M. L.; Frisoni G. Simple Kinetic Model for the Heterogeneous Enzymatic Hydrolysis of Natural Poly(3-Hydroxybutyrate). Macromolecules 1998, 31 (12), 3846–3851. 10.1021/ma980137y. [DOI] [Google Scholar]
- Pfaff L.; Breite D.; Badenhorst C. P. S.; Bornscheuer U. T.; Wei R.. Fluorimetric High-Throughput Screening Method for Polyester Hydrolase Activity Using Polyethylene Terephthalate Nanoparticles. In Methods in Enzymology; Elsevier, 2021; Vol. 648, pp 253–270. [DOI] [PubMed] [Google Scholar]
- Krause D.; Thörnig P. JURECA: Modular Supercomputer at Jülich Supercomputing Centre. JLSRF 2018, 4, A132. 10.17815/jlsrf-4-121-1. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.