

Abstract
Tropane derivatives are extensively used in medicine, but catalytic asymmetric methods for their synthesis are underexplored. Here, we report Rh-catalyzed asymmetric Suzuki–Miyaura-type cross-coupling reactions between a racemic N-Boc-nortropane-derived allylic chloride and (hetero)aryl boronic esters. The reaction proceeds via an unexpected kinetic resolution, and the resolved enantiopure allyl chloride can undergo highly enantiospecific reactions with N-, O-, and S-containing nucleophiles. The method was applied in a highly stereoselective formal synthesis of YZJ-1139(1), a potential insomnia treatment that recently completed Phase II clinical trials. Our report represents an asymmetric catalytic method for the synthesis of YZJ-1139(1) and related compounds.
Keywords: alkaloids, asymmetric catalysis, bicycles, rhodium, Suzuki−Miyaura coupling, kinetic resolution
Introduction
Molecules with N-methyl-8-aza-bicyclo[3.2.1]octane scaffolds, generally known as the tropane alkaloids, display a wide array of biological and pharmaceutical activities.1−3 Tropane derivatives, for example, cocaine (Scheme 1a) and scopolamine, are well-known for displaying psychoactive effects, and other tropane derivatives, including atropine (Scheme 1a), are used as anticholinergics and stimulants for treatment of neurological and psychiatric disorders such as Parkinson’s disease and depression.4−6 A 8-aza-bicyclo[3.2.1]octane (nortropane)-derived molecule YZJ-1139(1) (Scheme 1a) was also reported recently as an orexin receptor antagonist, which has completed Phase II clinical trials and may become a treatment for insomnia.7
Scheme 1. Tropane Derivatives and Works on Rh-Catalyzed Cross-Coupling Reactions of Cyclic Compounds.

Historically, tropane alkaloids had been extracted from plants, but their unique biological activities have inspired the development of synthetic routes to tropane derivatives. Willstätter’s first synthesis of cocaine8 and Robinson’s highly efficient double-Mannich approach toward tropinone9 are early milestones in this highly active field. Many strategies toward enantioenriched tropanes rely on the derivatization of natural tropane alkaloids, chiral resolution, and synthesis from the chiral pool.10
Given the fame of these molecules, and their importance to medicine, it is remarkable that stereoselective methods using achiral starting materials are limited.3 Current methods largely use one of two approaches: (1) desymmetrization of meso tropinone and its derivatives with stoichiometric chiral lithium amide bases,11−14 and (2) enantioselective synthesis of the tropane scaffold where the stereochemical information is introduced concomitant with the formation of the bicycle.15 Chiral auxiliary16−20 and asymmetric catalytic approaches are known for the latter.21−27
Previously, our group reported asymmetric Suzuki–Miyaura-type cross-coupling reactions with racemic mono- and bicyclic allyl chlorides (Scheme 1b).28−34 In these highly enantioselective transformations, both enantiomers of the starting material are converted into a single enantiomer of the product. Deracemization is believed to occur via the formation of a common pseudo-prochiral or meso Rh-π-allyl complex (DYKAT type II).35,36 We wondered if we could apply a related strategy to the catalytic asymmetric synthesis of sterically congested bicyclic N-heterocycles and hence develop an asymmetric cross-coupling approach to the nortropane scaffold (Scheme 1c).
Results and Discussion
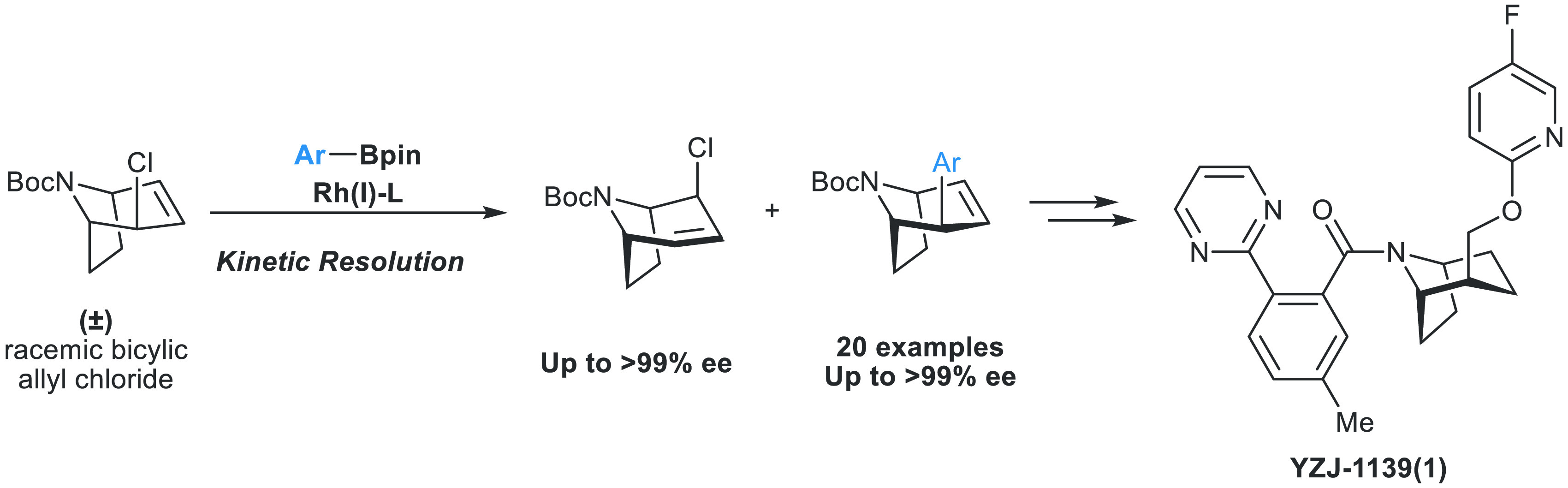
A suitable nortropane-derived allyl chloride (±)-1a was synthesized from N-Boc-nortropinone in five synthetic steps (see the Supporting Information). Using previously reported conditions for Rh-catalyzed Suzuki–Miyaura cross-couplings established by our group,31 the reaction of allyl chloride (±)-1a with phenyl boronic acid 2aa afforded 3a in 94% enantiomeric excess as a single diastereomer (>20:1), albeit only in 28% yield (Table 1, entry 1).
Table 1. Selected Optimization Experimentsa.


Rh[(cod)OH]2 (2.5 mol %), ligand (6 mol %), (±)-1a (0.2 mmol), 2aa or 2a, CsOH (50 wt % aq.), THF, 65 °C.
Isolated yield.
Enantiomeric excess determined by supercritical fluid chromatography (SFC) analysis on a chiral nonracemic stationary phase.
Diastereomeric ratio determined by the integration of 1H NMR spectra.
Numbers in brackets refer to the yield and ee of recovered allyl chloride.
We extensively examined the influence of temperature, solvent, base, boronic acid derivatives, catalyst loading, equivalents of reagents, and the use of additives (selected examples are presented in Table 1; for additional data see SI Tables S1 and S2). The protecting group on nitrogen and the leaving group of the nortropinone-derived substrate were investigated. We found that L1 was superior to related bidentate phosphine ligands regarding both reactivity and enantioselectivity (Table 1, entries 1–4). An increase in yield was observed by increasing both the equivalents of the base and the coupling partner (entry 5). Similar results were obtained using phenyl boronic pinacol ester 2a as the nucleophile (entry 6), and along with 3a, we also isolated enantiopure (>99% ee) allyl chloride (+)-1 in 21% yield.
Upon shortening the reaction time from overnight to 0.5 h, the yields of 3a were similar, but the yield of (+)-1a increased to 37% (entry 7) with a slight decrease in ee (97% ee). We attribute the decreased yield of (+)-1a during longer durations to the slower but competitive hydrolysis of 1a under the reaction conditions. Using less equivalents of boronic pinacol ester and base (entries 7 and 8) gave similar results but purification by column chromatography was easier. Here, using the pinacol ester gave superior results compared to the free boronic acid (entries 8 and 9); at 1 h reaction time, 3a was isolated in 50% yield (95% ee), and enantiopure (+)-1a was isolated in 37% yield (entry 10).
Changing the protecting group on nitrogen from N-Boc to methyl carbamate resulted in a higher yield at 63% and ee of 93% (entry 11). However, the diastereoselectivity decreased drastically from >20:1 to 5.9:1, likely due to steric reasons.
A highly selective kinetic resolution would intrinsically limit the obtained yields of these reactions to 50%, but we also speculated that catalyst deactivation or decomposition of the boronic ester could limit conversion. To help distinguish between these scenarios, we subjected (+)-1 (>99% ee) instead of racemic allyl chloride to our standard reaction conditions and allowed the reaction to occur overnight (Scheme 2a). Small amounts of the desired coupling product were obtained (7%), albeit to our surprise only with 61% ee. Additionally, some enantiopure allyl chloride (+)-1a (50%) was recovered, while the rest of the substrate was hydrolyzed to the corresponding allyl alcohol.
Scheme 2. Mechanistic Investigations and the Proposed Mechanism,

Rh-catalyzed Suzuki–Miyaura coupling reaction with (+)-1a and 2a.
Rh-catalyzed Suzuki–Miyaura coupling reaction with (+)-1a and biphep as ligand.
Rh-catalyzed Suzuki–Miyaura coupling reaction with (+)-1a and rac-Segphos as ligand.
Proposed mechanism.
Equilibration between two Rh-σ complexes.
This result indicates that, unlike the previous DYKAT process developed by our group, where both the enantiomers form a common symmetric Rh-π-allyl complex intermediate, when using L1, oxidative addition of (+)-1a either does not occur or does not give the same intermediate as enantiomer (−)-1a (Scheme 2d), and product formation via (+)-1a is slow.
To test if oxidative addition (−)-1a would result in the formation of meso π-complex, which would result in complete loss in stereochemical information upon oxidative addition, we performed a cross-coupling reaction with enantiopure allyl chloride (+)-1a with an achiral ligand biphep (Scheme 2b). Under these conditions, we obtained (+)-3a in 11 and 37% ee. The significant loss in ee in the reaction with biphep suggests that reductive elimination is at least partially enantio-determining and is controlled by the ligand when Segphos is used. We attribute the partial, but not complete, loss in stereochemical information in the experiment with biphep to either σ–π–σ isomerization mechanism (Scheme 2e), which occurs at a rate similar to reductive elimination, or to a lack of selectivity amongst different oxidative addition-type pathways when biphep is used.
Using a racemic mixture of Segphos as the ligand in the reaction with enantiopure (+)-1a, we obtained (+)-3a in 81% yield and 92% ee (Scheme 2c). In analogy to the combination of racemic allyl chloride and enantiopure ligand, this experiment shows the strong matched effect between (+)-1a and (R)-Segphos in the oxidative addition step (or (−)-1a and (S)-Segphos), most likely for steric reasons, which ultimately results in a highly selective kinetic resolution.
The partial DKYAT character of this transformation (Scheme 2a,e) does not allow for a meaningful quantification of s-factors (see SIp. S36).
A range of aryl boronic pinacol esters with electron-withdrawing and donating substituents at the para- and meta-positions yielded the desired coupling products typically in 40–50% yield (Scheme 3, 3a–3h, 3l–3o) and >94% ee as single diastereomers (>20:1 d.r.). These examples included various aryl halides, alkoxy groups, ester, and an aryl silane, which are useful intermediates for further reactions.
Scheme 3. Scope of the Reaction.
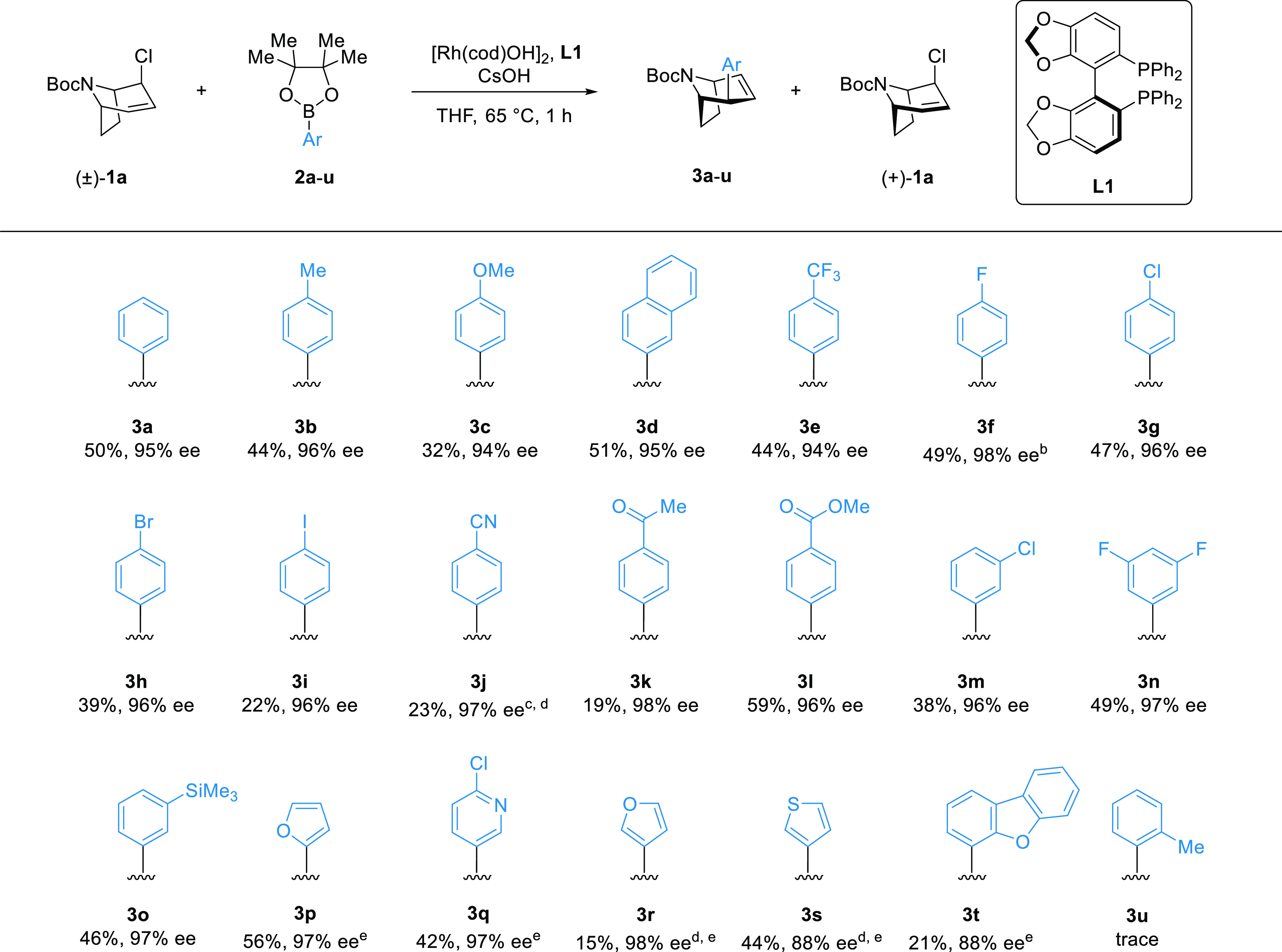
Rh[(cod)OH]2 (2.5 mol %), L1 (6 mol %), (±)-1a (0.2 mmol, 1.0 equiv), 2 (3.0 equiv), CsOH (50 wt % aq.; 2.0 equiv), THF (0.2 M), 65 °C, 1 h. All yields presented are isolated yields. Enantiomeric excess determined by supercritical fluid chromatography (SFC) analysis on a chiral nonracemic stationary phase. Single diastereomer (>20:1) obtained unless stated.
4 h.
Dioxane instead of THF, 80 °C overnight.
3j: 18:1 d.r., 3r: 17:1 d.r., 3s: 19:1 d.r.
2 h.
More challenging coupling partners featuring an iodide, cyano, or acetyl group (3i–3k) resulting in diminished yields but consistently high enantioselectivity.
We observed only trace product formation with 2-methylphenylboronic pinacol ester (2u), likely due to sterics and/or competitive protodeborylation.37
Heteroaryl boronic pinacol esters can be used. 2-Furanyl- and 2-chloropyridyl-boronic pinacol esters performed well with yields over 40% and high enantioselectivities (3p, 3q). 3-Furanylboronic pinacol esters gave only 15% yield, likely due to rapid protodeborylation—a common problem with heterocyclic boronic acids and esters.38
Interestingly, a few examples gave >50% yield (3l and 3p gave 59 and 56% yield, respectively), which we have briefly investigated but still do not fully understand, and the yield of 3p was found to further improve upon scale-up (see below).
The absolute and relative stereochemistries of the product and resolved allyl chloride (Schemes 3a and 4a) were determined by single-crystal X-ray diffraction of 3p and (+)-1a.39,40
Scheme 4. Scale-Up of the Cross-Coupling Reaction with 2p and Transformation of the Coupling Product and Enantioenriched Substrate,
Reaction at 1 g scale with 2-furanylboronic pinacol ester 2p.
Formal synthesis of YZJ-1139(1).
Enantiospecific transformation of enantiopure allyl chloride (+)-1a.
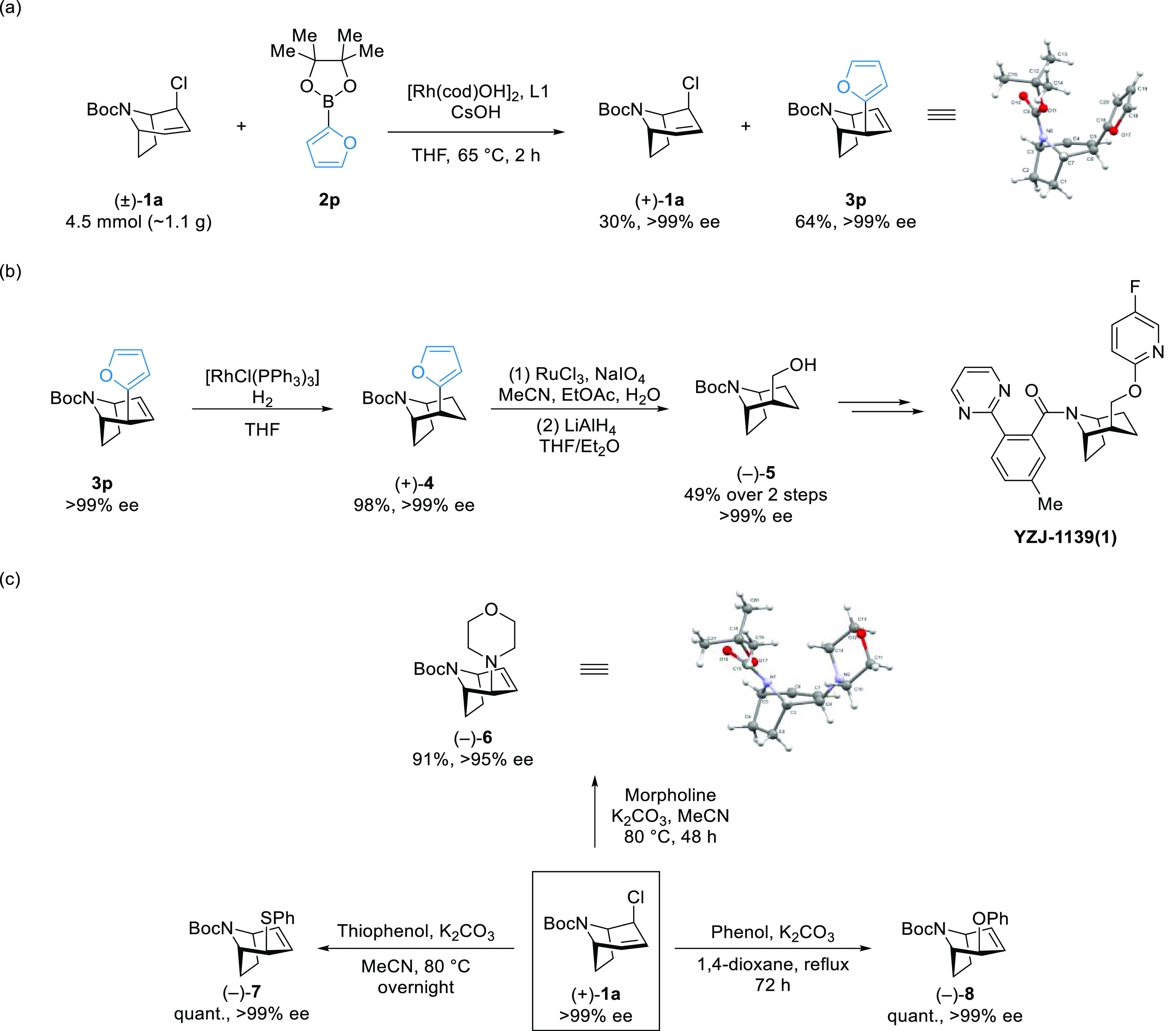
To demonstrate the synthetic utility of our method, we applied Rh-catalyzed Suzuki–Miyaura coupling with allyl chloride (±)-1a to formal synthesis of the orexin receptor antagonist YZJ-1129(1) (Scheme 4a,b),41 which recently passed Phase II clinical trials for the treatment of sleep disorders. To the best of our knowledge, the only other previously reported synthesis of YZJ-1129(1) was recently reported by a process chemistry group and relied on preparative high-performance liquid chromatography (HPLC) separation of the enantiomers or a chiral auxiliary approach.7
A gram-scale cross-coupling reaction between (±)-1a and 2-furanylboronic pinacol ester 2p afforded 3p in 64% isolated yield and >99% ee (Scheme 4a). The enantioenriched allyl chloride (+)-1a was isolated in 30% yield and >99% ee.
Reduction of 3p with Wilkinson’s catalyst gave (+)-4 in 98% yield and >99% ee. The furyl group was then converted into a hydroxymethyl group via a two-step oxidative cleavage/reduction protocol to give (−)-5 in 49% yield, a previously reported intermediate in the synthesis of YZJ-1129(1).
Previously, our group reported a Cu-catalyzed kinetic resolution reaction of a piperidine-derived allyl chloride, and the enantioenriched allyl chloride was used in enantiospecific substitution reactions with heteroatom-based nucleophiles,42 and we wondered if related substitutions with (+)-1a were possible.
The substitution with morpholine, thiophenol, and phenol gave (−)-6, (−)-7, and (−)-8 in excellent yield and enantiospecificity (Scheme 4c), respectively. Other enantiospecific substitution reactions with this substrate are likely possible. The absolute stereochemistry of (−)-6 was determined by single-crystal X-ray diffraction, and this, combined with the knowledge of the absolute configuration of the starting material, indicates that the substitution reaction proceeds via an syn-SN2′ pathway (Scheme 4c) The relative and absolute stereochemistry of (−)-7 and (−)-8 is assigned in analogy to (−)-6.
Conclusions
In summary, we developed an efficient kinetic resolution of a nortropane-derived allyl chloride via Rh(I)-catalyzed Suzuki–Miyaura cross-couplings. The reaction tolerates a range of different aryl- and heteroaryl boronic pinacol esters with synthetically useful functional groups in high enantioselectivity and diastereoselectivity. The coupling product with 2-furanyl pinacol ester was used in a formal asymmetric synthesis of YZJ-1129(1). Further, the resolved enantiopure allyl chloride can undergo enantiospecific syn-SN2′ reactions with O-, S-, and N-nucleophiles. Overall, this work provides access to a wide range of enantiomerically enriched tropane derivatives.
Acknowledgments
Financial support from the U.K. Engineering and Physical Sciences Research Council (EP/N022246/1) is gratefully acknowledged. F.W.G. is grateful to the National Research Fund, Luxembourg, for an AFR PhD Grant (11588566); the EPSRC Doctoral Training Partnership (DTP) for a studentship (EP/N509711/1); and Vertex Pharmaceuticals for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.2c02259.
Experimental procedure; characterization of substrates and products (NMR, IR, HRMS, optical rotation); 1H, 13C, and 19F NMR spectra; SFC traces and X-ray data (PDF)
Crystallographic data for (+)-1a, 3p, and (−)-6 have been deposited at the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 2156812, 2156553, and 2156805 and can be obtained viawww.ccdc.cam.ac.uk/data_request/cif (CIF)
Checkcif.pdf (PDF)
Author Contributions
Y.Z. performed all experiments. F.W.G. and S.P.F. conceived the project and supervised the work. K.E.C. performed and analyzed single-crystal X-ray diffraction experiments. Y.Z., F.W.G., and S.P.F. wrote the manuscript.
The authors declare the following competing financial interest(s): Oxford University Innovation has filed a patent application(PCT/GB2016/051612) with S.P.F. named as an inventor. Y.Z., F.W.G. and K.E.C. declare no competing financial interests.
Notes
Oxford University Innovation has filed a patent application (PCT/GB2016/051612) with S.P.F. named as an inventor. Y.Z. F.W.G., and K.E.C. declare no competing financial interests.
Dedication
Dedicated to the memory of Stephen A. Westcott.
Supplementary Material
References
- Fodor G.; Dharanipragada R. Tropane alkaloids. Nat. Prod. Rep. 1994, 11, 443–450. 10.1039/np9941100443. [DOI] [PubMed] [Google Scholar]
- Afewerki S.; Wang J.-X.; Liao W.-W.; Córdova A.. The Chemical Synthesis and Applications of Tropane Alkaloids. In The Alkaloids: Chemistry and Biology,Knölker H.-J., Ed.; Academic Press, 2019; Vol. 81, pp 151–233. [DOI] [PubMed] [Google Scholar]
- Rodriguez S.; Uria U.; Reyes E.; Prieto L.; Rodríguez-Rodríguez M.; Carrillo L.; Vicario J. L. Enantioselective construction of the 8-azabicyclo [3.2. 1] octane scaffold: application in the synthesis of tropane alkaloids. Org. Biomol. Chem. 2021, 19, 3763–3775. 10.1039/D1OB00143D. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G.; Gadzikowska M. Tropane alkaloids as medicinally useful natural products and their synthetic derivatives as new drugs. Pharmacol. Rep. 2008, 60, 439–463. 10.1002/CHIN.200940262. [DOI] [PubMed] [Google Scholar]
- Osbourn A. E.; Lanzotti V.. Plant-Derived Natural Products; Springer, 2009. [Google Scholar]
- Zimmerman J. L. Cocaine Intoxication. Crit. Care Clin. 2012, 28, 517–526. 10.1016/j.ccc.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Zeng Z.; Zhang J.; Jia M.; Wu B.; Cai X.; Zhang X.; Feng Y.; Ma Y.; Gao Q.; Fei Z. Development of a Scalable Route with Efficient Stereoisomer Control to YZJ-1139, an Orexin Receptor Antagonist. Org. Process Res. Dev. 2022, 26, 447–457. 10.1021/acs.oprd.1c00457. [DOI] [Google Scholar]
- Willstätter R.; Bode A. Ueberführung von Tropinon in r-Cocaïn. Ber. Dtsch. Chem. Ges. 1901, 34, 1457–1461. 10.1002/cber.19010340220. [DOI] [Google Scholar]
- Robinson R. LXIII.-A synthesis of tropinone. J. Chem. Soc., Trans. 1917, 111, 762–768. 10.1039/CT9171100762. [DOI] [Google Scholar]
- Pollini G. P.; Benetti S.; De Risi C.; Zanirato V. Synthetic Approaches to Enantiomerically Pure 8-Azabicyclo[3.2.1]octane Derivatives. Chem. Rev. 2006, 106, 2434–2454. 10.1021/cr050995+. [DOI] [PubMed] [Google Scholar]
- Majewski M.; Lazny R. Synthesis of pyranotropanes via enantioselective deprotonation strategy. Tetrahedron Lett. 1994, 35, 3653–3656. 10.1016/S0040-4039(00)73063-2. [DOI] [Google Scholar]
- Lee J. C.; Lee K.; Cha J. K. Enantioselective synthesis of unnatural (S)-(+)-cocaine. J. Org. Chem. 2000, 65, 4773–4775. 10.1021/jo000348c. [DOI] [PubMed] [Google Scholar]
- Zou M.-F.; Agoston G. E.; Belov Y.; Kopajtic T.; Katz J. L.; Newman A. H. Enantioselective synthesis of S-(+)-2β-carboalkoxy-3α-[bis (4-fluorophenyl) methoxy] tropanes as novel probes for the dopamine transporter. Bioorg. Med. Chem. Lett. 2002, 12, 1249–1252. 10.1016/S0960-894X(02)00155-5. [DOI] [PubMed] [Google Scholar]
- Simpkins N. S.; Weller M. Stereochemical Aspects of Organolithium Compounds. Top. Stereochem. 2010, 26, 1–52. 10.1002/9783906390628.ch1. [DOI] [Google Scholar]
- Simpkins N. S. Meldola Lecture. New stereoselective reactions in organic synthesis. Chem. Soc. Rev. 1990, 19, 335–354. 10.1039/cs9901900335. [DOI] [Google Scholar]
- Takahashi T.; Fujii A.; Sugita J.; Hagi T.; Kitano K.; Arai Y.; Koizumi T.; Shiro M. Asymmetric 1,3-dipolar cycloaddition of chiral sulphinylethenes with 1-methyl-3-oxido-pyridinium and some nitrones. Tetrahedron: Asymmetry 1991, 2, 1379–1390. 10.1016/S0957-4166(00)80034-8. [DOI] [Google Scholar]
- Davies H. M. L.; Huby N. J. S. Enantioselective synthesis of tropanes by reaction of rhodium-stabilized vinylcarbenoids with pyrroles. Tetrahedron Lett. 1992, 33, 6935–6938. 10.1016/S0040-4039(00)60899-7. [DOI] [Google Scholar]
- Rigby J. H.; Pigge F. C. Asymmetric Induction in the Metal-Promoted [6π + 2π] Cycloaddition of Azepines. Application to the Construction of Tropane Alkaloids and the Total Synthesis of (+)-Ferruginine. J. Org. Chem. 1995, 60, 7392–7393. 10.1021/jo00128a007. [DOI] [Google Scholar]
- Pham V. C.; Charlton J. L. Methyl (S)-Lactate as a Chiral Auxiliary in the Asymmetric Synthesis of Bao Gong Teng A. J. Org. Chem. 1995, 60, 8051–8055. 10.1021/jo00129a053. [DOI] [Google Scholar]
- Gerlinger C. K.; Krüger S.; Gaich T. Total Synthesis of Parvineostemonine by Structure Pattern Recognition: A Unified Approach to Stemona and Sarpagine Alkaloids. Chem. - Eur. J. 2018, 24, 3994–3997. 10.1002/chem.201800365. [DOI] [PubMed] [Google Scholar]
- Hiroya K.; Ogasawara K. Catalytic asymmetrization of meso-3, 7-bis-siloxycycloheptene by chiral rhodium (I)-binap [2, 2′-bis (diphenylphosphino)-1, 1′-binaphthyl] catalyst: the enantiocontrolled asymmetric synthesis of (−)-(S)-physoperuvine. J. Chem. Soc., Chem. Commun. 1995, 2205–2206. 10.1039/C39950002205. [DOI] [Google Scholar]
- Mans D. M.; Pearson W. H. Total synthesis of (+)-cocaine via desymmetrization of a meso-dialdehyde. Org. Lett. 2004, 6, 3305–3308. 10.1021/ol048777a. [DOI] [PubMed] [Google Scholar]
- Reddy R. P.; Davies H. M. L. Asymmetric Synthesis of Tropanes by Rhodium-Catalyzed [4 + 3] Cycloaddition. J. Am. Chem. Soc. 2007, 129, 10312–10313. 10.1021/ja072936e. [DOI] [PubMed] [Google Scholar]
- Narayan R.; Bauer J. O.; Strohmann C.; Antonchick A. P.; Waldmann H. Catalytic enantioselective synthesis of functionalized tropanes reveals novel inhibitors of hedgehog signaling. Angew. Chem., Int. Ed. 2013, 52, 12892–12896. 10.1002/anie.201307392. [DOI] [PubMed] [Google Scholar]
- Xu J. H.; Zheng S. C.; Zhang J. W.; Liu X. Y.; Tan B. Construction of tropane derivatives by the organocatalytic asymmetric dearomatization of isoquinolines. Angew. Chem., Int. Ed. 2016, 55, 11834–11839. 10.1002/anie.201605736. [DOI] [PubMed] [Google Scholar]
- Marcote D. C.; Varela I.; Fernández-Casado J.; Mascareñas J. L.; López F. Gold (I)-Catalyzed Enantioselective Annulations between Allenes and Alkene-Tethered Oxime Ethers: A Straight Entry to Highly Substituted Piperidines and aza-Bridged Medium-Sized Carbocycles. J. Am. Chem. Soc. 2018, 140, 16821–16833. 10.1021/jacs.8b10388. [DOI] [PubMed] [Google Scholar]
- Hu B.; Zhang X.; Mo Y.; Li J.; Lin L.; Liu X.; Feng X. Catalytic Asymmetric Tandem Cycloisomerization/[5 + 2] Cycloaddition Reaction of N-Aryl Nitrone Alkynes with Methyleneindolinones. Org. Lett. 2020, 22, 1034–1039. 10.1021/acs.orglett.9b04572. [DOI] [PubMed] [Google Scholar]
- Sidera M.; Fletcher S. P. Rhodium-catalysed asymmetric allylic arylation of racemic halides with arylboronic acids. Nat. Chem. 2015, 7, 935–939. 10.1038/nchem.2360. [DOI] [PubMed] [Google Scholar]
- Schäfer P.; Palacin T.; Sidera M.; Fletcher S. P. Asymmetric Suzuki–Miyaura coupling of heterocycles via Rhodium-catalysed allylic arylation of racemates. Nat. Commun. 2017, 8, 15762 10.1038/ncomms15762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J.; van Dijk L.; Goetzke F. W.; Fletcher S. P. Highly enantioselective rhodium-catalyzed cross-coupling of boronic acids and racemic allyl halides. Nat. Protoc. 2019, 14, 2972–2985. 10.1038/s41596-019-0209-8. [DOI] [PubMed] [Google Scholar]
- Goetzke F. W.; Mortimore M.; Fletcher S. P. Enantio- and Diastereoselective Suzuki–Miyaura Coupling with Racemic Bicycles. Angew. Chem., Int. Ed. 2019, 58, 12128–12132. 10.1002/anie.201906478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetzke F. W.; Dijk L.; Fletcher S. P. Catalytic Asymmetric Suzuki–Miyaura Couplings. PATAI’S Chem. Funct. Groups 2019, 1–54. [Google Scholar]
- Kučera R.; Goetzke F. W.; Fletcher S. P. An Asymmetric Suzuki–Miyaura Approach to Prostaglandins: Synthesis of Tafluprost. Org. Lett. 2020, 22, 2991–2994. 10.1021/acs.orglett.0c00745. [DOI] [PubMed] [Google Scholar]
- Hedouin G.; Hazra S.; Gallou F.; Handa S. The Catalytic Formation of Atropisomers and Stereocenters via Asymmetric Suzuki–Miyaura Couplings. ACS Catal. 2022, 12, 4918–4937. 10.1021/acscatal.2c00933. [DOI] [Google Scholar]
- Goetzke F. W.; Fletcher S. P. Additions to Racemates: A Strategy for Developing Asymmetric Cross-Coupling Reactions. Synlett 2021, 32, 1816–1825. 10.1055/s-0040-1706033. [DOI] [Google Scholar]
- van Dijk L.; Ardkhean R.; Sidera M.; Karabiyikoglu S.; Sari Ö.; Claridge T. D. W.; Lloyd-Jones G. C.; Paton R. S.; Fletcher S. P. Mechanistic investigation of Rh(i)-catalysed asymmetric Suzuki–Miyaura coupling with racemic allyl halides. Nat. Catal. 2021, 4, 284–292. 10.1038/s41929-021-00589-y. [DOI] [Google Scholar]
- Buter J.; Moezelaar R.; Minnaard A. J. Enantioselective palladium catalyzed conjugate additions of ortho-substituted arylboronic acids to β, β-disubstituted cyclic enones: total synthesis of herbertenediol, enokipodin A and enokipodin B. Org. Biomol. Chem. 2014, 12, 5883–5890. 10.1039/C4OB01085J. [DOI] [PubMed] [Google Scholar]
- Cox P. A.; Leach A. G.; Campbell A. D.; Lloyd-Jones G. C. Protodeboronation of heteroaromatic, vinyl, and cyclopropyl boronic acids: pH–rate profiles, autocatalysis, and disproportionation. J. Am. Chem. Soc. 2016, 138, 9145–9157. 10.1021/jacs.6b03283. [DOI] [PubMed] [Google Scholar]
- Parois P.; Cooper R. I.; Thompson A. L. Crystal structures of increasingly large molecules: meeting the challenges with CRYSTALS software. Chem. Cent. J. 2015, 9, 30 10.1186/s13065-015-0105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper R. I.; Thompson A. L.; Watkin D. J. CRYSTALS enhancements: dealing with hydrogen atoms in refinement. J. Appl. Crystallogr. 2010, 43, 1100–1107. 10.1107/S0021889810025598. [DOI] [Google Scholar]
- He H. Y.; Wu S. L.; Zhang Y.; Ma B.; Chen Y.; Wang Y. H.. et al. Piperidine Derivatives as Orexin Receptor Antagonist. US10,100,047,2015.
- Karabiyikoglu S.; Brethomé A. V.; Palacin T.; Paton R. S.; Fletcher S. P. Enantiomerically enriched tetrahydropyridine allyl chlorides. Chem. Sci. 2020, 11, 4125–4130. 10.1039/D0SC00377H. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.