

Abstract

Herein, we report a straightforward one-step procedure for modifying N-nucleophilic groups in the nucleobases of commercially available nucleoside phosphoramidites. This method involves the deprotonation of amide groups under phase-transfer conditions and subsequent reaction with electrophilic molecules such as alkyl halides or organic isocyanates. Using this approach, we obtained 10 different classes of modified nucleoside phosphoramidites suitable for the synthesis of oligonucleotides, including several noncanonical nucleotides found in natural RNA or DNA (e.g., m6A, i6A, m1A, g6A, m3C, m4C, m3U, m1G, and m2G). Such modification of nucleobases is a common mechanism for post-transcriptional regulation of RNA stability and translational activity in various organisms. To better understand this process, relevant cellular recognition partners (e.g., proteins) must be identified and characterized. However, this step has been impeded by limited access to molecular tools containing such modified nucleotides.
Introduction
The post-transcriptional modification of nucleobases is a common process in all domains of life. Noncanonical nucleotides were first observed in calf liver RNA hydrolysates in the early 1950s.1 To date, 143 modifications have been identified in various RNA molecules,2 whereas 47 modifications have been found in DNA.3 The chemical nature of these modifications varies from simple methyl group addition through the attachment of more complex molecules (e.g., amino acid derivatives, saccharides, and terpenes) to ring closure for tricyclic nucleobase formation.2 Most studies on RNA modification have focused on sequencing and mapping the whole transcriptome, which provides statistical information that can be difficult to correlate with the biological function.4 In some cases, such as for the most abundant N6-methyladenosine (m6A) mark, the biological effect depends on the structural context of the modification, which further complicates the task.5,6 Synthetic oligonucleotides with modified nucleobases have numerous applications in biological studies on natural cellular processes, such as elucidating the role of tRNA modification in codon recognition,7,8 characterizing the structures of nucleic acid binding proteins (e.g., epitranscriptomic readers and erasers),9,10 developing artificial RNA modification-specific deoxyribozymes,11 and creating fluorescent binding probes12 and isotopically labeled standards for MS analysis.13 However, systematic analyses of the chemical and biological properties of modified nucleic acids are hampered by limited access to nucleic acid fragments containing nucleotides with site-specific modifications. Recently, an elegant method for the ribozymatic methylation of adenosine at the N1 position was developed.14 Nonetheless, other modifications typically require traditional chemical synthesis.
The chemical synthesis of oligonucleotides is commonly achieved using the phosphoramidite method on a solid support.15 This efficient and inexpensive approach has been widely applied by the research and pharmaceutical communities since phosphoramidite building blocks became commercially available. However, the incorporation of nucleotides other than canonical A, C, G, T, and U usually requires the multistep synthesis of appropriate, commercially unavailable building blocks, which makes the process more laborious. The chemical properties of the nucleoside 3′-O-phoshoramidites and orthogonal protecting groups required for solid-phase synthesis interfere with most procedures used for nucleobase modification. As such, these modifications must be introduced early in the synthetic route, followed by base and sugar protection and phosphitylation.16
Notably, Kruse et al. realized the efficient and selective methylation of a fully protected 2′-O-methyladenosine phosphoramidite at the N6 position under phase-transfer conditions, providing quick access to the m6Am building block.17 This approach was based on previous work by the Sekine group on the alkylation of 2′,3′,5′-O,O,O-tri-tert-butyl-dimethylsilyl (TBDMS)-protected N6-acyladenosine anions generated in a two-phase NaOHaq/CH2Cl2 system in the presence of the phase-transfer catalyst Bu4NBr.18 Silyl-protected adenosine with various N6-protecting groups, including acetyl (Ac), phenoxyacetyl (Pac), and 4-nitrobenzoyl amides, was shown to react selectively with active alkylating agents such as methyl, benzyl, and allyl halides. As an exception, the N6-benzyladenosine derivative gave a mixture of N6 and N1 alkylation products.
Inspired by these studies, we investigated the scope of electrophiles compatible with this type of reaction and attempted to apply this approach to phosphoramidites of different nucleosides. We envisage that the generalization of this synthetic method will provide easy access to oligonucleotides containing several natural or unnatural modifications and allow for the introduction of various functional groups into nucleic acid fragments. Consequently, the molecular toolbox for creating structure or activity probes, affinity resins, aptamers, ribozymes, and conjugates with cellular delivery vehicles will be expanded.19
Results and Discussion
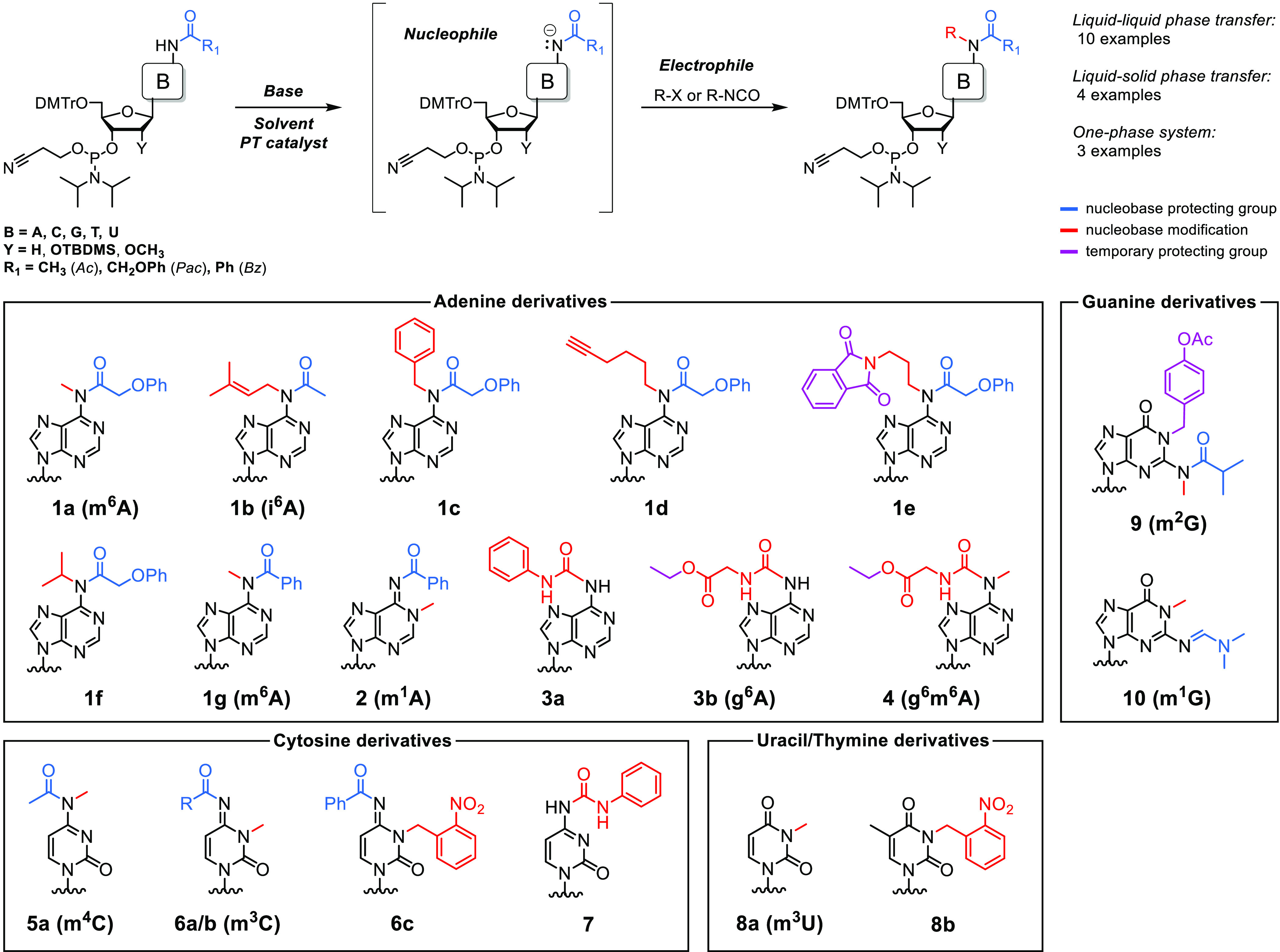
First, we verified whether fully protected adenosine phosphoramidite could be alkylated with electrophiles other than methyl iodide. We chose commercially available N6-acetyl and N6-phenoxyacetyl phosphoramidites because these protecting groups provided the best results for silyl-protected adenosine.18 Active alkylating agents such as benzyl and isopentenyl bromides reacted readily with N6-acetyl 2′-O-methyladenosine and N6-phenoxyacetyl-2′-O-TBDMS-adenosine phosphoramidites in 1 M NaOHaq/CH2Cl2 when an equimolar amount of Bu4NBr was used (full conversion of the starting material in 15–30 min). In this case, the fully protected N6-alkyladenosine phosphoramidites were the only observable product. Catalytic amounts of Bu4NBr also promoted the desired reaction, albeit at much lower rates, leading to competition from partial hydrolysis of the phosphoramidite moiety. Using this procedure (Path a, Scheme 1), we obtained phosphoramidites of naturally occurring adenosine derivatives, m6A (1a) and N6-isopentenyladenosine (i6A, 1b), as well as N6-benzyladenosine (Bn6A) (1c) in 59–80% yield (Table 1). Less active alkyl halides, such as 6-iodohex-1-yne, 3-bromopropylphthalimide, and 2-iodopropane, required much longer reaction times, which led to substantial hydrolysis of the phosphoramidite moiety. N6-Hexynyladenosine phosphoramidite (1d) was isolated in 56% yield, but the phthalimidopropyl and isopropyl derivatives were hydrolyzed before appreciable conversion was achieved. The conditions reported in the literature are then applicable only for modification with very reactive alkylating agents. To accelerate the formation of the desired product and limit hydrolysis, we switched to an anhydrous solid–liquid system with an organic solvent and a mixture of ground solid KOH and K2CO3 as the base.20 Under these conditions, the reaction rate was higher in toluene than in CH2Cl2 (complete conversion in 1 h vs 2–3 h). The optimal procedure provided amidites 1e and 1f in 48 and 45% yield, respectively (Table 1).
Scheme 1. Synthesis of Base-Modified Adenosine 3′-O-Phosphoramidites.

The chemical structures of compounds 1–4 are given in Table 1.
Table 1. Phosphoramidites of Base-Modified Nucleosides Synthesized in this Work.
| product | nucleophilea | electrophile | base | solvent(s) | phase-transfer catalyst | yieldb |
|---|---|---|---|---|---|---|
| 1a | AmPac | methyl iodide | 1 M NaOH (aq) | CH2Cl2/H2O | Bu4NBr | 79% |
| 1b | AAc | isopentenyl bromide | 1 M NaOH (aq) | CH2Cl2/H2O | Bu4NBr | 80% |
| 1c | AmPac | benzyl bromide | 1 M NaOH (aq) | CH2Cl2/H2O | Bu4NBr | 59% |
| 1d | AmPac | 6-iodohex-1-yne | 1 M NaOH (aq) | CH2Cl2/H2O | Bu4NBr | 56% |
| 1e | AmPac | 3-phthalimidopropyl bromide | KOH/K2CO3 (s) | toluene | Bu4NBr | 48% |
| 1f | AmPac | 2-iodopropane | KOH/K2CO3 (s) | toluene | Bu4NBr | 45% |
| 1g + 2 | ABz | methyl iodide | KOH/K2CO3 (s) | toluene | Bu4NBr | 62% + 29% |
| 3a | AAc | phenyl isocyanate | triethylamine | CH2Cl2 | 57% | |
| 3b | AAc | ethyl isocyanatoacetate | triethylamine | CH2Cl2 | 83% | |
| 4 | g6A (3b) | methyl iodide | 1 M NaOH (aq) | CH2Cl2/H2O | Bu4NBr | 70% |
| 5a + 6a | CAc | methyl iodide | 1 M NaOH (aq) | CH2Cl2/H2O | Bu4NBr | 43% + 25% |
| 6b | CBz | methyl iodide | 1 M NaOH (aq) | CH2Cl2/H2O | Bu4NBr | 75% |
| 6c | CBz | 2-nitrobenzyl chloride | KOH/K2CO3 (s) | toluene | Bu4NBr | 73% |
| 7 | CAc | phenyl isocyanate | triethylamine | CH2Cl2 | 42% | |
| 8a | Um | methyl iodide | 1 M NaOH (aq) | CH2Cl2/H2O | Bu4NBr | 89% |
| 8b | T | 2-nitrobenzyl chloride | KOH/K2CO3 (s) | toluene | Bu4NBr | 71% |
| 9 | GiBu | 4-(iodomethyl)phenyl acetate, methyl iodide | 1 M NaOH (aq) | CH2Cl2/H2O | Bu4NBr | 12% |
| 10 | Gdmf | methyl iodide | 1 M NaOH (aq) | CH2Cl2/H2O | Bu4NBr | 82% |
The protecting group of the exocyclic amine in the nucleoside phosphoramidite is indicated by the superscript, as defined by R1 in the abovementioned reaction scheme; the 2′-C substituent (Y in the abovementioned reaction scheme) is −H for DNA amidites, tert-butyldimethylsilyloxyl (−OTBDMS) for RNA amidites, and −OCH3 for 2′-O-methylRNA amidites (denoted by a subscript “m).”
Isolated yield (flash chromatography).
Aritomo et al. found that the phase-transfer-catalyzed (PTC) alkylation of N6-benzoyl-protected 2′,3′,5′-O,O,O-TBDMS-adenosine gave a mixture of N6 and N1 alkylation products, in contrast to N6-acetyl- and N6-phenoxyacetyl-protected compounds, which were alkylated only at the N6 position.18 We envisaged that isomeric product formation results from the mesomeric stabilization of the amide anion (Scheme 1), in which the negative charge is delocalized between two nitrogen atoms. As N1-methyladenosine (m1A) is also present in natural RNAs, we applied this finding to develop a simple synthetic route to m1A phosphoramidite. First, we checked whether this phenomenon was also observed for the alkylation of N6-benzoyl-protected adenosine phosphoramidite and, if so, whether the ratio of isomeric products depended on the reaction conditions. Indeed, the methylation of N6-benzoyl-protected adenosine phosphoramidite in NaOHaq/CH2Cl2 produced both m6A and m1A amidites in an 8:2 ratio. In contrast, in the KOH/K2CO3/toluene system, the distribution of isomeric products shifted slightly toward N1-substitution (∼7:3 m6A:m1A). Isomers 1h and 2 were isolated by flash chromatography, and their structures were confirmed by NMR. Consistent with the previous reports on nucleosides alkylation, we did not observe N1-substitution products for either phenoxyacetyl or acetyl-protected adenosine phosphoramidites.
To expand the scope of this method, we investigated the reaction of other types of electrophilic compounds with adenosine phosphoramidite under alkaline phase-transfer conditions. First, we evaluated representative Michael acceptors, namely, acrylonitrile, methyl cinnamate, and methyl propiolate. In all cases, the reaction proceeded more slowly and was accompanied by substantial degradation of the phosphoramidite. Although the desired products were identified in the reaction mixtures by electrospray ionization mass spectrometry (ESI-MS), their isolation was impractical.
Isocyanates, which are known to react with amines to form urea derivatives, were also tested as electrophiles. Phenyl isocyanate reacted instantaneously with protected adenosine phosphoramidite under phase-transfer conditions in the presence of either aqueous NaOH or solid KOH. However, thin-layer chromatography (TLC) analysis of the reaction mixture revealed multiple unidentified products. We envisaged that reducing the nucleophile concentration by using a milder base would limit the reaction to the isocyanate addition step. Indeed, urea derivative 3a was formed slowly when triethylamine was used as the base in a single-phase organic solvent (Path b, Scheme 1). Interestingly, for N6-benzoyl- and N6-phenoxyacetyladenosine phosphoramidites, the initial products reacted further to form the same final product, implying that the N6-amide bond was cleaved during the subsequent reactions. Further investigation revealed that the reaction of N6-acetyladenosine phosphoramidite also gave an analogous side product, although it only appeared after all the starting material was consumed (4–5 h). Mass spectrometry analysis showed that, in addition to acyl loss, a fragment with m/z = 28 was attached to the molecule, which could correspond to a carbonyl group. Under the investigated conditions, the only carbonyl group source was phenyl isocyanate, indicating that aniline was produced as a byproduct. Indeed, a peak at m/z = 94 was identified in the reaction mixture by ESI(+)-MS. A possible product is tricyclic adenosine derivative 3* (Path b*, Scheme 1),21 the formation of which would require the loss of the N6-acyl group to extend the aromatic system to the third ring. Optimized conditions with N6-acetyl-protected adenosine phosphoramidite provided amidite 3a in 4 h and the product was isolated in 57% yield (Table 1).
In contrast to the reactions with phenyl isocyanate, no side products were observed in the reactions with alkyl isocyanates, which are generally weaker electrophiles. This finding paves the way for the facile and efficient synthesis of an interesting class of compounds, carbamoyladenosine derivatives, which occur naturally in tRNAs at position 37.22 It has been postulated that such amino acid–RNA conjugates were present in the early Earth RNA–peptide world.23 As an example, we reacted N6-acetyl-protected adenosine phosphoramidite with commercially available ethyl isocyanatoacetate and then removed the acetyl group using methylamine. The resulting N6-glycinylcarbamoyl-adenosine (g6A) phosphoramidite 3b was isolated in 83% yield (Path b, Scheme 1). With this urea derivative in hand, we investigated selective alkylation at the N6 position to achieve both N6-carbamoylation and N6-methylation (e.g., m6t6A, another class of adenosine derivatives found in tRNAs).24 The reaction of compound 3b with methyl iodide under phase-transfer conditions proceeded rapidly to give 4 (Path c, Scheme 1), which was isolated in 70% yield.
Next, we examined analogous modification reactions for the phosphoramidites of another natural nucleoside, cytidine (Scheme 2). The N4-acetylcytidine amidite was methylated rapidly in NaOHaq/CH2Cl2, but N4-methylcytidine (m4C) 5a and N3-methylcytidine (m3C) 6a amidites were produced in a 63:37 ratio. Using the N4-benzoyl-protected cytidine derivative, N3-methylated compound 6b was obtained as the main product (15:85 m4C:m3C), which is consistent with the findings for adenosine (N4 of C is equivalent to N6 of A and N3 of C is equivalent to N1 of A). The ratio of isomeric products was significantly affected by the polarity of the solvent in solid–liquid systems. In the dimethylformamide (DMF)/tetrahydrofuran/CH2Cl2/toluene series, solvents with a high dielectric constant (i.e., DMF) promoted N3-alkylation, whereas those with a low dielectric constant (i.e., toluene) promoted N4-alkylation. Catalysts other than Bu4NBr, namely, tetrabutylammonium hydrogen sulfate, benzyltriethylammonium chloride, and Aliquat 336, did not improve the reaction yield and rate. To estimate the reactivity of the nucleophiles generated from cytidine phosphoramidites, we performed reactions with less active alkyl halides, namely, 3-phthalimidopropyl bromide and 2-iodopropane. The reactivity of 3-phthalimidopropyl bromide toward cytidine phosphoramidite was comparable to that of the adenosine amidite, whereas no product was observed in the reaction with the secondary halide (2-iodopropane).
Scheme 2. Synthesis of Base-Modified Pyrimidine 3′-O-Phosphoramidites.

The chemical structures of compounds 5–8 are given in Table 1.
In the case of uridine, PTC deprotonation has been reported for selective alkylation at the N3 position.25 Here, we found that this methodology was also applicable to uridine 3′-O-phosphoramidites (Scheme 2), providing N3-substituted U building blocks. As an example, we chose a naturally occurring modification of uridine, N3,2′-O-dimethyluridine (m3Um), which is found in the mRNA cap-4 structure of early eukaryotes such as Trypanosoma.26 Corresponding phosphoramidite 8a was formed in 25 min and isolated in 89% yield. Other N3 modifications of uridine and thymidine, except for N3-(3-amino-3-carboxypropyl)-uridine (acp3U),27 have limited applications in biological studies because they interfere with Watson–Crick base pairing. However, they may be useful for controlling oligonucleotide hybridization when photolabile substituents such as the 2-nitrobenzyl group are used.28 We obtained photoactivable derivative 8b in 71% yield by alkylation of thymidine phosphoramidite with 2-nitrobenzyl chloride (Table 1).
The final canonical nucleoside, guanosine, requires that the exocyclic amine group is protected to create phosphoramidite building blocks. N2-Acylated guanosines [isobutyryl (iBu), Ac, or Pac derivatives] contain two amide protons that can be abstracted by a base under phase-transfer conditions—one attached to the N1 atom and the other attached to N2. Although the N1 proton is more acidic than the N2 proton (Ka value difference of up to 10 orders of magnitude),29 selective methylation at the N1 position was challenging. An equimolar amount of MeI was insufficient for full conversion of the starting material, whereas excess MeI resulted in the formation of both mono- and dimethylated products (Scheme 3a). We envisaged that bulkier electrophiles and more hindered N2-protecting groups, such as isobutyryl (GiBu), would increase the yield of the single substitution reaction and allow asymmetric double substitution. As a proof of concept, we reacted N2-isobutyrylguanosine phosphoramidite with 2 equiv of 4-(iodomethyl)phenyl acetate (no reaction occurred with the corresponding chloride) and then with 5 equiv of methyl iodide. Expected N1-(4-acetoxybenzyl)-N2-methylguanosine derivative 9 was isolated from the reaction mixture in 12% yield. Thus, the use of a base-labile group for N1-alkylation provided easy access to oligonucleotides containing N2-modified guanosine. To simplify the synthesis of guanosine phosphoramidites monoalkylated selectively at the N1 position, we employed another commercially available guanosine amidite protected with the N2-[(dimethylamino)methylene] group (Gdmf), which has only one acidic proton on the nucleobase (Scheme 3b). Methylation in NaOHaq/CH2Cl2 was complete in 25 min, and the desired product 10 was isolated in 82% yield.
Scheme 3. Synthesis of Base-Modified Guanosine 3′-O-Phosphoramidites.

The chemical structures of compounds 9 and 10 are given in Table 1.
Finally, we investigated whether the standard conditions used for oligonucleotide cleavage and deprotection were compatible with the phosphoramidite derivatives (1–10) obtained using our approach. Some of these compounds or their analogues were previously synthesized using a standard approach (i.e., base modification, protection, and phosphitylation), and the resulting oligonucleotides did not require any special treatment. These previously evaluated derivatives include the phosphoramidites of m6A,17N4-methylcytidine (m4C),30N3-methyl-uridine (m3U),26N3-(3-amino-3-carboxypropyl)uridine (acp3U),27N6-threonyl-carbamoyladenosine (t6A),7N6-glycinylcarbamoyl-adenosine (g6A),23N4-carbamoylcytidines,31 and N1-methylguanosine (m1G).32 For oligonucleotides containing m1A, milder conditions (e.g., ammonium hydroxide at 25 °C) should be used because m1A is known to undergo Dimroth rearrangement under basic conditions to form m6A.33 These side reactions could be at least partially limited by carefully selecting the deprotection conditions.33−35 For base modifications that were not previously reported (i.e., those in 1b–f, 8c, and 9, as well as those in 3a,b, 4, and 7, which contain different protecting groups), we synthesized short oligonucleotides using a solid-phase approach. To ensure that the modified monomers were stable against every reagent used, we incorporated them in the first synthetic cycle, followed by another cycle with the unmodified phosphoramidite. In the literature, there are contradictory reports on the deprotection of m3C containing oligonucleotides;10a,35−37 therefore, we also included pm3CpG in our tests.
As expected, simple N6-alkyl adenosine derivatives 1b–d and 1f showed properties similar to those of m6A and were efficiently deprotected under standard conditions [e.g., 1:1 ammonium hydroxide /methylamine (AMA) mixture at room temperature for 2 h].38 The phthalimide protecting group in 1e has previously been removed from the ammonia-deprotected 2′-O-phthalimidopropyl oligonucleotide by additional treatment with methylamine.39 Here, we were able to fully deprotect the dinucleotide pA*pG prepared using amidite 1e in a one-step procedure using AMA at 37 °C for 3 h. N6-Carbamoyladenosines derived from compounds 3a,b and 4 were also stable under AMA treatment; however, the ethyl esters of 3b and 4 were converted into methylamides. This issue can be addressed by using different carboxyl protecting groups (such as trimethylsilylethyl esters)7 or different deprotection conditions (e.g., 1 M NaOH).40 Deprotection of the dinucleotide containing m3C (prepared using phosphoramidite 6a) with AMA (37 °C, 3 h) resulted in transamination with methylamine to produce N3,N4-dimethylcytidine derivative pm3,4CpG as the only product (as evidenced by MS and NMR analysis; see Supporting Information, compound 21), which is consistent with the most recent report.37 However, the desired pm3CpG (Supporting Information, compound 19) was efficiently prepared using aqueous ammonium hydroxide (RT, overnight) for cleavage and deprotection. Finally, we found that dinucleotide pNpG synthesized using phosphoramidite 9 (m2G) is deprotected readily with AMA to produce N1-(4-hydroxybenzyl)-N2-methylguanosine derivative and then, upon further incubation with AMA at 4 °C (overnight), it undergoes slow elimination of p-quinone methide to give Um2GU (compound 24).
To demonstrate the potential applications of the base-modified nucleoside phosphoramidites obtained in this work, we synthesized oligonucleotide analogues of mRNA 5′ end structures, namely, m6Am-modified cap-2 found in higher eukaryotes and cap-4 found in Trypanosoma.26,41 To this end, phosphoramidites 1a and 8a were utilized in the solid-phase synthesis of tri- and pentanucleotide 5′-phosphates (pm6AmpGmpG and pm6,6AmpAmpCmpm3UmpA, respectively) and then coupled with 7-methylguanosine 5′-diphosphate using the P-imidazolide activation strategy in solution.6 The final products 26 and 27 (Scheme 4) were purified by reversed-phase high-performance liquid chromatography and their structures were confirmed by high-resolution mass spectrometry.
Scheme 4. Chemical Structures of m6Am-Modified cap-2 (Compound 26) and cap-4 (Compound 27) Synthesized Using the Phosphoramidites Obtained in This Work.

Conclusions
In conclusion, we developed a one-step protocol for synthesizing nucleoside phosphoramidites with N-substituted nucleobases, which relies on the deprotonation of the amide moiety under phase-transfer conditions. This procedure was successfully applied to modify all five canonical nucleobases (adenine at the N6 and N1 positions, cytosine at the N4 and N3 positions, guanine at the N1 position, and thymine and uracil at the N3 position) with various alkylating agents (including methyl iodide and primary and secondary halides) in 40–89% yield, starting from commercially available phosphoramidites. Cytidine phosphoramidites were slightly less reactive in PTC alkylation than adenosine derivatives, resulting in the formation of two isomeric products. However, the product ratio was successfully shifted by changing the reaction conditions, allowing either isomer to be obtained as the major product. We also found that adenosine and cytosine phosphoramidites with N-protected nucleobases reacted with organic isocyanates (both alkyl and aryl) in the presence of triethylamine to form urea derivatives, which could be further alkylated under phase-transfer conditions to provide N-alkyl-N-carbamoyl derivatives. Many of the synthesized compounds (or their close structural analogues) are precursors to oligonucleotides containing natural modifications, which are very useful in biological studies on their structure and function. Our synthetic protocol is also suitable for synthesizing functionalized oligonucleotides, providing a powerful tool for obtaining molecular probes, affinity resins, and conjugates for diagnostic and therapeutic applications. This time- and cost-effective approach for phosphoramidite functionalization can also be applied to generate various modified synthetic RNA fragment libraries for high-throughput screening.
Experimental Section
General Information
Solvents, chemical reagents, and starting materials were acquired from commercial sources and used without further purification. Commercially available phosphoramidites were purchased from Biosearch Technologies or ChemGenes. Solid supports for oligonucleotide syntheses were purchased from GE Healthcare. DNA synthesis grade acetonitrile (<10 ppm of water) was used for the coupling reaction and for washing the solid support. All work-up and purification procedures were performed with reagent-grade solvents under an ambient atmosphere.
Analytical and Preparative Chromatography
TLC analysis was performed on precoated silica gel 60 Å on aluminum foil (Sigma-Aldrich) and visualized under a UV lamp (254 nm). The synthesized compounds were isolated by gel chromatography using the Biotage Selekt Flash Purification System with Biotage Sfär Silica cartridges (5, 10 g). Short oligonucleotides (11–26) were purified by ion-exchange chromatography on DEAE Sephadex A-25 (HCO3– form). After loading the column with the reaction mixture and washing it with deionized water, the products were eluted using a linear gradient of triethylammonium bicarbonate (TEAB) in water: 0–0.9 M for dinucleotides (12–16 and 19–22) and 0–1.2 M for trinucleotides (11, 17–18, and 23–25). The fractions containing the desired product were combined, concentrated under reduced pressure, and evaporated to dryness with repeated additions of 96% and then 99.8% ethanol to give a white solid of oligonucleotide triethylammonium salt. To facilitate NMR analysis, trinucleotides 11 and 17–26 were additionally purified by semi-preparative RP HPLC using a Gemini 5 μm NX-C18 LC column (110 Å, 150 × 10 mm, flow rate 5.0 mL/min) with linear gradient of MeCN in 0.05 M ammonium acetate buffer (pH 5.9) and UV detection at 254 nm. After repeated freeze-drying of the collected fractions, the products were isolated as ammonium salts forming white solids. Analytical RP HPLC was performed on a Gemini 3 μm NX-C18 LC column (110 Å, 150 × 4.6 mm, 3 μm, flow rate 1.0 mL/min) with linear gradient elution with 0.05 M ammonium acetate buffer pH 5.9 (buffer A) and 1:1v/v methanol/buffer A (buffer B): Method A: 0–100% in 15 min; Method B: 0–100% in 7.5 min.
Compound Characterization
NMR spectra were recorded at 25 °C with a Bruker Avance III HD spectrometer at 500.24 MHz (1H NMR) and 202.49 MHz (31P NMR) using 5 mm PABBO BB/19F-1H/D Z-GRD probe. The raw NMR data were processed using MestReNova v12.0.2-20910 Software. The 1H NMR chemical shifts were calibrated to CHCl3 (7.260 ppm) or D2O (4.790). For the calibration of 31P NMR chemical shifts, H3PO4 was used as an external standard. Signals were assigned based on correlation spectroscopy, heteronuclear single quantum coherence (1H-13C HSQC, 1H-31P HSQC), and optionally heteronuclear multiple bond correlation (HMBC) and 2D total correlation (1H-1H TOCSY) spectra. High-resolution mass spectra (HRMS) were recorded with a LTQ Orbitrap Velos (Thermo Scientific) spectrometer.
Chemical Syntheses of N-Substituted Nucleoside Phosphoramidites
General Procedure A (1a–d, 4, 5a, 6a,b, 8a, 9, and 10)
A nucleoside phosphoramidite (1.0 equiv) and an alkyl halide (2.0–10.0 equiv) were dissolved in dichloromethane (DCM) (to obtain 0.1 M amidite, 1 volume) and mixed with 1 volume of an aqueous solution of Bu4NBr (0.1 M, 1.0 equiv) and NaOH (1.0 M). The reaction mixture was stirred vigorously until the starting material was fully consumed, as indicated by TLC analysis. Then, the reaction mixture was partitioned between water (10 volumes) and diethyl ether (10 volumes), and the aqueous phase was extracted with ethyl acetate (10 volumes) three times. The organic layers were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was dissolved in DCM containing 0.5%v/v triethylamine, evaporated using silica gel, loaded into a solid sample loader, and purified by flash chromatography.
General Procedure B (1e–g, 2, 6c, and 8b)
To a 0.1 M solution of a nucleoside phosphoramidite (1.0 equiv) in toluene, an alkyl halide (2.0–20.0 equiv), Bu4NBr (1.0 equiv), and an equimolar mixture of ground solid KOH and K2CO3 (approximately 5 equiv each) were added. The reaction mixture was stirred vigorously until the starting material was fully consumed, as indicated by TLC analysis. Then, the reaction mixture was partitioned between water (10 volumes) and diethyl ether (10 volumes), and the aqueous phase was extracted with ethyl acetate (10 volumes) three times. The organic layers were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was dissolved in DCM containing 0.5%v/v triethylamine, evaporated using silica gel, loaded into a solid sample loader, and purified by flash chromatography.
General Procedure C (3a,b and 7)
A nucleoside phosphoramidite (1.0 equiv), alkyl/aryl isocyanate (8.0–10.0 equiv), and trimethylamine (1.0 equiv) were dissolved in DCM (to obtain 0.1 M amidite). The reaction mixture was stirred vigorously until the starting material was fully consumed, as indicated by TLC analysis. After adding a 33% solution of methylamine in ethanol (15.0 equiv), the reaction mixture was stirred for 30 min to remove the N6-acyl protecting group. Then, the reaction mixture was partitioned between water (10 volumes) and diethyl ether (10 volumes), and the aqueous phase was extracted with ethyl acetate (10 volumes) three times. The organic layers were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was dissolved in DCM containing 0.5%v/v triethylamine, evaporated using silica gel, loaded into a solid sample loader, and purified by flash chromatography.
N6-Methyladenosine Phosphoramidite [5′-O-DMT-2′-O-Me-m6APac] (1a)
Compound 1a was prepared according to procedure A using 1.00 g (1.09 mmol) of 5′-O-DMT-2′-O-Me-APac phosphoramidite and 272 μL (4.36 mmol, 4 equiv) of methyl iodide. The reaction was quenched after 30 min, and the product was isolated by flash chromatography (0 → 50% ethyl acetate in n-hexane with 0.5%v/v TEA in 30 min, 80 mL/min, Biotage Sfär HC 25 g column). The diastereomers were characterized separately and then combined to afford 1a (804 mg, 0.863 mmol, 79%) as a white solid. Diastereomer 1: TLC (hexane/ethyl acetate 1:1): Rf = 0.30; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.61 (s, 1H, H8), 8.26 (s, 1H, H2), 7.44 (m, 2H, ArH-2,6Ph-DMTr), 7.35–7.16 (m, 9H, ArH), 6.90 (m, 1H, ArH-4Pac), 6.82 (m, 4H, ArH-3,5MeOPh-DMTr), 6.71 (m, 2H, ArH-2,6Pac), 6.16 (m, 1H, H1′), 5.14 (s, 2H, CH2Pac), 4.68 (m, 1H, H3′), 4.58 (m, 1H, H2′), 4.43 (m, 1H, H4′), 3.78 (s, 6H, 2 × OCH3DMTr), 3.75 (s, 3H, CH3N6-Me), 3.73–3.55 (m, 5H, OCH2CH2CN, 2 × CHiPr, H5′), 3.49 (s, 3H, 2′-O-CH3), 3.39 (dd, 2JH,H = 10.7 Hz, 3JH,H = 3.9 Hz, 1H, H5″), 2.38 (t, 3JH,H = 6.7 Hz, 2H, OCH2CH2CN), 1.23–1.18 (m, 12H, CH3iPr) ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.0 (m, 1P, P) ppm; HRMS (ESI) m/z calcd for C50H59N7O9P+, [M + H]+: 932.41064, found: 932.41001; Diastereomer 2: TLC (hexane/ethyl acetate 1:1): Rf = 0.37; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.60 (s, 1H, H8), 8.20 (s, 1H, H2), 7.42 (d, 3JH,H = 7.2 Hz, 2H, ArH-2,6Ph-DMTr), 7.32 (d, 3JH,H = 8.8 Hz, 4H, ArH-2,6MeOPh-DMTr), 7.26 (t, 3JH,H = 7.2 Hz, 2H, ArH-3,5Ph-DMTr), 7.23–7.16 (m, 3H, ArH-4Ph-DMTr, ArH-3,5Pac), 6.90 (t, 3JH,H = 7.3 Hz, 1H, ArH-4Pac), 6.80 (d, 3JH,H = 8.8 Hz, 4H, ArH-3,5MeOPh-DMTr), 6.71 (d, 3JH,H = 7.6 Hz, 2H, ArH-2,6Pac), 6.18 (d, 3JH,H = 5.4 Hz, 1H, H1′), 5.14 (s, 2H, CH2Pac), 4.64 (m, 1H, H2′), 4.60 (m, 1H, H3′), 4.37 (m, 1H, H4′), 3.90 (m, 2H, OCH2CH2CN), 3.78 (s × 2, 6H, OCH3DMTr), 3.75 (s, 3H, CH3N6-Me), 3.61 (m, 2H, CHiPr), 3.53 (dd, 2JH,H = 10.7 Hz, 3JH,H = 3.7 Hz, 1H, H5′), 3.50 (s, 3H, 2′-O-CH3), 3.37 (dd, 2JH,H = 10.7 Hz, 3JH,H = 4.2 Hz, 1H, H5″), 2.64 (t, 3JH,H = 6.3 Hz, 2H, OCH2CH2CN), 1.19 (d, 3JH,H = 6.8 Hz, 6H, CH3iPr), 1.09 (d, 3JH,H = 6.8 Hz, 6H, CH3iPr) ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 150.4 (m, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C50H59N7O9P+ 932.41064, found: 932.41006; diastereomeric mixture: 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 170.7, 170.6, 158.8, 158.0, 158.0, 153.0, 152.9, 152.9, 152.8, 151.6, 151.6, 144.6, 144.5, 142.5, 142.5, 135.7, 135.6, 130.3, 130.2, 129.5, 128.4, 128.3, 128.0, 127.2, 127.2, 126.0, 126.0, 121.4, 121.4, 117.8, 117.6, 114.6, 114.6, 113.3, 87.1, 87.0, 86.9, 86.8, 84.0, 84.0, 82.6, 82.5, 82.1, 82.1, 77.4, 77.4, 77.2, 76.9, 71.3, 71.2, 70.9, 70.7, 68.9, 63.1, 62.7, 59.0, 59.0, 58.9, 58.9, 58.5, 58.5, 58.1, 58.0, 55.4, 55.4, 43.6, 43.5, 43.4, 43.3, 35.1, 24.8, 24.8, 24.7, 24.7, 24.7, 20.5, 20.5, 20.7, 20.3 ppm.
N6-Isopentenyladenosine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-i6AAc] (1b)
Compound 1b was prepared according to procedure A using 250 mg (0.270 mmol) of 5′-O-DMT-2′-O-TBDMS-AAc phosphoramidite and 156 μL (1.35 mmol, 5.0 equiv) of isopentenyl bromide. The reaction was quenched after 35 min, and the product was isolated by flash chromatography (0 → 60% ethyl acetate in n-hexane with 0.5%v/v TEA in 35 min, 40 mL/min, Biotage Sfär HC 10g column) to afford a mixture of diastereomers 1b (216 mg, 0.217 mmol, 80%) as a white solid. TLC (hexane/ethyl acetate 1:1): Rf = 0.51; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.69 (s, 1H, H8), 8.67 (s, 1H, H8), 8.30 (s, 1H, H2), 8.27 (s, 1H, H2), 7.47 (m, 4H, ArH-2,6Ph-DMTr), 7.36 (m, 8H, ArH-2,6MeOPh-DMTr), 7.30–7.20 (m, 6H, ArH-3,4,5Ph-DMTr), 6.82 (m, 8H, ArH-3,5MeOPh-DMTr), 6.13 (d, 3JH,H = 6.7 Hz, 1H, H1′), 6.06 (d, 3JH,H = 6.4 Hz, 1H, H1′), 5.22 (m, 2H, 2 × C=CHN6-isopent.), 5.04 (m, 2H, 2 × H2′), 4.84 (m, 4H, 2 × CH2N6-isopent.), 4.46 (m, 1H, H4′), 4.43–4.36 (m, 3H, 2 × H3′, H4′), 3.97 (m, 1H, OCH2CH2CN), 3.89 (m, 1H, OCH2CH2CN), 3.79 (overlapped s, 12H, 4 × OCH3DMTr), 3.71–3.53 (m, 8H, OCH2CH2CN, 2 × H5′, 4 × CHiPr), 3.36 (m, 1H, H5″), 3.33 (dd, 2JH,H = 10.7 Hz, 3JH,H = 3.8 Hz, 1H, H5″), 2.66 (m, 2H, OCH2CH2CN), 2.31 (m, 2H, OCH2CH2CN), 2.21 (s, 3H, CH3N6-Ac), 2.20 (s, 3H, CH3N6-Ac), 1.57 (s, 6H, 2 × CH3N6-isopent.), 1.56 (s, 6H, 2 × CH3N6-isopent.), 1.22–1.16 (m, 18H, 6 × CH3iPr), 1.06 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr), 0.72 (s, 18H, 2 × tBuTBDMS), −0.03 (s, 3H, CH3TBDMS), −0.05 (s, 3H, CH3TBDMS), −0.24 (s, 6H, 2 × CH3TBDMS) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 171.2, 158.7, 158.7, 153.7, 153.2, 152.2, 144.7, 144.6, 143.0, 142.9, 136.0, 135.8, 135.8, 135.6, 135.6, 130.3, 130.3, 130.2, 130.2, 128.4, 128.2, 128.1, 128.1, 128.0, 128.0, 127.1, 120.2, 117.4, 113.4, 113.3, 113.3, 88.4, 88.1, 87.0, 86.8, 84.6, 74.8, 74.8, 73.6, 73.6, 63.5, 63.3, 57.8, 57.6, 55.4, 55.4, 45.5, 45.5, 43.6, 43.5, 43.1, 43.0, 25.8, 25.7, 25.7, 24.9, 24.9, 24.8, 24.8, 24.7, 24.2, 20.6, 20.6, 20.3, 20.2, 18.1, 18.0, 18.0, −4.5, −4.6, −5.2 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.2 (s, 1P, P), 149.1 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C53H73N7O8PSi+ 994.50220, found: 994.50275;
N6-Benzyladenosine Phosphoramidite [5′-O-DMT-2′-O-Me-Bn6APac] (1c)
Compound 1c was prepared according to procedure A using 1.00 g (1.09 mmol) of 5′-O-DMT-2′-O-Me-APac phosphoramidite and 162 μL (1.36 mmol, 1.25 equiv) of benzyl bromide. The reaction was quenched after 35 min, and the product was isolated by flash chromatography (0 → 50% ethyl acetate in n-hexane with 0.5%v/v TEA in 30 min, 80 mL/min, Biotage Sfär HC 25g column) to afford a mixture of diastereomers of 1c (646 mg, 0.640 mmol, 59%) as a white solid. TLC (hexane/ethyl acetate 1:1): Rf = 0.58; 1H NMR (500 MHz, CDCl3, 25 °C): 8.60 (s, 1H, H8), 8.58 (s, 1H, H8), 8.26 (s, 1H, H2), 8.19 (s, 1H, H2), 7.42 (m, 4H, ArH-2,6Ph-DMTr), 7.35–7.12 (m, 28H, ArH), 6.89 (m, 2H, ArH), 6.81 (m, 8H, ArH), 6.63 (m, 4H, ArH), 6.15 (d, 3JH,H = 5.2 Hz, 1H, H1′), 6.13 (d, 3JH,H = 5.1 Hz, 1H, H1′), 5.65 (s, 4H, 2 × CH2N6-Bn), 5.13 (s, 4H, 2 × CH2Pac), 4.66 (m, 1H, H3′), 4.62–4.57 (m, 2H, H2′, H3′), 4.54 (m, 1H, H2′), 4.41 (m, 1H, H4′), 4.35 (m, 1H, H4′), 3.92 (m, 1H, OCH2CH2CN), 3.86 (m, 1H, OCH2CH2CN), 3.78–3.76 (overlapped s, 12H, 4 × OCH3DMTr), 3.73–3.55 (m, 7H, OCH2CH2CN, H5′, 4 × CHiPr), 3.52 (dd, 2JH,H = 10.6 Hz, 3JH,H = 3.8 Hz, H5′), 3.48 (s, 3H, CH32′-O), 3.47 (s, 3H, CH32′-O), 3.36 (m, 2H, 2 × H5″), 2.63 (t, 3JH,H = 6.3 Hz, 2H, OCH2CH2CN), 2.37 (t, 3JH,H = 6.3 Hz, 2H, OCH2CH2CN), 1.21–1.17 (m, 18H, 6 × CH3iPr), 1.08 (d, 3JH,H = 6.3 Hz, 3H, CH3iPr) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 170.7, 170.7, 158.8, 158.8, 157.9, 157.8, 152.9, 152.8, 152.2, 152.1, 151.8, 151.8, 144.6, 144.5, 142.6, 142.6, 137.4, 137.4, 135.7, 135.7, 135.6, 130.3, 130.3, 130.3, 129.5, 128.4, 128.4, 128.3, 128.0, 128.0, 128.0, 127.3, 127.2, 127.2, 126.6, 126.6, 121.5, 121.5, 117.8, 117.5, 117.4, 114.5, 114.5, 113.3, 87.1, 86.9, 86.9, 86.8, 84.0, 83.9, 82.5, 82.5, 71.3, 71.2, 70.7, 68.9, 68.9, 63.1, 62.6, 59.0, 58.9, 58.9, 58.9, 58.5, 58.5, 58.1, 58.0, 55.4, 55.4, 50.1, 43.6, 43.5, 43.4, 43.3, 24.8, 24.7, 24.7, 20.5, 20.5, 20.4, 20.3, 15.4.ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.0 (s, 1P, P), 150.4 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C56H63N7O9P+ 1008.44194, found: 1008.44298.
N6-Hexynyladenosine Phosphoramidite [5′-O-DMT-2′-O-Me-hex6APac] (1d)
Compound 1d was prepared according to procedure A using 315 mg (0.343 mmol) of 5′-O-DMT-2′-O-Me-APac phosphoramidite and 280 μL (2.06 mmol, 6 equiv) of 6-iodohex-1-yn. The reaction was quenched after 60 min, and the product was isolated by flash chromatography (0 → 50% ethyl acetate in n-hexane with 0.5%v/v TEA in 35 min, 40 mL/min, Biotage Sfär HC 10g column) to afford a mixture of diastereomers of 1d (192 mg, 0.192 mmol, 56%) as a white solid. TLC (hexane/ethyl acetate 1:1): Rf = 0.47; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.63 (s, 1H, H8), 8.62 (s, 1H, H8), 8.26 (s, 1H, H2), 8.19 (s, 1H, H2), 7.46–7.14 (m, 22H, ArH), 6.91–6.79 (m, 10H, ArH), 6.65 (d, 3JH,H = 5.4 Hz, 4H, ArH), 6.18 (d, 3JH,H = 5.3 Hz, 1H, H1′), 6.16 (d, 3JH,H = 5.0 Hz, 1H, H1′), 5.07 (s, 4H, 2 × CH2Pac), 4.69 (m, 1H, H3′), 4.64 (m, 1H, H3′), 4.58 (m, 2H, 2 × H2′), 4.42 (m, 2H, 2 × H4′), 4.36 (m, 4H, 2 × CH2C6-hex.), 3.93 (m, 1H, OCH2CH2CN), 3.86 (m, 1H, OCH2CH2CN), 3.78 (s, 12H, 4 × OCH3DMTr), 3.74–3.55 (m, 6H, OCH2CH2CN, 2 × H5′, 2 × CHiPr), 3.50 (s, 6H, 2 × CH32′-O), 3.50–3.46 (m, 2H, 2 × CHiPr), 3.38 (m, 2H, 2 × H5″), 2.64 (t, 3JH,H = 6.3 Hz, 2H, OCH2CH2CN), 2.38 (t, 3JH,H = 6.3 Hz, 2H, OCH2CH2CN), 2.14 (td, 3JH,H = 7.0 Hz, 4JH,H = 2.4 Hz, 4H, 2 × CH2C3-hex.), 1.85 (t, 4JH,H = 2.4 Hz, 2H, 2 × C≡CHC1-hex.), 1.71 (m, 4H, 2 × CH2C5-hex.), 1.53 (m, 4H, 2 × CH2C4-hex.), 1.20 (m, 18H, 6 × CH3iPr), 1.09 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 170.4, 170.4, 158.8, 157.9, 152.9, 152.9, 152.4, 152.3, 151.8, 144.6, 142.6, 142.6, 135.7, 135.7, 135.6, 130.3, 130.3, 129.5, 128.4, 128.3, 128.0, 127.2, 126.5, 126.5, 121.4, 117.6, 114.5, 113.3, 87.0, 86.9, 86.9, 86.8, 84.3, 84.0, 84.0, 82.1, 82.0, 71.3, 71.2, 68.9, 68.5, 66.0, 62.6, 58.5, 58.5, 58.1, 57.9, 55.4, 55.4, 46.8, 43.6, 43.5, 43.4, 43.3, 27.7, 25.7, 24.8, 24.7, 24.7, 20.4, 20.3, 18.2, 15.4 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.0 (s, 1P, P), 150.4 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C55H65N7O9P+ 998.45759, found: 998.45670.
N6-(3-Phthalimidopropyl)adenosine Phosphoramidite [5′-O-DMT-2′-O-Me-PhthNp6APac] (1e)
Compound 1e was prepared according to procedure B using 943 mg (1.03 mmol) of 5′-O-DMT-2′-O-Me-APac phosphoramidite and 549 mg (2.05 mmol, 2.0 equiv) of 3-phthalimidopropyl bromide. The reaction was quenched after 60 min, and the product was isolated by flash chromatography (0 → 50% ethyl acetate in n-hexane with 0.5%v/v TEA in 35 min, 40 mL/min, Biotage Sfär HC 10g column) to afford a mixture of diastereomers of 1e (601 mg, 0.544 mmol, 48%) as a white solid. TLC (hexane/ethyl acetate 1:1): Rf = 0.31; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.54 (s, 1H, H8), 8.52 (s, 1H, H8), 7.89 (s, 1H, H2), 7.80 (s, 1H, H2), 7.76 (m, 4H, ArHPhth-α), 7.62 (m, 4H, ArHPhth.β), 7.44 (m, 4H, ArH), 7.37–7.14 (m, 18H, ArH), 6.89 (m, 2H, ArH), 6.81 (m, 8H, ArH), 6.68 (d, 3JH,H = 8.2 Hz, 4H, ArH), 6.08 (d, 3JH,H = 5.1 Hz, 2H, 2 × H1′), 5.12 (s, 4H, 2 × CH2Pac), 4.63 (m, 2H, H3′, H2′), 4.56 (m, 2H, H3′, H2′), 4.42 (m, 5H, 2 × CH2C3_N6-prop., H4′), 4.35 (m, 1H, H4′), 3.89 (m, 2H, OCH2CH2CN), 3.77 (s, 12H, 4 × OCH3DMTr), 3.75 (m, 4H, 2 × CH2C1_N6-prop.), 3.72–3.48 (m, 8H, OCH2CH2CN, 2 × H5′, 4 × CHiPr), 3.47 (s, 6H, 2 × CH32′-O), 3.35 (m, 2H, 2 × H5″), 2.64 (t, 3JH,H = 5.9 Hz, 2H, OCH2CH2CN), 2.38 (t, 3JH,H = 6.3 Hz, 2H, OCH2CH2CN), 2.11 (p, 3JH,H = 6.9 Hz, 4H, 2 × CH2C2_N6-prop), 1.20 (m, 18H, 6 × CH3iPr), 1.09 (d, 3JH,H = 6.7 Hz, 6H, 2 × CH3iPr) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ = 170.6, 170.6, 168.4, 168.4, 158.7, 158.7, 157.9, 157.9, 152.9, 152.8, 152.0, 152.0, 151.6, 151.6, 144.7, 144.6, 142.4, 142.3, 135.7, 135.7, 135.7, 133.9, 133.9, 132.3, 130.3, 130.3, 130.2, 129.5, 128.3, 128.3, 128.0, 127.1, 127.1, 125.8, 125.7, 123.2, 121.4, 121.4, 117.8, 117.5, 114.5, 113.3, 87.0, 86.9, 86.8, 86.7, 84.0, 83.9, 82.1, 82.1, 81.8, 81.7, 71.4, 71.2, 70.8, 70.7, 69.1, 69.1, 63.2, 62.7, 59.0, 58.9, 58.9, 58.9, 58.5, 58.5, 58.1, 58.0, 55.4, 55.3, 45.2, 43.5, 43.4, 43.4, 43.3, 35.8, 35.8, 27.7, 24.8, 24.7, 24.7, 24.7, 20.5, 20.5, 20.3, 20.3 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.0 (s, 1P, P), 150.4 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C60H66N8O11P+ 1105.45832, found: 1105.46053.
N6-Dsopropyladenosine Phosphoramidite [5′-O-DMT-2′-O-Me-iPr6APac] (1f)
Compound 1f was prepared according to procedure B using 250 mg (0.272 mmol) of 5′-O-DMT-2′-O-Me-APac phosphoramidite and 544 μL (5.45 mmol, 20 equiv) of 2-iodopropane. The reaction was quenched after 2.5 h, and the product was isolated by flash chromatography (0 → 100% ethyl acetate in n-hexane with 0.5%v/v TEA in 35 min, 40 mL/min, Biotage Sfär HC 10g column) to afford a mixture of diastereomers of 1f (117 mg, 0.122 mmol, 45%) as a white solid. TLC (hexane/ethyl acetate 1:1): Rf = 0.60; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.74 (s, 1H, H8), 8.72 (s, 1H, H8), 8.28 (s, 1H, H2), 8.21 (s, 1H, H2), 7.43 (m, 4H, ArH-2,6Ph-DMTr), 7.33 (m, 8H, ArH-2,6MeOPh-DMTr), 7.29–7.20 (m, 6H, ArH-3,4,5Ph-DMTr), 7.13 (m, 4H, ArH-3,5Ph-Pac), 6.86 (m, 2H, ArH-4Ph-Pac), 6.81 (m, 8H, ArH-3,5MeOPh-DMTr), 6.56 (m, 4H, ArH-2,6Ph-Pac), 6.17 (d, 3JH,H = 5.3 Hz, 1H, H1′), 6.15 (d, 3JH,H = 5.1 Hz, 1H, H1′), 4.93 (m, 2H, 2 × CHN6_iPr), 4.69 (m, 1H, H3′), 4.68 (m, 4H, 2 × CH2Pac), 4.66–4.57 (m, 3H, H3′, 2 × H2′), 4.43 (m, 1H, H4′), 4.37 (m, 1H, H4′), 3.92 (m, 1H, OCH2CH2CN), 3.85 (m, 1H, OCH2CH2CN), 3.78 (s, 3H, OCH3DMTr), 3.78 (s, 3H, OCH3DMTr), 3.77 (s, 3H, OCH3DMTr), 3.77 (s, 3H, OCH3DMTr), 3.73–3.51 (m, 8H, OCH2CH2CN, 2 × H5′, 4 × CHiPr), 3.50 (s, 3H, CH32′-O), 3.50 (s, 3H, CH32′-O), 3.38 (m, 2H, 2 × H5″), 2.63 (t, 3JH,H = 6.3 Hz, 2H, OCH2CH2CN), 2.37 (t, 3JH,H = 6.2 Hz, 2H, OCH2CH2CN), 1.37 (m, 12H, 4 × CH3iPr), 1.23–1.17 (m, 18H, 6 × CH3iPr), 1.09 (d, 3JH,H = 6.7 Hz, 6H, 2 × CH3iPr) ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.0 (s, 1P, P), 150.4 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C52H63N7O9P+ 960.44194, found: 960.44371.
N6-Methyladenosine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-m6ABz] (1g) and N1-Methyladenosine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-m1ABz] (2)
Compounds 1g and 2 were prepared according to procedure B using 500 mg (0.506 mmol) of 5′-O-DMT-2′-O-TBDMS-ABz phosphoramidite and 315 μL (5.06 mmol, 10 equiv) of methyl iodide. The reaction was quenched after 30 min, and the products were isolated and separated by flash chromatography (0 → 50% ethyl acetate in n-hexane with 0.5%v/v TEA in 35 min, 40 mL/min, Biotage Sfär HC 10 g column) to afford a mixture of diastereomers of 1g (312 mg, 0.311 mmol, 62%) and a mixture of diastereomers of 2 (146 mg, 0.146 mmol, 29%) as white solids.
m6ABz: TLC (hexane/ethyl acetate 1:1): Rf = 0.50; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.44 (s, 1H, H8), 8.42 (s, 1H, H8), 8.14 (s, 1H, H2), 8.11 (s, 1H, H2), 7.43 (m, 8H, ArH), 7.33 (m, 8H, ArH-2,6MeOPh-DMTr), 7.24 (m, 8H, ArH), 7.13 (m, 4H, ArH), 6.80 (m, 8H, ArH-3,5MeOPh-DMTr), 6.03 (d, 3JH,H = 6.7 Hz, 1H, H1′), 5.97 (d, 3JH,H = 6.6 Hz, 1H, H1′), 4.99 (m, 2H, 2 × H2′), 4.42 (m, 1H, H4′), 4.35 (m, 3H, 2 × H3′, H4′), 3.95 (m, 1H, OCH2CH2CN), 3.86 (m, 1H, OCH2CH2CN), 3.78 (m, 18H, 4 × OCH3DMTr, 2 × CH3N6-Me), 3.71–3.50 (m, 8H, OCH2CH2CN, 2 × H5′, 4 × CHiPr), 3.30 (m, 2H, 2 × H5″), 2.63 (m, 2H, OCH2CH2CN), 2.29 (m, 2H, OCH2CH2CN), 1.20–1.15 (m, 18H, 6 × CH3iPr), 1.04 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr), 0.72 (s, 18H, 2 × tBuTBDMS), −0.07 (s, 3H, CH3TBDMS), −0.09 (s, 3H, CH3TBDMS), −0.32 (s, 3H, CH3TBDMS), −0.33 (s, 3H, CH3TBDMS) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 172.3, 158.7, 155.1, 155.1, 152.9, 152.9, 152.0, 144.7, 144.6, 142.7, 142.6, 136.4, 135.9, 135.8, 135.7, 135.6, 130.7, 130.3, 130.2, 130.2, 130.2, 128.8, 128.3, 128.2, 128.1, 128.0, 128.0, 128.0, 127.1, 126.9, 126.8, 117.7, 117.4, 113.4, 113.3, 113.3, 88.2, 88.0, 86.9, 86.7, 84.6, 84.2, 84.2, 75.2, 75.2, 74.6, 74.5, 73.6, 73.6, 72.8, 72.7, 63.4, 63.3, 59.0, 58.9, 57.8, 57.6, 55.4, 55.4, 43.6, 43.5, 43.1, 43.0, 36.0, 25.7, 25.7, 24.9, 24.9, 24.8, 24.8, 24.8, 24.7, 24.7, 20.6, 20.6, 20.2, 20.2, 18.0, 18.0, 15.4, −4.5, −4.5, −5.1, −5.1 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.2 (s, 1P, P), 149.0 (s, 1P, P) ppm; HRMS (ESI) m/z calcd for C54H69N7O8PSi+, [M + H]+: 1002.47090, found: 1002.47145.
m1ABz: TLC (hexane/ethyl acetate 1:1): Rf = 0.22; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.16 (m, 4H, ArH-2,6Bz), 7.85 (s, 1H, H8), 7.83 (s, 1H, H8), 7.82 (s, 1H, H2), 7.80 (s, 1H, H2), 7.50–7.48 (m, 10H, 2 × ArH-3,4,5Bz, 2 × ArH-2,6Ph-DMTr), 7.32 (m, 8H, 2 × ArH-2,6MeOPh-DMTr), 7.26–7.16 (m, 6H, 2 × ArH-3,4,5Ph-DMTr), 6.79 (m, 8H, 2 × ArH-3,5MeOPh-DMTr), 5.91 (d, 3JH,H = 6.1 Hz, 1H, H1′), 5.84 (d, 3JH,H = 6.0 Hz, 1H, H1′), 4.85 (m, 1H, H2′), 4.81 (m, 1H, H2′), 4.38 (m, 1H, H4′), 4.28 (m, 3H, 2 × H3′, H4′), 3.88 (m, 2H, OCH2CH2CN), 3.77 (s, 6H, 2 × OCH3DMTr), 3.76 (s, 6H, 2 × OCH3DMTr), 3.71 (s, 3H, CH3N1-Me), 3.71 (s, 3H, CH3N1-Me), 3.64–3.40 (m, 8H, OCH2CH2CN, 2 × H5′, 4 × CHiPr), 3.22 (m, 2H, 2 × H5″), 2.63 (t, 3JH,H = 6.5 Hz, 2H, OCH2CH2CN), 2.25 (m, 2H, OCH2CH2CN), 1.18–1.12 (m, 18H, 6 × CH3iPr), 1.00 (d, 3JH,H = 6.7 Hz, 6H, 2 × CH3iPr), 0.79 (s, 9H, tBuTBDMS), 0.79 (s, 9H, tBuTBDMS), −0.01 (s, 3H, CH3TBDMS), −0.02 (s, 3H, CH3TBDMS), −0.14 (s, 3H, CH3TBDMS), −0.14 (s, 3H, CH3TBDMS) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 177.2, 177.1, 171.3, 158.7, 158.6, 147.8, 147.7, 146.8, 146.7, 145.8, 144.8, 144.7, 139.0, 138.7, 136.0, 136.0, 135.9, 135.9, 135.7, 135.7, 131.9, 130.3, 130.3, 130.2, 129.9, 129.9, 128.3, 128.2, 128.1, 128.0, 128.0, 127.0, 122.9, 122.7, 117.7, 117.4, 113.3, 113.3, 113.3, 88.5, 88.1, 86.7, 86.5, 84.1, 83.7, 83.6, 75.6, 74.8, 74.7, 73.3, 73.2, 72.8, 72.7, 63.5, 63.4, 60.5, 59.0, 58.8, 57.8, 57.7, 55.4, 55.4, 43.6, 43.5, 43.1, 43.0, 37.0, 25.8, 25.8, 24.9, 24.8, 24.8, 24.8, 24.7, 24.7, 24.6, 21.2, 20.5, 20.5, 20.2, 20.1, 18.1, 18.0, 14.3, −4.5, −4.5, −4.5, −4.6, −4.8, −4.9 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 150.9 (s, 1P, P), 149.2 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C54H69N7O8PSi+ 1002.47090, found: 1002.47093.
N6-(N-Phenylcarbamoyl)adenosine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-PhNHCO6A] (3a)
Compound 3a was prepared according to procedure C using 250 mg (0.270 mmol) of 5′-O-DMT-2′-O-TBDMS-AAc phosphoramidite and 293 μL (2.70 mmol, 10 equiv) of phenyl isocyanate. After 3.5 h, the 33% solution of methylamine in ethanol (0.54 mL) was added. The product was isolated by flash chromatography (0 → 60% ethyl acetate in n-hexane with 0.5%v/v TEA in 60 min, 40 mL/min, Biotage Sfär HC 10g column) to afford a mixture of diastereomers of 3a (154 mg, 0.154 mmol, 57%) as a white solid. TLC (ethyl acetate): Rf = 0.77; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 11.73 (s, 1H, NHPhNHCO), 11.72 (s, 1H, NHPhNHCO), 8.51 (s, 1H, H8), 8.51 (s, 1H, H8), 8.22 (s, 1H, H2), 8.20 (s, 1H, H2), 8.04 (s, 1H, NHN6), 8.02 (s, 1H, NHN6), 7.64 (m, 2H, ArH-2,6PhNHCO), 7.49 (m, 4H, ArH-2,6Ph-DMTr), 7.40–7.33 (m, 12H, ArH-2,6MeOPh-DMTr, ArH-3,5PhNHCO), 7.33–7.22 (m, 6H, ArH-3,4,5Ph-DMTr), 7.12 (m, 2H, ArH-4PhNHCO), 6.83 (m, 8H, ArH-3,5MeOPh-DMTr), 6.09 (d, 3JH,H = 6.3 Hz, 1H, H1′), 6.03 (d, 3JH,H = 6.1 Hz, 1H, H1′), 5.05 (m, 2H, 2 × H2′), 4.45 (m, 1H, H4′), 4.43–4.38 (m, 2H, 2 × H3′), 4.36 (m, 1H, H4′), 3.96 (m, 1H, OCH2CH2CN), 3.88 (m, 1H, OCH2CH2CN), 3.79 (overlapped s, 12H, 4 × OCH3DMTr), 3.70–3.52 (m, 8H, OCH2CH2CN, 2 × H5′, 4 × CHiPr), 3.35 (m, 1H, H5″), 3.32 (dd, 2JH,H = 10.7 Hz, 3JH,H = 3.8 Hz, 1H, H5″), 2.65 (m, 2H, OCH2CH2CN), 2.30 (m, 2H, OCH2CH2CN), 1.22–1.16 (m, 18H, 6 × CH3iPr), 1.05 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr), 0.77 (s, 18H, 2 × tBuTBDMS), 0.00 (s, 3H, CH3TBDMS), −0.03 (s, 3H, CH3TBDMS), −0.18 (s, 3H, CH3TBDMS), −0.19 (s, 3H, CH3TBDMS) ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.0 (s, 1P, P), 149.2 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C53H68N8O8PSi+ 1003.46615, found: 1003.46713;
N6-Glycinylcarbamoyladenosine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-g6A] (3b)
Compound 3b was prepared according to procedure C using 500 mg (0.540 mmol) of 5′-O-DMT-2′-O-TBDMS-AAc phosphoramidite and 726 μL (6.47 mmol, 12 equiv) of ethyl isocyanatoacetate. After 7.5 h, the 33% solution of methylamine in ethanol (1.0 mL) was added. The product was isolated by flash chromatography (0 → 60% ethyl acetate in n-hexane with 0.5%v/v TEA in 35 min, 40 mL/min, Biotage Sfär HC 10g column) to afford mixture of diastereomers of 3b (452 mg, 0.446 mmol, 83%) as a white solid. TLC (hexane/ethyl acetate 1:1): Rf = 0.27; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 9.93 (m, 2H, NHGly), 8.46 (s, 1H, H8), 8.45 (s, 1H, H8), 8.18 (s, 1H, H2), 8.15 (s, 1H, H2), 7.96 (s, 2H, 2 × NH-6), 7.47 (m, 4H, ArH-2,6Ph-DMTr), 7.36 (m, 8H, ArH-2,6MeOPh-DMTr), 7.28 (m, 4H, ArH-3,5Ph-DMTr), 7.23 (m, 2H, ArH-4Ph-DMTr), 6.81 (m, 8H, ArH-3,5MeOPh-DMTr), 6.06 (d, 3JH,H = 6.4 Hz, 1H, H1′), 6.01 (d, 3JH,H = 6.1 Hz, 1H, H1′), 5.04 (m, 2H, 2 × H2′), 4.43 (m, 1H, H4′), 4.38 (m, 3H, 2 × H3′, H4′), 4.26 (q, 3JH,H = 7.1 Hz, 4H, CH2Et-Gly), 4.21 (m, 4H, CH2Gly-α), 3.95 (m, 1H, OCH2CH2CN), 3.87 (m, 1H, OCH2CH2CN), 3.79 (overlapped s, 12H, 4 × OCH3DMTr), 3.70–3.52 (m, 8H, OCH2CH2CN, 2 × H5′, 4 × CHiPr), 3.33 (m, 1H, H5″), 3.31 (m, 1H, H5′′), 2.65 (m, 2H, OCH2CH2CN), 2.30 (m, 2H, OCH2CH2CN), 1.31 (t, 3JH,H = 7.1 Hz, 6H, CH3Et-Gly), 1.21–1.15 (m, 18H, 6 × CH3iPr), 1.05 (d, 3JH,H = 6.9 Hz, 6H, 2 × CH3iPr), 0.76 (s, 18H, 2 × tBuTBDMS), −0.02 (s, 3H, CH3TBDMS), −0.05 (s, 3H, CH3TBDMS), −0.21 (s, 3H, CH3TBDMS), −0.22 (s, 3H, CH3TBDMS) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 171.1, 170.2, 169.4, 158.7, 157.3, 154.0, 153.9, 151.3, 150.6, 150.1, 144.7, 144.6, 141.8, 141.7, 135.9, 135.8, 135.6, 135.6, 130.3, 130.3, 130.2, 128.4, 128.3, 128.1, 128.0, 127.1, 121.0, 121.0, 117.7, 117.4, 113.3, 113.3, 113.3, 88.6, 88.3, 86.9, 86.8, 84.4, 74.8, 74.8, 73.5, 73.4, 63.3, 61.6, 61.5, 57.8, 57.7, 55.4, 55.4, 43.6, 43.5, 43.1, 43.0, 42.4, 42.3, 25.8, 25.7, 24.9, 24.9, 24.8, 24.8, 20.2, 20.2, 18.1, 18.0, 14.3, 14.3, −4.6, −4.6, −5.0 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.0 (s, 1P, P), 149.1 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C51H70N8O10PSi+ 1013.47163, found: 1013.47238.
N6-Glycinylcarbamoyl-N6-Methyladenosine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-g6m6A] (4)
Compound 4 was prepared according to procedure A using 200 mg (0.197 mmol) of 5′-O-DMT-2′-O-TBDMS-g6A phosphoramidite (3b) and 123 μL (1.97 mmol, 10 equiv) of methyl iodide. The reaction was quenched after 30 min, and the product was isolated by flash chromatography (0 → 60% ethyl acetate in n-hexane with 0.5%v/v TEA in 35 min, 40 mL/min, Biotage Sfär HC 10 g column) to afford 4 (141 mg, 0.137 mmol, 70%) as a white solid. TLC (hexane/ethyl acetate 1:1): Rf = 0.49; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 11.03 (m, 2H, NHGly), 8.47 (s, 1H, H8), 8.46 (s, 1H, H8), 8.19 (s, 1H, H2), 8.16 (s, 1H, H2), 7.47 (m, 4H, ArH-2,6Ph-DMTr), 7.36 (m, 8H, ArH-2,6MeOPh-DMTr), 7.28 (m, 4H, ArH-3,5Ph-DMTr), 7.23 (m, 2H, ArH-4Ph-DMTr), 6.81 (m, 8H, ArH-3,5MeOPh-DMTr), 6.12 (d, 3JH,H = 6.4 Hz, 1H, H1′), 6.06 (d, 3JH,H = 6.0 Hz, 1H, H1′), 5.01 (m, 2H, 2 × H2′), 4.43 (m, 1H, H4′), 4.37 (m, 3H, 2 × H3′, H4′), 4.25 (q, 3JH,H = 7.1 Hz, 4H, CH2Et-Gly), 4.20 (d, 3JH,H = 6.4 Hz, 4H, CH2Gly-α), 4.01 (s, 3H, CH3N6-Me), 4.00 (s, 3H, CH3N6-Me), 3.96 (m, 1H, OCH2CH2CN), 3.87 (m, 1H, OCH2CH2CN), 3.79 (overlapped s, 12H, 4 × OCH3DMTr), 3.71–3.52 (m, 8H, OCH2CH2CN, 2 × H5′, 4 × CHiPr), 3.30 (m, 2H, 2 × H5″), 2.65 (m, 2H, OCH2CH2CN), 2.29 (m, 2H, OCH2CH2CN), 1.31 (t, 3JH,H = 7.1 Hz, 6H, CH3Et-Gly), 1.20–1.15 (m, 18H, 6 × CH3iPr), 1.03 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr), 0.77 (s, 18H, 2 × tBuTBDMS), −0.01 (s, 3H, CH3TBDMS), −0.04 (s, 3H, CH3TBDMS), −0.17 (s, 3H, CH3TBDMS), −0.19 (s, 3H, CH3TBDMS) ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.0 (s, 1P, P), 149.1 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C52H72N8O10PSi+ 1027.48728, found: 1027.48746;
N4-Methylcytidine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-m4CAc] (5a) and N3-Methylcytidine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-m3CAc] (6a)
Compounds 5a and 6a were prepared according to procedure A using 250 mg (0.277 mmol) of 5′-O-DMT-2′-O-TBDMS-CAc phosphoramidite and 173 μL (2.77 mmol, 10.0 equiv) of methyl iodide. The reaction was quenched after 25 min, and the products were isolated and separated by flash chromatography (0 → 100% ethyl acetate in n-hexane with 0.5%v/v TEA in 30 min, 40 mL/min, Biotage Sfär HC 10g column) to afford mixture of diastereomers of 5a (108 mg, 0.118 mmol, 43%) and mixture of diastereomers of 6a (64 mg, 0.070 mmol, 25%) as white solids. m4CAc (5a): TLC (ethyl acetate): Rf = 0.48, 0.44; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.51 (d, 3JH,H = 7.6 Hz, 1H, H6), 8.42 (d, 3JH,H = 7.6 Hz, 1H, H6), 7.46 (m, 2H, ArH-2,6Ph-DMTr), 7.41 (m, 2H, ArH-2,6Ph-DMTr), 7.35 (m, 4H, ArH-2,6MeOPh-DMTr), 7.32–7.23 (m, 10H, ArH-3,4,5Ph-DMTr,ArH-2,6MeOPh-DMTr), 6.85 (m, 8H, ArH-3,5MeOPh-DMTr), 6.53 (d, 3JH,H = 7.6 Hz, 1H, H5), 6.36 (d, 3JH,H = 7.6 Hz, 1H, H5), 5.88 (d, 3JH,H = 1.7 Hz, 1H, H1′), 5.79 (s, 1H, H1′), 4.36 (m, 4H, 2 × H2′, 2 × H4′), 4.29 (m, 2H, 2 × H3′), 3.84 (m, 1H, OCH2CH2CN), 3.81 (s, 6H, 2 × OCH3DMTr), 3.80 (s, 6H, 2 × OCH3DMTr), 3.76–3.63 (m, 4H, 2 × H5′, 2 × OCH2CH2CN), 3.61–3.42 (m, 7H, OCH2CH2CN, 4 × CHiPr, 2 × H5″), 3.40 (s, 3H, CH3N4-Me), 3.38 (s, 3H, CH3N4-Me), 2.57 (t, 3JH,H = 6.3 Hz, 2H, OCH2CH2CN), 2.39 (m, 2H, OCH2CH2CN), 2.39 (s, 3H, CH3N4-Ac), 2.38 (s, 3H, CH3N4-Ac), 1.15–1.08 (m, 18H, 6 × CH3iPr), 0.96 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr), 0.92 (s, 9H, tBuTBDMS), 0.91 (s, 9H, tBuTBDMS), 0.29 (s, 6H, 2 × CH3TBDMS), 0.16 (s, 6H, 2 × CH3TBDMS) ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 150.6 (s, 1P, P), 148.8 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C48H66N5O9PSi 916.44402, found: 916.44443; m3CAc (6a): TLC (ethyl acetate): Rf = 0.65; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 7.81 (d, 3JH,H = 8.2 Hz, 1H, H6), 7.72 (d, 3JH,H = 8.2 Hz, 1H, H6), 7.41 (m, 2H, ArH-2,6Ph-DMTr), 7.36 (m, 2H, ArH-2,6Ph-DMTr), 7.33–7.24 (m, 16H, ArH-3,4,5Ph-DMTr,ArH-2,6MeOPh-DMTr), 6.84 (m, 8H, ArH-3,5MeOPh-DMTr), 5.96 (d, 3JH,H = 4.1 Hz, 1H, H1′), 5.88 (d, 3JH,H = 2.7 Hz, 1H, H1′), 5.81 (d, 3JH,H = 8.2 Hz, 1H, H5), 5.75 (d, 3JH,H = 8.2 Hz, 1H, H5), 4.34 (m, 1H, H2′), 4.31–4.25 (m, 4H, H2′, 2 × H3′, H4′), 4.22 (m, 1H, H4′), 3.92 (m, 1H, OCH2CH2CN), 3.81 (s, 6H, 2 × OCH3DMTr), 3.81 (s, 6H, 2 × OCH3DMTr), 3.78 (m, 1H, OCH2CH2CN), 3.69 (m, 1H, OCH2CH2CN), 3.64–3.51 (m, 7H, 2 × H5′, OCH2CH2CN, 4 × CHiPr), 3.42–3.36 (m, 2H, 2 × H5″), 3.36 (s, 3H, CH3N3-Me), 3.35 (s, 3H, CH3N3-Me), 2.64 (m, 2H, OCH2CH2CN), 2.39 (m, 2H, OCH2CH2CN), 2.19 (s, 3H, CH3N4-Ac), 2.18 (s, 3H, CH3N4-Ac), 1.15 (d, 3JH,H = 6.7 Hz, 18H, 6 × CH3iPr), 0.99 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr), 0.90 (s, 9H, tBuTBDMS), 0.89 (s, 9H, tBuTBDMS), 0.16 (s, 6H, CH3TBDMS), 0.15 (s, 6H, CH3TBDMS), 0.14 (s, 3H, CH3TBDMS), 0.12 (s, 3H, CH3TBDMS) ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 150.0 (s, 1P, P), 149.7 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C48H67N5O9PSi+ 916.44402, found: 916.44456.
N3-Methylcytidine Phosphoramidite (5′-O-DMT-2′-O-TBDMS-m3CBz) (6b)
Compound 6b was prepared according to procedure A using 500 mg (0.519 mmol) of 5′-O-DMT-2′-O-TBDMS-CBz phosphoramidite and 65 μL (1.04 mmol, 2.0 equiv) of methyl iodide. The reaction was quenched after 25 min, and the product was isolated and separated by flash chromatography (0 → 100% ethyl acetate in n-hexane with 0.5%v/v TEA in 30 min, 40 mL/min, Biotage Sfär HC 10 g column) to afford mixture of diastereomers of 6b (380 mg, 0.389 mmol, 75%) as white solids. TLC (ethyl acetate): Rf = 0.73; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.13 (m, 4H, 2 × ArH-2,6Ph-Bz), 7.88 (d, 3JH,H = 8.2 Hz, 1H, H6), 7.78 (d, 3JH,H = 8.2 Hz, 1H, H6), 7.52 (m, 2H, 2 × ArH-4Ph-Bz), 7.43 (m, 4H, 2 × ArH-3,5Ph-Bz), 7.39 (m, 2H, ArH-2,6Ph-DMTr), 7.35 (m, 2H, ArH-2,6Ph-DMTr), 7.31–7.23 (m, 14H, ArH-3,5Ph-DMTr,ArH-2,6MeOPh-DMTr), 7.20 (m, 2H, ArH-4Ph-DMTr), 6.82 (m, 8H, ArH-3,5MeOPh-DMTr), 6.10 (d, 3JH,H = 8.2 Hz, 1H, H5), 6.03 (d, 3JH,H = 8.2 Hz, 1H, H5), 6.00 (d, 3JH,H = 4.1 Hz, 1H, H1′), 5.91 (d, 3JH,H = 3.0 Hz, 1H, H1′), 4.37 (m, 1H, H2′), 4.34–4.25 (m, 4H, H2′, 2 × H3′, H4′), 4.24 (m, 1H, H4′), 3.93 (m, 1H, OCH2CH2CN), 3.80–3.77 (overlapped, 13H, OCH2CH2CN, 4 × OCH3DMTr), 3.70 (m, 1H, OCH2CH2CN), 3.62 (dd,2JH,H = 11.1 Hz, 3JH,H = 1.2 Hz, 1H, H5′), 3.60–3.51 (m, 12H, OCH2CH2CN, 4 × CHiPr, 2 × CH3N3-Me, H5′), 3.40 (dd,2JH,H = 11.1 Hz, 3JH,H = 2.1 Hz, 1H, H5″), 3.37 (dd,2JH,H = 11.1 Hz, 3JH,H = 2.7 Hz, 1H, H5″), 2.64 (m, 2H, OCH2CH2CN), 2.38 (m, 2H, OCH2CH2CN), 1.17–1.13 (m, 18H, 6 × CH3iPr), 0.99 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr), 0.91 (s, 9H, tBuTBDMS), 0.90 (s, 9H, tBuTBDMS), 0.17 (s, 6H, 2 × CH3TBDMS), 0.15 (s, 3H, CH3TBDMS), 0.13 (s, 3H, CH3TBDMS) ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 150.0 (s, 1P, P), 149.7 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C53H69N5O9PSi+ 978.45967, found: 978.46048;
N3-(2-Nitrobenzyl)cytidine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-(2-NO2-Bn)3CBz] (6c)
Compound 6c was prepared according to procedure B using 250 mg (0.259 mmol) of 5′-O-DMT-2′-O-TBDMS-CBz phosphoramidite and 445 mg (2.59 mmol, 10 equiv) of 2-nitrobenzyl chloride. The reaction was quenched after 30 min, and the product was isolated by flash chromatography (0 → 50% ethyl acetate in n-hexane with 0.5%v/v TEA in 30 min, 40 mL/min, Biotage Sfär HC 10 g column) to afford a mixture of diastereomers of 6c (207 mg, 0.188 mmol, 73%) as a pale yellow solid. TLC (ethyl acetate): Rf = 0.70; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.09 (m, 1H, ArH), 8.08 (m, 1H, ArH), 7.94 (d, 3JH,H = 8.3 Hz, 1H, H6C), 7.85 (d, 3JH,H = 8.2 Hz, 1H, H6C), 7.79 (m, 4H, ArH), 7.56 (m, 2H, ArH), 7.48–7.19 (m, 28H, ArH), 6.83 (m, 8H, 2 × ArH-3,5MeOPh-DMTr), 6.16 (d, 3JH,H = 8.3 Hz, 1H, H5C), 6.09 (d, 3JH,H = 8.2 Hz, 1H, H5C), 6.01 (d, 3JH,H = 4.2 Hz, 1H, H1′), 5.94 (d, 3JH,H = 3.2 Hz, 1H, H1′), 5.76 (m, 2H, CH2nBn), 5.75 (m, 2H, CH2nBn), 4.43 (m, 1H, H2′), 4.38 (m, 1H, H2′), 4.37–4.33 (m, 2H, 2 × H3′), 4.32 (m, 1H, H4′), 4.24 (m, 1H, H4′), 3.93 (m, 1H, OCH2CH2CN), 3.79 (m, 1H, OCH2CH2CN), 3.79 (s, 6H, 2 × OCH3DMTr), 3.78 (s, 6H, 2 × OCH3DMTr), 3.73 (m, 1H, OCH2CH2CN), 3.65–3.51 (m, 8H, OCH2CH2CN, 2 × H5′, 4 × CHiPr), 3.42 (m, 1H, H5″), 3.40 (m, 1H, H5′′), 2.64 (m, 2H, OCH2CH2CN), 2.40 (m, 2H, OCH2CH2CN), 1.17–1.13 (m, 18H, 6 × CH3iPr), 1.01 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr), 0.90 (s, 9H, tBuTBDMS), 0.88 (s, 9H, tBuTBDMS), 0.14 (s, 3H, CH3TBDMS), 0.14 (s, 3H, CH3TBDMS), 0.13 (s, 3H, CH3TBDMS), 0.12 (s, 3H, CH3TBDMS) ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 150.8 (s, 1P, P), 150.8 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C59H72N6O11PSi+ 1099.47605, found: 1099.47702.
N6-(N-Phenylcarbamoyl)cytidine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-PhNHCO4C] (7)
Compound 7 was prepared according to procedure C using 260 mg (0.288 mmol) of 5′-O-DMT-2′-O-TBDMS-CAc phosphoramidite and 250 μL (2.30 mmol, 8 equiv) of phenyl isocyanate. After 35 min, the 33% solution of methylamine in ethanol (0.52 mL) was added. The product was isolated by flash chromatography (0 → 75% ethyl acetate in n-hexane with 0.5%v/v TEA in 40 min, 40 mL/min, Biotage Sfär HC 10g column) to afford a mixture of diastereomers of 7 (119 mg, 0.122 mmol, 42%) as a white solid. TLC (ethyl acetate): Rf = 0.68, 0.59; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 11.49 (m, 2H, NHN4/PhNHCO), 10.79 (m, 2H, NHN4/PhNHCO), 8.53 (m, 1H, H6), 8.39 (m, 1H, H6), 7.64 (m, 4H, ArH), 7.45 (m, 2H, ArH), 7.40 (m, 2H ArH), 7.37–7.24 (m, 19H, ArH, H5), 7.21 (m, 1H, H5), 7.05 (m, 2H, ArH), 6.86 (m, 8H, ArH-3,5MeOPh-DMTr), 6.17 (s, 1H, H1′), 6.08 (s, 1H, H1′), 4.40 (m, 1H, H2′), 4.38–4.30 (m, 5H, H2′, 2 × H3′, 2 × H4′), 3.90 (m, 1H, OCH2CH2CN), 3.80 (overlapped s, 6H, 2 × OCH3DMTr), 3.79 (s, 6H, 2 × OCH3DMTr), 3.78 (m, 1H, OCH2CH2CN) 3.74–3.64 (m, 3H, 2 × H5′, OCH2CH2CN), 3.63–3.52 (m, 5H, OCH2CH2CN, 4 × CHiPr), 3.46 (m, 2H, 2 × H5″), 2.60 (m, 2H, OCH2CH2CN), 2.41 (t, 3JH,H = 6.5 Hz, 2H, OCH2CH2CN), 1.18–1.13 (m, 18H, 6 × CH3iPr), 1.00 (d, 3JH,H = 6.7 Hz, 6H, 2 × CH3iPr), 0.90 (s, 9H, tBuTBDMS), 0.89 (s, 9H, tBuTBDMS), 0.17 (s, 6H, 2 × CH3TBDMS), 0.13 (s, 3H, CH3TBDMS), 0.10 (s, 3H, CH3TBDMS) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 158.9, 158.9, 139.4, 138.5, 135.4, 135.2, 130.4, 130.3, 129.3, 128.7, 128.6, 128.2, 128.1, 127.4, 127.4, 123.9, 120.8, 120.0, 117.7, 117.5, 114.2, 113.5, 113.4, 87.5, 87.4, 55.4, 55.4, 43.5, 43.4, 43.3, 43.2, 34.0, 32.1, 29.8, 29.8, 29.8, 29.7, 29.5, 29.3, 29.1, 25.9, 25.9, 25.0, 25.0, 24.9, 24.8, 24.8, 24.7, 24.7, 22.8, 20.6, 20.6, 20.4, 20.3, 18.2, 14.3, −4.2, −4.2, −4.7, −4.8 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 150.3 (s, 1P, P), 149.9 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C52H68N6O9PSi+ 979.45492, found: 979.45708;
N3-Methyluridine Phosphoramidite [5′-O-DMT-2′-O-Me-m3Um] (8a)
Compound 8a was prepared according to procedure A using 250 mg (0.329 mmol) of 5′-O-DMT-2′-O-Me-U phosphoramidite and 205 μL (3.29 mmol, 10 equiv) of methyl iodide. The reaction was quenched after 25 min, and the product was isolated by flash chromatography (0 → 100% ethyl acetate in n-hexane with 0.5%v/v TEA in 30 min, 40 mL/min, Biotage Sfär HC 10 g column) to afford a mixture of diastereomers of 8a (227 mg, 0.293 mmol, 89%) as a white solid. TLC (ethyl acetate): Rf = 0.60; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.05 (d, 3JH,H = 8.1 Hz, 1H, H6U), 7.95 (d, 3JH,H = 8.1 Hz, 1H, H6U), 7.41 (m, 2H, ArHPh-DMTr), 7.36 (m, 2H, ArHPh-DMTr), 7.33–7.23 (m, 14H, ArH), 6.84 (m, 8H, 2 × ArH-3,5MeOPh-DMTr), 6.04 (d, 3JH,H = 2.7 Hz, 1H, H1′), 5.99 (d, 3JH,H = 1.8 Hz, 1H, H1′), 5.32 (d, 3JH,H = 8.1 Hz, 1H, H5U), 5.29 (d, 3JH,H = 8.1 Hz, 1H, H5U), 4.61 (m, 1H, H3′), 4.46 (m, 1H, H3′), 4.24 (m, 1H, H4′), 4.21 (m, 1H, H4′), 3.92 (m, 1H, H2′), 3.88 (m, 2H, H2′, OCH2CH2CN), 3.83 (m, 1H, OCH2CH2CN), 3.80 (s, 3H, OCH3DMTr), 3.80 (s, 3H, OCH3DMTr), 3.79 (s, 3H, OCH3DMTr), 3.79 (s, 3H, OCH3DMTr), 3.68–3.41 (m, 10H, OCH2CH2CN, 2 × H5′, 2 × H5′, 4 × CHiPr), 3.60 (s, 3H, CH3N3-Me), 3.60 (s, 3H, CH3N3-Me), 3.32 (s, 6H, 2 × CH32′-O), 2.64 (m, 2H, OCH2CH2CN), 2.40 (t, 3JH,H = 6.2 Hz, 2H, OCH2CH2CN), 1.19 (d, 3JH,H = 6.7 Hz, 6H, CH3iPr), 1.19 (d, 3JH,H = 6.7 Hz, 6H, CH3iPr), 1.16 (d, 3JH,H = 6.8 Hz, 6H, CH3iPr), 1.03 (d, 3JH,H = 6.8 Hz, 6H, CH3iPr) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 163.1, 163.1, 158.9, 158.9, 158.9, 151.2, 151.2, 144.4, 144.3, 137.8, 137.8, 135.4, 135.3, 135.2, 135.1, 130.4, 130.4, 128.5, 128.4, 128.1, 127.4, 127.4, 117.8, 117.6, 113.4, 113.3, 101.7, 101.6, 88.4, 88.4, 87.3, 87.1, 83.9, 83.8, 83.2, 83.2, 82.4, 82.3, 82.1, 82.1, 69.9, 69.8, 69.7, 69.7, 61.5, 60.8, 58.8, 58.8, 58.8, 58.7, 58.4, 58.4, 58.3, 58.1, 55.4, 55.4, 43.5, 43.4, 43.4, 43.3, 29.8, 27.7, 27.6, 24.8, 24.8, 24.8, 24.7, 24.7, 24.6, 20.5, 20.5, 20.4, 20.4 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 150.7 (s, 1P, P), 150.2 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C41H52N4O9P+ 775.34664, found: 775.34746.
N3-(2-Nitrobenzyl)thymidine Phosphoramidite [5′-O-DMT-(2-NO2-Bn)3T] (8b)
Compound 8b was prepared according to procedure B using 250 mg (0.336 mmol) of 5′-O-DMT-T phosphoramidite and 577 mg (3.36 mmol, 10 equiv) of 2-nitrobenzyl chloride. The reaction was quenched after 35 min, and the product was isolated by flash chromatography (0 → 100% ethyl acetate in n-hexane with 0.5%v/v TEA in 35 min, 40 mL/min, Biotage Sfär HC 10 g column) to afford a mixture of diastereomers of 8c (211 mg, 0.240 mmol, 71%) as a pale yellow solid. TLC (ethyl acetate): Rf = 0.67; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.04 (d, 3JH,H = 1.1 Hz, 1H, ArH-3nBn), 8.02 (d, 3JH,H = 1.2 Hz, 1H, ArH-3nBn), 7.73 (d, 3JH,H = 1.1 Hz, 1H, H6T), 7.68 (d, 3JH,H = 1.1 Hz, 1H, H6T), 7.52 (m, 2H, 2 × ArH-5nBn), 7.43–7.37 (m, 6H, 2 × ArH-4nBn, 2 × ArH-2,6Ph-DMTr), 7.33–7.27 (m, 12H, 2 × ArH-2,6MeOPh-DMTr, 2 × ArH-3,5Ph-DMTr), 7.27–7.20 (m, 4H, 2 × ArH-6nBn, 2 × ArH-4Ph-DMTr), 6.84 (m, 8H, 2 × ArH-3,5MeOPh-DMTr), 6.41 (m, 2H, 2 × H1′), 5.52 (m, 4H, 2 × CH2nBn), 4.66 (m, 2H, 2 × H3′), 4.18 (m, 1H, H4′), 4.14 (m, 1H, H4′), 3.80 (s, 6H, 2 × OCH3DMTr), 3.79 (s, 6H, 2 × OCH3DMTr), 3.77 (m, 2H, OCH2CH2CN), 3.68–3.45 (m, 8H, OCH2CH2CN, 2 × H5′, 4 × CHiPr), 3.34 (m, 2H, 2 × H5″), 2.61 (t, 3JH,H = 6.2 Hz, 2H, OCH2CH2CN), 2.56 (ddd, 2JH,H = 13.4 Hz, 3JH,H = 5.8 Hz, 3JH,H = 2.4 Hz, 1H, H2′), 2.49 (ddd, 2JH,H = 13.4 Hz, 3JH,H = 5.8 Hz, 3JH,H = 2.9 Hz, 1H, H2′), 2.41 (t, 3JH,H = 6.4 Hz, 2H, OCH2CH2CN), 2.35 (m, 2H, 2 × H2″), 1.48 (s, 6H, 2 × CH3T), 1.18–1.13 (m, 18H, 6 × CH3iPr), 1.05 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 163.5, 158.9, 158.9, 151.0, 151.0, 148.9, 144.4, 144.4, 135.5, 135.5, 135.4, 135.4, 134.5, 134.4, 133.6, 133.6, 132.5, 130.3, 130.3, 130.3, 128.4, 128.3, 128.1, 128.1, 128.1, 127.3, 127.3, 125.1, 125.1, 117.7, 117.5, 113.4, 113.4, 110.6, 110.6, 87.1, 87.1, 85.9, 85.9, 85.7, 85.6, 74.1, 74.0, 73.6, 73.5, 63.4, 63.2, 58.4, 58.3, 58.1, 55.4, 55.4, 43.5, 43.4, 43.4, 43.3, 41.8, 40.3, 40.3, 29.8, 24.8, 24.7, 24.7, 24.6, 24.6, 20.6, 20.5, 20.4, 20.3, 12.6, 12.6 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 148.9 (s, 1P, P), 148.5 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C47H55N5O10P+ 880.36811, found: 880.36806.
N1-(4-O-Acetyl)benzyl-N2-methylguanosine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-(4-OAc)Bn1m2GiBu] (9)
Compound 9 was prepared according to procedure A using 1.00 g (1.03 mmol) of 5′-O-DMT-2′-O-TBDMS-GiBu phosphoramidite and 569 mg (2.06 mmol, 2.0 equiv) of (iodomethyl)phenyl acetate. After 2.5 h, the aqueous fraction was removed and 320 μL (5.15 mmol, 5 equiv) of methyl iodide and a fresh aqueous solution of NaOH with Bu4NBr (1.0 equiv) was added to the organic phase. The reaction was quenched after 1 h, and the product was isolated by flash chromatography (0 → 50% ethyl acetate in n-hexane with 0.5%v/v TEA in 60 min, 40 mL/min, Biotage Sfär HC 10g column) and additionally purified by the second column chromatography (0 → 100% DCM in n-hexane with 0.5%v/v TEA in 30 min, 40 mL/min, Biotage Sfär HC 10 g column) to afford a mixture of diastereomers of 9 (140 mg, 0.124 mmol, 12%) as a white solid. TLC (ethyl acetate): Rf = 0.68; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.21 (s, 1H, H8), 8.17 (s, 1H, H8), 7.52 (m, 4H, ArHN1_AcOBn), 7.44 (m, 4H, ArH-2,6Ph-DMTr), 7.34 (m, 8H, ArH-2,6MeOPh-DMTr), 7.32–7.20 (m, 6H, ArH-3,4,5Ph-DMTr), 7.09 (m, 4H, ArHN1_AcOBn), 6.83 (m, 8H, ArH-3,5MeOPh-DMTr), 6.11 (d, 3JH,H = 6.7 Hz, 1H, H1′), 6.09 (d, 3JH,H = 6.9 Hz, 1H, H1′), 5.61 (m, 4H, CH2N1_AcOBn), 4.82 (dd, 3JH,H = 6.6 Hz, 3JH,H = 4.6 Hz, 1H, H2′), 4.74 (dd, 3JH,H = 6.8 Hz, 3JH,H = 5.0 Hz, 1H, H2′), 4.40 (m, 1H, H4′), 4.38 (m, 1H, H3′), 4.32 (m, 2H, H3′, H4′), 3.92 (m, 1H, OCH2CH2CN), 3.86 (m, 1H, OCH2CH2CN), 3.79–3.77 (overlapped s, 12H, 4 × OCH3DMTr), 3.69–3.50 (m, 7H, OCH2CH2CN, H5′, 4 × CHiPr), 3.45 (m, 1H, H5′), 3.40–3.36 (overlapped, 4H, CH3N2-Me, H5″), 3.35–3.28 (m, 3H, H5′′, 2 × CHiBu), 3.26 (dd, 2JH,H = 10.6 Hz, 3JH,H = 4.0 Hz, 1H, H5″), 3.12 (s, 3H, CH3G-dmf), 3.11 (s, 6H, 2 × CH3G-dmf), 2.61 (m, 2H, OCH2CH2CN), 2.29 (s, 6H, 2 × CH3AcOBn) 2.28 (m, 2H, OCH2CH2CN), 1.21–1.12 (m, 30H, 6 × CH3iPr, 4 × CH3iBu), 1.05 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr), 0.75 (s, 9H, tBuTBDMS), 0.72 (s, 9H, tBuTBDMS), 0.01 (s, 3H, CH3TBDMS), −0.01 (s, 3H, CH3TBDMS), −0.18 (s, 3H, CH3TBDMS), −0.21 (s, 3H, CH3TBDMS) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 169.5, 160.3, 158.8, 130.3, 130.2, 130.1, 129.4, 129.4, 129.2, 128.9, 128.8, 128.3, 128.2, 128.2, 128.2, 128.1, 127.3, 127.2, 121.9, 121.4, 113.5, 113.5, 113.4, 113.4, 68.2, 68.2, 55.4, 55.4, 43.6, 43.5, 25.7, 25.6, 24.9, 24.8, 24.8, 24.8, 21.3, 21.3, 20.3, 20.3, 20.3, 20.2, 18.1, 18.0, −4.5, −4.5, −5.0, −5.1 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 151.6 (s, 1P, P), 148.8 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C60H79N7O11PSi+ 1132.53390, found: 1132.53482.
N1-Methylguanosine Phosphoramidite [5′-O-DMT-2′-O-TBDMS-m1Gdmf] (10)
Compound 10 was prepared according to procedure A using 250 mg (0.262 mmol) of 5′-O-DMT-2′-O-TBDMS-Gdmf phosphoramidite and 163 μL (2.62 mmol, 10 equiv) of methyl iodide. The reaction was quenched after 25 min, and the product was isolated by flash chromatography (0 → 100% ethyl acetate in n-hexane with 0.5%v/v TEA in 30 min, 40 mL/min, Biotage Sfär HC 10 g column) to afford a mixture of diastereomers of 10 (208 mg, 0.215 mmol, 82%) as a white solid. TLC (ethyl acetate): Rf = 0.34; 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.52 (s, 1H, N=CH-NG-dmf), 8.47 (s, 1H, N=CH-NG-dmf), 7.86 (s, 1H, H8), 7.82 (s, 1H, H8), 7.44 (m, 4H, ArH-2,6Ph-DMTr), 7.33 (m, 8H, ArH-2,6MeOPh-DMTr), 7.28 (m, 4H, ArH-3,5Ph-DMTr), 7.22 (m, 2H, ArH-4Ph-DMTr), 6.81 (m, 8H, ArH-3,5MeOPh-DMTr), 6.02 (d, 3JH,H = 6.3 Hz, 1H, H1′), 5.96 (d, 3JH,H = 6.0 Hz, 1H, H1′), 4.71 (dd, 3JH,H = 6.3 Hz, 3JH,H = 4.7 Hz, 1H, H2′), 4.67 (dd, 3JH,H = 6.0 Hz, 3JH,H = 4.9 Hz, 1H, H2′), 4.37 (m, 1H, H4′), 4.34 (m, 1H, H3′), 4.28 (m, 2H, H3′, H4′), 3.90 (m, 2H, OCH2CH2CN), 3.79–3.77 (overlapped s, 12H, 4 × OCH3DMTr), 3.67 (s, 6H, 2 × CH3N1-Me), 3.66–3.50 (m, 7H, OCH2CH2CN, H5′, 4 × CHiPr), 3.42 (dd, 2JH,H = 10.6 Hz, 3JH,H = 2.6 Hz, 1H, H5′), 3.31 (dd, 2JH,H = 10.6 Hz, 3JH,H = 3.8 Hz, 1H, H5″), 3.26 (dd, 2JH,H = 10.6 Hz, 3JH,H = 4.0 Hz, 1H, H5″), 3.12 (s, 3H, CH3G-dmf), 3.11 (s, 6H, 2 × CH3G-dmf), 3.02 (s, 3H, CH3G-dmf), 2.65 (m, 2H, OCH2CH2CN), 2.29 (m, 2H, OCH2CH2CN), 1.19–1.14 (m, 18H, 6 × CH3iPr), 1.01 (d, 3JH,H = 6.8 Hz, 6H, 2 × CH3iPr), 0.81 (s, 9H, tBuTBDMS), 0.79 (s, 9H, tBuTBDMS), 0.01 (s, 3H, CH3TBDMS), 0.00 (s, 3H, CH3TBDMS), −0.12 (s, 3H, CH3TBDMS), −0.14 (s, 3H, CH3TBDMS) ppm; 13C{1H} NMR (126 MHz, CDCl3, 25 °C): δ = 158.7, 158.7, 157.5, 157.2, 157.1, 148.5, 148.3, 144.6, 144.5, 136.0, 135.8, 135.8, 135.6, 135.5, 130.3, 130.2, 130.1, 128.3, 128.2, 128.1, 128.1, 127.1, 120.2, 120.0, 117.6, 117.4, 113.4, 113.4, 113.4, 86.9, 86.7, 86.7, 86.4, 83.5, 83.5, 83.4, 76.6, 75.7, 75.7, 73.5, 73.4, 63.8, 63.5, 59.0, 58.9, 57.9, 57.7, 55.4, 55.4, 43.6, 43.5, 43.1, 43.0, 41.2, 41.0, 35.3, 35.2, 34.0, 30.1, 30.0, 29.8, 29.3, 29.1, 25.8, 25.8, 24.9, 24.8, 24.8, 24.7, 20.5, 20.4, 20.2, 20.2, 18.1, 18.1, 14.3, −4.5, −4.6, −4.8, −4.9 ppm; 31P NMR (202.5 MHz, CDCl3, 25 °C): δ = 150.7 (s, 1P, P), 149.6 (s, 1P, P) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C50H70N8O8PSi+ 969.48180, found: 969.48308.
Solid-Phase Synthesis of Oligonucleotides
General Procedure
Solid-phase syntheses of short oligonucleotides were performed in a 10 mL syringe equipped with frit and loaded with polystyrene support [ribo U 300 PrimerSupport 5G (298 μmol/g, GE Healthcare), ribo G 300 PrimerSupport 5G (308 μmol/g, GE Healthcare), or dC 350 PrimerSupport 5G (360 μmol/g, GE Healthcare)]. The typical synthesis scale was 15 μmol (based on the support loading provided by the manufacturer), but it could be easily scaled-up to ca. 200 μmol using this setup. The detritylation step was performed by passing 5 mL of 3% (v/v) trichloroacetic acid in DCM through the column. The solid support was washed with 5 mL of DNA synthesis grade acetonitrile (<10 ppm of H2O) and dried in a vacuum desiccator. In the coupling step, a 0.3 M solution of an appropriate phosphoramidite (3.0 equivalents) in anhydrous acetonitrile and a 1.5 volume of 0.3 M BTT Activator were shaken with the support for 30 min. Then the support was washed with 5 mL of acetonitrile and the phosphite triester was oxidized by passing 1.5 mL of 0.05 M iodine in pyridine/water 9:1v/v. To prepare the dinucleotide 5′-phosphates, the bis(2-cyanoethyl)-N,N-diisopropylphosphoramidite (3.0 equivalents, 0.3 M in acetonitrile + 1.5 volume of 0.3 M BTT Activator) was used in the last cycle and the detritylation step was omitted. After the last cycle of the synthesis, 2-cyanoethyl groups were removed by passing 5 mL of 20%v/v solution of diethylamine in acetonitrile. The support was dried in a vacuum desiccator and transferred to a 50 mL polypropylene tube, and the oligonucleotide was cleaved from the support using AMA (1 mL, 1:1v/v mixture of 33% ammonium hydroxide and 40% methylamine in water for 3 h at 37 °C (Eppendorf ThermoMixer C, 1000 rpm)***. The suspension was filtered, washed with water, evaporated to dryness, redissolved in water, and freeze-dried. The residue was dissolved in 20 μL of DMSO, followed by the addition of triethylamine (33 μL) and triethylammonium trihydrofluoride (TEA·3HF, 20 μL), and the resulting mixture was shaken for 3 h at 65 °C (Eppendorf ThermoMixer C, 1000 rpm). The reaction was quenched by addition of 0.05 M NaHCO3 in water (ca. 20 mL), and the pH was adjusted to 6–7 if necessary. A sample of the product for compound characterization was isolated by ion-exchange chromatography on DEAE Sephadex using a linear gradient of TEAB: 0–0.9 M for the dinucleotides and 0–1.2 M for the trinucleotides and evaporated to dryness with ethanol to give a white solid.
*** Oligonucleotide 14 (prepared using phosphoramidite 1e) was treated with AMA for 4 h at 37 °C (Eppendorf ThermoMixer C, 1000 rpm) to ensure complete aminolysis of phthalimide moiety. Oligonucleotide 24 prepared using phosphoramidite 9 (m2G) was deprotected with AMA for 3 h at 37 °C (Eppendorf ThermoMixer C, 1000 rpm) and then left at 4 °C overnight for complete elimination of 4-hydroxybenzyl substituent. Oligonucleotide 19 containing m3C was cleaved from the solid support and deprotected using 30–33% aqueous ammonium hydroxide to avoid N4-transamination with methylamine deprotection with AMA produced dinucleotide 21 (N3,N4-dimethylcytidine derivative p(m23,4C)pG) as the only product.
Ui6AU (11)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.38 (s, 1H, H8A2), 8.22 (s, 1H, H2A2), 7.77 (d, 3JH,H = 8.2 Hz, 1H, H6U), 7.76 (d, 3JH,H = 8.1 Hz, 1H, H6U), 6.10 (d, 3J = 3.6 Hz, 1H, H1′A2), 5.83 (d, 3JH,H = 4.0 Hz, 1H, H1′U3), 5.70 (m, 2H, H5U, H1′U1), 5.66 (d, 3JH,H = 8.1 Hz, 1H, H5U), 5.41 (m, 1H, CH=CN6-isopent), 4.80 (m, overlapped with HDO, 2H, H2′A2, H3′A2), 4.54 (m, 1H, H4′A2), 4.48 (m, 1H, H3′U1), 4.35–4.28 (m, 4H, H2′U1, H3′U3, H5′A2, H5′U3), 4.25–4.24 (m, 2H, H4′U3, H2′U3), 4.22–4.10 (m, 5H, H4′U1, H5″A2, H5″U3, CH2N6-isopent), 3.82–3.73 (m, 2H, H5′U1, H5″U1), 3.20 (q, 3JH,H = 7.3 Hz, 12H, CH2TEAH+), 1.76 (m, 6H, 2 × CH3N6-isopent), 1.28 (t, 3JH,H = 7.3 Hz, 18H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 0.17 (s, 1P,PA-U), 0.12 (s, 1P,PU-A); HRMS (ESI) m/z: [M + H]+ calcd for C33H42N9O20P2– 946.20268, found: 946.20454; physical description: white amorphous solid.
pBn6AmpG (12)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.40 (s, 1H, H8A), 8.13 (s, 1H, H2A), 7.93 (s, 1H, H8G), 7.41–7.35 (m, 4H, ArHBn), 7.31 (m, 1H, ArH-4Bn), 6.10 (d, 3JH,H = 5.2 Hz, 1H, H1′A), 5.83 (d, 3JH,H = 5.2 Hz, 1H, H1′G), 4.92 (m, 1H, H3′A), 4.82 (m, overlapped with HDO, 2H, CH2Bn), 4.73 (m, 1H, H2′G), 4.50–4.44 (m, 3H, H3′G, H2′A, H4′A), 4.34 (m, 1H, H4′G), 4.21 (m, 2H, H5′G, H5″G), 4.07 (m, 2H, H5′A, H5″A), 3.49 (s, 3H, CH32′-O-Me), 3.19 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 1.27 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 1.10 (s, 1P, P5′), 0.05 (s, 1P, PA-G) ppm; HRMS (ESI) m/z calcd for C28H33N10O14P2– [M-H]−: 795.1658, found: 795.16658; physical description: white amorphous solid.
phex6AmpG (13)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.38 (s, 1H, H8A), 8.14 (s, 1H, H2A), 7.91 (s, 1H, H8G), 6.09 (d, 3JH,H = 5.0 Hz, 1H, H1′A), 5.82 (d, 3JH,H = 5.1 Hz, 1H, H1′G), 4.92 (m, 1H, H3′A), 4.72 (m, 1H, H2′G), 4.50–4.43 (m, 3H, H3′G, H2′A, H4′A), 4.34 (m, 1H, H4′G), 4.21 (m, 2H, H5′G, H5″G), 4.07 (m, 2H, H5′A, H5″A), 3.59 (m, 2H, CH2-6hex), 3.50 (s, 3H, CH32′-O-Me), 3.20 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 2.33 (t, 4JH,H = 2.6 Hz, 1H, H1hex), 2.27 (td, 3JH,H = 7.0 Hz, 4JH,H = 2.6 Hz, CH2-3hex), 1.80 (m, 2H, CH2-5hex), 1.64 (m, 2H, CH2-4hex), 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 1.24 (s, 1P, P5′), 0.04 (s, 1P, PA-G) ppm; HRMS (ESI) m/z calcd For C27H35N10O14P2– [M-H]−: 785.1815, found: 785.18225; physical description: white amorphous solid.
pap6AmpApG (14)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.38 (s, 1H, H8A1), 8.14 (s, 1H, H8A2), 8.06 (s, 1H, Hpurine), 7.90 (s, 1H, Hpurine), 7.80 (s, 1H, Hpurine), 6.03 (d, 3JH,H = 3.9 Hz, 1H, H1′A1), 5.88 (d, 3JH,H = 3.1 Hz, 1H, H1′A2), 5.71 (d, 3JH,H = 5.4 Hz, 1H, H1′G3), 4.88 (m, 1H, H3′A1), 4.74 (m, 1H, H3′A2), 4.57 (m, 3H, H2′A1, H2′A2, H2′G3), 4.52 (m, 1H, H4′A2), 4.48 (m, 1H, H4′A1), 4.39 (m, 1H, H3′G3), 4.31–4.26 (m, 3H, H4′G3, H5′A2, H5′G3), 4.25–4.08 (m, 4H, H5″A2, H5′A1, H5″G3, H5″A1), 3.61–3.51 (m, 2H, CH2aminopropyl), 3.58 (s, 3H, CH32′-O-Me), 3.07 (s, 2H, CH2aminopropyl), 2.00 (m, 2H, CH2aminopropyl) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 1.96 (s, 1P, P5′), 0.14 (s, 1P, PA2-G3), −0.24 (s, 1P, PA1-A2) ppm; HRMS (ESI) m/z calcd for C34H46N16O20P3–, [M-H]−: 1091.22926, found: 1091.23012; physical description: white amorphous solid.
piPr6AmpG (15)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.39 (s, 1H, H8A), 8.16 (s, 1H, H2A), 7.93 (s, 1H, H8G), 6.09 (d, 3JH,H = 5.3 Hz, 1H, H1′A), 5.85 (d, 3JH,H = 5.3 Hz, 1H, H1′G), 4.93 (m, 1H, H3′A), 4.76 (m, overlapped with HDO, H2′G), 4.50–4.45 (m, 3H, H2′A, H3′G, H4′A), 4.35 (m, 1H, H4′G), 4.24–4.18 (m, H5′G, H5″G), 4.12–4.01 (m, 2H, H5′A, H5″A), 3.49–3.45 (m, 4H, CHiPr, CH32′-O-Me), 3.20 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 1.31 (m, 6H, 2 × CH3 iPr), 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 1.05 (s, 1P, P5′), 0.06 (s, 1P, PA-G) ppm; HRMS (ESI) m/z calcd for C24H33N10O14P2–, [M-H]−: 747.16584, found: 747.16699; physical description: white amorphous solid.
pPhNCO6ApG (16)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.47 (s, 1H, H8A), 8.43 (s, 1H, H2A), 7.70 (s, 1H, H8G), 7.28 (m, 2H, ArH-2,6PhNHCO), 7.20 (m, 2H, ArH-3,5PhNHCO), 6.99 (m, 1H, ArH-4PhNHCO), 5.94 (m, 1H, H1′A), 5.71 (d, 3JH,H = 4.2 Hz, 1H, H1′G), 4.78 (m, 2H, H2′A, H3′A), 4.53–4.47 (m, 2H, H2′G, H4′A), 4.40 (m, 1H, H3′G), 4.33–4.30 (m, 2H, H4′G, H5′G), 4.27–4.25 (m, 1H, H5′A), 4.17–4.12 (m, 2H, H5″A, H5″G) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 1.28 (s, 1P, P5′), 0.05 (s, 1P, PA-G) ppm; HRMS (ESI) m/z calcd for C27H30N11O15P2– [M-H]−: 810.14036, found: 810.14200; physical description: white amorphous solid.
Ug6AU (17)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.61 (s, 1H, H8A2), 8.60 (s, 1H, H2A2), 7.79 (d, JH,H = 8.1 Hz, 1H, H6U), 7.77 (d, 3JH,H = 8.1 Hz, 1H, H6U), 6.21 (d, 3JH,H = 3.6 Hz, 1H, H1′A2), 5.85 (d, 3JH,H = 4.3 Hz, 1H, H1′U3), 5.73 (d, 3JH,H = 8.1 Hz, 1H, H5U), 5.67 (d, 3JH,H = 4.2 Hz, 1H, H1′U1), 5.65 (d, 3JH,H = 8.1 Hz, 1H, H5U), 4.90–4.83 (m, 2H, H3′A2, H2′A2), 4.57 (m, 1H, H4′A2), 4.49 (m, 1H, H3′U1), 4.39–4.33 (m, 1H, H5′A2), 4.33–4.29 (m, 3H, H2′U1, H5′U3, H3′U3), 4.27–4.24 (m, 2H, H2′U3, H4′U3), 4.23–4.17 (m, 2H, H4′U1, H5″A2), 4.16–4.12 (m, 1H, H5″U3), 4.10 (s, 2H, CH2Gly-α) 3.78–3.75 (m, 2H, H5′U1, H5″U1), 3.20 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 2.79 (s, 3H, CH3CONHCH3), 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 0.18 (s, 1P,PA-U), 0.05 (s, 1P, PU-A); HRMS (ESI) m/z calcd for C32H40N11O22P2– [M-H]−: 992.18301, found: 992.18614; physical description: white amorphous solid.
Ug6m6AU (18)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.66 (s, 1H, H8A2), 8.65 (s, 1H, H2A2), 7.80 (d, 3JH–H = 8.1 Hz, 1H, H6U), 7.77 (d, 3JH–H = 8.1 Hz, 1H, H6U), 6.24 (d, 3JH–H = 3.5 Hz, 1H, H1′A2), 5.84 (d, 3JH–H = 4.2 Hz, 1H, H1′U3), 5.74 (d, 3JH–H = 8.1 Hz, 1H, H5U), 5.68 (d, 3JH–H = 4.3 Hz, 1H, H1′U1), 5.63 (d, 3JH–H = 8.1 Hz, 1H, H5U), 4.91–4.87 (m, 1H, H3′A2), 4.86–4.84 (m, 1H, H2′A2), 4.57 (m, 1H, H4′A2), 4.50 (m, 1H, H3′U1), 4.39–4.35 (m, 2H, H2′U1, H5′A2), 4.34–4.29 (m, 2H, H3′U3, H5′U3), 4.28–4.19 (m, 3H, H4′U3, H2′U3, H5″A2), 4.20–4.12 (m, 2H, H4′U1, H5″U3), 4.04 (s, 2H, CH2Gly-α), 3.76 (m, 2H, H5′U1, H5″U1), 3.72 (s, 3H, CH3N6-Me), 3.21 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 2.78 (s, 3H, CH3CONHCH3), 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 0.16 (s, 1P,PA-U), 0.05 (s, 1P, PU-A); HRMS (ESI) m/z: [M + H]+ calcd for C33H42N11O22P2– 1006.19866, found: 1006.20085; physical description: white amorphous solid.
pm3CpG (19)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.13 (d, 3JH,H = 8.1 Hz, 1H, H6C), 8.02 (s, 1H, H8G), 6.31 (d, 3JH,H = 8.0 Hz, 1H, H5C), 5.88 (d, 3JH,H = 5.9 Hz, 1H, H1′G), 5.81 (d, 3JH,H = 3.9 Hz, 1H, H1′C), 4.82 (m, overlapped with HDO, 1H, H2′G), 4.62 (m, 1H, H3′C), 4.49 (dd, 3JH,H = 5.3 Hz, 3JH,H = 3.7 Hz, 1H, H3′G), 4.39 (m, 1H, H4′C), 4.36–4.30 (m, 2H, H2′C, H4′G), 4.24–4.22 (m, 1H, H5′G), 4.19–4.09 (m, 2H, H5′C, H5″G), 4.09–4.02 (m, 1H, H5″C), 3.45 (s, 3H, CH3N3-Me), 3.20 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 0.90 (s, 1P, P5′), 0.19 (s, 1P, PC-G) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C20H27N8O15P2– 681.10766, found: 681.10921; physical description: white amorphous solid.
pm4CpG (20)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.04 (s, 1H, H8G), 7.94 (d, 3JH,H = 7.6 Hz, 1H, H6C), 5.94 (d, 3JH,H = 3.8 Hz, 1H, H1′C), 5.88–5.85 (m, 2H, H1′G, H5C), 4.75–4.73 (m, 1H, H2′G), 4.62 (m, 1H, H3′C), 4.54 (m, 1H, H3′G), 4.41 (m, 1H, H2′C), 4.35 (m, 2H, H4′G, H4′C), 4.24–4.16 (m, 2H, H5′G, H5″G), 4.12–4.08 (m, 1H, H5′C), 3.98–3.94 (m, 1H, H5″C), 3.20 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 2.84 (s, 3H, CH3N4-Me), 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 1P NMR (202.5 MHz, D2O, 25 °C): δ = 4.37 (s, 1P, P5′), 0.33 (s, 1P, PC-G) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C20H27N8O15P2– 681.10766, found: 681.10877; physical description: white amorphous solid.
pm23,4CpG (21)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.01 (s, 1H, H8G), 7.94 (d, 3JH,H = 8.1 Hz, 1H, H6C), 5.92 (d, 3JH,H = 8.1 Hz, 1H, H5C), 5.91 (d, 3JH,H = 3.7 Hz, 1H, H1′C), 5.87 (d, 3JH,H = 5.8 Hz, 1H, H1′G), 4.82 (m, 1H, H2′G), 4.73 (m, 1H, H3′C), 4.50 (dd, 3JH,H = 5.2, 3.7 Hz, 1H, H3′G), 4.35–4.32 (m, 2H, H4′C, H4′G), 4.18–4.16 (m, 2H, H5′G, H5″G), 4.12–4.07 (m, 2H, H2′C, H5′C), 4.06–4.02 (m, 1H, H5″C), 3.52 (s, 3H, CH3N3-Me), 3.20 (m, 9H, CH3N4-Me, 3 × CH2TEAH+), 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 1P NMR (202.5 MHz, D2O, 25 °C): δ = 1.01 (s, 1P, P5′), −0.02 (s, 1P, PC-G); HRMS (ESI) m/z: [M + H]+ calcd for C21H29N8O15P2– 695.12331, found: 695.12428; physical description: white amorphous solid.
pm3UmpG (22)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.31 (d, 3JH,H = 8.2 Hz, 1H, H6U), 8.00 (s, 1H, H8G), 6.44 (d, 3JH,H = 8.2 Hz, 1H, H5U), 5.86 (d, 3JH,H = 6.2 Hz, 1H, H1′G), 5.79 (d, 3JH,H = 3.3 Hz, 1H, H1′U), 4.81 (m, 1H, H2′G), 4.64 (m, 1H, H3′U), 4.49 (dd, 3JH,H = 5.2, 3JH,H = 3.4 Hz, 1H, H3′G), 4.38–4.41 (m, 2H, H2′U, H4′U), 4.33 (m, 1H, H4′G), 4.25–4.22 (m, 1H, H5′G), 4.20–4.13 (m, 2H, H5′U, H5″G), 4.11–4.01 (m, 1H, H5″U), 3.42 (s, 3H, CH32′-OMe), 3.20 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 3.15 (s, 3H, CH3N3-Me), 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 1.52 (s, 1P, P5′), 0.21 (s, 1P, PU-G) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C21H28N7O16P2– 696.10732, found: 696.10792; physical description: white amorphous solid.
d(G2nBn3TC) (23)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.01 (m, 1H, ArH-3nBn), 7.94 (s, 1H, H8G), 7.90 (d, 3JH,H = 7.7 Hz, 1H, H6C), 7.74 (d, 4JH,H = 0.9 Hz, 1H, H6T), 7.38 (m, 2H, ArH-4nBn, ArH-5nBn), 7.03 (m, 1H, ArH6-nBn), 6.27 (dd, 3JH,H = 7.9 Hz, 3JH,H = 6.2 Hz, 1H, H1′T), 6.15 (m, 2H, H1′C, H1′G), 6.06 (d, 3JH,H = 7.7 Hz, 1H, H5C), 5.33 (m, 2H, CH2nBnT), 4.91 (m, 2H, H3′G, H3′T), 4.52 (m, 1H, H3′C), 4.37 (m, 1H, H4′T), 4.30 (m, 1H, H4′G), 4.24 (ddd, 2JH,H = 11.4 Hz, 3JH,P = 4.4 Hz, 3JH,H = 2.2 Hz, 1H, H5′T), 4.18–4.10 (m, 3H, H5″T, H4′C, H5′C), 4.08 (m, 1H, H5″C), 3.80 (m, 2H, H5′G, H5″G), 3.20 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 2.75–2.63 (m, 2H, H2′G, H2″G), 2.55 (ddd, 2JH,H = 13.9 Hz, 3JH,H = 6.0 Hz, 3JH,H = 2.6 Hz, 1H, H2′T), 2.43–2.34 (m, 2H, H2″T, H2′C), 2.22 (m, 1H, H2″C), 1.89 (d,4JH,H = 0.9 Hz, 3H, CH35-T), 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = −0.06 (s, 1P, PT-C), −0.13 (s, 1P, PG-T) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C36H42N11O19P2– 994.21391, found: 994.21533; physical description: pale yellow amorphous solid.
Um2GU (24)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.10 (s, 1H, H8G), 7.85 (d, 3JH,H = 8.2 Hz, 1H, H6U), 7.77 (d, 3JH,H = 8.1 Hz, 1H, H6U), 5.98 (d, 3JH,H = 3.6 Hz, 1H, H1′G2), 5.90 (d, 3JH,H = 4.2 Hz, 1H, H1′U3), 5.80–5.78 (m, 2H, H1′U1, H5U), 5.76 (d, 3JH,H = 8.1 Hz, 1H, H5U), 4.94–4.88 (m, 2H, H2′G2, H3′G2), 4.52–4.47 (m, 2H, H4′G2, H3′U1), 4.34–4.28 (m, 5H, H2′U1, H2′U3, H3′U3, H5′G2, H5′U3), 4.26 (m, 1H, H4′U3), 4.24–4.21 (m, 1H, H5G2), 4.19–4.13 (m, 2H, H4′U1, H5U3), 3.69 (dd, 2JH,H = 12.9, 3JH,H = 3.8 Hz, 1H, H5′U1), 3.60 (dd, 2JH,H = 12.9, 3JH,H = 2.8 Hz, 1H, H5″U1), 3.21 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 2.97 (s, 3H, CH3N2-Me) 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = −0.76 (s, 2P, PU-G, PG-U) ppm; HRMS (ESI) m/z: [M + H]+ calcd for. C29H36N9O21P2– 908.15065, found: 908.15258; physical description: white amorphous solid.
Um1GU (25)
1H NMR (500 MHz, D2O, 25 °C): δ = 8.03 (s, 1H, H8G), 7.85 (d, 3J = 8.1 Hz, 1H, H6U), 7.78 (d, 3JH,H = 8.1 Hz, 1H, H6U), 5.91 (m, 2H, H1′U3, H1′G2), 5.75 (d, 3JH,H = 4.2 Hz, 1H, H1′U1), 5.73 (d, 3JH,H = 8.1 Hz, 1H, H5U), 5.72 (d, 3JH,H = 8.1 Hz, 1H, H5U), 4.85 (m, 1H, H3′G2), 4.76 (m, overlapped with HDO, 1H, overlapped, H2′G2), 4.51–4.45 (m, 2H, H3′U1, H4′G2), 4.35 (m, 1H, H2′U1), 4.32–4.30 (m, 2H, H3′U3, H5′G2), 4.29–4.26 (m, 3H, H2′U3, H4′U3, H5′U3), 4.22–4.19 (m, 2H, H4′U1, H5″G2), 4.17–4.12 (m, 1H, H5″U3), 3.74 (d, 3JH,H = 3.3 Hz, 2H, H5′U1, H5″U1), 3.43 (s, 3H, CH3N1-Me), 3.20 (q, 3JH,H = 7.3 Hz, 6H, CH2TEAH+), 1.28 (t, 3JH,H = 7.3 Hz, 9H, CH3TEAH+) ppm; 31P NMR (202.5 MHz, D2O, 25 °C): δ = 0.22 (s, 1P, PG-U), 0.12 (s, 1P, PU-G) ppm; HRMS (ESI) m/z: [M + H]+ calcd for. C29H36N9O21P2– 908.15065, found: 908.15245; physical description: white amorphous solid.
Synthesis of cap-2 and cap-4
The pm6AmpGmpG and pm6,6AmpAmpCmpm3UmpA oligonucleotides were synthesized and isolated as triethylammonium salts according to the procedure described above.
Cap-2: m7Gpppm6AmpGmpG (26)
Triethylammonium salt of pm6AmpGmpG (520 mOD, 13.3 μmol) was dissolved in DMSO (265 μL), and P-imidazolide of N7-methylguanosine 5′-diphosphate [m7GDP-Im]42 (16.7 mg, 26.5 μmol) and anhydrous ZnCl2 (36.1 mg, 265 μmol) were added. The mixture was stirred for ca. 48 h, and the reaction was quenched by addition of 6.2 mL of aqueous solution of EDTA (20 mg/mL) and NaHCO3 (10 mg/mL). The product was isolated by ion-exchange chromatography on DEAE Sephadex (gradient elution 0–1.2 M TEAB) and purified by semi-preparative RP HPLC (gradient elution 0–15% acetonitrile in 0.05 M ammonium acetate buffer pH 5.9) to afford—after evaporation and repeated freeze-drying from water—ammonium salt of trinucleotide 26 m7Gpppm6AmpGmpG (13.4 mg, 370 mOD, 8.39 μmol, 63%) as a white amorphous solid. HRMS (ESI) m/z: [M + H]+ calcd for C44H58N20O31P5– 1517.22704, found: 1517.22838.
Cap-4: m7Gpppm6,6AmpAmpCmpm3UmpA (27)
Triethylammonium salt of pm6,6AmpAmpCmpm3UmpA (206 mOD, 3.3 μmol) was dissolved in anhydrous DMF (132 μL) followed by the addition of imidazole (14.4 mg, 211 μmol), triethylamine (11 μL, 79 μmol), 2,2′-dithiodipyridine (17.4 mg, 79 μmol), and triphenylphospine (20.7 mg,79 μmol). After 5 h, the product was precipitated with a cold solution of NaClO4 (16.2 mg, 132 μmol) in acetonitrile (1.32 mL). The precipitate was centrifuged (6000 rpm, 6 min) in a 50 mL conical tube at 4 °C, washed with cold acetonitrile by centrifugation 3 times, and dried under reduced pressure. Thus obtained P-imidazolide was mixed with 7-methylguanosine 5′-diphosphate (30 mg, 33.0 μmol) in anhydrous DMSO (440 μL), followed by the addition of anhydrous ZnCl2 (72 mg, 528 μmol). The mixture was stirred for ca. 14 h, and the reaction was quenched by addition of 8.5 mL of aqueous solution of EDTA (20 mg/mL) and NaHCO3 (10 mg/mL). The product was isolated by ion-exchange chromatography on DEAE Sephadex using a linear gradient of TEAB (0–1.2 M) and purified by semi-preparative RP HPLC (gradient elution 0–15% acetonitrile in 0.05 M ammonium acetate buffer pH 5.9) to afford—after evaporation and repeated freeze-drying from water—ammonium salt of 27 m7Gpppm6,6AmpAmpCpm3UmpA (5.67 mg, 115 mOD, 1.88 μmol, 57%) as a white amorphous solid. HRMS (ESI) m/z: [M + H]+ calcd for C66H89N25O44P7– 2152.36640, found: 2152.36410;
Acknowledgments
This work was supported by the National Science Centre Poland (2019/33/B/ST4/01843 to J.J. and 2017/24/T/NZ1/00345 to M.W.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c01390.
NMR and HRMS spectra and HPLC profiles of oligonucleotides (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cohn W. E.; Volkin E. Nucleoside-5′-Phosphates from Ribonucleic Acid. Nature 1951, 167, 483–484. 10.1038/167483a0. [DOI] [Google Scholar]
- a McCown P. J.; Ruszkowska A.; Kunkler C. N.; Breger K.; Hulewicz J. P.; Wang M. C.; Springer N. A.; Brown J. A. Naturally occurring modified ribonucleosides. WIREs RNA 2020, 11, e1595 10.1002/wrna.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Boccaletto P.; Machnicka M. A.; Purta E.; Piątkowski P.; Bagiński B.; Wirecki T. K.; de Crécy-Lagard V.; Ross R.; Limbach P. A.; Kotter A.; Helm M.; Bujnicki J. M. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. 10.1093/nar/gkx1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood A. J.; Viner C.; Hoffman M. M. DNAmod: the DNA modification database. J. Cheminform. 2019, 11, 30. 10.1186/s13321-019-0349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt E. M.; Kietrys A. M.; Kool E. T. Chemical and structural effects of base modifications in messenger RNA. Nature 2017, 541, 339–346. 10.1038/nature21351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H.; Wei J.; He C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. 10.1016/j.molcel.2019.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski P. J.; Warminski M.; Kubacka D.; Ratajczak T.; Nowis D.; Kowalska J.; Jemielity J. The identity and methylation status of the first transcribed nucleotide in eukaryotic mRNA 5′ cap modulates protein expression in living cells. Nucleic Acids Res. 2020, 48, 1607–1626. 10.1093/nar/gkaa032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiec K.; Sochacka E. Efficient access to 3′-O-phosphoramidite derivatives of tRNA related N6-threonylcarbamoyladenosine (t6A) and 2-methylthio-N6-threonylcarbamoyladenosine (ms2t6A). RSC Adv. 2021, 11, 1992–1999. 10.1039/D0RA09803E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wein S.; Andrews B.; Sachsenberg T.; Santos-Rosa H.; Kohlbacher O.; Kouzarides T.; Garcia B. A.; Weisser H. A computational platform for high-throughput analysis of RNA sequences and modifications by mass spectrometry. Nat. Commun. 2020, 11, 926. 10.1038/s41467-020-14665-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Paulines M. J.; Wetzel C.; Limbach P. A. Using spectral matching to interpret LC-MS/MS data during RNA modification mapping. J. Mass Spectrom. 2019, 54, 906–914. 10.1002/jms.4456. [DOI] [PubMed] [Google Scholar]; (acccessed 2022/03/01).; c Murphy F. V.; Ramakrishnan V.; Malkiewicz A.; Agris P. F. The role of modifications in codon discrimination by tRNALysUUU. Nat. Struct. Mol. Biol. 2004, 11, 1186–1191. 10.1038/nsmb861. [DOI] [PubMed] [Google Scholar]; d Micura R.; Pils W.; Höbartner C.; Grubmayr K.; Ebert M.-O.; Jaun B. Methylation of the nucleobases in RNA oligonucleotides mediates duplex–hairpin conversion. Nucleic Acids Res. 2001, 29, 3997–4005. 10.1093/nar/29.19.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]; (acccessed 1/4/2022); e Micura R.; Pils W.; Grubmayr K. Bridged Cyclic Oligoribonucleotides as Model Compounds for Codon – Anticodon Pairing. Angew. Chem., Int. Ed. 2000, 39, 922–925. . [DOI] [PubMed] [Google Scholar]; f Stuart J. W.; Gdaniec Z.; Guenther R.; Marszalek M.; Sochacka E.; Malkiewicz A.; Agris P. F. Functional Anticodon Architecture of Human tRNALys3 Includes Disruption of Intraloop Hydrogen Bonding by the Naturally Occurring Amino Acid Modification, t6A. Biochemistry 2000, 39, 13396–13404. 10.1021/bi0013039. [DOI] [PubMed] [Google Scholar]; g Pütz J.; Florentz C.; Benseler F.; Giegé R. A single methyl group prevents the mischarging of a tRNA. Nat. Struct. Biol. 1994, 1, 580–582. 10.1038/nsb0994-580. [DOI] [PubMed] [Google Scholar]; h Müller F.; Escobar L.; Xu F.; Węgrzyn E.; Nainytė M.; Amatov T.; Chan C. Y.; Pichler A.; Carell T. A prebiotically plausible scenario of an RNA–peptide world. Nature 2022, 605, 279–284. 10.1038/s41586-022-04676-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li Y.; Bedi R. K.; Wiedmer L.; Huang D.; Śledź P.; Caflisch A. Flexible Binding of m6A Reader Protein YTHDC1 to Its Preferred RNA Motif. J. Chem. Theory Comput. 2019, 15, 7004–7014. 10.1021/acs.jctc.9b00987. [DOI] [PubMed] [Google Scholar]; b Relier S.; Ripoll J.; Guillorit H.; Amalric A.; Achour C.; Boissière F.; Vialaret J.; Attina A.; Debart F.; Choquet A.; Macari F.; Marchand V.; Motorin Y.; Samalin E.; Vasseur J. J.; Pannequin J.; Aguilo F.; Lopez-Crapez E.; Hirtz C.; Rivals E.; Bastide A.; David A. FTO-mediated cytoplasmic m6Am demethylation adjusts stem-like properties in colorectal cancer cell. Nat. Commun. 2021, 12, 1716. 10.1038/s41467-021-21758-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang X.; Wei L.-H.; Wang Y.; Xiao Y.; Liu J.; Zhang W.; Yan N.; Amu G.; Tang X.; Zhang L.; Guifang J. Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 2919. 10.1073/pnas.1820574116. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mauer J.; Luo X.; Blanjoie A.; Jiao X.; Grozhik A. V.; Patil D. P.; Linder B.; Pickering B. F.; Vasseur J.-J.; Chen Q.; Gross S. S.; Elemento O.; Debart F.; Kiledjian M.; Jaffrey S. R. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. 10.1038/nature21022. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Xu C.; Liu K.; Ahmed H.; Loppnau P.; Schapira M.; Min J. Structural Basis for the Discriminative Recognition of N6-Methyladenosine RNA by the Human YT521-B Homology Domain Family of Proteins. J. Biol. Chem. 2015, 290, 24902–24913. 10.1074/jbc.M115.680389. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Theler D.; Dominguez C.; Blatter M.; Boudet J.; Allain F. H. T. Solution structure of the YTH domain in complex with N6-methyladenosine RNA: a reader of methylated RNA. Nucleic Acids Res. 2014, 42, 13911–13919. 10.1093/nar/gku1116. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Zhu C.; Yi C. Switching Demethylation Activities between AlkB Family RNA/DNA Demethylases through Exchange of Active-Site Residues. Angew. Chem., Int. Ed. 2014, 53, 3659–3662. 10.1002/anie.201310050. [DOI] [PubMed] [Google Scholar]; h Siwek W.; Czapinska H.; Bochtler M.; Bujnicki J. M.; Skowronek K. Crystal structure and mechanism of action of the N6-methyladenine-dependent type IIM restriction endonuclease R.DpnI. Nucleic Acids Res. 2012, 40, 7563–7572. 10.1093/nar/gks428. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Yu B.; Hunt J. F. Enzymological and structural studies of the mechanism of promiscuous substrate recognition by the oxidative DNA repair enzyme AlkB. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14315. 10.1073/pnas.0812938106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rechkoblit O.; Delaney J. C.; Essigmann J. M.; Patel D. J. Implications for Damage Recognition during Dpo4-Mediated Mutagenic Bypass of m1G and m3C Lesions. Structure 2011, 19, 821–832. 10.1016/j.str.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hagelskamp F.; Borland K.; Ramos J.; Hendrick A. G.; Fu D.; Kellner S. Broadly applicable oligonucleotide mass spectrometry for the analysis of RNA writers and erasers in vitro. Nucleic Acids Res. 2020, 48, e41 10.1093/nar/gkaa091. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hou Y.; Sun J.; Wu B.; Gao Y.; Nie H.; Nie Z.; Quan S.; Wang Y.; Cao X.; Li S. CPSF30-L-mediated recognition of mRNA m6A modification controls alternative polyadenylation of nitrate signaling-related gene transcripts in Arabidopsis. Mol. Plant 2021, 14, 688–699. 10.1016/j.molp.2021.01.013. [DOI] [PubMed] [Google Scholar]
- a Liaqat A.; Sednev M. V.; Stiller C.; Höbartner C. RNA-Cleaving Deoxyribozymes Differentiate Methylated Cytidine Isomers in RNA. Angew. Chem., Int. Ed. 2021, 60, 19058–19062. 10.1002/anie.202106517. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liaqat A.; Stiller C.; Michel M.; Sednev M. V.; Höbartner C. N6-Isopentenyladenosine in RNA Determines the Cleavage Site of Endonuclease Deoxyribozymes. Angew. Chem., Int. Ed. 2020, 59, 18627–18631. 10.1002/anie.202006218. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sednev M. V.; Mykhailiuk V.; Choudhury P.; Halang J.; Sloan K. E.; Bohnsack M. T.; Höbartner C. N6-Methyladenosine-Sensitive RNA-Cleaving Deoxyribozymes. Angew. Chem., Int. Ed. 2018, 57, 15117–15121. 10.1002/anie.201808745. [DOI] [PubMed] [Google Scholar]; d Wachowius F.; Höbartner C. Probing Essential Nucleobase Functional Groups in Aptamers and Deoxyribozymes by Nucleotide Analogue Interference Mapping of DNA. J. Am. Chem. Soc. 2011, 133, 14888–14891. 10.1021/ja205894w. [DOI] [PubMed] [Google Scholar]
- a Kretschmer J.; Rao H.; Hackert P.; Sloan K. E.; Höbartner C.; Bohnsack M. T. The m6A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5′–3′ exoribonuclease XRN1. RNA 2018, 24, 1339–1350. 10.1261/rna.064238.117. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kellner S.; Seidu-Larry S.; Burhenne J.; Motorin Y.; Helm M. A multifunctional bioconjugate module for versatile photoaffinity labeling and click chemistry of RNA. Nucleic Acids Res. 2011, 39, 7348–7360. 10.1093/nar/gkr449. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang S.; Li Y.; Gan Y.; Zhou H.; Wang R. Labeling and quantitative analysis of i6A-incorporated RNA via In-situ azidation of prenyl functionality and click reaction. Tetrahedron Lett. 2022, 153873 10.1016/j.tetlet.2022.153873. [DOI] [Google Scholar]
- Dominissini D.; Nachtergaele S.; Moshitch-Moshkovitz S.; Peer E.; Kol N.; Ben-Haim M. S.; Dai Q.; Di Segni A.; Salmon-Divon M.; Clark W. C.; Zheng G.; Pan T.; Solomon O.; Eyal E.; Hershkovitz V.; Han D.; Doré L. C.; Amariglio N.; Rechavi G.; He C. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. 10.1038/nature16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheitl C. P. M.; Ghaem Maghami M.; Lenz A.-K.; Höbartner C. Site-specific RNA methylation by a methyltransferase ribozyme. Nature 2020, 587, 663–667. 10.1038/s41586-020-2854-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruthers M. H. The Chemical Synthesis of DNA/RNA - Our Gift to Science. J. Biol. Chem. 2013, 288, 1420–1427. 10.1074/jbc.X112.442855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamme M.; McKenzie L. K.; Sarac I.; Hollenstein M. Chemical methods for the modification of RNA. Methods 2019, 161, 64–82. 10.1016/j.ymeth.2019.03.018. [DOI] [PubMed] [Google Scholar]
- Kruse S.; Zhong S.; Bodi Z.; Button J.; Alcocer M. J. C.; Hayes C. J.; Fray R. A novel synthesis and detection method for cap-associated adenosine modifications in mouse mRNA. Sci. Rep. 2011, 1, 126. 10.1038/srep00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aritomo K.; Wada T.; Sekine M. Alkylation of 6-N-acylated adenosine derivatives by the use of phase transfer catalysis. J. Chem. Soc., Perkin Trans. 1 1995, 14, 1837–1844. 10.1039/P19950001837. [DOI] [Google Scholar]
- a Michel B. Y.; Dziuba D.; Benhida R.; Demchenko A. P.; Burger A. Probing of Nucleic Acid Structures, Dynamics, and Interactions With Environment-Sensitive Fluorescent Labels. Front. Chem. 2020, 8, 112. 10.3389/fchem.2020.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McKenzie L. K.; El-Khoury R.; Thorpe J. D.; Damha M. J.; Hollenstein M. Recent progress in non-native nucleic acid modifications. Chem. Soc. Rev. 2021, 50, 5126–5164. 10.1039/D0CS01430C. [DOI] [PubMed] [Google Scholar]; c Craig K.; Abrams M.; Amiji M. Recent preclinical and clinical advances in oligonucleotide conjugates. Expert Opin. Drug Delivery 2018, 15, 629–640. 10.1080/17425247.2018.1473375. [DOI] [PubMed] [Google Scholar]
- Gajda T.; Zwierzak A. Phase-Transfer-Catalysed N-Alkylation of Carboxamides and Sulfonamides. Synthesis 1981, 12, 1005–1008. 10.1055/s-1981-29683. [DOI] [Google Scholar]
- Bödeker J.; Courault K.; Köckritz A.; Köckritz P. Reaktionen von Heteroaryliminotriphenylphosphoranen mit Heterokumulenen: Synthese und Cycloadditionen α-N-Heteroaryl-substituierter Carbodiimide. J. Prakt. Chem. 1983, 325, 463–474. 10.1002/prac.19833250316. [DOI] [Google Scholar]; (acccessed 2021/08/17).
- Schweizer M. P.; Chheda G. B.; Baczynskyj L.; Hall R. H. Aminoacyl nucleosides. VII. N-(purin-6-ylcarbamoyl)threonine. A new component of transfer ribonucleic acid. Biochemistry 1969, 8, 3283–3289. 10.1021/bi00836a023. [DOI] [PubMed] [Google Scholar]
- Nainytė M.; Müller F.; Ganazzoli G.; Chan C.-Y.; Crisp A.; Globisch D.; Carell T. Amino Acid Modified RNA Bases as Building Blocks of an Early Earth RNA-Peptide World. Chem. – Eur. J. 2020, 26, 14856–14860. 10.1002/chem.202002929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S.; Miyauchi K.; Ikeuchi Y.; Thiaville P. C.; de Crécy-Lagard V.; Suzuki T. Discovery of the β-barrel–type RNA methyltransferase responsible for N6-methylation of N6-threonylcarbamoyladenosine in tRNAs. Nucleic Acids Res. 2014, 42, 9350–9365. 10.1093/nar/gku618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine M. General method for the preparation of N3- and O4-substituted uridine derivatives by phase-transfer reactions. J. Org. Chem. 1989, 54, 2321–2326. 10.1021/jo00271a015. [DOI] [Google Scholar]
- Leiter J.; Reichert D.; Rentmeister A.; Micura R. Practical Synthesis of Cap-4 RNA. ChemBioChem 2020, 21, 265–271. 10.1002/cbic.201900590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nainytė M.; Amatov T.; Carell T. Synthesis of an acp3U phosphoramidite and incorporation of the hypermodified base into RNA. Chem. Commun. 2019, 55, 12216–12218. 10.1039/C9CC06314E. [DOI] [PubMed] [Google Scholar]
- Chakrapani A.; Vaňková Hausnerová V.; Ruiz-Larrabeiti O.; Pohl R.; Krásný L.; Hocek M. Photocaged 5-(Hydroxymethyl)pyrimidine Nucleoside Phosphoramidites for Specific Photoactivatable Epigenetic Labeling of DNA. Org. Lett. 2020, 22, 9081–9085. 10.1021/acs.orglett.0c03462. [DOI] [PubMed] [Google Scholar]
- a Verdolino V.; Cammi R.; Munk B. H.; Schlegel H. B. Calculation of pKa Values of Nucleobases and the Guanine Oxidation Products Guanidinohydantoin and Spiroiminodihydantoin using Density Functional Theory and a Polarizable Continuum Model. J. Phys. Chem. B 2008, 112, 16860–16873. 10.1021/jp8068877. [DOI] [PubMed] [Google Scholar]; b Bordwell F. G.; Algrim D. Nitrogen acids. 1. Carboxamides and sulfonamides. J. Org. Chem. 1976, 41, 2507–2508. 10.1021/jo00876a042. [DOI] [Google Scholar]
- a Mao S.; Sekula B.; Ruszkowski M.; Ranganathan S. V.; Haruehanroengra P.; Wu Y.; Shen F.; Sheng J. Base pairing, structural and functional insights into N4-methylcytidine (m4C) and N4,N4-dimethylcytidine (m42C) modified RNA. Nucleic Acids Res. 2020, 48, 10087–10100. 10.1093/nar/gkaa737. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lu J.; Li N.-S.; Koo S. C.; Piccirilli J. A. Efficient Synthesis of N4-Methyl- and N4-Hydroxycytidine Phosphoramidites. Synthesis 2010, 2010, 2708–2712. 10.1055/s-0030-1258170. [DOI] [Google Scholar]
- Miyata K.; Kobori A.; Tamamushi R.; Ohkubo A.; Taguchi H.; Seio K.; Sekine M. Conformational Studies of 4-N-Carbamoyldeoxycytidine Derivatives and Synthesis and Hybridization Properties of Oligodeoxyribonucleotides Incorporating these Modified Bases. Eur. J. Org. Chem. 2006, 2006, 3626–3637. 10.1002/ejoc.200501006. [DOI] [Google Scholar]
- Höbartner C.; Kreutz C.; Flecker E.; Ottenschläger E.; Pils W.; Grubmayr K.; Micura R. The Synthesis of 2′-O-[(Triisopropylsilyl)oxy] methyl (TOM) Phosphoramidites of Methylated Ribonucleosides (m1G, m2G, m22G, m1I, m3U, m4C, m6A, m62A) for Use in Automated RNA Solid-Phase Synthesis. Monatsh. Chem. 2003, 134, 851–873. 10.1007/s00706-003-0592-1. [DOI] [Google Scholar]
- Tang Q.; Cai A.; Bian K.; Chen F.; Delaney J. C.; Adusumalli S.; Bach A. C.; Akhlaghi F.; Cho B. P.; Li D. Characterization of Byproducts from Chemical Syntheses of Oligonucleotides Containing 1-Methyladenine and 3-Methylcytosine. ACS Omega 2017, 2, 8205–8212. 10.1021/acsomega.7b01482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov S. N.; Rozenski J.; Efimtseva E. V.; Busson R.; Van Aerschot A.; Herdewijn P. Chemical incorporation of 1-methyladenosine into oligonucleotides. Nucleic Acids Res. 2002, 30, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]; PMC
- Mao S.; Haruehanroengra P.; Ranganathan S. V.; Shen F.; Begley T. J.; Sheng J. Base Pairing and Functional Insights into N3-Methylcytidine (m3C) in RNA. ACS Chem. Biol. 2021, 16, 76–85. 10.1021/acschembio.0c00735. [DOI] [PubMed] [Google Scholar]
- Mathivanan J.; Du J.; Mao S.; Zheng Y. Y.; Sheng J. Synthesis and Purification of N3-Methylcytidine (m3C) Modified RNA Oligonucleotides. Curr. Protoc. 2021, 1, e307 10.1002/cpz1.307. [DOI] [PubMed] [Google Scholar]
- Moreno S.; Flemmich L.; Micura R. Synthesis of N4-acetylated 3-methylcytidine phosphoramidites for RNA solid-phase synthesis. Monatsh. Chem. 2022, 153, 285–291. 10.1007/s00706-022-02896-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy M. P.; Hanna N. B.; Farooqui F. Fast cleavage and deprotection of oligonucleotides. Tetrahedron Lett. 1994, 35, 4311–4314. 10.1016/S0040-4039(00)73341-7. [DOI] [Google Scholar]
- Griffey R. H.; Monia B. P.; Cummins L. L.; Freier S.; Greig M. J.; Guinosso C. J.; Lesnik E.; Manalili S. M.; Mohan V.; Owens S.; Ross B. R.; Sasmor H.; Wancewicz E.; Weiler K.; Wheeler P. D.; Cook P. D. 2‘-O-Aminopropyl Ribonucleotides: A Zwitterionic Modification That Enhances the Exonuclease Resistance and Biological Activity of Antisense Oligonucleotides. J. Med. Chem. 1996, 39, 5100–5109. 10.1021/jm950937o. [DOI] [PubMed] [Google Scholar]
- Takahashi M.; Grajkowski A.; Cawrse B. M.; Beaucage S. L. Innovative 2′-O-Imino-2-propanoate-Protecting Group for Effective Solid-Phase Synthesis and 2′-O-Deprotection of RNA Sequences. J. Org. Chem. 2021, 86, 4944–4956. 10.1021/acs.joc.0c02773. [DOI] [PubMed] [Google Scholar]
- a Werner M.; Purta E.; Kaminska K. H.; Cymerman I. A.; Campbell D. A.; Mittra B.; Zamudio J. R.; Sturm N. R.; Jaworski J.; Bujnicki J. M. 2′-O-ribose methylation of cap2 in human: function and evolution in a horizontally mobile family. Nucleic Acids Res. 2011, 39, 4756–4768. 10.1093/nar/gkr038. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lewdorowicz M.; Yoffe Y.; Zuberek J.; Jemielity J.; Stepinski J.; Kierzek R.; Stolarski R.; Shapira M.; Darzynkiewicz E. Chemical synthesis and binding activity of the trypanosomatid cap-4 structure. RNA 2004, 10, 1469–1478. 10.1261/rna.7510504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska J.; Lewdorowicz M.; Zuberek J.; Grudzien-Nogalska E.; Bojarska E.; Stepinski J.; Rhoads R. E.; Darzynkiewicz E.; Davis R. E.; Jemielity J. Synthesis and characterization of mRNA cap analogs containing phosphorothioate substitutions that bind tightly to eIF4E and are resistant to the decapping pyrophosphatase DcpS. RNA 2008, 14, 1119–1131. 10.1261/rna.990208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.