

Graphical abstract
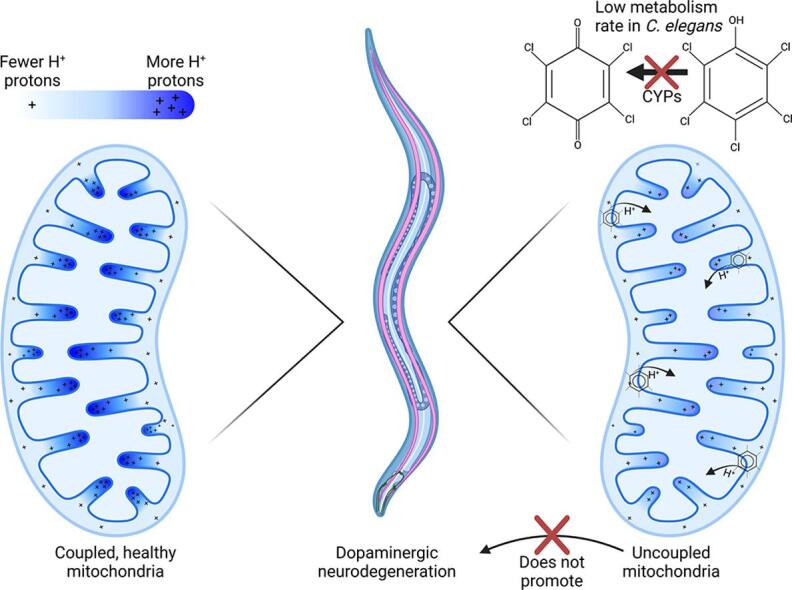
Keywords: Mitochondrial uncoupling, Dopaminergic neurodegeneration, Adverse outcome pathway, Redox tone, Caenorhabditis elegans
Highlights
-
•
Pentachlorophenol, not its redox-cycling derivatives, drive developmental delay.
-
•
Pentachlorophenol reduced steady-state ATP and led to a more-reduced cellular state.
-
•
Mitochondrial uncoupling did not cause dopaminergic neurodegeneration.
Abstract
Aims
Mitochondrial dysfunction is implicated in several diseases, including neurological disorders such as Parkinson’s disease. However, there is uncertainty about which of the many mechanisms by which mitochondrial function can be disrupted may lead to neurodegeneration. Pentachlorophenol (PCP) is an organic pollutant reported to cause mitochondrial dysfunction including oxidative stress and mitochondrial uncoupling. We investigated the effects of PCP exposure in Caenorhabditis elegans, including effects on mitochondria and dopaminergic neurons. We hypothesized that mild mitochondrial uncoupling by PCP would impair bioenergetics while decreasing oxidative stress, and therefore would not cause dopaminergic neurodegeneration.
Results
A 48-hour developmental exposure to PCP causing mild growth delay (∼10 % decrease in growth during 48 h, covering all larval stages) reduced whole-organism ATP content > 50 %, and spare respiratory capacity ∼ 30 %. Proton leak was also markedly increased. These findings suggest a main toxic mechanism of mitochondrial uncoupling rather than oxidative stress, which was further supported by a concomitant shift toward a more reduced cellular redox state measured at the whole organism level. However, exposure to PCP did not cause dopaminergic neurodegeneration, nor did it sensitize animals to a neurotoxic challenge with 6-hydroxydopamine. Whole-organism uptake and PCP metabolism measurements revealed low overall uptake of PCP in our experimental conditions (50 μM PCP in the liquid exposure medium resulted in organismal concentrations of < 0.25 μM), and no measurable production of the oxidative metabolites tetra-1,4-benzoquinone and tetrachloro-p-hydroquinone.
Innovation
This study provides new insights into the mechanistic interplay between mitochondrial uncoupling, oxidative stress, and neurodegeneration in C. elegans. These findings support the premise of mild uncoupling-mediated neuroprotection, but are inconsistent with proposed broad “mitochondrial dysfunction”-mediated neurodegeneration models, and highlight the utility of the C. elegans model for studying mitochondrial and neurotoxicity.
Conclusions
Developmental exposure to pentachlorophenol causes gross toxicological effects (growth delay and arrest) at high levels. At a lower level of exposure, still causing mild growth delay, we observed mitochondrial dysfunction including uncoupling and decreased ATP levels. However, this was associated with a more-reduced cellular redox tone and did not exacerbate dopaminergic neurotoxicity of 6-hydroxydopamine, instead trending toward protection. These findings may be informative of efforts to define nuanced mitochondrial dysfunction-related adverse outcome pathways that will differ depending on the form of initial mitochondrial toxicity.
Introduction
Mitochondrial dysfunction is associated with numerous human pathologies, including myopathies, cancer, and neurodegenerative diseases including Parkinson’s disease. Parkinson’s disease is characterized by a progressive loss of dopaminergic neurons, and has both familial (∼15 %, usually early-onset) and sporadic (∼85 %, usually late-onset) cases (Samii et al. 2004). Many familial Parkinson’s disease genes are involved in mitochondrial quality control, and environmental contributors are largely mitochondrial toxicants including pesticides such as rotenone and paraquat (Borsche et al. 2021). However, mitochondrial toxicity from different chemicals can take distinct forms, such as alterations to oxidative phosphorylation, cellular redox state, mitochondrial membrane potential, ATP levels, DNA homeostasis, lipid metabolism, and the tricarboxylic acid cycle (Dreier et al., 2019, Hallinger et al., 2020, Lehman-McKeeman, 2019, Meyer et al., 2018, Yang et al., 2021). Despite a clear link between mitochondrial dysfunction and Parkinson’s disease, it is unclear which particular mechanism(s) of mitochondrial dysfunction specifically drive cell death in dopaminergic neurons (Delp et al. 2021). This mechanistic detail is important to understand, because screening efforts demonstrate that a large number of chemicals appear to cause some form of mitochondrial dysfunction (Attene-Ramos et al., 2013, Datta et al., 2016, Will and Dykens, 2014, Wills, 2017). Therefore, understanding which specific mitochondrial mechanisms may lead to dopaminergic degeneration and Parkinson’s disease will allow more focused investigation of chemical contributors. Some proposed adverse outcome pathways (AOPs; conceptual tools describing sequential cause-effect relationships between an initial toxicant-cellular target interaction and ultimate disease outcomes) (Edwards et al. 2016) link specific mechanisms of mitochondrial toxicity, such as Complex I inhibition (Terron et al. 2018), to dopaminergic neurodegeneration. Others instead propose that the broad category of “mitochondrial dysfunction” leads to dopaminergic neurodegeneration (Cao et al. 2018). We have argued that it is unlikely that all forms of mitochondrial dysfunction have the same effect on dopaminergic neurodegeneration (Dreier et al. 2019), because the downstream outcomes of different forms of mitochondrial dysfunction vary. In particular, as described below, there is theoretical reason to hypothesize that mild mitochondrial uncoupling may be protective in some cases. However, empirical evidence for chemical uncoupling-mediated protection is limited, and this idea is not universally accepted.
2,3,4,5,6-Pentacholophenol (PCP) is a chlorine-substituted phenol that was introduced in the 1930′s as an industrial molluscicide, herbicide, and disinfectant (IUPAC, 1981, ATSDR., 2022). Prior to limitations that were imposed in the mid 1980′s in the United States and many other parts of the world (it is included in the Stockholm Convention), PCP was one of the most widely used wood preservation chemicals with as much as 50,000 metric tons being produced in 1981 (IUPAC, 1981, ATSDR., 2022). Entry of PCP to the environment has occurred through industrial waste disposal and dissipation from treated materials, causing contamination of water, air, and soil. PCP use has decreased in the United States and many other parts of the world, and PCP is susceptible to degradation from UV light and slow biotic metabolism in organisms and in the environment. However, its use remains significant in some parts of the world (Huo et al., 2022, World Health Organization, 1987), it is classified as a persistent organic pollutant indicating a high likelihood for persistence, and indeed urinary PCP is still regularly detected in the United States (Morgan et al., 2015, Morgan, 2015, National Biomonitoring Program, 2017) and German (Schmied-Tobies et al. 2021) populations. Thus, PCP remains a concern in the environment even in countries where its use has been significantly restricted. Further underscoring this is its continued inclusion in the United States’ Agency for Toxic Substances Disease Registry Substance Priority List that ranks pollutant importance based on environmental occurrence and potential health threats. PCP is #54 in the most recent, 2019 List (ATSDR, 2017). Reported non-acute effects include chloracne in workers who were occupationally exposed (Hryhorczuk et al. 1998), cancers (Cui et al. 2017), and gestational diabetes (Huo et al. 2022); additional health effects have been reviewed by the United States Environmental Protection Agency (U.S. EPA., 2010).
Perhaps more importantly, PCP is also structurally and functionally related to many other environmental pollutants, and thus its toxicological mechanisms may be translatable to other similarly structured chemicals. Chemical mitochondrial uncouplers uncouple proton pumping from ATP production, typically by dissipating the proton gradient in an unproductive fashion. This dissipation is often accomplished by shuttling protons downgradient across the inner mitochondria membrane, into the matrix. Chemicals that uncouple share the ability to gain and lose a proton, with pKa values that allow them to accept a proton in the intermembrane space and release it into the matrix, and are lipophilic enough (even when ionized, often via charge delocalization across aromatic rings) to diffuse across the inner membrane (Song and Villeneuve 2021). In addition to these general chemical characteristics, researchers have had some success in identifying toxicophores that identify uncouplers, informing in silico screening (Enoch et al., 2018, Naven et al., 2013); reviewed in (Song and Villeneuve 2021). The general (chemical-agnostic) action of chemical uncoupling has also been proposed as an initiating event in an AOP resulting in growth inhibition (Song and Villeneuve 2021). Development of AOPs for uncouplers will be useful; follow-up of screening efforts for mitochondrial toxicants are identifying a substantial number of those mitotoxicants as uncoupling agents (Hallinger et al., 2020, Wills et al., 2015, Xia et al., 2018).
There are two main mechanisms by which PCP exerts its toxicity. First is the direct effect of dissipation of the proton gradient of the mitochondrial membrane (Demine et al., 2019, Weinbach, 1957). ATP synthesis is driven by the electrochemical potential of this proton gradient; therefore, a leaky membrane wastes energy used to initially drive the protons across the membrane and limits the efficiency of ATP production. In extreme cases, ATP synthase may even work in reverse, consuming ATP to pump protons out of the matrix and maintain membrane potential. Sustained and/or high-concentration exposure to PCP can lead to reactive oxygen species (ROS) production and cell death (Xia et al. 2018). However, there is evidence that mild uncoupling can lead to decreases in mitochondrial membrane potential and the overall state of reduction of the electron transport chain, reducing mitochondrial production of ROS (Brand et al., 2004, Miwa and Brand, 2003), and thus be protective or beneficial under some circumstances. However, the idea of protection from mild uncoupling has been best supported in the context of endogenous uncoupling proteins, and is not uncontroversial (Shabalina and Nedergaard 2011). To our knowledge, potential protective effects have not been tested with PCP.
The second mechanism in PCP toxicity is redox cycling, resulting in the generation of ROS, by downstream metabolites of PCP (ATSDR 2022). Metabolism of PCP via cytochrome P450 enzymes produces tetrachloro-p-hydroquinone (TCHQ) and tetrachloro-1,4-benzoquinone (TCBQ) compounds through a series of quinone intermediates (Tsai et al. 2001). TCBQ exposure caused glutathione depletion (Fraser et al. 2019) and oxidative DNA damage (Dahlhaus et al. 1996) in cell culture, and glutathione depletion and induction of antioxidant genes in mouse liver (Xu et al. 2014a). Prolonged exposures to ROS can cause neurodegeneration, aging, carcinogenesis, and many other harmful effects (Di Meo et al. 2016).
In this study, we examined toxicity of pentachlorophenol and its metabolite TCBQ using the model organism Caenorhabditis elegans. We hypothesized that mild mitochondrial uncoupling by PCP would impair bioenergetics while decreasing ROS, and therefore would not cause dopaminergic neurodegeneration. To test this hypothesis, we first exposed developing C. elegans larvae to varying concentrations of PCP in order to identify the lowest observed adverse effect level (LOAEL) for growth. Then, using that concentration, we tested the ability of PCP to cause mitochondrial dysfunction and dopaminergic neurodegeneration. Finally, we tested the ability of the worms to generate the major metabolite of PCP, TCBQ. Together, our data provides evidence that mild mitochondrial uncoupling caused by PCP does not drive dopaminergic neurodegeneration.
Materials and Methods
Strains
The strains used in this study were N2 (Bristol wild-type strain) and JV2 (jrIs2 [rpl-17p::Grx1-roGFP2]), which were obtained from the Caenorhabditis Genetics Center, and BY200 (vtIs1[dat-1p::GFP]), which was a generous gift from Michael Aschner (Albert Einstein College of Medicine, Bronx, NY). BY200 and JV2 strains were back-crossed into the N2 wild-type strain at least 4 times.
C. elegans culture and PCP/TCBQ exposures
Populations of C. elegans were maintained on K-agar plates seeded with OP50 E. coli at 20 °C. Synchronized populations of nematodes were obtained by hypochlorite/NaOH treatment of gravid adults in order to isolate the eggs (Lewis and Fleming 1995). Embryos were allowed to hatch for between 14 and 16 h in K + medium, previously referred to as “complete K-medium” (Boyd et al. 2009). L1 larvae were then counted and transferred into exposure wells containing food (E. coli) and the chemical exposure. During exposure, a UV-sensitive strain of E. coli, UVRA, was treated with UV radiation before use as the food source in order to limit bacterial metabolism of the chemicals of interest (Meyer et al. 2010). After a 48 h exposure, the worms reached the L4 stage, and the various measurements were made.
Growth assays
Following a 48 h exposure of N2 wild-type worms to PCP, the size of animals in each dosing group was determined using the Copas Biosorter instrument, as previously described (Maurer et al. 2015). The Copas Biosorter measures worm size using time of flight (TOF), which correlates with the length of the worm. TOF data were plotted as a function of PCP concentration in order to determine the LOAEL.
ATP concentrations
ATP levels were measured using the CellTiter-Glo Luminescent Cell Viability Assay (Promega G7572), and normalized to protein content determined by the Pierce bicinchoninic acid assay (Thermo Scientific, Rockford, IL) (Palikaras and Tavernarakis 2016). Briefly, worms were collected and frozen down immediately at −80 °C. At the time of analysis, worm samples were boiled and spun down to remove debris. Aliquots of the worm extractions were used for ATP quantification and total protein determination. ATP content was normalized to total protein.
Seahorse respiration assays
Following a 48 h exposure to PCP at the minimally-growth-inhibiting concentration of 50 µM, worms were loaded into a Seahorse XFe24 microplate at a density of 75 worms/well, as described in our published protocol (Luz et al. 2015). The worms were then subjected to a modified version of the “mitochondrial stress test”. Briefly, basal OCR measurements are taken before injection of either 25 µM (final) carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, a mitochondrial uncoupler) to measure maximal respiration, or 20 µM N,N-dicyclohexylcarbodiimide (DCCD, an ATP synthase inhibitor), to determine ATP-linked respiration. After injection of either FCCP or DCCD, 14 measurements were taken before a final injection of 10 mM sodium azide to completely inhibit mitochondrial respiration and determine the non-mitochondrial OCR. Final parameters calculated include Basal Mitochondrial OCR (Basal OCR – non-mitochondrial OCR), Spare Capacity (also referred to as Reserve Capacity, Maximal OCR – Basal OCR), ATP-linked Respiration (Basal OCR – DCCD-inhibited OCR), and Proton Leak (DCCD-inhibited OCR – non-mitochondrial OCR). Seahorse experiments included 5 wells per treatment group and at least 3 experimental replicates.
Measurement of redox status with roGFP2-Grx1 sensor
The JV2 strain expressing cytosolic roGFP2-Grx1 in all somatic cells was used to estimate redox status. This sensor changes its structure and fluorescence properties according to the formation or absence of disulfide bridges, and is a proxy for reduced/oxidized glutathione in the cell. JV2 worms were exposed to PCP for 48 h, and then loaded into a 96-well black plate for fluorescence readings. Each well (n = 8 wells for both the control and exposed groups) contained 100 worms in 200 µL of K-medium. Eight additional wells were loaded with unexposed controls and treated with 1 % H2O2 as a positive control. The plate was incubated at room temperature for 15 min before loading into the plate reader (BMG Labtech FluoSTAR Omega, Cary, NC). Fluorescence values were measured at excitation wavelengths of 400 and 490 nm with a common emission wavelength of 510 nm. Unitless ratios were calculated as fluorescence at 400 nm excitation divided by fluorescence at 490 nm excitation. Increases in this ratio reflect a relative increase in oxidation (Albrecht et al. 2011b).
Dopaminergic neurodegeneration measurements
After exposure to PCP or the vehicle control for 48 h, worms were rinsed three times with K-medium and then divided into three groups. One group (K + control) was immediately placed on K-agar plates seeded with OP50 bacteria without further treatment. The second group (AA control) was exposed to 5 mM ascorbic acid, the vehicle control for 6-hydroxydopamine, for one hour. Ascorbic acid protects 6-hydroxydopamine (6-OHDA) from oxidative degradation during the duration of the exposure. The final group (6-OHDA) was exposed to 25 mM 6-OHDA in 5 mM ascorbic acid for one hour. After the acute one-hour exposure, AA- and 6-OHDA-exposed worms were rinsed three times with K-medium and placed on K-agar plates seeded with OP50 bacteria.
After 24 h of recovery in a 20 °C incubator, worms were picked to a 2 % agarose pad on glass slides and immobilized in 10 mM sodium azide. The GFP-labeled dopaminergic neurons were then imaged using a Keyence BZ-X700 All-in-One Fluorescent Microscope. Z-stacks were obtained in the EGFP channel with a 1 µm step size (30–40 images per stack) at 400x magnification, focusing on the four cephalic neurons in the head of the worm. Z-stacks were then cropped to a single worm and converted to a maximum Z-projection in ImageJ. Neuron damage was scored visually as described (Bijwadia et al. 2021), blinded to treatment, using the freely available scoring software we recently developed, Blinder (Cothren et al. 2018).
Uptake and metabolism quantifications
For uptake measurements, PCP exposures were carried out for 48 h as in other measurements. For each individual replicate, 600 worms were exposed to 50 µM PCP for 48 h. Following exposure, the liquid exposure was transferred to 15 mL centrifuge tubes and the worms were pelleted at 600×g. The supernatant was removed and saved for analysis. The worms were washed a total of 3 times as quickly as possible by adding 10 mL K-medium and centrifuging at 600×g. Following the last spin, the liquid was removed and the worm pellet was flash-frozen in liquid nitrogen. Frozen worm pellets were then allowed to warm to ambient lab temperature and spiked with internal standard (13C6-PCP, 200 µL of 2 µg/mL solution, Cambridge Isotope Laboratories, Cambridge, MA). Pellets were extracted in hexane (400 mL) via sonication for 15 min, two times, and combined. Dosing medium (0.9 mL and 0.1 mL of concentrated sulfuric acid) was spiked with internal standard (13C6-PCP, 200 µL of 2 µg/mL solution) extracted via liquid/liquid extraction with hexane (400 µL, two times). Extracts were analyzed by GCMS using an Agilent 7890A gas chromatography system, 5975 mass spectrometer, and 7693 auto sampler. Separation was completed using a 1 µL pulsed splitless injection at 200 °C at 11.45 psi on an HP5-MS 30 m × 250 µm × 0.25 µm at a 1.3 mL/min ultra pure helium flow rate. Oven temperature was held at 60 °C held for 1 min then ramped to 140 °C at 10 °C/min then ramped to 230 °C at 5 °C/min then ramped to 260 °C at 10 °C/min and held for 5 min. SIM parameters were as follows: 7 – 35 min: m/z 87.00, 147,00, 165.00, 166.00 172.00 247.00, 248.00, 265.00, 266.00 and 271.00.
Statistical methods
For growth experiments with multiple concentrations of PCP or TCBQ, a one-way ANOVA was used with Bonferroni-corrected multiple comparisons to determine significant differences in size at each concentration compared to the vehicle control. For analysis of growth inhibition across exposure concentrations and across nematode strains, we used a 2-way ANOVA. The Seahorse analysis was performed using a Student’s unpaired t-test. Pairwise analysis of the neurodegeneration data was performed with a Chi-squared analysis and Bonferroni correction for multiple comparisons. Error bars in all graphs represent standard error of the mean.
Results
PCP disrupts mitochondrial function
To be able to determine the effects of PCP on mitochondria and neurodegeneration in C. elegans without confounding effects of major growth delay but at concentrations at which at least mild organismal outcomes were observed, we first measured larval growth in the presence of varying exposure concentrations of PCP (Fig. 1A). We observed a concentration-dependent decrease in growth, with the highest concentrations causing growth arrest at the L1 stage (reflected in the plateau at ∼ 150 TOF). With this growth curve, we identified the lowest concentration causing a growth delay (LOAEL) as 45 µM. Based on this result, we chose 50 µM exposure concentrations for additional experiments. Because this concentration had an effect on growth, we can be confident that the negative results we observed (e.g., neurodegeneration) cannot be attributed to such limited uptake that no biological effect was observed. At the same time, because the growth effect was small, we can be confident that growth differences were not enough to confound measurements of mitochondrial function. We detected no difference in sensitivity to PCP of the different strains used in this study (Fig. 1B).
Fig. 1.

PCP causes growth delay at high concentrations, and decreases ATP and cellular oxidation at lower concentrations. Panel A, representative growth curve for one biological replicate with increasing concentrations of PCP. For experiments, L1-stage larvae were incubated with PCP for 48 h in liquid culture in 96-well plates. Following exposure, worms were loaded into a Copas Biosorter and Time of Flight (TOF) was measured as an indicator of worm length. Asterisks represent p < 0.0001 in one-way ANOVA with Bonferroni-corrected post-test compared to control. Panel B, compiled growth curve for three biological replicates in wild-type (N2) and two biological replicates in the other strains used in this study, BY200 and JV2. A 2-way ANOVA analysis showed a significant effect of exposure concentration (p < 0.0001), but not strain or interaction (p = 0.67 and 0.96, respectively). The 50 µM PCP dose used in all subsequent experiments is highlighted in green. Panel C, uptake data for PCP with wild-type (N2) worms. For experiments, incubations were prepared of UV-inactivated E. coli ± 50 µM PCP (first two bars, no worms present), and E. coli + worms ± 50 µM PCP. For incubations with worms, PCP was measured in the supernatant (External [PCP]) and in the lysed worm pellet after 3 brief washes with K-medium (Internal [PCP]). Panel D, ATP content in control and PCP-exposed worms. ATP content was determined using a luminescence-based ATP kit (see Methods) in wild-type (N2) worms after a 48 h exposure to 50 µM PCP. Asterisks represent p < 0.001, student’s unpaired t-test, 9 biological replicates. Panel E, normalized ratio of Grx1-roGFP2 fluorescence (JV2 strain) with excitation wavelengths of 400 nm / 470 nm after a 48 h exposure to 50 µM PCP or a 30 min exposure to 3 % H2O2 (positive control). Ratios were normalized to the vehicle control group. Shown are compiled data from > 3 biological replicates. Asterisks represent *, p < 0.05, **, p < 0.01, one-way ANOVA with Bonferroni-corrected post testing for significance compared to the vehicle control.
We next performed uptake experiments using LC/MS to determine the internal concentration of PCP, and observed that<1 % of PCP was taken up by the worms (Fig. 1C). After the 48 h incubation with 50 µM PCP, we measured ∼ 10 µM PCP in the supernatant. We detected an unidentified molecule with structural resemblance to PCP in the control (no PCP) groups that contained worms that was absent in the controls with no worms, suggesting it was produced by worms. The signal (unknown molecule + PCP) was present at a similar level in control and PCP-exposed worms. Additionally, in two of four replicates of internal concentration, levels were below the limit of detection (0.018 µM, shown as dashed line in Fig. 1C). Overall, these data suggest that uptake is very low and the relevant internal concentrations after an external exposure of 50 µM PCP are<0.25 µM. We note that our experimental design did not allow us to ascertain how much of the low uptake was the result of the toxicokinetic properties of C. elegans, versus extrinsic factors such as possible binding of the test compound to the plastic well surfaces.
We then measured whole-worm ATP content after 48 h PCP exposure (Fig. 1D). We observed a statistically significant ∼ 50 % decrease in ATP after PCP exposure, indicating that PCP was having a dramatic effect on energy metabolism in the worms. We also measured whole-worm redox status using the JV2 strain in which a cytosolic roGFP2-Grx1 sensor is expressed ubiquitously in somatic cells (Fig. 1E). This sensor permits in vivo estimation of the oxidized/reduced glutathione ratio via surface residues mutated and replaced with cysteine in positions that can form disulfide bonds (Back et al. 2012). The roGFP2-Grx1 fusion of glutaredoxin (Grx1) and roGFP2 (Albrecht et al. 2011a) allows the relative amount of GSSG and GSH to be determined by measuring the fluorescence at 400 nm and 490 nm respectively (Dooley et al. 2004). The ratio of fluorescence values for 400 nm/490 nm corresponds to the GSSG/GSH ratio inside the organism. We found significantly decreased oxidation (i.e., a more reduced state) in PCP-exposed worms, consistent with mitochondrial uncoupling resulting in decreased production of ROS.
To directly assess mitochondrial function, we then measured mitochondrial respiration in whole living worms after PCP exposure using the mitochondrial stress test (Fig. 2A) performed on a Seahorse XFe24 extracellular flux analyzer. Using the mitochondrial stress test, we observed no difference in basal OCR with PCP exposure (Fig. 2B). Interestingly, despite the significant decrease in ATP content (Fig. 1C), we observed no difference in ATP-linked OCR (Fig. 2C). However, we did observe a significant ∼ 30 % decrease in spare capacity, reflecting a decrease in maximal respiration (Fig. 2D). We also observed a substantial increase in proton leak (Fig. 2E). Together, these findings are consistent with mild mitochondrial uncoupling and indicate that at concentrations that cause only mildly delayed growth in the worms, PCP uncoupled mitochondria.
Fig. 2.

Exposure to PCP reduces spare respiratory capacity and increases proton leak in C. elegans. For experiments, L1-stage N2 (wild-type) worms were incubated with 50 µM PCP and UV-inactivated E. coli for 48 h. Following exposure, worms were loaded into a Seahorse XFe24 plate at a density of approximately 75 worms per well. Panel A, depiction of the concept of the mitochondrial stress test, indicating which portion of the measurements are used to calculate the parameters in panels B-E. Note that although injection of DCCD (ATP synthase inhibitor) and FCCP (uncoupler) are shown as sequential injections, for C. elegans experiments we run these inhibitors in separate wells as previously published (Luz et al. 2015). All measurements are normalized to the confirmed number of nematodes in each well (counted after the conclusion of the flux measurements). Panel B, Basal OCR represents the baseline OCR – non-mitochondrial respiration (azide-inhibited OCR). Panel C, ATP-linked OCR is calculated as the difference between baseline OCR and the DCCD-inhibited rate. Panel D, Spare capacity is calculated as the difference between maximal FCCP-induced OCR and the baseline OCR. Panel E, proton leak is calculated as the difference between DCCD-inhibited OCR and the non-mitochondrial azide-inhibited OCR. Data represent n = 5 wells per group per biological replicate and a total of three biological replicates (or six for basal OCR). *, p < 0.05, **, p < 0.01, student’s unpaired t-test.
PCP does not cause neurodegeneration or exacerbate 6OHDA-induced dopaminergic neurodegeneration
Dopaminergic neurons are highly energetic and rely heavily on mitochondria for ATP synthesis. This is one reason that dopaminergic neurons are thought to be particularly sensitive to mitochondrial toxicity (Surmeier 2018). Therefore, we wanted to test if PCP exposure, which we knew was decreasing ATP concentrations in the worms, would cause neurodegeneration on its own, or increase the dopaminergic neurodegeneration induced by 6-hydroxydopamine (6OHDA), a common Parkinson’s disease model utilized in our laboratory (Gonzalez-Hunt et al., 2014, Hartman et al., 2019). To observe neurodegeneration, we used the BY200 strain expressing GFP driven by the dat-1 (dopamine transporter) promoter (Fig. 3A) to visualize the four CEP neurons in the head of the worm. Each neuron is labeled by a bright cell body near the pharynx, and a single dendrite extending to the nose of the animal. Damage from neurotoxicants appears as bright spots, or “blebs” (Fig. 3B), and absence of continuous fluorescence, or “breaks” (Fig. 3C). In more severe damage, the entire dendrite or cell body may be missing. Following a 48 h incubation with UV-inactivated E. coli and vehicle or 50 µM PCP, we divided worms into three groups. One group we left in complete K-medium (Fig. 3D, “K+”), one group we exposed to the 6OHDA stabilizer ascorbic acid (“AA”), and the third group we exposed to 6OHDA dissolved in ascorbic acid (“6OHDA”). We observed no increase in neurodegeneration with PCP alone (Fig. 3D, no significant difference between K + groups in control and PCP-exposed worms). We also found that regardless of PCP exposure, 6OHDA caused neurodegeneration as reflected by an increase in the appearance of damage such as blebs and breaks and complete loss of dendrites. However, when compared to the control group, PCP-exposed worms were not more sensitive to 6OHDA. In fact, if anything, we saw the opposite: while not reaching the < 0.05 level of statistical significance, the data trended toward indicating protection (p = 0.073) from the damaging effects of 6OHDA.
Fig. 3.

PCP exposure does not cause neurodegeneration or exacerbate 6-hydroxydopamine-induced neurodegeneration. For experiments, 48-h exposures were carried out with 50 µM PCP and UV-inactivated E. coli. After exposure, worms were either plated immediately to K-agar plates to recover (K + group), or were immediately exposed to 25 mM 6-hydroxydopamine (6OHDA group), or its vehicle 5 mM ascorbic acid (AA group). The neurons were then visualized in the BY200 strain expressing GFP under the control of the dat-1 (dopamine transporter) promoter. We focused our analysis on the 4 cephalic neurons in the head, with bright cell bodies near the pharynx and dendrites extending to the nose (Panel A, intact neurons). Damage was scored as the presence of bright inclusions or “blebs” (Panel B) or “breaks” in the dendrite (Panel C). Panel D shows the compiled frequency of each score from 2 biological replicates and a total of 50 worms per group (200 neurons per group). P-value shown is based on a Chi-squared analysis.
Effects of PCP cannot be attributed to the redox-cycling metabolite TCBQ
Since PCP is metabolized in mammals to the redox-active metabolite TCBQ, we wanted to test if this metabolite could be contributing to the mitochondrial phenotypes observed in this study. To determine this, we first tested if we could detect the metabolite after exposing worms to 50 µM PCP for 48 h. Our limit of detection for this analysis was 0.41 µM. We could not detect any TCBQ in the worm pellet or the supernatant (data not shown), suggesting that any product formation was less than our limit of detection. We also exposed worms directly to TCBQ. We measured uptake of TCBQ after a 48 h exposure to 50 µM TCBQ and found similar levels in the worm pellet and supernatant (Fig. 4A). These levels were considerably lower than the original concentration, about 4 % of the starting concentration, suggesting either instability of TCBQ in the incubation mixture or sticking to the exposure well surfaces. We then tested growth inhibition of worms developmentally exposed to TCBQ for 48 h. We found that the worms were developmentally delayed beginning at the 10 µM concentration and were growth arrested at 50 µM (Fig. 4B). These results show that worms are more sensitive to growth inhibition from TCBQ compared to PCP, as lower concentrations are required to cause growth delay. Given that we did not detect any TCBQ in PCP-exposed worms, we do not expect that TCBQ is contributing to the phenotypes observed in this study. This is also supported by our observation of a more-reduced redox state after PCP exposure, as opposed to the more oxidized state that would be expected if the redox-cycling metabolite were driving cellular effects.
Fig. 4.

TCBQ is taken up into worms, but cannot explain the effects of PCP. Panel A, metabolism data for PCP. For experiments, incubations were prepared with E. coli + N2 worms ± 50 µM TCBQ. TCBQ was measured in the supernatant (External [TCBQ]) and in the lysed worm pellet after 3 brief washes with K-medium (Internal [TCBQ]). Panel B, growth curve for increasing concentrations of TCBQ. For experiments, L1-stage N2 larvae were incubated with TCBQ and UV-inactivated E. coli for 48 h in liquid culture in 6-well plates. Following exposure, worms (n > 40 per group) were placed on food-free K-agar plates and imaged using a stereomicroscope equipped with a camera. The worm volume was then calculated using the ImageJ plugin WormSizer (Moore et al. 2013). Experiments were performed in biological triplicate. **, p < 0.01, one-way ANOVA with Bonferroni-corrected multiple comparisons compared to control.
Discussion
In this study, we demonstrate that PCP exposure causes growth delay in C. elegans, and observe significant mitochondrial dysfunction even at exposure levels that cause minimal growth delay. The observed toxicity is consistent with uncoupling of mitochondria, characterized by decreases in organismal ATP levels, reduced spare respiratory capacity, and increased proton leak. However, despite the deleterious impacts on mitochondrial function, we did not observe PCP-induced neurodegeneration in the dopaminergic cephalic neurons, nor did PCP exacerbate dopaminergic neurotoxicity of 6-hydroxydopamine. Together, these findings contribute to a larger question about exactly what forms of mitochondrial toxicity may drive selective degradation of dopaminergic neurons, such as that observed in sporadic Parkinson’s disease. Specifically, these results are inconsistent with a role for mitochondrial uncoupling or its downstream consequence of energetic (ATP) depletion in driving neurodegeneration. This suggests that in the effort to determine which of the many chemicals that cause mitochondrial toxicity (Attene-Ramos et al., 2015, Attene-Ramos et al., 2013, Meyer et al., 2018, Meyer et al., 2013, Pereira et al., 2009, Wills et al., 2015) also contribute to dopaminergic neurodegeneration, it may be possibly to limit research effort to a subset of more specific forms of mitochondrial dysfunction, rather than considering all mitotoxicants. Additional research will be required to determine specifically which forms of mitochondrial toxicity, and which downstream effects, drive dopaminergic neurodegeneration. Illustrating the degree to which the specific form of mitochondrial dysfunction is critical, Delp et al. examined 21 different pesticides that inhibited Complex I, II, or III, and reported that some inhibitors of Complex I and Complex III, but no Complex II inhibitors, were toxic to dopaminergic cells in culture (Delp et al. 2021). Previous work by the same group (Delp et al. 2019) did not support a specific neurotoxicity of mitochondrial uncouplers.
Our observation of dramatically reduced growth in the presence of increasing concentrations of PCP fits with previous studies in other organisms. PCP causes growth delays in Saccharomyces cerevisiae, human neuroblastoma cells, zebrafish embryos, and Helix aspersa (Ehrlich et al., 1987, Fraser et al., 2019, Gomot-De Vaufleury, 2000, Xu et al., 2014b). The growth delay we observed in our study suggests that PCP causes similar developmental effects in C. elegans. Studies in Saccharomyces cerevisiae suggest that these mechanisms may be targeting RNA production and ribosome synthesis, either by PCP itself or via a downstream metabolite (Ehrlich et al. 1987). If PCP exposure acts via analogous pathways in the worm, it is possible that developing larvae may not be producing adequate amounts of protein and are suffering growth delays as a result. This observation is consistent with a recently-published AOP for mitochondrial uncoupling leading to growth inhibition (Song and Villeneuve 2021).
At a concentration causing minimal growth delay, PCP had dramatic effects on energy metabolism, including a significant, >50 % reduction in whole-worm ATP, and a ∼ 30 % decrease in spare respiratory capacity. Interestingly, the reduction in ATP was not reflected in a change in ATP-linked OCR. We would have anticipated that the loss of ATP would drive a higher demand for ATP production through the ETC, and therefore an increase in ATP-linked OCR. Uncoupling itself increases OCR, as observed with the intentional high-concentration addition of FCCP (Fig. 2A). The lack of increase may suggest that there was not sufficient substrate available to increase ATP-linked OCR, or that damage to the ETC prevented an increase in this rate. However, other aspects of mitochondrial respiration were more dramatically impacted by PCP, including spare capacity and proton leak.
Spare respiratory capacity, also known as respiratory reserve capacity, represents the amount that mitochondria are able to increase respiration under conditions of higher demand (Brand and Nicholls 2011). Our observation that the spare capacity of the exposed worms was reduced compared to the unexposed control worms indicates that the exposed animals are operating closer to their maximum OCR than the untreated worms. A reduced maximum OCR leads to a diminished ability for the worms to respond to other stressors by increasing their OCR.
Reduced spare capacity and increased proton leak are both consistent with PCP mitochondrial membrane uncoupling. This fits with a previous study in C. elegans demonstrating that uncouplers including FCCP and PCP synergize with the mitochondrial toxicant (Complex IV inhibitor) phosphine to enhance its toxicity (Valmas et al. 2008). Their results support a mechanism in which the uncouplers’ synergistic effect is mediated not through loss of ATP or changes to ROS, but rather directly through the mitochondrial membrane potential collapse. Our observation of a decreased roGFP glutathione ratio appears to be consistent this mechanism. Increased proton leak resulting in a lower mitochondrial membrane potential would cause a decrease in the average state of reduction of the electron transport chain complexes and therefore reduce electron leak to oxygen, which forms superoxide anion (Brookes, 2005, Miwa and Brand, 2003, Shabalina and Nedergaard, 2011). However, additional work would be required to prove that PCP-mediated decrease of state of reduction of the electron transport chain was responsible for the less-oxidized redox tone and possible protection against 6OHDA, and alternative explanations exist. For example, reduced mitochondrial membrane potential might reduce mitochondrial uptake of 6OHDA (to our knowledge, mechanisms of 6OHDA mitochondria uptake are not known, but many molecules are taken up by mitochondria in a membrane potential-dependent manner), or reduce activity of monoamine oxidase (inhibiting monoamine oxidase protects against 6OHDA (Salonen et al. 1996)).
In contrast to our findings, some previous studies have reported evidence of increased ROS or a more oxidized redox state after exposure to PCP. In bivalves, increased antioxidant gene expression was observed after exposure to 0.075–2 µM PCP (Xia et al. 2016). Similarly, rats fed PCP at a concentration of 40 mg/kg had significantly increased urinary 8-epi-prostaglandin F2α and increased serum AST (Wang et al. 2001). Another study in mice found increased 8-deoxyguanosine in liver nuclear DNA of PCP-fed mice, and found that PCP exacerbated dimethylnitrosamine-induced hepatocellular carcinoma (Umemura et al. 1999). However, these in vivo systems all included the capacity to bioactivate PCP to its redox-active metabolites TCBQ/TCHQ. In an in vitro system with limited metabolic capacity (splenocytes), another group reported minimal ROS production in PCP-exposed cells but dramatic ROS with TCHQ, suggesting that the metabolite is responsible for the oxidative stress (Chen et al. 2014). Our results with the biological sensor Grx1-roGFP2 showing a more reduced environment after PCP exposure, along with the lack of detection of TCBQ in PCP-exposed worms, support the finding that PCP itself does not drive oxidative stress, at least at relatively low, non-cytotoxic levels, and requires metabolic activation in order to exert this toxicity. It is possible that high levels of uncoupling could result in a more oxidative cellular environment by depleting reducing equivalents (NADPH) responsible for maintaining the normal reduced:oxidized ratio of glutathione; by very greatly accelerating electron flow, leading to an overall increase in electron leakage, despite presumably decreasing the proportion of electrons leaked; or by inducing general cellular toxicity.
It is somewhat surprising that we do not observe any metabolic activation of PCP by C. elegans. As we have recently reviewed (Hartman et al. 2021), worms do have the capacity to metabolize many xenobiotics and their genome encodes > 80 cytochrome P450 enzymes, more than exist in most mammals. Although CYP1A enzyme activity is notably absent in the genome (Harris et al. 2020), CYP3A4 activity is among the few mammalian P450 activities that have been directly observed in C. elegans (Harlow et al. 2018). It may be that CYP1A is more important for the production of these metabolites, or that the production of TCBQ/TCHQ was efficiently linked to further metabolism such as conjugation and did not reach the levels needed to observe oxidative stress. However, since we did not observe any substrate (parent PCP) depletion in the supernatant compared to controls lacking worms, it seems unlikely that substantial metabolism of PCP occurred during these exposures. The general observation of slow metabolism is in general in keeping with minimal metabolism of PCP by other organisms (U.S. EPA., 2010).
We did not observe any dopaminergic neurodegeneration from PCP exposure alone, and when worms were pre-treated with PCP before a neurotoxic challenge with 6-hydroxydopamine, we observed no exacerbation. This fits with previous literature showing neuroprotection from mitochondrial uncoupling through uncoupling proteins and small molecule uncouplers such as DNP (Cho et al., 2017, Kishimoto et al., 2020, Lee et al., 2017, Mattiasson et al., 2003, Wu et al., 2011), reviewed in (Geisler et al. 2017). There is also evidence of mild uncoupling being protective in other contexts, such as metabolic disease (Goedeke et al. 2022). We hypothesize that a critical aspect of the uncoupling-induced cellular protection is dose: under the right circumstances, mild uncoupling induces protection, while dramatic and prolonged uncoupling drives toxicity. From the perspective of mechanistic toxicology and development of adverse outcome pathways, additional work is required to define the degree of mitochondrial uncoupling that should be defined as “mitochondrial dysfunction.” Furthermore, we note that it is not entirely clear whether “protective” levels of uncoupling are exclusively beneficial, since there are likely trade-offs (e.g., decreased energetic efficiency) inherently present; these may manifest upon additional challenge, as observed by Valmas et al. (2008). The negative impact of those trade-offs are likely context-dependent (e.g., how limiting is energy availability) as well as concentration-dependent.
Finally, while we consider the principal value of this study to be to further mechanistic understanding of how sub-lethal mitochondrial uncoupling affects energetics, redox state, and neurodegeneration, it is interesting to compare the internal tissue values of PCP that we measured to those reported for human exposure. A 1989 publication (Cline et al., 1989) reported an average blood level of 4 µg/dL PCP in the general United States population, 42 µg/dL in residents of log homes treated with PCP (and children’s levels were 1.8x the concentration found in their parents), and averages of 8.3 to 5,760 µg/dL in blood of workers using PCP or PCP-treated materials in different occupations. Concentrations of roughly 1,600 µg/dL and above in blood were associated with poisoning, hyperpyrexia, and death in five workers (Wood et al., 1983). Based on these values, we might describe ∼ 5 µg/dL as “upper range of general population,” and ∼ 50 µg/dL as “high exposure,” blood levels. These correspond to ∼ 0.2 and 2 µM blood concentrations, which are similar to or somewhat above the worm-internal PCP concentrations we measured (∼0.15 µM; Fig. 1). Most PCP in human blood is expected to be bound to blood proteins, potentially limiting tissue distribution (ATSDR 2022), and PCP is poorly metabolized in people: ∼85 % of ingested PCP is excreted unchanged (U.S. EPA., 2010). However, PCP’s high lipophilicity and slow metabolism suggest that it could accumulate to higher levels in certain tissues in the context of chronic exposure. Data on relative tissue distribution in people is limited (ATDSR, 2022), but one study of autopsied individuals who had median concentrations of 2.3 µg/dL PCP in blood, found the highest levels in liver (0.067 µg/g), kidneys (0.043 µg/g), brain (0.047 µg/g) (Grimm et al., 1981). Tissue distribution within the worms is unknown.
Conclusions
In this study, we show that a developmental exposure to pentachlorophenol causes growth inhibition/arrest at high concentrations At a lower concentration only mildly inhibiting growth, it drove mitochondrial uncoupling, reduced spare respiratory capacity, increased proton leak, decreased organismal ATP, and caused a more-reduced cellular environment, yet did not impact dopaminergic neuronal morphology or exacerbate neurotoxicity from 6-hydroxydopamine. Together, these findings support a major role for uncoupling in PCP toxicity, do not support a major role for mild mitochondrial uncoupling in dopaminergic neurodegeneration, and further define the utility of C. elegans as a simple screening system for mechanistic studies of mitochondrial toxicity and neurotoxicity.
Authors’ Contributions
Z.R.M., J.H.H., A.S.J., K.A.H., and I.R. performed the experimental studies. J.N.M., P.L.F., and J.H.H. contributed to the design and analysis of the study. J.H.H. and Z.R.M. wrote the article. All authors analyzed the results, revised the article, and approved the final version of the article.
Funding Information
This work was supported by the National Institutes of Health Grant No P42-ES010356 (JNM), R01ES028218 (JNM), and K99-ES029552 (JHH).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank Latasha L. Smith for assistance with PCP neurodegeneration experiments.
Contributor Information
Zachary R. Markovich, Email: zmarkovich@ufl.edu.
Jessica H. Hartman, Email: hartmanj@musc.edu.
Ian T. Ryde, Email: ian.ryde@duke.edu.
Abigail S. Joyce, Email: abigail.joyce@duke.edu.
Patrick L. Ferguson, Email: lee.ferguson@duke.edu.
Joel N. Meyer, Email: joel.meyer@duke.edu.
References
- Albrecht S.C., Barata A.G., Großhans J., Teleman A.A., Dick T.P. In Vivo Mapping of Hydrogen Peroxide and Oxidized Glutathione Reveals Chemical and Regional Specificity of Redox Homeostasis: Cell Metabolism. Cell Metab. 2011;14:819–829. doi: 10.1016/j.cmet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Albrecht S.C., Barata A.G., Grosshans J., Teleman A.A., Dick T.P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab. 2011;14:19–829. doi: 10.1016/j.cmet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- ATSDR. 2017. The ATSDR 2019 Substance Priority List. https://www.atsdr.cdc.gov/spl/index.html#2019spl.
- ATSDR. 2022. Toxicological profile for pentachlorophenol. https://www.atsdr.cdc.gov/toxprofiles/tp51.pdf. [PubMed]
- Attene-Ramos M.S., Huang R., Sakamuru S., Witt K.L., Beeson G.C., Shou L., Schnellmann R.G., Beeson C.C., Tice R.R., Austin C.P., Xia M. Systematic study of mitochondrial toxicity of environmental chemicals using quantitative high throughput screening. Chem. Res. Toxicol. 2013;26:1323–1332. doi: 10.1021/tx4001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attene-Ramos M.S., Huang R., Michael S., Witt K.L., Richard A., Tice R.R., Simeonov A., Austin C.P., Xia M. Profiling of the Tox21 chemical collection for mitochondrial function to identify compounds that acutely decrease mitochondrial membrane potential. Environ. Health Perspect. 2015;123:49–56. doi: 10.1289/ehp.1408642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back P., De Vos W.H., Depuydt G.G., Matthijssens F., Vanfleteren J.R., Braeckman B.P. Exploring real-time in vivo redox biology of developing and aging Caenorhabditis elegans. Free Radical Biol. Med. 2012;52:850–859. doi: 10.1016/j.freeradbiomed.2011.11.037. [DOI] [PubMed] [Google Scholar]
- Bijwadia S.R., Morton K., Meyer J.N. Quantifying levels of dopaminergic neuron morphological alteration and degeneration in Caenorhabditis elegans. J. Vis. Exp. 2021 doi: 10.3791/62894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsche M., Pereira S.L., Klein C., Grünewald A. Mitochondria and Parkinson's Disease: Clinical, Molecular, and Translational Aspects. J. Parkinsons Dis. 2021;11:45–60. doi: 10.3233/JPD-201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd W.A., Smith M.V., Kissling G.E., Rice J.R., Snyder D.W., Portier C.J., Freedman J.H. Application of a mathematical model to describe the effects of chlorpyrifos on Caenorhabditis elegans development. PLoS ONE. 2009;4:e7024. doi: 10.1371/journal.pone.0007024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M.D., Buckingham J.A., Esteves T.C., Green K., Lambert A.J., Miwa S., Murphy M.P., Pakay J.L., Talbot D.A., Echtay K.S. Mitochondrial superoxide and aging: uncoupling-protein activity and superoxide production. Biochem. Soc. Symp. 2004:203–213. doi: 10.1042/bss0710203. [DOI] [PubMed] [Google Scholar]
- Brand M.D., Nicholls D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes P.S. Mitochondrial H+ leak and ROS generation: An odd couple. Free Radical Biol. Med. 2005;38:12–23. doi: 10.1016/j.freeradbiomed.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Cao F., Souders Ii C.L., Perez-Rodriguez V., Martyniuk C.J. Elucidating Conserved Transcriptional Networks Underlying Pesticide Exposure and Parkinson's Disease: A Focus on Chemicals of Epidemiological Relevance. Front. Genet. 2018;9:701. doi: 10.3389/fgene.2018.00701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.-M., Zhu B.-Z., Chen R.-J., Wang B.-J., Wang Y.-J., Ho Y.-S. The pentachlorophenol metabolite tetrachlorohydroquinone induces massive ROS and prolonged p-ERK expression in splenocytes, leading to inhibition of apoptosis and necrotic cell death. PLoS ONE. 2014;9(2):e89483. doi: 10.1371/journal.pone.0089483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I., Song H.O., Cho J.H. Mitochondrial Uncoupling Attenuates Age-Dependent Neurodegeneration in C. elegans. Mol. Cells. 2017;40:864–870. doi: 10.14348/molcells.2017.0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline R.E., Hill R.H., Phillips D.L., Needham L.L. Pentachlorophenol measurements in body fluids of people in log homes and workplaces. Arch. Environ. Contam. Toxicol. 1989;18:475–481. doi: 10.1007/BF01055012. [DOI] [PubMed] [Google Scholar]
- Cothren S.D., Meyer J.N., Hartman J.H. Blinded Visual Scoring of Images Using the Freely-available Software Blinder. Bio-protocol. 2018;8:e3103. doi: 10.21769/BioProtoc.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y., Liang L., Zhong Q., He Q., Shan X., Chen K., Huang F. The association of cancer risks with pentachlorophenol exposure: Focusing on community population in the areas along certain section of Yangtze River in China. Environ. Pollut. 2017;224:729–738. doi: 10.1016/j.envpol.2016.12.011. [DOI] [PubMed] [Google Scholar]
- Dahlhaus M., Almstadt E., Henschke P., Luttgert S., Appel K.E. Oxidative DNA lesions in V79 cells mediated by pentachlorophenol metabolites. Arch. Toxicol. 1996;70:457–460. doi: 10.1007/s002040050299. [DOI] [PubMed] [Google Scholar]
- Datta S., Sahdeo S., Gray J.A., Morriseau C., Hammock B.D., Cortopassi G. A high-throughput screen for mitochondrial function reveals known and novel mitochondrial toxicants in a library of environmental agents. Mitochondrion. 2016;31:79–83. doi: 10.1016/j.mito.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delp J., Funke M., Rudolf F., Cediel A., Bennekou S.H., van der Stel W., Carta G., Jennings P., Toma C., Gardner I., van de Water B., Forsby A., Leist M. Development of a neurotoxicity assay that is tuned to detect mitochondrial toxicants. Arch. Toxicol. 2019;93:1585–1608. doi: 10.1007/s00204-019-02473-y. [DOI] [PubMed] [Google Scholar]
- Delp J., Cediel-Ulloa A., Suciu I., Kranaster P., van Vugt-Lussenburg B.M., Munic Kos V., van der Stel W., Carta G., Bennekou S.H., Jennings P., van de Water B., Forsby A., Leist M. Neurotoxicity and underlying cellular changes of 21 mitochondrial respiratory chain inhibitors. Arch. Toxicol. 2021;95:591–615. doi: 10.1007/s00204-020-02970-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demine S., Renard P., Arnould T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells. 2019;8 doi: 10.3390/cells8080795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo S., Reed T.T., Venditti P., Victor V.M. Harmful and Beneficial Role of ROS. Oxid. Med. Cell. Longevity. 2016;2016:1–3. doi: 10.1155/2016/7909186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley C.T., Dore T.M., Hanson G.T., Jackson W.C., Remington S.J., Tsien R.Y. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- Dreier D.A., Mello D.F., Meyer J.N., Martyniuk C.J. Linking Mitochondrial Dysfunction to Organismal and Population Health in the Context of Environmental Pollutants: Progress and Considerations for Mitochondrial Adverse Outcome Pathways. Environ. Toxicol. Chem. 2019;38(8):1625–1634. doi: 10.1002/etc.4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S.W., Tan Y.M., Villeneuve D.L., Meek M.E., McQueen C.A. Adverse Outcome Pathways-Organizing Toxicological Information to Improve Decision Making. J. Pharmacol. Exp. Ther. 2016;356:170–181. doi: 10.1124/jpet.115.228239. [DOI] [PubMed] [Google Scholar]
- Ehrlich W., Mangir M., Lochmann E.R. The effect of pentachlorophenol and its metabolite tetrachlorohydroquinone on RNA, protein, and ribosome synthesis in Saccharomyces cells. Ecotoxicol. Environ. Saf. 1987;13:7–12. doi: 10.1016/0147-6513(87)90037-6. [DOI] [PubMed] [Google Scholar]
- Enoch S.J., Schultz T.W., Popova I.G., Vasilev K.G., Mekenyan O.G. Development of a Decision Tree for Mitochondrial Dysfunction: Uncoupling of Oxidative Phosphorylation. Chem. Res. Toxicol. 2018;31:814–820. doi: 10.1021/acs.chemrestox.8b00132. [DOI] [PubMed] [Google Scholar]
- Fraser D.L., Stander B.A., Steenkamp V. Cytotoxic activity of pentachlorophenol and its active metabolites in SH-SY5Y neuroblastoma cells. Toxicol. In Vitro. 2019;58:118–125. doi: 10.1016/j.tiv.2019.03.024. [DOI] [PubMed] [Google Scholar]
- Geisler J.G., Marosi K., Halpern J., Mattson M.P. DNP, mitochondrial uncoupling, and neuroprotection: A little dab'll do ya. Alzheimers Dement. 2017;13:582–591. doi: 10.1016/j.jalz.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedeke L., Murt K.N., Di Francesco A., Camporez J.P., Nasiri A.R., Wang Y., Zhang X.M., Cline G.W., de Cabo R., Shulman G.I. Sex- and strain-specific effects of mitochondrial uncoupling on age-related metabolic diseases in high-fat diet-fed mice. Aging Cell. 2022;21:e13539. doi: 10.1111/acel.13539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomot-De Vaufleury A. Standardized Growth Toxicity Testing (Cu, Zn, Pb, and Pentachlorophenol) with Helix aspersa. Ecotoxicol. Environ. Saf. 2000;46:41–50. doi: 10.1006/eesa.1999.1872. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hunt C.P., Leung M.C., Bodhicharla R.K., McKeever M.G., Arrant A.E., Margillo K.M., Ryde I.T., Cyr D.D., Kosmaczewski S.G., Hammarlund M., Meyer J.N. Exposure to mitochondrial genotoxins and dopaminergic neurodegeneration in Caenorhabditis elegans. PLoS ONE. 2014;9:e114459. doi: 10.1371/journal.pone.0114459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm H.G., Schellmann B., Schaller K.H., Gossler K. Pentachlorophenol concentrations in tissues and body fluids of normal persons (author's translation) Zentralblatt fur Bakteriologie, Mikrobiologie und Hygiene B. 1981;174:77–90. German. [PubMed] [Google Scholar]
- Hallinger D.R., Lindsay H.B., Paul Friedman K., Suarez D.A., Simmons S.O. Respirometric Screening and Characterization of Mitochondrial Toxicants Within the ToxCast Phase I and II Chemical Libraries. Toxicol. Sci. 2020;176:175–192. doi: 10.1093/toxsci/kfaa059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow P.H., Perry S.J., Stevens A.J., Flemming A.J. Comparative metabolism of xenobiotic chemicals by cytochrome P450s in the nematode Caenorhabditis elegans. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-31215-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J.B., Hartman J.H., Luz A.L., Wilson J.Y., Dinyari A., Meyer J.N. Zebrafish CYP1A expression in transgenic Caenorhabditis elegans protects from exposures to benzo[a]pyrene and a complex polycyclic aromatic hydrocarbon mixture. Toxicology. 2020;440:152473. doi: 10.1016/j.tox.2020.152473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman J.H., Gonzalez-Hunt C., Hall S.M., Ryde I.T., Caldwell K.A., Caldwell G.A., Meyer J.N. Genetic Defects in Mitochondrial Dynamics in Caenorhabditis elegans Impact Ultraviolet C Radiation- and 6-hydroxydopamine-Induced Neurodegeneration. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20133202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman J.H., Widmayer S.J., Bergemann C.M., King D.E., Morton K.S., Romersi R.F., Jameson L.E., Leung M.C.K., Andersen E.C., Taubert S., Meyer J.N. Xenobiotic metabolism and transport in Caenorhabditis elegans. J. Toxicol. Environ. Health B Crit. Rev. 2021;24:51–94. doi: 10.1080/10937404.2021.1884921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryhorczuk D.O., Wallace W.H., Persky V., Furner S., Webster J.R., Jr., Oleske D., Haselhorst B., Ellefson R., Zugerman C. A morbidity study of former pentachlorophenol-production workers. Environ. Health Perspect. 1998;106:401–408. doi: 10.1289/ehp.98106401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y., Wan Y., Huang Q., Wang A., Mahai G., He Z., Xu S., Xia W. Pentachlorophenol exposure in early pregnancy and gestational diabetes mellitus: A nested case-control study. Sci. Total Environ. 2022;831 doi: 10.1016/j.scitotenv.2022.154889. [DOI] [PubMed] [Google Scholar]
- IUPAC. 1981. Environmnetal Chemistry of Pentachlorophenol. In: D.G. Crosby (Ed), IUPAC Reports on Pesticides, University of California Davis, CA, USA, pp. 1051-1080.
- Kishimoto Y., Johnson J., Fang W., Halpern J., Marosi K., Liu D., Geisler J.G., Mattson M.P. A mitochondrial uncoupler prodrug protects dopaminergic neurons and improves functional outcome in a mouse model of Parkinson's disease. Neurobiol. Aging. 2020;85:123–130. doi: 10.1016/j.neurobiolaging.2019.09.011. [DOI] [PubMed] [Google Scholar]
- Lee Y., Heo G., Lee K.M., Kim A.H., Chung K.W., Im E., Chung H.Y., Lee J. Neuroprotective effects of 2,4-dinitrophenol in an acute model of Parkinson's disease. Brain Res. 2017;1663:184–193. doi: 10.1016/j.brainres.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Lehman-McKeeman L.D. In: Casarett & Doull's Toxicology: The Basic Science of Poisons. Klaassen C.D., editor. McGraw-Hill; United States of America: 2019. Mechanisms of Toxicity; pp. 65–126. [Google Scholar]
- Lewis J.A., Fleming J.T. In: Methods in Cell Biology. Epstein H.F., Shakes D.C., editors. Academic Press; 1995. Chapter 1 Basic Culture Methods; pp. 3–29. [PubMed] [Google Scholar]
- Luz A.L., Smith L.L., Rooney J.P., Meyer J.N. Seahorse Xfe 24 Extracellular Flux Analyzer-Based Analysis of Cellular Respiration in Caenorhabditis elegans. Curr. Protoc. Toxicol. 2015;66 doi: 10.1002/0471140856.tx2507s66. 25.27.21-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiasson G., Shamloo M., Gido G., Mathi K., Tomasevic G., Yi S., Warden C.H., Castilho R.F., Melcher T., Gonzalez-Zulueta M., Nikolich K., Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med. 2003;9:1062–1068. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- Maurer L.L., Ryde I.T., Yang X., Meyer J.N. Caenorhabditis elegans as a Model for Toxic Effects of Nanoparticles: Lethality, Growth, and Reproduction. Curr. Protoc. Toxicol. 2015;66 doi: 10.1002/0471140856.tx2010s66. 20.10.21-20.10.25. [DOI] [PubMed] [Google Scholar]
- Meyer J.N., Lord C.A., Yang X.Y., Turner E.A., Badireddy A.R., Marinakos S.M., Chilkoti A., Wiesner M.R., Auffan M. Intracellular uptake and associated toxicity of silver nanoparticles in Caenorhabditis elegans. Aquat. Toxicol. 2010;100:140–150. doi: 10.1016/j.aquatox.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Meyer J.N., Leung M.C., Rooney J.P., Sendoel A., Hengartner M.O., Kisby G.E., Bess A.S. Mitochondria as a target of environmental toxicants. Toxicol. Sci. 2013;134:1–17. doi: 10.1093/toxsci/kft102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J.N., Hartman J.H., Mello D.F. Mitochondrial Toxicity. Toxicol. Sci. 2018;162:15–23. doi: 10.1093/toxsci/kfy008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa S., Brand M.D. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem. Soc. Trans. 2003;31:1300–1301. doi: 10.1042/bst0311300. [DOI] [PubMed] [Google Scholar]
- Moore B.T., Jordan J.M., Baugh L.R. WormSizer: high-throughput analysis of nematode size and shape. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0057142. e57142-e57142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan M.K. Predictors of urinary levels of 2,4-dichlorophenoxyacetic acid, 3,5,6-trichloro-2-pyridinol, 3-phenoxybenzoic acid, and pentachlorophenol in 121 adults in Ohio. Int. J. Hyg. Environ. Health. 2015;218:479–488. doi: 10.1016/j.ijheh.2015.03.015. [DOI] [PubMed] [Google Scholar]
- Morgan M., Jones P., Sobus J. Short-term variability and predictors of urinary pentachlorophenol levels in Ohio preschool children. Int. J. Environ. Res. Public Health. 2015;12:800–815. doi: 10.3390/ijerph120100800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Biomonitoring Program, Centers fo Disease Control and Prevenetion. Pentachlorophenol biomonitoring summary. Webpage last reviewed April 7, 2017. Webpage accessed Jun 9, 2022. https://www.cdc.gov/biomonitoring/Pentachlorophenol_BiomonitoringSummary.html.
- Naven R.T., Swiss R., Klug-McLeod J., Will Y., Greene N. The development of structure-activity relationships for mitochondrial dysfunction: uncoupling of oxidative phosphorylation. Toxicol. Sci. 2013;131:271–278. doi: 10.1093/toxsci/kfs279. [DOI] [PubMed] [Google Scholar]
- Palikaras K., Tavernarakis N. Intracellular Assessment of ATP Levels in Caenorhabditis elegans. Bio Protoc. 2016;6(23) doi: 10.21769/BioProtoc.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira C.V., Moreira A.C., Pereira S.P., Machado N.G., Carvalho F.S., Sardão V.A., Oliveira P.J. Investigating drug-induced mitochondrial toxicity: a biosensor to increase drug safety? Curr. Drug Saf. 2009;4:34–54. doi: 10.2174/157488609787354440. [DOI] [PubMed] [Google Scholar]
- Salonen T., Haapalinna A., Heinonen E., Suhonen J., Hervonen A. Monoamine oxidase B inhibitor selegiline protects young and aged rat peripheral sympathetic neurons against 6-hydroxydopamine-induced neurotoxicity. Acta Neuropathol. 1996;91:466–474. doi: 10.1007/s004010050453. [DOI] [PubMed] [Google Scholar]
- Samii A., Nutt J.G., Ransom B.R. Parkinson's disease. Lancet. 2004;363:1783–1793. doi: 10.1016/S0140-6736(04)16305-8. [DOI] [PubMed] [Google Scholar]
- Schmied-Tobies M.I.H., Murawski A., Schmidt L., Rucic E., Schwedler G., Apel P., Göen T., Kolossa-Gehring M. Pentachlorophenol and nine other chlorophenols in urine of children and adolescents in Germany - Human biomonitoring results of the German Environmental Survey 2014–2017 (GerES V) Environ. Res. 2021;196:110958. doi: 10.1016/j.envres.2021.110958. [DOI] [PubMed] [Google Scholar]
- Shabalina I.G., Nedergaard J. Mitochondrial (‘mild’) uncoupling and ROS production: physiologically relevant or not? Biochem. Soc. Trans. 2011;39:1305–1309. doi: 10.1042/BST0391305. [DOI] [PubMed] [Google Scholar]
- Song Y., Villeneuve D.L. AOP Report: Uncoupling of Oxidative Phosphorylation Leading to Growth Inhibition via Decreased Cell Proliferation. Environ. Toxicol. Chem. 2021;40:2959–2967. doi: 10.1002/etc.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier D.J. Determinants of dopaminergic neuron loss in Parkinson's disease. FEBS J. 2018;285:3657–3668. doi: 10.1111/febs.14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terron A., Bal-Price A., Paini A., Monnet-Tschudi F., Bennekou S.H., Leist M., Schildknecht S. An adverse outcome pathway for parkinsonian motor deficits associated with mitochondrial complex I inhibition. Arch. Toxicol. 2018;92:41–82. doi: 10.1007/s00204-017-2133-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C.H., Lin P.H., Waidyanatha S., Rappaport S.M. Characterization of metabolic activation of pentachlorophenol to quinones and semiquinones in rodent liver. Chem. Biol. Interact. 2001;134:55–71. doi: 10.1016/s0009-2797(00)00318-5. [DOI] [PubMed] [Google Scholar]
- U.S. EPA. 2010. IRIS Toxicological Review of Pentachlorophenol (Final Report). U.S. Environmental Protection Agency, Washington, DC, EPA/635/R-09/004F. https://cfpub.epa.gov/ncea/risk/hhra/recordisplay.cfm?deid=230890.
- Umemura T., Kai S., Hasegawa R., Sai K., Kurokawa Y., Williams G.M. Pentachlorophenol (PCP) produces liver oxidative stress and promotes but does not initiate hepatocarcinogenesis in B6C3F1 mice. Carcinogenesis. 1999;20:1115–1120. doi: 10.1093/carcin/20.6.1115. [DOI] [PubMed] [Google Scholar]
- Valmas N., Zuryn S., Ebert P.R. Mitochondrial uncouplers act synergistically with the fumigant phosphine to disrupt mitochondrial membrane potential and cause cell death. Toxicology. 2008;252:33–39. doi: 10.1016/j.tox.2008.07.060. [DOI] [PubMed] [Google Scholar]
- Wang Y.-J., Lee C.-C., Chang W.-C., Liou H.-B., Ho Y.-S. Oxidative stress and liver toxicity in rats and human hepatoma cell line induced by pentachlorophenol and its major metabolite tetrachlorohydroquinone. Toxicol. Lett. 2001;122(2):157–169. doi: 10.1016/s0378-4274(01)00361-7. [DOI] [PubMed] [Google Scholar]
- Weinbach E.C. Biochemical basis for the toxicity of pentachlorophenol. Proc. Natl. Acad. Sci. U. S. A. 1957;43:393–397. doi: 10.1073/pnas.43.5.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will Y., Dykens J. Mitochondrial toxicity assessment in industry–a decade of technology development and insight. Expert Opin. Drug Metab. Toxicol. 2014;10:1061–1067. doi: 10.1517/17425255.2014.939628. [DOI] [PubMed] [Google Scholar]
- Wills L.P. The use of high-throughput screening techniques to evaluate mitochondrial toxicity. Toxicology. 2017;391:34–41. doi: 10.1016/j.tox.2017.07.020. [DOI] [PubMed] [Google Scholar]
- Wills L.P., Beeson G.C., Hoover D.B., Schnellmann R.G., Beeson C.C. Assessment of ToxCast Phase II for Mitochondrial Liabilities Using a High-Throughput Respirometric Assay. Toxicol. Sci. 2015;146:226–234. doi: 10.1093/toxsci/kfv085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood S., Rom W.N., White G.L., Jr., Logan D.C. Pentachlorophenol poisoning. J. Occup. Med. 1983;25:527–530. [PubMed] [Google Scholar]
- World Health Organization, International Programme on Chemical Safety. 1987. Pentachlorophenol. https://apps.who.int/iris/handle/10665/38414.
- Wu Y.N., Munhall A.C., Johnson S.W. Mitochondrial uncoupling agents antagonize rotenone actions in rat substantia Nigra dopamine neurons. Brain Res. 2011;1395:86–93. doi: 10.1016/j.brainres.2011.04.032. [DOI] [PubMed] [Google Scholar]
- Xia X., Hua C., Xue S., Shi B., Gui G., Zhang D., Wang X., Guo L. Response of selenium-dependent glutathione peroxidase in the freshwater bivalve Anodonta woodiana exposed to 2,4-dichlorophenol,2,4,6-trichlorophenol and pentachlorophenol. Fish Shellfish Immunol. 2016;55:499–509. doi: 10.1016/j.fsi.2016.06.012. [DOI] [PubMed] [Google Scholar]
- Xia M., Huang R., Shi Q., Boyd W.A., Zhao J., Sun N., Rice J.R., Dunlap P.E., Hackstadt A.J., Bridge M.F., Smith M.V., Dai S., Zheng W., Chu P.H., Gerhold D., Witt K.L., DeVito M., Freedman J.H., Austin C.P., Houck K.A., Thomas R.S., Paules R.S., Tice R.R., Simeonov A. Comprehensive Analyses and Prioritization of Tox21 10K Chemicals Affecting Mitochondrial Function by in-Depth Mechanistic Studies. Environ. Health Perspect. 2018;126 doi: 10.1289/EHP2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D., Hu L., Xia X., Song J., Li L., Song E., Song Y. Tetrachlorobenzoquinone induces acute liver injury, up-regulates HO-1 and NQO1 expression in mice model: the protective role of chlorogenic acid. Environ. Toxicol. Pharmacol. 2014;37:1212–1220. doi: 10.1016/j.etap.2014.04.022. [DOI] [PubMed] [Google Scholar]
- Xu T., Zhao J., Hu P., Dong Z., Li J., Zhang H., Yin D., Zhao Q. Pentachlorophenol exposure causes Warburg-like effects in zebrafish embryos at gastrulation stage. Toxicol. Appl. Pharmacol. 2014;277:183–191. doi: 10.1016/j.taap.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Yang H., van der Stel W., Lee R., Bauch C., Bevan S., Walker P., van de Water B., Danen E.H.J., Beltman J.B. Dynamic Modeling of Mitochondrial Membrane Potential Upon Exposure to Mitochondrial Inhibitors. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.679407. [DOI] [PMC free article] [PubMed] [Google Scholar]