

Abstract
Metaproteomics has been increasingly utilized for high-throughput characterization of proteins in complex environments and has been demonstrated to provide insights into microbial composition and functional roles. However, significant challenges remain in metaproteomic data analysis, including creation of a sample-specific protein sequence database. A well-matched database is a requirement for successful metaproteomics analysis, and the accuracy and sensitivity of PSM identification algorithms suffer when the database is incomplete or contains extraneous sequences. When matched DNA sequencing data of the sample is unavailable or incomplete, creating the proteome database that accurately represents the organisms in the sample is a challenge. Here, we leverage a de novo peptide sequencing approach to identify the sample composition directly from metaproteomic data. First, we created a deep learning model, Kaiko, to predict the peptide sequences from mass spectrometry data and trained it on 5 million peptide–spectrum matches from 55 phylogenetically diverse bacteria. After training, Kaiko successfully identified organisms from soil isolates and synthetic communities directly from proteomics data. Finally, we created a pipeline for metaproteome database generation using Kaiko. We tested the pipeline on native soils collected in Kansas, showing that the de novo sequencing model can be employed as an alternative and complementary method to construct the sample-specific protein database instead of relying on (un)matched metagenomes. Our pipeline identified all highly abundant taxa from 16S rRNA sequencing of the soil samples and uncovered several additional species which were strongly represented only in proteomic data.
Keywords: de novo sequencing, deep learning model, metaproteomics, soil microbiome
1. Introduction
The soil microbiome is responsible for carrying out many functions that are important on a global scale, including cycling of carbon and other nutrients and support of plant growth. Over the last few decades, high-throughput sequencing technologies have made great strides in revealing the soil microbial community composition in a variety of soil habitats and how those communities are impacted by environmental change. Amplicon sequencing has revealed that soil and sediment microorganisms have a very high diversity, much more so than other ecosystems.1 In addition, metagenome sequencing has proven to be an extremely useful tool for determining not only the composition of soil microbiomes but also their putative functions. However, not all genes detected in a metagenome survey are actively expressed, and significant challenges remain in understanding the biological functions that are carried out by active members of the soil microbiome. Other meta-omics technologies, such as metatranscriptomics and metaproteomics, have helped to close this current knowledge gap. Metatranscriptomics provides information on community transcription and is often used as a proxy for assigning metabolically active members of a soil microbiome. However, metatranscriptomics can only provide a snapshot of gene expression at the moment of sampling. A significant amount of post-transcriptional regulation affects protein abundance and activity.2 Therefore, metaproteomics provides an essential layer of information about microbiome activity by revealing which proteins are actually produced and have passed transcriptional and translational regulation points.3
Despite the promise of metaproteomics for elucidating functions of elusive soil microorganisms, significant challenges remain.4−6 An important assumption in most mass spectrometry proteomics identification algorithms is that the set of potential proteins is known, and thus a database of these protein sequences is a typical requirement.7−9 In environmental samples, however, obtaining an accurate catalog of organisms and their proteins is a challenge, as it is not possible to know the organisms present in the sample beforehand. Amplicon and metagenome sequencing of a matched sample is often used to identify community membership; however, many species might not be observable by sequencing.10−15 Alternatively, some metaproteomics algorithms attempt to identify species from an exhaustive sequence database like the 113 million protein sequences in NCBI’s RefSeq.16 A final method is a two-step search of the data, where the first step identifies the organisms present and the second step utilizes a database of proteins from organisms identified in the first step. Most commonly, the first step is done with a very large sequence database and a database search algorithm.17−19 If using this two-step strategy, it is essential to identify the organisms present in a sample with taxonomic precision, so that databases include as few species as possible. Therefore, methods for constructing an optimal metaproteomics database are an area of significant interest.6,9
Here we present a new method to generate a protein database directly from metaproteomic data using de novo mass spectrometry20 to identify species from the annotated peptides and then gather full proteomic databases for these organisms. As currently available software tools for de novo identification were not sufficiently accurate for environmental samples, we first needed to train a new deep learning model with a library of diverse organisms. To achieve this, we mined the extensive data archive housed in the Environmental and Molecular Sciences Laboratory resulting in a training data set including spectra from 55 bacteria across 9 phyla. After confirming that the new model could successfully identify organisms from soil isolates and synthetic communities, we applied the model to metaproteomics samples. Using a metaproteomics data set from Kansas soil, our pipeline identified all abundant taxa identified in traditional 16S data as well as identifying new abundant organisms in the soil. Using the identified organisms, we reanalyzed the metaproteomics data and identified differential metabolic functions between species in the microbiome.
2. Methods
2.1. Data Generation for Kaiko
Organism Choice
The most important aspect of selecting organisms from which to create MS/MS spectra used for training the machine learning algorithm is the diversity of the peptide sequence. In order for the model to generalize and become broadly useful to any organism in any environmental niche, the diversity of sequences must be sufficient for the model to learn general principles. For this reason, we chose a large number of phylogenetically diverse organisms as opposed to extensive LC-MS/MS of a more limited number of organisms. As such, the exact organism is less important than their contribution to diversity. To be explicit, although we chose bacteria commonly found in soils, we could have chosen any organisms from any environment as long as the sufficient number of diverse peptide/spectrum matches was generated. Our goal was 1 million unique peptides. We identified organisms which could be accurately sourced (e.g., available from ATCC) and had a curated proteome available in public repositories like Uniprot and Genbank. We made a special effort to gather bacteria from different phyla, as the bacterial phylal radiation event happened 3.5 billion years ago and therefore would be most likely to provide sufficient sequence diversity.
Cell Culture and Sample Preparation
The growth, sample preparation, and data collection were reported previously.21 Cells were harvested by centrifuging at 3500 × g for 5 min at room temperature and washed twice with 5 mL PBS by centrifuging at the same conditions. Cells were lysed in a Bullet Blender (Next Advance) for 4 min at speed 8 in 200 μL of 100 mM ammonium bicarbonate (NH4HCO3) and approximately 100 μL 0.1 mm zirconia/silica beads at 4 °C. Lysates were transferred into clean tubes, and the remaining beads were washed with 200 μL of 100 mM NH4HCO3. The supernatants from the washing step were collected and combined with the cell lysate. Resulting protein extract was assayed by bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, San Jose, CA) following manufacturer’s instructions. Aliquots of 300 μg of proteins were denatured and reduced using 8 M urea and 5 mM DTT, and incubated at 60 °C for 30 min with 850 rpm shaking. Samples were then diluted 10-fold in 100 mM NH4HCO3, and CaCl2 was added to a final concentration of 1 mM using a 1 M stock. Trypsin was added at 1/50 of the protein concentration, and the digestion was carried out for 3 h at 37 °C. Digestion products were desalted in 1 mL C18 cartridges (50 mg beads, Strata, Phenomenex). Cartridges were activated with 3 mL of methanol and equilibrated with 2 mL of 0.1% TFA before loading the samples. After sample loading, the cartridges were washed with 4 mL of 5% acetonitrile (ACN)/0.1% TFA and peptides were eluted with 1 mL of 80% ACN/0.1% TFA. Peptides were dried in a vacuum centrifuge, resuspended in water, and assayed using a BCA assay. Peptide concentrations were normalized to 0.1 μg/μL before randomization and analysis by liquid chromatography–tandem mass spectrometry (LC-MS/MS).
LC-MS/MS Data Acquisition
The data acquisition was performed as previously described in detail21 using a Waters nanoEquity UPLC system (Millford, MA) coupled with a Q Exactive Plus mass spectrometer from Thermo Fisher Scientific (San Jose, CA). The LC was configured to load the sample first on a solid-phase extraction (SPE) column followed by separation on an analytical column. 500 ng of peptides were loaded into the SPE column (5 cm × 360 μm OD × 150 μm ID fused silica capillary tubing (Polymicro, Phoenix, AZ), packed with 3.6 μm Aeries C18 particles (Phenomenex, Torrance, CA), and the separation was carried out in a capillary column (70 cm × 360 μm OD × 75 μm ID packed with 3 μm Jupiter C18 stationary-phase particles (Phenomenex). The elution was performed at a 300 nL/min flow rate and the following gradient of acetonitrile (ACN) in water, both containing 0.1% formic acid: 1–8% ACN solvent in 2 min; 8–12% ACN in 18 min; 12–30% ACN in 55 min; 30–45% ACN in 22 min; 45–95% ACN in 3 min; hold for 5 min in 95% ACN and 99–1% ACN in 10 min. Eluting peptides were directly analyzed in the mass spectrometer by electrospray using etched silica fused tips.22 Full MS spectra were acquired at a scan range of 400–2000 m/z and a resolution of 35,000 at m/z 400. Tandem mass spectra were collected for the top 12 most intense ions with ≥2 charges using high-collision energy (HCD) fragmentation from the collision with N2 at a normalized collision energy of 30% and a resolution of 17,500 at m/z 400. Each parent ion was targeted once for fragmentation and then dynamically excluded for 30 s.
Peptide Identification for Training/Testing the Kaiko Model
In the training and test sets, the true source/taxonomy of each sample is known. To create the ground truth of spectrum identifications, we used the correct organism’s protein sequence database and annotated spectra with the MSGF+ algorithm, as previously described.21 PSM results from MSGF+ were filtered using a q-value threshold of 0.001. The PSMs passing this filter were considered the ground truth for the deep neural network training and testing. Because our use of this data is for de novo spectrum annotation, we limited peptides/spectrum matches further to exclude peptides longer than 30 residues, as these were unlikely to have complete peptide fragment peaks, which are important for a de novo solution. We also filtered peptides with a precursor mass of >3000 Da. After filtering, the total number of distinct peptides was 1,013,498 from 5,116,305 spectra. Peptide sequences are highly specific to each organism, and the overlap between organisms was very low. Except for the pairs of organisms within the same genus or species (i.e., the two different strains of B. subtilis or the two different species within Bifidobacterium), the average amount of shared peptides between any two organisms was ∼0.17%. These arise from highly conserved proteins like EF-Tu or RpoC for which peptides can be found conserved across phyla.
2.2. Training Kaiko
Codebase
Kaiko is based on DeepNovo, a deep neural network algorithm for peptide/spectrum matching.23 We downloaded the source code for DeepNovo (https://github.com/nh2tran/DeepNovo) and its pretrained model, which is publicly available at https://drive.google.com/open?id=0By9IxqHK5MdWalJLSGliWW1RY2c. As described below, we modified the original DeepNovo codebase, keeping with Python 2.7 and TensorFlow 1.2 as used in the original. First, we modified the codebase to accept multiple input files for training and testing. Our training and testing data came from over 250 mass spectrometry files, but the original DeepNovo was designed for only a single input file. Therefore, we added extra command-line options (e.g., --multi_decode and --multi_train) and the associated wrapper methods to allow for multifile execution. A second change was done to avoid rebuilding the Cython codes on every parameter adjustment. For this, we replaced the Cython with the python numba package without any loss of performance and speed. Finally, we changed the code for spectral modeling based on domain knowledge. Specifically, we corrected the mass calculation of doubly charged ions and changed the bins used for isotopic profiles within the ion-CNN model. Our new model and software are available at https://github.com/PNNL-Comp-Mass-Spec/Kaiko.
We trained multiple models for Kaiko, which differed primarily in the number of peptides/spectra used during training: ∼300 K spectra, 1 M spectra, 2 M spectra, 3 M spectra, and the final models trained with all spectra (see Figures S1 and S2). When training the final model on the full data set, we adjusted the learning rate to 10–4 rather than using the default value (10–3) of AdamOptimizer in DeepNovo. Training our final model requires very significant computational resources and time. With the hardware used in this project, training took ∼12 h per epoch; our final model was achieved after 60 epochs. All training and testing were performed on PNNL’s Marianas cluster, a machine learning platform that is part of PNNL’s Institutional Computing. System specifications on the nodes used in this training were as follows: Dual Intel Broadwell E5–2620 v4 @ 2.10 GHz CPUs (16 cores per node), 64 GB 2133 MHz DDR4 memory, and Dual NVIDIA P100 12GB PCI-e based GPUs.
Experimental Design and Statistical Rationale
Given that Deep Neural Networks are very sensitive to overfit during the training procedure, we anticipated that a very large amount of data would be required to make useful models. The original DeepNovo was trained on 50,000 spectra, and we believed that a significantly larger amount of data would be necessary. As described below in Assessing Progress, we were able to quickly determine that a model with only 300,000 spectra was overfit. We therefore determined that we would aim for 5,000,000 spectra representing about 1,000,000 peptides in order to have sufficient data for training the very large neural networks that comprise Kaiko. During training we were able to determine that this number was more than sufficient to produce a generalized model that did not overfit to training data. Spectra included in the training, validation, and testing set are assessed as described above in the Data Generation section.
Assessing Progress
The training regimen for deep learning is pragmatically broken up into several rounds of iteration over the training data, called epochs. During each epoch, a minibatch stochastic optimization was employed, in which each batch of 128 spectra is randomly chosen and training proceeds on each batch one at a time. The model is trained by updating the parameters within the neural network (weights and biases) after each batch is compared to the true labels. While training, the error associated with the model can be calculated as a cross-entropy loss for the probabilities of correctly predicting the amino acid letters on the training data. After each batch, we also randomly sample 15,000 spectra from the validation data set (∼1% of total testing data) and compute the loss error, which we call the validation error. Importantly, model performance on this validation set is not used to update the model parameters; we simply use it to independently evaluate model performance and make a checkpoint to track the best models. The training and validation errors after each batch for 20 epochs of training are shown in Figure S2.
By comparing the training and validation errors, we clearly see when the model has started to overfit. This happens when the training error crosses over (becomes smaller than) the validation error and continues to decrease as the validation error levels off. This is a result of the model learning specific features of the training data that are not generalizable. In models built with more than 3 million spectra, no overfitting is seen yet; models built with less than 3 million spectra quickly overfit to the training data.
2.3. Comparing Kaiko to Other De Novo Tools
To compare the performance of Kaiko to state-of-the-art de novo tools, we analyzed all files in the testing data sets using DeepNovo,23 PEAKS,24 and Novor.25 As mentioned above, we used a pretrained model for the DeepNovo to predict peptide sequences for the test files using a “decode” option. PEAKS Studio version 8.5 was run using the default data refinement options on mzML formatted data.26De novo settings were as follows: precursor error tolerance - 20 ppm, fragment ion error tolerance - 0.02 Da. Oxidation of methionine was set as a variable modification. For Novor, the spectral files were converted from mzML to Mascot generic format (MGF) using MSConvert.27 Novor version 1.05 was run using the following settings: fragmentation - HCD, massAnalyzer - FT, precursor error tolerance - 20 ppm, fragment ion error tolerance - 0.02 Da. Oxidation of methionine was set as a variable modification. All other settings were left at their defaults. Only the best peptide spectrum match was used in the evaluation. Please refer to https://github.com/PNNL-Comp-Mass-Spec/Kaiko_Publication/analysis/for_novor and /for_peaks for specific implementation details.
2.4. Assigning Taxonomy to Unknown Samples
Proteomics data from six bacterial soil isolates was acquired using the same sample preparation and LC-MS/MS methods as described above. The isolates are from the natural isolate collection at the Kristen DeAngelis laboratory at the University of Massachusetts Amherst, and researchers at PNNL were blinded to the identity of these isolates until after both data generation and analysis were finished. Kaiko’s top-scoring peptide sequence for each spectrum was used for species identification. We filtered these peptide/spectrum matches to include only the top 25% according to Kaiko’s quality prediction score. We then exclude sequences shorter than 10 and longer than 17 residues. The resulting sequences were used to search the Uniref100 protein database (https://www.uniprot.org/uniref/) using DIAMOND28 to identify an organism(s) containing that peptide sequence. Only database matches of 100% were retained for species prediction. Taxon scoring then proceeded using a two-pass procedure. In the first pass, for each peptide sequence, all taxa possessing a 100% match were assigned 1 hit, such that multiple taxa were often assigned a hit from a single peptide sequence. Taxa were then ranked by the total number of hits assigned. In the second pass, hits were only assigned to the highest-ranking taxon with a 100% match to each predicted sequence. In this way, scoring is assigned to the candidate most likely to be correct.
For the analysis of the simplified human intestinal microbiota (SIHUMIx) synthetic community,29 we downloaded proteomic data from PRIDE accession PXD017035, http://ftp.ebi.ac.uk/pride-archive/2020/02/PXD017035/. All data sets corresponding to a transit time of 12 h from day 1 to 15 were downloaded and analyzed using the same method as above to determine eight bacterial strains present in the synthetic community.
2.5. Metaproteomics Data Analysis
Sample Preparation from Soils
Kansas prairie soil was quickly thawed and weighed into 10 g aliquots in 50 mL methanol/chloroform compatible tubes (Genesee Scientific, San Diego, CA) along with 10 mL of 0.9–2.0 mm stainless steel beads, 0.1 mm zirconia beads, and 0.1 mm garnet beads. All beads had previously been washed with chloroform and methanol and dried in a fume hood. Protein extraction occurred using a modified method of the Folch extraction30 specifically for soil called Soil MPLex (Metabolite, protein, lipid extraction).31 Here, 4 mL of ice-cold ultrapure “Type 1” water (Millipore, Billerica, MA) was added to each sample and transferred to an ice bucket in a fume hood. Using a 25 mL glass serological pipet, −20 °C 2:1 chloroform:methanol (v/v) (Sigma-Aldrich, St. Louis, MO) was added to the sample in a 5:1 ratio over sample volume (20 mL) and vigorously mixed (by vortexing). The tubes were attached to a 50 mL tube vortex-attachment and horizontally mixed for 10 min at 4 °C and placed inside a −80 °C freezer for 5 min. Using a probe sonicator (model FB505, Thermo Fisher Scientific, Waltham, MA) inside a fume hood, each sample was sonicated with a 6 mm probe (20 kHz fixed ultrasonic frequency) at 60% of the maximum amplitude for 30 s on ice, allowed to cool on ice, and then sonicated once more. Samples were allowed to cool for 5 min at −80 °C and then mixed for 60 s and centrifuged at 4500 × g for 10 min at 4 °C. The upper aqueous phase was removed, and the interphase containing proteins that partitioned between the methanol and chloroform phases was collected into a separate tube and precipitated through the addition of 5 mL of −20 °C 100% methanol. Following methanol addition, the tube was mixed and then centrifuged at 4500 × g for 5 min at 4 °C in order to pellet the proteins. The supernatant was decanted, and the protein pellet dried upside down. Meanwhile, the bottom organic phase was removed, and 5 mL of −20 °C 100% methanol was added to the bottom debris pellet, mixed, and centrifuged at 4500 × g for 5 min at 4 °C. The supernatant was removed, and the protein pellet was dried upside down. Protein pellets from both the debris and the interphase were frozen and lyophilized for 2 h.
Proteins from the interphase were solubilized by addition of 10 mL of SDS-Tris buffer containing 4% sodium dodecyl sulfate (SDS), 100 mM dl-dithiothreitol (DTT) in 100 mM Tris-HCl, pH 8.0 (Sigma-Aldrich, St Louis, MO), briefly probe sonicated at 20% amplitude, and then incubated on a lab tube rotator for 30 min at 300 rpm, 50 °C. Proteins from the debris pellet were solubilized in 20 mL of SDS buffer, horizontally vortexed for 10 min to lyse any remaining intact cells, and then combined with the interphase proteins and mixed on the rotator assembly for the time remaining (approximately 20 min). Following mixing, the tubes were centrifuged at 4500 × g for 10 min, and the supernatants from each tube were combined into a single 50 mL tube. The proteins were precipitated by adding up to 25% trichloroacetic acid (TCA; Sigma-Aldrich, St. Louis, MO), mixed, and placed at −20 °C overnight. The proteins were thawed and centrifuged at 4500 × g at 4 °C for 10 min to collect the precipitated proteins. The supernatant was gently decanted, and the protein pellet washed through addition of 2 mL of −20 °C acetone, mixed, and then placed at −80 °C for 5 min. Proteins were pelleted by centrifugation for 10 min at 4500 × g at 4 °C. The acetone was removed by gently decanting, and the wash step was repeated 2 more times. The washed pellet was then air-dried by inverting the tube. After drying, 100–200 μL of SDS-Tris buffer was added; the solution was transferred into 1.5 mL tubes and incubated at 95 °C for 5 min, then cooled at 4 °C for 10 min. The samples were centrifuged at 15,000 × g for 10 min to pellet any remaining debris and transferred into fresh 1.5 mL tubes in preparation for digestion using the Filter-Aided-Sample-Preparation (FASP) digestion method.32 For protein digestion, up to 30 μL of proteins in SDS-Tris buffer were transferred to a 30,000 Da molecular weight cut off (MWCO) 500 μL spin filter provided in the Expedeon FASP kit (Expedeon LTD, Cambridgeshire, UK) along with 400 μL of 8 M urea solution. The spin filter was centrifuged at 14,000 × g for 30 min. The waste was removed from the collection tubes, and 400 μL of 8 M urea solution was added to each sample and centrifuged as described above, then repeated for a total of 3 urea additions. 400 μL of 25 mM NH4HCO3, pH 8, was added and centrifuged as described above, then repeated for a total of 2 ammonium bicarbonate washes. The spin column was transferred into a freshly labeled collection tube, and 75 μL of NH4HCO3 was added to the filter along with 4 μL of 1 μg/μL molecular grade trypsin (Thermo Fisher, Waltham, MA), then incubated at 37 °C for 3 h. After digestion, 40 μL of NH4HCO3 was added to the sample and centrifuged at 14,000 × g for 20 min. Another 40 μL of NH4HCO3 was added to the top of the filter, mixed, and centrifuged again for 10 min. The filter was discarded, and the collected peptides were treated with potassium chloride (KCl) in order to ensure all the SDS was removed.33 To accomplish this, potassium chloride was added to the peptides in NH4HCO3 resulting in a final concentration of 2 M KCl, and then mixed and allowed to rest for 10 min at room temperature. To pellet the SDS, the peptide solution containing NH4HCO3 and KCl was centrifuged at 14,000 × g for 10 min. The supernatant was transferred to a fresh tube without disturbing the SDS pellet, and salts were removed using a microspin C18 column according to the manufacturer’s instructions (the Nest Group, Inc., Southborough, MA). Peptides from the aliquots of 10 g of soil were combined to generate a single peptide sample. A bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, Waltham, MA) was performed to determine the peptide concentration.
The peptide sample was separated with a commercial Waters (Milford, MA) XBridge 5 μm particle size C18 column (4.6 mm i.d. × 250 mm length) with an attached 20-mm-long × 4.6-mm-i.d. guard column. Fractionation was performed at 0.5 mL/min using an Agilent 1100 series HPLC system (Agilent Technologies, Santa Clara, CA) with two mobile phases: (A) 10 mM NH4HCO2 (pH 10.0) and (B) 10 mM NH4HCO2 (pH 10.0) with acetonitrile (10:90). A six-step gradient was adjusted over 120 min by replacing mobile phase A with B according to (1) 100–95% over the first 10 min, (2) 95–65% from minutes 10 to 70, (3) 65–30% from minutes 70 to 85, (4) then maintained mobile phase A at 30% from minutes 85 to 95, (5) re-equilibrated with 100% mobile phase A from minute 95 to 105, and (6) mobile phase A held at 100% until minute 120. Fractions were collected every 1.25 min (96 fractions over the entire gradient) with every 24th fraction combined for a total of 24 final fractions (rows of a 96 well plate were pooled by every other row). All fractions were dried under vacuum and suspended in 25 μL H2O. A final BCA assay was done on the fractions, and each was diluted to 0.1 μg/μL for LC-MS/MS analysis. (See LC-MS/MS data acquisition methods above.)
Analyzing 16S rRNA Amplicon Sequences
16S rRNA gene amplicon sequencing data was downloaded from https://osf.io/4uvj7/, performed using the protocol developed by the Earth Microbiome Project.1 Please refer to the previous studies34,35 for 16S rRNA gene amplicon sequencing in detail. The 16S rRNA amplicon sequences were first reprocessed by Hundo pipeline36 (v1.2.8), a command line interface work comprising a set of existing software together with validated custom methods derived from QIIME.37 In brief, the sequences were first quality filtered to remove the adaptors and contaminated reads from Phix genomes by BBDuk2.38 The passing reads were merged and checked for chimera, which were subjected to clustering into OTUs by VSEARCH39 using the default parameters. The abundance of each OUT was estimated by the read coverage of the OUT representative sequences (VSEARCH). In comparison to the Silva database40 implemented in Hundo, the NCBI database was reported with a higher confidence of lineage assignment to lower taxonomic levels.41 The de-replicated representative sequences of each OUT were then annotated following the same workflow coded in Hundo with modifications and using NCBI 16S Refseq database (https://www.ncbi.nlm.nih.gov/refseq/targetedloci/16S_process/, accessed on April 9, 2020) instead. The top 25 hits of each OUT representative sequence were kept and screened for those with percent identity higher than 85% and bit score greater than 125. For OTUs with more than one qualified hits, we will perform the lowest common ancestor (LCA) algorithm using an R package, taxize.42 OTUs with only one qualified hit adopted the lineage of the hits, and the rest were left unclassified.
Constructing Protein Database for Metaproteomic Data Analysis with Kaiko
Raw mass spectrometry files were converted to the PSI open format mzML26 using msConvert,27 which were converted to MGF files compatible with the Kaiko model. After performing Kaiko prediction, as used for assigning taxa to the unknown samples, we used Kaiko’s top 25% scoring peptide sequences predicted from each sample to identify the most likely candidate organisms using DIAMOND over the Uniref100 database. The protein database was constructed by aggregating all the reference sequences associated with the top 100 bacterial organisms from the Uniref100 into a single fasta (8.2GB).
Peptide Identification and Functional Analysis with the Constructed Database
Against the protein database constructed from the Kaiko prediction, MSGF+ was performed to identify peptide sequences with the false discovery rate (FDR) cutoff. The search parameters and values or settings were as follows: PrecursorMassTolerance, 20.0 ppm; IsotopeErrorRange, (−1, 1); TargetDecoyAnalysis, true; FragmentationMethod, as written in the spectrum; InstrumentID, 0; Enzyme, Tryp; NumTolerableTermini, 2; MinPeptideLength, 6; MaxPeptideLength, 50; MinCharge, 2; MaxCharge, 5; and NumMatchesPerSpec, 1. PSM results from MSGF+ were filtered using MSnID43 (v1.20.0). Filters based on the cleavage patterns for the trypsin were applied, e.g., nuIrregCleavages==0 and numMissCleavages<=2. Optimizing the MS/MS filter was applied to achieve the maximum number of identifications within a given FDR upper limit threshold. The Nelder–Mead method was employed for parameter optimization (MS-GF:SpecEValue and absParentMassErrorPPM), and for 1% peptide FDR, SpecEValue ≤1.0e-11, and 11 ppm mass window with the ppm offset adjustment were determined. For functional annotation for metaproteomics, Unipept44 (v4.6.3, https://unipept.ugent.be/datasets, accessed on May 23, 2022) was used with “Equate I and L”, “Filter duplicate peptides”, and “Advanced missed cleavage handling” options via Unipept Desktop (v1.2.5).
2.6. Data Availability
The mass spectrometry proteomics data for this benchmark set are split into two separate depositions, for the training and testing data sets, respectively. The training data set consists of spectra from 51 organisms and has been deposited to the ProteomeXchange Consortium via the PRIDE45 partner repository with the data set identifier PXD010000. The testing data set consists of spectra for 4 organisms and has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD010613. The metaproteomics data set has been deposited to the MassIVE Repository with the accession identifier MSV000086336.
3. Results
3.1. New Model for De Novo MS/MS Identification
Using a large and environmentally diverse set of mass spectrometry proteomics data, we sought to improve on peptide/spectrum identification where no protein sequence database is available. We adapted a deep neural network structure23 and trained a new model called Kaiko, after the Japanese deep ocean submersible used to explore the Marianas Trench. For training and validation, we used 4,604,540 spectra and 927,316 peptides from 51 distinct bacteria (Figure 1A, Table S1). After training, we evaluated the accuracy of Kaiko for PSM identification against spectra in the test data set consisting of spectra from four additional organisms not used in model training (511,765 spectra and 90,048 peptides). Kaiko achieved an average accuracy of 33% over all testing files and organisms, a significant improvement over other de novo algorithms (Figure 1B). When considering the top five spectrum annotations, average accuracy exceeded 41%.
Figure 1.

Training, validation, and testing of a new de novo peptide identification algorithm. (A) Bacteria represented in training and testing data and shown in a phylogenetic tree built from the multiple sequence alignment of rplB is shown for all organisms in the training (white nodes) and testing (red nodes) data sets. The size of the node is scaled to represent the number of spectra used. (B) Accuracy of spectrum annotation for four de novo spectrum annotation tools. (C) For each peptide sequence length, the accuracy of spectrum annotation is shown for each of the four algorithms.
Deep neural networks, like Kaiko, require very large training data sets for parameter optimization. The proteomics data used in training, validation, and testing come from 55 distinct bacteria and contain more than 1 million unique peptides (see Methods). The specific bacteria chosen for this set of 55 is less important than their contribution to diversity of peptide sequences, which is required for the model to fully generalize. Although we used microbes frequently found in soils, this does not constrict Kaiko’s utility to only soil microbes or only bacteria. After training, we demonstrate that the Kaiko model achieves generality (Figures S1 and S2) and is not limited by taxonomic divisions or environmental niche. Training Kaiko with an extensive diversity of peptide sequences allowed the model to successfully identify organisms in taxonomic phyla where no training data existed (see section 3.2).
We looked at model performance as a function of peptide length (Figure 1C). Most algorithms performed well with short peptides, length <8. Unfortunately, these peptides are infrequent in bottom-up proteomics data samples (Figure S3). Kaiko exhibited significantly improved accuracy at all lengths, but especially for the most common peptide lengths (10–15 residues), where it achieved an accuracy of ∼30–60%. We note that Kaiko had high accuracy at very long peptide lengths of 15 and above. Although these peptides are extremely difficult to annotate de novo, they are valuable for predicting phylogeny, as the long sequences are more likely to be uniquely mapped to a small taxonomy range.
3.2. Organism Identification via De Novo Proteomics
Proteomics identification of natural bacterial isolates often requires de novo spectrum annotation. In section 3.1, Kaiko was trained and tested on the ability to identify a correct PSM. To demonstrate the ability of our deep learning-based algorithm to annotate spectra from an unknown organism and also to accurately identify the unknown organism, we obtained bottom-up proteomics data from six microbes isolated from soil and attempted to identify the sample. For each sample, we annotated the proteomics data with Kaiko and used DIAMOND28 to identify the closest sequences in the UniProt database46 (see Methods). We then plotted the organisms that had the most matching spectra and inferred the organism for the sample.
For four samples, a matched proteome database became public during our investigation; however, this was still blinded from our analysis. In each of these cases, we identified the exact species as the source of the sample (Table S2). This included two Verrucomicrobia: Opitutus sp. GAS368 and Verrucomicrobium sp. GAS474. The other two isolates with a matched genome were from the order Rhizobiales: Afipia sp. GAS231 and Rhizobiales bacterium GAS188. The Afipia sample also contained spectra which mapped to neighboring Bradyrhizobium species, which could be from shared gene content, contamination, or previously unidentified coculturing. We emphasize that Kaiko’s training/validation data did not include any organisms from the phylum Verrucomicrobia; thus, the correct identification of these two newly isolated microbes demonstrates that the model properly generalized and is not limited to the taxonomy of its training data.
For two samples, there is no sequenced genome, a very realistic situation for environmental samples. To evaluate the proteomics-based organism identification, we attempted to derive the true sample identity by 16S sequencing. Isolate 02 cannot be definitively assigned to a genus within NCBI’s taxonomy based on 16S sequencing, but it is close to multiple genera within the family Acidobacteriaceae. In Uniprot, there is no genome or associated proteome for this organism. Therefore, the species attribution from our pipeline will simply be to the nearest organism (which might be quite distant, as the 16S data indicates that the closest match is most likely a different genus). Using Kaiko’s peptide annotations, we identified two potential candidates for the sample: Acidobacterium capsulatum and Silvibacterium bohemicum (both Acidobacteriaceae). However, both species had significantly fewer peptide hits matching their proteome and, therefore, were weaker matches than expected. This weak attribution and splitting between organisms within the same family are consistent with the isolate’s ambiguous taxonomic assignment. The final sample, Isolate 01, was suggested to be a Gemmobacter by 16S sequencing. Peptide hits from Kaiko identified this sample as Rhodobacter sp. 24-YEA-8, which is within the same family as Gemmobacter (Rhodobacteraceae). With the difficulties surrounding bacterial taxonomic classification and the uncertainty of species designation, this is still a close match.
As a final demonstration of Kaiko’s ability to accurately identify organisms, we processed proteomics data from a synthetic community of known composition—the Simplified Human Intestinal Microbiota or SIHUMIx,29 which consisted of eight bacteria and represented common metabolic activities found in the human gut. Using our pipeline, we identified seven of the eight organisms as an exact match. A final organism with the average relative abundance of 0.08%, Clostridium butyricum was only resolved at the level of genus. Thus, even in multiorganism mixtures, Kaiko can confidently identify organisms directly from proteomic data—even when the organisms are from a taxonomic division or environmental niche that was not part of Kaiko’s training set.
3.3. Building a Protein Database without Metagenomics
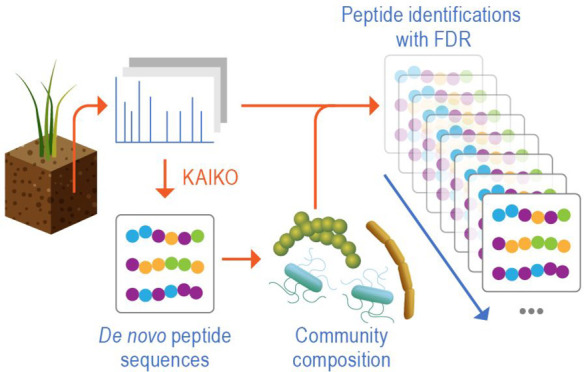
In metaproteomics data analysis, constructing a protein sequence database is a critical component for protein identification,47,48 as identification sensitivity suffers as database size increases.49 Given the success of Kaiko in identifying unknown organisms, we derive the organisms present in a sample directly from Kaiko’s analysis of metaproteomics data, thus enabling confident peptide identification when the genomic information is limited or unavailable (Figure 2). This set of organisms and their respective proteins can then be used in a second-pass database search to take advantage of a database algorithm’s natural sensitivity advantage.50
Figure 2.

Overview of the metaproteomics data analysis leveraging de novo spectrum identification based on the Kaiko model. Peptides are identified using Kaiko and used to infer community composition (steps 1–3). In step 4, the spectra are reanalyzed using a database search algorithm, e.g., MSGF+, and the protein sequence database created in step 3. This yields a final list of peptide identifications which can be used for functional analysis.
To demonstrate this de novo-based metaproteomics pipeline, we analyzed metaproteomic data acquired from pooled samples of native soils collected in three sites located in Kansas.34,51 To identify species, the Kaiko model and DIAMOND were employed to determine the most dominant organisms, and whole proteomes were retrieved from UniProt (see Methods). 6410 unique taxa IDs were identified in total, and 224 taxa had more than 5 matched peptides. These taxa included well-known bacterial phylotypes consistently detected as a core component of soil ecosystem such as Proteobacteria, Actinobacteria, Acidobacteria, Planctomycetes, Chloroflexi, Verrucomicrobia, Bacteroidetes, Gemmatimonadetes, Firmicutes, and Armatimonadetes.52,53 In addition, our pipeline revealed globally abundant fungal classes such as Agaricomycetes, Sordariomycetes, Eurotiomycetes, Leotiomycetes, and Mortierellomycetes.53,54 This identification of fungal components of the soil microbiome demonstrates a significant advantage of metaproteomics over 16S metagenomics which is strictly limited to bacteria.
To evaluate the taxa annotation from the Kaiko model, we also identified taxa using 16S rRNA data from the same samples (see Methods). 243 unique taxa IDs were determined for 3,693 OTUs. All of the highly abundant phyla detected by 16S were also detected by Kaiko (Table 1). Several phyla uniquely found by Kaiko are known to be present in environmental soils.45,55−59 For example, Candidatus Rokubacteria is distributed globally in diverse terrestrial ecosystems, including soils and the rhizosphere,55 and Candidatus Tectomicrobia has also been detected in soils.45
Table 1. Relative Abundance of the Top 20 Bacterial Phyla Detected from 16S and Kaikoa.
| Phylum | Read counts by 16S | Peptide counts by Kaiko | Relative read counts % total reads at the phylum level | Relative Peptide counts % By Kaiko at the phylum level |
|---|---|---|---|---|
| Proteobacteria* | 40778 | 4903 | 34.6 | 38.1 |
| Actinobacteria* | 16501 | 3949 | 14.0 | 30.7 |
| Acidobacteria* | 18562 | 1010 | 15.7 | 7.8 |
| Firmicutes* | 6761 | 634 | 5.7 | 4.9 |
| Chloroflexi* | 767 | 479 | 0.7 | 3.7 |
| Bacteroidetes* | 9712 | 467 | 8.2 | 3.6 |
| Planctomycetes* | 11427 | 321 | 9.7 | 2.5 |
| Candidatus Rokubacteria* | - | 266 | - | 2.1 |
| Verrucomicrobia* | 11841 | 237 | 10.0 | 1.8 |
| Cyanobacteria | 489 | 162 | 0.4 | 1.3 |
| Gemmatimonadetes* | 869 | 61 | 0.7 | 0.5 |
| Nitrospirae* | 18 | 44 | - | 0.3 |
| Candidatus Tectomicrobia* | - | 43 | - | 0.3 |
| Deinococcus-Thermus | - | 32 | - | 0.2 |
| Spirochaetes | - | 32 | - | 0.2 |
| Elusimicrobia | - | 15 | - | 0.1 |
| Tenericutes | 99 | 15 | 0.1 | 0.1 |
| Armatimonadetes | 75 | 13 | 0.1 | 0.1 |
| Ignavibacteriae | 16 | 6 | 0.01 | 0.05 |
| Chlamydiae | 2 | 4 | 0.00 | 0.03 |
A dash in the table represents the corresponding phylum was “not detected”. The asterisk (*) indicates that some taxa in the corresponding phyla are used to construct the protein DB.
To construct the protein database from the identified organisms, we selected the 100 most abundant bacterial taxa, resulting in a protein database containing 17,448,135 protein clusters (UniRef sequences) from 12 bacterial phyla. We note that the 100 taxa identified by proteome data consist of 91 species, 1 genus, 7 strains, and the remaining 1 had no phylogenetic rank. Unfortunately, the 16S taxa annotations were often resolved only to a phylum or class level; relatively few taxa from 16S data were able to be narrowly identified at the level of genus or species. Therefore, using a 16S data set to create a metaproteomic database would include hundreds of species within a broad taxonomic category, such as phylum, and would dramatically increase the size of the protein sequence database and significantly reduce the sensitivity of the proteomics database tools.
3.4. Soil Metaproteomic Data Analysis
Using the protein database generated by Kaiko, we reanalyzed the mass spectra from the soil samples using the database search tool MSGF+ and identified 20,089 unique peptides from 23,381 PSMs with 1% peptide FDR (see Methods). We performed functional annotations with these identified peptides using Unipept44 and found 1059 Enzyme Commission (EC) numbers matched to 7161 peptides (36%). Functions in the top 20 EC numbers (Table S3) included various enzymatic functions for transcription and translation, energy production, and signaling. 618 EC numbers were mapped to KEGG metabolic pathways, extensively covering carbohydrate and amino acid metabolism, as well as the metabolism of cofactors, vitamins, and xenobiotics.
Among identified peptides, 10,784 peptides were highly conserved sequences and therefore were assigned to bacterial phyla. 2972 of these phyla-affiliated peptides were linked to 578 EC numbers (Figure S4). These highly conserved peptides were assigned to ubiquitous bacterial functions commonly detected across most phyla, such as DNA-directed RNA polymerase (EC:2.7.7.6), H(+)-transporting two-sector ATPase (EC:7.1.2.2), with a cytochrome as acceptor (EC:1.1.2.-), glutamine synthetase (EC: 6.3.1.2), polyribonucleotide nucleotidyltransferase (EC:2.7.7.8), superoxide dismutase (EC:1.15.1.1), and protein-synthesizing GTPase (EC:3.6.4.-). In particular, EC numbers of highly ranked peptide counts were mainly detected in abundant phyla (Proteobacteria, Actinobacteria, and Acidobacteria), and functional information was biased by the common and abundant proteins.
We next examined the mapped EC numbers to identify metabolic functions for specific taxa (Figure 3). By mapping the taxonomic affiliation of the enzymatic reactions within metabolic pathways, it was possible to determine which metabolic pathways were shared or unique among the represented phyla. EC numbers involved in carbon metabolism were often found in organisms from multiple phyla and represent basic functions from glycolysis, carbon fixation, the TCA cycle, etc. Enzymes and metabolic functions for 362 EC numbers were represented by only a single phylum and are shown with different colors in Figure 3. The two most abundant phyla detected in the metaproteomics data were Proteobacteria and Actinobacteria. It is clear from the functional mapping of peptides that these two phyla utilize distinct metabolic routes. For example, purine metabolism contains numerous enzymes which are exclusively found in either Actinobacteria or Proteobacteria (Figure S5). Significant divergence between these two dominant taxa was also seen in enzymes related to amino acid metabolism.
Figure 3.

Distribution of bacterial functions in the metabolic pathway map. Several metabolic steps are shared among multiple phyla (dark gray). Other colors indicate unique EC numbers and their associated metabolic function found only in a specific phylum.
Finally, we examined the peptides and biological functions associated with species unique to the Kaiko database, i.e., species not found in the 16S rRNA sequences. 92 peptides were identified in Candidatus Rokubacteria and mapped to EC numbers. Biological functions associated with seven EC numbers were exclusive to Candidatus Rokubacteria: thioredoxin-dependent peroxiredoxin (EC:1.11.1.24), pyrroloquinoline-quinone synthase (EC:1.3.3.11), thioredoxin-disulfide reductase (EC:1.8.1.9, selenocompound metabolism), 3-oxoadipate enol-lactonase (EC:3.1.1.24, benzoate degradation), inositol-phosphate phosphatase (EC:3.1.3.25, inositol phosphate metabolism and streptomycin biosynthesis), 1,4-dihydroxy-2-naphthoyl-CoA synthase (EC:4.1.3.36, Ubiquinone and other terpenoid-quinone biosynthesis), and nicotinate phosphoribosyltransferase (EC:6.3.4.21, nicotinate and nicotinamide metabolism).
4. Discussion and Conclusion
Although genome and metagenome sequencing have greatly expanded the number of species that contain a sequenced genome and therefore an annotated proteome, there are still significant practical and financial barriers that prevent laboratories from always having an assembled and well-annotated genome for samples taken from nature. Yet, metaproteome spectrum identification tools rely on a protein sequence database. Therefore, tools which can create a proteome database for environmental samples without requiring sequencing data are a significant benefit to the microbiome community. One option for creating a proteome database without using sequencing data utilizes a de novo interpretation of metaproteomics data to identify organisms present in the sample. A significant drawback of current de novo tools is their poor performance on spectra from diverse organisms (see Figure 1). Algorithms which are only exposed to a limited number of organisms,20,23 or those that focus only on human data,25 will be inadequate when faced with the vast sequence diversity of microbial proteins found in soil and environmental samples.
To assist in the analysis of metaproteomic data, we have created a pipeline for generating the proteome sequence database directly from the metaproteomic data. A key element in our pipeline is a new de novo spectrum annotation tool, Kaiko, which has significantly improved accuracy compared to other de novo algorithms. This improvement comes from a deliberate focus on training the algorithm with mass spectrometry data from dozens of diverse environmental bacteria. Moreover, our training data set size is dramatically larger than comparable de novo tools in terms of the number of peptides and spectra, which was essential for overcoming an overfit model. We evaluated Kaiko by using it to identify the taxonomy of bacterial soil isolates, including samples from phyla where no training data existed. Thus, it is better equipped for evaluating metaproteomics data where identifying spectra from diverse organisms is essential. Indeed, our algorithm was able to confidently identify numerous fungi from metaproteomics data, despite having never been trained on eukaryotic peptides. Thus, we believe that Kaio will work well for any organism.
When using Kaiko as part of our database generation pipeline to identify soil community composition, we were able to identify all abundant species from 16S data, and also new species with significant proteomic evidence which were not seen in the sequencing data. Indeed, 5 of the top 16 taxa (>30%) identified in the metaproteomics data were not identified in sequencing data. These “hidden microbes” represent bacteria that are known to play an important role in community metabolism and function,55 including secondary metabolite biosynthesis60,61 as seen in our Candidatus Rokubacteria data.
A second significant advantage of inferring community composition directly from metaproteome data is the level of taxon specificity. Using metaproteome data, we could narrow taxon identification to species or strain (98%). However, taxa identified using 16S data for these same samples frequently were only able to distinguish broad taxonomic levels. Unfortunately, spectrum identification algorithms generally suffer a significant sensitivity loss when working with large protein databases.49 Therefore, methods which specify community composition in broad taxonomic terms will yield poor results, compared to a method which is able to narrowly define organisms present in the community.
As metaproteomics data analysis continues to mature, progress will happen in multiple areas, e.g., more sensitive peptide ID algorithms, improved protein inference for multiorganism mapped peptides, and functional analysis of pathways with multiple participating organisms. However, a central feature in all of this work is the original identification of spectra, and currently the best algorithms require a protein database. Thus, the creation of a protein sequence database is a pivotal step in metaproteomics data analysis. The most important future improvement in creating a protein sequence database will come from greater coverage and greater specificity in the identification of community membership. De novo proteomics offers one avenue for this, which is independent of advances made in sequencing technologies. Improving the accuracy of de novo tools, especially with regard to diverse environmental sequences, will be a significant benefit to metaproteomics. We envision continued improvements in peptide identification will be obtainable in the near future with larger and more diverse training data sets becoming available, and increasingly sophisticated learning models, furthering the utility of this approach for database building.
Acknowledgments
The authors thank Court Corley and Nathan Hodas (PNNL) for insightful discussions. We thank Kristen DeAngelis and Grace Pold (University of Massachusetts Amherst) for natural isolate samples. Funding for this project was provided by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, Early Career Research Program (to SHP and KEBJ) and PNNL’s Deep Learning for Scientific Discovery initiative. Proteomics data used in this manuscript were generated in the Environmental Molecular Science Laboratory, a DOE national scientific user facility at Pacific Northwest National Laboratory (PNNL) in Richland, WA. Battelle operates PNNL for the DOE under contract DE-AC05-76RLO01830.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.2c00334.
Figure S1. Improvement of deep neural networks with more training data. Figure S2. Training and validation errors. During the epochs of learning for the deep neural network, progress is measured by evaluating the accuracy of spectrum annotation. Figure S3. Distribution of peptide lengths used for training and testing the Kaiko model. Figure S4. Heatmap of the peptide counts for the most common functions over the diverse phyla. Columns and rows in the heatmap represent the phyla and EC numbers, respectively. Figure S5. Taxa-specific peptides for enzymes in purine metabolism at the phylum level. Table S1. LC/MS data files for training and testing the Kaiko model. Table S2. For each of the six natural isolates, replicate proteomics data was annotated with Kaiko and high-rank taxa were identified. Table S3. Top 20 of EC numbers most frequently matched from the unique peptides using Unipept 4.6.3 with the identified peptide sequences. (PDF)
Supplementary data. (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Thompson L. R.; Sanders J. G.; McDonald D.; Amir A.; Ladau J.; Locey K. J.; Prill R. J.; Tripathi A.; Gibbons S. M.; Ackermann G.; Navas-Molina J. A.; Janssen S.; Kopylova E.; Vázquez-Baeza Y.; González A.; Morton J. T.; Mirarab S.; Xu Z. Z.; Jiang L.; Haroon M. F.; Kanbar J.; Zhu Q.; Song S. J.; Kosciolek T.; Bokulich N. A.; Lefler J.; Brislawn C. J.; Humphrey G.; Owens S. M.; Hampton-Marcell J.; Berg-Lyons D.; McKenzie V.; Fierer N.; Fuhrman J. A.; Clauset A.; Stevens R. L.; Shade A.; Pollard K. S.; Goodwin K. D.; Jansson J. K.; Gilbert J. A.; Knight R.; Agosto Rivera J. L.; Al-Moosawi L.; Alverdy J.; Amato K. R.; Andras J.; Angenent L. T.; Antonopoulos D. A.; Apprill A.; Armitage D.; Ballantine K.; Bárta J.; Baum J. K.; Berry A.; Bhatnagar A.; Bhatnagar M.; Biddle J. F.; Bittner L.; Boldgiv B.; Bottos E.; Boyer D. M.; Braun J.; Brazelton W.; Brearley F. Q.; Campbell A. H.; Caporaso J. G.; Cardona C.; Carroll J. L.; Cary S. C.; Casper B. B.; Charles T. C.; Chu H.; Claar D. C.; Clark R. G.; Clayton J. B.; Clemente J. C.; Cochran A.; Coleman M. L.; Collins G.; Colwell R. R.; Contreras M.; Crary B. B.; Creer S.; Cristol D. A.; Crump B. C.; Cui D.; Daly S. E.; Davalos L.; Dawson R. D.; Defazio J.; Delsuc F.; Dionisi H. M.; Dominguez-Bello M. G.; Dowell R.; Dubinsky E. A.; Dunn P. O.; Ercolini D.; Espinoza R. E.; Ezenwa V.; Fenner N.; Findlay H. S.; Fleming I. D.; Fogliano V.; Forsman A.; Freeman C.; Friedman E. S.; Galindo G.; Garcia L.; Garcia-Amado M. A.; Garshelis D.; Gasser R. B.; Gerdts G.; Gibson M. K.; Gifford I.; Gill R. T.; Giray T.; Gittel A.; Golyshin P.; Gong D.; Grossart H. P.; Guyton K.; Haig S. J.; Hale V.; Hall R. S.; Hallam S. J.; Handley K. M.; Hasan N. A.; Haydon S. R.; Hickman J. E.; Hidalgo G.; Hofmockel K. S.; Hooker J.; Hulth S.; Hultman J.; Hyde E.; Ibáñez-Álamo J. D.; Jastrow J. D.; Jex A. R.; Johnson L. S.; Johnston E. R.; Joseph S.; Jurburg S. D.; Jurelevicius D.; Karlsson A.; Karlsson R.; Kauppinen S.; Kellogg C. T. E.; Kennedy S. J.; Kerkhof L. J.; King G. M.; Kling G. W.; Koehler A. V.; Krezalek M.; Kueneman J.; Lamendella R.; Landon E. M.; Lanede Graaf K.; LaRoche J.; Larsen P.; Laverock B.; Lax S.; Lentino M.; Levin I. I.; Liancourt P.; Liang W.; Linz A. M.; Lipson D. A.; Liu Y.; Lladser M. E.; Lozada M.; Spirito C. M.; MacCormack W. P.; MacRae-Crerar A.; Magris M.; Martín-Platero A. M.; Martín-Vivaldi M.; Martínez L. M.; Martínez-Bueno M.; Marzinelli E. M.; Mason O. U.; Mayer G. D.; McDevitt-Irwin J. M.; McDonald J. E.; McGuire K. L.; McMahon K. D.; McMinds R.; Medina M.; Mendelson J. R.; Metcalf J. L.; Meyer F.; Michelangeli F.; Miller K.; Mills D. A.; Minich J.; Mocali S.; Moitinho-Silva L.; Moore A.; Morgan-Kiss R. M.; Munroe P.; Myrold D.; Neufeld J. D.; Ni Y.; Nicol G. W.; Nielsen S.; Nissimov J. I.; Niu K.; Nolan M. J.; Noyce K.; O’Brien S. L.; Okamoto N.; Orlando L.; Castellano Y. O.; Osuolale O.; Oswald W.; Parnell J.; Peralta-Sánchez J. M.; Petraitis P.; Pfister C.; Pilon-Smits E.; Piombino P.; Pointing S. B.; Pollock F. J.; Potter C.; Prithiviraj B.; Quince C.; Rani A.; Ranjan R.; Rao S.; Rees A. P.; Richardson M.; Riebesell U.; Robinson C.; Rockne K. J.; Rodriguezl S. M.; Rohwer F.; Roundstone W.; Safran R. J.; Sangwan N.; Sanz V.; Schrenk M.; Schrenzel M. D.; Scott N. M.; Seger R. L.; Seguinorlando A.; Seldin L.; Seyler L. M.; Shakhsheer B.; Sheets G. M.; Shen C.; Shi Y.; Shin H.; Shogan B. D.; Shutler D.; Siegel J.; Simmons S.; Sjöling S.; Smith D. P.; Soler J. J.; Sperling M.; Steinberg P. D.; Stephens B.; Stevens M. A.; Taghavi S.; Tai V.; Tait K.; Tan C. L.; Taş N.; Taylor D. L.; Thomas T.; Timling I.; Turner B. L.; Urich T.; Ursell L. K.; Van Der Lelie D.; Van Treuren W.; Van Zwieten L.; Vargas-Robles D.; Thurber R. V.; Vitaglione P.; Walker D. A.; Walters W. A.; Wang S.; Wang T.; Weaver T.; Webster N. S.; Wehrle B.; Weisenhorn P.; Weiss S.; Werner J. J.; West K.; Whitehead A.; Whitehead S. R.; Whittingham L. A.; Willerslev E.; Williams A. E.; Wood S. A.; Woodhams D. C.; Yang Y.; Zaneveld J.; Zarraonaindia I.; Zhang Q.; Zhao H. A Communal Catalogue Reveals Earth’s Multiscale Microbial Diversity. Nature 2017, 551 (7681), 457–463. 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C.; Marcotte E. M. Insights into the Regulation of Protein Abundance from Proteomic and Transcriptomic Analyses. Nat. Rev. Genet. 2012, 13 (4), 227–232. 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmes P.; Bond P. L. Metaproteomics: Studying Functional Gene Expression in Microbial Ecosystems. Trends Microbiol 2006, 14 (2), 92–97. 10.1016/j.tim.2005.12.006. [DOI] [PubMed] [Google Scholar]
- van den Bossche T.; Arntzen M. Ø.; Becher D.; Benndorf D.; Eijsink V. G. H.; Henry C.; Jagtap P. D.; Jehmlich N.; Juste C.; Kunath B. J.; Mesuere B.; Muth T.; Pope P. B.; Seifert J.; Tanca A.; Uzzau S.; Wilmes P.; Hettich R. L.; Armengaud J. The Metaproteomics Initiative: A Coordinated Approach for Propelling the Functional Characterization of Microbiomes. Microbiome 2021, 9 (1), 243. 10.1186/s40168-021-01176-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Figeys D. Perspective and Guidelines for Metaproteomics in Microbiome Studies. J. Proteome Res. 2019, 18 (6), 2370–2380. 10.1021/acs.jproteome.9b00054. [DOI] [PubMed] [Google Scholar]
- Jouffret V.; Miotello G.; Culotta K.; Ayrault S.; Pible O.; Armengaud J. Increasing the Power of Interpretation for Soil Metaproteomics Data. Microbiome 2021, 9 (1), 195. 10.1186/s40168-021-01139-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng J. K.; McCormack A. L.; Yates J. R. An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom. 1994, 5 (11), 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Mass Spectrometry-Based Proteomics. Nature 2003, 422 (6928), 198–207. 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Timmins-Schiffman E.; May D. H.; Mikan M.; Riffle M.; Frazar C.; Harvey H. R.; Noble W. S.; Nunn B. L. Critical Decisions in Metaproteomics: Achieving High Confidence Protein Annotations in a Sea of Unknowns. ISME J. 2017, 11 (2), 309–314. 10.1038/ismej.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao D. P.; Zhou Q.; Chen C. Y.; Quan Z. X. Coverage Evaluation of Universal Bacterial Primers Using the Metagenomic Datasets. BMC Microbiology 2012, 12 (1), 1–8. 10.1186/1471-2180-12-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe A. C.; Jansson J. K.; Malfatti S. A.; Tringe S. G.; Tiedje J. M.; Brown C. T. Tackling Soil Diversity with the Assembly of Large, Complex Metagenomes. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (13), 4904–4909. 10.1073/pnas.1402564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-R L. M.; Konstantinidis K. T. Nonpareil: A Redundancy-Based Approach to Assess the Level of Coverage in Metagenomic Datasets. Bioinformatics 2014, 30 (5), 629–635. 10.1093/bioinformatics/btt584. [DOI] [PubMed] [Google Scholar]
- Poretsky R.; Rodriguez-R L. M.; Luo C.; Tsementzi D.; Konstantinidis K. T. Strengths and Limitations of 16S RRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS One 2014, 9 (4), e93827. 10.1371/journal.pone.0093827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira-Marques J.; Hout A.; Ferreira R. M.; Weber M.; Pinto-Ribeiro I.; Van Doorn L. J.; Knetsch C. W.; Figueiredo C. Impact of Host DNA and Sequencing Depth on the Taxonomic Resolution of Whole Metagenome Sequencing for Microbiome Analysis. Frontiers in Microbiology 2019, 10 (JUN), 1277. 10.3389/fmicb.2019.01277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-R L. M.; Konstantinidis K. T. Estimating Coverage in Metagenomic Data Sets and Why It Matters. ISME J. 2014, 8 (11), 2349–2351. 10.1038/ismej.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. K. R.; Jung T.; Thuy-Boun P. S.; Wang A. Y.; Yates J. R.; Wolan D. W. ComPIL 2.0: An Updated Comprehensive Metaproteomics Database. J. Proteome Res. 2019, 18 (2), 616–622. 10.1021/acs.jproteome.8b00722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap P.; Goslinga J.; Kooren J. A.; Mcgowan T.; Wroblewski M. S.; Seymour S. L.; Griffin T. J. A Two-Step Database Search Method Improves Sensitivity in Peptide Sequence Matches for Metaproteomics and Proteogenomics Studies. PROTEOMICS 2013, 13 (8), 1352–1357. 10.1002/pmic.201200352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Ning Z.; Mayne J.; Moore J. I.; Li J.; Butcher J.; Deeke S. A.; Chen R.; Chiang C.-K.; Wen M.; Mack D.; Stintzi A.; Figeys D. MetaPro-IQ: A Universal Metaproteomic Approach to Studying Human and Mouse Gut Microbiota. Microbiome 2016, 4 (1), 31. 10.1186/s40168-016-0176-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Deeke S. A.; Ning Z.; Starr A. E.; Butcher J.; Li J.; Mayne J.; Cheng K.; Liao B.; Li L.; Singleton R.; Mack D.; Stintzi A.; Figeys D. Metaproteomics Reveals Associations between Microbiome and Intestinal Extracellular Vesicle Proteins in Pediatric Inflammatory Bowel Disease. Nat. Commun. 2018, 9 (1), 2873. 10.1038/s41467-018-05357-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank A.; Pevzner P. PepNovo: De Novo Peptide Sequencing via Probabilistic Network Modeling. Anal. Chem. 2005, 77 (4), 964–973. 10.1021/ac048788h. [DOI] [PubMed] [Google Scholar]
- Nakayasu E. S.; Burnet M. C.; Walukiewicz H. E.; Wilkins C. S.; Shukla A. K.; Brooks S.; Plutz M. J.; Lee B. D.; Schilling B.; Wolfe A. J.; Müller S.; Kirby J. R.; Rao C. V.; Cort J. R.; Payne S. H. Ancient Regulatory Role of Lysine Acetylation in Central Metabolism. mBio 2017, 8 (6), 1. 10.1128/mBio.01894-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly R. T.; Page J. S.; Luo Q.; Moore R. J.; Orton D. J.; Tang K.; Smith R. D. Chemically Etched Open Tubular and Monolithic Emitters for Nanoelectrospray Ionization Mass Spectrometry. Anal. Chem. 2006, 78 (22), 7796–7801. 10.1021/ac061133r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran N. H.; Zhang X.; Xin L.; Shan B.; Li M. De Novo Peptide Sequencing by Deep Learning. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (31), 8247–8252. 10.1073/pnas.1705691114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B.; Zhang K.; Hendrie C.; Liang C.; Li M.; Doherty-Kirby A.; Lajoie G. PEAKS: Powerful Software for Peptide de Novo Sequencing by Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2003, 17 (20), 2337–2342. 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- Ma B. Novor: Real-Time Peptide de Novo Sequencing Software. J. Am. Soc. Mass Spectrom. 2015, 26 (11), 1885–1894. 10.1007/s13361-015-1204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E. MzML: A Single, Unifying Data Format for Mass Spectrometer Output. PROTEOMICS 2008, 8 (14), 2776–2777. 10.1002/pmic.200890049. [DOI] [PubMed] [Google Scholar]
- Kessner D.; Chambers M.; Burke R.; Agus D.; Mallick P. ProteoWizard: Open Source Software for Rapid Proteomics Tools Development. Bioinformatics 2008, 24 (21), 2534–2536. 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B.; Xie C.; Huson D. H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12 (1), 59–60. 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- Schäpe S. S.; Krause J. L.; Engelmann B.; Fritz-Wallace K.; Schattenberg F.; Liu Z.; Müller S.; Jehmlich N.; Rolle-Kampczyk U.; Herberth G.; von Bergen M. The Simplified Human Intestinal Microbiota (SIHUMIx) Shows High Structural and Functional Resistance against Changing Transit Times in In Vitro Bioreactors. Microorganisms 2019, 7 (12), 641. 10.3390/microorganisms7120641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J.; Lees M.; Stanley G.H. S. A SIMPLE METHOD FOR THE ISOLATION AND PURIFICATION OF TOTAL LIPIDES FROM ANIMAL TISSUES. J. Biol. Chem. 1957, 226 (1), 497–509. 10.1016/S0021-9258(18)64849-5. [DOI] [PubMed] [Google Scholar]
- Nicora C. D.; Burnum-Johnson K. E.; Nakayasu E. S.; Casey C. P.; White R. A.; Chowdhury T. R.; Kyle J. E.; Kim Y. M.; Smith R. D.; Metz T. O.; Jansson J. K.; Baker E. S. The MPLEx Protocol for Multi-Omic Analyses of Soil Samples. JoVE (Journal of Visualized Experiments 2018, 2018 (135), e57343. 10.3791/57343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Universal Sample Preparation Method for Proteome Analysis. Nat. Methods 2009, 6 (5), 359–362. 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Zhou J. Y.; Dann G. P.; Shi T.; Wang L.; Gao X.; Su D.; Nicora C. D.; Shukla A. K.; Moore R. J.; Liu T.; Camp D. G.; Smith R. D.; Qian W. J. Simple Sodium Dodecyl Sulfate-Assisted Sample Preparation Method for LC-MS-Based Proteomics Applications. Anal. Chem. 2012, 84 (6), 2862–2867. 10.1021/ac203394r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy Chowdhury T.; Lee J.-Y.; Bottos E. M.; Brislawn C. J.; White R. A.; Bramer L. M.; Brown J.; Zucker J. D.; Kim Y.-M.; Jumpponen A.; Rice C. W.; Fansler S. J.; Metz T. O.; McCue L. A.; Callister S. J.; Song H.-S.; Jansson J. K. Metaphenomic Responses of a Native Prairie Soil Microbiome to Moisture Perturbations. mSystems 2019, 4 (4), 1. 10.1128/mSystems.00061-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R. A.; Bottos E. M.; Roy Chowdhury T.; Zucker J. D.; Brislawn C. J.; Nicora C. D.; Fansler S. J.; Glaesemann K. R.; Glass K.; Jansson J. K. Moleculo Long-Read Sequencing Facilitates Assembly and Genomic Binning from Complex Soil Metagenomes. mSystems 2016, 1 (3), 1. 10.1128/mSystems.00045-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.; Zavoshy N.; Brislawn C. J.; McCue L. A. Hundo: A Snakemake Workflow for Microbial Community Sequence Data. PeerJ Preprints 2018, 1. 10.7287/PEERJ.PREPRINTS.27272V1. [DOI] [Google Scholar]
- Caporaso J. G.; Kuczynski J.; Stombaugh J.; Bittinger K.; Bushman F. D.; Costello E. K.; Fierer N.; Pẽa A. G.; Goodrich J. K.; Gordon J. I.; Huttley G. A.; Kelley S. T.; Knights D.; Koenig J. E.; Ley R. E.; Lozupone C. A.; McDonald D.; Muegge B. D.; Pirrung M.; Reeder J.; Sevinsky J. R.; Turnbaugh P. J.; Walters W. A.; Widmann J.; Yatsunenko T.; Zaneveld J.; Knight R. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7 (5), 335–336. 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BBMap: A Fast, Accurate, Splice-Aware Aligner (Conference) | OSTI.GOV. https://www.osti.gov/biblio/1241166 (accessed 2021–11–12).
- Rognes T.; Flouri T.; Nichols B.; Quince C.; Mahé F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ. 2016, 2016 (10), e2584. 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C.; Pruesse E.; Yilmaz P.; Gerken J.; Schweer T.; Yarza P.; Peplies J.; Glöckner F. O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41 (D1), D590–D596. 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balvočiute M.; Huson D. H. SILVA, RDP, Greengenes, NCBI and OTT - How Do These Taxonomies Compare?. BMC Genomics 2017, 18 (2), 1–8. 10.1186/s12864-017-3501-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain S. A.; Szöcs E. Taxize: Taxonomic Search and Retrieval in R. F1000Res. 2013, 2, 191. 10.12688/f1000research.2-191.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bioconductor - MSnID. https://www.bioconductor.org/packages/release/bioc/html/MSnID.html (accessed 2021–11–12).
- Gurdeep Singh R.; Tanca A.; Palomba A.; Van Der Jeugt F.; Verschaffelt P.; Uzzau S.; Martens L.; Dawyndt P.; Mesuere B. Unipept 4.0: Functional Analysis of Metaproteome Data. J. Proteome Res. 2019, 18 (2), 606–615. 10.1021/acs.jproteome.8b00716. [DOI] [PubMed] [Google Scholar]
- Wilson M. C.; Mori T.; Rückert C.; Uria A. R.; Helf M. J.; Takada K.; Gernert C.; Steffens U. A. E.; Heycke N.; Schmitt S.; Rinke C.; Helfrich E. J. N.; Brachmann A. O.; Gurgui C.; Wakimoto T.; Kracht M.; Crüsemann M.; Hentschel U.; Abe I.; Matsunaga S.; Kalinowski J.; Takeyama H.; Piel J. An Environmental Bacterial Taxon with a Large and Distinct Metabolic Repertoire. Nature 2014, 506 (7486), 58–62. 10.1038/nature12959. [DOI] [PubMed] [Google Scholar]
- Bateman A.; Martin M. J.; Orchard S.; Magrane M.; Agivetova R.; Ahmad S.; Alpi E.; Bowler-Barnett E. H.; Britto R.; Bursteinas B.; Bye-A-Jee H.; Coetzee R.; Cukura A.; Silva A.; Da; Denny P.; Dogan T.; Ebenezer T. G.; Fan J.; Castro L. G.; Garmiri P.; Georghiou G.; Gonzales L.; Hatton-Ellis E.; Hussein A.; Ignatchenko A.; Insana G.; Ishtiaq R.; Jokinen P.; Joshi V.; Jyothi D.; Lock A.; Lopez R.; Luciani A.; Luo J.; Lussi Y.; MacDougall A.; Madeira F.; Mahmoudy M.; Menchi M.; Mishra A.; Moulang K.; Nightingale A.; Oliveira C. S.; Pundir S.; Qi G.; Raj S.; Rice D.; Lopez M. R.; Saidi R.; Sampson J.; Sawford T.; Speretta E.; Turner E.; Tyagi N.; Vasudev P.; Volynkin V.; Warner K.; Watkins X.; Zaru R.; Zellner H.; Bridge A.; Poux S.; Redaschi N.; Aimo L.; Argoud-Puy G.; Auchincloss A.; Axelsen K.; Bansal P.; Baratin D.; Blatter M. C.; Bolleman J.; Boutet E.; Breuza L.; Casals-Casas C.; de Castro E.; Echioukh K. C.; Coudert E.; Cuche B.; Doche M.; Dornevil D.; Estreicher A.; Famiglietti M. L.; Feuermann M.; Gasteiger E.; Gehant S.; Gerritsen V.; Gos A.; Gruaz-Gumowski N.; Hinz U.; Hulo C.; Hyka-Nouspikel N.; Jungo F.; Keller G.; Kerhornou A.; Lara V.; Le Mercier P.; Lieberherr D.; Lombardot T.; Martin X.; Masson P.; Morgat A.; Neto T. B.; Paesano S.; Pedruzzi I.; Pilbout S.; Pourcel L.; Pozzato M.; Pruess M.; Rivoire C.; Sigrist C.; Sonesson K.; Stutz A.; Sundaram S.; Tognolli M.; Verbregue L.; Wu C. H.; Arighi C. N.; Arminski L.; Chen C.; Chen Y.; Garavelli J. S.; Huang H.; Laiho K.; McGarvey P.; Natale D. A.; Ross K.; Vinayaka C. R.; Wang Q.; Wang Y.; Yeh L. S.; Zhang J. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49 (D1), D480–D489. 10.1093/NAR/GKAA1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer R.; Schallert K.; Zoun R.; Becher B.; Saake G.; Benndorf D. Challenges and Perspectives of Metaproteomic Data Analysis. J. Biotechnol. 2017, 261, 24–36. 10.1016/j.jbiotec.2017.06.1201. [DOI] [PubMed] [Google Scholar]
- Xiao J.; Tanca A.; Jia B.; Yang R.; Wang B.; Zhang Y.; Li J. Metagenomic Taxonomy-Guided Database-Searching Strategy for Improving Metaproteomic Analysis. J. Proteome Res. 2018, 17 (4), 1596–1605. 10.1021/acs.jproteome.7b00894. [DOI] [PubMed] [Google Scholar]
- Jagtap P.; Goslinga J.; Kooren J. A.; Mcgowan T.; Wroblewski M. S.; Seymour S. L.; Griffin T. J. A Two-Step Database Search Method Improves Sensitivity in Peptide Sequence Matches for Metaproteomics and Proteogenomics Studies. PROTEOMICS 2013, 13 (8), 1352–1357. 10.1002/pmic.201200352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzihradszky K. F.; Chalkley R. J. Lessons in de Novo Peptide Sequencing by Tandem Mass Spectrometry. Mass Spectrom. Rev. 2015, 34 (1), 43–63. 10.1002/mas.21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure R. S.; Lee J. Y.; Chowdhury T. R.; Bottos E. M.; White R. A.; Kim Y. M.; Nicora C. D.; Metz T. O.; Hofmockel K. S.; Jansson J. K.; Song H. S. Integrated Network Modeling Approach Defines Key Metabolic Responses of Soil Microbiomes to Perturbations. Sci. Rep. 2020, 10 (1), 1–9. 10.1038/s41598-020-67878-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Baquerizo M.; Oliverio A. M.; Brewer T. E.; Benavent-González A.; Eldridge D. J.; Bardgett R. D.; Maestre F. T.; Singh B. K.; Fierer N. A Global Atlas of the Dominant Bacteria Found in Soil. Science (1979) 2018, 359 (6373), 320–325. 10.1126/SCIENCE.AAP9516. [DOI] [PubMed] [Google Scholar]
- Starke R.; Jehmlich N.; Bastida F. Using Proteins to Study How Microbes Contribute to Soil Ecosystem Services: The Current State and Future Perspectives of Soil Metaproteomics. Journal of Proteomics 2019, 198, 50–58. 10.1016/j.jprot.2018.11.011. [DOI] [PubMed] [Google Scholar]
- Tedersoo L.; Bahram M.; Põlme S.; Kõljalg U.; Yorou N. S.; Wijesundera R.; Ruiz L. V.; Vasco-Palacios A. M.; Thu P. Q.; Suija A.; Smith M. E.; Sharp C.; Saluveer E.; Saitta A.; Rosas M.; Riit T.; Ratkowsky D.; Pritsch K.; Põldmaa K.; Piepenbring M.; Phosri C.; Peterson M.; Parts K.; Pärtel K.; Otsing E.; Nouhra E.; Njouonkou A. L.; Nilsson R. H.; Morgado L. N.; Mayor J.; May T. W.; Majuakim L.; Lodge D. J.; Lee S.; Larsson K. H.; Kohout P.; Hosaka K.; Hiiesalu I.; Henkel T. W.; Harend H.; Guo L. D.; Greslebin A.; Grelet G.; Geml J.; Gates G.; Dunstan W.; Dunk C.; Drenkhan R.; Dearnaley J.; De Kesel A.; Dang T.; Chen X.; Buegger F.; Brearley F. Q.; Bonito G.; Anslan S.; Abell S.; Abarenkov K. Global Diversity and Geography of Soil Fungi. Science (1979) 2014, 346 (6213), 1256688. 10.1126/SCIENCE.1256688. [DOI] [PubMed] [Google Scholar]
- Becraft E. D.; Woyke T.; Jarett J.; Ivanova N.; Godoy-Vitorino F.; Poulton N.; Brown J. M.; Brown J.; Lau M. C. Y.; Onstott T.; Eisen J. A.; Moser D.; Stepanauskas R. Rokubacteria: Genomic Giants among the Uncultured Bacterial Phyla. Frontiers in Microbiology 2017, 8 (NOV), 2264. 10.3389/fmicb.2017.02264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Wang J.; Ye Z.; Zhang T.; Qu L.; Li J. Soil Property and Plant Diversity Determine Bacterial Turnover and Network Interactions in a Typical Arid Inland River Basin, Northwest China. Frontiers in Microbiology 2019, 10, 2655. 10.3389/fmicb.2019.02655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogwu M. C.; Srinivasan S.; Dong K.; Ramasamy D.; Waldman B.; Adams J. M. Community Ecology of Deinococcus in Irradiated Soil. Microbial Ecology 2019, 78 (4), 855–872. 10.1007/s00248-019-01343-5. [DOI] [PubMed] [Google Scholar]
- Li H. Y.; Wang H.; Wang H. T.; Xin P. Y.; Xu X. H.; Ma Y.; Liu W. P.; Teng C. Y.; Jiang C. L.; Lou L. P.; Arnold W.; Cralle L.; Zhu Y. G.; Chu J. F.; Gilbert J. A.; Zhang Z. J. The Chemodiversity of Paddy Soil Dissolved Organic Matter Correlates with Microbial Community at Continental Scales. Microbiome 2018, 6 (1), 1–16. 10.1186/s40168-018-0561-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J.; Yin Y.; Zhu W.; Zhou Y. Variations in Soil Bacterial Community Diversity and Structures among Different Revegetation Types in the Baishilazi Nature Reserve. Frontiers in Microbiology 2018, 9 (NOV), 2874. 10.3389/fmicb.2018.02874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crits-Christoph A.; Diamond S.; Butterfield C. N.; Thomas B. C.; Banfield J. F. Novel Soil Bacteria Possess Diverse Genes for Secondary Metabolite Biosynthesis. Nature 2018, 558 (7710), 440–444. 10.1038/s41586-018-0207-y. [DOI] [PubMed] [Google Scholar]
- Hug L. A.; Thomas B. C.; Sharon I.; Brown C. T.; Sharma R.; Hettich R. L.; Wilkins M. J.; Williams K. H.; Singh A.; Banfield J. F. Critical Biogeochemical Functions in the Subsurface Are Associated with Bacteria from New Phyla and Little Studied Lineages. Environmental Microbiology 2016, 18 (1), 159–173. 10.1111/1462-2920.12930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data for this benchmark set are split into two separate depositions, for the training and testing data sets, respectively. The training data set consists of spectra from 51 organisms and has been deposited to the ProteomeXchange Consortium via the PRIDE45 partner repository with the data set identifier PXD010000. The testing data set consists of spectra for 4 organisms and has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD010613. The metaproteomics data set has been deposited to the MassIVE Repository with the accession identifier MSV000086336.