

Abstract
BACKGROUND:
Chimeric antigen receptor (CAR) T cells achieve response and durable remission in patients with relapsed/refractory (R/R) B cell malignancies. Following collection of patient T cells, chemotherapy (“bridging chemotherapy”) is utilized during the manufacture of CAR T cells. However, the optimal bridging chemotherapy has yet to be defined.
OBJECTIVE(S):
Our objective in this study was to report clinical outcomes following bridging chemotherapy in a cohort of pediatric/young adult patients with R/R B cell acute lymphoblastic leukemia (B-ALL) treated with CAR T cells.
STUDY DESIGN:
This retrospective study included patients enrolled on clinical trial NCT01860937 or referred to MSKCC for commercial CAR T cell therapy (tisagenlecleucel). Bridging chemotherapy (given after T cell collection and prior to CAR T cell infusion) was defined as high intensity if myelosuppression was expected for > 7 days. Outcome comparison analyses were performed in high versus low intensity bridging chemotherapy, 1 versus ≥ 2 cycles of bridging chemotherapy, disease burden at start of bridging chemotherapy, disease burden at start of bridging chemotherapy with chemotherapy intensity, tumor debulking by bridging chemotherapy, and disease burden pre-lymphodepleting chemotherapy (LDC) for CAR T cell treatment.
RESULTS:
The outcomes of this analysis showed that the incidence of grade ≥ 3 infection was significantly higher (94% versus 56%, p=0.019) and overall survival (OS) was significantly lower (HR 3.73, 95% CI: 1.39–9.97, p=0.006) in patients who received ≥ 2 cycles versus 1 cycle of bridging chemotherapy. No difference in incidence was found for cytokine release syndrome (CRS) (p>0.99) or neurotoxicity (NTX)/immune effector cell-associated neurotoxicity syndrome (ICANS) (p=0.70). Disease burden at start of bridging chemotherapy, disease burden prior to LDC, and tumor debulking by bridging chemotherapy also did not significantly affect outcomes after CAR T cell therapy in this cohort.
CONCLUSIONS:
Patients receiving ≥ 2 cycles of bridging chemotherapy have higher rates of infection and lower OS but no difference in CAR-specific toxicity. Clinicians should carefully consider the use of additional cycles of chemotherapy during the bridging period as it delays treatment with CAR T cells and increases the risk of infectious complications.
Keywords: tisagenlecleucel, B-cell acute lymphoblastic leukemia, CAR T cell therapy, immunotherapy, CD19 antigen, bridging chemotherapy
INTRODUCTION:
The outcomes for children/young adults with relapsed/refractory (R/R) B cell acute lymphoblastic leukemia (B-ALL) remain dismal.1, 2 Response and durable remission have been achieved with autologous CD19-specific chimeric antigen receptor (CAR) T cells, thereby creating new opportunities for long term survival in this patient population.3, 4 Following collection of patient T cells, CAR T cell manufacturing requires several weeks before the release of a qualified product. Chemotherapy is typically utilized during this bridging period (“bridging chemotherapy”) with a goal of halting or slowing disease progression. Optimal bridging chemotherapy has yet to be defined and varies among providers and clinical trial protocols. Further, bridging chemotherapy can lead to toxicities in this heavily pretreated and often chemotherapy-refractory patient population precluding subsequent cellular therapy.
It has been shown that a lower disease burden at time of CAR T cell infusion is correlated with improved long-term survival and decreased treatment-related complications such as cytokine release syndrome (CRS) and neurotoxicity (NTX)/immune effector cell-associated neurotoxicity syndrome (ICANS).5–7 Thus, there may be a benefit to intensive bridging chemotherapy if disease burden can be effectively reduced prior to CAR T cell therapy. However, recent studies in adult and smaller pediatric cohorts have demonstrated that high-intensity bridging chemotherapy is associated with greater infectious complications without improving long-term outcomes.8–11 In this retrospective study, we reviewed the bridging treatment strategies and clinical outcomes in a cohort of pediatric/young adult R/R B-ALL patients treated with CD19-specific CAR T cells.
PATIENTS AND METHODS:
Patients < 26 years of age with successful T cell collection and who received bridging chemotherapy on clinical trial NCT01860937 or referred for commercial CAR T cell therapy (tisagenlecleucel) from 2013 through 2019 were included in this study. Both the initial study protocol and the retrospective analysis were reviewed and approved by the Memorial Sloan Kettering Cancer Center Institutional Review Board. Bridging chemotherapy was defined as any chemotherapy given after T cell collection and prior to lymphodepleting chemotherapy (LDC) for CAR T cell therapy. If patients were not infused, the bridging period ended at time of patient’s death, progression of disease, or decision to pursue alternative treatment. The bridging regimen was defined as high intensity if myelosuppression was expected for more than seven days (Table 1). This analysis also included the number of cycles of bridging chemotherapy given prior to infusion (1 versus ≥ 2). Outcome comparison analyses were performed in high versus low intensity bridging chemotherapy groups, number of cycles of bridging chemotherapy received, disease burden at start of bridging chemotherapy, disease burden at start of bridging chemotherapy with chemotherapy intensity, tumor debulking by bridging chemotherapy, and disease burden pre-LDC.
Table 1.
Bridging regimens stratified by low versus high intensity.
| Low Intensity (Myelosuppression ≤ 7 days) +/− intrathecal chemotherapy | High Intensity (Myelosuppression > 7 days) +/− intrathecal chemotherapy |
|---|---|
|
VPL Vincristine Asparaginase Prednisone (or dexamethasone) |
VPLD Vincristine Asparaginase Prednisone (or dexamethasone) Daunorubicin |
|
VPL (with addition of one or more agents below) +/− Mercaptopurine (daily) +/− Methotrexate (weekly, PO) +/− Hydroxyurea |
VPL (with addition of one or more agents below) +/− Anthracycline +/− Cyclophosphamide +/− Etoposide +/− Thioguanine +/− Cytarabine +/− Targeted therapy |
|
DFCI 11-001 Continuation Vincristine Prednisone Mercaptopurine (Daily × 14 days) Methotrexate IV (Day 8, 15) |
Cyclophosphamide + Etoposide14 +/− Targeted therapy +/− Cytarabine +/− Asparaginase |
|
Targeted Therapy (single agent) Inotuzumab Tyrosine Kinase Inhibitor |
UK-R3 (AALL1331 – Block 1)15 Mitoxantrone Asparaginase Vincristine Dexamethasone |
|
High Dose Cytarabine (3 grams/m2/dose × 4 or 8 doses) +/− Asparaginase +/− Hydroxyurea |
|
|
Clo-21816 Clofarabine Cyclophosphamide Etoposide |
|
|
TVTC17 Topotecan Vinorelbine Thiotepa Clofarabine |
|
|
Clofarabine (single agent) +/− Bortezomib |
|
|
AALL1131 Interim Maintenance I High-dose Methotrexate Vincristine Mercaptopurine |
|
|
CCG-1901 Consolidation I High Dose Cytarabine (3gm/m2/dose × 2 doses) Methotrexate (200mg/m2/dose × 1 dose) Vincristine × 2 doses Prednisone × 7 days +/− Asparaginase × 1 dose |
|
| Etoposide (single agent) |
Adverse events of CRS, NTX/ICANS, and infections were captured for all treated patients until 90 days after CAR T cell infusion unless disease relapse, alternative therapy administration, or death occurred earlier. CRS was graded according to the American Society for Transplantation and Cellular Therapy (ASTCT) consensus CRS grading.12 NTX was assessed according to National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), v4.03 until the adoption of the ASTCT immune effector cell-associated neurotoxicity syndrome (ICANS) grading system in 2019.12 Infectious complications were reported during the bridging period and after CAR T cell infusion and graded as per NCI CTCAE, v5.0. Infectious complications included bacteremia, pneumonia, fungal infections, viral infections, and septic shock requiring intensive care unit admission.
Overall survival (OS) was defined as time from T cell collection (for non-infused patients) or infusion (for patients infused with CAR T cells) until death. Event-free survival (EFS) was defined as time until death or relapse, with allogeneic hematopoietic stem cell transplant (allo-HCT) used as a competing risk. Patients alive (alive without relapse for EFS) were censored at their last follow-up. Complete remission (CR) was defined as < 5% bone marrow blasts, absence of circulating blasts, and no extramedullary sites of disease. A negative status for minimal residual disease (MRD) was defined as less than 0.01% bone marrow blasts as assessed by multiparameter flow cytometry. High disease burden was defined by ≥ 5% bone marrow lymphoblasts, any peripheral blood lymphoblasts, CNS3 status, or non-CNS extramedullary site of disease. Low disease burden was defined by the absence of morphologic or flow cytometry detectable disease or detectable bone marrow or CNS disease not meeting high disease burden criteria and without extramedullary disease. Tumor debulking by bridging chemotherapy was defined as ≥ 5% decrease in bone marrow blasts by morphology or flow cytometry, achievement of MRD negative CR, and/or clearance of peripheral blood blasts.
Comparisons across groups were performed using either Fisher’s exact tests or Chi-squared tests of independence. OS was estimated using Kaplan Meier estimator. Univariable Cox proportional hazards models were used to evaluate the relationship between relevant covariates and survival. A 28-day landmark analysis was used to evaluate difference in OS between CR and non-CR patients, as the response status was known at 28 days. EFS was estimated using a sub-distribution hazard function and covariates were examined using cause-specific hazard ratios (HR). All analyses were done in R v4.0.0.
Data sharing statement:
Requests for de-identified individual participant data can be made beginning 12 months after publication and for up to 36 months after publication. De-identified individual participant data reported in the manuscript will be shared under the terms of a Data Use Agreement and may only be used for approved proposals. Requests may be made to: crdatashare@mskcc.org.
RESULTS:
A total of 51 patients were screened, 48 of whom had successful T cell collection and received bridging chemotherapy, and 35 of whom proceeded to CAR T cell infusion (Figure 1 and Table 2). The median time from apheresis to start of bridging chemotherapy was 3 days (range 0 to 37 days). The median follow-up after T cell collection was 46.9 months (range 1.1 to 64.6 months). Thirteen patients were not infused because they proceeded to allo-HCT (n=7), died from infection (n=4), died from progressive disease (n=1), or had a manufacturing failure (n=1). OS in the 13 non-infused patients was 53% at 24-months (95% CI: 31–89%). In the entire cohort (n=48), tumor debulking was associated with high intensity bridging chemotherapy as compared to low intensity (41% versus 0%, p=0.020) but was not associated with cycles of bridging chemotherapy (1 versus ≥ 2, p=0.26) or pre-bridge disease burden (p=0.17). Intensity of bridging chemotherapy (p=0.12) and number of cycles of therapy (p=0.075) were not associated with infection in the entire cohort.
Figure 1. Patient flow diagram for all patients screened for study.
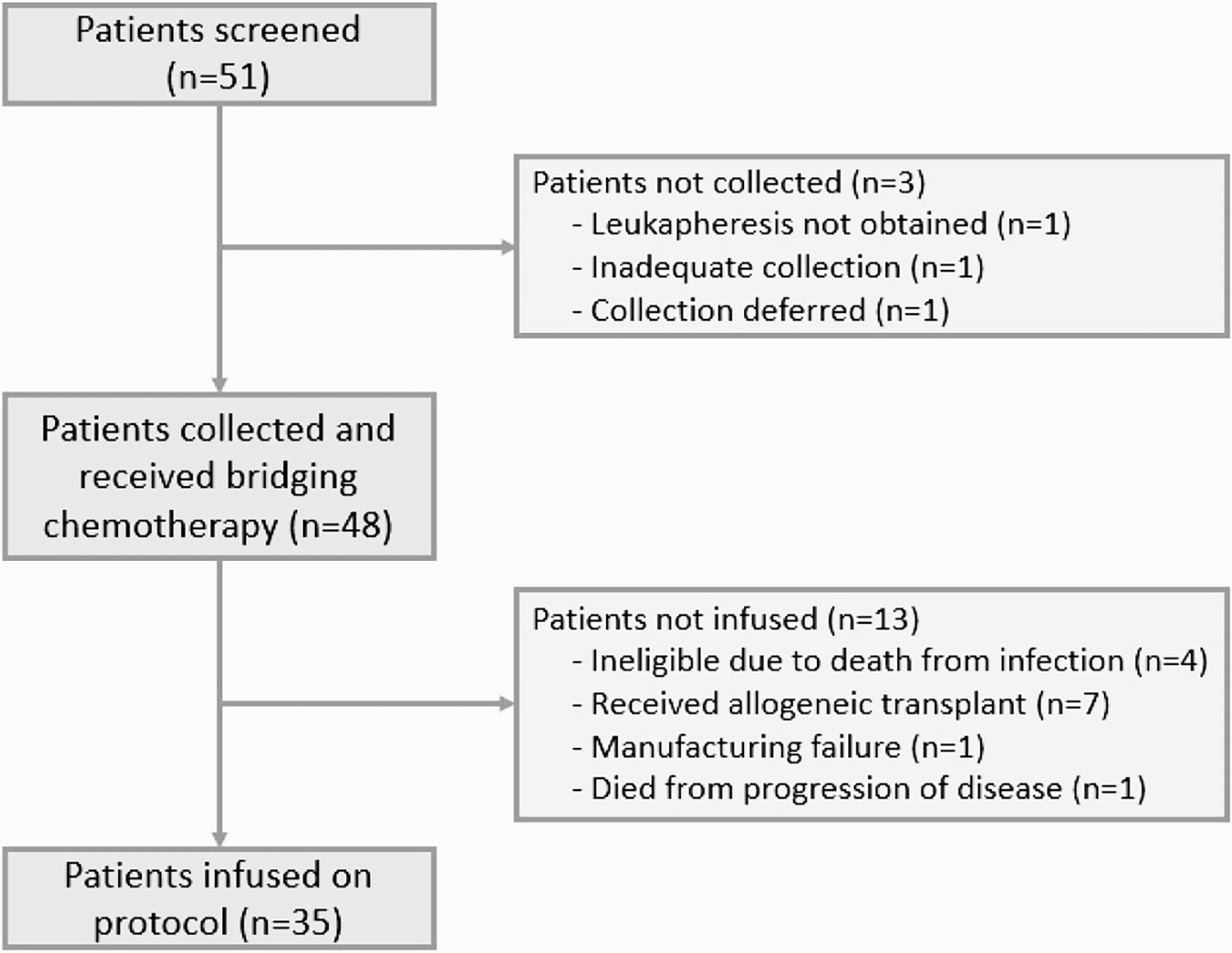
Table 2.
Baseline patient characteristics.
| Characteristic | N = 48 |
|---|---|
| Age at Collection (in years; N=48) | 12.8 (0.8, 25.6) |
| Gender (N=48) | |
| Female | 21 (44%) |
| Male | 27 (56%) |
| CAR Type (N=48; intention to treat) | |
| 19–28z | 38 (79%) |
| 19-BBz/Tisa | 10 (21%) |
| Age at Infusion (in years) (N=35) | 13.5 (1.1, 25.8) |
| Time from Collection to Infusion (in days; N=35) | 54 (32, 663) |
| 19–28z | 60 (32, 663) |
| 19-BBz/Tisa | 45 (35, 63) |
| 1 Month Response CR (N=35) | 27 (82%) |
| No Response | 6 (18%) |
| Not evaluable (death under 30 days) | 2 |
| 1 Month Response CR + MRD Negative (N=35) | 25 (76%) |
| No Response | 8 (24%) |
| Not evaluable (death under 30 days) | 2 |
| Infection Post-CAR (N=35) | |
| Grade 0–2 | 20 (59%) |
| Grade 3+ | 14 (41%) |
| Unknown | 1 |
| Transplant (N=48; intention to treat) | 27 (56%) |
| Post-bridge | 7 |
| Post-CAR | 20 |
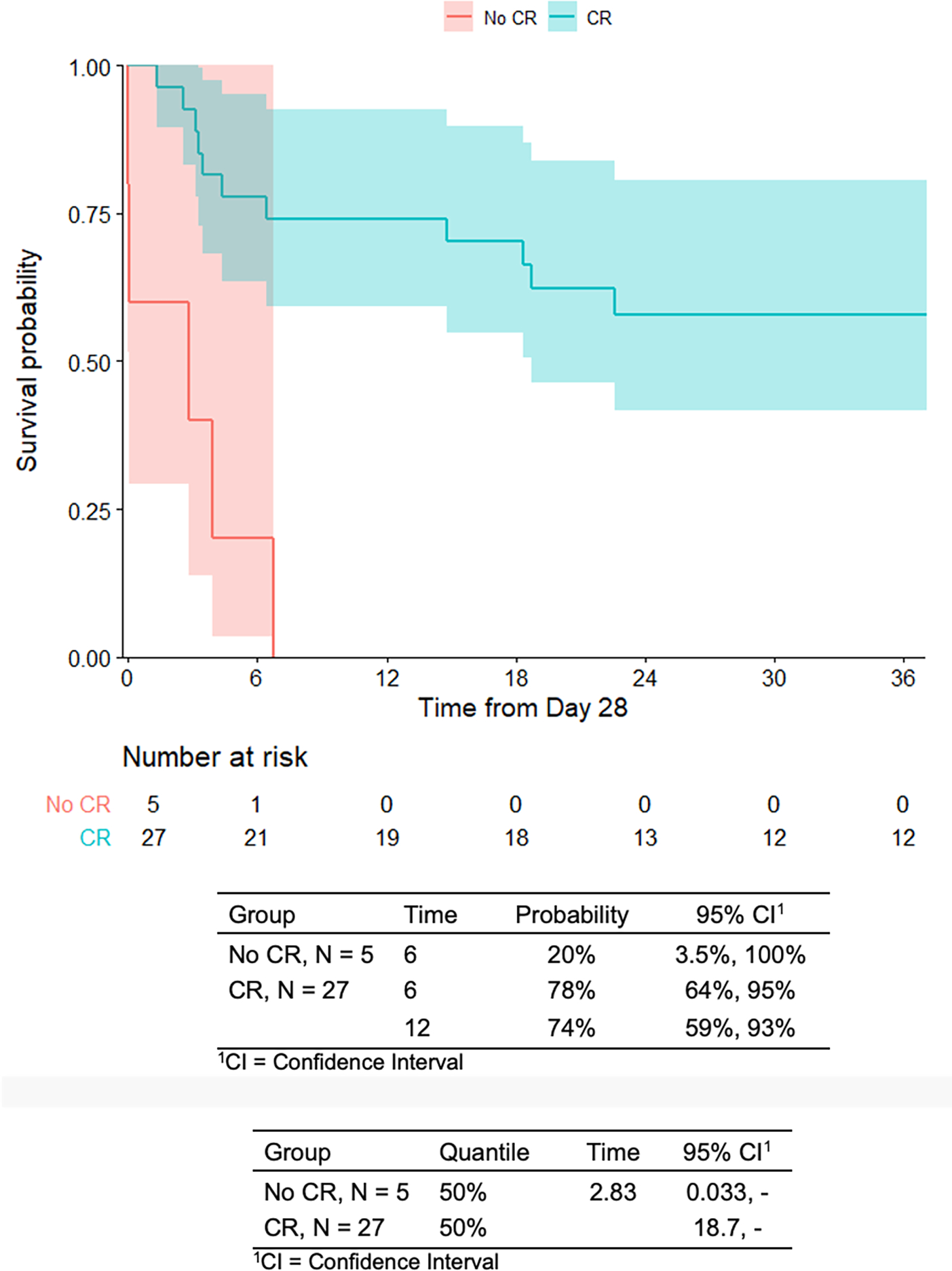
For patients who received CAR T cell therapy (n=35), the median age at time of infusion was 13.5 years (range 1.1 to 25.8 years) and median time from collection to infusion was 54 days (range 32 to 663 days). OS in the 35 infused patients was 45% at 24-months (95% CI: 31–65%) (Figure 2A). The overall day 28 CR rate was 82% (27/33 evaluable patients), of which 93% were MRD negative as measured by flow cytometry. Notably, OS in the patients who had a CR at day 28 (n=27) was 58% at 24-months post-response (95% CI: 42–81%) as compared to no survivors by 24-months in the patients who did not have a CR at day 28 (n=6) (Figure 2B). There was no significant difference in CR rate between patients who received 19–28z versus 19-BBz/tisagenlecleucel (p=0.30) or high versus low intensity bridging chemotherapy (p>0.99). Complicating interpretation of response to CAR T cell therapy in this cohort are the various LDC regimens used, which we have previously published as impacting response following 19–28z CAR T cells.3 There were no differences in CRS and NTX/ICANS between patients who received high or low intensity bridging chemotherapy. Grade ≥ 3 infections occurred in 80% (28/35) of patients who received CAR T cell therapy. This included infection after bridging chemotherapy alone (n=14), after CAR T cell therapy alone (n= 3), or both (n=11). Of these 28 patients with grade ≥ 3 infections, 9 patients received bridging regimens containing high-dose cytarabine and 8 patients received clofarabine-containing bridging regimens. An identifiable source of infection (some patients experienced more than one infection) was demonstrated in 6/9 patients in the high-dose cytarabine cohort (bacterial n=5, viral n=3, fungal n=1), in 7/8 patients in the clofarabine cohort (bacterial n=7, viral n=4, fungal n=3), and in 10/11 of the remaining patients with grade ≥ 3 infections (bacterial n=8, viral n=9, fungal n=1).
Figure 2. Overall survival curves.
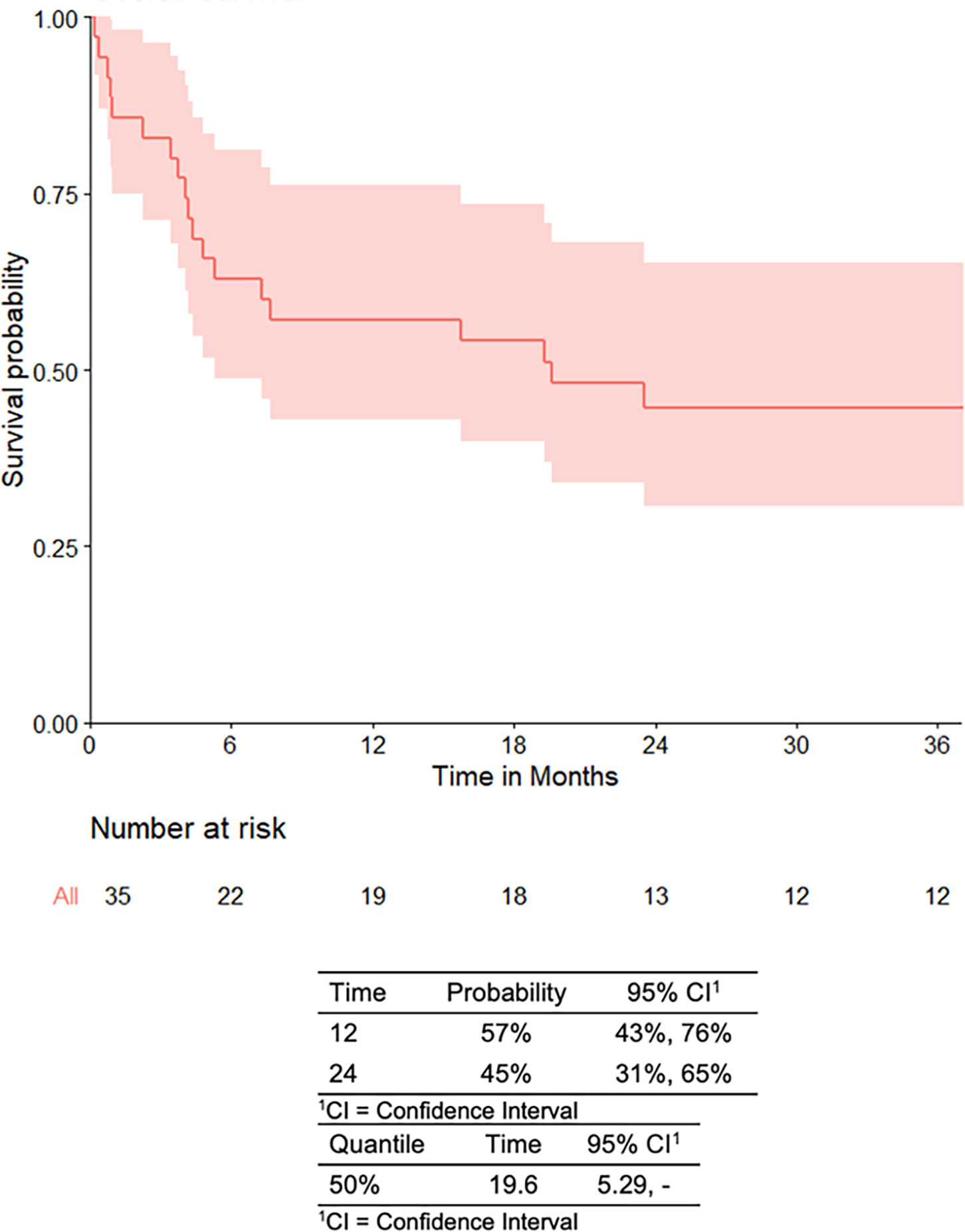
A. Infused patients.
Start time used is date of infusion.
B. Infused patients stratified by D28 response.
To assess the post-response survival, the OS time is defined as the time from 28 days post-infusion to the time of death. A landmark analysis is done in which patients who died before 28 days are excluded from the analysis (n=1 who did not achieve CR).
In the cohort of patients who received CAR T cell therapy (n=35), 54.3% (n=19) of patients received 1 cycle of bridging chemotherapy and 45.7% (n=16) of patients received ≥2 cycles of bridging chemotherapy (Figure 3). Rationale for patients who received ≥2 cycles of bridging chemotherapy (n=16) included awaiting CAR T cell production (n=3), treating physician preference (n=3), and disease progression (n=10). The median number of days from collection to infusion for patients who received 1 cycle of bridging chemotherapy was 47 days versus 66 days for patients who receive ≥ 2 cycles of bridging chemotherapy. Day 28 MRD-negative CR rates (56% versus 94%, p=0.017) as well as OS (HR 3.73, 95% CI: 1.39–9.97, p=0.006) were significantly lower in the ≥ 2 cycles versus 1 cycle of bridging chemotherapy cohorts while the incidence of grade ≥ 3 infection was significantly higher (Table 3). Specifically, patients who received ≥ 2 cycles of bridging chemotherapy had more grade ≥ 3 infections during the bridging period (94% versus 56%, p=0.019). There was no difference in CRS (p>0.99) and NTX/ICANS (p=0.70) between patients who received 1 cycle versus ≥ 2 cycles of bridging chemotherapy. Death from disease progression was higher in patients who received ≥2 cycles (62.5%, 10/16) versus 1 cycle (15.8%, 3/19). Thus, ≥ 2 cycles of bridging therapy was associated with an increased rate of infections during the bridging period and lower MRD response rates following CAR T cell therapy.
Figure 3. Flow diagram of all patients who received ≥ 2 cycles of bridging chemotherapy.
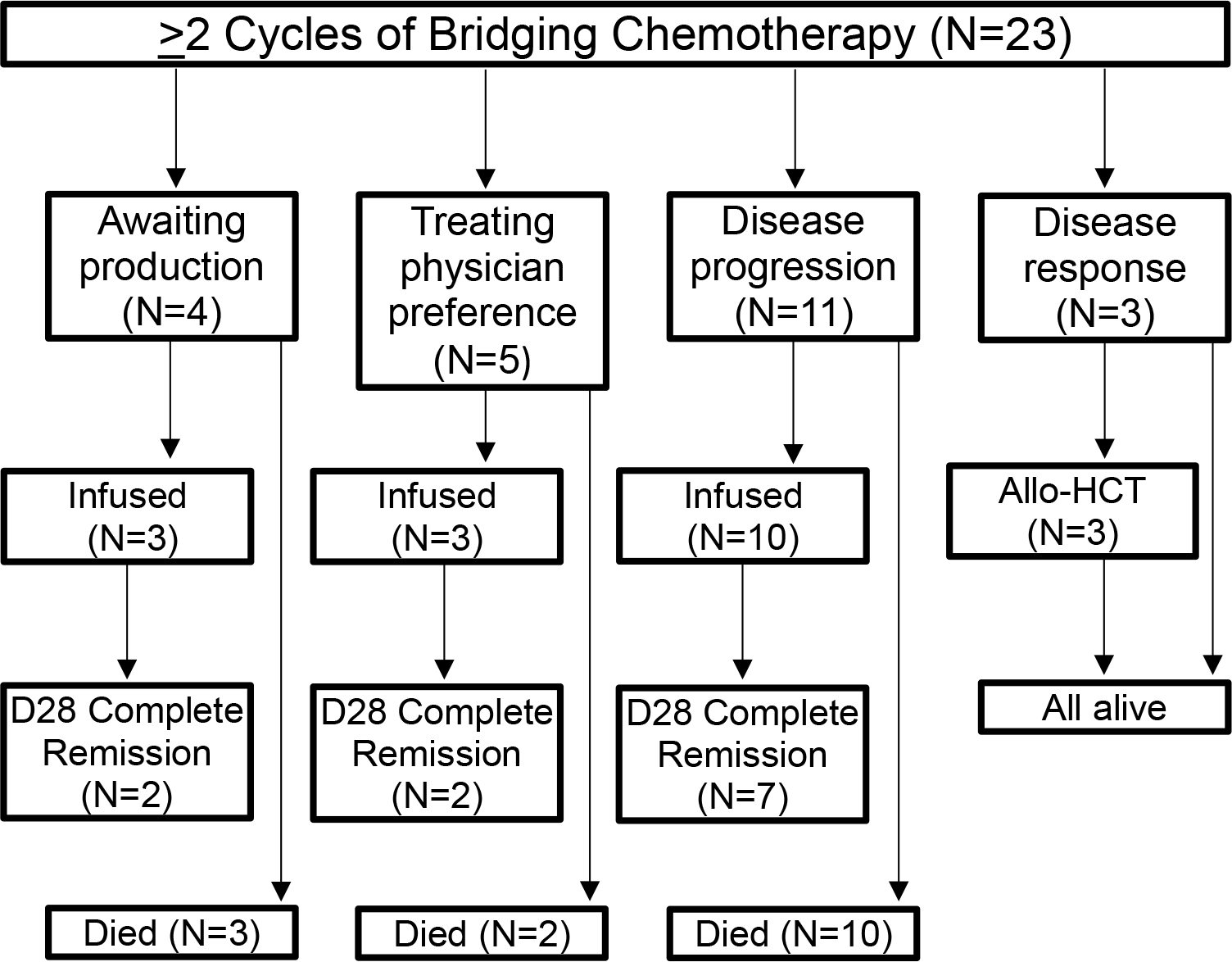
Table 3.
Characteristics and outcomes of patients infused with CAR T cells (n=35) grouped by 1 (n=19) versus ≥ 2 (n=16) cycles of bridging chemotherapy.
| Characteristic | 1 Cycle, n = 191 | 2+ Cycles, n = 161 | p-value2 |
|---|---|---|---|
| Age at Diagnosis | 0.85 | ||
| <1 or >=10 | 9 (47%) | 9 (56%) | |
| >=1 to <10 | 10 (53%) | 7 (44%) | |
| Ethnicity | 0.86 | ||
| Black/Hispanic | 6 (32%) | 6 (38%) | |
| Other | 1 (5%) | 1 (6%) | |
| White | 12 (63%) | 9 (56%) | |
| Initial Risk Stratification | 0.31 | ||
| High Risk | 10 (53%) | 12 (75%) | |
| Standard Risk | 9 (47%) | 4 (25%) | |
| CAR Type | 0.46 | ||
| 19–28z | 13 (68%) | 13 (81%) | |
| 19-BBz | 6 (32%) | 3 (19%) | |
| Median Days from Collection to Infusion | 47 | 66 | 0.022 |
| 1 Month Response CR | 16/17* (94%) | 11 (69%) | 0.085 |
| 1 Month Response CR MRD Negative | 16/17* (94%) | 9 (56%) | 0.017 |
| Grade 3+ Infection During Bridge | 10/18* (56%) | 15 (94%) | 0.019 |
| Grade 3+ Infection After CAR T Cells | 5/18* (28%) | 9 (56%) | 0.18 |
| Grade 3+ Infection Overall (During Bridging or After CAR) | 12/18* (67%) | 16 (100%) | 0.02 |
| Transplant Post-CAR | 12 (63%) | 8 (50%) | |
| Relapse Post-CAR | 6 (32%) | 12 (75%) | |
| Cause of Death | |||
| Infection | 2 (33%) | 0 (0%) | |
| Progression of disease | 3 (50%) | 10 (77%) | |
| Treatment related mortality | 1 (17%) | 3 (23%) | |
| Unknown | 13 | 3 |
n (%)
Pearson’s Chi-squared test; Fisher’s exact test; Wilcoxon rank sum test
Information not available for all patients; denominator specifies number of evaluable patients
In the cohort of patients who received CAR T cell therapy (n=35), analysis of disease burden and tumor debulking showed no statistical significance for outcomes. Disease burden at the start of bridging chemotherapy was divided into high disease burden (n=23) versus low disease burden (n=12). There was no significant difference in day 28 CR, day 28 MRD-negative CR, CRS, NTX/ICANS, or infections (Table 4). Additionally, patients were grouped by disease burden at start of bridging chemotherapy with intensity of bridging chemotherapy received: high disease burden and high intensity bridging chemotherapy (n=18), high disease burden and low intensity bridging chemotherapy (n=5), low disease burden and high intensity bridging chemotherapy (n=10), and low disease burden and low intensity chemotherapy (n=2). The low disease burden and low intensity chemotherapy cohort was excluded from the analysis because of small sample size. There was no significant difference in day 28 CR, day 28 MRD-negative CR, CRS, NTX/ICANS, or infections (Table 5). Patients were divided into high disease burden at start of bridging therapy and high disease burden at time of CAR infusion (n=15), high disease burden at start of bridging therapy and low disease burden at time of CAR infusion (n=8), low disease burden at start of bridging therapy and low disease burden at time of CAR infusion (n=8), and low disease burden at start of bridging therapy and high disease burden at time of CAR infusion (n=4). There was no significant difference in day 28 CR, day 28 MRD-negative CR, CRS, NTX/ICANS, or infections (Table 6). Lastly, disease burden prior to LDC was divided into high disease burden (n=19) versus low disease burden (n=16). There was no significant difference in day 28 CR, day 28 MRD-negative CR, CRS, NTX/ICANS, or infections (Table 7).
Table 4.
Analysis of disease burden at the start of bridging chemotherapy on outcomes.
| Outcome | Low tumor burden, n = 121 | High tumor burden, n = 231 | p-value2 |
|---|---|---|---|
| 1 Month Response CR | 10/11* (91%) | 17/22* (77%) | 0.64 |
| 1 Month Response CR MRD Negative | 9/11* (82%) | 16/22* (73%) | 0.69 |
| Grade 3+ Infection During Bridge | 7/11* (64%) | 18 (78%) | 0.43 |
| Grade 3+ Infection After CAR T Cells | 3/11* (27%) | 11 (48%) | 0.29 |
| Grade 3+ Infection Overall (During Bridging or After CAR) | 9/11* (82%) | 19 (83%) | >0.99 |
| Grade 3+ CRS | 2 (17%) | 5 (22%) | >0.99 |
| Grade 3+ NTX/ICANS | 2 (17%) | 6 (26%) | 0.69 |
n (%)
Pearson’s Chi-squared test; Fisher’s exact test
Information not available or not applicable for all patients; denominator specifies number of evaluable patients
Table 5.
Analysis of disease burden at start of bridging chemotherapy with intensity of bridging chemotherapy received on outcomes.
| Outcome | High disease, high intensity bridging, n = 181 | High disease, low intensity bridging, n = 51 | Low disease, high intensity bridging, n = 101 | p-value2 |
|---|---|---|---|---|
| 1 Month Response CR | 13/17* (76%) | 4 (80%) | 9 (90%) | 0.83 |
| 1 Month Response CR MRD Negative | 12/17* (71%) | 4 (80%) | 8 (80%) | 0.86 |
| Grade 3+ Infection During Bridge | 15 (83%) | 3 (60%) | 7/9* (78%) | 0.53 |
| Grade 3+ Infection After CAR T Cells | 9 (50%) | 2 (40%) | 2/9* (22%) | 0.37 |
| Grade 3+ Infection Overall (During Bridging or After CAR) | 16 (89%) | 3 (60%) | 8/9* (89%) | 0.32 |
| Grade 3+ CRS | 5 (28%) | 0 (0%) | 1 (10%) | 0.40 |
| Grade 3+ NTX/ICANS | 6 (33%) | 0 (0%) | 2 (20%) | 0.29 |
n (%)
Pearson’s Chi-squared test; Fisher’s exact test
Information not available or not applicable for all patients; denominator specifies number of evaluable patients
Table 6.
Analysis of response to bridging chemotherapy on outcomes.
| Outcome | High disease pre-bridge → high disease pre-CAR, n = 151 | High disease pre-bridge → low disease pre-CAR, n = 81 | Low disease pre-bridge → low disease pre-CAR, n = 81 | Low disease pre-bridge → high disease pre-CAR, n = 41 | p-value2 |
|---|---|---|---|---|---|
| 1 Month Response CR | 11 (73%) | 6/7* (86%) | 8 (100%) | 2/3* (67%) | 0.39 |
| 1 Month Response CR MRD Negative | 11 (73%) | 5/7* (71%) | 8 (100%) | 1/3* (33%) | 0.093 |
| Grade 3+ Infection During Bridge | 12 (80%) | 6 (75%) | 5 (63%) | 2/3* (67%) | 0.89 |
| Grade 3+ Infection After CAR T Cells | 8 (53%) | 3 (38%) | 1 (13%) | 2/3* (67%) | 0.22 |
| Grade 3+ Infection Overall (During Bridging or After CAR) | 13 (87%) | 6 (75%) | 6 (75%) | 3/3* (100%) | 0.72 |
| Grade 3+ CRS | 4 (27%) | 1 (13%) | 0 (0%) | 2 (50%) | 0.16 |
| Grade 3+ NTX/ICANS | 3 (20%) | 3 (38%) | 2 (25%) | 0 (0%) | 0.69 |
n (%)
Pearson’s Chi-squared test; Fisher’s exact test
Information not available or not applicable for all patients; denominator specifies number of evaluable patients
Table 7.
Analysis of disease burden pre-lymphodepleting chemotherapy on outcomes.
| Outcome | Low tumor burden, n = 161 | High tumor burden, n = 191 | p-value2 |
|---|---|---|---|
| 1 Month Response CR | 14/15* (93%) | 13/18* (72%) | 0.19 |
| 1 Month Response CR MRD Negative | 13/15* (87%) | 12/18* (73%) | 0.24 |
| Grade 3+ Infection After CAR T Cells | 4 (25%) | 10/18* (56%) | 0.071 |
| Grade 3+ CRS | 1 (6%) | 6 (32%) | 0.10 |
| Grade 3+ NTX/ICANS | 5 (31%) | 3 (16%) | 0.42 |
n (%)
Pearson’s Chi-squared test; Fisher’s exact test
Information not available or not applicable for all patients; denominator specifies number of evaluable patients
Six patients received prior targeted therapies: blinatumomab only (n=1), SGN-19A only (n=1), inotuzumab only (n=1), blinatumomab and inotuzumab (n=2), or blinatumomab, inotuzumab, and CD22-targeted CAR T cells (n=1). Three of these patients were ultimately infused with CAR T cells. All three infused patients achieved CR at day 28, relapsed after CAR T cell treatment, and ultimately died. All three patients received high intensity bridging chemotherapy, one out of the three patients received ≥ 2 cycles of bridging chemotherapy, and none of the three infused patients ultimately went on to receive HCT.
DISCUSSION:
This cohort of children/young adult patients with R/R B-ALL treated with CAR T cells demonstrates that receiving ≥ 2 cycles of bridging chemotherapy is associated with higher rates of infections, but no difference in post-CAR T cell infusion toxicities (CRS or NTX/ICANS) and lower OS. Tumor debulking has previously been considered a goal of bridging chemotherapy as pre-treatment disease burden has been correlated with improved response, durability of disease control, and reduced toxicity.5–7 However, we suggest that selection of a bridging regimen should favor limiting the number of cycles to minimize complications and maximize response.
Disease progression was the driving rationale for administering additional cycles of bridging chemotherapy in both the entire cohort and the infused cohort. This highlights our institutional practice of attempting disease control prior to cellular therapy with additional cycles of bridging chemotherapy. When excluding the 3 infused patients with production delays in the infused cohort, we note an inferior day 28 CR (62% versus 94%) and OS (23% versus 68%) for patients who received ≥ 2 cycles of bridging chemotherapy as compared to those receiving 1 cycle of bridging chemotherapy. Additional cycles of bridging chemotherapy were not associated with an improvement in pre-treatment disease burden as 31% (5/16) of patients who received 1 cycle bridging therapy had effective tumor debulking which was similar to the 26% (5/19) of patients who received ≥2 cycles of bridging chemotherapy. Therefore, additional cycles of bridging chemotherapy are not an effective method in reducing tumor burden and may negatively affect patient outcome. The worse outcomes in patients who received ≥ 2 lines of bridging chemotherapy suggest that distinct disease biology necessitating different bridging chemotherapy strategies may be driving the outcomes. To that end, our analysis suggests that a second cycle of bridging chemotherapy is a poor choice for patients with biologically difficult-to-treat leukemia because it does not ultimately improve outcomes and delays treatment with CAR T cells. Further study of disease characteristics in patients who required ≥ 2 cycles of bridging chemotherapy may yield potential therapeutic improvement, but that is not provided by additional cycles of chemotherapy. Our cohort of infused patients also demonstrates the complexity of CAR T cell therapy as 4 patients received additional bridging chemotherapy due to production delays. This complexity can be obviated with the use of off-the-shelf allogeneic CAR T cell products.
This study is limited by small sample size, inclusion of two CAR T cell products, and diverse LDC regimens. Analysis of outcomes following CAR T cells in pediatric and young adult R/R B-ALL patients have demonstrated decreased outcomes (response, EFS, and OS) in patients with high pre-treatment disease burden compared to patients with lower/no disease burden.13 Our current analysis is limited by heterogeneity and therefore analysis of long-term outcomes based on pre-treatment disease burden is not feasible. Another limitation is that although this analysis demonstrates an inferior MRD-negative CR rate and OS in patients who received ≥2 cycles of bridging chemotherapy, this analysis is underpowered for a multivariable analysis of confounding factors such as number of prior remissions or refractory status, prior HCT, age, NCI risk category, and cytogenetics. Further studies are warranted to prospectively evaluate the role of bridging chemotherapy in CAR T cell therapy with a more homogeneous patient population. Despite these limitations, our data suggests clinicians should carefully consider the negative impact that additional cycles of bridging chemotherapy will have on patients. This includes delaying CAR T cell treatment, increasing infectious complications, and decreasing OS.
HIGHLIGHTS:
Bridging chemotherapy is used during the manufacture of CAR T cells
Grade ≥ 3 infection is higher in patients following ≥ 2 cycles of bridging therapy
Overall survival is lower following ≥ 2 cycles of bridging therapy
Pre-treatment disease burden is not reduced with ≥ 2 cycles of bridging therapy
ACKNOWLEDGEMENTS:
We acknowledge support of the NCI Cancer Center Support Grant P30 CA008748. We thank Joseph Olechnowicz for editorial assistance. All authors critically reviewed and provided final approval of the submitted manuscript. SS and KJC had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
FINANCIAL DISCLOSURE STATEMENT:
KP discloses patent or royalties (Neximmune, Inc.). JHP discloses consulting or advisory roles (Adaptive Biotechnologies; Amgen; Kite Pharma; Novartis; Pfizer) and research funding (Genentech/Roche; Juno Therapeutics). JJB discloses consulting or advisory roles (Avrobio; Advanced Clinical; Bluerock; Omeros; Takeda; Race Oncology). NAK discloses equity interest (Amgen; Johnson and Johnson; Merck; Pfizer) and research funding (Jazz Pharmaceuticals). KJC discloses consulting or advisory role (Novartis; Mesoblast) and research funding (Juno Therapeutics; Novartis; Celegene). The remaining authors declare no conflicts of interest relevant to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- 1.Sun W, Malvar J, Sposto R, et al. Outcome of children with multiply relapsed B-cell acute lymphoblastic leukemia: a therapeutic advances in childhood leukemia & lymphoma study. Leukemia. 2018;32:2316–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reismüller B, Peters C, Dworzak MN, et al. Outcome of children and adolescents with a second or third relapse of acute lymphoblastic leukemia (ALL): a population-based analysis of the Austrian ALL-BFM (Berlin-Frankfurt-Münster) study group. J Pediatr Hematol Oncol. 2013;35:e200–204. [DOI] [PubMed] [Google Scholar]
- 3.Curran KJ, Margossian SP, Kernan NA, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. 2019;134:2361–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pasquini MC, Hu Z-H, Curran K, et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Advances. 2020;4:5414–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park JH, Rivière I, Gonen M, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378:449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perica K, Flynn J, Curran KJ, et al. Impact of bridging chemotherapy on clinical outcome of CD19 CAR T therapy in adult ALL. Journal of Clinical Oncology. 2019;37:2520–2520. [Google Scholar]
- 9.Gupta S, Alexander S, Zupanec S, et al. High Vs. Low-Intensity Bridging Chemotherapy in Children with Acute Lymphoblastic Leukemia Awaiting Chimeric Antigen Receptor T-Cell Therapy: A Population-Based Study from Ontario, Canada. Blood. 2018;132:1410–1410. [Google Scholar]
- 10.Pinnix CC, Gunther JR, Dabaja BS, et al. Bridging therapy prior to axicabtagene ciloleucel for relapsed/refractory large B-cell lymphoma. Blood Adv. 2020;4:2871–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perica K, Flynn J, Curran KJ, et al. Impact of bridging chemotherapy on clinical outcome of CD19 CAR T therapy in adult acute lymphoblastic leukemia. Leukemia. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee DW, Santomasso BD, Locke FL, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25:625–638. [DOI] [PubMed] [Google Scholar]
- 13.Schultz LM, Baggott C, Prabhu S, et al. Disease Burden Impacts Outcomes in Pediatric and Young Adult B-Cell Acute Lymphoblastic Leukemia after Commercial Tisagenlecleucel: Results from the Pediatric Real World CAR Consortium (PRWCC). Blood. 2020;136:14–15. [Google Scholar]
- 14.Carceller F, Hirsch SG, Khabra K, et al. High-dose etoposide and cyclophosphamide in adults and children with primary refractory and multiply relapsed acute leukaemias: The Royal Marsden experience. Leukemia Research. 2019;85:106217. [DOI] [PubMed] [Google Scholar]
- 15.Parker C, Waters R, Leighton C, et al. Effect of mitoxantrone on outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): an open-label randomised trial. Lancet. 2010;376:2009–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu AP, Lee V, Li CK, Ha SY, Chiang AK. Refractory acute lymphoblastic leukemia in Chinese children: bridging to stem cell transplantation with clofarabine, cyclophosphamide and etoposide. Ann Hematol. 2016;95:501–507. [DOI] [PubMed] [Google Scholar]
- 17.Shukla N, Kobos R, Renaud T, Steinherz LJ, Steinherz PG. Phase II trial of clofarabine with topotecan, vinorelbine, and thiotepa in pediatric patients with relapsed or refractory acute leukemia. Pediatr Blood Cancer. 2014;61:431–435. [DOI] [PubMed] [Google Scholar]