

Abstract
Background
Metastatic castration-resistant prostate cancers (mCRPC) are enriched for DNA repair gene defects (DRD) that can be susceptible to synthetic lethality through inhibition of poly(adenosine diphosphate-ribose) polymerase (PARP). We evaluated the efficacy and safety of the PARP inhibitor niraparib in patients with mCRPC and DRD who progressed on prior androgen signaling inhibitor and taxane therapy.
Methods
In this multicentre, open-label, single-arm, phase 2 study, patients ≥18 years of age with histologically confirmed mCRPC (mixed histology accepted, with the exception of the small cell pure phenotype) and DRD (assessed in blood, tumour tissue, or saliva) with progression on a prior next-generation androgen signaling inhibitor and a taxane per RECIST 1.1/PCWG3 and an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of ≤2 were eligible. For final study analysis, all patients who received at least one dose of study drug were included in the safety analysis population; patients with germline pathogenic or somatic biallelic pathogenic alterations in BRCA1 or BRCA2 (BRCA cohort) or biallelic alterations in other prespecified DRD (non-BRCA cohort) were included in the efficacy analysis population. Enrolled patients received niraparib 300mg orally once daily until treatment discontinuation, death, or study termination. The primary endpoint was objective response rate (ORR) in patients with BRCA alterations and measurable disease (measurable BRCA cohort). ClinicalTrials.gov number, NCT02854436.
Findings
Between September 28, 2016 and June 26, 2020, 289 patients were enrolled, of whom 182 (63·0%) had ≥3 systemic therapies for prostate cancer. A total of 223 patients were included in the BRCA (n=142) and non-BRCA (n=81) cohorts. At final analysis, ORR in the measurable BRCA cohort (n=76) was 34·2% (95% CI 23·7–46·0) with a median follow-up of 10·0 months (interquartile range: 6·6–13·3) and a median (95% CI) duration of response of 5·55 months (95% CI 3·91–7·20). In the safety analysis population, the most common all-grade treatment-emergent adverse events were nausea (169 [58·5%] of 289), anaemia (156 [54·0%] of 289), and vomiting (111 [38·4%] of 289); the most common grade ≥3 events were haematologic (anaemia, 95 [32·9%] of 289; thrombocytopaenia, 47 [16·3%] of 289; neutropaenia, 28 [9·7%] of 289). Of 134 (46·4%) of 289 patients with ≥1 serious treatment-emergent adverse event, the most common were also haematologic (thrombocytopaenia, 17 [9·3%] of 289; anaemia, 13 [4·5%] of 289). Haematologic events were manageable with dose interruptions, reductions, and/or appropriate supportive measures. Two adverse events with fatal outcome (1 patient with urosepsis in BRCA cohort; 1 patient with sepsis in non-BRCA cohort) were deemed possibly related to niraparib treatment.
Interpretation
Niraparib is tolerable and has anti-tumour activity in heavily pre-treated patients with mCRPC and DRD, particularly in those with BRCA alterations.
Funding
Janssen Research & Development.
Introduction
Patients with metastatic castration-resistant prostate cancer (mCRPC) who have progressed on a next-generation androgen signaling inhibitor and taxane chemotherapy (docetaxel and/or cabazitaxel) have limited treatment options.1,2 DNA repair gene defects (DRD), seen in approximately 12–23% of tumours in patients with metastatic prostate cancer when considering both germline and somatic alterations,3–5 are associated with cancer development, aggressiveness, and progression.6 These DRD-altered tumours are not only relatively frequent in metastatic disease, but are also associated with poor prognosis and potential resistance to systemic therapies.7 Developing more effective treatments to improve survival for these patients is a critical unmet need.
Cancers with DRD, particularly those with defects in homologous recombination repair, are highly sensitive to the blockade of DNA single-strand break repair via inhibition of the poly(adenosine diphosphate-ribose) polymerase (PARP) family of nuclear proteins, which are involved in single-strand DNA break repair.8 PARP inhibitors, such as olaparib, rucaparib, and talazoparib, have been studied in patients with mCRPC and DRD in prior phase 2 and 3 studies.9–13 Olaparib is approved in the European Union for breast cancer gene (BRCA)1/2-mutated mCRPC14 and by the United States Food and Drug Administration (FDA) for patients with mCRPC and DRD progressing after enzalutamide and/or abiraterone treatment,15 while rucaparib is approved for patients with BRCA1/2-mutated mCRPC who have been treated with an androgen signaling inhibitor therapy and a taxane.16
Niraparib, a potent and highly selective inhibitor of PARP-1 and PARP-2, is FDA approved for the maintenance treatment of select patient populations with ovarian, fallopian tube, and primary peritoneal cancers.17,18 Here, we report the final efficacy and safety results of a multicentre, open-label, phase 2 study of niraparib in patients with mCRPC and tumour DRD whose disease had progressed on androgen signaling inhibitor therapy and taxane chemotherapy (docetaxel and/or cabazitaxel).
Methods
Study design and participants
GALAHAD is a multicentre, open-label, phase 2 study at 115 sites in 15 countries that consisted of the following phases: prescreening, screening, treatment, follow-up, and long-term extension. The study protocol is available in the appendix (p 24).
Male patients at least 18 years of age with histologically confirmed mCRPC (mixed histology was acceptable, with the exception of the small cell pure phenotype, which was excluded) were eligible if they had a predefined DRD and disease progression on an androgen signaling inhibitor and taxane chemotherapy (docetaxel and/or cabazitaxel). Disease progression was defined as progression of metastatic prostate cancer in the setting of castrate levels of testosterone ≤50 ng/dL on a gonadotropin releasing hormone analog (GnRHa) or with history of bilateral orchiectomy at study entry, with progression specifically defined as prostate-specific antigen (PSA) progression or radiographic progression of soft tissue by RECIST 1.1 or bone disease by Prostate Cancer Working Group 3 (PCWG3) criteria.19,20 Patients were also required to have an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of ≤2. Patients with measurable and non-measurable disease were enrolled. Exclusion criteria included prior treatment with a PARP inhibitor or platinum-based chemotherapy regimen, known history or current diagnosis of myelodysplastic syndrome (MDS)/acute myeloid leukaemia (AML), and others. Independent Ethics Committees/Institutional Review Boards of each participating institution approved this study, and all patients provided written informed consent. Full details on the eligibility criteria can be found in the study protocol, which is included in the appendix (p 24).
Patients were prescreened using a blood (Resolution Bioscience) or tumour tissue (Foundation Medicine21) sample for evaluation of DRD alterations. The Resolution Bioscience assay, ‘Resolution HRD’, is a targeted hybrid capture, next-generation sequencing (NGS) assay that detects single-nucleotide variants, indels, and copy number variation (including homozygous deletions) in genes involved in homologous recombination repair using cell-free DNA from plasma. The specific genes of interest for DRD consisted of eight candidates including ATM serine/threonine kinase (ataxia telangiectasia mutated; ATM), BRCA1, BRCA2, BRCA1 interacting helicase 1 (BRCA1 interacting protein C-terminal helicase 1 gene; BRIP1), checkpoint kinase 2 (CHEK2); FA complementation group A (Fanconi anaemia complementation group A gene; FANCA); histone deacetylase 2 (HDAC2); and partner and localizer of BRCA2 (PALB2); in addition, three genes that are commonly mutated in mCRPC tumours were evaluated, namely androgen receptor (AR), cyclin dependent kinase inhibitor 2A (CDKN2A), and tumour protein 53 (TP53). This assay can also identify patients with monoallelic and biallelic pathogenic alterations in the genes of interest. Patients were eligible to enter the screening phase if a deleterious germline or somatic alteration was found in at least one of the following genes: BRCA1, BRCA2, FANCA, PALB2, CHEK2, BRIP1, HDAC2, or ATM. Patients were considered DRD-positive if they had an alteration with known pathogenic consequences including homozygous deletions, rearrangements, and nonsense, missense, frame shift, and splice-site mutations. After an updated biomarker logic that distinguished between biallelic and monoallelic DRD was developed, patients who had been enrolled with monoallelic or non-pathogenic DRD were excluded from the final analysis per approved protocol amendment (Figure 1). As such, only patients with germline pathogenic or biallelic pathogenic alterations in BRCA1 or BRCA2 (BRCA cohort) or other pre-specified non-BRCA genes (non-BRCA cohort) were included in the final analysis. Additional details on testing methods are available in the appendix (p 4).
Figure 1. Trial profile.

DRD=DNA repair gene defect. BRCA=breast cancer gene.
Procedures
All patients received niraparib 300 mg in the form of 100-mg capsules (manufactured for this study by Quotient Sciences; Boothwyn, PA, USA) starting on day 1 of cycle 1 (once daily dosing, with a cycle defined as 28 days) until treatment discontinuation due to disease progression, unacceptable toxicity or adverse events (AEs), diagnosis of MDS/AML, investigator decision in the best interest of the patient, patient withdrawal of consent, death, or study termination. Monitoring for need of dose adjustments or interruptions (eg, with laboratory measurements) was at the discretion of the investigator based on the severity of the AEs experienced. Patients who were not surgically castrate continued regularly prescribed GnRHa.
During the treatment phase, study visits occurred weekly for the first month, biweekly for the second month, and monthly thereafter. Computed tomography (CT)/magnetic resonance imaging (MRI) and technetium-99m bone scans were performed during screening, every 8 weeks for 24 weeks, and then every 12 weeks thereafter. Circulating tumour cell (CTC) counts were assessed at every cycle through cycle 7 and then at the end-of-treatment visit. PSA assessments were conducted every 4 weeks through cycle 7 and then every three cycles thereafter.
The follow-up phase began after completion of the treatment phase. If a patient discontinued treatment without radiographic progression, imaging was conducted every 12 weeks (± 2 weeks) until radiographic progression was documented. The long-term extension phase began after completion of the primary analysis, at which point patients could elect to discontinue treatment or continue niraparib until disease progression. Throughout the study, AEs were evaluated and graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE, version 4·03 or later). Appropriate supportive measures to address AEs could be administered at the discretion of investigators per institutional standards of care. Based on interactions with health authorities, the study concluded as planned per the protocol after specified enrollments were met and subsequent study evaluations completed. A full schedule of study assessments and procedures is available in the protocol (appendix p 24).
Outcomes
The final primary endpoint was investigator-assessed objective response rate (ORR defined as the proportion of patients achieving a confirmed partial response [PR] or complete response [CR] as defined by RECIST 1·119 per sums of target tumour lesion diameters, with no evidence of bone progression on bone scan per PCWG3 criteria20) in patients with BRCA alterations and measurable disease (measurable BRCA cohort). The primary endpoint was amended early on in this study from a composite response endpoint to ORR to comply with feedback from health authorities, and hence the primary efficacy analysis included only these subjects with measurable disease; additional details regarding this amendment are available in the study protocol (appendix p 24). Subjects with non-measurable disease were still included in the study to increase the size of the safety database and to also assess the activity of niraparib in this population.
Secondary efficacy endpoints included ORR in patients with non-BRCA alterations and measurable disease; CTC response (CTC0) defined as CTC=0 per 7·5 mL blood at 8 weeks post-baseline in patients with CTC>0 (1 or more) at baseline22,23; overall survival (OS; time from enrollment to death from any cause); radiographic progression-free survival (rPFS; time from enrollment to radiographic progression or death from any cause, whichever occurred first); time from enrollment to radiographic progression; time to PSA progression; time to symptomatic skeletal event (SSE; defined as time from enrollment to tumour-related spinal cord compression, radiation to bone to relieve skeletal symptoms, surgery to bone or need for tumour-related orthopaedic surgical intervention, or symptomatic or pathologic fracture); and duration of objective response (defined as time from CR or PR to radiographic progression of disease, unequivocal clinical progression, or death, whichever occurred first). Prespecified exploratory endpoints included the composite response rate (CRR), defined as either an objective response for patients with measurable disease, CTC conversion (defined as CTC count ≥5 per 7·5 mL blood at baseline and <5 per 7·5 mL blood post-therapy nadir), or a ≥50% decline in PSA (PSA50). Both CTC conversion and PSA50 were also assessed separately as prespecified exploratory analyses.
Safety evaluations included incidence, intensity, and types of AEs as well as deaths. AEs were classified as treatment-emergent AEs if they were reported on or after the date of first dose until 30 days (inclusive) after the last dose of study drug. Drug-related AEs were determined by investigators if they were considered related to the study drug.
Statistical analysis
Statistical analysis followed Simon’s two-stage design for phase 2 single-arm clinical trials.24 Specifically for the non-BRCA cohort, a futility analysis for ORR based on this design was implemented, where Simon’s stage 1 was assessed after approximately 14 patients with measurable disease were evaluated for ORR with at least one post-treatment scan and a confirmatory scan; enrollment was to be terminated for this cohort if two or fewer responses were observed in the first stage. Otherwise, enrollment was specified to proceed to the second stage with a total of 45 subjects enrolled for the 2 stages combined, and the null hypothesis was to be rejected if 10 or more responses were observed. For the primary endpoint, the null hypothesis of ORR ≤15% was tested against the alternate hypothesis of ORR ≥32%. With approximately 120 patients with biomarker-positive measurable disease (75 BRCA and 45 non-BRCA) planned for enrollment, the study had over a 90% power to show that the lower limit of the 95% confidence interval (CI) for the primary endpoint of ORR exceeded 15% in the measurable BRCA cohort. Efficacy of niraparib was to be declared if the lower bound of the two-sided 95% exact CI for ORR was >15% in this cohort. For the secondary endpoint of ORR in patients with non-BRCA alterations, the null hypothesis of ORR ≤15% was tested against the alternate hypothesis of ORR ≥32%, with a 1-sided α of 0.05 and power of 80%.
Objective response in soft tissue disease was evaluated in both BRCA and non-BRCA patients with measurable disease within the efficacy analysis population of patients who fulfilled the final biomarker logic criteria for this study; however, as prespecified, the primary endpoint was specifically evaluated in the measurable BRCA cohort, and final primary endpoint analysis was planned for approximately 6 months after the last patient with measurable disease in the BRCA cohort was enrolled. For analysis of ORR, patients who discontinued treatment prior to any efficacy assessments were considered non-responders; patients with no imaging available for a particular study visit were considered not evaluable for that visit, and patients without valid baseline data were considered not evaluable. Anti-tumour activity, such as PSA response, CTC conversion or response, and CRR, was analysed separately for the BRCA and non-BRCA cohorts, with corresponding criteria for identification of non-responders and non-evaluable patients based on discontinuation or availability of laboratory measurements. Response rates were calculated along with exact two-sided 95% CIs. Time-to-event endpoints were summarized by Kaplan-Meier curves, and median times and 95% CIs were calculated. Post-hoc analyses of efficacy included ORR by baseline characteristics of interest (such as in subgroups of patients with visceral disease at baseline or patients with ≥3 prior lines of therapy), specific evaluation of stable disease (defined as neither sufficient decrease in target lesions to qualify for PR, nor sufficient increase to qualify for progressive disease, with respect to smallest sum of target lesion diameters while on study), and biomarker analyses for non-DRD alterations of interest were also performed using available data. Analyses of additional study outcomes, such as the prespecified calculation of treatment compliance in the form of relative dose intensity (RDI), are described in further detail in the appendix (p 4).
Safety and treatment compliance were analysed in the safety analysis population (ie, all patients who received at least one dose of the study drug). AEs and serious AEs were reported and summarised. Sensitivity analyses with censoring rules were to be conducted if warranted. Additional details from the statistical analysis plan are available in supplementary materials. Statistical analyses were performed using SAS (version 15.1).
This study was registered with ClinicalTrials.gov (NCT02854436) and with the European Clinical Trials database (EudraCT 2016-002057-38) and was conducted according to Good Clinical Practice guidelines and the Declaration of Helsinki.
Role of the funding source
The sponsor and employees of the sponsor of this study participated in the study design, data collection, data analysis, and data interpretation, with writing and editorial assistance also funded by the sponsor. All authors participated in the writing process and provided critical input. All authors had full access to all data for the study and had final responsibility for the decision to submit for publication.
Results
A total of 385 patients were screened for this study; of these, 289 (75·1% of 385) patients were enrolled between September 28, 2016 and June 26, 2020 and hence included in the safety analysis population, and 223 (77·2% of 289) patients were included in the efficacy analysis population based on DRD eligibility from the validated biomarker logic (142 BRCA [139 biallelic, three monoallelic germline pathogenic]; 81 non-BRCA; Figure 1). Sixty-six patients (22·8% of 289) with monoallelic or non-pathogenic DRD from the safety analysis were not included in the final analysis of efficacy. Additional information on the prescreening results is available in the appendix (p 5). For patients with measurable disease, 76 with BRCA alterations (primary efficacy population) and 47 with non-BRCA alterations were enrolled, which fulfilled the numbers required per study sample size estimation for evaluation of ORR. The types and frequencies of genotypes observed in the efficacy analysis are reported in the appendix (p 10).
Based on the Simon’s two-stage design, enrollment proceeded through both stages to fulfill the estimated sample size requirements. At the clinical cutoff date of January 26, 2021, 271 (93·8%) of 289 patients had discontinued treatment, with reasons summarized in the appendix (p 11). Seven (9·2%) of 76 patients in the primary efficacy cohort (BRCA with measurable disease) discontinued therapy prior to their first study evaluation due to either progressive disease (n=4), urosepsis leading to death (n=1), or withdrawal of consent (n=1 due to fear of COVID-19; n=1 received Radium-223 due to bone disease burden [a predefined treatment discontinuation criterion]). The median treatment duration was 6·47 months (interquartile range [IQR]: 3·25–9·40) months in the BRCA cohort and 3·55 months (IQR: 1·81–5·55) in the non-BRCA cohort. Dose adjustments and interruptions are summarized in the appendix (p 12), with most treatment dose reductions due to an AE.
Patients were heavily pre-treated and exhibited advanced disease in both efficacy cohorts (Table 1). Nearly all patients had bone metastases and, in the primary efficacy population (measurable BRCA cohort), approximately 40% (30 of 76 patients) had visceral disease: 30·3% (23 of 76) patients had liver metastasis, and 17·1% (13 of 76) patients had lung metastasis. In addition, a majority of patients had nodal disease.
Table 1:
Baseline characteristics of patients in the efficacy analysis population
|
BRCA (n = 142) |
Measurable BRCA (n = 76) |
Non-BRCA (n = 81) |
|
|---|---|---|---|
| Age, years, median (IQR) | 67·0 (63·0 – 73·0) | 66·0 (62·0 – 73·0) | 70·0 (66·0 – 75·0) |
| Body weight, kg, mean (SD) | 82·7 (15·5) | 80·5 (13·4) | 79·9 (16·1) |
| Race, n (%) | |||
| White | 101 (71·1) | 57 (75·0) | 54 (66·7) |
| Asian | 9 (6·3) | 6 (7·9) | 6 (7·4) |
| Black or African American | 5 (3·5) | 3 (3·9) | 0 (0·0) |
| Other | 3 (2·1) | 0 (0·0) | 2 (2·5) |
| Multiple | 1 (0·7) | 0 (0·0) | 2 (2·5) |
| Not reported | 11 (7·7) | 5 (6·6) | 8 (9·9) |
| Unknown | 12 (8·5) | 5 (6·6) | 9 (11·1) |
| PSA at baseline, ng/mL, median (IQR) | 141·5 (41·0 – 512·4) | 197·0 (40.1 – 653.9) | 161·7 (43·7 – 611·1) |
| Patients with alterations in a single gene, n (%) a | 0 | 9 (18·0%) | 9 (11·8%) |
| BRCA1 | 4 (2·8) | 3 (3·9) | ·· |
| BRCA2 | 127 (89·4) | 69 (90·1) | ·· |
| ATM | ·· | ·· | 37 (45·7) |
| BRIP1 | ·· | ·· | 1 (1·2) |
| CHEK2 | ·· | ·· | 5 (6·2) |
| FANCA | ·· | ·· | 18 (22·2) |
| HDAC2 | ·· | ·· | 8 (9·9) |
| PALB2 | ·· | ·· | 0 (0) |
| ECOG-PS score, n (%) | |||
| 0 | 48 (33·8) | 25 (32·9) | 18 (22·2) |
| 1 | 78 (54·9) | 44 (57·9) | 47 (58·0) |
| 2 | 16 (11·3) | 7 (9·2) | 16 (19·8) |
| Extent of disease progression at study entry, n (%) | |||
| Bone | 127 (89·4) | 61 (80·3) | 79 (97·5) |
| Visceral | 33 (23·2) | 30 (39·5) | 20 (24·7) |
| Liver | 24 (16·9) | 23 (30·3) | 13 (16·0) |
| Lung | 15 (10·6) | 13 (17·1) | 10 (12·3) |
| Lymph node | 79 (55·6) | 67 (88·2) | 39 (48·1) |
| Soft tissue | 22 (15·5) | 21 (27·6) | 16 (19·8) |
| Disease status, n (%) | |||
| Measurable | 76 (53·5) | 76 (100·0) | 47 (58·0) |
| Non-measurable | 66 (46·5) | 0 (0) | 34 (42·0) |
| Gleason score at diagnosis, n (%) b | |||
| <8 | 39 (28·9) | 20 (27·3) | 26 (33·8) |
| ≥8 | 96 (71·1) | 53 (72·6) | 51 (66·2) |
| Prior therapies for prostate cancer, n (%) c | |||
| 2 | 59 (41·5) | 29 (38·2) | 22 (27·2) |
| 3 | 54 (38·0) | 31 (40·8) | 31 (38·3) |
| 4 | 21 (14·8) | 12 (15·8) | 19 (23·5) |
| 5 | 7 (4·9) | 3 (3·9) | 9 (11·1) |
| 6 | 1 (0·7) | 1 (1·3) | 0 (0) |
| Prior androgen signaling inhibitor therapy, n (%) | |||
| 1 | 97 (68·3) | 51 (67·1) | 45 (55·6) |
| 2 | 44 (31·0) | 25 (32·9) | 31 (38·3) |
| 3 | 1 (0·7) | 0 (0) | 5 (6·2) |
| Prior taxane-based chemotherapy, n (%) | |||
| 1 | 100 (70·4) | 51 (67·1) | 41 (50·6) |
| 2 | 42 (29·6) | 25 (32·9) | 40 (49·4) |
Patient numbers and percentages may not add up to 100% as some patients had more than one gene alterations. All patients with PALB2 had co-occurring alterations and are thus not listed here.
n=135 in BRCA cohort with Gleason score at initial diagnosis; n=73 in BRCA measurable disease cohort with Gleason score at initial diagnosis; n=77 in non-BRCA cohort with Gleason score at initial diagnosis.
Number of androgen signaling inhibitors, taxane-based chemotherapies, cytotoxic chemotherapies, and other therapies received. Specifically, prior therapies could include taxane-based chemotherapy for metastatic prostate cancer with evidence of disease progression, or next-generation androgen signaling inhibitor therapy for either metastatic prostate cancer with evidence of disease progression or non-metastatic castration-resistant prostate cancer with evidence of subsequent metastasis.
BRCA=breast cancer gene. IQR=interquartile range. SD=standard deviation. PSA=prostate-specific antigen. ATM=ataxia telangiectasia mutated. BRIP1=BRCA1 interacting protein C-terminal helicase 1 gene. CHEK2=checkpoint kinase 2 gene. FANCA=Fanconi anaemia complementation group A gene. HDAC2=histone deacetylase 2 gene. ECOG PS=Eastern Cooperative Oncology Group performance status.
At baseline, 182 of 289 (63·0%) patients in the safety analysis population had received ≥3 prior systemic therapies for metastatic prostate cancer; approximately one third (94 of 289) had received two prior androgen signaling inhibitor therapies, and 107 of 289 patients had received two prior taxane-based chemotherapies. Demographics and baseline characteristics of the safety analysis population are also available in the appendix (p 13).
The final efficacy results for the overall BRCA cohort (with median follow-up of 10·1 months [IQR: 7·5–13·4]) are shown in Table 2, Table 3, and the appendix (p 6, 7, 14).
Table 2:
Objective response rates
| Response | Measurable BRCAa (n = 76) |
Measurable non-BRCA (n = 47) |
|---|---|---|
|
ORR, n (%)
(95% CI) |
26 (34·2) (23·7 – 46·0) |
5 (10·6) (3·5 – 23·1) |
| CR, n (%) | 2 (2·6) | 0 |
| PR, n (%) | 24 (31·6) | 5 (10·6) |
Primary efficacy analysis cohort. ORR in measurable non-BRCA patients was a secondary efficacy endpoint.
BRCA=breast cancer gene. ORR=objective response rate. CI=confidence interval. CR=complete response. PR=partial response.
Table 3:
Secondary and exploratory efficacy endpoints for niraparib in patients with mCRPC
| Endpoint |
BRCA (n = 142) |
Measurable BRCA (n = 76) |
Non-BRCA (n = 81) |
|---|---|---|---|
| CTC response (CTC0) b , n/N (%) | 31/131 (23·7) | 18/71 (25.4) | 6/71 (8·5) |
| OS, mo, median (95% CI) | 13·01 (11·04 – 14·29) | 10·87 (9·49 – 13·77) | 9·63 (8·05 – 13·44) |
| rPFS, mo, median (95% CI) | 8·08 (5·55 – 8·38) | 5·52 (5·29 – 7·59) | 3·71 (1·97 – 5·49) |
| Time to radiographic progression, mo, median (95% CI) | 8·08 (5·75 – 8·97) | 5·55 (5·36 – 8·08) | 3·78 (2·00 – 5·55) |
| Time to PSA progression, mo, median (95% CI) | 5·13 (4·60 – 5·59) | 5·55 (4·60 – 8·31) | 3·65 (2·83 – 3·71) |
| Time to SSE, mo, median (95% CI) | 13·80 (10·41 – NE) | 13·80 (9·07 – NE) | 10·35 (8·18 – NE) |
| DOR, mo, median (95% CI) | 6·28 (3·65 – 9·23) | 5·55 (3·91 – 7·20) | 5·16 (2·14 – NE) |
|
CRRa, n/N (%)
(95% CI) |
82/142 (57·7) (49·2 – 66·0) |
46/76 (60·5) (48·7 – 71·6) |
12/81 (14·8) (7·9 – 24·5) |
|
CTC conversionc, n/N (%)
(95% CI) |
55/117 (47·0) (37·7 – 56·5) |
28/64 (43·8) (31·4 – 56·7) |
9/60 (15·0) (7·1 – 26·6) |
|
PSA50, n/N (%)
(95% CI) |
61/142 (43·0) (34·7 – 51·5) |
31/76 (40·8) (29·7 – 52·7) |
4/81 (4·9) (1·4 – 12·2) |
Defined as either an objective response for patients with measurable disease, CTC conversion (defined as CTC count ≥5 per 7·5 mL blood at baseline and <5 per 7·5 mL blood post-therapy nadir), or PSA50.
Defined per protocol and SAP as CTC=0 per 7·5 mL blood at 8 weeks post-baseline in patients with baseline CTC >0.
Among patients with baseline CTC ≥5.
mCRPC=metastatic castration-resistant breast cancer. BRCA=breast cancer gene. CTC=circulating tumour cells. CTC0=CTC count >0 at baseline to 0/7·5 mL blood at week 8. OS=overall survival. CI=confidence interval. rPFS=radiographic progression-free survival. PSA=prostate-specific antigen. SSE=symptomatic skeletal event. NE=not estimable. DOR=duration of objective response. CRR=composite response rate. CTC conversion=CTC count ≥5/7·5 mL blood at baseline to <5/7·5 mL blood at nadir. PSA50=≥50% decline in prostate-specific antigen.
The primary endpoint, ORR per protocol in the measurable BRCA cohort, was 34·2% (26 of 76 patients; Table 2) with a median follow-up of 10·0 months (IQR: 6·6–13·3). Of the 76 patients in the measurable BRCA cohort, 35 (46·1%) patients had at least a partial response by RECIST 1.1 (≥30% decrease of maximum change from baseline in the sum of longest target lesion diameters) relative to baseline (appendix p 6). The median duration of objective response in this cohort was 5·55 months (95% CI 3·91–7·20), of which 8 of 26 responses (30·8%) were ongoing at the time of data cutoff.
Median rPFS in the overall BRCA cohort was 8·08 months (95% CI 5·55–8·38; Figure 2A), with 87 (61·3%) events at the time of data cutoff. Median OS was 13·01 months (95% CI 11·04–14·29; Figure 2B), with 88 (62·0%) events and 12-month and 24-month event-free rates (95% CI) of 56·4% (47·2–64·6%) and 15·2% (7·7–25·1%), respectively. Approximately one quarter of patients achieved CTC0 (Table 3); meanwhile, 85 (59·9%) of 142 patients and 42 (55·3%) of 76 patients experienced PSA progression in the BRCA and measurable BRCA cohorts, respectively, and 46 (32·4%) of 142 patients and 23 (30·3%) of 76 patients had a documented SSE in these cohorts, respectively.
Figure 2. Radiographic progression-free survival and overall survival in the BRCA and non-BRCA cohorts. (A) Radiographic progression-free survival. (B) Overall survival.
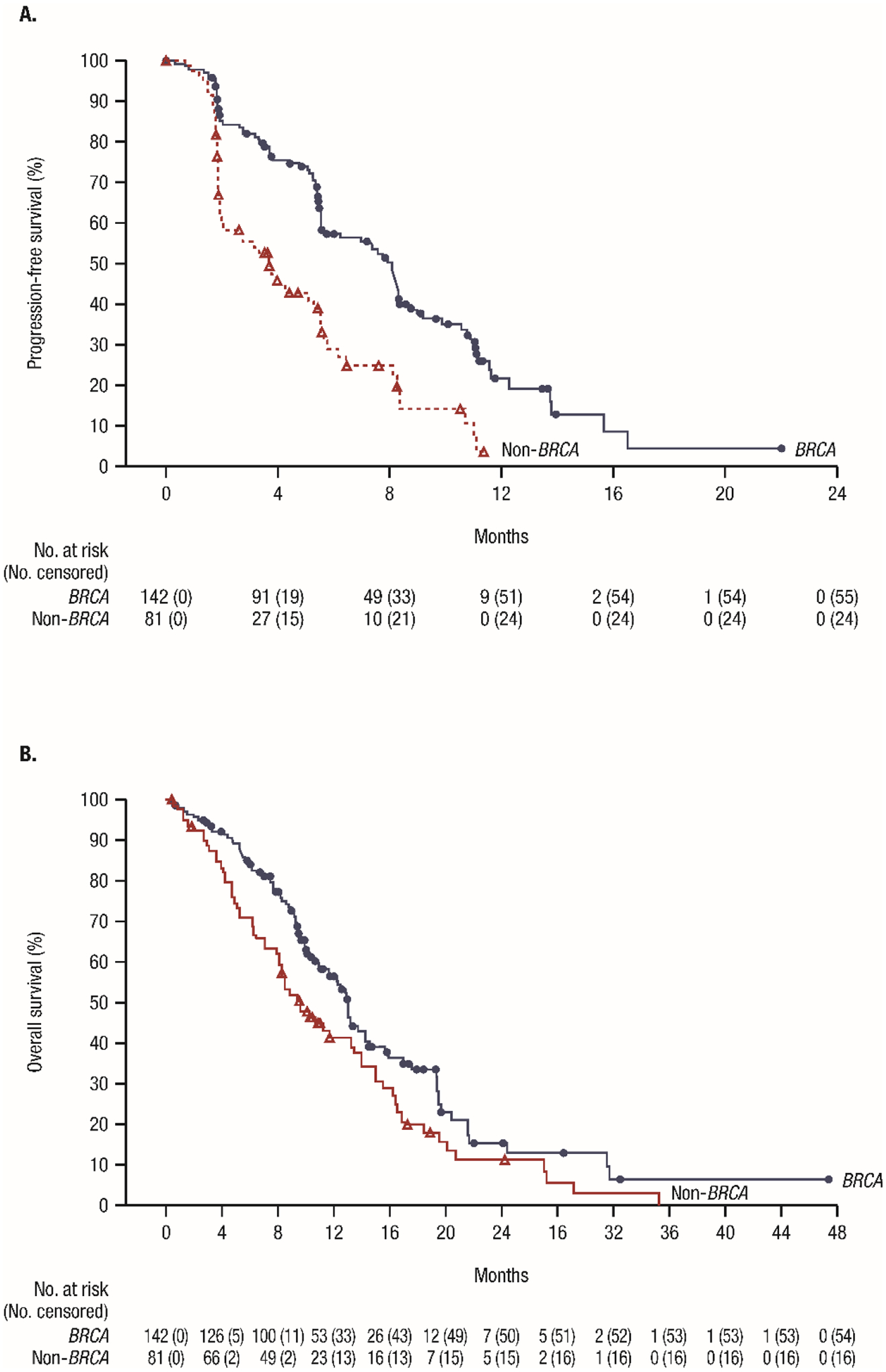
Symbols represent censored patients.
BRCA=breast cancer gene.
The CRR in the overall BRCA cohort is also presented in Table 3. More than 40% achieved PSA50 and CTC conversion, and approximately two-thirds of evaluable patients experienced a decrease in PSA levels from baseline (Table 3 and appendix p 7). Similar outcomes in CRR were obtained in the BRCA cohort for patients with measurable and non-measurable disease (appendix p 14).
Of note, two patients (both with biallelic alterations) in the measurable BRCA cohort achieved a CR. One with visceral (adrenal) and nodal disease at baseline maintained the CR for 9·7 months, and a second with nodal disease at baseline experienced a CR that persisted for 9·5 months based on imaging despite having received only 2·1 months of treatment that was discontinued due to an AE of anaemia. Of the 30 patients who had visceral disease at baseline, 11 (36·7%) achieved an objective response. Furthermore, of the 20 patients who experienced stable disease for >6 months in this study, 14 (70%) belonged to the BRCA cohort. A total of 16 of the 76 (21·1%) patients continued treatment after radiographic progression with no unequivocal clinical progression because they were considered to still be benefitting from therapy.
In the non-BRCA cohort, with a median follow-up of 8·6 months (IQR: 4·8–14·0), objective response was achieved in 5 (10·6%) of 47 patients with measurable disease (Table 2). The median duration of objective response was 5·16 months (95% CI 2·14–not estimable), none of which is ongoing. The corresponding maximum changes in the sum of target tumour lesion diameters from baseline are presented in the appendix (p 6).
Median rPFS in the non-BRCA cohort was 3·71 months (95% CI 1·97–5·49; Figure 2A), with 57 (70·4%) events at the time of data cutoff. OS was 9·63 months (95% CI 8·05–13·44; Figure 2B) with 65 (80·2%) events and 12-month and 24-month event-free rates (95% CI) of 41·3% (30·0–52·2%) and 11·1% (4·4–21·2%), respectively. Less than 10% (6 of 71 evaluable patients) achieved CTC0 in this cohort; 39 (48.1%) of 81 patients had PSA progression in this analysis, and 19 (23.5%) of 81 patients had a documented SSE.
The CRR in the non-BRCA cohort is also presented in Table 3, for which the maximum change in PSA from baseline is also presented in the appendix (p 7). Among the non-BRCA patients with non-measurable disease, four (11.8%) of 34 demonstrated response by either PSA50 or CTC conversion (appendix p 14).
Of note, two (12·5%) of 16 patients with visceral disease and four (80·0%) of five patients who had received ≥3 lines of therapy experienced objective response in the non-BRCA cohort. Six patients in this cohort experienced stable disease for >6 months. A total of 15 out of 47 (31·9%) patients continued treatment after radiographic progression with no unequivocal clinical progression.
Notably, 11 of 142 (7·7%) patients in the BRCA cohort and seven of 81 (8·6%) patients in the non-BRCA cohort had co-expression of 2 or more eligible DRD biomarkers. Moreover, in addition to being DRD positive, 162 of 220 (73·6%) with available plasma DNA results had alterations in the AR and 49 of 220 (22·3%) alterations in TP53, of which 42 of 220 (19·1%) had both TP53 and AR alterations. These additional alterations in plasma DNA were comparably distributed between the BRCA and non-BRCA cohorts, and no significant difference in ORR was observed in subgroups of patients with or without co-occurring alterations.
Safety results are summarised in Table 4 and the appendix (p 15–18, p 21–23). Nearly all patients in the safety population experienced at least one treatment-emergent AE. The most common events (any grade) were nausea (58·5%; 169 of 289), anaemia (54·0%; 156 of 289), and vomiting (38·4%; 111 of 289). Of the grade ≥3 treatment-emergent AEs reported in 217 (75·1%) of 289 patients, most were haematologic, with both the most common grade ≥3 AEs overall and grade ≥3 AEs of special interest being anaemia (32·9%; 95 of 289), thrombocytopaenia (16·3%; 47 of 289), and neutropaenia (9·7%; 28 of 289). These events were manageable with treatment interruptions, dose reductions, and/or supportive measures such as blood transfusions. One patient experienced neutropaenic sepsis. The most common non-haematologic grade 3/4 treatment-emergent AEs were fatigue and nausea. A total of 134 (46·4%) of 289 patients had ≥1 serious treatment-emergent AE, with the most common again being haematologic (17 [9·3%] of 289 with thrombocytopaenia; 13 (4·5%) of 289 with anaemia). Similarly, of 43 (14·9%) of 289 patients with drug-related serious treatment-emergent AE, the most common were thrombocytopaenia (4 [2·8%] of 142 in BRCA and 7 [8·6%] of 81 in non-BRCA) and anaemia (3 [2·1%] of 142 in BRCA and 3 [3·7%] of 81 in non-BRCA). The most common AEs overall in the BRCA and non-BRCA cohorts are also summarised in the appendix (p 18).
Table 4:
Treatment-emergent adverse events
| Events, n (%) | Safety analysis set (N=289)a,b |
|||
|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 5 | |
| Nausea | 154 (53·3) | 15 (5·2) | 0 | 0 |
| Vomiting | 101 (34·9) | 10 (3·5) | 0 | 0 |
| Constipation | 95 (32·9) | 5 (1·7) | 1 (0·3) | 0 |
| Fatigue | 87 (30·1) | 19 (6·6) | 0 | 0 |
| Decreased appetite | 85 (29·4) | 8 (2·8) | 0 | 0 |
| Anaemia | 61 (21·1) | 92 (31·8) | 2 (0·7) | 1 (0·3) |
| Thrombocytopaenia | 52 (18·0) | 24 (8·3) | 23 (8·0) | 0 |
| Neutropaenia | 27 (9·3) | 17 (5·9) | 11 (3·8) | 0 |
A total of n=288 patients had one or more recorded TEAEs.
Data presented for grade 1–2 TEAEs with a combined incidence of ≥20% reported or any higher-grade (grades 3, 4, and/or 5) TEAEs with an incidence of >5% reported.
TEAEs=treatment-emergent adverse events.
The estimated RDI for each cohort is presented in the appendix (p 20). In the safety population, 128 of 289 (44·3%) patients had an AE leading to a dose reduction: consistent with the above findings, the majority (87 of 128, or 68·0%) were haematologic (p 21), which were again manageable with said dose reductions, interruptions, and/or appropriate supportive measures (administered at the discretion of investigators per institutional standards of care). Among those evaluated for efficacy, 70 (94·6%) of 74 patients and 37 (92·5%) of 40 patients with dose reductions in the BRCA and non-BRCA cohorts, respectively, had dose reductions due to AEs versus other reasons; 103 of 223 (46·2%) patients received at least one transfusion (platelets or packed red blood cells), and the incidence of transfusions was similar between the BRCA and non-BRCA cohorts. Other supportive measures included colony-stimulating factors which were administered to 12 of 289 (4·2%) patients, and erythropoietin which was administered to 15 of 289 (5·2%) patients. A total of 37 (12·8%) of 289 patients were deemed to have discontinued treatment due to drug-related toxicities (treatment-emergent AEs deemed related to study drug). The most common drug-related toxicities leading to discontinuation were thrombocytopaenia (in 7 patients with BRCA alterations and 3 patients with non-BRCA alterations) and anaemia (in 6 patients with BRCA alterations and 1 patient with non-BRCA alteration).
AEs with fatal outcome are summarised in the appendix (p 22). Of these, two events (1 patient with urosepsis in the BRCA cohort; 1 patient with sepsis in the non-BRCA cohort) were deemed possibly related to niraparib treatment. A total of 208 (72·0%) of 289 patients died during the study; reasons for deaths are further summarized in the appendix (p 23). Sensitivity analysis for COVID-19 was the only sensitivity analysis warranted, but there was only one patient with a COVID-19 related adverse event (non-serious) and one death due to COVID-19 in this study (appendix p 23).
Discussion
The results of this multicentre, open-label, phase 2 study establish the anti-tumour activity of niraparib in patients with mCRPC and DRD who have progressed on both androgen signaling inhibitors and taxanes. Treatment with niraparib was manageable, and adverse events observed were consistent with the known safety profile of niraparib with no new safety signals.
The activity of niraparib in the measurable BRCA cohort is noteworthy given the heavily pre-treated end-stage patient population with relatively few therapeutic options. This is especially remarkable considering the high percentage of patients with visceral metastasis, in particular to the liver, that is strongly associated with poor survival,25 the high percentage of patients with ≥3 lines of prior therapy, and that there were incidences of patients in the BRCA cohort achieving CR. Further evidence of benefit was shown by the proportion of patients experiencing stabilization of disease for 6 months or more in both the BRCA and non-BRCA cohorts, a clinically meaningful finding given this heavily pretreated advanced disease state. Also noteworthy was that patients in both cohorts, some with multiple poor prognostic features, continued to derive clinical benefit from niraparib after radiographic progression (in line with the importance of considering the totality of a patient’s disease before discontinuing a drug rather than strictly at the first evidence of progression in any site, as recommended by PCWG321). Objective response rate, median rPFS and OS, and CRR were numerically greater in the BRCA cohort, including a median rPFS time that was approximately double that in the non-BRCA cohort. Interestingly, CRR was largely comparable between the subgroups of patients with measurable versus non-measurable disease in both cohorts (particularly BRCA). Taken together, these results extend the evidence of efficacy of PARP inhibitors in mCRPC patients with DRD-altered tumours whose disease has progressed on approved life-prolonging therapies, while further highlighting the importance of biologic profiling of an individual patient’s disease in mCRPC.8,21 Of note, this trial is the first to prospectively test the CTC0 endpoint, which has been shown to strongly associate with survival.
The dose of niraparib used in this study (300 mg oral; once daily dosing) was chosen based on the previously evaluated pharmacokinetics, clinical activity, and safety profile of niraparib and is the approved dose for patients with ovarian cancer.17,26 As expected, grade ≥3 treatment-emergent adverse events were largely haematologic and manageable with supportive measures (such as blood transfusions and growth factor treatment), dose interruptions, and dose reductions. Rates of treatment interruptions and reductions were relatively higher than the previously reported rates for other PARP inhibitors.9–13 This is in line with the more advanced disease stage of patients in GALAHAD, who all had progressed on androgen signaling inhibitor therapy and taxane chemotherapy and also tended to be on later lines of therapy; with a majority having received ≥3 lines of prior therapy. Moreover and importantly, the present study applied more stringent dose interruption criteria (for instance, skipping a dose was also considered as dose interruption). Thus, the finding that at least half of the patients in both the BRCA and non-BRCA cohorts maintained the full dose (300 mg) of niraparib throughout the study supports the overall tolerability of this regimen. RDI was also generally high, including in the primary efficacy cohort, and was numerically higher for those who achieved objective response in that cohort.
The clinical activity of niraparib demonstrated in this study’s specific patient population (noting particular efficacy observed in the BRCA cohort) can be further contextualized by results of studies of other PARP inhibitors that point to a benefit for both minimally and heavily pretreated patients with mCRPC and select DRD alterations. Of note, in the phase 3 PROfound study that evaluated patients treated with the PARP inhibitor olaparib versus those treated with physician’s choice of an androgen signaling inhibitor, olaparib treatment resulted in a median rPFS of 7·4 months vs 3·6 months (hazard ratio [HR], 0·34; p <0·001), median OS of 19·1 months vs 14·7 months (HR, 0·69; p = 0·02 at final analysis), and confirmed ORR of 33% (28/84) vs 2% (1/43; odds ratio, 20·86; 95% CI: 4·18–379·18; p <0·001) with the control treatment, respectively, in a cohort of patients with alterations in BRCA1, BRCA2, or ATM.10,11 Rucaparib, which was investigated in the phase 2 TRITON-2 study, reported ORR and median time to PSA progression of 47·5% and 6·5 months, respectively, with better response seen in patients with BRCA alterations compared with alterations in other genes such ATM or cyclin-dependent kinase 12 (CDK12).12,13 In the phase 2 TALAPRO-1 study, patients with select gene alterations reported to sensitize to PARP inhibitors were enrolled and treated with talazoparib. ORR was 29·8% (31/104; 95% CI: 21·2–39·6) with talazoparib treatment after a median follow-up of 16·4 months.9
Limitations of the present study include that some patients in this study developed progressive disease prior to completing their first disease evaluation, consistent with the advanced stage and aggressiveness of the disease in the enrolled population. Also, tissue-based assays rely on sufficient high-quality biopsy samples, which may be difficult to obtain, and further challenges ensue when archival samples are found to be unsuitable or unavailable for analysis. In this study, a commercially available tissue-based assay was initially used to select patients, but due to the aforementioned challenges, a blood-based assay was subsequently implemented, with early prescreening rates and biomarker logic finalization then addressed during a brief study pause. As such, a considerable number of patients in GALAHAD were prescreened using the latter assay type. Blood-based assays may offer valuable data, especially for metastatic prostate cancer for which tumour biopsies are challenging to acquire and biopsies are largely limited to bone that has significant issues with quality of materials. However, there are also limitations to liquid biopsies. Given the lack of parallel NGS of corresponding white blood cells in this study, clonal hematopoietic alterations of indeterminate potential (CHIP) may have presented as a biological confounding factor.27 In addition, blood-based assays may show false negative results in blood samples with low plasma circulating tumour DNA content, especially for mutations that are difficult to detect such as homozygous deletions. Conversely, circulating tumour DNA assays may select for patients with higher percentages of circulating tumour DNA, which was previously found to be a predictive factor for a poorer prognosis.28 With this said, the number of patients enrolled for efficacy evaluation in GALAHAD (n=223 with specifically germline pathogenic/biallelic DRD alterations, of n=4218 total with any biomarker sample submitted) represents an incidence of DRD alterations that is within expectations. Finally, 22·3% of the GALAHAD study population exhibited TP53 alterations, which are also associated with overall poor prognosis.
Additional studies, including those designed to evaluate niraparib-based regimens in appropriate biomarker-identified populations at earlier stages of disease, are underway to expand on the present findings. The phase 3 MAGNITUDE study evaluates niraparib in combination with abiraterone acetate plus prednisone as first-line therapy in patients with metastatic castration-resistant disease with or without DRD.29 The phase 3 AMPLITUDE study will also evaluate niraparib in combination with abiraterone acetate plus prednisone in a biomarker-selected population with metastatic castration-sensitive disease.30
In conclusion, these results suggest that niraparib has promising clinical activity with a manageable safety profile when administered as a monotherapy for treatment-refractory mCRPC with BRCA alterations and or select non-BRCA alterations. Such findings also underscore the need for and importance of molecular testing to inform management along with continued research to establish treatment paradigms with appropriately targeted therapies for patients with prostate cancer. Efforts to investigate and better understand predictive markers and signatures of both response and resistance to treatment with PARP inhibitors such as niraparib are needed to guide therapy selection and optimize treatment outcomes.
Supplementary Material
Research in Context.
Evidence before this study
At the time that the GALAHAD study was designed, docetaxel and the androgen signaling inhibitors abiraterone acetate and enzalutamide were the only established treatment options for patients with metastatic castration-resistant prostate cancer, but there remained a subset of patients who either did not initially respond or became refractory to these agents and for whom no approved therapeutic options were available. Given that genomic instability is a hallmark of cancer that has been documented in this patient population, we evaluated the medical literature by searching PubMed through August 31, 2016 with (castration) AND (resistant) AND (“prostatic neoplasm” OR “prostate cancer”) AND (“genomic instability” OR “DNA repair” OR “DNA repair defect”) as search terms of interest with no additional restrictions (eg, to English language publications only), yielding 33 results. Only two of these were clinical trial publications, which presented results from an early phase 1 study of veliparib and the phase 2 TOPARP study of olaparib—both PARP inhibitors—in this disease setting. Veliparib reported limited efficacy, but the high response rate observed among patients with specified DNA repair gene defects in the TOPARP study provided a compelling rationale for assessing the clinical activity and safety of other such agents in a genetically-select patient population. Authors also contributed appropriate citations of importance not detected by the original search strategy or that were found in subsequent literature, with emphasis on randomised clinical trials, systematic reviews, meta-analyses, and prospective observational studies. Taken together, findings indicated that PARP inhibitors exhibit notable activity in cancers with DNA repair gene defects. In articles published after the GALAHAD study’s initiation, further clinical activity has since been reported in metastatic castration-resistant prostate cancers with select DNA repair gene defects for the PARP inhibitors olaparib, rucaparib, and talazoparib (for which key primary publications are cited in the present work) but not yet for niraparib, which is also a potent and highly selective PARP inhibitor with established efficacy and tolerability in other cancers.
Added value of this study
To our knowledge, the GALAHAD study is the first to demonstrate the anti-tumour activity of niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects who previously progressed on both androgen signaling inhibitors and taxanes, with notable efficacy particularly in the cohort of patients with defects in breast cancer gene (BRCA)1/2.
Implications of all the available evidence
This final analysis of the GALAHAD study suggests that in patients with heavily pre-treated metastatic castration-resistant disease, niraparib may offer promising clinical activity with a manageable safety profile. These findings motivate the further assessment of niraparib alone or in combination with other agents to improve treatment options and underscore importance of biologic disease profiling in informing treatment.
Acknowledgments
This study was funded by Janssen Research & Development, LLC. We thank the patients who participated in this study, along with their families, and the study teams who were involved at each participating institution. Statistical analysis support was provided by Eugene Zhu and George Wang of Janssen Research & Development (Raritan, NJ, USA). Medical writing and editorial assistance were provided by Michelle Kwon, PhD, and Danyang Zhou, PharmD, of Cello Health Communications/MedErgy (Yardley, PA, USA), which was provided in accordance with Good Publication Practice (GPP3) guidelines and funded by Janssen Research & Development.
Funding:
This study was funded by Janssen Research & Development.
Declaration of interests
MRS has received grants and personal fees from Bayer, Amgen, Janssen, and Lilly; and has received personal fees from Astellas Pharma, Novartis, and Pfizer.
HIS has a leadership role in Asterias Biotherapeutics; stock and other ownership interests in Asterias Biotherapeutics; served in a consulting or advisory role in Ambry Genetics Corporation, Konica Minolta Inc, Amgen, Bayer, ESSA, Janssen Biotech Inc, Janssen Research & Development, Menarini Silicon Biosystems, Sanofi Aventis, WIRB-Copernicus Group Research Funding - Epic Sciences (Inst), Illumina (Inst), Innocrin Pharma (Inst), Janssen (Inst), Menarini Silicon Biosystems (Inst), Thermo Fisher Scientific Biomarkers (Inst); personal fees from Amgen, Asterias Biotherapeutics, Bayer, ESSA, Konica Minolta Inc, Menarini Silicon Biosystems, Prostate Cancer Foundation, Sanofi Aventis, and WIRB-Copernicus Group.
SS has received grants from Amgen, Endocyte, and Genentech; has received grants and personal fees from AstraZeneca and Merck; and has received personal fees from Bristol Myers Squibb and Merck Serono.
EE has received research grant support, honoraria, and personal fees from and served on advisory boards for Sanofi, Janssen, Astellas, Tolmar, and Bayer.
PNL Jr. has served in a consulting or advisory role for Janssen and received research grant support from Aragon Pharmaceuticals, Janssen Biotech Inc, TRACON Pharmaceuticals, Merck, Pharmacyclics, Incyte, and Taiho Pharmaceuticals.
EY has received grants from Bayer, Dendreon, Merck, Pharmacyclics, Seagen Inc., Daiichi-Sankyo, Taiho, Blue Earth, and Lantheus; and has received personal fees from Amgen, Astrazeneca, Bayer, Clovis, Dendreon, Janssen, Merck, Pharmacyclics, Seagen Inc., QED, Sanofi, Abbvie, Advanced Accelerator Applications, Exelixis.
DJG has received personal fees from the American Association for Cancer Research, Axess Oncology, Capio Biosciences, Constellation Pharma, EMD Serono, Flatiron, Ipsen, Merck Sharp & Dohme, Michael J. Hennessey Association, Millennium Medical Publishing, Modra Pharma, Myovant Sciences, Inc., NCI Genitourinary, Nektar Therapeutics, Physician Education Resource, Propella TX, RevHealth, LLC, and UroGPO; has received grants and personal fees from Astellas, AstraZeneca, Bristol Myers Squibb, and Pfizer; has received personal fees and non-financial support from Bayer H/C Pharmaceuticals and UroToday; has received grants from Calithera and Novartis; has received grants, personal fees, and non-financial support from Exelixis, Inc., Sanofi, and Janssen Pharma.
KNC has received grants from Janssen during the conduct of the study; has received grants and personal fees from AstraZeneca, Bayer, Astellas, Novartis, Pfizer, Point Biopharma, Roche, and Sanofi; and has received personal fees from Daiichi Sankyo, Merck, and Bristol Myers Squibb.
FS has received grants, personal fees, and non-financial support from Janssen during the conduct of the study; and has received grants, personal fees, and non-financial support from AstraZeneca, Astellas, Pfizer, Bayer, Myovant, Sanofi, and Novartis.
OS has nothing to disclose.
DO has received grants, personal fees, and non-financial support from AstraZeneca, Bayer, Janssen, and Pfizer; has received personal fees from Clovis, Daiichi Sankyo, and MSD; and has received non-financial support from Astellas, F. Hoffman-LaRoche, Genentech, and Ipsen.
DCD has received research support from the US Department of Defense, the American Society of Clinical Oncology, the Prostate Cancer Foundation, Stand Up to Cancer, Janssen Research & Development, Astellas, Medivation, Agensys, Genentech, and CreaTV.
GEM, BME, KAU, PF, and ALG are employees of Janssen Research & Development. GEM owns stocks with Janssen. XZ was an employee of Janssen at the time of this study.
KF has received personal fees from AAA, Astellas, Curevac, and Sanofi; has received grants and personal fees from AstraZeneca, Bayer, Janssen, MSD, and Orion; and has received grants from Bristol Myers Squibb and Pfizer.
Data sharing statement
The full study protocol is available for download in the accompanying appendix.
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu. De-identified patient-level data will be made available to qualified researchers upon request, after signing of a data transfer agreement with Janssen Research & Development. Requests for data sharing, including a research proposal, can also be made to the corresponding author.
References
- 1.Gillessen S, Attard G, Beer TM, et al. Management of patients with advanced prostate cancer: report of the Advanced Prostate Cancer Consensus Conference 2019. Eur Urol 2020; 77(4): 508–47. [DOI] [PubMed] [Google Scholar]
- 2.Sartor O, de Bono J, Chi KN, et al. Lutetium-177–PSMA-617 for metastatic castration-resistant prostate cancer. N Engl J Med 2021; 385(12): 1091–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016; 375(5): 443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tukachinsky H, Madison RW, Chung JH, et al. Genomic analysis of circulating tumor DNA in 3,334 patients with advanced prostate cancer identifies targetable BRCA alterations and AR resistance mechanisms. Clin Cancer Res 2021; 27(11): 3094–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015; 161(5): 1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schiewer MJ, Knudsen KE. DNA damage response in prostate cancer. Cold Spring Harb Perspect Med 2019; 9(1): a030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warner EW, Yip SM, Chi KN, Wyatt AW. DNA repair defects in prostate cancer: impact for screening, prognostication and treatment. BJU Int 2019; 123(5): 769–76. [DOI] [PubMed] [Google Scholar]
- 8.Ratta R, Guida A, Scotte F, et al. PARP inhibitors as a new therapeutic option in metastatic prostate cancer: a systematic review. Prostate Cancer Prostatic Dis 2020; 23(4): 549–60. [DOI] [PubMed] [Google Scholar]
- 9.de Bono JS, Mehra N, Scagliotti GV, et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): an open-label, phase 2 trial. Lancet Oncol 2021; 22(9): 1250–64. [DOI] [PubMed] [Google Scholar]
- 10.de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med 2020; 382(22): 2091–102. [DOI] [PubMed] [Google Scholar]
- 11.Hussain M, Mateo J, Fizazi K, et al. Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med 2020; 383(24):2345–57. [DOI] [PubMed] [Google Scholar]
- 12.Abida W, Campbell D, Patnaik A, et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase II TRITON2 study. Clin Cancer Res 2020; 26(11): 2487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abida W, Patnaik A, Campbell D, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol 2020; 38(32): 3763–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.AstraZeneca AB. Lynparza hard capsules—summary of product characteristics; 2019.
- 15.LYNPARZA® (olaparib) [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals; May 2020. [Google Scholar]
- 16.RUBRACA® (rucaparib) [package insert]. Boulder, CO: Clovis Oncology; May 2020. [Google Scholar]
- 17.Washington CR, Moore KN. PARP inhibitors in the treatment of ovarian cancer: a review. Curr Opin Obstet Gynecol 2021; 33(1): 1–6. [DOI] [PubMed] [Google Scholar]
- 18.ZEJULA™ (niraparib) [package insert]. Waltham, MA: TESARO; March 2017. [Google Scholar]
- 19.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45(2): 228–47. [DOI] [PubMed] [Google Scholar]
- 20.Scher HI, Morris MJ, Stadler WM, et al. Trial design and objectives for castration-resistant prostate cancer: updated recommendations from the prostate cancer clinical trials working group 3. J Clin Oncol 2016; 34(12): 1402–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.US Food and Drug Administration. Premarket Approval (PMA). 2017. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=p170019. Accessed 8/23/21.
- 22.Heller G, McCormack R, Kheoh T, et al. Circulating tumor cell number as a response measure of prolonged survival for metastatic castration-resistant prostate cancer: a comparison with prostate-specific antigen across five randomized phase III clinical trials. J Clin Oncol 2018; 36(6): 572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lorente D, Olmos D, Mateo J, et al. Decline in circulating tumor cell count and treatment outcome in advanced prostate cancer. Eur Urol 2016; 70(6): 985–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simon R Optimal two-stage designs for phase II clinical trials. Control Clin Trials 1989; 10(1): 1–10. [DOI] [PubMed] [Google Scholar]
- 25.Halabi S, Lin CY, Kelly WK, et al. Updated prognostic model for predicting overall survival in first-line chemotherapy for patients with metastatic castration-resistant prostate cancer. J Clin Oncol 2014; 32(7): 671–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones P, Wilcoxen K, Rowley M, Toniatti C. Niraparib: a poly(ADP-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. J Med Chem 2015; 58(8): 3302–14. [DOI] [PubMed] [Google Scholar]
- 27.Jensen K, Konnick EQ, Schweizer MT, et al. Association of clonal hematopoiesis in DNA repair genes with prostate cancer plasma cell-free DNA testing interference. JAMA Oncol 2021; 7(1): 107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maurice-Dror C, Fonseca N, Herberts C, Fan W, Wyatt AW, Chi KN. Circulating tumor DNA fraction (ctDNA%) to independently predict for clinical outcomes in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol 2021; 39(Suppl 15): 5049. [Google Scholar]
- 29.Chi KN, Rathkopf DE, Attard G, et al. A phase III randomized, placebo-controlled, double-blind study of niraparib plus abiraterone acetate and prednisone versus abiraterone acetate and prednisone in patients with metastatic prostate cancer (NCT03748641). J Clin Oncol 2020; 38(Suppl 6): TPS257. [Google Scholar]
- 30.Rathkopf DE, Chi KN, Olmos D, et al. AMPLITUDE: A study of niraparib in combination with abiraterone acetate plus prednisone (AAP) versus AAP for the treatment of patients with deleterious germline or somatic homologous recombination repair (HRR) gene-altered metastatic castration-sensitive prostate cancer (mCSPC). J Clin Oncol 2021; 39(Suppl 6): TPS176. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The full study protocol is available for download in the accompanying appendix.
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu. De-identified patient-level data will be made available to qualified researchers upon request, after signing of a data transfer agreement with Janssen Research & Development. Requests for data sharing, including a research proposal, can also be made to the corresponding author.