

Abstract
CCN1/Cyr61 is a dynamically expressed matricellular protein that serves regulatory functions in multiple tissues. Previous studies from our laboratory demonstrated that CCN1 regulates bone maintenance. Using an osteoblast and osteocyte conditional knockout mouse model (Ccn1OCN), we found a significant decrease in trabecular and cortical bone mass in vivo, in part through suppression of Wnt signaling since the expression of the Wnt antagonist sclerostin (SOST) is increased in osteoblasts lacking CCN1. It has been established that parathyroid hormone (PTH) signaling also suppresses SOST expression in bone. We therefore investigated the interaction between CCN1 and PTH-mediated responses in this study. We find that loss of Ccn1 in osteoblasts leads to impaired responsiveness to anabolic intermittent PTH treatment in Ccn1OCN mice in vivo and in osteoblasts from these mice in vitro. Analysis of Ccn1OCN mice demonstrated a significant decrease in parathyroid hormone receptor-1 (PTH1R) expression in osteoblasts in vivo and in vitro. We investigated the regulatory role of a non-canonical integrin-binding domain of CCN1 because several studies indicate that specific integrins are critical to mechanotransduction, a PTH-dependent response, in bone. These data suggest that CCN1 regulates the expression of PTH1R through interaction with the αvβ3 and/or αvβ5 integrin complexes. Osteoblasts that express a mutant form of CCN1 that cannot interact with αvβ3/β5 integrin demonstrate a significant decrease in mRNA and protein expression of both PTH1R and αv integrin. Overall, these data suggest that the αvβ3/β5-binding domain of CCN1 is required to endow PTH signaling with anabolic activity in bone cells.
Keywords: CCN1/CYR61, INTEGRIN, MECHANOTRANSDUCTION, OSTEOBLASTS, PTH, PTH1R
Introduction
The CCN protein family consists of six structurally related matricellular proteins that play essential roles in multiple tissues during development, immune function, and regenerative biology. Moreover, disruption in the expression or function of multiple CCN proteins is associated with diseases such as arthritis, cancer, and fibrosis. CCN proteins do not have structural roles but rather serve regulatory functions within tissues. CCN1 is best known as a proangiogenic factor; Ccn1-null mice exhibit embryonic lethality as a result of placental vascular insufficiency and compromised vessel integrity.(1) However, CCN1 regulates proliferation, differentiation, senescence, apoptosis, adhesion, and extracellular matrix (ECM) synthesis in a variety of tissues.(2) For example, previous work from our laboratory demonstrated that CCN1 is expressed in bone lining cells, cuboidal osteoblasts, trabecular osteocytes, and cortical osteocytes.(3) Disruption of CCN1 in osteoblasts results in diminished trabecular/cortical bone formation accompanied by decreased WNT signaling. This reduction in Wnt activity was correlated with elevated expression of the Wnt antagonist sclerostin (SOST), and with elevated expression of MEF2C, the direct activator of Sost transcription. However, the mechanisms by which CCN1 regulates its anabolic effects in bone remain unclear.
The canonical Wnt and parathyroid hormone (PTH) pathways are intertwined in their anabolic effects on bone. Intermittent PTH administration stimulates bone formation by activating canonical Wnt signaling through inhibition of expression of MEF2C, thus leading to reduced expression of SOST.(4–6) The finding that loss of CCN1 led to elevated MEF2C expression(3) therefore suggested a potential role for CCN1 in mediating the effects of anabolic PTH. Several other features of intermittent PTH anabolic action in bone also suggested a potential role for CCN1 as a mediator of PTH action. For example, PTH signaling is necessary for the anabolic effect of mechanical loading on bone,(7) and Ccn1 expression is directly activated by mechanical signals in vascular cell types.(8–10) Moreover, αvβ3 and β1 integrins expressed by osteocytes/osteoblasts are required for load-induced bone formation,(11) and CCN1 can interact with αvβ3, αvβ5, or α6β1 integrin complexes to activate distinct signaling pathways.(12–14) Therefore, the aim of this work was to determine if CCN1 mediates anabolic effects in bone as a transducer and/or regulator of PTH signaling.
Materials and Methods
Reagents and materials
hPTH(1–34) acetate salt was acquired from Bachem Inc (Hauptstrasse, Switzerland). Recombinant full-length murine CCN1 was purchased from Abnova, Inc. (Taipei, Taiwan). Anti-CCN1 antibody was from R&D Systems, Inc (Minneapolis, MN, USA); anti-PTH1R antibody was from R&D Systems; anti-GFP antibody was from Invitrogen, Inc. (Carlsbad, CA, USA); and anti-GAPDH antibody was from Cell Signaling, Inc. (Danvers, MA, USA). Forskolin was purchased from Qiagen (Germantown, MD, USA). Prunetin was purchased from Qiagen. Retroviral vectors for Ccn1 and Pth1r overexpression was acquired from Addgene (Cambridge, MA, USA).
Vertebrate animals
Ccn1/Cyr61-floxed mice were a gift from Dr Lester Lau.(15) To produce bone-specific knockout mice, Ccn1-floxed mice were crossed with mice expressing Cre recombinase driven by an osteocalcin promoter (OCN-Cre; Tg(BGLAP-Cre)1Clem; Jackson Laboratory, Bar Harbor, ME, USA) to generate Ccn1fx/fx;OCN-Cre (hereafter referred to as Ccn1OCN) mice. Ccn1fx/fx mice were of the C57Bl/6J background, and OCN-Cre was on a B6.FVB background. Ccn1 reporter mice were obtained from the Mutant Mouse Regional Resource Center Repository. They are a BAC transgenic line in which green fluorescent protein (GFP) is expressed under the control of the Ccn1eGFP locus.(16) All animals were treated in accordance with the National Institutes of Health guidelines for care and use of animals and approved by the UCLA Institutional Animal Care and Use Committee.
Histology and immunostaining
Femurs and tibias were dissected and fixed with 4% paraformal-dehyde (PFA) overnight at 4°C, then decalcified in 19% EDTA for 2 weeks. The tissues were dehydrated through a graded ethanol series and embedded in paraffin. Longitudinal 5-μm paraffin sections were analyzed either by histology, immunohistochemistry (IHC), or immunofluorescence (IF). All histology and IHC/IF images were analyzed on a fluorescent microscope Model BX60F (Olympus Optical Co., Tokyo, Japan) equipped with a digital camera (Model 01-RET-OEM-F-CLR-12; QImaging, Surrey, Canada). Photomicrographs were taken with a Nikon Ti-DH Microscope. Images were processed in ImageJ (http://imagej.nih.gov/ij/).
Micro-computed tomography (μCT) analysis
Bone parameters were quantified on femurs or tibias from male mutant and control mice by μCT (Skyscan1172; Bruker MicroCT, Kontich, Belgium) using CTAn (v. 1.14.4) and CTVol (v. 2.2) software. The microradiography unit was set to an energy level of 55 kVp, intensity of 181 μA, and 900 projections; specimens were scanned at a 10-μm voxel resolution. A three-dimensional reconstruction was generated with NRecon software (Bruker MicroCT) from the set of scans. Bone mineral density was determined by converting grayscale values to mineral density values using a density calibration curve from the scanned phantom and then averaging mineral content values over all regions of interest (ROI) for trabecular bone (volumetric BMD [vBMD]) or over all bone voxels for cortical bone (tissue BMD [tBMD]). The ROI of trabecular bone was defined as the area between 1 mm and 3 mm from the growth plate in the metaphyseal region of distal femurs. The ROI of cortical bone was defined as 0.75-mm segments of the femoral middle diaphysis. ROIs were drawn automatically and trabecular regions were assessed for bone mineral density (BMD), bone volume (BV), bone volume fraction (bone volume/total volume [BV/TV]), trabecular thickness (Tb. Th), and trabecular number (Tb.N). The midshaft cortical bone volume and thickness (Ct.Th), endocortical perimeter (Ec.Pn), and periosteal perimeter (Ps.Pm) values were analyzed. In the mechanical loading assay, the ROI was defined as an identical square area between 1 mm and 3 mm from the growth plate of proximal tibia. All abbreviations and nomenclature are standardized according to previously published guidelines.(17)
Mechanical strength measurement (mechanical loading)
An Instron (model # 5564, Instron, High Wycombe, UK) platform fitted with a 2-kN load cell was used to conduct cyclical loading on Ccn1OCN mice and their WT littermates, which were anesthetized by isoflurane. The four-point mechanical loading was set up as 2 mm between the upper two points and 10 mm between the bottom two points of the four-point load cell. We initiated a 9-Newton pre-load force to keep the limb (left tibia) in place, and then started the cyclical load at a 2-Hertz frequency. The moving distance of the load cell was 0.9 mm and 1 min/d for 12 days. The contralateral tibia is the unloaded control.
Bone histomorphometry
Cortical bone apposition rate and mineral surface was measured on 3-month-old mice. Mice were injected intraperitoneally with calcein (10 μg/g of body weight; Sigma-Aldrich, St. Louis, MO, USA) two times at 7-day intervals, and euthanized 2 days after the last injection. Femurs were dissected and fixed in 4% PFA overnight, then embedded in methyl methacrylate. Longitudinal sections (5 μm) of the femur were prepared and stained with toluidine blue. Cortical dynamic histomorphometry was performed where femurs were sectioned horizontally, and the bone mineralizing surface percentage (MS/BS) and mineral apposition rate (MAR) were determined by quantifying calcein-labeled surfaces, bone surface, and mean separation between calcein double labels using ImageJ. Bone formation rate (BFR/BS) is the volume of mineralized bone formed per unit time and per unit bone surface and was calculated by multiplying MS/BS and MAR. All abbreviations and nomenclature are standardized according to previously published guidelines.(18)
Total RNA extraction and qRT-PCR
For in vivo samples, femurs and tibias were dissected and soft tissue was removed. Bones were then cut to remove both 3-mm termini, and the bone marrow was flushed out with cold PBS until the cortical bone became white. The resulting cortical bone was flash-frozen with liquid nitrogen, homogenized with a grinder, and resuspended in 1 mL Trizol (Thermo Fisher Scientific, Waltham, MA, USA). Total RNA was isolated by the phenol-chloroform method and converted to cDNA. Total RNA was converted to cDNA using the RevertAid First Strand cDNA Synthesis Kit following the manufacturer’s instruction (Thermo Fisher Scientific). cDNA was amplified and quantified using Maxima SYBR Green qPCR master mix (Thermo Fisher Scientific). Analysis of relative gene expression was done using the 2-ΔΔCT method and results were normalized to the housekeeping gene GAPDH. All the primers used in the study are listed in Table 1.
Table 1.
qRT-PCR Primer List
| Gene | Primer sequences (5′−3′) |
|---|---|
| mCCN1 | CTGCAGCAAAACTCAGCCCT |
| CACAGGGTCTGCCTTCTGAC | |
| mNurr1 | TGAATGAAGAGAGCGGACAA |
| CGAGGAAATTAAAGGTGGACAG | |
| mPTH1R | TCCTTCTCTGCTGCCCAGT |
| GGTGCAGCAGGAAAATCTGT | |
| Integrin-αv | CGTCCTCCAGGATGTTTCTCC |
| TCCAAACCACTGGTGGGACT | |
| Integrin-β3 | GCTCATTGGCCTTGCTACTC |
| CCCGGTAGGTGATATTGGTG | |
| mGAPDH | CTTTGGCATTGTGGAAGGGC |
| CAGGGATGATGTTCTGGGCA |
Western blot analysis
Immunoblot analyses were performed to detect PTH1R levels in MC3T3 cells. In brief, cells were washed in PBS and then proteins were solubilized with RIPA buffer containing protease inhibitor. Extracts (30 μg) were electrophoresed in a 10% SDS-polyacrylamide gel and blotted onto a polyvinylidene difluoride membrane. Immunoblot detection was performed with anti-PTH1R and anti-GAPDH antibody using an ECL substrate (Thermo Fisher Scientific).
Elisa
ELISA assay was conducted on the collected cell culture medium using a commercially available kit (Cell Signaling) to detect the cellular cAMP levels following the kit instructions.
CRISPR and viral vectors
Lentiviral vector lentiCRISPRv2 was obtained from Addgene (Addgene, Watertown, MA, USA; #52961). We designed guide RNA (gRNA) 5′CUGCGCUAAACAACUCAACG for Ccn1 knockout in MC3T3-E1 cells and cloned these into lentiCRISPRv2 using restriction endonuclease BsmBI. To produce lentivirus, the plasmid lentiCRISPRv2-mCCN1 was co-transfected into HEK293T cells with the packaging plasmids pVSVG (Addgene #8454) and psPAX2 (Addgene #12260). After transfection, MC3T3-E1-Crispr-Cas9-mCCN1 cells were selected with puromycin (2 μg/mL, Sigma-Aldrich) for 5 days. Multiple clones were isolated. Data from a representative clone with low levels of Ccn1 expression as measured by qPCR were used. Retroviral vector pQCXIH (Clontech, Mountain View, CA; #631516) was used to generate pQCXIH-mCCN1-E61E and pQCXIH-mCCN1-E61E-D125A (E61E, agg(E) to aag(E), to mutate PAM sequence of gRNA-mCCN1) to overexpress CCN1 and CCN1D125A. Cells were selected with hygromycin (100 μg/mL, Sigma-Aldrich). Retroviral vector pLNCX2 (Clontech #631503) was used to generate pLNCX2-mPTH1R for PTH1R overexpression and cells were selected with geneticin (1 mg/mL, Sigma-Aldrich).
Flow cytometry
MC3T3-Ctrl and MC3T3-Ccn1Δ cells were seeded at 50,000 cells/well in six-well plates and cultured for 24 hours. Cells were scraped out of the wells and pelleted in a centrifuge at 300g for 10 minutes. The cells were blocked with 0.5% BSA and 2% FBS in 1x PBS for 30 minutes at room temperature. Integrin αv surface expression was measured using monoclonal antibody (ab179475) that was diluted 1/500 in blocking buffer. The blocked cells were incubated with the antibody solution for 30 minutes on ice. The cells were washed in 1x PBS and stained with a secondary antibody conjugated FITC (BD554020). The cells were washed in cold PBS and analyzed with the LSRII IMED at Janis V Giorgi flow cytometry core at UCLA. Isotype control IgG antibody (ab172730) was stained simultaneously to show staining was specific.
Statistical analysis
In this study, the data represent results from at least three independent experiments and are presented as the mean ± standard deviation (SD). Statistical significance between experimental and control groups was compared by two-tailed Student’s t test. For experiments with more than two parameters, one-way and two-way analysis of variance (ANOVA) followed by Tukey’s post hoc analysis was used. A value of p < 0.05 was considered significant.
Results
Ccn1OCN mice have attenuated bone anabolic response to intermittent PTH
Multiple studies in humans and animal models demonstrate that intermittent PTH promotes bone formation in vivo.(19–21) To investigate whether CCN1 plays a role in intermittent PTH-mediated bone formation, we subcutaneously injected 3-month-old Ccn1OCN mice and WT littermates with hPTH(1–34) at a dose of 80 μg/kg/d, 5 days per week for a total of 4 weeks and assessed the anabolic activity in bone relative to vehicle injections. We showed previously(3) that Ccn1 is expressed at the highest levels in bone lining cells and mature osteoblasts and at somewhat lower levels in trabecular and cortical osteocytes. Expression was not observed in bone marrow stroma, sinusoids, or other vascular elements in bone. We also demonstrated efficient ablation of Ccn1 in bone cell types in Ccn1OCN mice.(3) μCT analyses of transverse sections in the epiphyseal region in trabecular bone showed decreased trabecular and cortical bone mineral density in Ccn1OCN mice treated with intermittent PTH compared with WT mice (Fig. 1A–D). A significant decrease in trabecular bone mineral density (vBMD) was detected in Ccn1OCN mice relative to WT mice with or without intermittent PTH treatment (Fig. 1B); furthermore, whereas trabecular vBMD increased in WT mice treated with intermittent PTH, no increase was observed in Ccn1OCN mice (Fig. 1A, B). Because of high data variation, there is no significance between groups but a trend toward increased BV/TV in WT versus Ccn1OCN mice in response to intermittent PTH (Fig. 1B). μCT analysis of cortical bone confirmed previous findings that Ccn1OCN mice exhibit decreased BMD and thickness compared with WT littermates(3) (Fig. 1C, D). Furthermore, although intermittent PTH led to a significant increase in both tBMD and cortical bone thickness in WT mice, no response was observed in Ccn1OCN mice (Fig. 1C, D). Similarly, the ratios of endocortical perimeter and periosteal perimeter (Ec. Pm/Ps.Pm) of the femoral midshaft did not change in Ccn1OCN mice in response to intermittent PTH, which is consistent with Fig. 1D, indicating no response of cortical bone in Ccn1OCN mice (Fig. 1E). Calcein labeling assays of the endocortical bone surface indicated a decreased mineral apposition rate in Ccn1OCN mice compared with WT littermates and showed that Ccn1OCN mice failed to respond to intermittent PTH (Fig. 1F, G). These data indicate that CCN1 is required for intermittent PTH-induced anabolic activity in bone in vivo.
Fig 1.
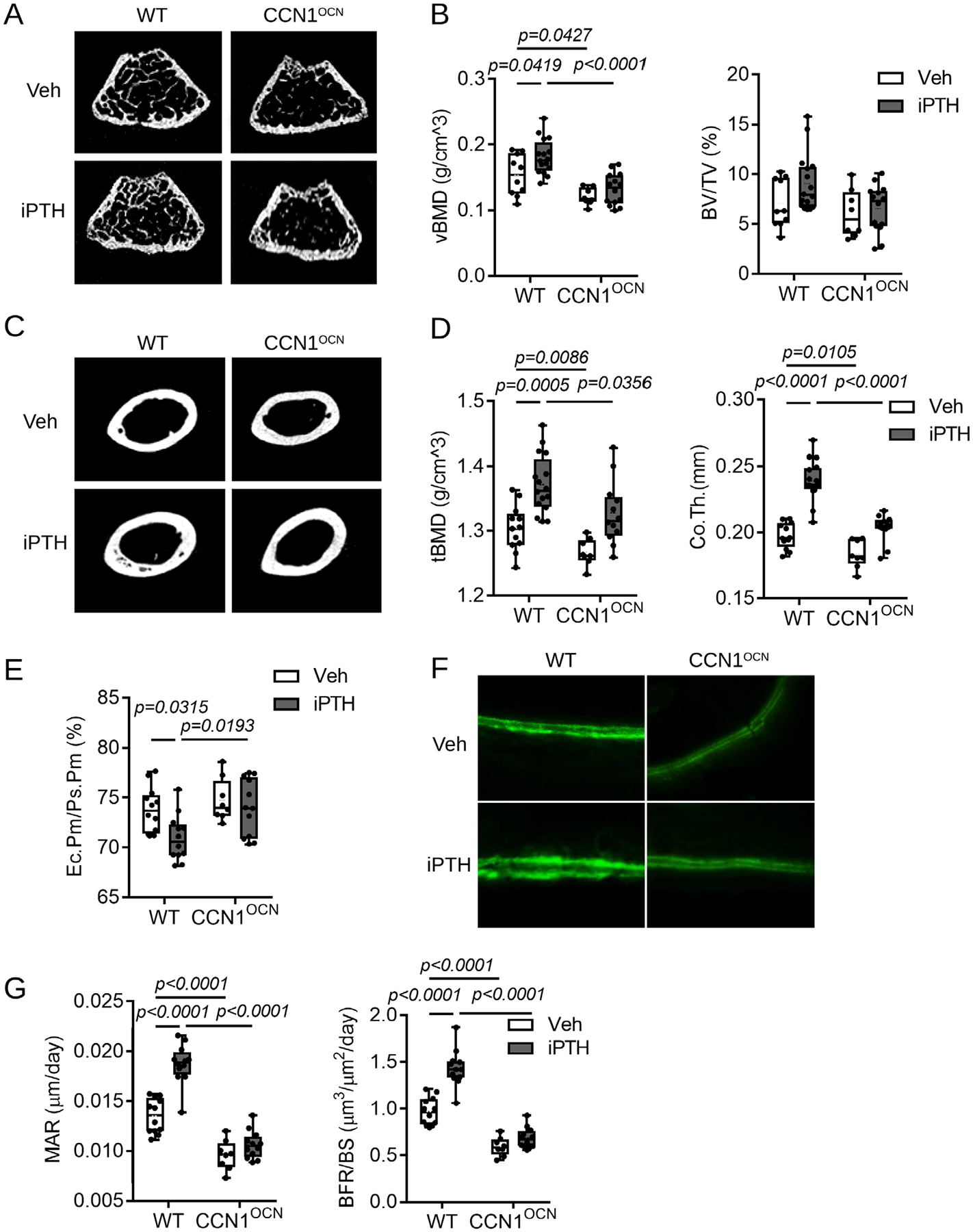
Disruption of Ccn1 in mature osteoblasts prevents responsiveness to intermittent PTH in vivo. Three-month-old male Ccn1OCN mice and WT littermates were subcutaneously injected with intermittent PTH ([1–34] fragment 80 μg/kg/d, 5 d/wk for 4 weeks), and (A) representative cross-sectional images from trabecular bone were analyzed by μCT. (B) Quantitative analysis indicated that BMD was unresponsive to intermittent PTH treatment in the trabecular bone of Ccn1OCN mice relative to WT littermates. (C) μCT analysis of cortical bone in WT and Ccn1OCN littermates. (D) A significant decrease was detected in tissue BMD (tBMD) and cortical thickness (Co.Th) in Ccn1OCN mice compared with WT littermates, and Ccn1OCN mice failed to respond to intermittent PTH relative to WT littermates. (E) The ratio of endocortical perimeter (Ec.Pm) and periosteal perimeter (Ps.Pm) of femoral midshaft demonstrated that Ccn1OCN mice failed to respond to intermittent PTH relative to WT littermates. (F, G) Calcein labeling studies revealed that the cortical mineral apposition rate is attenuated in Ccn1OCN mice treated with intermittent PTH relative to WT littermates. Calcein was ip injected 2 times with a 1-week interval. Error bars represent ±SD. Two-way ANOVA followed by the Bonferroni post test. n = 8–16/genotype.
The PTH signaling pathway in osteoblasts requires CCN1 in vitro
A number of primary response genes are activated by PTH in MC3T3-E1 osteoblastic cells.(22–24) We therefore investigated whether the effects of CCN1 on responsiveness to PTH were direct by treating MC3T3-E1 cells with varying doses of PTH. As reported previously, one of the PTH primary response genes, Nurr1, which is involved in osteoblastic differentiation,(25,26) was increased 1 hour post-PTH treatment (10−8 mol/L) by qPCR (Fig. 2A). We therefore generated Ccn1Δ MC3T3-E1 cells using CRISPR/Cas9 (Fig. 2B) and compared Nurr1 expression in response to increasing concentrations of PTH at 1 hour post-treatment in Ccn1Δ MC3T3-E1 and control MC3T3-E1 cells (Fig. 2C). These studies revealed decreased Nurr1 mRNA expression levels relative to control cells at all tested concentrations and indicated a significant role of CCN1 in PTH signaling (Fig. 2B). Parathyroid hormone receptor-1 (PTH1R) is a G protein coupled receptor (GPCR). As such, it regulates adenylate cyclase activity, which subsequently controls cyclic adenosine monophosphate (cAMP) levels in cells. cAMP levels were elevated in control MC3T3-E1 cells in response to PTH, but no change was observed in Ccn1Δ cells treated with PTH, providing further evidence that the inability of PTH to induce Nurr1 expression in Ccn1Δ cells reflects a direct impairment of PTH signaling (Fig. 2D).
Fig 2.

CCN1 regulates the expression of Nurr1 mediated by PTH in osteoblasts. (A) Time course of Nurr1 mRNA expression was analyzed in MC3T3-E1 cells at 0, 1, 2, and 4 hours of PTH treatment (n = 3). (B) Protein levels of CCN1 in MC3T3-ctrl and MC3T3-Ccn1KO cells analyzed by Western blot (n = 3). (C) MC3T3-ctrl and MC3T3-Ccn1KO cells analyzed by Western blot (n = 3). (C) MC3T3-ctrl and MC3T3-Ccn1KO cells were treated with increasing concentrations of PTH for 1 hour and Nurr1 mRNA levels were determined by qPCR. MC3T3-ctrl and MC3T3-Ccn1KO cells were treated with either vehicle or PTH (10−8 Mol/L) for 1 hour and (D) cellular cAMP concentrations were measured by ELISA (n = 6). (E) PTH (80 μg/kg) was subcutaneously injected into WT and Ccn1OCN mice for 1 hour and Nurr1 mRNA expression was measured in the calvaria bone by qPCR (n = 6). (F) MC3T3-ctrl and MC3T3-Ccn1KO cells were treated with either vehicle, PTH (10−8 Mol/L, 1 hour), Forskolin (FSK, 10−5 Mol/L, 1 hour), and Prunetin (10−6 Mol/L, 1 hour) and the mRNA levels of Nurr1 were measured by qPCR. (G) PTH and vehicle-treated MC3T3-E1 cells; Nurr1 and Ccn1 mRNA levels were measured by qPCR (n = 3). (H) PTH (80 μg/kg) was subcutaneously injected into WT and Ccn1OCN mice for 3 hours and Ccn1 mRNA levels were measured in calvarial bone by qPCR (n = 6). The in vitro results are triplicated in each of three independent experiments (n = 3). Error bars represent ±SD, compared with control or WT; two-tailed Student’s t test for B, C, and G; two-way ANOVA followed by the Bonferroni post test for results of D, E, F, and H).
To confirm that the decreased responsiveness to PTH that we observed in Ccn1OCN mice in vivo (Fig. 1) is due to an effect on osteoblast lineage cells, we measured Nurr1 mRNA levels in isolated calvarial osteoblasts obtained from Ccn1OCN or control littermate mice subcutaneously injected with PTH for 1 hour. Nurr1 was expressed at a low level in calvarial osteoblasts obtained from Ccn1OCN and control littermates injected with vehicle. In contrast, robust induction of Nurr1 was observed within 1 hour of PTH treatment in control mice, but a greatly attenuated response was found in calvarial osteoblasts from Ccn1OCN mice (Fig. 2E). This suggests that CCN1 utilizes similar mechanisms to enable responsiveness to PTH in osteoblasts in vitro and in vivo.
As shown in Fig. 2D, cAMP production in response to PTH was impaired in Ccn1Δ osteoblasts. To confirm that loss of CCN1 impairs PTH responsiveness through a cAMP/PKA pathway, we treated Ccn1Δ and control MC3T3-E1 osteoblasts with forskolin (FSK), an activator of adenylate cyclase. As expected, FSK induced robust expression of Nurr1 in both Ccn1Δ and control cells (Fig. 2F), confirming that CCN1 is required for adenylate cyclase activation in response to PTH. The GPCR GPR30 has also been shown to exert anabolic effects in bone through production of cAMP, and the polyphenol prunetin is a selective agonist for GPR30.(27,28) Prunetin did not induce expression of Nurr1 in Ccn1Δ or control MC3T3-E1 osteoblasts (Fig. 2F), indicating that CCN1 acts specifically through the PTH-PTH1R signaling axis.
Because CCN1 is crucial to convey PTH anabolic activity, a question arises as to whether PTH regulates CCN1 expression. To address this question, we assessed Nurr1 and Ccn1 mRNA expression by qPCR under PTH stimulation in vitro (MC3T3-E1 cells) and in vivo (Ccn1OCN mice). The data revealed that Ccn1 mRNA levels increased when treated with PTH for 3 hours in vitro (Fig. 2G), and a fivefold increase in Ccn1 expression was observed in vivo (Fig. 2H). This indicates that CCN1 is both a PTH target and a regulator of PTH signaling in osteoblasts.
CCN1 regulates the expression of the PTH1R in vivo and in vitro
As the activation of adenylate cyclase (AC) by PTH occurs directly via the PTH receptor (PTH1R), the finding that FSK treatment enables similar levels of induction of Nurr1 in control MC3T3-E1 and Ccn1Δ cells suggests that CCN1 may regulate PTH signaling at the level of PTH1R expression or activity. To test this possibility, we harvested femurs from Ccn1OCN mice and control littermates. We analyzed Pth1r mRNA levels in the cortical bone by qPCR (Fig. 3A) and PTH1R protein levels by immunohistochemistry (Fig. 3B) and found a significant decrease in Pth1r mRNA levels in Ccn1OCN mice and diminished protein levels in Ccn1OCN relative to control littermates. To model these in vivo observations into an in vitro system, we analyzed PTH1R expression in Ccn1Δ MC3T3-E1 osteoblasts and found a significant decrease in Pth1r mRNA expression levels compared with control cells (Fig. 3C). To determine if decreased levels of PTH1R mRNA is cell autonomous to osteoblasts, we isolated calvarial osteoblasts from Ccn1fl/fl mice and infected the cells with adenovirus that contained either a GFP or Cre. The osteoblasts that expressed Cre-recombinase demonstrated a significant decrease in Pth1r mRNA expression levels relative to osteoblasts expressing the GFP control (Fig. 3D). These data collectively show that in the absence of CCN1, the expression of PTH1R is downregulated in osteoblasts in vivo and in vitro.
Fig 3.

Loss of CCN1 decreases the mRNA and protein expression of PTH1R in osteoblasts. (A) Pth1r mRNA expression was significantly downregulated in the femurs of Ccn1OCN mice (n = 6). (B) PTH1R was detected by immunostaining in osteocytes and PTH1R+ cell numbers were quantified in the cortical bone of Ccn1OCN mice and WT littermates (n = 6). (C) MC3T3-Ccn1KO cells demonstrate a significant downregulation of Pth1r mRNA levels in vitro. (D) Adenovirus expressing Cre-recombinase significantly decreased the mRNA expression of Ccn1 and Pth1r in osteoblasts from the calvaria of Ccn1fl/fl mice compared with the adenovirus expressing GFP control. (E) Treatment of MC3T3-Ccn1KO cells with recombinant CCN1 (rhCCN1) (100 ng/mL) for 24 hours restored the relative Pth1r mRNA expression levels and (F) protein levels of PTH1R to the levels that are found in MC3T3-ctrl cells when analyzed by qPCR and Western blot, respectively. (G) Pth1r was overexpressed in MC3T3-ctrl and MC3T3-Ccn1KO cells with retroviral vector. The cells were treated with either vehicle or PTH (10−8 Mol/L) for 1 hour, after which (H) Nurr1 mRNA expression levels were measured via qPCR and (I) cellular cAMP concentrations were quantified by ELISA. The in vitro results were performed in triplicate in each of three independent experiments. (J) Microscopic images of mechanically loaded femurs from Ccn1eGFP reporter mice demonstrated increased Ccn1eGFP expression in the loaded cortical bone relative to the contralateral unloaded bones. CCN1 protein expression levels were examined by immunohistochemistry in the femurs of WT mice. (K) The mRNA levels of Ccn1, Pth1r, Nurr1, and Integrin αvβ3 in cortical bone of WT mice were examined by qPCR (n = 6). Error bars represent ±SD. The p values were shown and compared with control or WT (two-tailed Student’s t test for A, C, D, J, and K; one-way ANOVA followed by the Tukey post hoc test for E, two-way ANOVA followed by the Tukey test for results of G, H, and I).
We examined the effects of recombinant CCN1 (rhCCN1) and overexpression of PTH1R in control and Ccn1Δ MC3T3-E1 osteoblasts to investigate the relationship between CCN1 and levels of PTH1R expression. Ccn1Δ cells treated with rhCCN1 for 24 hours expressed significantly higher levels of Pth1r mRNA levels compared with Ccn1Δ cells alone, and treatment with rhCCN1 recovered Pth1r mRNA levels to those found in control MC3T3-E1 cells (Fig. 3E). In accordance, Pth1r mRNA levels were associated with PTH1R protein levels (Fig. 3F). Given that Pth1r mRNA and protein levels are diminished in Ccn1Δ cells, we investigated whether PTH-mediated signaling could be rescued in Ccn1Δ cells that overexpress PTH1R. We therefore infected Ccn1Δ cells with either an empty retroviral vector (Vector) or one encoding PTH1R driven by a CMV promoter (Fig. 3G). Control and Ccn1Δ cells infected with either Vector or PTH1R retrovirus were then treated with vehicle or PTH for 1 hour. Nurr1 mRNA and cAMP levels were measured by qPCR and ELISA, respectively. In control MC3T3-E1 cells, overexpression of PTH1R led to a robust increase in Nurr1 mRNA expression and cAMP levels relative to Vector control cells. In Ccn1Δ cells, PTH treatment did not induce Nurr1 mRNA expression or cAMP levels, but a significant increase in both measurements was observed in Ccn1Δ cells overexpressing PTH1R and treated with PTH (Fig. 3H, I). Collectively these data indicate that CCN1 regulates PTH1R expression and function in osteoblasts. A caveat of the experiments using rhCCN1 is that as a matricellular protein, CCN1 may zhave distinct effects in soluble versus matrix-bound forms. Although soluble rhCCN1 was able to rescue PTH1R expression in Ccn1Δ cells (Fig. 3H) and our in vitro results are consistent with the in vivo findings in Fig. 2, further studies would be required to discriminate matrix-bound versus soluble activities of CCN1.
Mechanical forces play a critical role in skeletal development and tissue homeostasis. αvβ3 integrin complexes located on the surface of osteoblast lineage cells are a key component in sensing mechanical loads and converting them into biochemical signals in the cell.(11) Since previous studies reported that PTH signaling is essential for the anabolic response of bone to mechanical load,(7,29) we examined whether PTH signaling and CCN1 levels are increased in response to loading. First, we localized CCN1 during cyclical loading of the tibia of 3-month-old Ccn1eGFP reporter mice. Increased expression of Ccn1eGFP and CCN1-positive cell number were observed by immunohistochemistry within cortical bone relative to unloaded controls (Fig. 3J). Next, the expression of Ccn1 and Pth1r is increased with mechanical loading (Fig. 3K). The elevated expression of Pht1r correlated with elevated expression of the downstream target gene Nurr1 (Fig. 3K). Mechanosensory responses in bone require integrin αvβ3.(29,30) Therefore, to strengthen these findings, we examined expression of integrin αV and β3 (which act as both mechanotransduction sensors and CCN1 receptors). Expression levels were found to be increased (Fig. 3K). Although the mechanism remains rather unclear, the findings suggested CCN1 is important to both the ability to sense mechanical loading in bone and the ability to enable responsiveness to PTH.
The integrin-binding domain of CCN1 modulates PTH1R expression and signaling pathway
CCN proteins trigger cellular functions through binding to distinct integrin complexes on the cell surface. Several of these binding domains have been identified in CCN1, including a site that is required for interaction with αvβ3 and αvβ5 integrins.(13) The αvβ3-binding motif in CCN1 is known to be essential to osteoblastogenesis and angiogenesis. Furthermore, αvβ3 integrins promote osteoblast proliferation and differentiation.(30–34) Therefore, we examined the role of αvβ3 integrin complexes in CCN1-mediated activation of PTH1R signal transduction pathways in bone.
To determine whether CCN1 regulates the PTH-PTH1R-Nurr1 mRNA signaling pathway via an integrin-dependent mechanism, MC3T3-E1 control and Ccn1Δ cells were seeded and cultured for 24 hours, after which αv and β3 mRNA levels were measured by qPCR. αv levels were significantly reduced in Ccn1Δ cells relative to control MC3T3-E1 cells, but β3 levels remained unchanged (Fig. 4A). To confirm that this change in αv mRNA expression corresponded with the αv protein levels on the cell surface, we performed flow cytometry. The average frequency of αv on the cell surface of Ccn1Δ cells was significantly decreased compared with control cells (Fig. 4B). The decreased surface expression of αv is expected to lead to decreased signaling through αvβ3. However, because integrin signaling is also regulated by cell localization, matrix engagement, conformational status, and binding partners, elucidation of the precise impact of CCN1-mediated regulation of αv expression on integrin-mediated signaling requires further investigation.
Fig 4.

The RGD motif of CCN1 that mediates its interaction with the αvβ3 or αvβ5 integrin complex is required for CCN1-mediated Pth1r mRNA expression. The relative mRNA expression levels of (A) αV and β3 in MC3T3-ctrl and MC3T3-CCN1KO cells were measured by qPCR. (B) The αV integrin protein levels were measured in MC3T3-ctrl and MC3T3-CCN1KO cells by flow cytometry (n = 6). MC3T3-ctrl and MC3T3-CCN1KO cells were conditioned with or without the presence of Integrin inhibitor (Cilengitide) for 24 hours and the relative mRNA expression of (C) Pth1r was measured by qPCR. MC3T3-ctrl and MC3T3-CCN1KO cells were treated with either vehicle or PTH (10−8 mol/L), conditioned with or without the presence of Cilengitide, and the relative mRNA expression levels of (D) Nurr1 were measured by qPCR. (E) Site-directed mutagenesis of adenosine (A) into a cytosine (C) in the CCN1 retroviral plasmid was performed, and the resulting Retro-CCN1D125A was expressed in MC3T3-CCN1KO cells (generated by CRISPR/Cas) to produce MC3T3-CCN1D125A cells. (F) Protein levels of CCN1 relative to GAPDH in MC3T3-CCN1KO, MC3T3-CCN1D125A, and control cell lines were analyzed by Western blot. (G) The MC3T3-CCN1KO, MC3T3-Retro-CCN1, MC3T3-CCN1D125A, MC3T3-Retro-PTH1R, and control cell lines were treated with either vehicle or PTH for 1 hour and mRNA expression of Nurr1 and (H) Pth1r was analyzed by qPCR. The results are triplicated in each of three independent experiments (n = 3). Cilengitide, 10−6 mol/L for 24 hours and PTH 10−8 mol/L for 1 hour. Error bars represent ±SD. p values were shown and compared with control (two-tailed Student’s t test for A, B; one-way ANOVA followed by the Tukey post hoc test for F; two-way ANOVA followed by the Tukey test for C, D, G, and H).
To ascertain the impact that αvβ3 integrin complexes have on the ability of CCN1 to promote PTH signaling, we assessed PTH1R and Nurr1 mRNA expression levels in MC3T3-E1 control and Ccn1Δ cells in the presence of the αvβ3/β5 integrin inhibitor cilengitide. Control cells treated with cilengitide demonstrated significantly decreased levels of Pth1r mRNA expression relative to cells treated with vehicle (Fig. 4C). As discussed above, Pth1r expression was decreased in Ccn1Δ cells compared with control cells. Treatment with cilengitide had less effect on Pth1r expression in Ccn1Δ cells (Fig. 4C). To confirm that the downregulation of Pth1r mRNA expression was associated with decreased response to PTH, we measured the Nurr1 mRNA expression levels of MC3T3-ctrl and MC3T3-Ccn1Δ cells in the presence of cilengitide by qPCR. MC3T3-ctrl cells treated with cilengitide demonstrated significantly decreased levels of Nurr1 mRNA expression levels compared with treatment of vehicle, and no change was observed in MC3T3-Ccn1Δ treated with or without cilengitide (Fig. 4D). Taken together, these findings suggest that integrins αvβ3 and/or αvβ5 are required for expression of PTH1R.
To test whether the ability of CCN1 to engage integrins αvβ3 and/or αvβ5 is required for CCN1 to regulate PTH1R expression and activity, we compared the ability of WT CCN1 and a mutated form of CCN1 (CCN1-D125A) that is unable to bind to these integrins.(13) Site-directed mutagenesis was used to engineer a retroviral plasmid that encoded CCN1-D125A (Fig. 4E). We measured the response to PTH in Ccn1Δ MC3T3-E1 cells infected with retroviral constructs overexpressing either WT CCN1 or CCN1-D125A. Western blot analysis revealed that similar levels of expression of WT and CCN1-D125A were achieved (Fig. 4F). PTH treatment of Ccn1Δ MC3T3-E1 cells overexpressing WT CCN1 led to a significant induction in Nurr1 mRNA expression levels relative to Ccn1Δ cells transduced with empty vector (Fig. 4G). These data show that the overexpression of CCN1 rescues the ablated response to PTH in Ccn1Δ cells. However, overexpression of CCN1-D125A in Ccn1Δ cells did not rescue responsiveness of PTH, indicating that the ability of CCN1 to bind to αvβ3 and/or αvβ5 integrins is essential for CCN1-mediated responsiveness to PTH (Fig. 4G). Overexpression of PTH1R in Ccn1Δ cells infected with empty vector or Ccn1Δ cells overexpressing CCN1-D125A rescued responsiveness to PTH, indicating that the impairment in PTH responsiveness in CCN1-D125A-expressing cells occurs at the level of PTH receptor expression (Fig. 4H). We therefore monitored Pth1r mRNA levels in response to CCN1 or CCN1-D125A expression. No significant changes in Pth1r levels were found in WT MC3T3-E1 cells or in Ccn1Δ cells exposed to PTH (Fig. 4H). Moreover, although overexpression of WT CCN1 in Ccn1Δ cells rescued Pthr1 expression to levels found in control MC3T3-E1 cells, overexpression of CCN1-D125A in Ccn1Δ cells did not rescue Pth1r expression (Fig. 4H). These data collectively suggest that the interaction of CCN1 with αvβ3 and/or αvβ5 complexes regulates expression of PTH1R.
Discussion
Previous studies from our laboratory demonstrated that disruption of CCN1 decreases anabolic activity in bone by affecting osteoblast commitment and activity and by impairing angiogenesis. These effects were related to decreased activity of the Wnt pathway, likely due to increased expression of the Wnt antagonist SOST.(3) Given the central role of PTH in suppressing SOST expression and our previously identified role for CCN1 as a suppressor of SOST expression, we examined the impact of CCN1 on PTH signaling in the current study. There are divergent effects of PTH on bone metabolism by PTH, depending on whether the hormone is increased chronically (catabolic effect) or intermittently as achieved by pharmacological daily injections (anabolic effect).(35) In pathological conditions, such as severe primary hyperparathyroidism, or secondary hyperparathyroidism because of vitamin D or calcium deficiency, chronic increase of PTH is characterized by enhanced bone loss due to unbalanced bone remodeling associated with increased generation and activity of osteoclasts.(36) In contrast, intermittent PTH daily injection can rapidly increase proliferation of osteoblast precursors, inhibit osteoblast apoptosis, and reactivate bone lining cells to become matrix synthesizing osteoblasts.(37) Although the effects of PTH injections on bone have been extensively studied, the underlying mechanisms regulating PTH receptor signaling remain unclear. Many of the anabolic effects of PTH are due to its ability to directly inhibit Sost transcription.(6,38,39) Furthermore, the finding that mice lacking Pth1r in DMP1-expressing osteoblasts and osteocytes revealed a deficient response to mechanical loading or intermittent PTH injection,(7) indicating that PTH1R in osteocyte is required for bone anabolism induced by mechanical signals in addition to direct PTH injection.
Here, we show that osteoblast lineage cells in Ccn1OCN mice exhibit an attenuated response to intermittent PTH in vivo and in vitro and to mechanical load in vivo. Specifically, we show that disruption of CCN1 decreases the expression of PTH1R and thereby attenuates PTH-mediated signaling in osteoblasts, which corresponds with diminished bone properties in vivo. Furthermore, the results reported here show that mechanical loading induces the expression of CCN1 and integrin αvβ3 in bone. Thus, CCN1 is a mechanosensitive gene that is required for the ability of PTH to transduce the effects of mechanical load in bone. CCN1 is known to be directly activated by load in various tissues.(8–10) It will be of interest in future studies to identify the mechanisms by which load induces CCN1 expression in bone.
Our results suggest that a key function for CCN1 is to induce PTH1R expression. Previous studies reported that BMP2 and ER stress promotes the expression of Pth1r in osteoblasts and renders these cells responsive to PTH;(40) the skeletal resistance to PTH in the setting of chronic kidney disease is related to a decrease in Pth1r mRNA levels.(41) Moreover, mice with deletion of PTH1R in osteoblasts have disrupted trabecular bone formation,(42) and osteocytic PTH1R signaling is required for PTH/mechanical loading-induced bone anabolism.(7) These findings show that regulation of PTH1R levels has important physiological and pathophysiological roles in bone. Although the precise mechanisms by which CCN1 induces Pth1r expression in bone are unknown, the ability of CCN1 to mediate this effect appears to involve integrin αvβ3 and/or αvβ5 complexes. This conclusion is supported by multiple lines of evidence that a direct role for αvβ3 in mechanotransduction, a PTH-dependent process.(31,43–45)
Disruption of CCN1 in osteoblasts and osteocytes revealed decreased αv integrin mRNA and protein expression. Furthermore, the finding that the D125A variant of CCN1 that is unable to bind to αv integrin leads to reduced expression of Pth1r and failure of responsiveness to PTH raises the likelihood that CCN1 exerts its effects through direct binding of αv integrin. The mechanisms by which CCN1 might mediate αvβ3 action and by which αvβ3 regulates PTH1R levels in PTH/mechanotransduction in bone are not clear. However, Berry and colleagues(43) reported that JunB regulates Pth1r mRNA levels in cementoblasts, which have osteoblast characteristics. JunB1 is an AP-1 transcription factor and a classical downstream effector of integrins. Cowles and colleagues(44) reported that integrin-mediated signaling regulates expression of AP-1 transcription factors in osteobasts. We speculate that CCN1 may promote integrin signaling via increased expression of αvβ3 and by acting as a direct αvβ3 ligand. Elevated integrin signaling promoted by CCN1 may subsequently lead to elevated Pth1r expression at least in part via increased AP-1 activity. Further research will be required to test the validity of this speculation.
Given that members of the CCN family have structural homology, it is possible that other members of the CCN family of proteins may induce the expression of Pth1r in bone in an integrin-dependent manner. Previous studies show that CCN4 is highly expressed in bone and also plays an important role in bone maintenance.(45) Ccn4−/− mice exhibit decreased bone thickness, mineral density, and biomechanical strength. Therefore, CCN1 and CCN4 may have additive effects on bone development and experiments that address the link between both the matricellular proteins and PTH-mediated bone formation are warranted.
Furthermore, our finding that CCN1 is required for sensitivity to PTH is reminiscent of previous studies showing that the matricellular protein periostin also acts in an integrin αvβ3-dependent manner to transduce PTH and mechanical signals in bone.(46,47) Periostin is expressed primarily in the periosteum, but PTH exerts only a modest anabolic activity in the periosteum relative to the endocortical bone.(48) CCN1 is expressed in endosteal cells (Fig. 3J). While the patterns of CCN1 and periostin expression in bone are distinct, both proteins are expressed in osteocytes. Although it is unknown whether periostin mediates any of its effects through regulation of PTH1R expression, our studies indicate that multiple matricellular proteins that interact with αvβ3 may play a central role in the ability of bone to respond to PTH and to sense mechanical load. Our study was performed using OCN-Cre, which targets mature osteoblasts in addition to osteocytes. Further studies that investigate the role of CCN1 specifically in osteocytes may elucidate whether the CCN1, which is induced by mechanical load and which is required for responsiveness to PTH, is also required for the ability of PTH to mediate the anabolic response to mechanical load in bone.
Acknowledgments
This work was supported by NIH/NIAMS grants R01 AR052686 and R21 AR071734 to KML, and T32AR059033 to EWK. Salary support for SM is provided by a Senior Research Career Scientist Award from the US Department of Veterans Affairs.
Footnotes
Disclosures
International Fibrodysplasia Ossificans Progressiva (IFOP) grant review panel (KML). All authors state that they have no conflicts of interest.
References
- 1.Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol. 2002;22(24):8709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lau LF. CCN1/CYR61: the very model of a modern matricellular protein. Cell Mol Life Sci. 2011;68(19):3149–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao G, Huang BL, Rigueur D, et al. CYR61/CCN1 regulates Sclerostin levels and bone maintenance. J Bone Miner Res. 2018;33(6):1076–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo J, Liu M, Yang D, et al. Suppression of Wnt signaling by Dkk1 attenuates PTH-mediated stromal cell response and new bone formation. Cell Metab. 2010;11(2):161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leupin O, Kramer I, Collette NM, et al. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res. 2007;22(12):1957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37(2):148–58. [DOI] [PubMed] [Google Scholar]
- 7.Delgado-Calle J, Tu X, Pacheco-Costa R, et al. Control of bone anabolism in response to mechanical loading and PTH by distinct mechanisms downstream of the PTH receptor. J Bone Miner Res. 2017;32 (3):522–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanna M, Liu H, Amir J, et al. Mechanical regulation of the proangiogenic factor CCN1/CYR61 gene requires the combined activities of MRTF-A and CREB-binding protein histone acetyltransferase. J Biol Chem. 2009;284(34):23125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaqour B, Whitbeck C, Han JS, et al. Cyr61 and CTGF are molecular markers of bladder wall remodeling after outlet obstruction. Am J Physiol Endocrinol Metab. 2002;283(4):E765–74. [DOI] [PubMed] [Google Scholar]
- 10.Tamura I, Rosenbloom J, Macarak E, Chaqour B. Regulation of Cyr61 gene expression by mechanical stretch through multiple signaling pathways. Am J Physiol Cell Physiol. 2001;281(5):C1524–32. [DOI] [PubMed] [Google Scholar]
- 11.Geoghegan IP, Hoey DA, McNamara LM. Integrins in osteocyte biology and mechanotransduction. Curr Osteoporos Rep. 2019;17(4):195–206. [DOI] [PubMed] [Google Scholar]
- 12.Chen N, Chen CC, Lau LF. Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin α6β1 and cell surface heparan sulfate proteoglycans. J Biol Chem. 2000;275(32):24953–61. [DOI] [PubMed] [Google Scholar]
- 13.Chen N, Leu SJ, Todorovic V, Lam SC, Lau LF. Identification of a novel integrin αvβ3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J Biol Chem. 2004;279(42): 44166–76. [DOI] [PubMed] [Google Scholar]
- 14.Leu SJ, Chen N, Chen CC, et al. Targeted mutagenesis of the angiogenic protein CCN1 (CYR61). Selective inactivation of integrin α6β1-heparan sulfate proteoglycan coreceptor-mediated cellular functions. J Biol Chem. 2004;279(42):44177–87. [DOI] [PubMed] [Google Scholar]
- 15.Kim KH, Chen CC, Monzon RI, Lau LF. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol. 2013;33(10):2078–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heintz N Gene expression nervous system atlas (GENSAT). Nat Neurosci. 2004;7(5):483. [DOI] [PubMed] [Google Scholar]
- 17.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25(7):1468–86. [DOI] [PubMed] [Google Scholar]
- 18.Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28(1):2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19): 1434–41. [DOI] [PubMed] [Google Scholar]
- 20.Lindsay R, Zhou H, Cosman F, Nieves J, Dempster DW, Hodsman AB. Effects of a one-month treatment with PTH(1–34) on bone formation on cancellous, endocortical, and periosteal surfaces of the human ilium. J Bone Miner Res. 2007;22(4):495–502. [DOI] [PubMed] [Google Scholar]
- 21.Iida-Klein A, Lu SS, Cosman F, Lindsay R, Dempster DW. Effects of cyclic vs. daily treatment with human parathyroid hormone (1–34) on murine bone structure and cellular activity. Bone. 2007;40(2):391–8. [DOI] [PubMed] [Google Scholar]
- 22.Choi H, Magyar CE, Nervina JM, Tetradis S. Different duration of parathyroid hormone exposure distinctively regulates primary response genes Nurr1 and RANKL in osteoblasts. PLoS One. 2018;13(12): e0208514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qin L, Qiu P, Wang L, et al. Gene expression profiles and transcription factors involved in parathyroid hormone signaling in osteoblasts revealed by microarray and bioinformatics. J Biol Chem. 2003;278 (22):19723–31. [DOI] [PubMed] [Google Scholar]
- 24.Kulkarni NH, Halladay DL, Miles RR, et al. Effects of parathyroid hormone on Wnt signaling pathway in bone. J Cell Biochem. 2005;95 (6):1178–90. [DOI] [PubMed] [Google Scholar]
- 25.Tetradis S, Bezouglaia O, Tsingotjidou A. Parathyroid hormone induces expression of the nuclear orphan receptor Nurr1 in bone cells. Endocrinology. 2001;142(2):663–70. [DOI] [PubMed] [Google Scholar]
- 26.Lee MK, Choi H, Gil M, Nikodem VM. Regulation of osteoblast differentiation by Nurr1 in MC3T3-E1 cell line and mouse calvarial osteoblasts. J Cell Biochem. 2006;99(3):986–94. [DOI] [PubMed] [Google Scholar]
- 27.Khan K, Pal S, Yadav M, et al. Prunetin signals via G-protein-coupled receptor, GPR30(GPER1): stimulation of adenylyl cyclase and cAMP-mediated activation of MAPK signaling induces Runx2 expression in osteoblasts to promote bone regeneration. J Nutr Biochem. 2015;26(12):1491–501. [DOI] [PubMed] [Google Scholar]
- 28.Torre E Molecular signaling mechanisms behind polyphenol-induced bone anabolism. Phytochem Rev. 2017;16(6):1183–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wozniak M, Fausto A, Carron CP, Meyer DM, Hruska KA. Mechanically strained cells of the osteoblast lineage organize their extracellular matrix through unique sites of αvβ3-integrin expression. J Bone Miner Res. 2000;15(9):1731–45. [DOI] [PubMed] [Google Scholar]
- 30.Thi MM, Suadicani SO, Schaffler MB, Weinbaum S, Spray DC. Mechanosensory responses of osteocytes to physiological forces occur along processes and not cell body and require αvβ3 integrin. Proc Natl Acad Sci U S A. 2013;110(52):21012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson SK, Stewart JP, Bam R, et al. CYR61/CCN1 overexpression in the myeloma microenvironment is associated with superior survival and reduced bone disease. Blood. 2014;124(13):2051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su JL, Chiou J, Tang CH, et al. CYR61 regulates BMP-2-dependent osteoblast differentiation through the αvβ3 integrin/integrin-linked kinase/ERK pathway. J Biol Chem. 2010;285(41):31325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee DY, Li YS, Chang SF, et al. Oscillatory flow-induced proliferation of osteoblast-like cells is mediated by αvβ3 and beta1 integrins through synergistic interactions of focal adhesion kinase and Shc with phosphatidylinositol 3-kinase and the Akt/mTOR/p70S6K pathway. J Biol Chem. 2010;285(1):30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haugh MG, Vaughan TJ, McNamara LM. The role of integrin αvβ3 in osteocyte mechanotransduction. J Mech Behav Biomed Mater. 2015;42:67–75. [DOI] [PubMed] [Google Scholar]
- 35.Silva BC, Costa AG, Cusano NE, Kousteni S, Bilezikian JP. Catabolic and anabolic actions of parathyroid hormone on the skeleton. J Endocrinol Invest. 2011;34(10):801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma YL, Cain RL, Halladay DL, et al. Catabolic effects of continuous human PTH (1–38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology. 2001;142(9):4047–54. [DOI] [PubMed] [Google Scholar]
- 37.Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40(6):1434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bellido T, Ali AA, Gubrij I, et al. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146(11):4577–83. [DOI] [PubMed] [Google Scholar]
- 39.O’Brien CA, Plotkin LI, Galli C, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One. 2008; 3(8):e2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tohmonda T, Yoda M, Mizuochi H, et al. The IRE1α-XBP1 pathway positively regulates parathyroid hormone (PTH)/PTH-related peptide receptor expression and is involved in pth-induced osteoclastogenesis. J Biol Chem. 2013;288(3):1691–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Disthabanchong S, Hassan H, McConkey CL, Martin KJ, Gonzalez EA. Regulation of PTH1 receptor expression by uremic ultrafiltrate in UMR 106-01 osteoblast-like cells. Kidney Int. 2004; 65(3):897–903. [DOI] [PubMed] [Google Scholar]
- 42.Qiu T, Xian L, Crane J, et al. PTH receptor signaling in osteoblasts regulates endochondral vascularization in maintenance of postnatal growth plate. J Bone Miner Res. 2015;30(2):309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berry JE, Pettway GJ, Cordell KG, et al. JunB as a potential mediator of PTHrP actions: new gene targets Ephrin B1 and VCAM-1. Oral Dis. 2008;14(8):713–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cowles EA, Brailey LL, Gronowicz GA. Integrin-mediated signaling regulates AP-1 transcription factors and proliferation in osteoblasts. J Biomed Mater Res. 2000;52(4):725–37. [DOI] [PubMed] [Google Scholar]
- 45.Maeda A, Ono M, Holmbeck K, et al. WNT1-induced secreted protein-1 (WISP1), a novel regulator of bone turnover and WNT signaling. J Biol Chem. 2015;290(22):14004–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonnet N, Standley KN, Bianchi EN, et al. The matricellular protein periostin is required for sost inhibition and the anabolic response to mechanical loading and physical activity. J Biol Chem. 2009;284 (51):35939–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonnet N, Conway SJ, Ferrari SL. Regulation of beta catenin signaling and parathyroid hormone anabolic effects in bone by the matricellular protein periostin. Proc Natl Acad Sci U S A. 2012;109(37): 15048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lindsay R, Cosman F, Zhou H, et al. A novel tetracycline labeling schedule for longitudinal evaluation of the short-term effects of anabolic therapy with a single iliac crest bone biopsy: early actions of teriparatide. J Bone Miner Res. 2006;21(3):366–73. [DOI] [PubMed] [Google Scholar]