

SUMMARY
mRNA function is influenced by modifications that modulate canonical nucleobase behavior. We show that a single modification mediates distinct impacts on mRNA translation in a position-dependent manner. While cytidine acetylation (ac4C) within protein-coding sequences stimulates translation, ac4C within 5’UTRs impacts protein synthesis at the level of initiation. 5’UTR acetylation promotes initiation at upstream sequences, competitively inhibiting annotated start codons. Acetylation further directly impedes initiation at optimal AUG contexts: ac4C within AUG-flanking Kozak sequences reduced initiation in base-resolved transcriptome-wide HeLa results, and in vitro utilizing substrates with site-specific ac4C incorporation. Cryo-EM of mammalian 80S initiation complexes revealed ac4C in the −1-position adjacent to an AUG start codon disrupts an interaction between C and hypermodified t6A at nucleotide 37 of the initiator tRNA. These findings demonstrate the impact of RNA modifications on nucleobase function at a molecular level and introduce mRNA acetylation as a factor regulating translation in a location-specific manner.
Keywords: epitranscriptome, mRNA, translation, NAT10, acetylcytidine
Graphical Abstract

In Brief (eTOC blurb)
mRNA modifications alter canonical nucleobase behavior. Arango et al. show that a single modification, ac4C, influences mRNA translation in a position dependent manner. ac4C within coding sequences promotes elongation, while 5’UTR ac4C inhibits initiation through the generation of repressive structures and through direct modulation of tRNAiMet interactions.
INTRODUCTION
Post-transcriptional modification of the nucleic acids within protein-coding messenger RNAs (mRNAs) is emerging as a major contributor to the regulation of gene expression. Known as the ‘epitranscriptome,’ mRNA modifications influence diverse aspects of mRNA metabolism, including pre-mRNA splicing, mRNA structure, transport, stability and translation (Franco and Koutmou, 2022; Wiener and Schwartz, 2021). In addition, select modifications are reversible, allowing for temporal and stimulus responsive modulation of mRNA function (Wiener and Schwartz, 2021). We recently identified N4-acetylcytidine (ac4C) as an mRNA modification that is catalyzed by the enzyme N-acetyltransferase 10 (NAT10) (Arango et al., 2018). Thousands of ac4C sites were detected within HeLa mRNA, the majority of which occurred within coding sequences (CDS) (Arango et al., 2018). Mechanistically, the N4-acetyl group of ac4C generates an intramolecular hydrogen bond that enforces a cytidine conformation that, in turn, stabilizes Watson-Crick base-pairing with guanosine (Kumbhar et al., 2013; Parthasarathy et al., 1978; Stern and Schulman, 1978). CDS ac4C thus enhances translation elongation through improving interactions with cognate tRNAs, thereby inhibiting co-translational decay pathways that would otherwise occur at non-optimal codon contexts (Arango et al., 2018).
While the association between CDS ac4C and translation elongation was robust, the sum of our previous data did not explain the strong positional bias observed in ac4C mapping. In brief, ac4C accumulated towards the 5’ ends of transcripts in proximity of translation start sites. ac4C was additionally enriched in the 5’ untranslated regions (5’UTRs) of substrates wherein a relationship to overall mRNA expression was not observed (Arango et al., 2018). Though designated as untranslated, elements within 5’UTRs interact with the translational machinery in multiple manners to achieve critical roles in the regulation of translation (Hinnebusch et al., 2016; Leppek et al., 2018). Initiation factors assemble at the 5’ end of mRNAs through interaction with the 7-methylguanosine (m7G) cap and are joined by the 40S ribosomal subunit in association with the initiator methionine tRNA (tRNAiMet) (Hinnebusch et al., 2016; Leppek et al., 2018). The resulting pre-initiation complex (PIC) scans in the 3’ direction until a start codon, typically an AUG, is encountered through association with the tRNAiMet anticodon loop (Hinnebusch et al., 2016). Start codon recognition allows the 60S ribosomal subunit to join and mature 80S ribosomes capable of entering the elongation phase of translation are formed (Hinnebusch et al., 2016). Beyond providing a platform for ribosome assembly, 5’UTR elements exert both positive and negative influences on start codon selection (Hinnebusch et al., 2016; Leppek et al., 2018). If the nucleotide context surrounding the first AUG is unfavorable and lacks a consensus ‘Kozak’ sequence, the ribosome may fail to assemble (Kozak, 1986). Conversely, the ribosome may assemble at imperfect ‘near-cognate’ codons that differ from the canonical AUG at a single nucleotide. In addition, 5’UTR structures influence initiation in nuanced ways, such as obstructing ribosome access to downstream start codons, and/or increasing PIC residency at non-optimal start codons associated with upstream open-reading frames (uORFs) (Manjunath et al., 2019). uORF translation, in turn, competitively limits downstream protein synthesis (Kozak, 1990).
Here we examine ac4C function within 5’UTRs and determine a distinctive role in the regulation of translation initiation. mRNAs with 5’UTR ac4C showed increased initiation upstream of expected locations and a concomitant decrease at annotated start codon in vitro and in vivo. The negative influence of ac4C on otherwise optimal initiation contexts was mechanistically attributed to altered base-pairing, both through the generation of repressive structures and through direct modulation of favorable interactions associated with cytidine in AUG-flanking Kozak sequences. Through these combined functions, 5’UTR acetylation is demonstrated to potently regulate the proteome at the level of initiation. Altogether, our results identify decisive location specific roles for ac4C in mRNA translation and provide unexpected insight into the molecular underpinnings defining Kozak sequence optimality.
RESULTS
5’UTR and CDS ac4C mediate opposing influences on mRNA translation
We previously established acRIP-seq as a method for identification of acetylated regions through antibody-based enrichment of fragmented RNA. acRIP-seq in wildtype and CRISPR-Cas9 ablated NAT10−/− cells revealed more than 4000 discretely acetylated regions in HeLa cells (Arango et al., 2018). The majority of ac4C sites were found within coding sequences, wherein a role in promoting mRNA stability through enhanced translation was determined. ac4C further displayed a pronounced 5’ localization bias, though the relevance was unclear. While ~22% of ac4C sites occurred with 5’UTRs, loss of ac4C from these locations had no overall influence on the expression of substrate mRNAs (Arango et al., 2018). Likewise, whereas ac4C(+) regions within CDS were generally characterized by reduced codon optimality as compared to analogous ac4C(−) regions, this trend was not observed for translation initiation site (TIS) proximal ac4C (Figure 1A). acRIP-seq enriches from ~200 base pair (bp) mRNA fragments and is thereby imprecise for discrete mapping to generally short 5’UTRs, raising the possibility of a distinctive role for 5’ enriched ac4C unrelated to translation elongation.
Figure 1: 5’UTR and CDS ac4C exert distinct influences on mRNA translation.

(A) ac4C RIP-seq peak locations relative to annotated TIS in HeLa. Inset depicts median Codon Stability Coefficients (CSCs) for the codons within each distance bin, comparing ac4C(−) to ac4C(+) mRNA regions.
(B) Firefly luciferase mRNA was in vitro transcribed in the presence of CTP or ac4CTP and in vitro translated in RRL. Firefly translation was monitored through luminescence. Mean±SEM, n=3. p=Two-way ANOVA.
(C) As in (B) but including mutations that removed all cytidines from the 5’ or 3’UTR. Mean±SEM, n=3. p=t-test.
(D) Nanoluciferase (NanoLuc) mRNA with localized ac4C restricted to the CDS or 5’UTR was generated through in vitro transcription of a 5’ and 3’ mRNA fragment followed by splint ligation. Additional in vitro capping and polyadenylation of splint ligated NanoLuc was performed for 5’UTR acetylated mRNA.
(E) In vitro translation of NanoLuc mRNA with site-specific CDS or 5’UTR ac4C. NanoLuc translation was monitored through luminescence. Mean±SEM, n=4. p=Two-way ANOVA.
(F) Capped and polyadenylated NanoLuc mRNA translation in RRL (left) and transfected HeLa cells (right). Unmodified Firefly mRNA was co-transfected for simultaneous monitoring of NanoLuc and Firefly translation through dual luminescence. Mean±SEM of NanoLuc normalized by Firefly activity, n=5. p=Two-way ANOVA (left) or t-test (right).
To directly assess the influence of ac4C within the 5’UTR versus CDS, we turned to in vitro systems for controlled analysis of mRNA translation. Luciferase mRNA was in vitro transcribed in the presence of either CTP or ac4CTP for uniform incorporation into the CDS and UTRs (Figures 1B and S1A). Subsequent translation in rabbit reticulocyte lysates (RRL) showed the expected accumulation of Luciferase activity and plateau at steady state for ac4C(−) mRNA (Figure 1B). In contrast, ac4C(+) Luciferase mRNA was characterized by an initial lag followed by robust stimulation of translation, suggestive of both repressive and stimulatory ac4C activities (Figure 1B). Importantly, removal of all 5’UTR cytidines through mutation relieved the early lag in Luciferase production associated with the ac4C(+) mRNA while translation of ac4C(−) Luciferase was unaffected (Figures 1C and S1A). Likewise, mutation of 3’UTR cytidines did not influence Luciferase production in an ac4C-dependent manner (Figure 1C). Thus, the early repression of translation observed for acetylated mRNA is at least partially attributed to ac4C presence in the 5’UTR.
To explicitly resolve location-specific ac4C effects, we generated NanoLuciferase (NanoLuc) mRNA through splint ligation to introduce ac4C at defined residues within the 5’UTR or CDS. A short 5’ mRNA fragment encompassing the 5’UTR and portion of the CDS was in vitro transcribed in the presence of CTP or ac4CTP (Figures 1D and S1B–S1D). This sequence lacked cytidines other than a short stretch upstream of the TIS within the 5’UTR or a comparable distance downstream of the TIS within the CDS. Separately, a 3’ mRNA fragment encompassing the remainder of the NanoLuc CDS and 3’UTR was transcribed in the presence of CTP only (Figure 1D). The mRNA fragments were joined through splint ligation and used in RRL in vitro translation (Figures 1E, S1D and S1E). Monitoring of protein synthesis through NanoLuc luminescence established a direct and location specific role for ac4C in mRNA translation: acetylation of six tandem codons downstream of the TIS robustly stimulated translation, whereas incorporation of the same sequence within the 5’UTR inhibited protein output (Figure 1E). NanoLuc translation was not detected in the absence of fragment ligation, validating that the results specifically relate to the ligated product (Figure S1F). To ensure that the inhibitory influence of 5’UTR ac4C is not exclusive to cap-independent translation, we further generated capped and polyadenylated splint ligated mRNA (Figures S1G and S1H). The resulting mRNA was translated in vitro and in vivo through transfection into HeLa cells along with unmodified Luciferase mRNA to control for general discrepancies in transfection and translation (Figure S1I). Normalized luminescence associated with capped and polyadenylated mRNA was reduced in the presence of 5’UTR ac4C in vitro and in vivo, corroborating the inhibitory role of ac4C in this context (Figure 1F).
Base-resolution mapping of ac4C
The mRNA reporters point to discrete roles for 5’UTR versus CDS ac4C in translation initiation and elongation, respectively. To test this premise in cellular substrates, we required a method for base-resolution mapping of TIS proximal ac4C. The importance of accurate feature mapping is exemplified in the in vitro results, wherein repositioning an acetylated sequence 130 nucleotides from the 5’UTR to CDS elicited opposing impacts on translation (Figure 1E). To achieve base-resolution mapping, we employed sodium borohydride (NaBH4) reduction of ac4C to tetrahydro-ac4C, which induces C to T mismatches during first strand cDNA synthesis (RedaC:T) (Figure 2A) (Thomas et al., 2018). Sanger sequencing of cDNA from NaBH4 treated HeLa total RNA showed ~50% C and T detection at uniformly acetylated 18S rRNA nucleotide 1842, indicative of incomplete conversion (Figure 2A). Nevertheless, mismatches were reduced to background levels in parallel-treated RNA from NAT10−/− HeLa, highlighting comparative profiling in wildtype and NAT10−/− cells as an effective method to determine ac4C locations in cellular RNA (Figure 2A).
Figure 2: Base-resolved mapping of ac4C in HeLa mRNA.

(A) RedaC:T-seq protocol and pipeline for determination of ac4C sites through NaBH4-induced C:T mismatches.
(B) Distribution of C:T mismatches in 18S rRNA from wildtype (WT, red) and NAT10−/− (dark blue) HeLa. Positions 1337 and 1842 correspond to known ac4C sites.
(C) Frequency of all mismatch types observed in RedaC:T-seq prior to filtering relative to untreated or NAT10−/− NaBH4-treated controls
(D) Volcano plot depicting the change in mismatch ratio vs. significance comparing wildtype and NAT10−/− HeLa RedaC:T-seq results. Color coding indicates significant differences (FDR adj p≤0.05, Fisher’s exact test, ≥5 fold).
(E) Cumulative frequency plot of C:T mismatch frequency distribution in wildtype and NAT10−/− RedaC:T-seq reads.
(F) Median local RedaC:T-seq coverage at all detected ac4C sites in wildtype HeLa as compared to untreated control.
(G) Browser views of example start codon proximal RedaC:T-seq defined ac4C sites showing: (top) wildtype HeLa acRIP-seq density (Arango et al., 2018), (middle) wildtype and NAT10−/− HeLa RedaC:T-seq, and (bottom) total and poly(A) HeLa ac4C-seq (Sas-Chen et al., 2020).
(H) Sequencing depth at RedaC:T-seq defined ac4C sites in the current study and previous HeLa ac4C-seq.
(I) Metagene plot of transcript coverage in HeLa RedaC:T-seq (red) and reanalysis of published HeLa ac4C-seq (purple). Solid lines show all mRNAs while dashed lines reflect coverage for RedaC:T-defined acetylated mRNAs.
To map ac4C transcriptome-wide, we coupled RedaC:T to paired-end Illumina sequencing in ribodepleted total RNA from wildtype and NAT10−/− HeLa cells (Figure 2A). High depth sequencing was performed to ensure robust conditions for mismatch detection in less abundant mRNAs (Table S1). Inspection of residual 18S rRNA reads indicated a ~25% C:T mismatch rate at 100% acetylated nucleotide 1842, whereas ~15% conversion was detected at ~80% acetylated nucleotide 1337 (Taoka et al., 2018) (Figures 2B and S2A). In contrast, mismatch accumulation was not observed in non-acetylated 28S rRNA reads (Figure S2B). These results support RedaC:T as a reliable method for relative stoichiometry assessment and establish ~25% as the upper limit for mismatch detection. Importantly, read coverage was evenly distributed surrounding the known ac4C locations, indicative that tetrahydro-ac4C did not obstruct reverse transcription (Figure S2A). 18S rRNA mismatches were reduced to background in NAT10−/− HeLa RedaC:T-seq, demonstrating the robustness of the method for identifying NAT10-catalyzed ac4C (Figure 2B).
To identify ac4C sites in mRNA, we enacted a stepwise computational pipeline that rejects nucleotide variants found in untreated HeLa input and NaBH4-treated NAT10−/− HeLa RNA-seq (Figure 2A). Based on a basal Illumina sequencing error rate of ~0.8%, mismatches that were detected at ≥1% in HeLa input RNA-seq and/or did not show ≥10x coverage in the NaBH4-treated sequencing results were omitted as likely artifacts. Through these measures, we identified >57,000 NaBH4-induced nucleotide variants in HeLa RNA (Table S2, Figure S2C). In support of specificity, ~2/3 of the identified sites represented C>T mismatches and greater than 90% showed a complete absence of variant calls in untreated input (Figures 2C and S2C). Variants were evenly distributed across reads and showed no evidence of positional detection biases (Figure S2D).
We next compared the wildtype defined variants to NAT10−/− HeLa RedaC:T-seq to discriminate ac4C from other NaBH4 related activities on RNA that alter behavior in sequencing. As NaBH4 can also react with other modified RNA species, this is an essential step in defining ac4C sites that is facilitated through the non-redundant nature of NAT10-catalyzed ac4C (Arango et al., 2018; Cerutti et al., 1968; Enroth et al., 2019; Macon and Wolfenden, 1968). In support of this approach, C:T mismatches were substantially elevated in wildtype vs. NAT10−/− HeLa RedaC:T-seq mRNA reads prior to filtering (Figures 2D and S2E). Fisher’s Exact Test (FET) followed by false discovery rate (FDR) correction was performed to define high confidence variants that significantly differ between the two cellular conditions (Table S2). In total, 7,851 cytidines in mRNA were >5 fold enriched in C:T mismatches in wildtype relative to NAT10−/− HeLa results (Figure 2D). Consistent with the 18S results, C:T conversion rates plateaued at a maximum value of ~25% and we did not observe a loss of read depth surrounding the defined ac4C sites (Figures 2E and 2F). Mismatch rates at RedaC:T-defined ac4C sites were substantially ablated in NAT10−/− cells with 71% of locations registering a complete absence of mismatches in NAT10−/− results (Figure 2E, Table S2). The aggregate ac4C:C ratio at all cytidines with greater than 10x coverage in HeLa RedaC:T-seq was 0.016%, consistent with mass spectrometry measurements of 0.01-0.1% ac4C:C in human poly(A) RNA (Arango et al., 2018; Dong et al., 2016; Guo et al., 2020) (Table S2). These analyses endorse RedaC:T-seq as a robust method for base-resolved ac4C determination within substrate mRNAs through comparative profiling of wildtype and NAT10 ablated results.
RedaC:T-seq enabled accurate assignment of TIS-proximal ac4C to the 5’UTR vs. CDS. While TRMT6 and FAM8A1 are characterized by acRIP-seq peaks that span the 5’UTR/CDS boundary, RedaC:T-seq unambiguously designates ac4C to the 5’UTR and CDS, respectively (Figure 2G). C:T mismatches within the wildtype HeLa results were encompassed within the previously determined acRIP-seq peaks, and mismatches were not observed in NAT10−/− RedaC:T-seq results, supporting the accuracy of the method for base-resolved ac4C determination at a genic level. However, a recent study using a similar strategy involving sodium cyanoborohydride (NaCNBH3) reduction and sequencing (termed ac4C-seq) did not detect ac4C in HeLa RNA (Sas-Chen et al., 2020). To reconcile this inconsistency, we reanalyzed the HeLa ac4C-seq results and found depth to be a major factor driving the discrepancy. We achieved ~35x increased mRNA base coverage in wildtype HeLa RedaC:T-seq as compared to the published ac4C-seq (Table S1). This distinction is driven by overall read recovery and sequencing read length (28-37 bp single-end for ac4C-seq vs. 125 bp paired-end for RedaC:T-seq). As a result, the majority of called RedaC:T sites did not reach a minimum threshold of 10 reads in ac4C-seq (Figures 2G and 2H). Compounding matters for 5’ enriched ac4C, pronounced attrition towards the 5’ ends of sequenced transcripts was observed in the ac4C-seq (Figure 2I). In total, of the >400 HeLa 5’UTR ac4C sites determined through RedaC:T-seq, none reached sufficient power for detection in the published HeLa ac4C-seq as determined through simulation-based analysis (Figure S2F). Nevertheless, though not statistically significant in ac4C-seq due to low-depth, we detected concordant examples of CDS and 3’UTR localized ac4C for which RedaC:T- and ac4C-seq produced overlapping C:T mismatches in wildtype HeLa mRNA that were not observed in NAT10−/− results (Figure S2G). acRIP-RT-qPCR provided independent validation through an orthogonal method (Figure S2H). Of note, higher depth ac4C-seq pursued in HEK293 with overexpression of NAT10 and the tRNA adaptor THUMPD1 identified ac4C in mRNA (Sas-Chen et al., 2020). NAT10 levels are comparatively low in HEK293 and overexpression results in cytosolic accumulation, where THUMPD1 is also found (Figures S2I and S2J). These findings point to NAT10 level as a factor driving mRNA acetylation in human cells. Overlap of ac4C mapping in HeLa acRIP and RedaC:T-seq, and loss of signal in NAT10−/− cells establish the validity of NaBH4 reduction for base-level ac4C determination.
Increased upstream initiation in the presence of 5’UTR ac4C
With a precision map of ac4C in HeLa, we next sought to determine the positional relationship to mRNA translation. As in acRIP-seq, the bulk of ac4C sites occurred within the CDS with several hundred locations mapping exclusively to the 5’UTR (Figure S3A and Table S2) (Arango et al., 2018). Base-resolved ac4C was evenly distributed across CDS, while 5’ and 3’UTR sites accumulated proximal to translation start and stop codons (Figure 3A), providing a rationale for enrichment at these locations in HeLa acRIP-seq (Arango et al., 2018). The general pattern of ac4C across transcripts was preserved when considering all sites, or just the top 25% of mismatch rates (Figures 3A, S3B, and S3C). RedaC:T defined ac4C showed strong functional concordance to acRIP-seq based associations including enhanced mRNA stability in the presence of ac4C, and decreased translation efficiency (TE) in response to ac4C loss from the CDS as gauged through Ribo-seq in wildtype and NAT10−/− HeLa performed in the presence of cycloheximide (CHX) (Figures 3B and 3C) (Arango et al., 2018). Through RedaC:T, we further determined that sensitivity to NAT10 loss is enhanced with increasing number of CDS ac4C per mRNA (Figures 3B, 3C, and S3A). Importantly, ac4C loss from unambiguously assigned 5’UTR locations through NAT10 ablation had no influence on TE, bolstering the premise of a distinctive function.
Figure 3. Acetylated 5’UTRs display increased upstream translation initiation.
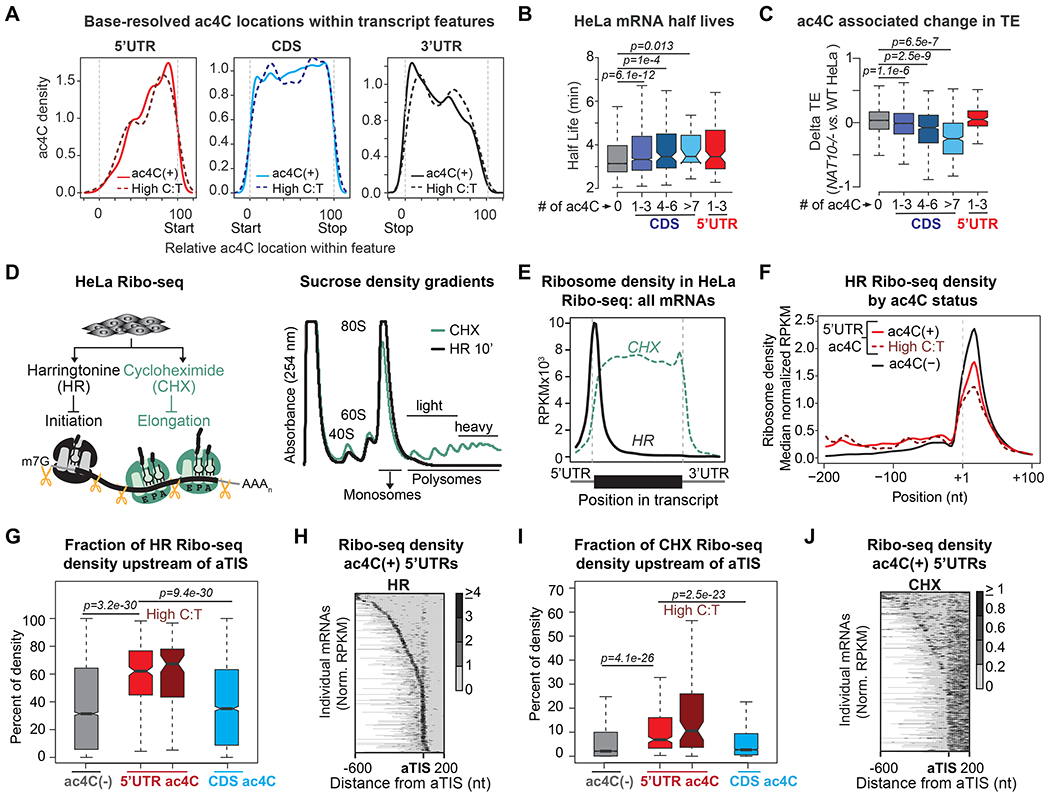
(A) Density plots depicting HeLa RedaC:T-seq mismatch rates segregated by transcript feature. Solid lines represent all ac4C sites exceeding the basal threshold (≥1.25% C:T), while dashed lines represent the top quartile of C:T mismatch rate (‘High C:T’, throughout).
(B) HeLa mRNA half-lives segregated by transcript feature and number of ac4C sites. p=Wilcoxon test.
(C) Difference in translation efficiency (TE) for NAT10−/− vs. wildtype HeLa cells segregated by transcript feature and number of ac4C sites. p=Wilcoxon test.
(D) Absorbance at 254 nm in sucrose density gradient fractions from HeLa cells treated with harringtonine (HR) or cycloheximide (CHX).
(E) Metagene plot of read density from HeLa HR and CHX Ribo-seq, RPKM: reads per kilobase per million mapped reads.
(F) HR normalized Ribo-seq RPKMs segregated by ac4C status in a range of −200bp to +100bp from annotated TIS. Medians of binned values within the indicated transcript groups are plotted.
(G) Fraction of HR RPF density localizing to 5’UTRs of mRNAs segregated by ac4C location. p=Wilcoxon test.
(H) Heatmap of HR ribosome protected fragment (RPF) density in 5’UTR acetylated mRNAs, normalizing each row by total reads within a −600 to +200bp window.
(I) CHX RPF density, as in (G). p=Wilcoxon test.
(J) Heatmap of CHX RPF density in 5’UTR acetylated mRNAs, ordered as in (H).
To specifically address the influence of 5’UTR ac4C on translation initiation, we performed Ribo-seq in the presence of Harringtonine (HR). While CHX accesses vacant E-sites and thus halts ribosomes engaged in productive elongation, HR defines locations of translation initiation through binding A-sites in the 60S ribosomal subunit prior to joining 40S (Fresno et al., 1977; Garreau de Loubresse et al., 2014) (Figure 3D). For comparison, whereas actively translating polysomes are visible in sucrose density centrifugation following CHX treatment in HeLa, 10 minutes of HR allowed elongating ribosomes to run-off and only ‘trapped’ 80S initiation complexes (ICs) remained (Figure 3D). RNase I footprinting followed by isolation and sequencing of 80S-protected mRNA fragments yielded the anticipated fragment sizes and strong concordance between replicates (Figures S3D to S3G). As expected, mapping of HR Ribo-seq reads showed robust enrichment over annotated TIS (aTIS), while CHX resulted in an accumulation of reads over protein-coding regions in HeLa metagene analyses (Figure 3E).
HeLa HR Ribo-seq results were next segregated according to ac4C status and examined for read density surrounding the aTIS. Summarized HR Ribo-seq density across transcripts uncovered a prominent accumulation upstream of the aTIS and reciprocal reduction at expected initiation sites for mRNAs with 5’UTR ac4C as compared to ac4C(−) mRNAs (Figure 3F). This relationship was exclusive to 5’UTR ac4C: the proportion of HR Ribo-seq reads mapping to annotated 5’UTRs with overlapping ac4C was significantly elevated as compared to both ac4C(−) and CDS acetylated controls (Figure 3G). Upstream initiation was further increased if only considering 5’UTR ac4C sites in the highest mismatch bin, suggestive of a functional relationship (Figures 3F and 3G). These observations were corroborated through an independent computational tool that analyzes initiation site locations and usage via statistical modeling of Ribo-seq reads (Ribo-TISH), demonstrating that the association between 5’UTR ac4C and upstream initiation is robust to computational method (Figure S3H) (Zhang et al., 2017). To gain insight into the source of the 5’ shift in ribosome density, we examined HR Ribo-seq signal per 5’UTR acetylated mRNA and found the dominant initiation site for the majority of mRNAs occurred upstream of the aTIS (Figure 3H). Heat map rows are organized by distance to the dominant TIS with the mRNA sequence shown in gray and thus demonstrate that upstream initiation on ac4C(+) 5’UTRs bears no positional relationship to the aTIS nor the 5’ end of the interrogated mRNAs (Figure 3H). The overall proportion of mRNAs with dominant upstream translation initiation sites (upTIS) was significantly elevated for 5’UTR ac4C(+) as compared to CDS ac4C(+) and ac4C(−) mRNAs (Figure S3I).
To gain confidence that the identified upTIS represent bona fide initiation sites, we further examined HeLa CHX Ribo-seq results at ac4C(+) 5’UTRs. In support of productive initiation, cumulative CHX Ribo-seq density at annotated 5’UTRs was enriched in the presence of 5’UTR ac4C as compared to CDS ac4C and ac4C(−) mRNAs (Figure 3I). Moreover, a heat map of per mRNA CHX Ribo-seq ordered as in the HR results showed a similar pattern of enrichment, confirming that some portion of the initiating ribosomes detected in HR Ribo-seq proceed to active elongation (Figure 3J). As would be expected at non-canonical initiation sites, upTIS-associated CHX Ribo-seq signal was generally discontinuous to the downstream protein-coding sequences, suggestive of short polypeptide synthesis related to uORFs (Figure 3J). Likewise, summarized CHX Ribo-seq centered on HR defined upTIS of 5’ UTR acetylated mRNAs revealed pronounced signal overlap at the detected initiation sites with attenuating 3’ read density (Figure S3J). These analyses reveal an unexpected enrichment of non-canonical upstream initiation in the presence of 5UTR ac4C.
ac4C positioning relative to HeLa mRNA TIS
Co-occurrence of 5’UTR ac4C and non-canonical initiation in HeLa reinforces a unique function unrelated to translation elongation. In support of a causative role in upstream initiation, ac4C sites were enriched for overlapping HR Ribo-seq signal as compared to random chance (Figure 4A). However, overlap occurred at both upTIS and aTIS, and approximately one half of 5’UTR ac4C sites existed outside of an initiating ribosome footprint (Figure 4B). To gain contextual insight, we examined for sequence features that could inform on ac4C involvement in translation initiation. Though initiation typically occurs at the first AUG encountered during PIC scanning, the sequence surrounding canonical TIS or impediments such as secondary structures within 5’UTRs can facilitate initiation at suboptimal cryptic AUG or near cognate codons (Kearse and Wilusz, 2017; Kozak, 1986; 1990; Leppek et al., 2018) (Figure 4C). Empirical mapping of initiating P-site codons from HeLa HR Ribo-seq recovered ~23,000 upstream and annotated TIS, in-line with previous reports (Lauria et al., 2018; Lee et al., 2012) (Figures S4A, S4B and Table S3). In support of the established annotations, P-sites associated with aTIS were overwhelmingly AUG, whereas detected upTIS were AUG-depleted and most often occurred at the near-cognate CUG (Figures 4D and S4C, Table S3). P-site codon distribution was generally preserved for mRNAs with 5’UTR ac4C, though an increase in CUG and concomitant decrease in AUG initiation was observed at upstream locations, particularly when ac4C directly overlapped a ribosome-protected fragment (RPF) (Figures 4D and S4D). Consistent with their non-canonical status, detected upTIS lacked features associated with consensus Kozak-sequences and AUG-associated upTIS were uniformly discontinuous by frame or stop codon to the downstream protein-coding ORF (Figures 4E and S4E, Tables S3). In contrast, detected aTIS of 5’UTR ac4C(+) mRNAs were uniformly AUG-initiating and showed a similar enrichment for surrounding Kozak-preferred nucleotides as aTIS of ac4C(−) mRNAs (Figures 4D, 4E, and S4E).
Figure 4. Positional relationship between ac4C and translation initiation in base-resolved mapping.
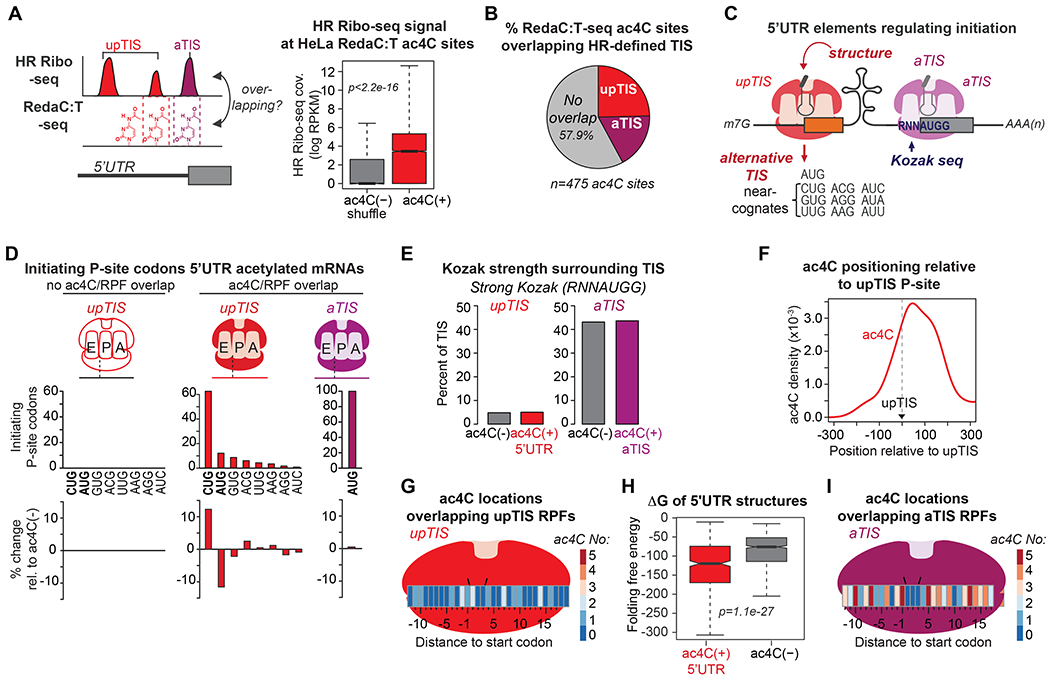
(A) Boxplot of log HR Ribo-seq read density overlapping 5’UTR ac4C sites compared to random 5’UTR locations of ac4C(−) transcripts in HeLa mRNA. p=Wilcoxon test.
(B) The portion of HeLa ac4C sites overlapping an HR Ribo-seq RPF associated with upstream (upTIS) or annotated (aTIS) initiation.
(C) Representative 5’UTR elements influencing translation initiation.
(D) Mapping of utilized start codons in HeLa HR Ribo-seq for 5’UTR ac4C(+) and ac4C(−) mRNAs. The difference in codon composition for 5’UTR ac4C(+) as compared to ac4C(−) mRNAs is also shown (bottom).
(E) Percent of TIS with a strong Kozak sequence, by ac4C status.
(F) Smoothed histogram density plot depicting ac4C distribution relative to dominant upTIS P-sites.
(G)Heat map summarizing ac4C abundance overlapping initiating ribosomes at specific distances to dominant upTIS.
(H) Boxplot of folding free energy (ΔG) of 5’UTRs by ac4C status. p=Wilcoxon test.
(I) Heat map as in (G) for aTIS.
A main advantage of coupling RedaC:T-seq to HR Ribo-seq is the ability to spatially relate these parameters at base-resolution. Focusing on upTIS, ac4C was substantially enriched downstream of upTIS that represented the dominant initiation site of the associated mRNA, irrespective of start codon identity (Figures 4F and S4F). This bias in localization was observed both in absolute numbers and relative distance to the upTIS P-site codon (Figure 4F). However, direct ac4C overlap at dominant upTIS RPFs was rare, representing only ~8% of cases (Figure S4F, Table S3). These findings suggest an indirect role for ac4C in upstream initiation that is potentially mediated from downstream locations. Supporting this notion, ac4C(+) 5’UTRs are more likely to generate structures that could inhibit PIC scanning as compared to 5’UTRs of ac4C(−) mRNAs (Figure 4H) (Ringner and Krogh, 2005). In contrast to upTIS, ac4C was prominently observed overlapping initiating ribosomes at aTIS (Figure 4I). In total, 83 mRNAs were characterized by direct ac4C overlap within an aTIS RPF, with numerous additional sites existing immediately outside this narrow 31 nucleotide range (Table S3). Specific examination of ac4C positioning within canonical initiating ribosomes revealed considerable accumulation at numerous locations, including multiple key Kozak residues (Figure 4I). These base-resolved analyses provide a dedicated template from which to test the direct impact of 5’UTR ac4C on translation initiation.
ac4C directly represses canonical translation initiation in vitro
Guided by in vivo defined principles, we returned to in vitro systems to assess the site-specific influence of ac4C on translation initiation. Based on ac4C positioning within 5’UTRs and the relative impact on translation in HeLa, we addressed ac4C function from downstream of an out-of-frame ‘upTIS’ and when directly overlapping an in-frame ‘aTIS’ associated with the NanoLuc CDS (Figure 5A). As in Figure 1, ac4C was incorporated into specific 5’UTR locations through in vitro transcription of an otherwise C-less 5’ mRNA fragment in the presence of ac4CTP or CTP, followed by splint ligation to an unmodified 3’ mRNA fragment (Figures 5A and S5A). Translation was pursued in RRL, and in transfected wildtype and NAT10−/− HeLa cells.
Figure 5. Position-specific influence of ac4C on translation initiation in mRNA reporters.

(A) ac4C was incorporated into specific 5’UTR locations in NanoLuc mRNA through in vitro transcription and splint ligation as in Fig. 1D. NanoLuc translation was achieved in RRL or in transfected wildtype and NAT10−/− HeLa cells.
(B) NanoLuc mRNA containing ac4C or C within a structured uORF upstream of a consensus AUG start codon was generated through splint ligation and confirmed through Northern blot/phosphorimagery (left). Uncapped mRNAs were in vitro translated in RRL. NanoLuc activity was normalized by mRNAs levels (middle), Mean±SEM, n=3. p=Two-way ANOVA (middle). Capped and polyadenylated NanoLuc mRNAs were transfected into wildtype and NAT10−/− HeLa cells along with unmodified Firefly luciferase mRNA for in vivo translation. NanoLuc activity was normalized by Firefly luciferase (right), Mean±SEM, n=3 for HeLa WT, n=8 for NAT10−/−. p=t-test.
(C) As in (B), but with NanoLuc mRNA containing C or ac4C in positions −1 and −2 of the Kozak sequence surrounding a consensus AUG start codon. Mean±SEM, n=5 for in vitro translation, n=5 for HeLa WT, n=4 for NAT10−/− p=Two-way ANOVA, in vitro results. p=t-test, in vivo results.
(D) As in (B), but with NanoLuc mRNA containing C or ac4C directly over three tandem CUG start codons. Mean±SEM, n=3 for in vitro translation, n=4 for HeLa WT. n=4 for NAT10−/−. p=Two-way ANOVA, in vitro results. p=t-test, in vivo results.
To test the indirect influence on upstream initiation, ac4C was introduced downstream of a non-consensus ‘weak’ AUG codon associated with a short open-reading frame. NanoLuc translation was derived from an in-frame downstream ‘aTIS’ AUG that would be competitively restricted by initiation at the upTIS (Figures 5B and S5B). Considering that ac4C promotes mRNA structures and 5’UTR structures influence initiation through altered accessibility/ribosome scanning kinetics (Kozak, 1990; 2005; Sas-Chen et al., 2020), we further explored the contribution of structure to ac4C function on upTIS through variable introduction of a proximal complementary sequence (Figure 5B, Table S1). In vitro translation of NanoLuc was strongly inhibited by 5’UTR ac4C in a structured context, while ac4C had no influence on translation associated with the unstructured 5’UTR (Figures 5B and S5B). The inhibitory influence of structured 5’UTR ac4C was also observed in capped and polyadenylated mRNA, both in RRL and upon transfection into wildtype and NAT10−/− HeLa (Figure 5B and S5C). These results provide a rationale for ac4C enrichment downstream of non-canonical upTIS in HeLa (Figures 4F and S4E) and point to obstruction of ribosome scanning as a potential mechanism competitively limiting access to optimal downstream aTIS.
Combined RedaC:T- and HR Ribo-seq analysis further implicated a role for ac4C in the immediate vicinity of utilized start codons. This was principally observed at aTIS-adjacent locations associated with Kozak sequences, while direct near-cognate acetylation was rare with only two cases of CUG acetylation detected in HeLa (Figures 4G and 4I). We accordingly generated reporters to test the basis of both ac4C enrichment and depletion from overlapping contexts in the transcriptome-wide results. To examine Kozak acetylation, we introduced ac4C at 5’UTR positions −1 and −2 relative to an AUG ‘aTIS’ start codon in-frame with NanoLuc cDNA (Figure 5C, Table S1). Strikingly, acetylation of these Kozak residues, wherein cytidine is normally favorable to initiation, substantially inhibited translation as compared to unmodified NanoLuc mRNA (Figure 5C) (Simonetti et al., 2020). This reduction was observed in RRL +/− capping and polyadenylation, and upon transfection of capped and polyadenylated mRNA into wildtype and NAT10−/− HeLa cells (Figure 5C and S5D). In contrast, introduction of ac4C at position 1 of a CUG start codon that was in-frame to downstream NanoLuc mRNA robustly stimulated translation in vitro and upon transfection into wildtype and NAT10−/− HeLa cells as compared to ac4C(−) control mRNA (Figures 5D and S5E). The near-cognate reporter lacked 5’UTR AUGs that could otherwise competitively impact downstream translation (Table S1). Though direct near-cognate acetylation is uncommon in HeLa (Figure 4G), these results illustrate how a one nucleotide shift in ac4C can fundamentally alter initiation and raise the possibility that CUG acetylation is selected against in cells to avoid aberrant translation. Together, these analyses involving mRNA reporters with site-specific ac4C conclusively establish a role in initiation that is achieved independently of a specific cellular context shaped by NAT10 activities that are unrelated to mRNA acetylation.
5’UTR acetylation regulates translation initiation in vivo
In combining our observations from in vivo mapping and in vitro translation, an intriguing dual role for ac4C in promoting recognition of non-optimal upstream codons while repressing canonical AUG is revealed. To test this model in cells, we examined how loss of ac4C from 5’UTRs influences translation initiation in conditions of NAT10 ablation. Accordingly, HR Ribo-seq was executed and analyzed in NAT10−/− HeLa cells exactly as performed for wildtype cells (Figures S6A–C). Previous characterization of NAT10−/− HeLa ruled out gross pleiotropic effects in translation (Arango et al., 2018). On the whole, the distribution of initiating ribosomes across mRNAs defined as 5’UTR acetylated in wildtype cells was roughly preserved in NAT10−/− HR Ribo-seq (Figure 6A). However, the overall change in HR Ribo-seq density was statistically enriched for mRNAs with 5’UTR ac4C as compared to ac4C(−) controls with clear differences observed at major upTIS and aTIS (Figures 6A and Figure S6D).
Figure 6. ac4C within 5’UTRs and overlapping Kozak sequences inhibits canonical translation initiation in vivo.

(A) Heatmap of the change in initiating ribosome density in NAT10−/− vs. wildtype HeLa for mRNAs with ac4C(+) 5’UTRs within a −600 to +200bp window. Transcripts with major initiation occurring at an upTIS vs. aTIS are indicated.
(B) Boxplot of the change in HR Ribo-seq density at major upTIS of ac4C(+) 5’UTRs in NAT10−/− vs. wildtype HeLa, separated based on relative ac4C positioning as indicated. Results from upTIS of ac4C(−) mRNAs are shown as control. Wilcoxon test=n.s.
(C) As in (B) but plotting the change in HR Ribo-seq density at major aTIS, as indicated. p=Wilcoxon test.
(D) Browser views of abundance normalized RedaC:T- and HR Ribo-seq, and Western blots of ac4C(−) and 5’UTR ac4C(+) mRNAs, as indicated.
(E) Mfold prediction of PXN and KRT80 RNA structures surrounding detected ac4C sites.
(F) Representative images at 0 and 24 hrs after scratch infliction in confluent wildtype and NAT10−/− HeLa cultures.
(G) Biological pathway enrichment for genes with 5’UTR ac4C. p=Fisher’s exact test and Benjamini-Hochberg correction.
(H) Propidium iodide staining and flow cytometry at the indicated times after release from double thymidine block in wildtype and NAT10−/− HeLa cells.
Through examining the impact of ac4C modulation on relative TIS distribution in HeLa cellular mRNA, we gain in vivo support for location-specific 5’UTR functions. Focusing on upTIS, NAT10 ablation led to a general though moderate reduction in HR Ribo-seq signal at major upTIS of ac4C(+) 5’UTRs as compared to ac4C(−) controls (Figure 6B). This trend was maintained from upstream and downstream ac4C locations, though absolute frequency was increased for the latter (Figures 4F and 6B). mRNAs with ac4C overlapping a dominant upTIS represented an exception wherein a directional change in HR Ribo-seq density was not observed, perhaps related to competing functions associated with adjacent vs. direct TIS acetylation (Figures 5C, 5D and 6B). These findings support a role for ac4C in upstream initiation from neighboring positions within 5’UTRs (Figure 5B). In contrast, a direct role for ac4C in modulating adjacent start codon recognition is evident in results comparing aTIS initiation in wildtype and NAT10−/− HeLa. When examining all aTIS of mRNAs with ac4C(+) 5’UTRs, the overall change in initiation associated with NAT10 ablation did not significantly differ from ac4C(−) mRNAs (Figure 6C). However, when considering mRNAs with ac4C overlapping a major initiating ribosome at the aTIS, a generalized shift to increased ribosome density was observed in response to NAT10 ablation (Figure 6C). This effect is enhanced when considering ac4C immediately flanking the P-site AUG within the defined Kozak range, including at mRNA nucleotides that generate intermolecular contacts in 48S ICs (Figures 6C and S6E) (Simonetti et al., 2020). As consensus Kozak sequences are generally C-rich (Kozak, 1986; 1987), these results suggest that ac4C neutralizes otherwise favorable interactions between AUG-proximal cytidine in mRNA and elements within scanning ribosomes.
Importantly, we were able to readily identify HeLa mRNAs that endorse an ac4C function in translation initiation and were also detected as differentially initiating through Ribo-TISH (Table S3). The mRNAs encoding paxillin (PXN) and keratin 80 (KRT80) are characterized by a 5’UTR ac4C site located in a predicted mRNA structure occurring between a minor HR Ribo-seq determined upTIS and the aTIS (Figures 6D and 6E). Loss of ac4C from these mRNAs through NAT10 ablation produced a shift in HR Ribo-seq density to the aTIS and increased protein output, while ac4C(−) GAPDH was unaffected. UpTIS promoted by ac4C thus competitively restrict aTIS initiation and ac4C may further directly occlude start codons through thermodynamic stabilization of encompassing stem loop structures, as shown for KRT80 (Figure 6D and 6E). With respect to the consequence on HeLa function, PXN and KRT80 promote cell migration (Li et al., 2018; Lopez-Colome et al., 2017). Consistent with de-repression of these factors, confluent NAT10−/− cells displayed increased migration in a scratch assay as compared to wildtype HeLa (Figures 6F and S6F). While RNAi against PXN generally inhibited cell proliferation, KRT80 depletion eliminated the enhanced migration of NAT10−/− cells without impacting cell viability (Figures S6G–I), bolstering the involvement of NAT10 5’UTR mRNA substrates in the observed phenotype.
Consistent with a repressive role for ac4C within Kozak sequences, ac4C loss from the −1 position relative to the AUG start codon of the transcription factor IRF1 and chromatin modifier KDM4B led to a substantial increase in aTIS HR Ribo-seq density and enhanced protein output (Figure 6D). The relatively modest increase in IRF1 protein is influenced by protein turnover, as shown through IRF1 accumulation upon proteasome inhibition (Figure S6J). Cell-cycle and transcriptional regulators, including KDM4B and IRF1, were overall enriched in the cohort of 5’UTR acetylated mRNAs, implicating a possible function for 5’UTR ac4C in the regulation of gene expression and cellular proliferation (Figure 6G and Table S2) (Dou et al., 2014; Wilson and Krieg, 2019). Indeed, a general shift in the cell cycle is a main phenotypic change observed in NAT10−/− as compared to wildtype HeLa (Figure 6H). Moreover, re-expression of full-length NAT10 cDNA, but not a mutant lacking the RNA helicase domain, rescued proliferation in NAT10−/− cells (Figures S6K and S6L). These results examining the change in initiation in response to NAT10 ablation clearly define a role for 5’UTR ac4C in the regulation of translation and hint at a function in modulating cellular behavior in response to specific stimuli.
ac4C in mRNA modulates interaction with t6A in tRNAiMet
Throughout the analyses presented herein, the repressive influence of ac4C within Kozak sequences has remained constant. While ac4C function on upTIS is likely achieved through generating an obstacle to ribosome scanning, how ac4C directly affects initiation adjacent to start codons remained unclear. To gain mechanistic insight, we performed cryo-EM with a short in vitro transcribed mRNA fragment encompassing a 5’UTR and sufficient CDS sequence to allow for IC assembly (Figure 7A and S7A). This mRNA fragment lacked cytidines other than at the Kozak favored −1 position relative to an AUG start codon [C(−1)], wherein a repressive function for ac4C was observed in vitro and in vivo (Figures 5C, 6C, and 6E). Translation was performed in HR-treated RRL and 80S ribosomes were isolated and subjected to cryogenic freezing (Figure S7A). To ensure sample homogeneity, RNA isolated from parallel-processed samples was analyzed through RT-qPCR and high-throughput Illumina sequencing (Figure S7B, Table S3). By either metric, our input RNA was >4,000 fold enriched as compared to hemoglobin mRNAs, which are the most abundant transcripts in reticulocytes, indicating an endogenous mRNA contamination level of <0.025% (Goh et al., 2007).
Figure 7. Acetylation of cytidine at Kozak nucleotide (−1) structurally alters intermolecular interaction with tRNAiMet t6A in 80S initiation complex cryo-EM.

(A) Mammalian 80S ICs were formed on in vitro transcribed mRNA fragments containing a single C (gold throughout) or ac4C (purple throughout) in the −1 position immediately adjacent to an AUG start codon. Cryo-EM was performed on purified complexes for structural determination.
(B) Overall cryo-EM map of the ac4C(−1) 80S IC.
(C) Zoomed in view of the model fitted tRNAiMet and mRNA interface.
(D) Detailed local maps of fitted models focused at the codon:anticodon interface.
(E-F) Zoomed in view comparing tRNA t6A(37) interaction with mRNA C(−1) versus ac4C(−1). A strong 2.1 Å hydrogen bond is observed between the C(−1) ribose 2’OH and t6A carboxyl side chain. Acetylation weakens this interaction to 3.7Å through a shift at the ribose.
(G) Schematic of molecular interactions between t6A and C versus ac4C.
To resolve the influence of (−1) ac4C on translation initiation, cryo-EM grids were prepared by vitrification followed by screening and micrograph collection. Particle picking was guided by 2D classes generated from high contrast negative stained images (Figure S7C). Through multiple rounds of 2D and 3D classification followed by particle subtraction, ~53K and 107K well-defined 80S complexes were obtained from the ac4C(−) and ac4C(+) templates, respectively (Figure S7D). Iterative contrast transfer function (CTF) and particle polishing resulted in overall maps of 3.1 Å [C(−1)] and 2.8 Å [ac4C(−1)]. Focused refinements centered on the mRNA produced 2.8 Å local maps that improved density surrounding ac4C(−1), while multibody refinement enhanced resolution at other subregions (Figures S7D–E). Best-fit tRNA conformations were determined through available cryo-EM and NMR structures, and ac4C(−1) mRNA conformations were referenced from previously published cryo-EM structures (Sas-Chen et al., 2020; Simonetti et al., 2020; Stuart et al., 2000). The overall refined maps from both datasets contain densities corresponding to initiator tRNA (tRNAiMet) engaged in the P-site and mRNA traversing the ribosomal channel (Figures 7B, 7C, S7F, and S7G). The produced maps closely conform with previously solved structures of late-stage mammalian 48S ICs and elongation competent 70S ICs determined through kinetic cryo-EM analysis in bacteria, whereas notable distinctions are seen in comparisons to early 70S ICs and elongating 80S particles (Figures S7H and S7I) (Kaledhonkar et al., 2019; Shen et al., 2021; Simonetti et al., 2020). In further support of successful IC capture, electron density attributed to the P-site tRNA is apparent, while the A- and E-sites remain vacant (Figure S7I) (Kaledhonkar et al., 2019; Shen et al., 2021). Local density maps show clear contacts between tRNAiMet, mRNA and ribosomal subunits: the P-site tRNA anticodon loop is positioned within the decoding center engaged with mRNA, while the acceptor arm is bound to 28S rRNA (Figures 7C and S7G). Additional side chain density at tRNA adenine 37 is consistent with the presence of hypermodified t6A at this location in tRNAiMet (Figure 7D) (Parthasarathy et al., 1977; Stuart et al., 2000).
To determine whether and how ac4C impacts IC assembly, we directly compared the cryo-EM structures derived from unmodified C(−1)AUG vs. modified ac4C(−1)AUG mRNA. Though the overall structures were nearly identical, the tRNA-mRNA interface represented a notable exception (Figures 7D and 7E). While Watson-Crick base-pairing between the tRNA and mRNA anticodon: codon is maintained in both complexes, the presence of ac4C at the −1 position disturbs interaction with t6A in the tRNA (Figures 7E and S7J). Consistent with the published mammalian 48S IC structure, densities surrounding unmodified C in the −1 mRNA position and t6A of the tRNA are in close contact, evident of a physical interaction (Figures 7D and 7E) (Brown et al., 2018; Simonetti et al., 2020). In specific, a 2.1 Å hydrogen bond is observed between the carboxyl side chain (CO2-) of t6A and 2’-hydroxyl (OH) of the cytosine ribose (Figures 7E and 7F). In contrast, the distance between ac4C and t6A is overall increased to 3.7 Å in the analogous comparison involving acetylated mRNA, resulting in structural maps that are inconsistent with intermolecular hydrogen bonding (Figures 7E and 7F). Though the mechanistic basis is unclear, acetylation appears to shift the cytosine base away from 18S rRNA G1207 and towards mRNA nucleotide G(~2) (Figure S7K). Consequently, the ac4C 2’-OH moves away from tRNA nucleotide 37 and t6A adopts a structure that closely aligns with its solution NMR structure, further indicative of ablated hydrogen-bonding (Figure S7L) (Stuart et al., 2000). Thus, the presence of a single acetyl group adjacent to an AUG start codon perceptibly altered intermolecular interactions within initiating ribosomes. In total, these cryo-EM results provide a mechanistic rationale for the repressive role of ac4C within Kozak sequences through disrupting otherwise stabilizing interactions with t6A in tRNAiMet.
DISCUSSION
The analyses presented herein document the direct positional influence of mRNA acetylation in translation. While CDS ac4C robustly stimulates translation elongation, 5’UTR ac4C regulates translation initiation with exquisite locational specificity that is achieved directly and indirectly: ac4C immediately adjacent to a strong AUG start codon inhibited translation, whereas ac4C downstream of a weak upTIS promoted its use. This tendency was observed in transcriptome-wide results and confirmed through in vitro translation involving site-specific ac4C incorporation. 5’UTR ac4C was accordingly associated with widespread alternative initiation encompassing non-canonical sequence contexts in HeLa with demonstrated physiological relevance in the regulation of gene expression. ac4C further stimulated translation when overlapping a CUG near-cognate codon in vitro, though detection of ac4C in this context was exceedingly rare in vivo. These results highlight a unifying biological principle for 5’UTR ac4C in promoting non-optimal initiation while inhibiting canonical TIS.
Aberrant upstream initiation highlights a central challenge for the initiation machinery: the appropriate discrimination of start codons associated with protein-coding ORFs from closely related sequences, often including upstream AUGs (Hinnebusch, 2017). To ensure fidelity in start codon selection, intermolecular interactions between tRNA, mRNA, rRNA, and PIC associated initiation factors work in concert to guard against inappropriate initiation (Hinnebusch, 2017). However, initiation is ultimately dictated by the kinetics and thermodynamics of tRNA/mRNA base-pairing (Kolitz et al., 2009). In this manner, factors that promote residency at upTIS, such as partial interactions at near-cognate codons and structural impediments, can stimulate initiation at otherwise non-optimal contexts (Hinnebusch et al., 2016). A common theme amongst these activities is a reliance on RNA:RNA interactions, highlighting a potential basis for ac4C involvement through modulation of base-pairing dynamics.
Mechanistically, the positional influence of ac4C in translational regulation is rooted in molecular forces that alter the conformation of cytosine and associated intermolecular interactions. In specific, ac4C locks cytosine in an unusual ‘proximal’ conformation that enhances base-pairing with guanosine (Kumbhar et al., 2013; Parthasarathy et al., 1978; Taniguchi et al., 2018). As a result, ac4C increases the thermostability of Watson:Crick interactions involving cytosine, including RNA:RNA and RNA:DNA duplexes (Bartee et al., 2022; Sas-Chen et al., 2020; Wada, 1998). With respect to the current study, this has bearing both on the indirect and direct role of ac4C in initiation. Indirectly, RNA:RNA interactions involving 5’UTR ac4C within structured contexts obstruct access to downstream aTIS, synthesizing observations that 5’UTR structures impede ribosome scanning and ac4C promotes RNA structures into a tangible impact on translation initiation at upstream locations (Kozak, 1990; Leppek et al., 2018). Directly, ac4C mitigated the otherwise stimulatory influence of cytidine within Kozak sequences through modulating intermolecular contacts between scanning ribosomes and mRNA nucleotides. The importance to start codon discrimination is underscored by observations that single nucleotide polymorphisms (SNPs) within 5’UTR portions of Kozak sequences increase susceptibility to diseases such as cancer and autoimmune disorders (Cheng et al., 2016; Jacobson et al., 2005; Norder et al., 2019). Our findings showing a repressive function for ac4C within Kozak sequences is particularly notable within this physiological framework. Surprisingly, we also found that direct acetylation of cytidine within a CUG near cognate codon enhanced initiation in vitro. Though not a major mechanism of upstream initiation in HeLa, these observations provide further mechanistic insight into ac4C function in translation. Start codon sequence deviation is most often observed at the first nucleotide, with CUG representing the most utilized near-cognate codon followed by GUG (Kearse and Wilusz, 2017; Kozak, 1989). This is in contrast to coding sequences, wherein A-site tRNAs tolerate ‘wobble’ variations in the third nucleotide within an mRNA codon (Crick, 1966). We previously determined that ac4C was particularly stimulatory to translation elongation when occurring within mRNA codon wobble sites (Arango et al., 2018). These findings point to a distinctive role for ac4C in stabilizing mRNA:tRNA wobble. It will be of high interest to determine how mRNA acetylation influences intermolecular behavior in initiating versus elongating ribosomes in future studies.
Cryo-EM analysis of the 80S IC formed on acetylated versus unmodified mRNA provided critical insight into the mechanism by which ac4C modulates Kozak function. Acetylation of the exocyclic amino hydrogen of cytidine induced a conformational change that perturbed an intermolecular interaction at the codon: anticodon interface. In specific, acetylation disrupted hydrogen bonding between the 2’OH of the cytosine ribose ring and carboxyl side chain of hypermodified t6A at tRNAiMet nucleotide 37. The importance of t6A interaction with mRNA nucleotide −1 is edified by studies in yeast involving t6A biosynthetic pathway mutants wherein loss of t6A shifted initiation to upstream non-AUG codons (Thiaville et al., 2016). Our observations documenting reduced aTIS initiation when ac4C is present within Kozak sequences is reminiscent of these results in yeast and provides a mechanistic rationale for the repressive role of ac4C in translation initiation. Of note, the impact of cytidine modification within Kozak sequences is unlikely to be restricted to mRNA C(−1): mammalian 48S cryo-EM also revealed weak interactions between ribosomal protein eS26 and mRNA C(−4) , eIF1A and mRNA positions +3 to +7, and ribosomal protein uS3 and mRNA position +10 (Simonetti et al., 2020). Overall, these cryo-EM results document the complexity introduced through simultaneous modification of interacting nucleobases in tRNA and mRNA and raise the prospect of extensive direct regulation of translation through a diversity of RNA modifications.
It is tempting to speculate that 5’UTR mRNA acetylation is an evolved mechanism to regulate protein expression through a tunable means that is distinguished from the constitutive factors that participate in canonical initiation, including the otherwise static mRNA sequence. Indeed, ac4C levels in poly(A) RNA have been shown to vary between organisms, cell types, and in response to viral infection or oxidative stress, suggestive of dynamic regulation (Guo et al., 2020; Levy et al., 2020; McIntyre et al., 2018; Tardu et al., 2019; Tsai et al., 2020). Though the mechanisms guiding ac4C distribution within mRNA remain unknown, overall NAT10 abundance within cells is a likely contributing factor with potential pathophysiological relevance: NAT10 overexpression correlates with poor prognosis in a variety of cancers (Li et al., 2017; Tan et al., 2018; Yang et al., 2021). Consistent with our observations in HeLa linking NAT10-catalyzed ac4C to cell cycle and migration (Figure 6), NAT10 has further been directly linked to enhanced cell proliferation, cell cycle progression, and tumor invasion in cancer cells, including a recent report implicating COL5A1 mRNA acetylation in cancer metastasis (Liu et al., 2022; Yang et al., 2021; Zhang et al., 2021). Of note, though HeLa cells tolerate NAT10 ablation, NAT10 is an essential gene in a variety of cellular settings (Tshemiak et al., 2017). While generally attributed to NAT10 function in ribosomal rRNA acetylation, a recent study employing snoRNA deletion and cross-evolutionary analysis established that 18S rRNA acetylation is not required for cell survival nor function (Marie-Line et al., 2021). Likewise, loss of tRNA acetylation through THUMPD1 deletion does not impact cell viability (Tsherniak et al., 2017). Together, these observations point to mRNA as a key substrate for essential NAT10 functions that basally support cell proliferation but can result in tumorigenesis when left unchecked. Intriguingly, while ac4C within Kozak sequences repressed translation, direct CUG acetylation stimulated initiation (Figure 5). Whether near-cognate acetylation emerges in conditions of NAT10 up-regulation in cancers remains to be seen.
The contrasting behavior of ac4C within 5’UTRs and coding sequences raises important implications regarding its potential use in future mRNA therapeutics. The repressive function of ac4C within 5’UTRs may antagonize the positive influence exerted from CDS locations, as we observed for fully acetylated Luciferase mRNA (Figure 1). Indeed, a recent report exploring the use of ac4C in mRNA therapeutics found an unexpected reduction in total protein output associated with ac4C(+) as compared to ac4C(−) GFP mRNA (Nance et al., 2021). This study involved in vitro transcribed mRNA that introduced ac4C throughout the UTRs and CDS, including direct overlap at the Kozak sequence flanking the GFP start codon (Nance et al., 2021). Thus, reduced GFP production from the ac4C(+) mRNA suggests that the repressive function within the 5’UTR superseded the stimulatory influence from the CDS during the sampling window. These findings demonstrate the critical importance of considering location-specific effects of mRNA modifications when ascribing cellular functions and in therapeutic development.
In sum, in pursuing location-based analyses of mRNA acetylation, we uncover distinctive roles in translation that display remarkable positional sensitivity. Within coding sequences, ac4C promotes translation elongation, whereas within 5’UTRs, ac4C generally enhances upstream initiation while reciprocally repressing annotated TIS. We thus document a spectrum of possible impacts on translation that is achieved through a direct influence on RNA-RNA interactions. With respect to initiation, 5’UTR ac4C alters base-pairing interactions that promote fidelity, including an otherwise stabilizing interaction with t6A of tRNAiMet when adjacent to a strong start codon. ac4C thus provides molecular insight into Kozak sequence function and exposes unexpected nuances when RNA modifications meet. These findings document the powerful impact of post-transcriptional mRNA modifications in the regulation of protein production and identify a role for the epitranscriptome in the regulation of alternative initiation.
Limitations of the Study
This study utilizes a variety of approaches to document a role for mRNA acetylation in translation in vitro and in vivo. However, the in vivo investigations were limited to HeLa cells and their NAT10−/− derivative. Accordingly, whether 5’UTR acetylation represents a global regulatory mechanism of translation initiation remains to be determined. In addition, while RedaC:T-seq is an efficient method for localization of ac4C within substrate RNAs, incomplete C to T conversion presents a barrier to absolute stoichiometry measurements. Improved methods will be required to quantitively assess ac4C distribution at base-resolution.
STAR*METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shalini Oberdoerffer (shalini.oberdoerffer@nih.gov).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RedaC:T-seq, HR Ribo-seq, and Rabbit Reticulocyte RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Cryo-EM data have been deposited at PDB and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse IgG-HRP antibody | GE Healthcare | Cat#:NA931; RRID: AB_772210 |
| Anti-rabbit IgG-HRP antibody | Cell Signaling Technology | Cat#:7074S; RRID: AB_2099233 |
| Mouse monoclonal anti-GAPDH (6C5) antibody | Santa Cruz Biotechnology | Cat#:sc-32233; RRID: AB_627679 |
| Rabbit monoclonal anti-ac4C antibody | Abcam | Cat#:ab25215; RRID: AB_2827750 |
| Rabbit monoclonal anti-m7G antibody | MBL International Corporation | Cat#:RN016M; N/A |
| Rabbit Monoclonal IgG Isotype Control | Cell Signaling Technology | Cat#:3900S; RRID: AB_1550038 |
| Rabbit polyclonal anti-IRF1 antibody | Abcam | Cat#:ab191032; RRID: AB_2904575 |
| Rabbit polyclonal anti-KDM4B antibody | Cell Signaling Technology | Cat#:8639S; RRID: AB_11140642 |
| Rabbit polyclonal anti-KRT80 antibody | ProteinTech | Cat#:16835-1-AP; RRID: AB_1851273 |
| Rabbit polyclonal anti-LMNA antibody | Bethyl Laboratories | Cat#:A303-433A; RRID: AB_10951693 |
| Rabbit polyclonal anti-PXN antibody | ProteinTech | Cat#:10029-1-Ig; RRID: AB_513929 |
| Rabbit polyclonal anti-NAT10 antibody | ProteinTech | Cat#:13365-1-AP; RRID: AB_2148944 |
| Rabbit polyclonal anti-THUMPD1 antibody | Bethyl Laboratories | Cat#:A304-643AT; RRID: AB_2620838 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| 32P-γATP | PerkinElmer | Cat#:BLU502A100UC |
| 32P-αUTP | PerkinElmer | Cat#:BLU007H250UC |
| AMPure XP beads | Beckam Coulter | Cat#:A63881 |
| Acid-Phenol:Chloroform, pH 4.5 | ThermoFisher Scientific | Cat#:AM9722 |
| Bovine Calf Serum (BCS) | HyClone | Cat#:SH30073.03 |
| Bovine Serum Albumin (BSA) | Roches | Cat#:03117332001 |
| Bradford Reagent | BioRad | Cat#:5000205 |
| CIAP | Promega | Cat. #:M2825 |
| Cycloheximide | Sigma-Aldrich | Cat#:C7698 |
| DMEM | ThermoFisher Scientific | Cat#:11995073 |
| DNase I | Sigma-Aldrich | Cat#:4716728001 |
| DNAse-free RNAse A | ThermoFisher Scientific | Cat#:EN0531 |
| E. coli Poly(A) Polymerase | New England Biolabs | Cat#:M0276S |
| ECL Western Blotting Substrate | Promega | Cat#:W1001 |
| Halt™ protease inhibitor cocktail | ThermoFisher Scientific | Cat#:78429 |
| Harringtonine | Abcam | Cat#:ab141941 |
| Illustra™ MicrosSpin™ G-50 Columns | GE Healthcare | Cat#:27-5330-01 |
| L-glutamine | ThermoFisher Scientific | Cat#:25030149 |
| Linear acrylamide | ThermoFisher Scientific | Cat#:AM9520 |
| Lipofectamine 3000 | ThermoFisher Scientific | Cat#:L3000008 |
| Murine RNAse inhibitor | New England Biolabs | Cat#:M0314 |
| N4-acetylcytidine triphosphate (ac4CTP) | Trilink | Custom synthesis |
| Protease Inhibitor Mini Tablets, EDTA-free | ThermoFisher Scientific | Cat#:32955 |
| Preadenylated universal miRNA cloning linker | New England Biolabs | Cat#:S1315S |
| ProSignal Pico ECL Reagent | Genesee Scientific | Cat#:20-300B |
| Propidium iodide | Roche | Cat#:11348639001 |
| Protein G Magnetic Beads | New England Biolabs | Cat#:S1430S |
| Proteinase K, Molecular Biology Grade | New England Biolabs | Cat#:P8107S |
| RNA Clean & concentrator-5 | Zymo Research | Cat#:R1013 |
| RNA Spike-In mix | ThermoFisher Scientific | Cat#:4456740 |
| RNAse A | Millipore | Cat#:111199115001 |
| RNase I | ThermoFisher Scientific | Cat#:EN0601 |
| Superase-In | ThermoFisher Scientific | Cat#:AM2694 |
| T4 Polynucleotide kinase (PNK) | New England Biolabs | Cat#:M0201 |
| T4 RNA ligase 2 | New England Biolabs | Cat#:M0239S |
| TRIzol Reagent | ThermoFisher Scientific | Cat#:15596026 |
| Trypan blue | Lonza | Cat#:17-942E |
| Turbo™ DNAse I | ThermoFisher Scientific | Cat#:AM2239 |
| Critical Commercial Assays | ||
| human/mouse/rat Ribo-Zero Gold rRNA removal | Illumina | Cat#:MRZG12324 |
| LightCycler 480 SYBR Green I Master | Roche | Cat#:04887352001 |
| Luciferase assay reagent | Promega | Cat#:E1500 |
| MEGAscript T7 Transcription Kit | ThermoFisher Scientific | Cat#:AM1334 |
| Nano-Glo® Luciferase Assay System | Promega | Cat#:N1120 |
| Nano-Glo Dual-Luciferase Reporter System | Promega | Cat#:N1610 |
| NEBNext® Magnesium RNA Fragmentation buffer | New England Biolabs | Cat#:E6150 |
| NEBNext® rRNA Depletion Kit | New England Biolabs | Cat#:E6310L |
| NEBNext® UltraII™ Directional RNA Library Prep Kit | New England Biolabs | Cat#:E7770S |
| NorthernMax kit | ThermoFisher Scientific | Cat#:AM1940 |
| NorthernMax Prehyb/Hyb buffer | ThermoFisher Scientific | Cat#:AM8677 |
| ONE-Glo EX Reagent | Promega | Cat#:E8110 |
| PowerUp SYBR Green Master Mix | ThermoFisher Scientific | Cat#:A25777 |
| ProSignal Pico ECL Reagent | Genesee Scientific | Cat#:20-300B |
| Q5 Hot Start High-Fidelity | New England Biolabs | Cat#:M0493S |
| Rabbit Reticulocyte Lysate | Promega | Cat#:L4960 |
| SuperScript IV First-Stranded Synthesis System | ThermoFisher Scientific | Cat#:18091050 |
| SuperSignal ELISA Femto Maximum Sensitivity Substrate | ThermoFisher Scientific | Cat#:37075 |
| TransIT-mRNA Transfection Kit | Mirus | Cat#:MIR2250 |
| Vaccinia Capping System | New England Biolabs | Cat#:M2080S |
| Deposited Data | ||
| Raw and processed RedaC:T-seq data | This Study | GEO: GSE162043 |
| Raw and processed HR Ribo-seq-seq data | This Study | GEO: GSE162043 |
| Rabbit Reticulocyte RNA-seq | This Study | GEO: GSE162043 |
| Cryo-EM data | This Study | ac4C 80s ribosome: PDB:7UCK and EMDB:EMD-26445 AUG 80s ribosome: PDB:7UCJ and EMDB:EMD-26444 |
| Experimental Models: Cell Lines | ||
| Human: HeLa | ATCC | Cat#:CCL-2; RRID: CVCL_0030 |
| Human: NAT10+/+ | Arango et. al. 2018 | N/A |
| Human: Flp-In Trex 293 | ThermoFisher Scientific | Cat. #:R71007 RRID: CVCL_U427 |
| Human: Flp-In Trex 293 NAT10-FL | Arango et. al. 2018 | N/A |
| Oligonucleotides | ||
| ON-TARGETplus siRNAs against PXN | Dharmacon | Cat.#:L-005163-00-0005 |
| ON-TARGETplus siRNAs against KRT80 | Dharmacon | Cat.#:L-018941-02-0005 |
| ON-TARGETplus siRNAs Control | Dharmacon | Cat.#:D-001810-10 |
| Oligonucleotide sequences used in this study | This Study | Table S1 |
| Recombinant DNA | ||
| pNL1.1PGK | Promega | Cat#:N1441 |
| pcDNA/FRT | ThermoFisher Scientific | Cat#:V6010-20 |
| pcDNA/FRT-NAT10-FL | Arango et al., 2018 | |
| pcDNA/FRT-NAT10-ΔHel | Arango et al., 2018 | |
| Software and Algorithms | ||
| BEDtools | Quinlan, 2014 | RRID:SCR_006646 |
| Bowtie2 v.00.14 | Langmead and Salzberg, 2012 | RRID:SCR_016368 |
| Cutadapt v 2.0 | Martin, 2011 | RRID:SCR_011841 |
| DeepTools | Ramírez et al., 2014 | RRID:SCR_016366 |
| DESeq2 | Love et al., 2014 | RRID:SCR_017673 |
| Fastx_clipper v.0.014 | http://hannonlab.cshl.edu/fastx_toolkit/ | |
| Hisat2 v2.2.1.0 | Kim et al., 2015 | RRID:SCR_015530 |
| HTSeq v 0.6.1p1 | Anders et al., 2015 | RRID:SCR_005514 |
| ImageJ | National Center for Microscopy and Imaging Research | RRID:SCR_001935 |
| ImageLab | BioRad | http://www.biorad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| MACS2 | Zhang et al., 2008 | RRID:SCR_013291 |
| MFOLD v3.6 | Zuker, 2003 | RRID:SCR_008543 |
| Mpileup v 1.10 | Li et al., 2009 | RRID:SCR_002105 |
| PRISM 7 | GraphPad Software, Inc. | RRID:SCR_002798 |
| Relion v 3.0 | Zivanov et al., 2018 | RRID:SCR_016274 |
| Ribo-TISH | Zhang et al., 2017 | https://github.com/zhpn1024/ribotish |
| Rstudio v 3.6 | Rstudio | RRID:SCR_000432 |
| STAR v. 2.5.4.a | Dobin et al., 2013 | RRID:SCR_019993 |
| Tophat2 v.2.1.1 | Trapnell et al., 2009 | RRID:SCR_013035 |
| ViennaRNA tool v 2.4.18 | Lorenz et al., 2011 | https://www.tbi.univie.ac.at/RNA/# |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Parental (wildtype) HeLa cells (Human cervix carcinoma, female) were purchased from ATCC (Cat. #: CCL-2). HeLa cells with CRISPR-Cas9 mediated ablation of NAT10 were previously generated and validated (Arango et al., 2018). Parental Flp-In 293T-REx (Human embryonic kidney, female) were purchased from ThermoFisher Scientific (Cat. #: R71007). Generation and validation of 293T-Rex cells stably overexpressing NAT10 was previously reported (Arango et al., 2018). Wildtype HeLa, NAT10−/− HeLa, Flip-In 293T-REx, and 293T-REx cells overexpressing NAT10 were cultured in Dulbeccos’ Modified Eagle Medium (DMEM, ThermoFisher Scientific, Cat. #: 10313021) containing 25 mM glucose, 1 mM sodium pyruvate and supplemented with 2 mM L-glutamine (ThermoFisher Scientific, Cat. #: 25030149) and 10% bovine calf serum (BCS, HyClone, Cat. #: SH30073.03), in the absence of antibiotics.
METHOD DETAILS
In vitro transcription
Firefly luciferase, NanoLuciferase (NanoLuc), and 5’UTR DNA templates (see Table S1 for oligonucleotide sequence) were in vitro transcribed using the MEGAscript T7 Transcription Kit (ThermoFisher Scientific, Cat. #: AM1334), according to the manufacturer’s instructions. For modified transcripts, ac4CTP (custom nucleotide, Trilink) replaced CTP in the reaction mix. Validation of ac4CTP incorporation in RNA probes was assessed by dot blots.
Generation of C-less 5’-UTRs and C-less 3’UTR in Firefly luciferase mRNAs
Plasmid DNA encoding Firefly luciferase mRNA under the control of the T7 RNA polymerase promoter (pLGENB1) was used as DNA template in PCR reactions. C-less 5’UTR Firefly luciferase DNA templates were generated by high fidelity PCR using Q5 Hot Start High-Fidelity 2X Master Mix (NEB, Cat. #: M0494), and a forward primer specific for the first 20 nt of the Firefly luciferase CDS with an overhang corresponding to the T7 promoter and a 5’UTR lacking cytidines (See Table S1 for oligonucleotide sequence). The reverse primer annealed to the last 20 nucleotides (nt) of the 3 ’UTR.
C-less 3’UTR Firefly luciferase DNA templates were generated by high fidelity PCR using the Q5 Hot Start High-Fidelity 2X Master Mix (NEB, Cat. #: M0494), and a reverse primer annealing to the last 20 nt of the Firefly luciferase CDS with an overhang containing the stop codon and a 20 nt C-less 3’UTR. The forward primer annealed to the T7 promoter and the first 20 nt of the 5’UTR of Firefly luciferase CDS (See Table S1 for oligonucleotide sequence). PCR amplicons were purified using AMPure XP beads (Beckman Coulter, Cat. #:A63881) and used for in vitro transcription.
Splint ligations of NanoLuc mRNAs
Oligonucleotides (see Table S1 for oligonucleotide sequence) containing 5’UTRs and a translation initiation site (TIS) were in vitro transcribed in the presence of CTP or ac4CTP. Fragments were separated in 10% Tris-Borate-EDTA-Urea polyacrylamide gels (TBU-PAGE), visualized by UV shadowing and specific bands were excised using a razor blade. RNA was extracted using gel extraction buffer [200 mM sodium acetate, 1 mM EDTA, 0.25% (w/v) SDS] and isopropanol precipitation. 3’ ends containing NanoLuc CDS and lacking a TIS were in vitro transcribed in the presence of CTP using plasmid pNL1.1PGK (Promega Cat#: N1441) as template (see Table S1 for primer sequence). The resulting NanoLuc CDS RNA was gel-purified from 6% TBU-PAGE gels and dephosphorylated using 0.5 U/μl of Calf Intestinal Alkaline Phosphatase (CIAP, Promega, Cat. #: M2825) and 5’ phosphorylated with T4 Polynucleotide Kinase (PNK, New England Biolabs, NEB, Cat. #: M0201).
For splint ligations of uncapped mRNAs, equimolar amounts (10 pmol) of 5’ends, 3’ends and antisense DNA oligonucleotide (see Table S1 for oligonucleotide sequences) were mixed, denatured at 95°C for 2 min and ramped down to room temperature for annealing. Ligations were performed using RNA ligase 2 (NEB, Cat. #: M0239L) for 4 hr at 37 °C according to the manufacturer’s suggestions. Reactions were treated with Turbo DNase I (ThermoFisher Scientific, 30 min at 37°C) and purified using RNA Clean & Concentrator-5 (Zymo Research).
For splint ligations of capped and polyadenylated mRNAs, gel-purified 5’ fragments were capped using 0.5 mM GTP, 1 mM S-adenosyl-methionine (SAM), and 10 U of the Vaccinia Capping Enzyme, whereas 3’ NanoLuc fragments were polyadenylated using 1 mM ATP and 5 U of E. coli poly(A) polymerase. Fragments were repurified with RNA Clean & Concentrator-5 (Zymo Research) and used in splint ligations with equimolar amounts (10 pmol) of 5’ends, 3’ends and antisense DNA oligonucleotide, as described above.
To verify the efficiency of splint ligations, 30 ng of ligated RNAs were separated by 6% TBU-PAGE and transferred to Amersham Hybond-N+ membranes (GE Healthcare) using 0.5X TBE buffer at 0.3 A overnight. Membranes were rinsed with PBS and crosslinked twice with 120 mJ/cm2 in the UV254nm Stratalinker 2400 (Stratagene). Hybridization was performed with radiolabeled probes specific for the 5’UTR of each construct (see Table S1 for sequence) at 37°C overnight in NorthernMax Prehyb/Hyb buffer (ThermoFisher Scientific, Cat #: AM8677), washed twice in low stringency buffer, followed by exposure to a phosphorimager. Equivalent amounts of splinted RNA were used for in vitro translation assays. For splint ligation related to Figures 1E and S1D, 3’ ends containing NanoLuc CDS were in vitro transcribed in the presence of UTP[α-32P]. RNA purification and splint ligation was performed as described above.
In vitro translation assay
Translation of Firefly mRNA was achieved through incubating 180 ng of acetylated or unmodified mRNA templates with 31.5 μl of rabbit reticulocyte lysate (Promega) in a final volume of 45 μl at 30°C. After specific reaction incubation time points, a 5 μl aliquot was removed, diluted with 95 μl of 1 mg/ml BSA and immediately frozen on dry ice until all time points had been collected. For Firefly luciferase assays, 20 μl of diluted sample was mixed with 80 μl of Luciferase assay reagent (Promega, Cat. #: E1483) in 96-well flat white plates.
In vitro translation of splint ligated NanoLuc mRNAs was performed by incubating equivalent amounts of C- or ac4C-containing mRNA templates (250-350 ng) with 25 μl of rabbit reticulocyte lysate (Promega) in a total volume of 35 μl at 30°C. After specific reaction incubation time points, a 5 μl aliquot was removed, diluted with 95 μl of 1 μg/mL BSA and immediately frozen on dry ice until all time points had been collected. For NanoLuc assays, 10 μl of diluted sample was mixed with 20 μl PBS, 49 μl of NanoGlo buffer and 1 μl of NanoGlo substrate (Promega, Cat. #: N1120) in 96-well flat white plates. Relative light units (RLU) were immediately measured for 1 sec in a luminescence plate reader. Total RNA was obtained from 70 μl aliquots of diluted lysate using the TRIzol Reagent (ThermoFisher Scientific, Cat. #: 15596018). Total RNA was reverse transcribed using the Superscript IV system (ThermoFisher Scientific, Cat. #18091050) and gene specific primers for NanoLuc mRNAs as well as 18S rRNA (Table S1). NanoLuc light units were normalized by mRNA levels.
Transfection of splint ligated RNAs
HeLa cells were seeded the day before transfection at a density of 20,000 cells in 100 μl of complete DMEM medium using 96-well plates. Cells were transfected with 100 ng of capped and polyadenylated splint ligated C- or ac4C-containing NanoLuc mRNA and co-transfected with 100 ng of capped and polyadenylated unmodified Firefly luciferase. Transfection reactions were prepared using the TransIT-mRNA Transfection Kit (Mirus) following the manufacturer’s instructions for 96-well plates. Following addition of the transfection complexes, cells were incubated for 6 hrs at 37°C. For luminescence detection, culture medium was discarded, wells were washed twice with PBS, and cells were lysed in 100 μl 1X lysis Buffer (Promega) containing 40 U of murine RNAse inhibitor (New England Biolabs). Firefly and NanoLuc activities were measured using the Nano-Glo Dual-Luciferase Reporter System and following the manufacturer’s instructions (Promega). Since Firefly luciferase is the invariable mRNA template, it was used to normalize NanoLuc activity.
Dot blots
Dot blots were performed to verify ac4C incorporation and capping of in vitro transcribed mRNAs. Briefly, 30-50 ng RNA was denatured at 65°C for 5 min, immediately placed on ice for 1 min and loaded onto Hybond-N+ membranes. Membranes were crosslinked twice with 150 mJ/cm2 in the UV254nm Stratalinker 2400 (Stratagene), blocked with 5% non-fat milk in 0.1% Tween- 20 PBS (PBST) for 30 min at room temperature, and probed for 1-2 hr with rabbit anti-ac4C (1:1000, clone NCI-184-128, Cat. # ab25215, Abcam) or rabbit anti-m7G (1:1000, Cat. # RN016M, MBL International Corporation) antibodies in 1% non-fat milk at room temperature. Membranes were next washed three times with 0.1% PBST, incubated with HRP-conjugated secondary anti-rabbit IgG in 1% non- fat milk (1:10000 dilution, Cell Signaling Technology) at room temperature for 1 hr, washed three times with 0.1% PBST and developed with the SuperSignal ELISA Femto Maximum Sensitivity Substrate (ThermoFisher Scientific).
NaBH4 treatment and RedaC:T-seq library preparation
Total RNA was purified from wildtype and NAT10−/− HeLa cells using TRIzol Reagent followed by treatment with Turbo™ DNase I. DNase-treated Total RNA (1 μg) was ribodepleted using the NEBNext® rRNA Ribodepletion step (NEB, Cat. #: E7850) following the manufacturer’s instructions. Ribodepleted RNA was treated with 100 mM NaBH4 in a 20 μl volume at 55 °C in the dark for 1 hr. This step reduces ac4C to tetrahydroacetylcytidine and fragments RNA to ~100-200 bp (Thomas et al., 2018). During the incubation time, tube lids were opened every 10 min to release pressure from NaBH4-induced bubbling. NaBH4-treated RNA was precipitated using 0.3M sodium acetate [pH 5.5], 15 μg/ml linear acrylamide (carrier) and 2.5X ethanol. Illumina libraries were constructed from two biological replicates using the NEBNext® UltraII™ Directional RNA Library Prep Kit for Illumina® (NEB, Cat. #: E7760) following manufacturer’s suggestions with the reverse transcription step performed at 48 °C. Libraries were multiplexed on an Illumina HiSeq2500 instrument using TruSeq V4.0 chemistry and sequenced for 126 cycles in paired-end mode. In total, ~200 million mRNA reads were recovered per replicate (Table S1).
acRIP and RT-qPCR
Acetylated RNA Immunoprecipitation (acRIP) was performed as previously described (Arango et al., 2019). Briefly, poly(A) RNA from parental and NAT10−/− HeLa cells was fragmented using the NEBNext Magnesium RNA Fragmentation buffer for 4 min at 94°C. Fragmented poly(A) RNA (4 μg) was immunoprecipitated with 1 μg anti-ac4C (clone EPRNCI-184-128, Cat. #: ab252215, Abcam), or IgG Isotype control (clone DA1E, Cat. #: 3900S, Cell Signaling Technologies) pre-coupled to Protein G Magnetic beads (New England Biolabs) and immunoprecipitated for 4 hrs at 4°C in 100 μl of acRIP buffer containing PBS, 0.05% Triton X-100, 0.1% BSA, and 40U murine RNase inhibitor (New England Biolabs). After immunoprecipitations, beads were washed five times in acRIP buffer and elution of RNA was carried out by RNase-free Proteinase K (2 U, New England Biolabs) digestion in 100 μl buffer containing 50 mM Tris-HCl [pH7.5], 75 mM NaCl, 6.25 mM EDTA and 1% SDS for 1hr at 37°C. RNA was extracted by Phenol:Chloroform [pH 4.5] and ethanol precipitation using 0.3M sodium acetate [pH 5.5] and 15 μg/ml linear acrylamide. Immunoprecipitated RNA was reverse transcribed using random hexamers and the Superscript IV system following the manufacturer’s suggestions (ThermoFisher Scientific) and quantified through qPCR using Applied Biosystems PowerUp SYBR Green Master Mix (ThermoFisher Scientific) in a QuantStudio 3 Instrument (ThermoFisher Scientific). Data are represented as percentage of input. The primers used for RT-qPCR primers are described in Supplementary Table S1.
HR Ribo-seq
Wildtype and NAT10−/− HeLa cells (2 x 106 cells) were seeded in 10 cm tissue culture dishes in 10 ml of complete DMEM medium and grown for 48 hr at 37°C, 5% CO2. Harringtonine (HR, Abcam Cat. #: ab141941, dissolved at 2 mg/ml in DMSO) was added to the medium at a final concentration of 2 μg/ml. Cells were incubated for 10 min at 37°C, 5% CO2 in HR containing medium. Control cells were incubated with the same amount of DMSO for 10 min at 37°C, 5% CO2. After each incubation period, CHX was added to the medium at a final concentration of 100 μg/ml, culture medium was immediately aspirated, plates were placed on top of liquid nitrogen and transferred to dry ice. Lysis buffer (500 μl, 10 mM Tris-HCl [pH 7.4], 5 mM MgCl2, 100 mM KCl, 1% Triton X-100, 2 mM DTT, 100 μg/ml CHX) was added to plates and cells were scraped into lysis buffer. Cell lysates were passed through a 26-G needle 10 times and clarified by spinning at 20,000 x g for 10 min at 4 °C. OD260 was measured in the Nanodrop-1000 and ~250 μl of lysates were loaded on a 10%–45% sucrose gradient, previously prepared in gradient buffer (100 mM Tris-HCl [pH 7.5], 75 mM KCl, 1.5 mM MgCl2) using a BioComp Gradient Master (Short Run, Sucr 10-45% SW41, wt15t). Gradients were spun at 37,000 rpm in a SW 41 Ti rotor for 2 hr at 4°C. Absorbance at 260 nm was recorded using a BioComp Fractionation System and a Triax Flow Cell model FC-1 for 260 nm scans. 450 μl of each lysate was treated with 0.3 U/μl RNase I for 40 min at room temperature, while the remaining lysate was flash frozen in liquid nitrogen for later total RNA isolation. Following RNase I digestion (ThermoFisher Scientific, Cat. #: EN0601), 5 μl of Superase-In (ThermoFisher Scientific, Cat. #: AM2694) was added to each sample, and OD260 was measured in the Nanodrop 1000. ~300 μl of lysates was loaded on a 10%–45% sucrose gradient previously prepared in gradient buffer (100 mM Tris-HCl [pH 7.5], 75 mM KCl, 1.5 mM MgCl2) using a BioComp Gradient Master (Short Run, Sucr 10-45% SW41, wt15t). Gradients were spun at 37,000 rpm in a SW 41 Ti rotor for 2 hr at 4°C. Fractions were collected using a BioComp Density Gradient Fractionation system. Ribosome footprinted RNA was precipitated from fractions containing 80S monosomes using 2 volumes of 100% ethanol followed by extraction using TRIzol Reagent. Ribosomal RNA was depleted with the human/mouse/rat Ribo-Zero Gold rRNA removal kit (Illumina).
Input RNA was obtained from 130 μl aliquots of lysate (without RNase I digestion) using TRIzol Reagent. Total RNA was treated with Turbo DNase I (ThermoFisher Scientific, 30 min at 37°C). RNA Spike-In mix (ThermoFisher Scientific, Cat. #: 4456740) was added to 5 γg of DNase I-treated total RNA according to the manufacturer’s instructions, and ribosomal RNA was depleted using the human/mouse/rat Ribo-Zero Gold rRNA removal kit. Next, total RNA samples were fragmented in alkaline fragmentation buffer (1 mM EDTA, 50 mM Na2CO3, 50 mM NaHCO3, [pH 9.2] for 40 min at 95°C, as we described previously (Arango et al., 2018). Fragments between 26 and 34 nucleotides in size were gel purified from input and footprinted RNA samples and used for library preparation as described previously (Arango et al., 2018). Eight samples were multiplexed and sequenced on an Illumina NextSeq instrument for 75 cycles in single-end mode (Table S1).
Western blot
Cells were seeded at a density of 1 x 105 cells/ml in 6-well plates. After 48 hrs, cells were rinsed with 1 ml PBS once, detached with 0.05% trypsin for 5 min at 37 °C and pelleted by centrifugation at 3,000 rpm for 5 min at 4 °C. Cells were rinsed with cold PBS and lysed in NP40 lysis buffer containing 0.1 % (v/v) NP40, 150 NaCl, 2 mM EDTA, 1 mM NaF, 0.5 mM fresh DTT and 1% Halt™ protease inhibitor cocktail (ThermoFisher Scientific, Cat. #: 78430), followed by Bioruptor sonication for three cycles of 30 sec on/off at low setting (Bioruptor Twin, Diagenode). For the blots shown in Figure S2I, cells were lysed in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% Sodium deoxycholate, 0.1% SDS, 50 mM TRIS (pH 7.4) and 1% Halt™ protease inhibitor cocktail). Cell lysates were cleared by centrifugation at 13,000 rpm for 5 min at 4 °C and protein concentration was quantified using the Bradford reagent (BioRad, Cat. #: 5000201). Equal amounts of protein (25 μg) were loaded on 4-12% Bis-Tris gels, separated using NuPAGE MOPS SDS running buffer (ThermoFisher Scientific, Cat. #: NP0001) and transferred onto 0.2 μm nitrocellulose membranes using Tris-Glycine buffer (250 mM Tris [pH 6.8], 1.92 M Glycine, 20% methanol) for 75 min at 0.25 A.
Membranes were blocked with 5% milk in 0.05% Tween-20 PBS buffer and incubated in a solution containing 1% milk in 0.05% Tween-20 PBS buffer with primary antibodies as follows: rabbit anti-IRF1 (1:2000 dilution, Cat. #: ab191032, Abcam), rabbit anti-KDM4B (1:2000 dilution, clone D7E6, Cat. #: 8639S, Cell Signaling Technologies), rabbit anti-PXN (1:2000 dilution, Cat. #: 10029-1-Ig, ProteinTech), rabbit anti-KRT80 (1:2000 dilution, Cat. #: 16835-1-AP, ProteinTech), rabbit anti-THUMPD1 (1:2500, Cat. #: A304-643AT), rabbit anti-LMNA (1:2500, Cat. #: A303-433A, Bethyl Laboratories Inc.), and mouse anti-GAPDH (1:2500 dilution, clone 6C5, Cat. #: sc-32233, Santa Cruz Biotechnology). After overnight incubation at 4 °C, membranes were washed 3 times in 0.05% Tween-20 PBS buffer, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies; anti-mouse IgG (1:5000 dilution, GE Healthcare, Cat. #: NA931), or anti-rabbit IgG (1:10000 dilution, Cell Signaling Technology, Cat. #: 7074). Western blots were visualized by enhanced chemiluminescence using the ProSignal Pico ECL Reagent (Genesee Scientific, Cat. #: 20-300B) or the ECLWestern Blotting Substrate (Promega). Chemiluminescence was detected using the ChemiDoc Imaging System (BioRad) or the LiCOR Odyssey Fc (LiCOR).
Subcellular Fractionation
Cells were seeded at a density of 2.5 x 105 cells/ml in 10-cm dishes. After 48 hrs of culture, cells were rinsed with 5 ml PBS once, detached with 0.05% trypsin for 5 min at 37 °C and pelleted by centrifugation at 330 x g for 5 min at 4 °C. Pellets were resuspended in 1 ml of cold PBS, and centrifuged again at 720 x g in a microcentrifuge for 5 min at 4 °C. Pellets were resuspended in 500 μl cold hypotonic buffer containing 10 mM Hepes, pH 7.9, 1.5 mM MgCl2, 10 mM KC1, 0.5 mM DTT, and 1X protease inhibitor EDTA-free cocktail (Cat. #: PIA32965, ThermoFisher Scientific). Cells were homogenized by 20 strokes using a 2 ml Dounce homogenizer. Lysates were centrifuged at 720 x g in a microcentrifuge for 5 min at 4 °C. Pellets were kept for nuclei extractions while supernatants were transferred to a new 1.5 ml microcentrifuge tube and used for cytoplasm collection. Cytoplasmic fraction was then centrifuged at 20,000 x g in a microcentrifuge for 5 min at 4 °C. Supernatants were transferred to a new 1.5 ml tube and protein concentration in the cytoplasm fraction was quantified using the Bradford reagent. To extract nuclei, pellets were resuspended in 300 μl of cold S1 buffer containing 0.25M sucrose, 10 mM MgCl2, and 1X protease inhibitor EDTA-free cocktail and layered on top of 300 μl of S2 buffer containing 0.35M sucrose, 0.5 mM MgCl2, and 1X protease inhibitor EDTA-free cocktail. Sucrose cushion was centrifuged at 2,160 x g in a microcentrifuge for 5 min at 4 °C. After discarding supernatants, pellets were washed with 500 μl cold PBS and resuspended in 100 μl of cold lysis buffer containing 10 mM Tris-HCl pH 7.5, 1 mM EDTA, 1 mM MgCl2, 150 mM NaCl, 1% Triton-X, and 1X protease inhibitor EDTA-free cocktail. Nuclei were then sonicated in the BioRuptor (Diagenode) with 5 pulses of 30 sec on/off in high mode. Nuclear lysates were quantified using the Bradford reagent. Cytoplasmic and Nuclear fractions were analyzed by Western blot (see section Western Blots).
Cell cycle analysis
Cells were treated with 2 mM thymidine (Sigma-Aldrich) for 15 hr (first blockage), rinsed twice with PBS and incubated in complete medium for 9 hr before treating with 2 mM thymidine for additional 15 hr (second blockage). Cells were rinsed twice in PBS and incubated in complete medium for 0, 6, and 12 hrs before harvesting by trypsinization. Cells were washed with PBS prior to fixation in 70% ethanol, followed by two additional PBS washes. Fixed cells were next stained with propidium iodide (50 μg/ml; Roche) containing 0.2 mg/ml DNase-free RNase (Sigma-Millipore) for 30 min at room temperature and immediately analyzed by a BD FACSCalibur using the BD Cell Quest Pro software (BD biosciences).
Scratch assay
Wildtype and NAT10−/− HeLa cells were cultured to confluency in 12-well plates (three well per cell lines) in 10% BCS DMEM medium. A scratch was applied with a 200 μl tip and the medium was changed to 0.5% BCS DMEM. Three images per well were recorded at time 0 and again 24 hrs after the scratch. The area of the scratch was measured using ImageJ in three images per well at time 0 and again at 24 hrs and the relative area was the average of all the images in three biological replicates.
To evaluate the role of PXN and KRT80 in NAT10-dependent wound healing, cells were seeded at a density of 2 x 105 cell/ml in 6-well plates and cultured for 24 hr. Cells were transfected using 9 μl Lipofectamine RNAiMax and 20 nM of Dharmacon ON-TARGETplus siRNAs against PXN (Cat. #: L-005163-00-0005), KRT80 (Cat. #: L-018941-02-0005), or a scramble control (Cat. #: D-001810-10). Seventy-two hours after transfection, cells were re-transfected with siRNAs, cultured for 24 hrs, harvested and seeded for the scratch assay as described above. Efficiency of silencing was verified by Western blot as described in the section Western blots.
NAT10 Reconstitutions
Wildtype and NAT10−/− HeLa cells were cultured in 6-well plates in 10% BCS DMEM medium and transfected with 2 μg of vector control plasmid, plasmid encoding full-length NAT10 (NAT10-FL), or plasmid encoding NAT10 lacking the RNA helicase domain (Δhel) using the Lipofectamine 3000 reagent (ThermoFisher Scientific). We previously reported the generation of pcDNA/FRT-NAT10-FL and pcDNA/FRT-NAT10-Δhel plasmids (Arango et al., 2018). Seventy-two hours after transfections, cells were counted under the light microscope using Trypan blue exclusion as a viability marker. Expression of NAT10-FL and NAT10-Δhel was verified by Western blot and RT-qPCR. For the latter, total RNA was reverse transcribed using random hexamers and the Superscript IV system following the manufacturer’s suggestions (ThermoFisher Scientific). qPCR was performed using primers against NAT10 and GAPDH for normalization purposes (see Table S1 for primer sequences). qPCR was run using the Applied Biosystems PowerUp SYBR Green Master Mix in a QuantStudio 3 Instrument (ThermoFisher Scientific).
Sample preparation for Cryo-EM
Synthetic mRNAs were designed to contain a single cytidine preceding a consensus AUG start codon (see Table S1 for RNA sequences). C- or ac4C-containing mRNAs were generated by in vitro transcription in the presence of CTP or ac4CTP. mRNAs were separated in 10% Tris-Borate-EDTA-Urea polyacrylamide (TBU-PAGE) gels and visualized by UV shadowing. Specific bands were excised using a razor blade, and RNA was extracted using gel extraction buffer [200 mM sodium acetate, 1 mM EDTA, 0.25% (w/v) SDS] and isopropanol precipitation. Purified RNAs were capped in vitro using 0.5 mM GTP, 1 mM S-adenosyl-methionine (SAM), and 10 El of the Vaccinia Capping Enzyme (New England Biolabs). Capped mRNAs were repurified using the RNA Clean & Concentrator-5 system (Zymo Research). Acetylation and capping efficiencies were verified by dot blot (see the section Dot blots).
To capture 80S translation initiation complexes, 140 μl of reticulocyte lysates was preincubated with 20 ng/μl Harringtonine for 10 min at 30°C, followed by addition of 800 ng mRNA (pre-heated at 65°C for 2 min) in a total volume of 200 μl. Translation reactions were then incubated for 10 min at 30°C. To stop the reactions, tubes were placed on ice, followed by addition of 800 μl of cold resuspension buffer (50 mM Hepes-KOH, pH 7.4-7.6, 100 mM KOAc, 5 mM Mg(OAc)2, 2 ng/μl Harringtonine, 2 mM DTT). 1 ml solutions containing the initiation complexes were pipetted onto a sucrose cushion (1.5 ml 15% sucrose on top of 0.5 ml 25% sucrose) and pelleted by high-speed centrifugation at 100,000 rpm (TLA 100.3 rotor) for 1 hr at 4 °C. Supernatants were completely removed, and pellets were resuspended in 100 μl of cold resuspension buffer. Absorbance at 260 nm was measured in the Nanodrop and ~7-10 Abs units were used for cryo-EM analysis.
To confirm the presence of the synthetic mRNA in the initiation complexes, total RNA was isolated from resuspended ribosome pellets with TRIzol Reagent and subjected to RT-qPCR analysis and deep-sequencing. For RT-qPCR, RNA was reverse transcribed using random hexamers and the Superscript IV reverse transcriptase system, following the manufacturer’s suggestions (ThermoFisher Scientific). cDNA was analyzed by qPCR using LightCycler® 480 SYBR Green I Master mix and primers that were specific to either the synthetic mRNA, HBB (beta-globin) mRNA, HBA (alpha-globin) mRNA, or 18S rRNA. The list of primers is described in Table S1. qPCR was performed in a Roche LightCycler® 96 Instrument. To further accurately identify all mRNA types present in the ribosome pellets, ribodepleted RNA isolated from purified initiation complexes was also sequenced in a MiSeq instrument in paired-end mode at 75 bp read lengths.
Negatively stained grid preparation
Negatively stained EM grids were prepared from the purified samples at a concentration of 0.08 mg/μl. 3 μl of diluted sample was applied to carbon-coated 200 Cu mesh grid (Electron Microscopy Sciences, Inc.) with pretreated glow discharge at 15mA for 15 seconds by a Pelco easiGlow Glow discharge cleaning system (Ted Pella, Inc.) and left as such for 20 seconds. 3 μl of 0.7% (w/v) uranyl formate was added for 5 seconds followed by blotting using Whatman No. 1 qualitative filter paper. This staining process was repeated another 4 times. Samples were imaged on Talso 120C TEM (ThermoFisher Scientific) that operated in 120 kV integrated with 4k x 4k CCD. Micrographs were collected using SerialEM software (Mastronarde, 2005) with magnification of 36,000 x that resulted in a pixel size of 2.94 Å at the specimen plane.
Cryo-EM sample preparation and imaging
To gain structural insight, cryo-EM analysis was performed using purified 80S initiation complexes assembled on C(−1)AUG or ac4C(−1)AUG mRNAs. Frozen hydrated grids were prepared by vitrification using Vitrobot Mark IV (ThermoFisher Scientific) operated at 4°C and 98% relative humidity. 3 ml of 0.4 mg/ml sample was applied to Quantifoil R 1.2/1.3 Cu200 mesh grids (Quantifoil Micro Tools GmbH, Jena, Germany) which were previously glow discharged at 15 mA for 5 seconds using a PELCO easiGlow Glow Discharge Cleaning System (Ted Pella, Inc., Redding, CA). After a wait time of 5 seconds, grids were blotted for 2 seconds with blot force 0 and plunged into liquid ethane. Vitrified grids were preserved under liquid nitrogen until imaging. Images were collected from frozen hydrated samples at liquid nitrogen temperature using a Titan Krios TEM operated at 300 kV and equipped with a Gatan K2 direct electron detector (Gatan, Inc., Pleasanton, CA). Images were acquired in counting mode using SerialEM software (Mastronarde, 2005) at a nominal defocus of −2 to −3 μm and a nominal magnification of 18,000x, resulting in a pixel size of 1.358 Å/pixel. Images were collected by image shift 3x3 (multi-shot) as dose-fractionated movies of 40 frames with exposure time of 9.2 second and a total dose of 50 e−/Å2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Codon optimality around TIS
Codon optimality was examined using the Codon Stability Coefficient (CSC), computed for each codon through the correlation coefficient of the mRNA half-life and the proportion of the given codon (Presnyak et al., 2015). HeLa CSC values were obtained from Forrest et al., 2020 (Forrest et al., 2020). Gene subsets were generated based on ac4C summit occurrence in 80 codon intervals from the annotated translation initiation site. In each interval, mean CSC values were calculated for each gene for the codons within the respective window. Intervals corresponding to the beginning, middle, and end of coding sequences were compared. The intervals were compared to mean CSC values for ac4C(−) genes calculated the same way.
RedaC:T-seq analysis
Raw reads were pre-processed to remove low quality bases and adapter sequences using cutadapt (Martin, 2011). We aligned to the human genome (hg19) using STAR (v 2.5.4.a (Dobin et al., 2013)) in local alignment mode, allowing only five mismatches per read. Specific relevant parameters included: [ --outFilterMultimapNmax 10 --outFilterMultimapScoreRange 1 -- outFilterScoreMin 10 --outFilterMismatchNmax 5 --clip5pNbases 6 --clip3pNbases 6 -- alignEndsType Local ]. During alignment, RedaC:T-seq reads were trimmed of the initial six nucleotides to remove artifacts related to random hexamers annealing. This step was performed because we noted an increased mutational frequency adjacent to C in the first 6 nt, suggesting that hexamers may bind at inappropriate locations. The priming sequence would thus lead to artifactual miscalls at locations that were not technically a component of the cDNA. Separate alignments to 18S rRNA, 28S rRNA and tRNA sequences were performed to specifically analyze reads originating from these features. To maximize sequencing depth for the untreated wildtype HeLa comparison, untreated Total RNA-seq samples were pooled (Table S1, untreated RNA-seq), yielding depth exceeding that of the treated samples. The identity of each nucleotide at each genomic position was extracted using samtools mpileup on pooled replicate alignment files, including a minimum base quality -Q 20, the BAQ (Base Alignment Quality) and supplying a -f indexed reference fasta file (Li et al., 2009). Low quality bases, secondary alignments, and reads with more than five mismatches were excluded from the analysis. Specific relevant parameters included: [-A -R -Q20 -C0 -d 100000 --ff UNMAP,SECONDARY,QCFAIL,DUP ]. Results were then tabulated using the mpileup2readcounts script (https://github.com/IARCbioinfo/mpileup2readcounts). The specific command of this script used was [mpileup2readcounts 0 −5 true 0 0 ]. As control, we queried RedaC:T-seq for mismatches in 18S rRNA and observed a 0.25 C:T mismatch rate in C:1842 (100% stoichiometry) and 0.15 C:T mismatch rate in C:1337 (~80% stoichiometry in HeLa) with a significant reduction of mismatches in NAT10−/− cells. To identify NaBH4-sensitive sites, we applied the following filtering parameters: a coverage depth ≥ 10 reads: at least four reads containing mismatched T; > 90% decrease in mismatches in untreated wildtype HeLa compared to NaBH4 treatment; a mismatch rate higher than 0.0125, which corresponds to ~5% relative stoichiometry compared to 100% in 18S rRNA C:1842; a significant decrease in mismatches in NAT10−/− treated with NaBH4 compared to wildtype HeLa treated with NaBH4 (Fisher’s exact test p < 0.05) with a False Discovery Rate (FDR) adjustment with the Benjamini-Hochberg approach of padj < 0.05; and at least five-fold greater mismatch rate in wildtype over NAT10−/−. C:T mismatches that passed the above criteria were classified as ac4C sites (Table S2). To assess position-specific biases of C:T mismatches within sequencing reads, we extracted the position within the read of these mismatches at called ac4C positions. This was done using the jvarkit utilities (https://github.com/lindenb/jvarkit). ac4C:C was estimated by comparing total detected C>T mismatches to the sequencing depth at reference C positions. Only reference C positions assayed with at least 10x depth, and unambiguously assigned to transcripts, were used. This yields and estimate of (304,772/1,910,594,167) = 0.016% (Table S2).
Comparison to ac4C-seq
We compared detection of C:T mismatches in RedaC:T-seq to published data from the ac4C-seq protocol (Sas-Chen et al., 2020). Given the lower number of raw reads and shorter read lengths in the ac4C-seq dataset (Table S1), we processed these data with a depth-preserving and sensitive alignment approach. Raw reads were aligned to the hg19 genome using hisat2 (v2.1.0, (Kim et al., 2015), with default alignment parameters and supplying splice site locations from Ensembl release 75. Coverage and mismatch comparisons to RedaC:T-seq were performed on these alignment files, rather than on stringently filtered post-alignment summaries, extracting coverage at relevant locations using Bedtools (Quinlan, 2014). To examine for potential RT effects or other local coverage biases, RedaC:T-seq coverage values were divided by the coverage sum within a 40 nt window, and the median was calculated across sites. For analysis of statistical power to detect mismatch rate differences, we used the power.fisher.test function (v. 1.4.36) in the statmod package (Gordon Smyth, https://CRAN.R-proiect.org/package=statmod) supplying mismatch rates observed in RedaC:T-seq HeLa WT and NAT10−/− samples, and coverage in either RedaC:T-Seq or ac4C-seq. Given the power estimation in a value from 0 to 1, we characterize a value >= 0.50 as sufficient power, or having enough coverage to have at least a 50% chance of detecting significance at alpha=0.05.
mRNA half-lives and translation efficiency (TE)
Data for mRNA half-lives and TE are taken from Arango et al., 2018. The latter measure is derived from CHX Ribo-seq. CHX traps ribosomes in the process of elongation. Subsequent RNase I footprinting and sucrose density centrifugation allows for isolation and sequencing of ribosome-protected mRNA fragments (RPFs) (Ingolia et al., 2009). In this manner, TE can be gauged through RPF relative to overall mRNA abundance (Ingolia et al., 2009).
Gene definitions based on ac4C position
In cases where it was necessary to specifically interrogate the functional consequences of ac4C by gene feature (e.g., translation efficiency (TE) and mRNA half-lives), we generated sets of genes with distinct ac4C locations by feature exclusivity. Otherwise, genes were categorized as ac4C plus in the 5’UTR or in the CDS (Table S2) nonexclusively based on RedaC:T-seq locations. The UCSC canonical gene annotation (hg19 assembly) was used to define all regions. Distributions of translation efficiency (TE) and mRNA half-life are plotted as boxplots using the R statistical environment. These data were extracted from (Arango et al., 2018) and grouped by our acetylation location and frequency.
HR Ribo-seq Analysis
To remove low quality sequence, primer, and adapter, reads were trimmed using the fastx_clipper command in the fastx toolkit (v 0.0.14) (http://hannonlab.cshl.edu/fastx_toolkit/). After trimming, reads were stringently filtered for the presence of ribosomal RNA using a two-step process. First, we performed local alignments to the ribosomal repeating subunit (gb|U13369.1) in very sensitive mode using bowtie2 (v.2.3.5.1) (Langmead and Salzberg, 2012). Second, we mapped remaining reads using Hisat2 (v2.2.1.0) (Kim et al., 2015) , explicitly allowing for mismatches at the first nucleotide position. Trimmed and filtered reads were then aligned to a reference composed of canonical transcript sequence from the UCSC genome browser in the hg19 assembly, using bowtie2 (v.2.3.5.1) (Langmead and Salzberg, 2012). With the above routine, we successfully explain >93% of reads from ribosomal protected fragments in Harringtonine treated Ribo-seq.
Normalized (RPKM) coverage tracks in bigwig format were generated from Ribo-seq alignments using the DeepTools bamCoverage command (Ramírez et al., 2014). Density summaries presented are from pooled replicates. Metagenes (representing signal across genes in differently sized UTR and CDS in a common range) were generated for Ribo-seq data with the computeMatrix function within deeptools (Lauria et al., 2018), as previously described (Arango et al., 2018). To compare read densities over translation start and stop codons without compressing differently sized regions, we used computeMatrix with the reference-point option. Matrices are computed +/− 600bp from the reference point, with N/As reported where the feature does not extend to that range. From computed matrices, we normalized the data in one of two ways: to compare between genes in these and other plots, we normalized Ribo-seq read densities to mRNA abundance in Inputs, as described in (Arango et al., 2018); to compare the relative location in features within the same gene, we normalized local density to the sum of density values within the computed matrix. Read densities are displayed in heatmaps using the pheatmap function (https://CRAN.R-proiect.org/package=pheatmap).
We assessed the general redistribution of initiating ribosome density by adapting the Jensen-Shannon Divergence (JSD) metric to Ribo-seq density profiles. To assess for initiating ribosome density differences between HeLa WT and NAT10−/− cells, we compared within binned non-normalized HR Ribo-seq density profiles in upstream regions including the aTIS. We converted ribosome densities to probability distributions and compared HeLa WT to NAT10−/− using the JSD function from the philentropy Bioconductor package (Drost, 2018). To make the comparisons more equal within gene sets, we report results for gene sets that have equivalent minimum read densities in the upstream plus aTIS window.
To assess the effect of acetylation by precise position relative to the TIS, we defined regions surrounding the TIS from the most frequently observed HR Ribo-seq RPF length and 5’ offset. RedaC:T-seq ac4C sites within this range of the TIS (12 nt upstream to 19 nt downstream) were considered overlapping the RPF context of the initiating ribosome and examined in detail. Changes in TIS use were assayed by quantifying the overlapping HR Ribo-seq reads within this 31 nt range surrounding the TIS and comparing to total HR Ribo-seq reads per transcript, for wildtype and NAT10−/− HeLa conditions. Changes in initiation for ac4C within the Kozak sequence range as related to relevant tRNA or ribosomal contacts are also made. Where indicated, these comparisons are made on initiation sites where HR Ribo-seq is robustly detected (major TIS).
Rabbit reticulocyte lysate RNA-Seq
We quantified transcript abundance in rabbit reticulocyte RNA-Seq data using Kallisto (v.0.48) (Bray et al., 2016). Refseq transcript annotations and sequence were downloaded from the UCSC browser (oryCun2 assembly) and supplied to the kallisto index command. For each sample, transcripts were then quantified in units of transcripts per million (TPM) using kallisto quant and results were compiled at the gene level using DESeq2/tximport (Soneson et al., 2015). To filter for mRNA TPMs, we selected RefSeq identifiers without the non-coding “NR_” accession prefix.
P-site analysis
We used a multi-step process to identify initiation sites at the codon level from HR Ribo-seq data (Figure S4A). First-pass P-site locations were called from Ribo-seq alignment files with the RiboWaltz package (Lauria et al., 2018). This tool defines P-site offsets for a given read length based on the position of annotated TIS sites within reads of the same length, applying this offset position to all reads. We adjusted P-site positions to proximal AUG or CUG, when the initial predictions were mapped to non-AUG/CUG codons, within a range of 3bp. Next, we resolved transcript regions with multiple cognate / near cognate initiation sites with an empirical probability metric. Clusters of proximal P-sites were identified with Bedtools, and for each site, a probability score was generated as the number of reads supporting each site times the frequency of the codon in first-pass P-site locations. In each cluster, the top-scoring site was selected to represent the cluster, and its abundance was recalculated from alignment coverage.
Ribo-TISH
For an orthogonal analysis to confirm the association between ac4C and upstream initiation, we used the Ribo-TISH tool (Zhang et al., 2017). To use this tool, we re-aligned trimmed Ribo-seq reads using STAR with parameters specified in the manual (aligning in end-to-end mode, and reporting required flags using --outSAMattributes All). We inspected quality and generated offset parameter files using the ribotish quality command, using a fragment length range parameter of 24,38 and the –t option for initiation-enriched data (“TI-Seq”). Next, we used the ribotish predict command, which uses a negative-binomial model to fit TI-Seq background, to report initiation sites. For this command, we specified –-harr for Harringtonine treated data, and –alt to report near-cognate initiation codons. Finally, we used the ribotish tisdiff command to test for differential TIS activity between WT HeLa and NAT10−/−, from the preceding tisdiff results. This test was performed in both differential TIS activity mode, and differential TIS efficiency mode (supplying RNA counts from untreated control samples). Results are in Table S3.
Kozak sequence analysis
Translation start sequence contexts were assessed relative to canonical start sites using the Kozak motif (Kozak, 1987). To generate a quantitative measure of Kozak motif, we adapted the scoring system of the preTIS program (Reuter et al., 2016). Sequence surrounding each detected TIS was extracted, and scored according the sequence content in the −3 and +4 positions (Grant et al., 2011). Specifically, each P-site and TIS is identified as strong Kozak if it has A or G at −3 and G at +4 (RNNAUGG). Visualization of Kozak sequence patterns was accomplished through sequence logos. The relevant sequence contexts were extracted and position rate matrices (PWMs) of nucleotide frequency by position were calculated. Logos were drawn in R with the seqLogo package v1.60 (https://bioconductor.org/packages/release/bioc/html/seqLogo.html). Logos depicting enrichment/depletion of sequence context were made with the Logolas package (Dey et al., 2018).
Functional annotation analysis
Inference of functionality of gene subsets was performed by analysis of annotations via the Enrichr web server (Chen et al., 2013), and using the Molecular Signatures Datasets (Table S2).
Computational mRNA structure prediction
To analyze whether ac4C locations resided within RNA structures, we overlayed RedaC:T-defined ac4C sites with computational mRNA structure predictions. Fasta sequence of transcript 5’UTRs was extracted, including an additional 20bp of sequence downstream of the annotated translation start. We generated structure plots using MFOLD v3.6 (Zuker, 2003), selecting the highest probability structure reported, and forcing local structure formation (parameters NA=RNA MAX=1 MAXBP=40). To identify locations within stem loops, we employed the Dot-Bracket notation of the ViennaRNA tool v 2.4.18 to parse structure by location (Lorenz et al., 2011). Local structure visualizations were performed by running MFOLD on only relevant sequence regions of interest.
Structure determination
To determine 80S initiating ribosome structures, 24,970 particles were extracted from 100 negative staining micrographs to generate initial 2D classes and initial 3D models in Relion3.0 (Zivanov et al., 2018). The best 12 2D class averages from the negative staining dataset were used as template to guide particle picking by Gautomatch (https://www2.mrc-lmb.cam.ac.uk/download/gautomatch-056/). This generated 1,163k raw particles from the ac4C(−1)AUG (acetylated) dataset and 756K raw particles from the C(−1)AUG (unmodified) dataset for follow-up processing in Relion3.0. Particles were extracted with box size of 480 and rescaled to 240 for multiple rounds of 2D classifications and supervised 3D classifications with the reference of the 3D initial model generated previously from the negative staining dataset. After removing ill-defined particles and contaminants by extensive 2D and 3D classifications, 456k ac4C(−1)AUG and 462K C(−1)AUG well-defined ribosome particles were left for 3D refinement with the reference from the best class in previous 3D classifications. Particles were then re-extracted and rescaled to bin1 from the run data of the 3D refinement. Supervised 3D classifications without alignment were performed to distinguish initiating ribosomes from other unwanted conformations (Figure S7D). 178K ac4C(−1) and 160K C(−1) particles harboring initiation complexes were selected for refinement which generated maps below 4 angstroms (unfiltered) with weak densities on the P-site tRNA. Particle subtractions were then performed on these particles with mask keeping signals for P-site tRNA and densities surrounding mRNA region. Focused 3D classifications without alignment were performed to enrich particles with the best P-site tRNA densities. 108K ac4C(−1) and 53K C(−1) particles were selected for final 3D refinement. Iterative rounds of CTF refinements and particle polishing were performed to generate overall maps of C(−1) initiating ribosomes at a resolution of 3.1 Å and ac4C(−1)AUG at 2.8 Å (Figure S7D). Focused local refinement centered on the mRNA-tRNA interface was then applied to further improve the quality of the interested region for the ac4C(−1) dataset (Figure S7E). Multibody refinement was performed on ac4C(−1) to improve other regions to Nyquist. Resolution and map quality was estimated by gold standard FSC (Fourier shell correlation) curves and local resolution in Relion 3.0 (Figure S7E).
Model building and structure refinement:
Major rRNA and proteins PDB from 6MTB (Brown et al., 2018) were fitted into the maps of either C(−1)AUG or ac4C(−1)AUG rabbit 80S initiation complexes with mRNA mutated to our designed sequence (see Table S1 for sequence) and P-site tRNA mutated to 6YAN (Simonetti et al., 2020) in Coot (Emsley and Cowtan, 2004). The conformation of t6A(37) in the ac4C(−1) AUG dataset was referenced from a solution NMR structure (PDB: 1FEQ) (Stuart et al., 2000). ac4C(−1) mRNA conformations were referenced from the previously published cryo-EM structures (PDB ID: 6SKF) (Sas-Chen et al., 2020). Inference on A- and E-sites occupancy used the following structures: (PDB ID: 6O9J vs. 6O9K and 7R81) (Kaledhonkar et al., 2019; Shen et al., 2021). Structures were manually refined in Coot and automatically refined and validated in Phenix by real space refinement (Table S3) (Afonine et al., 2012).
Supplementary Material
Table S1: Compiled information on experiments and data generated. Sequencing experiments summary, sequencing depth details, and list of oligonucleotides used in this study. Related to Figures 1, 2, 3, 5, and 7.
Table S2: RedaC:T-Seq data and results. Contains candidate variants (all mismatch types), RedaC:T-seq determined ac4C locations, summary ac4C information compiled by gene, and functional annotation associated with acetylated transcripts. Related to Figures 2, 3, 4, and 6.
Table S3: Compiled information on Ribo-seq and initiation analysis. Contains all detected P-sites, compiled information about translation initiation sites (TIS) and proximity to ac4C, compiled translation initiation results from Ribo-TISH, transcript abundances, rabbit reticulocyte lysates, and refinement and validation statistics for the two local structures. Related to Figures 3, 4, 6, and 7.
Video S1: Rotation of the codon:anticodon interface for the C(−1) mRNA. Related to Figure 7C.
Video S2: Rotation of the codon:anticodon interface for the ac4C(−1) mRNA. Related to Figure 7C.
Video S3: Rotation of the t6A:C(−1) interface. Related to Figure 7E.
Video S4: Rotation of the t6A:ac4C(−1) interface. Related to Figure 7E.
Highlights.
mRNA acetylation impacts translation in a position-dependent manner.
5’UTR ac4C promotes upstream initiation and inhibits canonical start codons.
ac4C within Kozak sequences structurally alters interaction with tRNAiMet.
ac4C within mRNA coding sequences enhances translation.
ACKNOWLEDGMENTS
We thank Dr. J. Coller (JHU), Dr. L. Passmore and Dr. E. P. Absmeier (Univ. of Cambridge), and Dr. H. Beiki (NCI) for protocol sharing and insightful discussions. We thank M. Prigge and A. Chiappetta for technical assistance and members of the CCR Sequencing Facility (NCI, Frederick, MD) for Illumina sequencing services. This study utilized the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). This work is supported by the Intramural Research Program of NIH, Center for Cancer Research. National Cancer Institute. D.A. was supported by NCI K99/R00 grant: 4R00CA245035.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68, 352–367. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arango D, Sturgill D, Alhusaini N, Dillman AA, Sweet TJ, Hanson G, Hosogane M, Sinclair WR, Nanan KK, Mandler MD, et al. (2018). Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 175, 1872–1886 e1824. 10.1016/j.cell.2018.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arango D, Sturgill D, and Oberdoerffer S (2019). Immunoprecipitation and Sequencing of Acetylated RNA. Bio Protoc 9, e3278. 10.21769/BioProtoc.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartee D, Nance KD, and Meier JL (2022). Site-Specific Synthesis of N(4)-Acetylcytidine in RNA Reveals Physiological Duplex Stabilization. J Am Chem Soc 144, 3487–3496. 10.1021/jacs.1c11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Brown A, Baird MR, Yip MC, Murray J, and Shao S (2018). Structures of translationally inactive mammalian ribosomes. Elife 7. 10.7554/eLife.40486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti P, Kondo Y, Landis WR, and Witkop B (1968). Photoreduction of uridine and reduction of dihydrouridine with sodium borohydride. J Am Chem Soc 90, 771–775. 10.1021/ja01005a039. [DOI] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Liu YF, Su CW, Su SC, Chen MK, Yang SF, and Lin CW (2016). Functional genetic variant in the Kozak sequence of WW domain-containing oxidoreductase (WWOX) gene is associated with oral cancer risk. Oncotarget 7, 69384–69396. 10.18632/oncotarget.12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick FH (1966). Codon--anticodon pairing: the wobble hypothesis. J Mol Biol 19, 548–555. [DOI] [PubMed] [Google Scholar]
- Dey KK, Xie D, and Stephens M (2018). A new sequence logo plot to highlight enrichment and depletion. BMC Bioinformatics 19, 473. 10.1186/s12859-018-2489-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Niu L, Song W, Xiong X, Zhang X, Zhang Z, Yang Y, Yi F, Zhan J, Zhang H, et al. (2016). tRNA modification profiles of the fast-proliferating cancer cells. Biochem Biophys Res Commun. 10.1016/j.bbrc.2016.05.124. [DOI] [PubMed] [Google Scholar]
- Dou L, Liang HF, Geller DA, Chen YF, and Chen XP (2014). The regulation role of interferon regulatory factor-1 gene and clinical relevance. Hum Immunol 75, 1110–1114. 10.1016/j.humimm.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Drost H (2018). Philentropy: Information Theory and Distance Quantification with R. Journal of Open Source Software. Journal of Open Source Software 3, 765. 10.21105/joss.00765. [DOI] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Enroth C, Poulsen LD, Iversen S, Kirpekar F, Albrechtsen A, and Vinther J (2019). Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res 47, e126. 10.1093/nar/gkz736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest ME, Pinkard O, Martin S, Sweet TJ, Hanson G, and Coller J (2020). Codon and amino acid content are associated with mRNA stability in mammalian cells. PLoS One 15, e0228730. 10.1371/journal.pone.0228730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco MK, and Koutmou KS (2022). Chemical modifications to mRNA nucleobases impact translation elongation and termination. Biophys Chem 285, 106780. 10.1016/j.bpc.2022.106780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno M, Jimenez A, and Vazquez D (1977). Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem 72, 323–330. 10.1111/j.1432-1033.1977.tb11256.x. [DOI] [PubMed] [Google Scholar]
- Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, and Yusupov M (2014). Structural basis for the inhibition of the eukaryotic ribosome. Nature 513, 517–522. 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- Goh SH, Josleyn M, Lee YT, Danner RL, Gherman RB, Cam MC, and Miller JL (2007). The human reticulocyte transcriptome. Physiol Genomics 30, 172–178. 10.1152/physiolgenomics.00247.2006. [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Shi X, Wang H, Ye L, Tong X, Yan K, Ding N, Chen C, Zhang H, and Xue X (2020). Epitranscriptomic N4-Acetylcytidine Profiling in CD4(+) T Cells of Systemic Lupus Erythematosus. Front Cell Dev Biol 8, 842. 10.3389/fcell.2020.00842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG (2017). Structural Insights into the Mechanism of Scanning and Start Codon Recognition in Eukaryotic Translation Initiation. Trends Biochem Sci 42, 589–611. 10.1016/j.tibs.2017.03.004. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG, Ivanov IP, and Sonenberg N (2016). Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416. 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson EM, Concepcion E, Oashi T, and Tomer Y (2005). A Graves’ disease-associated Kozak sequence single-nucleotide polymorphism enhances the efficiency of CD40 gene translation: a case for translational pathophysiology. Endocrinology 146, 2684–2691. 10.1210/en.2004-1617. [DOI] [PubMed] [Google Scholar]
- Kaledhonkar S, Fu Z, Caban K, Li W, Chen B, Sun M, Gonzalez RL Jr., and Frank J (2019). Late steps in bacterial translation initiation visualized using time-resolved cryo-EM. Nature 570, 400–404. 10.1038/s41586-019-1249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, and Wilusz JE (2017). Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev 31, 1717–1731. 10.1101/gad.305250.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolitz SE, Takacs JE, and Lorsch JR (2009). Kinetic and thermodynamic analysis of the role of start codon/anticodon base pairing during eukaryotic translation initiation. RNA 15, 138–152. 10.1261/rna.1318509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1986). Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 44, 283–292. 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- Kozak M (1987). An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res 15, 8125–8148. 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1989). Context effects and inefficient initiation at non-AUG codons in eucaryotic cell-free translation systems. Mol Cell Biol 9, 5073–5080. 10.1128/mcb.9.11.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1990). Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc Natl Acad Sci U S A 87, 8301–8305. 10.1073/pnas.87.21.8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (2005). Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361, 13–37. 10.1016/j.gene.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Kumbhar BV, Kamble AD, and Sonawane KD (2013). Conformational preferences of modified nucleoside N(4)-acetylcytidine, ac4C occur at “wobble” 34th position in the anticodon loop of tRNA. Cell Biochem Biophys 66, 797–816. 10.1007/s12013-013-9525-8. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria F, Tebaldi T, Bernabo P, Groen EJN, Gillingwater TH, and Viero G (2018). riboWaltz: Optimization of ribosome P-site positioning in ribosome profiling data. PLoS Comput Biol 14, e1006169. 10.1371/journal.pcbi.1006169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Liu B, Lee S, Huang SX, Shen B, and Qian SB (2012). Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci U S A 109, E2424–2432. 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppek K, Das R, and Barna M (2018). Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol 19, 158–174. 10.1038/nrm.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy MJ, Montgomery DC, Sardiu ME, Montano JL, Bergholtz SE, Nance KD, Thorpe AL, Fox SD, Lin Q, Andresson T, et al. (2020). A Systems Chemoproteomic Analysis of Acyl-CoA/Protein Interaction Networks. Cell Chem Biol 27, 322–333 e325. 10.1016/j.chembiol.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Liu X, Liu Y, Liu X, Wang R, Liao J, Wu S, Fan J, Peng Z, Li B, and Wang Z (2018). Keratin 80 promotes migration and invasion of colorectal carcinoma by interacting with PRKDC via activating the AKT pathway. Cell Death Dis 9, 1009. 10.1038/s41419-018-1030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Liu X, Jin K, Lu M, Zhang C, Du X, and Xing B (2017). NAT10 is upregulated in hepatocellular carcinoma and enhances mutant p53 activity. BMC Cancer 17, 605. 10.1186/s12885-017-3570-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Zhang Y, Qiu L, Zhang S, Meng Y, Huang C, Chen Z, Zhang B, and Han J (2022). Uncovering N4-Acetylcytidine-Related mRNA Modification Pattern and Landscape of Stemness and Immunity in Hepatocellular Carcinoma. Front Cell Dev Biol 10, 861000. 10.3389/fcell.2022.861000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Colome AM, Lee-Rivera I, Benavides-Hidalgo R, and Lopez E (2017). Paxillin: a crossroad in pathological cell migration. J Hematol Oncol 10, 50. 10.1186/s13045-017-0418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol Biol 6, 26. 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macon JB, and Wolfenden R (1968). 1-Methyladenosine. Dimroth rearrangement and reversible reduction. Biochemistry 7, 3453–3458. 10.1021/bi00850a021. [DOI] [PubMed] [Google Scholar]
- Manjunath H, Zhang H, Rehfeld F, Han J, Chang TC, and Mendell JT (2019). Suppression of Ribosomal Pausing by eIF5A Is Necessary to Maintain the Fidelity of Start Codon Selection. Cell Rep 29, 3134–3146 e3136. 10.1016/j.celrep.2019.10.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie-Line B-C, Aurélie Q, Supuni TG, Thomas JM, Aldema S-C, Sunny S, Célia P-C, Laurence V, Patrick B, Lafontaine DLJ, et al. (2021). Probing small ribosomal subunit RNA helix 45 acetylation across eukaryotic evolution. bioRxiv, 2021.2011.2030.470322. 10.1101/2021.11.30.470322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. 10.14806/ej.17.1.200 pp. 10-12. [DOI] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- McIntyre W, Netzband R, Bonenfant G, Biegel JM, Miller C, Fuchs G, Henderson E, Arra M, Canki M, Fabris D, and Pager CT (2018). Positive-sense RNA viruses reveal the complexity and dynamics of the cellular and viral epitranscriptomes during infection. Nucleic Acids Res. 10.1093/nar/gky029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance KD, Gamage ST, Alam MM, Yang A, Levy MJ, Link CN, Florens L, Washburn MP, Gu S, Oppenheim JJ, and Meier JL (2021). Cytidine acetylation yields a hypoinflammatory synthetic messenger RNA. Cell Chemical Biology. 10.1016/j.chembiol.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norder H, Twagirumugabe T, Said J, Tian Y, Tang KW, and Lindh M (2019). High Frequency of Either Altered Pre-Core StartCodon or Weakened Kozak Sequence in the CorePromoter Region in Hepatitis B Virus A1 Strainsfrom Rwanda. Genes (Basel) 10. 10.3390/genes10030182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy R, Ginell SL, De NC, and Chheda GB (1978). Conformation of N4-acetylcytidine, a modified nucleoside of tRNA, and stereochemistry of codon-anticodon interaction. Biochem Biophys Res Commun 83, 657–663. [DOI] [PubMed] [Google Scholar]
- Parthasarathy R, Ohrt JM, and Chheda GB (1977). Modified nucleosides and conformation of anticodon loops: crystal structure of t6A and g6A. Biochemistry 16, 4999–5008. 10.1021/bi00642a010. [DOI] [PubMed] [Google Scholar]
- Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, and Coller J (2015). Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124. 10.1016/j.cell.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR (2014). BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Current Protocols in Bioinformatics 47, 11.12.11–34. 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Research 42, W187–191. 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter K, Biehl A, Koch L, and Helms V (2016). PreTIS: A Tool to Predict Non-canonical 5′ UTR Translational Initiation Sites in Human and Mouse. PLoS Comput Biol 12, e1005170. 10.1371/journal.pcbi.1005170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringner M, and Krogh M (2005). Folding free energies of 5′-UTRs impact post-transcriptional regulation on a genomic scale in yeast. PLoS Comput Biol 1, e72. 10.1371/journal.pcbi.0010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sas-Chen A, Thomas JM, Matzov D, Taoka M, Nance KD, Nir R, Bryson KM, Shachar R, Liman GLS, Burkhart BW, et al. (2020). Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping. Nature 583, 638–643. 10.1038/s41586-020-2418-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Su Z, Yang K, Wu C, Becker T, Bell-Pedersen D, Zhang J, and Sachs MS (2021). Structure of the translating Neurospora ribosome arrested by cycloheximide. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2111862118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetti A, Guca E, Bochler A, Kuhn L, and Hashem Y (2020). Structural Insights into the Mammalian Late-Stage Initiation Complexes. Cell Rep 31, 107497. 10.1016/j.celrep.2020.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521. 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern L, and Schulman LH (1978). The role of the minor base N4-acetylcytidine in the function of the Escherichia coli noninitiator methionine transfer RNA. J Biol Chem 253, 6132–6139. [PubMed] [Google Scholar]
- Stuart JW, Gdaniec Z, Guenther R, Marszalek M, Sochacka E, Malkiewicz A, and Agris PF (2000). Functional anticodon architecture of human tRNALys3 includes disruption of intraloop hydrogen bonding by the naturally occurring amino acid modification, t6A. Biochemistry 39, 13396–13404. 10.1021/bi0013039. [DOI] [PubMed] [Google Scholar]
- Tan Y, Zheng J, Liu X, Lu M, Zhang C, Xing B, and Du X (2018). Loss of nucleolar localization of NAT10 promotes cell migration and invasion in hepatocellular carcinoma. Biochem Biophys Res Commun 499, 1032–1038. 10.1016/j.bbrc.2018.04.047. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Miyauchi K, Sakaguchi Y, Yamashita S, Soma A, Tomita K, and Suzuki T (2018). Acetate-dependent tRNA acetylation required for decoding fidelity in protein synthesis. Nat Chem Biol. 10.1038/s41589-018-0119-z. [DOI] [PubMed] [Google Scholar]
- Taoka M, Nobe Y, Yamaki Y, Sato K, Ishikawa H, Izumikawa K, Yamauchi Y, Hirota K, Nakayama H, Takahashi N, and Isobe T (2018). Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 10.1093/nar/gky811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardu M, Jones JD, Kennedy RT, Lin Q, and Koutmou KS (2019). Identification and Quantification of Modified Nucleosides in Saccharomyces cerevisiae mRNAs. ACS Chem Biol 14, 1403–1409. 10.1021/acschembio.9b00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiaville PC, Legendre R, Rojas-Benitez D, Baudin-Baillieu A, Hatin I, Chalancon G, Glavic A, Namy O, and de Crecy-Lagard V (2016). Global translational impacts of the loss of the tRNA modification t(6)A in yeast. Microb Cell 3, 29–45. 10.15698/mic2016.01.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JM, Briney CA, Nance KD, Lopez JE, Thorpe AL, Fox SD, Bortolin-Cavaille ML, Sas-Chen A, Arango D, Oberdoerffer S, et al. (2018). A Chemical Signature for Cytidine Acetylation in RNA. J Am Chem Soc 140, 12667–12670. 10.1021/jacs.8b06636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K, Jaguva Vasudevan AA, Martinez Campos C, Emery A, Swanstrom R, and Cullen BR (2020). Acetylation of Cytidine Residues Boosts HIV-1 Gene Expression by Increasing Viral RNA Stability. Cell Host Microbe 28, 306–312 e306. 10.1016/j.chom.2020.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada TK, Kawahara A, Sekine S, M. (1998). Synthesis and Properties of Oligodeoxyribonucleotides Containing 4-N-Acetylcytosine Bases. Tetrahedron Letters 39, 6987–6910. [Google Scholar]
- Wiener D, and Schwartz S (2021). The epitranscriptome beyond m(6)A. Nat Rev Genet 22, 119–131. 10.1038/s41576-020-00295-8. [DOI] [PubMed] [Google Scholar]
- Wilson C, and Krieg AJ (2019). KDM4B: A Nail for Every Hammer? Genes (Basel) 10. 10.3390/genes10020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Wu T, Zhang J, Liu J, Zhao K, Sun W, Zhou X, Kong X, and Shi J (2021). Prognostic and Immunological Role of mRNA ac4C Regulator NAT10 in Pan-Cancer: New Territory for Cancer Research? Front Oncol 11, 630417. 10.3389/fonc.2021.630417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, He D, Xu Y, Hou J, Pan BF, Wang Y, Liu T, Davis CM, Ehli EA, Tan L, et al. (2017). Genome-wide identification and differential analysis of translational initiation. Nat Commun 8, 1749. 10.1038/s41467-017-01981-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jing Y, Wang Y, Tang J, Zhu X, Jin WL, Wang Y, Yuan W, Li X, and Li X (2021). NAT10 promotes gastric cancer metastasis via N4-acetylated COL5A1. Signal Transduct Target Ther 6, 173. 10.1038/s41392-021-00489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31, 3406–3415. 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Compiled information on experiments and data generated. Sequencing experiments summary, sequencing depth details, and list of oligonucleotides used in this study. Related to Figures 1, 2, 3, 5, and 7.
Table S2: RedaC:T-Seq data and results. Contains candidate variants (all mismatch types), RedaC:T-seq determined ac4C locations, summary ac4C information compiled by gene, and functional annotation associated with acetylated transcripts. Related to Figures 2, 3, 4, and 6.
Table S3: Compiled information on Ribo-seq and initiation analysis. Contains all detected P-sites, compiled information about translation initiation sites (TIS) and proximity to ac4C, compiled translation initiation results from Ribo-TISH, transcript abundances, rabbit reticulocyte lysates, and refinement and validation statistics for the two local structures. Related to Figures 3, 4, 6, and 7.
Video S1: Rotation of the codon:anticodon interface for the C(−1) mRNA. Related to Figure 7C.
Video S2: Rotation of the codon:anticodon interface for the ac4C(−1) mRNA. Related to Figure 7C.
Video S3: Rotation of the t6A:C(−1) interface. Related to Figure 7E.
Video S4: Rotation of the t6A:ac4C(−1) interface. Related to Figure 7E.
Data Availability Statement
RedaC:T-seq, HR Ribo-seq, and Rabbit Reticulocyte RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Cryo-EM data have been deposited at PDB and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse IgG-HRP antibody | GE Healthcare | Cat#:NA931; RRID: AB_772210 |
| Anti-rabbit IgG-HRP antibody | Cell Signaling Technology | Cat#:7074S; RRID: AB_2099233 |
| Mouse monoclonal anti-GAPDH (6C5) antibody | Santa Cruz Biotechnology | Cat#:sc-32233; RRID: AB_627679 |
| Rabbit monoclonal anti-ac4C antibody | Abcam | Cat#:ab25215; RRID: AB_2827750 |
| Rabbit monoclonal anti-m7G antibody | MBL International Corporation | Cat#:RN016M; N/A |
| Rabbit Monoclonal IgG Isotype Control | Cell Signaling Technology | Cat#:3900S; RRID: AB_1550038 |
| Rabbit polyclonal anti-IRF1 antibody | Abcam | Cat#:ab191032; RRID: AB_2904575 |
| Rabbit polyclonal anti-KDM4B antibody | Cell Signaling Technology | Cat#:8639S; RRID: AB_11140642 |
| Rabbit polyclonal anti-KRT80 antibody | ProteinTech | Cat#:16835-1-AP; RRID: AB_1851273 |
| Rabbit polyclonal anti-LMNA antibody | Bethyl Laboratories | Cat#:A303-433A; RRID: AB_10951693 |
| Rabbit polyclonal anti-PXN antibody | ProteinTech | Cat#:10029-1-Ig; RRID: AB_513929 |
| Rabbit polyclonal anti-NAT10 antibody | ProteinTech | Cat#:13365-1-AP; RRID: AB_2148944 |
| Rabbit polyclonal anti-THUMPD1 antibody | Bethyl Laboratories | Cat#:A304-643AT; RRID: AB_2620838 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| 32P-γATP | PerkinElmer | Cat#:BLU502A100UC |
| 32P-αUTP | PerkinElmer | Cat#:BLU007H250UC |
| AMPure XP beads | Beckam Coulter | Cat#:A63881 |
| Acid-Phenol:Chloroform, pH 4.5 | ThermoFisher Scientific | Cat#:AM9722 |
| Bovine Calf Serum (BCS) | HyClone | Cat#:SH30073.03 |
| Bovine Serum Albumin (BSA) | Roches | Cat#:03117332001 |
| Bradford Reagent | BioRad | Cat#:5000205 |
| CIAP | Promega | Cat. #:M2825 |
| Cycloheximide | Sigma-Aldrich | Cat#:C7698 |
| DMEM | ThermoFisher Scientific | Cat#:11995073 |
| DNase I | Sigma-Aldrich | Cat#:4716728001 |
| DNAse-free RNAse A | ThermoFisher Scientific | Cat#:EN0531 |
| E. coli Poly(A) Polymerase | New England Biolabs | Cat#:M0276S |
| ECL Western Blotting Substrate | Promega | Cat#:W1001 |
| Halt™ protease inhibitor cocktail | ThermoFisher Scientific | Cat#:78429 |
| Harringtonine | Abcam | Cat#:ab141941 |
| Illustra™ MicrosSpin™ G-50 Columns | GE Healthcare | Cat#:27-5330-01 |
| L-glutamine | ThermoFisher Scientific | Cat#:25030149 |
| Linear acrylamide | ThermoFisher Scientific | Cat#:AM9520 |
| Lipofectamine 3000 | ThermoFisher Scientific | Cat#:L3000008 |
| Murine RNAse inhibitor | New England Biolabs | Cat#:M0314 |
| N4-acetylcytidine triphosphate (ac4CTP) | Trilink | Custom synthesis |
| Protease Inhibitor Mini Tablets, EDTA-free | ThermoFisher Scientific | Cat#:32955 |
| Preadenylated universal miRNA cloning linker | New England Biolabs | Cat#:S1315S |
| ProSignal Pico ECL Reagent | Genesee Scientific | Cat#:20-300B |
| Propidium iodide | Roche | Cat#:11348639001 |
| Protein G Magnetic Beads | New England Biolabs | Cat#:S1430S |
| Proteinase K, Molecular Biology Grade | New England Biolabs | Cat#:P8107S |
| RNA Clean & concentrator-5 | Zymo Research | Cat#:R1013 |
| RNA Spike-In mix | ThermoFisher Scientific | Cat#:4456740 |
| RNAse A | Millipore | Cat#:111199115001 |
| RNase I | ThermoFisher Scientific | Cat#:EN0601 |
| Superase-In | ThermoFisher Scientific | Cat#:AM2694 |
| T4 Polynucleotide kinase (PNK) | New England Biolabs | Cat#:M0201 |
| T4 RNA ligase 2 | New England Biolabs | Cat#:M0239S |
| TRIzol Reagent | ThermoFisher Scientific | Cat#:15596026 |
| Trypan blue | Lonza | Cat#:17-942E |
| Turbo™ DNAse I | ThermoFisher Scientific | Cat#:AM2239 |
| Critical Commercial Assays | ||
| human/mouse/rat Ribo-Zero Gold rRNA removal | Illumina | Cat#:MRZG12324 |
| LightCycler 480 SYBR Green I Master | Roche | Cat#:04887352001 |
| Luciferase assay reagent | Promega | Cat#:E1500 |
| MEGAscript T7 Transcription Kit | ThermoFisher Scientific | Cat#:AM1334 |
| Nano-Glo® Luciferase Assay System | Promega | Cat#:N1120 |
| Nano-Glo Dual-Luciferase Reporter System | Promega | Cat#:N1610 |
| NEBNext® Magnesium RNA Fragmentation buffer | New England Biolabs | Cat#:E6150 |
| NEBNext® rRNA Depletion Kit | New England Biolabs | Cat#:E6310L |
| NEBNext® UltraII™ Directional RNA Library Prep Kit | New England Biolabs | Cat#:E7770S |
| NorthernMax kit | ThermoFisher Scientific | Cat#:AM1940 |
| NorthernMax Prehyb/Hyb buffer | ThermoFisher Scientific | Cat#:AM8677 |
| ONE-Glo EX Reagent | Promega | Cat#:E8110 |
| PowerUp SYBR Green Master Mix | ThermoFisher Scientific | Cat#:A25777 |
| ProSignal Pico ECL Reagent | Genesee Scientific | Cat#:20-300B |
| Q5 Hot Start High-Fidelity | New England Biolabs | Cat#:M0493S |
| Rabbit Reticulocyte Lysate | Promega | Cat#:L4960 |
| SuperScript IV First-Stranded Synthesis System | ThermoFisher Scientific | Cat#:18091050 |
| SuperSignal ELISA Femto Maximum Sensitivity Substrate | ThermoFisher Scientific | Cat#:37075 |
| TransIT-mRNA Transfection Kit | Mirus | Cat#:MIR2250 |
| Vaccinia Capping System | New England Biolabs | Cat#:M2080S |
| Deposited Data | ||
| Raw and processed RedaC:T-seq data | This Study | GEO: GSE162043 |
| Raw and processed HR Ribo-seq-seq data | This Study | GEO: GSE162043 |
| Rabbit Reticulocyte RNA-seq | This Study | GEO: GSE162043 |
| Cryo-EM data | This Study | ac4C 80s ribosome: PDB:7UCK and EMDB:EMD-26445 AUG 80s ribosome: PDB:7UCJ and EMDB:EMD-26444 |
| Experimental Models: Cell Lines | ||
| Human: HeLa | ATCC | Cat#:CCL-2; RRID: CVCL_0030 |
| Human: NAT10+/+ | Arango et. al. 2018 | N/A |
| Human: Flp-In Trex 293 | ThermoFisher Scientific | Cat. #:R71007 RRID: CVCL_U427 |
| Human: Flp-In Trex 293 NAT10-FL | Arango et. al. 2018 | N/A |
| Oligonucleotides | ||
| ON-TARGETplus siRNAs against PXN | Dharmacon | Cat.#:L-005163-00-0005 |
| ON-TARGETplus siRNAs against KRT80 | Dharmacon | Cat.#:L-018941-02-0005 |
| ON-TARGETplus siRNAs Control | Dharmacon | Cat.#:D-001810-10 |
| Oligonucleotide sequences used in this study | This Study | Table S1 |
| Recombinant DNA | ||
| pNL1.1PGK | Promega | Cat#:N1441 |
| pcDNA/FRT | ThermoFisher Scientific | Cat#:V6010-20 |
| pcDNA/FRT-NAT10-FL | Arango et al., 2018 | |
| pcDNA/FRT-NAT10-ΔHel | Arango et al., 2018 | |
| Software and Algorithms | ||
| BEDtools | Quinlan, 2014 | RRID:SCR_006646 |
| Bowtie2 v.00.14 | Langmead and Salzberg, 2012 | RRID:SCR_016368 |
| Cutadapt v 2.0 | Martin, 2011 | RRID:SCR_011841 |
| DeepTools | Ramírez et al., 2014 | RRID:SCR_016366 |
| DESeq2 | Love et al., 2014 | RRID:SCR_017673 |
| Fastx_clipper v.0.014 | http://hannonlab.cshl.edu/fastx_toolkit/ | |
| Hisat2 v2.2.1.0 | Kim et al., 2015 | RRID:SCR_015530 |
| HTSeq v 0.6.1p1 | Anders et al., 2015 | RRID:SCR_005514 |
| ImageJ | National Center for Microscopy and Imaging Research | RRID:SCR_001935 |
| ImageLab | BioRad | http://www.biorad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| MACS2 | Zhang et al., 2008 | RRID:SCR_013291 |
| MFOLD v3.6 | Zuker, 2003 | RRID:SCR_008543 |
| Mpileup v 1.10 | Li et al., 2009 | RRID:SCR_002105 |
| PRISM 7 | GraphPad Software, Inc. | RRID:SCR_002798 |
| Relion v 3.0 | Zivanov et al., 2018 | RRID:SCR_016274 |
| Ribo-TISH | Zhang et al., 2017 | https://github.com/zhpn1024/ribotish |
| Rstudio v 3.6 | Rstudio | RRID:SCR_000432 |
| STAR v. 2.5.4.a | Dobin et al., 2013 | RRID:SCR_019993 |
| Tophat2 v.2.1.1 | Trapnell et al., 2009 | RRID:SCR_013035 |
| ViennaRNA tool v 2.4.18 | Lorenz et al., 2011 | https://www.tbi.univie.ac.at/RNA/# |