

Abstract
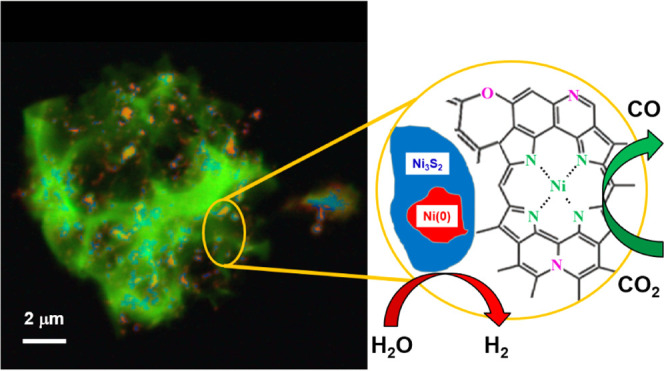
Atomically dispersed metal–nitrogen–carbon (M–N–C) materials are a class of electrocatalysts for fuel cell and electrochemical CO2 reduction (CO2R) applications. However, it is challenging to characterize the identity and concentration of catalytically active species owing to the structural heterogeneity of M–N–C materials. We utilize scanning transmission X-ray microscopy (STXM) as a correlative spectromicroscopy approach for spatially resolved imaging, identification, and quantification of structures and chemical species in mesoscale regions of nickel–nitrogen–carbon (Ni–N–C) catalysts, thereby elucidating the relationship between Ni content/speciation and CO2R activity/selectivity. STXM results are correlated with conventional characterization approaches relying on either bulk average (X-ray absorption spectroscopy) or spatially localized (scanning transmission electron microscopy with electron energy loss spectroscopy) measurements. This comparison illustrates the advantages of soft X-ray STXM to provide spatially resolved identification and quantification of active structures in Ni–N–C catalysts. The active site structures in these catalysts are identified to be atomically dispersed NiNx/C sites distributed throughout entire catalyst particles. The NiNx/C sites were notably demonstrated by spectroscopy to possess a variety of chemical structures with a spectroscopic signature that most closely resembles nickel(II) tetraphenylporphyrin molecules. The quantification and spatial distribution mapping of atomically dispersed Ni active sites achieved by STXM address a target that was elusive to the scientific community despite its importance in guiding advanced material designs.
Keywords: nickel−nitrogen−carbon catalysts, CO2 electroreduction, X-ray absorption spectroscopy, soft X-ray STXM, STEM-EELS
1. Introduction
Nanostructured carbon materials consisting of metal–nitrogen–carbon (M–N–C) species are a promising class of materials for electrocatalytic applications.1−9 M–N–C materials have attracted great attention as they can be synthesized by straightforward methods that include pyrolyzing a mixture of nitrogen, carbon, and transition-metal-containing precursors, along with their ability to elegantly combine the benefits of heterogeneous and homogeneous catalysts.10,11 On the heterogeneous side, the catalytically active sites (presumed to be atomically dispersed metal ions coordinated by nitrogen dopants in a MNx/C configuration12,13 that is similar in structure to many homogeneous catalysts14,15) are embedded in a solid carbon-based framework, so downstream separation of the catalyst from the products is not necessary. Furthermore, the carbon support is highly conductive and no catalyst regeneration is needed, as required for metal-centered homogeneous molecular catalysts that need oxidation or reduction back to their “active” state. On the homogeneous side, M–N–C catalysts in principle can have 100% metal utilization and well-defined active site structures to provide good catalytic activity and selectivity.
M–N–C electrocatalysts first came into prominence with Fe–N–C materials demonstrated as oxygen reduction catalysts in polymer electrolyte membrane fuel cells.16,17 To date, Fe–N–C materials are the most active nonplatinum group metal catalysts for oxygen reduction in acidic electrolytes due to the presence of an atomically dispersed FeNx/C active structure that has been identified by a variety of spectroscopy and microscopy techniques.18−22 More recently, M–N–C research has expanded to other areas, including catalysis for electrochemical CO2 reduction (CO2R).23 In particular, Ni–N–C electrocatalysts have shown promise for electrochemical CO2 reduction to CO.24,25 Scientists have postulated that these catalysts are composed of atomically dispersed NiNx/C active sites, with a local structure similar to those of Ni(II) porphyrin (such as Ni tetraphenylporphyrin, NiTPP) or nickel(II) phthalocyanine (such as octabutoxy Ni phthalocyanine, O-NiPc), as depicted in Scheme 1a,b.12,25 The postulated NiNx/C sites can offer a unique structure for CO2 adsorption and activation, which reduces the energy barrier for electrochemical CO2 reduction and exhibits high intrinsic CO2 reduction activity and selectivity for CO generation.26,27 Although atomically dispersed MNx/C moieties have been deemed important for electrocatalytic applications, the identification and characterization of active site structures in M–N–C catalysts prepared by pyrolysis techniques are challenging due to the highly heterogeneous nature of these materials in terms of compositions, phases, and structures that are present.
Scheme 1. Structures of Reference Species and Ni–N–C Samples.

(a) Nickel(II) tetraphenylporphyrin (NiTPP), (b) octabutoxy nickel(II) phthalocyanine (O-NiPc), and (c) schematic diagram of the Ni–N–C structure.
Many techniques have been used to characterize M–N–C electrocatalysts with the objective of elucidating the structures and properties of catalytically active sites to provide mechanistic insight and inform advanced catalyst designs. Time-of-flight secondary ion mass spectrometry (ToF-SIMS) was the first characterization technique to directly identify species consisting of coordinated Fe, N, and C in oxygen reduction catalysts.28,29 X-ray absorption spectroscopy (XAS) has also been used widely to characterize the local structure of transition metals and nitrogen atoms in M–N–C catalysts, leading to the elucidation of MNx/C coordination environments.30−33 Scanning transmission electron microscopy (STEM) has been used to image single metal atoms in M–N–C catalyst samples. State-of-the-art atomic resolution transmission electron microscopy (TEM) and electron energy loss spectroscopy (TEM-EELS) have provided analytical confirmation of the identity of the single metal atom and provided some insights into the local chemical environment.34−36
Prior to this work, characterization using either bulk-averaged or very spatially localized measurements has (i) verified the presence of atomically dispersed MNx/C sites and (ii) measured the average properties of M–N–C samples and correlated them with the activity/ selectivity/performance of the overall catalyst. Opportunity exists to leverage mesoscale spatial resolution spectroscopy techniques, such as soft X-ray scanning transmission X-ray microscopy (STXM), that will enable a comprehensive understanding of (i) the distribution, content, and properties of different species present in M–N–C catalysts; (ii) how these factors differ with different synthetic strategies; and (iii) the impact of these physicochemical properties on electrocatalytic performance. This comprehensive insight into the heterogeneous compositions and structures of M–N–C materials would help guide the design and optimization of improved performance catalysts for carbon dioxide reduction (CO2R) and other important electrocatalytic reactions.37 STXM is a synchrotron-based technique that provides precise chemical speciation through near-edge X-ray absorption fine structure (NEXAFS) spectroscopy38 and quantitative, chemically selective imaging with sub-40 nm spatial resolution. Here, we use STXM,39−41 along with XAS and TEM-EELS, to provide a spatially resolved, holistic understanding of the chemical species and structures present in M–N–C materials. Section S1 and Table S1 present a comparison of the advantages and limitations of ToF-SIMS, STEM/EELS, STXM, and XAS.
The samples studied were a series of pyrolyzed, polymer-derived Ni–N–C catalysts that were investigated previously for their oxygen reduction reaction performance and chemical composition.42 STXM image sequences (stacks) were used to collect spatially resolved NEXAFS spectra at the C 1s, N 1s, O 1s, Ni 2p, and S 2p edges of Ni–N–C materials prepared with different synthetic parameters. These stacks were compared and fit using NEXAFS spectra collected from suitable reference compounds. The Ni 2p results showed the presence of metallic Ni (shown as Ni(0)), as well as trinickel disulfide (Ni3S2) and NiNx/C species. The N 1s and C 1s spectra provided evidence that the local structure of NiNx/C is closer to that of NiTPP than nickel(II) phthalocyanine, which was the coordination speculated in previous reports.12,25 Detailed analysis of STXM data on the Ni–N–C catalysts provided quantitative maps of the amount and spatial distributions of the NiNx/C sites, Ni(0), and Ni3S2 on an oxidized carbonaceous matrix, as shown in Scheme 1c. The NiNx/C structures, which are the active CO2R catalytic sites, are widely distributed throughout the catalyst structure. The amount of the catalytic NiNx/C sites in the various Ni–N–C samples was determined from quantitative analysis of the STXM data and used to explain the relative activity and selectivity for CO2 reduction to CO for different Ni–N–C materials. These insights can be used to inform the design of next-generation M–N–C catalysts. In addition, our work shows that STXM is an excellent tool for electrocatalyst studies since it overcomes the limitations of bulk/average or overly localized techniques. This advantage is particularly important in studies such as M–N–C catalyst development, where the materials are intrinsically highly heterogeneous. This ex situ study thereby validates STXM as a powerful tool for characterization of electrocatalyst materials and is an essential step toward in situ electrochemical STXM measurements43 that our team is currently performing.
2. Experimental Methods
2.1. Material Synthesis
The synthesis of the Ni–N–C catalysts has been reported previously.42 Briefly, the Ni–N–C catalysts were prepared by mixing aniline (1 mL, Sigma-Aldrich), cyanamide (2 g, Sigma-Aldrich), and nickel chloride hexahydrate (NiCl2·6H2O, Sigma-Aldrich) in a 1.5 M HCl solution (200 mL, ACS reagent). This solution was mixed by magnetic stirring, followed by the addition of ammonium persulfate (1.5 g, Sigma-Aldrich) as an oxidant for the polymerization of aniline to polyaniline. The water was evaporated by heating the solution to 80 °C and leaving it stirring overnight. The resulting product was pyrolyzed at 900 °C in Ar for 1 h, acid-washed to remove labile Ni compounds, and then pyrolyzed a second time at 900 °C in Ar for 3 h. The acid washing procedure was to remove any surface exposed Ni particles that would be active for the hydrogen evolution reaction under CO2R conditions.25 The second pyrolysis was to remove any labile residues that remain after the acid wash procedure. In the synthesis, the amount of NiCl2·6H2O was varied to prepare two different Ni–N–C catalyst materials: sample A, Ni–N–C-high (1.2 g of NiCl2·6H2O), and sample B, Ni–N–C-low (0.08 g of NiCl2·6H2O). A comparison sample N–C (Ni-free) was prepared without the addition of any nickel precursor. Ni3S2/C samples were prepared by mixing 20 wt % commercial Ni3S2 (Sigma-Aldrich) particles and 80 wt % high-surface-area Vulcan XC-72 (VC, FuelCellStore) powders in a ratio of 20–80 wt %, respectively.
2.2. Catalyst Testing
The performance of the Ni–N–C-low, Ni–N–C-high, N–C, and Ni3S2/C materials for electrochemical CO2R was tested to gain insight into how the structures and chemical components of the Ni–N–C catalysts impacted the catalytic activity and selectivity. Ten milligrams of material was suspended in an ink including 1.5 mL of isopropyl alcohol and 0.5 mL of H2O with 110 μL of Nafion (Sigma-Aldrich, 5 wt % Nafion in ethanol) and sonicated for 30 min. The suspended ink was drop-cast onto graphite foil (Fisher Scientific) and dried at 70 °C for 2 h, leading to an electrode loading of 1 mg/cm2. The catalyst-coated electrodes were used as the working electrode for electrochemical CO2 reduction and tested in a custom-built two-compartment cell that has been reported previously.44,45 The geometric area of the working electrode was 5.7 cm2. The electrolyte compartments of the working and counter electrode were filled with 0.1 M KHCO3 (10 mL), and CO2 was flown continuously through the catholyte and anolyte chambers at 20 sccm throughout the course of the experiment. A Pt foil was used as the counter electrode and an Ag/AgCl electrode was used as the reference electrode. The Ag/AgCl reference electrode was calibrated vs a home-made reversible hydrogen electrode (RHE). All potentials reported in this manuscript are in terms of vs RHE. The catholyte and anolyte chambers were separated by an ion exchange membrane (Selemion AMV, AGC Inc.). Catalysts were tested by chronoamperometric measurements at various electrochemical potentials in the CO2-saturated 0.1 M KHCO3. Ni–N–C catalyst materials are known to selectively produce only gas-phase products (CO and H2),25,46,47 so gas chromatography (GC) was used for product detection and quantification; liquid product quantification was not performed. A 1 mL sample of the effluent gas from the cathode compartment of the electrochemical cell was fed to a gas chromatography (GC) unit (supplier, SRI; model, 8610C in the multigas configuration #5, equipped with flame ionization and thermal conductivity detectors) for product identification and quantitation to determine faradic efficiencies (FEs). While our setup can also collect liquid products for subsequent analysis by either nuclear magnetic resonance spectroscopy or high-performance liquid chromatography, this was not done in this study as M–N–C catalysts produce almost exclusively gas-phase product, and the faradic efficiency (FE) values were close to 100%.
2.3. STXM Measurements
STXM imaging and spectroscopy were performed using the spectromicroscopy beamline (SM) 10ID1 at the Canadian Light Source (CLS, Saskatoon, Canada). Details of the beamline48 and STXM,49 along with operating procedures40 have been presented elsewhere. Briefly, the monochromatic X-ray beam was focused to a ∼40 nm spot by a Fresnel zone (ZP) plate (Applied Nanotools Ltd.). The sample was positioned at the focal point of the X-ray beam, and STXM images were measured by (x,y) raster-scanning the sample while recording the transmitted X-ray intensity pixel by pixel in a single-photon counting mode using a phosphor/photomultiplier.50 Spectroscopic information was obtained by recording image sequences (also known as stacks51). The energy scale was calibrated by recording spectra of standard gases Ne,52 N2,53 SF6,5454 and CO2.5353 All STXM data was analyzed using aXis2000 software.55 Detailed information about sample preparation for STXM, a summary of samples investigated, energy-scale calibration, and STXM data analysis can be found in Sections S2–S5, Supporting Information.
2.4. STEM/EELS and XAS Measurements
The catalyst materials were characterized by field-emission transmission electron microscopy (TEM), high-resolution aberration-corrected transmission electron microscopy (HRTEM), and high-angle, annular dark-field scanning transmission electron microscopy (HAADF-STEM) imaging along with EELS using an FEI Titan 80-300 HB microscope at the Canadian Centre of Electron Microscopy (CCEM, McMaster University). The FEI Titan 80-300 HB microscope with an XFEG Source was operated at 200 kV with two hexapole Cs correctors (CEOS) using a 19.1 mrad convergence angle STEM probe and a Fischione HAADF detector. When doing the EELS, the Gatan Quantum GIF with a Gatan K2 Summit direct electron detection camera was used with 55.0 mrad collection semiangle, 0.4 nm/pixel, 0.25 eV/channel dispersion, and 10 ms exposure.
Ensemble-averaged (limited spatial resolution) X-ray absorption spectroscopy (XAS) at the Ni 2p and N 1s edges was performed using the spherical grating monochromator (SGM) beamline 11ID1 at the CLS. To minimize radiation damage, a quick scan protocol was used, with multiple measurements (30 for Ni 2p and 10 for the N 1s) from adjacent 250 μm × 250 μm areas of each sample. The total electron yield (TEY), total fluorescence yield (TFY), and partial fluorescence yield (PFY) signals were measured simultaneously. In all cases, the fluorescence yield spectra were highly distorted by absorption saturation of both incident and fluorescence intensities. Partial ion yields (PFYs) and inverse PFY spectra were also explored, without significant improvement. The reason is that either the Ni–N–C particles were too thick (see the comparison of A1 and A2) or particles overlap in the samples used for the XAS study, where multiple layers of the powder were pressed into the In foil. This demonstrates another major advantage of STXM—the ability to select and measure particles that are sufficiently thin to avoid spectral distortion from absorption saturation. Such particles are also thinner and thus the catalytically active surface sites contribute more than in larger particles. The XAS-TEY results are compared with STXM-derived NEXAFS spectra of the same material to provide an ensemble-averaged confirmation of the STXM results.
3. Results
3.1. Electrochemical CO2 Reduction Properties
The results of evaluation of the catalytic performance of our Ni–N–C catalysts for electrochemical CO2 reduction in CO2-saturated 0.1 M KHCO3 are displayed in Figure 1. At an electrode potential of −0.67 V vs RHE, the Ni–N–C-low sample showed a current density of −3.9 mA/cm2 (Figure 1a) and a CO faradic efficiency of 88% (Figure 1b). At all potentials investigated between −0.67 and −0.94 V vs RHE, the faradic efficiency of Ni–N–C-low toward CO was >60% and toward H2 was <25% (Figure 1c), with a decrease in selectivity to CO and an increase in selectivity to H2 observed at more negative electrode potentials. The Ni–N–C-high sample showed the similar catalytic performance to the Ni–N–C-low sample but with relatively lower activity and lower faradic efficiency (Figure 1d). These trends, including high CO selectivity that diminishes at more negative potentials, are consistent with previous reports on Ni–N–C catalysts and have been attributed to the presence of atomically dispersed NiNx/C active sites.56,57 This notion is supported by the electrochemical performance of the N–C (Ni-free) sample, which was found to produce negligible amounts of CO under electrochemical CO2R conditions (Figure 1b). We also measured the electrochemical CO2R performance of Ni3S2/C as a reference compound, as we had previously identified that the synthesized Ni–N–C materials contain Ni3S2 and metallic Ni particles.42 While metallic Ni would be expected to exclusively produce H2 under CO2 reduction conditions,58,59 it was prudent to determine what impact, if any, the Ni3S2 species have on CO2 reduction activity and selectivity. We tested commercial Ni3S2 particles supported on Vulcan XC-72 carbon black (Ni3S2/C). This sample demonstrated significantly increased current densities in comparison to Ni–N–C-low at all electrode potentials evaluated (Figure 1a). However, near-negligible amounts of CO were detected (Figure 1b), indicating that Ni3S2/C was selective toward the hydrogen evolution reaction (Figure 1c) and not CO2R. This is consistent with a previous report on transition-metal sulfide catalysts, which were found to be selective only for hydrogen evolution owing to the steep kinetic barrier for a proton–electron transfer to COads intermediate species.60 It should be noted that less than 100% of total faradic efficiencies were measured for N–C and Ni3S2/C samples. This is attributed to the low current densities that result in H2 concentrations in the reactor effluent of <50 ppm, which is lower than the reliable concentration for detection by the thermal conductivity detector in the gas chromatograph used to measure H2. Taken together, the electrochemical CO2R testing results indicate that Ni is essential for producing catalysts that are selective for CO production, but the resulting active site species are not Ni3S2 or metallic Ni particles.
Figure 1.

Evaluation of activity and selectivity for electrochemical CO2 reduction by Ni–N–C-low, Ni–N–C-high, Ni3S2/C, and N–C (a catalyst synthesized without Ni). (a) Overall geometric current density. (b) Faradic efficiency toward CO production. (c) Faradic efficiency toward H2 production. (d) Comparison of CO and H2 production at −0.67 V vs RHE. Note that in some instances significantly less than 100% faradic efficiencies were reported owing to low current densities that resulted in H2 concentrations in the effluent from the reactor below the detection limit of the thermal conductivity detector.
3.2. STEM/EELS
Results from a HAADF-STEM and STEM-EELS study of the Ni–N–C-low sample are shown in Figure 2. At low magnifications (Figure 2a), the material was found to consist of a carbonaceous matrix decorated with bright particles. At high magnifications (Figures 2b and S3a), single-atom bright spots (indicating a Z-number higher than carbon) were observed throughout the graphitic carbonaceous matrix. These bright spots are interpreted as Ni atoms coordinated to nitrogen atoms within the carbonaceous matrix (in the form of NiNx/C), as reported previously,12,25 although without atomic-scale EELS imaging this cannot be stated conclusively. Higher-magnification STEM imaging of a particle shows a core–shell structure (Figure 2c). EELS mapping of sulfur (Figure 2d) and nickel (Figure 2e) indicates that the center of the particle is Ni and the outer shell is nickel-sulfide-based species (Figure 2c). EELS mapping of carbon, nitrogen, and oxygen (Figure S3b) indicates there is a small amount of oxygen in both the particles and the nitrogen-doped carbonaceous matrix. To further understand the chemical environment of Ni, EELS spectra of both the carbonaceous matrix and particle regions are shown in Figure S3c. However, due to the limited energy resolution of the EELS spectra, which is typically ∼1 eV, the chemical species cannot be identified. Therefore, in this work, STXM-derived XAS spectra (energy resolution ∼0.1 eV) are used to clarify the chemical composition of Ni–N–C electrocatalysts and probe the single-atom catalyst hypothesis.
Figure 2.
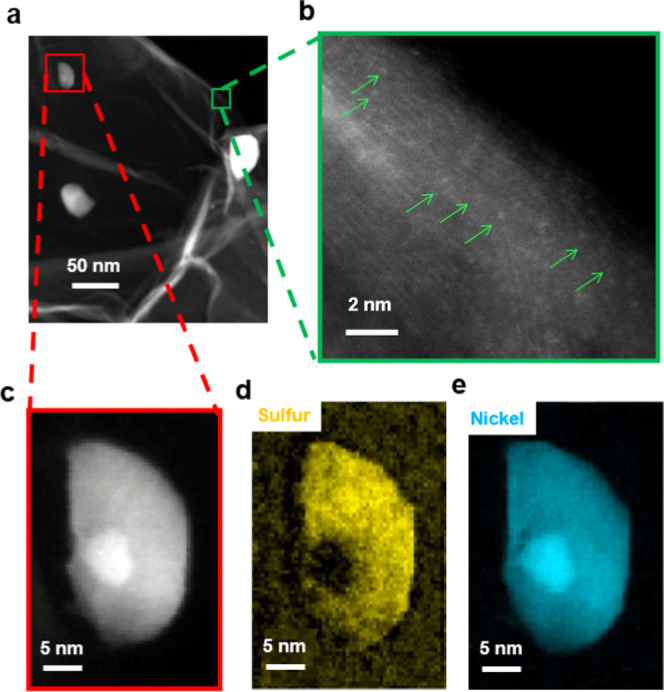
Analysis of the Ni–N–C-low sample by transmission electron microscopy and electron energy loss spectroscopy mapping. (a) Lower-resolution high-angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) image. (b) Higher-resolution HAADF-STEM image showing graphitic planes, with individual bright (high Z-number) atoms indicated by green arrows. (c) STEM image of a particle showing a core–shell structure. STEM electron energy loss spectroscopy (EELS) mapping of (d) sulfur and (e) nickel in this particle.
3.3. STXM
3.3.1. Ni 2p STXM of Ni–N–C-High on SiNx Windows (A1 and A1a Regions)
Our previous research42 showed that the Ni–N–C samples (Ni–N–C-high, Ni–N–C-low) used in this study consist of Ni-containing particles distributed throughout a nitrogen-doped carbonaceous matrix, with an increasing quantity of particles observed when higher amounts of nickel chloride were used in the synthesis. The presence of Ni-based particles following acid leaching is likely due to two reasons. First, Ni-based particles can catalyze the formation of a graphitic shell during pyrolysis, which protects them from being exposed to acid.25 Second, we have found the Ni3S2 to be stable through an acid wash, so the Ni3S2 shell on the Ni-based particles could prevent their removal during acid washing.42
STXM was used to study a Ni–N–C-high particle on a SiNx window, denoted region A1 (see Figure S1a–c). STXM transmission images at photon energies of 848 eV (pre-Ni 2p absorption edge) and 852.7 eV (peak of the Ni 2p3/2 absorption of Ni metal) are shown in Figure 3a,b, respectively. The Ni–N–C-high materials consist of particles ranging in size from tens of nanometers to ∼0.5 μm, distributed across the carbonaceous matrix. The heterogeneous morphology observed in the Ni–N–C-high sample is similar to previous TEM observations of the same42 and similar Ni–N–C materials.12,25 However, as shown in the previous section, TEM is limited in its ability to provide speciation. Therefore, we have probed these catalysts by STXM spectromicroscopy to perform chemical speciation and investigate the spatial distributions of the species present.
Figure 3.

STXM analysis of a Ni–N–C-high structure (A1 region). (a) Transmission image at 848 eV (Ni 2p pre-edge). (b) Transmission image at 852.7 eV (Ni 2p3/2 peak). (c) Difference in the optical density, ΔOD = OD852.7 – OD848. (d) Average of 143 OD images (Ni 2p stack) recorded in the A1a region (yellow rectangle in (c)). (e) Masks used to select spectra of the particles (light red) and matrix (light green).
Both the particles and carbonaceous matrix in the Ni–N–C-high show lower contrast at 848 eV, below the onset of the Ni 2p absorption edge (Figure 3a), than at 852.7 eV, the peak of Ni 2p3/2 absorption (Figure 3b). The change in contrast with photon energy for both morphologies indicates that Ni atoms are present throughout the entire structure. At a photon energy of 848 eV, only large particles are observed throughout the carbonaceous matrix. This is because at 848 eV (Figure 3a) the energy is not high enough to excite the Ni 2p electrons, and therefore, the contrast arises from thickness and composition. At 852.7 eV (Figure 3b), the large particles are much darker and numerous smaller particles appear with high contrast, indicating that the Ni in these particles is in the form of Ni metal.
The STXM transmission images were converted to an OD scale, as shown in Figure S4 and Section S7. Figure 3c displays the difference between the OD images at 852.7 eV (Figure S4d) and 848 eV (Figure S4b), which is referred to as a stack map (ΔOD). Such stack maps identify regions of the sample containing Ni, with the net absorption by Ni directly indicated by the ΔOD value (Figure 3c). Although the highest ΔOD values (OD852.7 – OD848) are observed at the particles, there is an increase in Ni 2p3/2 absorption throughout the entire Ni–N–C-high structure, including the carbonaceous matrix. However, on a per-area basis, the Ni 2p3/2 signal is considerably lower in the carbonaceous matrix than that in the discrete particles.
To investigate the chemical identity and composition of the particles and species in the carbonaceous matrix of the Ni–N–C-high catalyst, a complete Ni 2p stack (143 images from 836 to 929 eV) was recorded. Details of the experimental parameters for each measurement contributing to the figures shown in the paper or the Supporting Information are listed in Table S2. The stack was measured from a thin region of the catalyst particle indicated by the yellow rectangle in Figure 3c (region A1a). Figure 3d displays the average X-ray absorption image—i.e., the average of all images in the Ni 2p stack, after alignment and conversion to OD. The grayscale of Figure 3d is the average OD
From Figure 3d,
this A1a region can be divided into two morphologies:
particles ( , showing high Ni 2p absorption) and the
carbonaceous matrix (
, showing high Ni 2p absorption) and the
carbonaceous matrix ( , showing a relative low
Ni 2p absorption).
These regions are indicated in Figure 3e. The Ni 2p spectra of the Ni–N–C-high
catalyst averaged over the particles (light red) and carbonaceous
matrix (light green) are shown in Figure 4a. The spectra for both the particles and
carbonaceous matrix are dominated by the Ni 2p3/2 →
3d (852–855 eV) and Ni 2p1/2 → 3d (868–872
eV) resonance regions. Compared with the Ni 2p spectrum of the particles,
the carbonaceous matrix has a weaker Ni signal but a higher pre-edge
signal relative to the Ni resonance peak intensities. The Ni 2p3/2 → 3d signal of the carbonaceous matrix consists
of two peaks of similar intensities at 853.1 and 854.2 eV, respectively.
In contrast, the Ni 2p3/2→3d signal of the particles
is shifted to lower energy and consists of the main peak at 852.7
eV, with a shoulder at 854.2 eV. Since Ni 2p → 3d resonances
shift to higher energy as the oxidation state of Ni increases,61−64 these observations suggest that the Ni in the carbonaceous matrix
has a higher oxidation state than that of the Ni in the particles.
, showing a relative low
Ni 2p absorption).
These regions are indicated in Figure 3e. The Ni 2p spectra of the Ni–N–C-high
catalyst averaged over the particles (light red) and carbonaceous
matrix (light green) are shown in Figure 4a. The spectra for both the particles and
carbonaceous matrix are dominated by the Ni 2p3/2 →
3d (852–855 eV) and Ni 2p1/2 → 3d (868–872
eV) resonance regions. Compared with the Ni 2p spectrum of the particles,
the carbonaceous matrix has a weaker Ni signal but a higher pre-edge
signal relative to the Ni resonance peak intensities. The Ni 2p3/2 → 3d signal of the carbonaceous matrix consists
of two peaks of similar intensities at 853.1 and 854.2 eV, respectively.
In contrast, the Ni 2p3/2→3d signal of the particles
is shifted to lower energy and consists of the main peak at 852.7
eV, with a shoulder at 854.2 eV. Since Ni 2p → 3d resonances
shift to higher energy as the oxidation state of Ni increases,61−64 these observations suggest that the Ni in the carbonaceous matrix
has a higher oxidation state than that of the Ni in the particles.
Figure 4.

Analysis of the Ni 2p stack of region A1a of the Ni–N–C-high sample. (a) Ni 2p X-ray absorption spectra (XAS) of the particles and the carbonaceous matrix of region A1a of the Ni–N–C-high sample. (b) XAS spectra of Ni(0) NiTPP, O-NiPc, and Ni3S2 on absolute optical density (OD1) scales. Note the scaling used to optimize the visibility of the spectral features. Quantitative component maps of (c) Ni(0), (d) NiTPP, and (e) Ni3S2 derived from an SVD fit. The grayscale indicates the thickness based on the use of absolute OD1 reference spectra. (f) Rescaled color-coded composite of the Ni(0) (dark red), NiTPP (dark green), and Ni3S2 (dark blue) component maps.
To interpret the Ni 2p spectra of the Ni–N–C-high catalyst, the spectra of several reference materials were measured by STXM, including metallic Ni, NiTPP, Ni3S2, NiO, NiCO3, NiSO4(NH4)2SO4, and O-NiPc (Figure S5). A comparison of the Ni 2p spectra of the Ni–N–C-high catalyst to those of the reference spectra indicates that the main peak at 852.7 eV in the spectra of the particles is due to metallic Ni, which is identified as Ni(0) in the following. The position of the peak at 853.1 eV observed in the particles matches that of the main peak in the spectra of Ni3S2, NiO, NiCO3, and NiSO4(NH4)2SO4. Previous X-ray diffraction (XRD) and TEM42 results showed that there is Ni3S2 in the Ni–N–C samples due to the reaction of Ni compounds with the polymerization oxidant, (NH4)2S2O8. Therefore, the peak at 853.1 eV is assigned to Ni3S2 particles. The peak at 854.2 eV aligns with the main peak in the Ni 2p spectra of both NiTPP and O-NiPc. NiTPP and O-NiPc are transition-metal-centered macrocyclic molecules in which Ni(II) ions are coordinated by four nitrogen ligands, with a structure similar to those proposed for atomically dispersed NiNx/C catalysts.12,25 However, the peaks at 854.2 eV in the spectra of the particle and matrix regions are much broader than the main peak in NiTPP and O-NiPc (Figure S5). This strongly suggests that there is a range of chemical environments of the NiNx/C single-atom catalyst sites in the Ni–N–C sample, not only the well-defined NiN4/C structure in NiTPP and O-NiPc (Scheme 1a,b). On the one hand, NiNx/C sites could exist in various forms. NiN/C, NiN2/C, NiN3/C, and NiN4/C have been proposed for the Ni–N–C materials.12 On the other hand, although the first coordination shell is similar in NiTPP and O-NiPc, there is ∼0.1 eV difference in the positions of their main 2p3/2 peak (Table S4). This energy variation has also been observed between Ni(II) phthalocyanine and other Ni(II) porphyrins.64,65 This indicates that the second coordination shell (eight carbon atoms and four nitrogen atoms around the NiN4 center in O-NiPc vs 12 carbon atoms around the NiN4 center in NiTPP and other Ni(II) porphyrins, Scheme 1a,b) affects the chemical and thus catalytic properties of the Ni atoms in NiNx/C sites. Therefore, in the highly heterogeneous Ni–N–C materials, variation in the surrounding chemical environment of different NiNx/C sites likely leads to the broad peak at 854.2 eV.
The reference spectra
of Ni(0), NiTPP, O-NiPc, and Ni3S2 used for
analyzing the Ni 2p spectra of the catalysts
via STXM measurements are displayed on the OD1 intensity scale in Figure 4b. OD1 scales, which
indicate the optical density that 1 nm thickness of a material at
a specified density has at specific photon energy, provide quantitative
analysis. A comparison of these reference spectra to the spectra of
the particles and carbonaceous matrix (Figure 4a) indicates that the particle regions in
the Ni–N–C-high sample are primarily comprised of Ni(0)
along with Ni3S2 and NiNx/C-type sites, whereas the carbonaceous matrix region is
primarily comprised of NiNx/C-type sites,
along with lesser amounts of Ni(0) and Ni3S2. To verify the composition of the particle and carbonaceous matrix
regions, as well as their distribution throughout the Ni–N–C-high
sample, a Ni 2p stack of region A1a was analyzed using
several different sets of reference spectra. Initially, the internal
spectra of the particles and matrix (Figure 4a) were used. The particle and carbonaceous
matrix component maps derived from that analysis (Figure S6) identified the particle and matrix regions more
precisely than using thresholding of the average Ni 2p 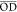 (Figure 3e). While the threshold approach does group spectrally
similar regions and provides a statistically meaningful analysis,
there is considerable subjectivity with respect to identifying the
correct threshold to use for the analysis. To make connections to
the spectroscopy of materials with known composition, we have used
the high-quality OD1 spectra of the Ni-containing reference species
shown in Figure 4b.
Due to the similar structure and almost identical energy position,
the Ni 2p spectrum of either NiTPP or O-NiPc can be used as the external
reference spectrum in the analysis process. Eventually, we selected
the NiTPP for analysis because subsequent STXM studies showed that
the N 1s and C 1s spectra of NiTPP are closer than O-NiPc to that
of the NiNx/C sites in the Ni–N–C
materials (see the Discussion Section). In
the fitting procedure, the NiTPP reference spectrum was modified by
adding a constant, which accounts for X-ray absorption in the Ni 2p
region by carbon- and nitrogen-containing species not directly bonded
to Ni.
(Figure 3e). While the threshold approach does group spectrally
similar regions and provides a statistically meaningful analysis,
there is considerable subjectivity with respect to identifying the
correct threshold to use for the analysis. To make connections to
the spectroscopy of materials with known composition, we have used
the high-quality OD1 spectra of the Ni-containing reference species
shown in Figure 4b.
Due to the similar structure and almost identical energy position,
the Ni 2p spectrum of either NiTPP or O-NiPc can be used as the external
reference spectrum in the analysis process. Eventually, we selected
the NiTPP for analysis because subsequent STXM studies showed that
the N 1s and C 1s spectra of NiTPP are closer than O-NiPc to that
of the NiNx/C sites in the Ni–N–C
materials (see the Discussion Section). In
the fitting procedure, the NiTPP reference spectrum was modified by
adding a constant, which accounts for X-ray absorption in the Ni 2p
region by carbon- and nitrogen-containing species not directly bonded
to Ni.
Figure 4c–e shows the quantitative thickness maps of the Ni(0), NiTPP, and Ni3S2 components derived by fitting the Ni 2p stack. Because OD1 reference spectra were used (Section S5), the intensity grayscale indicted on each component map is an estimate of the thickness in nanometer of the indicated species at each pixel. This does not mean that the NiTPP material in the matrix of the Ni–N–C-high sample is a block of that thickness. Rather, it means that the Ni 2p X-ray absorption by NiNx/C sites in the Ni–N–C-high sample is equivalent to the Ni 2p X-ray absorption of a certain thickness of NiTPP at its standard, bulk density. Figure 4f shows a rescaled color-coded composite of the Ni(0), NiTPP, and Ni3S2 component maps, which shows the spatial distribution of the three species relative to each other. In agreement with the qualitative conclusion derived from the fit to the internal spectra of particles and matrix (Figure S6), the analysis presented in Figure 4 shows that the Ni metal is mainly in the particles, but it is also present at a lower level throughout the matrix. Ni3S2 is mainly in the shell of the particles and the matrix adjacent to the particles. The NiTPP structure, which is interpreted as NiNx/C sites, is mainly dispersed throughout the carbonaceous matrix but is also present at enhanced levels adjacent to some of the particles.
The dominant areas of each of the Ni(0), NiTPP, and Ni3S2 components were identified by threshold-masking the component maps (Figure 4c–e), as shown in Figure S7a. The component-specific masks were used to extract the average spectrum of the Ni(0), NiTPP, and Ni3S2 regions. These spectra were then curve-fit to the external reference spectra to explore the quality of this spectroscopic analysis. Figure S7b compares the internal spectra of each of the masked components to the reference spectra of Ni(0), NiTPP, and Ni3S2. The quality of the curve fit analysis (Figure S7c–e and Table S5) supports the validity of the analysis, as shown in Figure 4. Since the external reference spectra are on quantitative OD1 intensity scales, the curve fit results produce a quantitative estimate of the amount of each reference species (Ni(0), NiTPP, and Ni3S2) in each of the masked regions. In each case, while the component specified is the largest contributor, the other two components also contribute to varying degrees. This reflects the fact that this is a very heterogeneous material and that each of the Ni species is present in all areas, without a fixed ratio or concentration. This quantitative analysis confirms that the particle regions contain a large amount of Ni(0) (average thickness of 30 ± 3 nm), while the matrix contains a very large amount of NiTPP-like species (average thickness of 160 ± 10% nm), which are the NiNx/C single-atom catalyst sites.
STXM spectromicroscopy of region A1a was also performed at the N 1s, C 1s, and O 1s edges. However, most of this region was too thick for X-ray transmission at these lower energies, leading to spectral distortion from absorption saturation. Thus, thinner regions were selected for further measurements to avoid spectral distortions arising from absorption saturation. It is also noted that there was significant spectral distortion from absorption saturation at the N 1s edge for samples supported on SiNx windows, even when measuring very thin areas with low OD. This was likely due to the N present in the SiNx membrane. To overcome this problem, subsequent analysis was performed on Ni–N–C catalyst particles deposited on formvar-coated 3 mm TEM Cu grids.
3.3.2. Ni 2p STXM of Ni–N–C-High on TEM Grids (A2 Region)
Figure 5a shows an STXM image collected at 852.7 eV from region A2, a thin area of the Ni–N–C-high catalyst on a formvar-coated Cu TEM grid (Figure S1d–f). STXM stacks were measured at the Ni 2p, S 2p, C 1s, N 1s, and O 1s edges in this area. Details of the acquisition parameters of each of these stacks are listed in Table S2. The Ni(0), Ni3S2, and NiTPP reference spectra (Figure 4b) were used to fit the Ni 2p stack of region A2. The component maps for Ni(0), NiTPP, and Ni3S2 from that analysis are displayed in Figure S8. Figure 5b presents the rescaled color-coded composite of the Ni(0), NiTPP, and Ni3S2 component maps. The spatial distribution of these components in region A2 is similar to that found in region A1a, with the carbonaceous matrix dominated by NiTPP, the dense regions of the particles dominated by Ni metal, and Ni3S2 adjacent to or colocated with the Ni metal. The Ni(0), NiTPP, and Ni3S2 component maps were threshold-masked (see the insets of Figures 5c and S9a) and Ni 2p spectra of the Ni(0), NiTPP, and Ni3S2 regions were generated (see Figures 5c and S9b).
Figure 5.
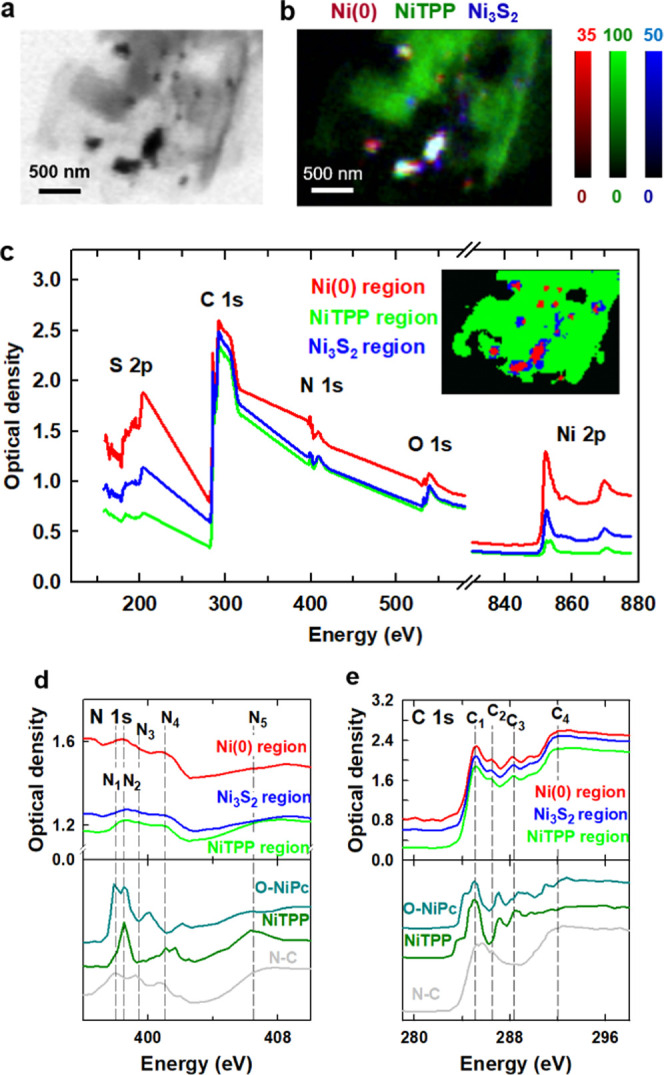
Ni 2p, N 1s, and C 1s results for region A2 of the Ni–N–C-high sample. (a) STXM transmission image at 852.7 eV. (b) Rescaled color-coded composite of the Ni(0) (dark red), NiTPP (dark green), and Ni3S2 (dark blue) component maps derived by a fit of the spectra displayed in Figure 4b to a Ni 2p stack. (c) S 2p, C 1s, N 1s, O 1s, and Ni 2p spectra of Ni(0), NiTPP, and Ni3S2 regions. The areas from Ni(0) (red), NiTPP (green), and Ni3S2 (blue) regions were generated are indicated in the inset. (d) N 1s spectra of the Ni(0), NiTPP, and Ni3S2 regions compared to the N 1s spectra of O-NiPc, NiTPP, and N–C (Ni-free catalyst). (e) C 1s spectra of the Ni(0), NiTPP, and Ni3S2 regions compared to the C 1s spectra of O-NiPc, NiTPP, and N–C (Ni-free).
The averaged spectra for these areas and their fits to the external reference spectra are shown in Figure S9c–e with the results summarized in Table S6. The Ni 2p stack analysis indicates that the composition and distribution of Ni species in region A2 are similar to those in region A1a (Figure 4f), which supports the coexistence of Ni(0), NiTPP, and Ni3S2 throughout the different morphologies of the Ni–N–C-high sample.
3.3.3. N 1s, C 1s, O 1s, and S 2p STXM of Ni–N–C-High on TEM Grids (A2 Region)
In addition to the Ni 2p spectra, Figure 5c plots the S 2p, C 1s, N 1s, and O 1s spectra of the Ni(0), NiTPP, and Ni3S2 regions. The stacks at each absorption edge were combined and coaligned to extract spectra of each edge from exactly the same areas. An expanded presentation of the N 1s spectra of the masked areas is given in Figure 5d, along with the reference spectra for O-NiPc, NiTPP, and N–C, which is the nickel-free material synthesized using the same method as for the Ni–N–C catalysts but without the addition of NiCl2·6H2O. The bonding configurations of the nitrogen atoms can be distinguished by the energy position of the absorption peaks observed in the N 1s spectra. The three distinct peaks, N1 at 398.0 eV, N3 at 399.4 eV, and N4 at 401.0 eV in the N–C sample, are assigned to N 1s → π* transitions of pyridinic, pyrrolic, and graphitic N, respectively.66,67 There is a fifth, broad peak, labeled N5, in the range of 406–409 eV, which is attributed to N 1s → σ*C–N transitions. In the N 1s spectra of the three areas of the Ni–N–C-high sample, two broad peaks are observed at 398.5 eV (N2) and 401.5 eV (N4), indicating that higher amounts of Ni lead to different structures of the N-doped carbon materials. According to other spectral studies,25 the single broad N2 peak, which is located between the pyridinic (N1) and pyrrolic (N3) peaks, exists because the abundant pyrrolic N functionalities coordinating with Ni atoms cause unsaturated pyrrolic nitrogen sites, thus forming a broad convoluted pyridinic/pyrrolic peak, N2.
The N2 feature is also observed in the N 1s spectra of NiTPP and O-NiPc. From comparisons with the spectra of reference compounds, the N 1s spectra of the three areas of the Ni–N–C-high sample are close to that of NiTPP, indicating the NiNx/C species has a structure similar to NiTPP. Especially for the Ni(0) and Ni3S2 regions, the N 1s spectra exhibit sharper N 1s → π* features (N2 and N4) and relatively lower N 1s → σ* transitions (N5). The spectra of NiTPP and O-NiPc showed similar trends with a higher intensity of the N 1s → π* than N 1s → σ* transitions, as well as the same peaks (N2 and N4) at a similar energy position as in the N 1s spectra of the Ni–N–C-high samples. The N 1s spectrum of the NiTPP region in Ni–N–C-high is similar to that of both NiTPP and N–C. We interpret these observations as follows:
-
(1)
Some N species in the Ni–N–C-high samples are N atoms doped into the carbonaceous matrix created by the pyrolysis of the N-containing polymers. This is confirmed by similar N 1s spectra observed for N–C and the NiTPP region, which is the matrix region in Ni–N–C-high, and strong graphitic N peak (N4).66,67 The signal of atomically dispersed NiNx/C species in the Ni–N–C-high samples is convoluted to a degree with the signal from N doped within the carbon materials, causing the broad N2 peak to be less obvious in the NiTPP region.
-
(2)
The observation of the N 1s → π* peaks (N2 and N4) and lower intensity of the N 1s→ σ* transitions (N5) in the spectrum of the Ni(0) area, along with a comparison to the O-NiPc and NiTPP reference spectra, suggests that the NiNx/C sites likely aggregate to an extent in the particle regions, which is consistent with the NiNx/C distribution from the Ni 2p component maps—see Figures 4 and S8 and the quantitative component amounts derived from the Ni 2p spectra (Figures S7, S9 and Tables S5, S6).
-
(3)
The N 1s spectra of the NiNx/C sites, NiTPP, and O-NiPc are different from each other, indicating the existence of a variety of NiNx/C structures. The N atoms in the NiNx/C sites of Ni–N–C-high are closer to those in NiTPP than O-NiPc.
Figure 5e presents the C 1s spectra of the three regions of the Ni–N–C-high sample, in comparison to the C 1s spectra of NiTPP, O-NiPc, and N–C. The C1 (285 eV) and C4 (292 eV) spectral features observed in Ni–N–C-high are attributed to C 1s excitations to π* and σ* graphitic states, respectively.66,67 The strong graphitic-like signal indicates that all areas of the catalyst are highly graphitized. The C3 features in the 287–290 eV region are at positions consistent with C 1s → π*C=N transitions in porphyrins and other compounds with C=N bonds.65,68 These features are particularly clear in the NiTPP and O-NiPc reference spectra, on account of their well-defined structure. Corresponding transitions are observed in the spectra of all regions in Ni–N–C-high but not in the N–C (Ni-free) sample. This indicates that the nitrogen–carbon chemical environment in Ni–N–C-high, NiTPP, and O-NiPc are similar, which is consistent with an involvement of single Ni atom sites, as postulated for the NiNx/C structure. The C2 feature at 286.5 eV, attributed to C 1s → π*C=O transitions, is observed in the spectra of the Ni–N–C-high and N–C samples, indicating the existence of oxygen-containing species in the carbonaceous matrix.
The O 1s spectra for Ni–N–C-high are shown in Figure S10a along with the O 1s reference spectra of N–C and NiO. The O 1s spectra of both Ni–N–C-high and N–C are generally similar, with a sharp O 1s → π*C=O transition (O1) at 532 eV and a broad band at around 540 eV, which is likely the overlap of σ*C–O and σ*C=O peaks (O3). The spectral features of NiO are significantly different, with a sharp peak observed at 534.0 eV (O2) and a broad peak observed at around 540.0 eV. These distinct spectral differences indicate that there is little or no NiO in the Ni–N–C-high catalysts, and the O 1s signal observes likely arises from oxygen-containing unsaturated functional species in the carbonaceous matrix.
The S 2p spectra of Ni–N–C-high are shown in Figure S10b along with the S 2p spectra from the Ni3S2 reference sample. The features at 167.0 eV (S1) and 181.0 eV (S2) in the S 2p spectra show there is S in the Ni–N–C-high sample, with spectral similarity to Ni3S2, an observation consistent with the presence of Ni3S2 in the catalyst as identified previously by XRD.42 The S 2p features are the strongest in the Ni metal and Ni3S2 dominant areas, consistent with our analysis of the Ni 2p stacks.
In summary, for the Ni–N–C-high catalyst sample, the spectral information presented is consistent with a structure consisting of (1) a carbonaceous matrix containing NiNx/C sites distributed throughout a nitrogen-doped and oxygen functionalized graphitic carbonaceous matrix and (2) dense Ni-metal rich particles surrounded by Ni3S2. A representation of the structure for the Ni–N–C-high samples is shown in Scheme 1c.
3.3.4. STXM of Ni–N–C-Low on TEM Grids (B1 and B2 Regions)
To investigate how the morphology and species present in the catalyst depend on the amount of Ni used during synthesis, STXM imaging and spectroscopy at the C 1s, N 1s, O 1s, and Ni 2p edges were performed on the Ni–N–C-low sample (details in Table S2). STXM transmission images at a photon energy of 852.7 eV (above the Ni 2p absorption edge) of two different Ni–N–C-low catalyst particles (regions B1 and B2) are shown in Figure 6a,c, respectively. Ni–N–C-low was found to have very few (in many cases, no) particles throughout the carbonaceous matrix. This is consistent with previous reports, whereby when lower Ni content is used during catalyst synthesis, fewer Ni-rich particles are formed.25,42Figure S11 presents the Ni(0), NiTPP, and Ni3S2 component maps derived from fitting the Ni 2p stack of region B1 to the spectra of the reference compounds (Figure 4b). The rescaled, color-coded composites of the Ni(0), NiTPP. and Ni3S2 component maps are shown in Figure 6b,d. Masks for the regions of the catalyst dominated by Ni(0), NiTPP, and Ni3S2 signals were derived by threshold-masking the component maps for each species in region B1 (Figure S12a). These masks were then used to extract the Ni 2p, N 1s, O 1s, and C 1s spectra of the Ni(0), NiTPP, and Ni3S2 regions of B1 (Figure S13), which were fit to the reference spectra (Figure S12b–e). Based on the Ni 2p, N 1s, O 1s, and C 1s spectra of the Ni metal, NiTPP, and Ni3S2 dominant areas in Ni–N–C-low, the chemical composition of each area in the Ni–N–C-low sample was found to be similar to that in the Ni–N–C-high sample, showing that Ni–N–C electrocatalysts with different Ni contents have a related structure (Scheme 1c) but with fewer Ni-containing particles in the Ni–N–C-low sample. Although the particles consisted of Ni metal and Ni3S2, the structure is different from the core–shell structure shown in Figure 2. We speculated that this is because the low spatial resolution of STXM (∼40 nm) and low X-ray signal for Ni metal/Ni3S2 in a small region made it difficult to completely separate the Ni metal and Ni3S2 regions to show the core–shell structure of the particles. The amount of each chemical species present in the matrix and particle areas of both materials was determined quantitatively and directly compared in the next section.
Figure 6.
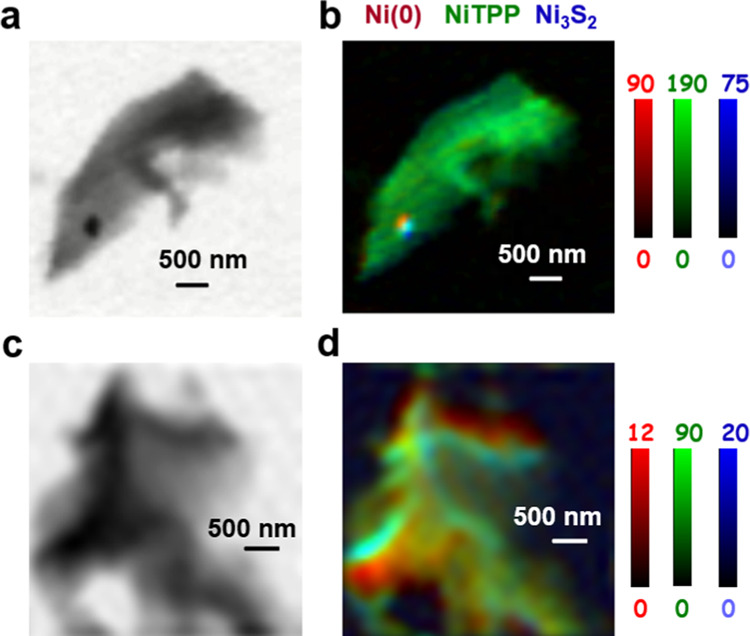
Results for Ni 2p analysis of regions B1 and B2 of the Ni–N–C-low sample. (a) STXM transmission image at 852.7 eV of region B1. (b) Rescaled color-coded composite of the Ni(0) (dark red), NiTPP (dark green), and Ni3S2 (dark blue) component maps for region B1. (c) STXM transmission image at 852.7 eV of region B2. (d) Rescaled color-coded composite for B2.
3.4. Quantitative Results from Ni 2p and C 1s Stack Analyses
An advantage of STXM is that the amount of various chemical species present in any region of a sample can be estimated when quantitative OD1 reference spectra are used in the analysis. The average thicknesses of Ni(0), NiTPP, and Ni3S2 for nine different regions of the catalyst (five from Ni–N–C-high—A1a and A2–A5—and four from Ni–N–C-low—B1–B4) were determined by curve-fitting the average spectra to the quantitative OD1 reference spectra shown in Figure 4b. These results are summarized in Figures S14 and S15, while the stack details for each region of the catalysts that were characterized are listed in Table S2. The volumes of each Ni species were calculated from the average thickness and the geometric cross-sectional area of the catalyst regions. Subsequently, the volumes were converted to mass using the density of the corresponding Ni species. Since Ni(0) and Ni3S2 are well-defined species, it is straightforward to determine the molar amounts for these two species. However, it was more of a challenge to estimate the amount of NiNx/C, the NiTPP-like Ni sites, since NiTPP itself is not present in the pyrolyzed Ni–N–C materials. Therefore, the calculated volume and mass of the NiTPP refers to the amount of NiTPP that would have the same X-ray absorption as the NiNx/C species. The molar amount of Ni in NiNx/C sites was taken to be that of NiTPP, derived from the mass and volume of NiTPP determined from the curve fit and the area in the NiTPP component maps. To quantitatively compare the amounts of NiNx/C, Ni(0), and Ni3S2 in the Ni–N–C samples, the mole fraction of the Ni atoms present in each of these species was calculated. Furthermore, to understand the absolute content of NiNx/C sites, the atomic ratio of the Ni atoms in NiNx/C to the total number of C atoms in the Ni–N–C samples was estimated using the same methods. In this way, although the Ni loading (the amount of all Ni atoms) is different in the different Ni–N–C materials, the concentration of NiNx/C species in different areas and different samples was determined by comparing the absolute content of NiNx/C sites to the total number of C atoms. The calculated results for the nine areas are summarized in Table S7. More details of the calculations are presented in Sections S11 and S12.
The atomic fractions of Ni(0), NiNx/C, and Ni3S2 determined from the Ni 2p data and the ratio of the number of Ni atoms in NiNx/C sites to the total number of C atoms in the Ni–N–C-high samples in regions A1a–A5 of the Ni–N–C-high sample are displayed in Figure S14. The corresponding fractions and ratios for regions B1–B4 of the Ni–N–C-low samples are displayed in Figure S15. The proportions of Ni atoms in Ni–N–C-high and Ni–N–C-low samples that are present in the Ni(0), NiNx/C, and Ni3S2 domains, averaged over all regions, are presented as bar graphs in Figure 7a. For Ni–N–C-high, 16 ± 5, 69 ± 7, and 15 ± 5 mol % of the total number of Ni atoms in the catalyst are present in the Ni(0), NiNx/C, and Ni3S2 domains, respectively, In the Ni–N–C-low sample, the average content of Ni(0) and Ni3S2 is reduced from 16 to 12 ± 4 mol % and from 15 to 8 ± 3%, respectively, while the mole fraction of NiNx/C is increased from 69 to 80 ± 8%. The estimated uncertainties include contributions from statistical variation and systematic errors, the largest of which is likely the degree to which the reference materials actually correspond to the species present in the catalysts.
Figure 7.
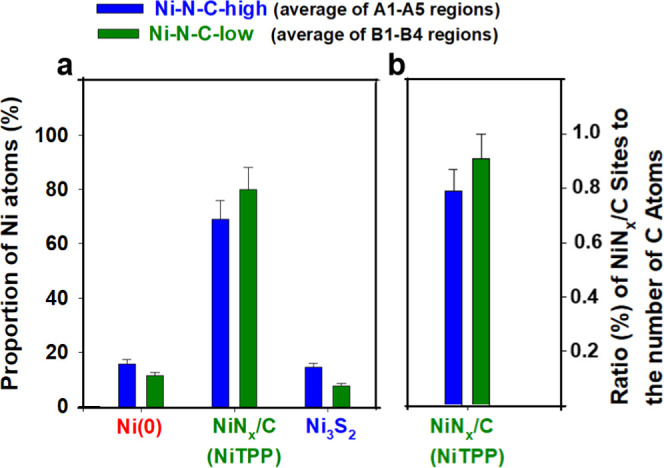
Group bar chart representing the quantitative distribution of different Ni-sites averaged over the five regions of the Ni–N–C-high and four regions of the Ni–N–C-low sample. (a) Total proportion of Ni atoms (out of all the Ni atoms in the sample) present in the domains shown on the x-axis, for both catalysts investigated. (b) Ratio of the total number of NiNx/C sites present in the catalyst divided by the total number of carbon atoms present in the catalyst, providing a quantitative gauge of NiNx/C species concentration. Values are averaged over the five regions of the Ni–N–C-high and four regions of the Ni–N–C-low sample. The error bars take into account statistical and estimated systematic uncertainties.
Despite differences in the relative mole fraction of Ni atoms present in NiNx/C between Ni–N–C-high and Ni–N–C-low, it is advantageous to be able to gauge the quantitative content of NiNx/C in the catalyst materials. To accomplish this, the atomic ratio of the number of Ni atoms present in NiNx/C to the total number of C atoms in the Ni–N–C-high and Ni–N–C-low was determined. We define this as the “absolute ratio (%) of NiNx/C sites”. As presented in Figure 7b, relative to the Ni–N–C-high sample, the Ni–N–C-low sample has a slightly higher amount of NiNx/C sites, with an average value of 0.91 ± 0.09% for the absolute ratio (%) of NiNx/C sites, compared with an average of 0.79 ± 0.08% for the Ni–N–C-high sample. Therefore, the absolute number of NiNx/C sites increased when using a lower Ni loading during catalyst synthesis, while the absolute amounts of Ni metal and Ni3S2 greatly decreased. In addition, the high heterogeneity of Ni–N–C catalysts can be confirmed by the different quantitative results in each region, as shown in Figures S14 and S15. This heterogeneity has often been overlooked by other bulk average and spatially localized measurements. This is especially prominent for Ni–N–C-high, which demonstrates a relatively large fluctuation in the Ni atomic fractions of Ni(0), NiNx/C, and Ni3S2 and the absolute ratio (%) of NiNx/C sites in the different regions of the catalyst in comparison to Ni–N–C-low. This observation indicates that the presence of dense Ni(0)/Ni3S2 particles contributes to increasing the heterogeneity of Ni–N–C materials. The resulting high degree of heterogeneity therefore introduces challenges for quantitative estimation of the chemical species present in Ni–N–C-high materials because the calculated results are dependent on the region of the catalyst that is characterized. Although averaging results from multiple regions improves the statistical precision, the preference to select relatively thin catalyst particles to avoid absorption saturation (such as the selection of the A1a region in A1) could result in the calculated amounts of Ni(0) and Ni3S2 being lower than what is actually present in Ni–N–C-high. Thus, the calculated proportions of different species, more specifically the Ni atomic fraction of the NiNx/C species and the absolute ratio (%) of NiNx/C Sites, are likely overestimated in Ni–N–C-high, and therefore, the differences between Ni–N–C-high and Ni–N–C-low are likely more pronounced than what is shown in Figure 7a,b.
To make sure our quantitative analysis is reliable, we have compared the spectroscopic results of STXM to ensemble-averaged XAS-TEY spectra—see Section S14, Supporting Information. The similarity of the TEY-XAS and STXM results confirms that, despite measuring an extremely small volume, STXM can measure the average properties of heterogeneous materials. However, a key advantage of STXM is that it probes the chemistry of individual catalyst particles with sub-50 nm spatial resolution, even when the species of interest (i.e., NiNx/C) contribute only a portion of the overall spectral features. For example, consider the Ni–N–C-high data shown in Figure S17a. When interpreting the TEY-XAS data, the Ni3S2 contribution at 853.1 eV could be easily overlooked in an ensemble-averaged XAS study as it is largely overshadowed by the neighboring Ni(0) peak at 852.7 eV. On the contrary, with the spatial resolution of STXM it is possible to identify and differentiate these individual chemical species, which allows for mapping of their chemical signals and distributions throughout the catalyst structure. After using Ni 2p STXM stacks to characterize the spatial distribution of specific species, stacks at other edges can be measured over the same area, thereby gaining a more complete understanding of the chemical properties and how they vary throughout the catalyst (e.g., Figures 5 and S13). More importantly, based on this comprehensive understanding, enabled by location-dependent spectral interpretation, the amount of various chemical species present in the catalysts can be quantified (e.g., Figures 7, S14, and S15). Achieving this level of insight is not possible using ensemble-averaged measurements such as X-ray absorption spectroscopy with limited or no spatial resolution. This comparison demonstrates the significant advantage of STXM for generating fundamental insight into catalyst structures and properties that are essential for guiding advanced material designs.
4. Discussion
These STXM results provide spatially resolved spectral insight into the various chemical species present in these two heterogeneous Ni–N–C catalysts. Metallic Ni(0), Ni3S2, and atomically dispersed NiNx/C species, structurally similar to NiTPP (and O-NiPc), were identified, spatially mapped, and quantified. The atomically dispersed NiNx/C species were found to be present throughout the entire carbonaceous matrix of the catalyst structure. The observation of these NiNx/C sites is consistent with previous investigations of Ni–N–C catalysts using various techniques including XAS without spatial resolution30−33 and STEM-EELS.12−17 STXM provides an opportunity to image and quantify the distribution of the various Ni-containing species over mesoscale regions of the catalyst structure. The electrochemical CO2 reduction testing of the Ni–N–C-low catalyst demonstrated a high selectivity toward CO. Based on the fact that Ni3S2 and Ni(0) selectively produce hydrogen under CO2 reduction conditions, the electrochemical results, combined with the STXM analysis, indicate that the activity of the Ni–N–C electrocatalyst for CO2 reduction to CO arises from the NiNx/C sites present throughout the entire carbonaceous matrix. The calculated fraction of NiNx/C sites in the two Ni–N–C catalysts with different Ni contents showed that, as the Ni content in the Ni–N–C electrocatalyst decreased, the amount of NiNx/C sites increased. This is desirable from an electrocatalytic CO2R standpoint, as an increased concentration of NiNx/C provides increased activity and selectivity toward CO production.
A comparison of the Ni 2p, N 1s, and C 1s spectra of the Ni–N–C materials to those of O-NiPc and NiTPP reference compounds showed that a variety of NiNx/C structures exist in the Ni–N–C catalysts. In the Ni 2p spectra of O-NiPc and NiTPP, the main peaks observed at 854.1 and 854.2 eV, respectively, are quite sharp (Figure S5) due to the well-defined local coordination of Ni in these pure reference compounds. For Ni–N–C (Figure 4), the peak at 854.2 eV is quite broad, likely indicating the existence of NiNx/C sites with different chemical environments. The high energy of the Ni 2p → 3d transition for NiNx/C sites is because of a decrease in the effective number of Ni 3d electrons due to the strong Ni 3d–N 2p covalent bonding.64 With different numbers of N atoms bonding to Ni (NiN/C, NiN2/C, NiN3/C, and NiN4/C have all been proposed for the Ni–N–C materials12), the electronic structure of the Ni atom would also vary, causing a shift in the peak location around a mean NiN4/C peak energy position (∼854.2 eV), thereby forming a broad peak.69 Similarly, different numbers of C atoms in the second shell around the Ni atom in NiNx/C will affect the Ni 3d electron distributions. For example, the 8 C and 4 N second-shell atoms in O-NiPc will shift the Ni 2p →3d transition to a higher energy than the 12 second-shell C atoms in NiTPP, as is evidenced by a 0.1 eV higher energy in O-NiPc than NiTPP. A comparison of the Ni 2p, N 1s, and C 1s spectra of the NiNx/C sites to those of O-NiPc and NiTPP indicates that the local coordination of Ni in the NiNx/C sites is closer to NiTPP than O-NiPc (Scheme 1c). An important and challenging question then arises: what is the impact of different possible Ni–Nx/C sites on catalytic activity?. While it is out of the scope of this work, the scientific community could potentially work toward elucidating this information by methods, such as (1) systematic investigations of well-defined model systems such as synthesized Ni-based macrocycles, although structural and electronic differences between such model compounds and catalytic structures in Ni–N–C materials must be taken into account; (2) preparation of structurally homogeneous Ni–N–C catalyst consisting of a single identity of active site structures, that comes with a wide array of synthetic challenges; and (3) computational simulations, such as by density functional theory calculations to understand how the active site structure and surrounding environment of Ni–Nx/C sites impact catalytic activity by tuning adsorption energies with reactive intermediates (i.e., adsorbed *CO)
These results clearly show the power of STXM to perform spatially resolved, mesoscale studies of the morphology and chemical structures using precise spectroscopic characterization at various edges, thereby identifying the chemical species, mapping their spatial distribution, and quantitatively estimating their content. One of the drawbacks of STXM is that measurements are conducted on individual micron or sub-micron catalyst particles, which, although larger than the sub-nm regions probed by STEM, still may not capture all of the heterogeneity of the global catalyst structure such as that provided by ensemble-averaged XAS measurements that can measure thousands of particles at a time. This limitation can be mitigated by conducting measurements on multiple regions of the catalyst and performing statistical analyses. However, this may not always be necessary as we showed that the Ni 2p spectra collected by STXM on a single catalyst particle had only very subtle differences from bulk-averaged spectra collected by TEY-XAS. Therefore, STXM measurements can largely overcome the issues associated with very spatially localized measurements, such as the nanometer-scale measurements conducted by STEM and related analytical techniques (i.e., EELS). Another limitation of STXM is the spatial resolution, which is much lower than that provided by TEM. To bridge the gap, soft X-ray ptychography70−74 can be used to significantly improve the spatial resolution of STXM. Ptychography measurements at metal 2p edges in the 600–1100 eV energy range are able to achieve sub-10 nm spatial resolution,70−72 with a record spatial resolution of 3 nm.70 Based on very recent work at Soleil,75 ptychography at energies below 500 eV is now also possible, enabling ptychographic measurements of Ni–N–C catalysts at the N 1s and C 1s edges. Our initial exploration of applying ptychography to catalysts has shown that a spatial resolution of ∼10 nm can be achieved with a similar measurement time to conventional STXM. As an indication of the resolution gains that ptychography can provide, Figure S19 presents a ptychographic amplitude image at 852.7 eV of the A2 region of the Ni–N–C-high sample, along with an evaluation of its spatial resolution. In addition to ptychography, we are performing in situ STXM characterization of these electrocatalyst materials using a microfluidic flow-electrochemical system developed by our team.43,76 This will allow determination of the chemical species present and their spatial distribution in catalyst materials under relevant operating conditions, thereby providing better mechanistic understanding and guiding new catalyst designs.77
Overall, this work demonstrates that STXM is a very effective tool for studying catalysts such as Ni–N–C, where their highly heterogeneous nature leads to difficulty in characterization by nonspatially resolved methods. The combination of approaches used in this work has led to an improved fundamental understanding of the CO2–reduction active species in this Ni–N–C CO2R electrocatalyst.
5. Conclusions
Gas product analysis measurements showed that lower concentrations of Ni used in the synthesis of Ni–N–C catalysts provide a higher activity for CO2 reduction and higher selectivity for CO production. To understand this seemingly contradictory observation, we have carried out a comprehensive correlative multi-characterization study. The electrocatalytic properties suggested the existence of atomic NiNx/C sites. STEM-HAADF imaging showed the presence of atomically dispersed high Z elements (likely Ni) in the carbonaceous matrix of Ni–N–C samples. STXM was applied to further investigate the identity and concentrations of chemical species (especially NiNx/C sites) present. These materials were found to be highly heterogeneous, consisting of Ni(0) and Ni3S2 particles embedded in a carbonaceous matrix with NiNx/C sites dispersed throughout. The chemical environments, amounts, and spatial distributions of Ni species present in samples prepared with two different Ni loadings were evaluated quantitatively by analyzing sequences of X-ray absorption images at the Ni 2p, N 1s, C 1s, O 1s, and S 2p edges using reference X-ray absorption spectra of Ni(0), NiTPP, O-NiPc, and Ni3S2. The results show that, as the Ni loading increases, the amount of Ni(0) and Ni3S2 species increases but the amount of NiNx/C sites decreases. This explains why the Ni–N–C sample prepared with low Ni loading has better electrocatalytic CO2R performance. In addition, the spectra of the Ni–N–C catalysts showed the existence of a variety of NiNx/C structures, with the measured spectra being more similar to the spectra of NiTPP than O-NiPc reference compounds. The results provide insight into how the structures, compositions, and spatial distributions of chemical species present in these Ni–N–C electrocatalysts might be modified to achieve higher activity and improved selectivity for CO production via electrochemical CO2R. This is the first report of using STXM for characterizing atomically dispersed M–N–C catalysts. It has demonstrated the capabilities and advantages of soft X-ray STXM for characterizing atomically dispersed single-atom catalytic sites. In combination with other methods, STXM can generate fundamental insights to guide the design of improved performance Ni–N–C CO2R catalysts.
Acknowledgments
The authors thank Prof. Daniel Leznoff from Simon Fraser University for providing the reference sample of octa α-octabutoxy Ni phthalocyanine (O-NiPc). This research was supported by the Natural Sciences and Engineering Research Council (NSERC) Discovery program, and the National Research Council of Canada (NRC) Materials for Clean Fuels Challenge program. Measurements were performed using the ambient STXM on beamline 10ID1 and the SGM spectrometer on beamline 11ID1 at the Canadian Light Source (CLS). CLS is supported by the CFI Major Facilities Access program and the University of Saskatchewan. The authors thank staff scientists Jian Wang and Jay Dynes at CLS for their assistance and support of the 10ID1 beamline and STXMs and Jay Dynes for measuring the TEY spectra using the SGM beamline. Electron microscopy imaging was performed at the Canadian Centre for Electron Microscopy (CCEM) at McMaster University. The authors thank Natalie Hamada for her assistance in electron microscopy measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.2c01255.
Additional experimental details (images of samples, summary of measurements), data analysis methods, further supporting results (STXM, (S)TEM, and ensemble-averaged total electron yield X-ray absorption spectroscopy), quantitative analysis details, and ptychography results (PDF)
Author Present Address
§ Electrical and Computer Engineering, University of Victoria, Victoria, British Columbia, Canada V8P 5C2
The authors declare no competing financial interest.
Notes
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplementary Material
References
- Ju W.; Bagger A.; Hao G.-P.; Varela A. S.; Sinev I.; Bon V.; Roldan Cuenya B.; Kaskel S.; Rossmeisl J.; Strasser P. Understanding Activity and Selectivity of Metal-Nitrogen-Doped Carbon Catalysts for Electrochemical Reduction of CO2. Nat. Commun. 2017, 8, 944 10.1038/s41467-017-01035-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirth A. A.; Varnell J. A.; DiAscro A. M. Nonprecious Metal Catalysts for Oxygen Reduction in Heterogeneous Aqueous Systems. Chem. Rev. 2018, 118, 2313–2339. 10.1021/acs.chemrev.7b00335. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Xia Z.; Dai L. Carbon-Based Electrocatalysts for Advanced Energy Conversion and Storage. Sci. Adv. 2015, 1, e1500564 10.1126/sciadv.1500564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z.; Yang W.; Gu Y.; Liao T.; Sun Z. Metal-Nitrogen-Doped Carbon Materials as Highly Efficient Catalysts: Progress and Rational Design. Adv. Sci. 2020, 7, 2001069 10.1002/advs.202001069. [DOI] [Google Scholar]
- Chen M.-X.; Zhu M.; Zuo M.; Chu S.-Q.; Zhang J.; Wu Y.; Liang H.-W.; Feng X. Identification of Catalytic Sites for Oxygen Reduction in Metal/Nitrogen-Doped Carbons with Encapsulated Metal Nanoparticles. Angew. Chem. 2020, 132, 1644–1650. 10.1002/ange.201912275. [DOI] [PubMed] [Google Scholar]
- Jaouen F.; Proietti E.; Lefèvre M.; Chenitz R.; Dodelet J.-P.; Wu G.; Taek Chung H.; Marie Johnston C.; Zelenay P. Recent Advances in Non-Precious Metal Catalysis for Oxygen - Reduction Reaction in Polymer Electrolyte Fuel Cells. Energy Environ. Sci. 2011, 4, 114–130. 10.1039/C0EE00011F. [DOI] [Google Scholar]
- Xia C.; Qiu Y.; Xia Y.; Zhu P.; King G.; Zhang X.; Wu Z.; Kim J. Y.; Cullen D. A.; Zheng D.; Li P.; Shakouri M.; Heredia E.; Cui P.; Alshareef H. N.; Hu Y.; Wang H. General Synthesis of Single-Atom Catalysts with High Metal Loading Using Graphene Quantum Dots. Nat. Chem. 2021, 13, 887–894. 10.1038/s41557-021-00734-x. [DOI] [PubMed] [Google Scholar]
- Franco F.; Rettenmaier C.; Sang Jeon H.; Cuenya B. R. Transition Metal-Based Catalysts for the Electrochemical CO2 Reduction: From Atoms and Molecules to Nanostructured Materials. Chem. Soc. Rev. 2020, 49, 6884–6946. 10.1039/D0CS00835D. [DOI] [PubMed] [Google Scholar]
- Li J.; Jiang Y.; Wang Q.; Xu C.-Q.; Wu D.; Banis M. N.; Adair K. R.; Doyle-Davis K.; Meira D. M.; Finfrock Y. Z.; Li W.; Zhang L.; Sham T.-K.; Li R.; Chen N.; Gu M.; Li J.; Sun X. A General Strategy for Preparing Pyrrolic-N4 Type Single-Atom Catalysts via Pre-Located Isolated Atoms. Nat. Commun. 2021, 12, 6806 10.1038/s41467-021-27143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela A. S.; Kroschel M.; Leonard N. D.; Ju W.; Steinberg J.; Bagger A.; Rossmeisl J.; Strasser P. PH Effects on the Selectivity of the Electrocatalytic CO2 Reduction on Graphene-Embedded Fe–N–C Motifs: Bridging Concepts between Molecular Homogeneous and Solid-State Heterogeneous Catalysis. ACS Energy Lett. 2018, 3, 812–817. 10.1021/acsenergylett.8b00273. [DOI] [Google Scholar]
- Qu Y.; Li Z.; Chen W.; Lin Y.; Yuan T.; Yang Z.; Zhao C.; Wang J.; Zhao C.; Wang X.; Zhou F.; Zhuang Z.; Wu Y.; Li Y. Direct Transformation of Bulk Copper into Copper Single Sites via Emitting and Trapping of Atoms. Nat. Catal. 2018, 1, 781–786. 10.1038/s41929-018-0146-x. [DOI] [Google Scholar]
- Koshy D. M.; Landers A. T.; Cullen D. A.; Ievlev A. V.; Meyer H. M. III; Hahn C.; Bao Z.; Jaramillo T. F. Direct Characterization of Atomically Dispersed Catalysts: Nitrogen-Coordinated Ni Sites in Carbon-Based Materials for CO2 Electroreduction. Adv. Energy Mater. 2020, 10, 2001836 10.1002/aenm.202001836. [DOI] [Google Scholar]
- Xie X.; He C.; Li B.; He Y.; Cullen D. A.; Wegener E. C.; Kropf A. J.; Martinez U.; Cheng Y.; Engelhard M. H.; Bowden M. E.; Song M.; Lemmon T.; Li X. S.; Nie Z.; Liu J.; Myers D. J.; Zelenay P.; Wang G.; Wu G.; Ramani V.; Shao Y. Performance Enhancement and Degradation Mechanism Identification of a Single-Atom Co–N–C Catalyst for Proton Exchange Membrane Fuel Cells. Nat. Catal. 2020, 3, 1044–1054. 10.1038/s41929-020-00546-1. [DOI] [Google Scholar]
- Sharma P.; Kumar S.; Tomanec O.; Petr M.; Zhu Chen J.; Miller J. T.; Varma R. S.; Gawande M. B.; Zbořil R. Carbon Nitride-Based Ruthenium Single Atom Photocatalyst for CO2 Reduction to Methanol. Small 2021, 17, 2006478 10.1002/smll.202006478. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Wang Q.; Si R.; Song Z.; Lin X.; Banis M. N.; Adair K.; Li J.; Doyle-Davis K.; Li R.; Liu L.-M.; Gu M.; Sun X. New Insight of Pyrrole-Like Nitrogen for Boosting Hydrogen Evolution Activity and Stability of Pt Single Atoms. Small 2021, 17, 2004453 10.1002/smll.202004453. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Higgins D.; Yu A.; Zhang L.; Zhang J. A Review on Non-Precious Metal Electrocatalysts for PEM Fuel Cells. Energy Environ. Sci. 2011, 4, 3167–3192. 10.1039/c0ee00558d. [DOI] [Google Scholar]
- Chung H. T.; Cullen D. A.; Higgins D.; Sneed B. T.; Holby E. F.; More K. L.; Zelenay P. Direct Atomic-Level Insight into the Active Sites of a High-Performance PGM-Free ORR Catalyst. Science 2017, 357, 479–484. 10.1126/science.aan2255. [DOI] [PubMed] [Google Scholar]
- Zitolo A.; Goellner V.; Armel V.; Sougrati M.-T.; Mineva T.; Stievano L.; Fonda E.; Jaouen F. Identification of Catalytic Sites for Oxygen Reduction in Iron- and Nitrogen-Doped Graphene Materials. Nat. Mater. 2015, 14, 937–942. 10.1038/nmat4367. [DOI] [PubMed] [Google Scholar]
- Bezerra C. W. B.; Zhang L.; Lee K.; Liu H.; Marques A. L. B.; Marques E. P.; Wang H.; Zhang J. A Review of Fe–N/C and Co–N/C Catalysts for the Oxygen Reduction Reaction. Electrochim. Acta 2008, 53, 4937–4951. 10.1016/j.electacta.2008.02.012. [DOI] [Google Scholar]
- Jia Q.; Ramaswamy N.; Tylus U.; Strickland K.; Li J.; Serov A.; Artyushkova K.; Atanassov P.; Anibal J.; Gumeci C.; Barton S. C.; Sougrati M.-T.; Jaouen F.; Halevi B.; Mukerjee S. Spectroscopic Insights into the Nature of Active Sites in Iron–Nitrogen–Carbon Electrocatalysts for Oxygen Reduction in Acid. Nano Energy 2016, 29, 65–82. 10.1016/j.nanoen.2016.03.025. [DOI] [Google Scholar]
- Jia Q.; Ramaswamy N.; Hafiz H.; Tylus U.; Strickland K.; Wu G.; Barbiellini B.; Bansil A.; Holby E. F.; Zelenay P.; Mukerjee S. Experimental Observation of Redox-Induced Fe–N Switching Behavior as a Determinant Role for Oxygen Reduction Activity. ACS Nano 2015, 9, 12496–12505. 10.1021/acsnano.5b05984. [DOI] [PubMed] [Google Scholar]
- Kramm U. I.; Herranz J.; Larouche N.; Arruda T. M.; Lefèvre M.; Jaouen F.; Bogdanoff P.; Fiechter S.; Abs-Wurmbach I.; Mukerjee S.; Dodelet J.-P. Structure of the Catalytic Sites in Fe/N/C-Catalysts for O2 Reduction in PEM Fuel Cells. Phys. Chem. Chem. Phys. 2012, 14, 11673–11688. 10.1039/c2cp41957b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela A. S.; Ju W.; Strasser P. Molecular Nitrogen–Carbon Catalysts, Solid Metal Organic Framework Catalysts, and Solid Metal/Nitrogen-Doped Carbon (MNC) Catalysts for the Electrochemical CO2 Reduction. Adv. Energy Mater. 2018, 8, 1802905 10.1002/aenm.201802905. [DOI] [Google Scholar]
- Li X.; Bi W.; Chen M.; Sun Y.; Ju H.; Yan W.; Zhu J.; Wu X.; Chu W.; Wu C.; Xie Y. Exclusive Ni–N4 Sites Realize Near-Unity CO Selectivity for Electrochemical CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 14889–14892. 10.1021/jacs.7b09074. [DOI] [PubMed] [Google Scholar]
- Koshy D. M.; Chen S.; Lee D. U.; Stevens M. B.; Abdellah A. M.; Dull S. M.; Chen G.; Nordlund D.; Gallo A.; Hahn C.; Higgins D. C.; Bao Z.; Jaramillo T. F. Understanding the Origin of Highly Selective CO2 Electroreduction to CO on Ni,N-Doped Carbon Catalysts. Angew. Chem., Int. Ed. 2020, 59, 4043–4050. 10.1002/anie.201912857. [DOI] [PubMed] [Google Scholar]
- Yang H. B.; Hung S.-F.; Liu S.; Yuan K.; Miao S.; Zhang L.; Huang X.; Wang H.-Y.; Cai W.; Chen R.; Gao J.; Yang X.; Chen W.; Huang Y.; Chen H. M.; Li C. M.; Zhang T.; Liu B. Atomically Dispersed Ni(I) as the Active Site for Electrochemical CO2 Reduction. Nat. Energy 2018, 3, 140–147. 10.1038/s41560-017-0078-8. [DOI] [Google Scholar]
- Yasri N. G.; Al-Attas T. A.; Hu J.; Golam Kibria M. Electropolymerized Metal-Protoporphyrin Electrodes for Selective Electrochemical Reduction of CO2. Catal. Sci. Technol. 2021, 11, 1580–1589. 10.1039/D0CY02150D. [DOI] [Google Scholar]
- Lefèvre M.; Dodelet J. P.; Bertrand P. O2 Reduction in PEM Fuel Cells: Activity and Active Site Structural Information for Catalysts Obtained by the Pyrolysis at High Temperature of Fe Precursors. J. Phys. Chem. B 2000, 104, 11238–11247. 10.1021/jp002444n. [DOI] [Google Scholar]
- Lefèvre M.; Dodelet J. P.; Bertrand P. Molecular Oxygen Reduction in PEM Fuel Cells: Evidence for the Simultaneous Presence of Two Active Sites in Fe-Based Catalysts. J. Phys. Chem. B 2002, 106, 8705–8713. 10.1021/jp020267f. [DOI] [Google Scholar]
- Zhao S.; Cheng Y.; Veder J.-P.; Johannessen B.; Saunders M.; Zhang L.; Liu C.; Chisholm M. F.; De Marco R.; Liu J.; Yang S.-Z.; Jiang S. P. One-Pot Pyrolysis Method to Fabricate Carbon Nanotube Supported Ni Single-Atom Catalysts with Ultrahigh Loading. ACS Appl. Energy Mater. 2018, 1, 5286–5297. 10.1021/acsaem.8b00903. [DOI] [Google Scholar]
- Wang C.; Zhang H.; Wang J.; Zhao Z.; Wang J.; Zhang Y.; Cheng M.; Zhao H.; Wang J. Atomic Fe Embedded in Carbon Nanoshells–Graphene Nanomeshes with Enhanced Oxygen Reduction Reaction Performance. Chem. Mater. 2017, 29, 9915–9922. 10.1021/acs.chemmater.7b03100. [DOI] [Google Scholar]
- Tylus U.; Jia Q.; Strickland K.; Ramaswamy N.; Serov A.; Atanassov P.; Mukerjee S. Elucidating Oxygen Reduction Active Sites in Pyrolyzed Metal–Nitrogen Coordinated Non-Precious-Metal Electrocatalyst Systems. J. Phys. Chem. C 2014, 118, 8999–9008. 10.1021/jp500781v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Wang Z.; Zhu Z. The Inherent Kinetic Electrochemical Reduction of Oxygen into H2O on FeN4-Carbon: A Density Functional Theory Study. J. Power Sources 2014, 255, 65–69. 10.1016/j.jpowsour.2014.01.008. [DOI] [Google Scholar]
- Egerton R. F.; Watanabe M. Characterization of Single-Atom Catalysts by EELS and EDX Spectroscopy. Ultramicroscopy 2018, 193, 111–117. 10.1016/j.ultramic.2018.06.013. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Yang Y.; Schimmenti R.; Murray E.; Peng H.; Wang Y.; Ge C.; Jiang W.; Wang G.; DiSalvo F. J.; Muller D. A.; Mavrikakis M.; Xiao L.; Abruña H. D.; Zhuang L. A Completely Precious Metal–Free Alkaline Fuel Cell with Enhanced Performance Using a Carbon-Coated Nickel Anode. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2119883119 10.1073/pnas.2119883119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Mohd Adli N.; Shan W.; Wang M.; Zachman M. J.; Hwang S.; Tabassum H.; Karakalos S.; Feng Z.; Wang G.; Li Y. C.; Wu G. Atomically Dispersed Single Ni Site Catalysts for High-Efficiency CO2 Electroreduction at Industrial-Level Current Densities. Energy Environ. Sci. 2022, 15, 2108–2119. 10.1039/D2EE00318J. [DOI] [Google Scholar]
- Shi Q.; Hwang S.; Yang H.; Ismail F.; Su D.; Higgins D.; Wu G. Supported and Coordinated Single Metal Site Electrocatalysts. Mater. Today 2020, 37, 93–111. 10.1016/j.mattod.2020.02.019. [DOI] [Google Scholar]
- Stöhr J.NEXAFS Spectroscopy, Springer Series in Surface Science; Springer, 1992; Vol. 25. [Google Scholar]
- Ade H.; Hitchcock A. P. NEXAFS Microscopy and Resonant Scattering: Composition and Orientation Probed in Real and Reciprocal Space. Polymer 2008, 49, 643–675. 10.1016/j.polymer.2007.10.030. [DOI] [Google Scholar]
- Hitchcock A. P.Soft X-ray Imaging and Spectromicroscopy. In Handbook of Nanoscopy; John Wiley & Sons, Ltd., 2012; pp 745–791. [Google Scholar]
- Hitchcock A. P. Soft X-ray Spectromicroscopy and Ptychography. J. Electron Spectrosc. Relat. Phenom. 2015, 200, 49–63. 10.1016/j.elspec.2015.05.013. [DOI] [Google Scholar]
- Shahcheraghi L.; Zhang C.; Lee H.-J.; Cusack-Striepe M.; Ismail F.; Abdellah A.; Higgins D. C. Identifying Activity and Selectivity Trends for the Electrosynthesis of Hydrogen Peroxide via Oxygen Reduction on Nickel–Nitrogen–Carbon Catalysts. J. Phys. Chem. C 2021, 125, 15830–15840. 10.1021/acs.jpcc.1c03775. [DOI] [Google Scholar]
- Prabu V.; Obst M.; Hosseinkhannazer H.; Reynolds M.; Rosendahl S.; Wang J.; Hitchcock A. P. Instrumentation for in Situ Flow Electrochemical Scanning Transmission X-Ray Microscopy (STXM). Rev. Sci. Instrum. 2018, 89, 063702 10.1063/1.5023288. [DOI] [PubMed] [Google Scholar]
- Kuhl K. P.; Cave E. R.; Abram D. N.; Jaramillo T. F. New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. 10.1039/c2ee21234j. [DOI] [Google Scholar]
- Higgins D.; Landers A. T.; Ji Y.; Nitopi S.; Morales-Guio C. G.; Wang L.; Chan K.; Hahn C.; Jaramillo T. F. Guiding Electrochemical Carbon Dioxide Reduction toward Carbonyls Using Copper Silver Thin Films with Interphase Miscibility. ACS Energy Lett. 2018, 3, 2947–2955. 10.1021/acsenergylett.8b01736. [DOI] [Google Scholar]
- Ismail F.; Abdellah A.; Lee H.-J.; Sudheeshkumar V.; Alnoush W.; Higgins D. C. Impact of Nickel Content on the Structure and Electrochemical CO2 Reduction Performance of Nickel–Nitrogen–Carbon Catalysts Derived from Zeolitic Imidazolate Frameworks. ACS Appl. Energy Mater. 2022, 5, 430–439. 10.1021/acsaem.1c02522. [DOI] [Google Scholar]
- Leverett J.; Yuwono J. A.; Kumar P.; Tran-Phu T.; Qu J.; Cairney J.; Wang X.; Simonov A. N.; Hocking R. K.; Johannessen B.; Dai L.; Daiyan R.; Amal R. Impurity Tolerance of Unsaturated Ni-N-C Active Sites for Practical Electrochemical CO2 Reduction. ACS Energy Lett. 2022, 7, 920–928. 10.1021/acsenergylett.1c02711. [DOI] [Google Scholar]
- Kaznatcheev K. V.; Karunakaran C.; Lanke U. D.; Urquhart S. G.; Obst M.; Hitchcock A. P. Soft X-Ray Spectromicroscopy Beamline at the CLS: Commissioning Results. Nucl. Instrum. Methods Phys. Res., Sect. A 2007, 582, 96–99. 10.1016/j.nima.2007.08.083. [DOI] [Google Scholar]
- Kilcoyne A. L. D.; Tyliszczak T.; Steele W. F.; Fakra S.; Hitchcock P.; Franck K.; Anderson E.; Harteneck B.; Rightor E. G.; Mitchell G. E.; Hitchcock A. P.; Yang L.; Warwick T.; Ade H. Interferometer-Controlled Scanning Transmission X-Ray Microscopes at the Advanced Light Source. J. Synchrotron Radiat. 2003, 10, 125–136. 10.1107/S0909049502017739. [DOI] [PubMed] [Google Scholar]
- Fakra S.; Kilcoyne A. L. D.; Tyliszczak T. Scintillator Detectors for Scanning Transmission X-ray Microscopes at the Advanced Light Source. AIP Conf. Proc. 2004, 705, 973–976. 10.1063/1.1757959. [DOI] [Google Scholar]
- Jacobsen C.; Wirick S.; Flynn G.; Zimba C. Soft X-Ray Spectroscopy from Image Sequences with Sub-100 nm Spatial Resolution. J. Microsc. 2000, 197, 173–184. 10.1046/j.1365-2818.2000.00640.x. [DOI] [PubMed] [Google Scholar]
- Hitchcock A. P.; Brion C. E. Neon K-Shell Excitation Studied by Electron Energy-Loss Spectroscopy. J. Phys. B: At. Mol. Phys. 1980, 13, 3269–3273. 10.1088/0022-3700/13/16/023. [DOI] [Google Scholar]
- Ma Y.; Chen C. T.; Meigs G.; Randall K.; Sette F. High-Resolution K-Shell Photoabsorption Measurements of Simple Molecules. Phys. Rev. A 1991, 44, 1848–1858. 10.1103/PhysRevA.44.1848. [DOI] [PubMed] [Google Scholar]
- Sodhi R. N. S.; Brion C. E. Reference Energies for Inner Shell Electron Energy-Loss Spectroscopy. J. Electron Spectrosc. Relat. Phenom. 1984, 34, 363–372. 10.1016/0368-2048(84)80050-X. [DOI] [Google Scholar]
- Hitchcock A. P.aXis2000 Is Written In Interactive Data Language (IDL). It Is Available Free for Noncommercial Use From http://unicorn.mcmaster.ca/aXis2000.html (accessed June 20, 2022).
- Varela A. S.; Ju W.; Bagger A.; Franco P.; Rossmeisl J.; Strasser P. Electrochemical Reduction of CO2 on Metal-Nitrogen-Doped Carbon Catalysts. ACS Catal. 2019, 9, 7270–7284. 10.1021/acscatal.9b01405. [DOI] [Google Scholar]
- Hu X.-M.; Hval H. H.; Bjerglund E. T.; Dalgaard K. J.; Madsen M. R.; Pohl M.-M.; Welter E.; Lamagni P.; Buhl K. B.; Bremholm M.; Beller M.; Pedersen S. U.; Skrydstrup T.; Daasbjerg K. Selective CO2 Reduction to CO in Water Using Earth-Abundant Metal and Nitrogen-Doped Carbon Electrocatalysts. ACS Catal. 2018, 8, 6255–6264. 10.1021/acscatal.8b01022. [DOI] [Google Scholar]
- Nitopi S.; Bertheussen E.; Scott S. B.; Liu X.; Engstfeld A. K.; Horch S.; Seger B.; Stephens I. E. L.; Chan K.; Hahn C.; Nørskov J. K.; Jaramillo T. F.; Chorkendorff I. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. 10.1021/acs.chemrev.8b00705. [DOI] [PubMed] [Google Scholar]
- Kuhl K. P.; Hatsukade T.; Cave E. R.; Abram D. N.; Kibsgaard J.; Jaramillo T. F. Electrocatalytic Conversion of Carbon Dioxide to Methane and Methanol on Transition Metal Surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. 10.1021/ja505791r. [DOI] [PubMed] [Google Scholar]
- Landers A. T.; Fields M.; Torelli D. A.; Xiao J.; Hellstern T. R.; Francis S. A.; Tsai C.; Kibsgaard J.; Lewis N. S.; Chan K.; Hahn C.; Jaramillo T. F. The Predominance of Hydrogen Evolution on Transition Metal Sulfides and Phosphides under CO2 Reduction Conditions: An Experimental and Theoretical Study. ACS Energy Lett. 2018, 3, 1450–1457. 10.1021/acsenergylett.8b00237. [DOI] [Google Scholar]
- Lu M.; Wang J.; Fang H.; Hu Y.; Zhou J. Unexpected Phase Separation in Li1–x Ni0.5Mn1.5O4 within a Porous Composite Electrode. Chem. Commun. 2018, 54, 4152–4155. 10.1039/C8CC01866A. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Zhang B.; De Luna P.; Liang Y.; Comin R.; Voznyy O.; Han L.; García de Arquer F. P.; Liu M.; Dinh C. T.; Regier T.; Dynes J. J.; He S.; Xin H. L.; Peng H.; Prendergast D.; Du X.; Sargent E. H. Theory-Driven Design of High-Valence Metal Sites for Water Oxidation Confirmed Using in Situ Soft X-Ray Absorption. Nat. Chem. 2018, 10, 149–154. 10.1038/nchem.2886. [DOI] [PubMed] [Google Scholar]
- Lenser C.; Lu Q.; Crumlin E.; Bluhm H.; Yildiz B. Charge Transfer Across Oxide Interfaces Probed by in Situ X-Ray Photoelectron and Absorption Spectroscopy Techniques. J. Phys. Chem. C 2018, 122, 4841–4848. 10.1021/acs.jpcc.7b10284. [DOI] [Google Scholar]
- Krasnikov S. A.; Preobrajenski A. B.; Sergeeva N. N.; Brzhezinskaya M. M.; Nesterov M. A.; Cafolla A. A.; Senge M. O.; Vinogradov A. S. Electronic Structure of Ni(II) Porphyrins and Phthalocyanine Studied by Soft X-Ray Absorption Spectroscopy. Chem. Phys. 2007, 332, 318–324. 10.1016/j.chemphys.2006.12.015. [DOI] [Google Scholar]
- Kera S.; Casu M. B.; Schöll A.; Schmidt T.; Batchelor D.; Rühl E.; Umbach E. High-Resolution Inner-Shell Excitation Spectroscopy of H2-Phthalocyanine. J. Chem. Phys. 2006, 125, 014705 10.1063/1.2212406. [DOI] [PubMed] [Google Scholar]
- Yang H. B.; Miao J.; Hung S.-F.; Chen J.; Tao H. B.; Wang X.; Zhang L.; Chen R.; Gao J.; Chen H. M.; Dai L.; Liu B. Identification of Catalytic Sites for Oxygen Reduction and Oxygen Evolution in N-Doped Graphene Materials: Development of Highly Efficient Metal-Free Bifunctional Electrocatalyst. Sci. Adv. 2016, 2, e1501122 10.1126/sciadv.1501122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J.; Deng J.-J.; Mao B.-H.; Xie T.; Sun X.-H.; Mou Z.-G.; Hong C.-H.; Yang P.; Wang S.-D. Probing Solid State N-Doping in Graphene by X-Ray Absorption near-Edge Structure Spectroscopy. Carbon 2012, 50, 335–338. 10.1016/j.carbon.2011.08.046. [DOI] [Google Scholar]
- Yabuta H.; Uesugi M.; Naraoka H.; Ito M.; Kilcoyne A. L. D.; Sandford S. A.; Kitajima F.; Mita H.; Takano Y.; Yada T.; Karouji Y.; Ishibashi Y.; Okada T.; Abe M. X-ray Absorption near Edge Structure Spectroscopic Study of Hayabusa Category 3 Carbonaceous Particles. Earth, Planets Space 2014, 66, 156 10.1186/s40623-014-0156-0. [DOI] [Google Scholar]
- Ikeno H.; Groot F. M. F.; de Stavitski E.; Tanaka I. Multiplet Calculations of L2,3 X-ray Absorption near-Edge Structures for 3d Transition-Metal Compounds. J. Phys.: Condens. Matter 2009, 21, 104208 10.1088/0953-8984/21/10/104208. [DOI] [PubMed] [Google Scholar]
- Shapiro D. A.; Yu Y.-S.; Tyliszczak T.; Cabana J.; Celestre R.; Chao W.; Kaznatcheev K.; Kilcoyne A. L. D.; Maia F.; Marchesini S.; Meng Y. S.; Warwick T.; Yang L. L.; Padmore H. A. Chemical Composition Mapping with Nanometre Resolution by Soft X-Ray Microscopy. Nat. Photonics 2014, 8, 765–769. 10.1038/nphoton.2014.207. [DOI] [Google Scholar]
- Pfeiffer F. X-Ray Ptychography. Nat. Photonics 2018, 12, 9–17. 10.1038/s41566-017-0072-5. [DOI] [Google Scholar]
- Liu D.; Shadike Z.; Lin R.; Qian K.; Li H.; Li K.; Wang S.; Yu Q.; Liu M.; Ganapathy S.; Qin X.; Yang Q.-H.; Wagemaker M.; Kang F.; Yang X.-Q.; Li B. Review of Recent Development of In Situ/Operando Characterization Techniques for Lithium Battery Research. Adv. Mater. 2019, 31, 1806620 10.1002/adma.201806620. [DOI] [PubMed] [Google Scholar]
- Shapiro D. A.; Babin S.; Celestre R. S.; Chao W.; Conley R. P.; Denes P.; Enders B.; Enfedaque P.; James S.; Joseph J. M.; Krishnan H.; Marchesini S.; Muriki K.; Nowrouzi K.; Oh S. R.; Padmore H.; Warwick T.; Yang L.; Yashchuk V. V.; Yu Y.-S.; Zhao J. An Ultrahigh-Resolution Soft x-Ray Microscope for Quantitative Analysis of Chemically Heterogeneous Nanomaterials. Sci. Adv. 2020, 6, eabc4904 10.1126/sciadv.abc4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.; Yuan H.; Casagrande T.; Shapiro D.; Yu Y.-S.; Enders B.; Lee J. R. I.; van Buuren A.; Biener M. M.; Gammon S. A.; Baumann T. F.; Hitchcock A. P. 4D Imaging of ZnO-Coated Nanoporous Al2O3 Aerogels by Chemically Sensitive Ptychographic Tomography: Implications for Designer Catalysts. ACS Appl. Nano Mater. 2021, 4, 621–632. 10.1021/acsanm.0c02924. [DOI] [Google Scholar]
- Mille N.; Yuan H.; Vijayakumar J.; Stanescu S.; Swaraj S.; Desjardins K.; Favre-Nicolin V.; Belkhou R.; Hitchcock A. P. Ptychography at the Carbon K-Edge. Commun. Mater. 2022, 3, 8 10.1038/s43246-022-00232-8. [DOI] [Google Scholar]
- Zhang C.; Ingino P.; Obst M.; Shahcheraghi L.; Yuan H.; Eraky H.; Wang J.; Higgins D.; Hitchcock A. P. In-Situ Soft X-Ray Spectromicroscopy Characterization of Electrochemical Processes. ECS Meet. Abstr. 2020, MA2020-02, 3176. 10.1149/MA2020-02623176mtgabs. [DOI] [Google Scholar]
- Handoko A. D.; Wei F.; Jenndy; Yeo B. S.; Seh Z. W. Understanding Heterogeneous Electrocatalytic Carbon Dioxide Reduction through Operando Techniques. Nat. Catal. 2018, 1, 922–934. 10.1038/s41929-018-0182-6. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.