

Abstract
Background:
Common genetic variance in apolipoprotein E (APOE), β-glucocerebrosidase (GBA), microtubule-associated protein tau (MAPT), and α-synuclein (SNCA) has been linked to cognitive decline in Parkinson’s disease (PD), although studies have yielded mixed esults.
Objectives:
To evaluate the effect of genetic variants in APOE, GBA, MAPT, and SNCA on cognitive decline and risk of dementia in a pooled analysis of six longitudinal, non-selective, population-based cohorts of newly diagnosed PD patients.
Methods:
1002 PD patients, followed for up to 10 years (median 7.2 years), were genotyped for at least one of APOE-ε4, GBA mutations, MAPT H1/H2, or SNCA rs356219. We evaluated the effect of genotype on the rate of cognitive decline (Mini-Mental State Examanation, MMSE) using linear mixed models and the development of dementia (diagnosed using standardized criteria) using Cox regression; multiple comparisons were accounted for using Benjamini–Hochberg corrections.
Results:
Carriers of APOE-ε4 (n = 281, 29.7%) and GBA mutations (n = 100, 10.3%) had faster cognitive decline and were at higher risk of progression to dementia (APOE-ε4, HR 3.57, P < 0.001; GBA mutations, HR 1.76, P = 0.001) than non-carriers. The risk of cognitive decline and dementia (HR 5.19, P < 0.001) was further increased in carriers of both risk genotypes (n = 23). No significant effects were observed for MAPT or SNCA rs356219.
Conclusions:
GBA and APOE genotyping could improve the prediction of cognitive decline in PD, which is important to inform the clinical trial selection and potentially to enable personalized treatment
Keywords: Parkinson’s disease, dementia, cognitive decline, APOE, GBA
Introduction
Patients with Parkinson’s disease (PD) are more likely to experience cognitive decline than healthy older adults.1 Problems with cognition affect patients’ ability to work and function independently, placing them at higher risk of poor quality of life and nursing home placement.2 The evolution of cognitive symptoms in PD is very heterogeneous1 and impedes the recruitment of relevant participants into clinical trials. Hence, the means to identify patients at high risk of cognitive deficits would significantly improve the design and costs of future trials.
Genetic factors are candidate predictors of cognitive decline and dementia in PD (PDD), although heterogeneity in the design of published studies has contributed to inconsistent findings.3 Among the strongest candidates are variants in the apolipoprotein E (APOE), β-glucocerebrosidase (GBA), microtubule-associated protein tau (MAPT), and α-synuclein (SNCA) loci. The ε4 allele of APOE (APOE-ε4) is the strongest genetic risk factor for sporadic Alzheimer’s disease (AD)4 and the top hit in genome-wide association studies for dementia with Lewy bodies (DLB),5 and several large studies showed its impact on the progression of cognitive decline in PD.6–8 GBA mutations are the commonest genetic risk factors for PD,9 and the risk of dementia in carriers of GBA mutations is modulated by the type of mutation.10 The H1 MAPT haplotype has been linked to tauopathies including AD and also to PD, and common variants in SNCA are established risk factors for sporadic PD.11,12 Some studies suggest that variants in SNCA and MAPT might also affect the cognitive decline in PD,13,14 although results are inconsistent.7,15,16
Despite extensive literature on the impact of genetic variants in APOE, GBA, MAPT, and SNCA on cognitive progression in PD, large studies with prospective follow-up from the time of PD diagnosis are scarce and many studies track patients solely from a clinical environment. In this work we establish the significance of the APOE, GBA, MAPT, and SNCA loci on global cognitive decline and the development of dementia over the natural course of PD in the Parkinson’s Incidence Cohorts Collaboration (PICC). Together the six longitudinal, population-based European cohorts form a large sample of patients with deeply characterized disease progression up to 10 years from the time of diagnosis with PD.
Methods
Subjects
We used data from PICC, a project pooling data from six PD population-based cohorts in Northern Europe, each designed to collect demographic and clinical data at the point of diagnosis and during prospective follow-up. Each cohort is summarized in Supplementary Table 1 and has been described in detail: Cambridgeshire Incidence of Parkinson’s disease from General Practitioner to Neurologist (CamPaIGN),17 Incidence of Cognitive Impairment in Cohorts with Longitudinal Evaluation-PD (ICICLE-PD),18 New Parkinson Patient in Umeå (NYPUM),19 ParkWest,20 Parkinsonism: Incidence, Cognition and Non-motor heterogeneity in Cambridgeshire (PICNICS),21 and Parkinsonism Incidence in Northeast Scotland (PINE).22 Briefly, patients were diagnosed with idiopathic PD using UK Parkinson’s Disease Society Brain Bank criteria without using family history as an exclusion criterion. Only those with a confirmed clinical diagnosis at their last clinical visit or autopsy are eligible for the PICC study, which currently has 1107 patients (1035 [93.5%] incident cases); of these, DNA or genetic data were available for 1002 (932 [93.0%] incident cases).
Demographic and Clinical Assessment
Data acquisition has been described in detail.17–22 Age at PD diagnosis, age at symptom onset, and age at baseline were defined as the age when PD diagnosis was made, at first self-reported motor symptoms, and at inclusion in the study, respectively. Patients are reassessed at regular follow-up visits (Supplementary Fig. 1). Home visits and/or telephone follow-up were offered to minimize attrition bias. The progression and severity of parkinsonism were evaluated using the Hoehn and Yahr scale23 and the Unified Parkinson’s Disease Rating Scale (UPDRS)24 Part III (CamPaIGN, NYPUM, ParkWest, PINE) or the Movement Disorders Society-UPDRS25 (MDS-UPDRS) Part III (ICICLE-PD, PICNICS). UPDRS-III scores were converted into MDS-UPDRS-III as described.26 Global cognitive function was assessed using the Mini-Mental State Examination27 (MMSE) scale. Dementia was diagnosed according to Diagnostic and Statistical Manual of Mental Disorders, 4th Edition28 (PINE, CamPaIGN, PICNICS) or Movement Disorder Society criteria29 (ICICLE-PD, NYPUM, ParkWest) (Supplementary Table 2).
Genetic Data Collection
Genetic data were available or acquired for this study for APOE, GBA, MAPT, and SNCA using a combination of whole exome sequencing, genotyping arrays, or targeted genotyping using TaqMan genotyping assays. Full details are given in Supplementary methods and the final genetic data set is summarized in Supplementary Table 3.
Statistical Analysis
For primary analysis, patients were grouped by genotype based on previous studies: APOE, carriers of ε4 allele versus non-carriers6; GBA, carriers of any GBA mutation versus non-carriers6; MAPT, carriers of H1/H1 versus H2 haplotype14; and SNCA rs356219, carriers of GG genotype versus A-allele.30 Primary analyses were corrected for multiple comparisons using the Benjamini–Hochberg false discovery rate (FDR) method at FDR < 0.05. For secondary analysis, carriers of APOE-ε4 were subdivided into carriers of one or two ε4 alleles, and GBA carriers were subdivided into carriers of PD risk or mild mutations, severe mutations, or variants of unknown significance. The classification of GBA mutations was based on pathogenicity in Gaucher disease (GD)31 and PD (Supplementary Table 4).
Baseline genotype-group comparisons were performed in IBM SPSS 26.0 (Armonk, NY, USA) using t-tests, Mann–Whitney U tests, or χ2 tests as appropriate, followed by multivariate linear regression for significant findings: age variables were compared with adjustment for study cohort and sex. Median (interquartile range [IQR]) follow-up time and cumulative proportion of dementia were estimated using the Kaplan–Meier method.
Linear mixed-effects regression models were performed in STATA 16.0 using repeated measurements of total MMSE. We performed transformation as described by Philipps et al32 to minimize bias due to the ceiling/floor effect and curvilinearity of the raw MMSE score. Time in study, genotype, and an interaction between these were included as fixed effects. Analyses were adjusted for study, age at baseline, sex, and education as fixed effects. All models had patients’ IDs as random intercepts and random slope of time. Marginal predictions of the decline in MMSE were based on fully adjusted models.
Development of PDD was evaluated using Cox regression in R 4.0.4 (package survival). The date of PDD onset was computed as the midpoint between the study visit at which dementia was diagnosed and the preceding visit, or as the midpoint between the first record of PDD diagnosis in clinical records or death certificates and the preceding study visit. Patients were censored due to death, loss to follow-up, or last recorded visit. Models were adjusted for confounders: age at baseline, sex, and education. In models including only confounders, Akaike Information Criterion was used to decide on the form of continuous confounders (original, natural logarithm [log] or square root [sqrt] transformed), and interaction with time t (t, sqrt(t), log(t), or log(t + 20)) for variables violating the proportional hazard assumption. If significant, time interaction was added also for genetic variables in the fully adjusted models. PDD-free survival was visualised in Kaplan–Meier plots (package survminer). The potential confounding effect of death was investigated using FineGray models allowing for competing risk of death before developing PDD. Data transformed by finegray function was analyzed by weighted Cox regression models adjusted for confounders and time interactions as in the Cox models. All models were stratified for study cohort.
Standard Protocol and Informed Consent
All participants signed written informed consent and regional ethical committees approved each study.
Results
Study Population
The study population is summarized in detail in Supplementary Table 1. A total of 1002 patients were included in the pooled analysis: 139 from CamPaIGN, 146 from ICICLE-PD, 133 from NYPUM, 189 from ParkWest, 250 from PICNICS, and 145 from PINE. The proportion of males was 61.0% (n = 611) and the mean age at diagnosis was 69.1 ± 9.8 years. The median disease duration from diagnosis at baseline was 0.1 years (IQR 0.0–0.2). Patients were followed up for a maximum of 10 years, with a median of 7.2 years (IQR 6.7–10.0). During follow-up, 344 (34.3%) patients died and 177 (17.7%) patients dropped out from the study for reasons other than death. Each of the 1002 patients in the study was genotyped for at least one of APOE, GBA, MAPT, or SNCA loci, and 928 (92.6%) had genotype information for all loci (Table 1 and Supplementary Table 3).
TABLE 1.
Comparison of baseline demographic and clinical characteristics with respect to the APOE, GBA, MAPT, and SNCA genotype
| Clinical variables | APOE-ε4 non-carriers | APOE-ε4 carriers | GBA non-carriers | GBA carriers | MAPT H2 | MAPT H1/H1 | SNCA rs356219-A | SNCA rs356219-GG |
|---|---|---|---|---|---|---|---|---|
| N (%) | 676 (70.6) | 282 (29.4) | 876 (89.6) | 102 (10.4) | 298 (30.3) | 684 (69.7) | 784 (81.2) | 182 (18.8) |
| Male, N (%) | 411 (60.8) | 172 (61.0) | 533 (60.8%) | 65 (63.7%) | 180 (60.4) | 418 (61.1) | 475 (60.6) | 114 (62.6) |
| Age at diagnosis, years, mean (SD) | 69.9 (9.8) | 67.5 (9.4) | 69.4 (9.7) | 66.8 (9.7) | 69.8 (10.2) | 68.9 (9.6) | 69.3 (9.8) | 68.6 (9.7) |
| Age at baseline, years, mean (SD) | 69.7 (9.8) | 67.3 (9.4) | 69.2 (9.8) | 66.6 (9.7) | 69.5 (10.2) | 68.5 (9.6) | 69.0 (9.7) | 68.5 (9.6) |
| Age at motor symptom onset, years, mean (SD) | 68.1 (9.9) | 65.7 (9.5) | 67.6 (9.9) | 65.3 (9.4) | 67.8 (10.3) | 67.0 (9.7) | 67.5 (9.8) | 67.0 (9.8) |
| Positive family history, N (%) | 73 (10.9) | 37 (13.4) | 99 (11.5%) | 14 (13.9%) | 40 (13.7%) | 72 (10.7%) | 81 (10.5) | 28 (15.6) |
| Education, years, mean (SD) | 11.7 (3.5) | 12.1 (3.5) | 11.6 (3.4) | 11.8 (3.5) | 11.7 (3.4) | 11.9 (3.5) | 11.8 (3.5) | 11.8 (3.3) |
| MMSE score, median (IQR) | 29.0 (3.0) | 29.0 (2.0) | 29.0 (3.0) | 29.0 (3.0) | 29.0 (3.0) | 29.0 (2.0) | 29.0 (3.0) | 29.0 (2.0) |
| MDS-UPDRS III, mean (SD) | 31.6 (13.3) | 29.8 (12.3) | 31.2 (13.2) | 30.4 (11.6) | 32.0 (13.7) | 30.6 (12.8) | 30.7 (12.8) | 32.5 (14.1) |
| Hoehn and Yahr, median (IQR) | 2.0 (1.0) | 2.0 (1.0) | 2.0 (1.0) | 2.0 (1.0) | 2.0 (1.0) | 2.0 (1.0) | 2.0 (1.0) | 2.0 (0.6) |
Between-group differences marked in bold if P < 0.05 in adjusted analysis (multivariate linear regression as described in Methods).
Abbreviations: N, number; SD, standard deviation; IQR, interquartile range; MMSE, Mini-Mental State Examination; MDS-UPDRS III, Movement Disorder Society Unified Parkinson’s Disease Rating Scale Part III; GBA, glucocerebrosidase gene; APOE, apolipoprotein E gene; MAPT, microtubule-associated protein tau gene; SNCA, alpha synuclein gene.
Younger Age at PD Diagnosis in Carriers of APOE-ε4 and GBA Mutations
Carriers of APOE-ε4 or any GBA mutation were significantly younger at the time of PD diagnosis compared with non-carriers in both unadjusted analysis (Table 1) and after adjustment for sex and study cohort (APOE-ε4, β, −2.31; 95% CI, −3.58 to −1.04; P < 0.001; GBA carriers, β, −2.89; 95% CI, −4.82 to −0.96; P = 0.003). Similarly, APOE-ε4 and GBA were associated with younger age at onset of first motor symptoms (APOE-ε4, β, −2.13; 95% CI, −3.43 to −0.83; P = 0.001; GBA carriers, β, −2.46; 95% CI, −4.38 to −0.53; P = 0.012) and at inclusion in the study (APOE-ε4, β, −2.39; 95% CI, −3.67 to −1.11; P < 0.001; GBA carriers, β, −2.95; 95% CI, −4.86 to −1.04, P = 0.003), compared to non-carriers. We did not observe any significant differences in age at PD diagnosis for the MAPT H1/H2 or rs356219 groups.
Faster Cognitive Decline in Carriers of APOE-ε4 and GBA Mutations
The rate of annual decline in MMSE score was assessed in linear mixed effects models using data from up to 4477 visits. Both APOE-ε4 and GBA carriers had a faster rate of annual decrease in scores (Table 2, Fig. 1A and B). For GBA carriers, this remained significant after we excluded 6 carriers with variants of unknown severity (interaction with time, β, −1.19; 95% CI, −2.05 to −0.33; P = 0.007) or 278 patients screened for selected GBA variants using TaqMan and RFLP assays (β, −1.18; 95% CI, −2.16 to −0.19; P = 0.019). The estimated drop in MMSE score over 10 years from diagnosis was from around 29 to 24 points for carriers of APOE-ε4, and from 29 to 23 points for those carrying any GBA mutation, while non-carriers of either APOE-ε4 or GBA mutations were predicted to decline only to 26 points. An association between rs356219-GG and a faster decline in MMSE score (Table 2; Fig. 1D) marginally missed the threshold for significance after correction for multiple comparisons. No effect on global cognitive decline was shown for the MAPT haplotype (Table 2; Fig. 1C).
TABLE 2.
Association between the genotype and the annual change in Mini–Mental State Examination (MMSE) score
| Genotype | Genotype carriers, N (%)a | Main effectb β (95% CI) | P | Interaction with timeb β (95% CI) | P |
|---|---|---|---|---|---|
| APOE-ε4 | |||||
| ε4 Non-carriers | 661 (70.3) | Ref. | Ref. | ||
| ε4 Carriers | 279 (29.7) | −1.07 (−2.90–0.75) | 0.249 | −0.81 (−1.40–−0.21) | 0.008 |
| APOE-ε4 subgroupsc | |||||
| Carriers of one ε4 allele | 259 (27.6) | −1.25 (−3.12–0.63) | 0.192 | −0.81 (−1.42–−0.20) | 0.009 |
| Carriers of two ε4 alleles | 20 (2.1) | 1.17 (−4.67–7.00) | 0.695 | −0.76 (−2.69–1.18) | 0.443 |
| GBA | |||||
| Non-carriers | 862 (89.7) | Ref. | Ref. | ||
| GBA mutation carriersd | 99 (10.3) | −0.07 (−2.77–2.62) | 0.958 | −1.28 (−2.12–−0.44) | 0.003 |
| GBA subgroupsc | |||||
| Risk or mild mutation carriers | 79 (8.3) | 0.97 (−2.00–3.94) | 0.523 | −1.14 (−2.06–−0.22) | 0.015 |
| Severe mutation carriers | 14 (1.5) | −7.79 (−14.66–0.91) | 0.026 | −1.43 (−3.63–0.77) | 0.202 |
| MAPT | |||||
| H2 carriers | 293 (30.4) | Ref. | Ref. | ||
| H1/H1 carriers | 671 (69.6) | −0.03 (−1.81–1.76) | 0.978 | −0.09 (−0.68–0.50) | 0.766 |
| SNCA rs356219 | |||||
| A-allele carriers | 771 (81.3) | Ref. | Ref. | ||
| GG carriers | 177 (18.7) | −0.46 (−2.58–1.65) | 0.668 | −0.72 (−1.41–−0.03) | 0.041 |
| APOE-ε4 and GBA subgroups | |||||
| Non-carriers of APOE-ε4 and GBA mutations | 574 (62.2) | Ref. | Ref. | ||
| Carriers of both APOE-ε4 and GBA mutations | 23 (2.5) | 0.83 (−4.70–6.37) | 0.769 | −2.87 (−4.46–−1.28) | < 0.001 |
Models adjusted for study cohort, age at baseline, sex and education.
Numbers include participants with information for education and Mini-Mental State Examination (MMSE).
Main effect indicates the effect of genotype on the intercept and the interaction with time indicates the effect of genotype on slope (annual change in MMSE score). MMSE scores were transformed before analysis as outlined in Methods.
The models of APOE-ε4 or GBA subgroups include non-carriers of APOE-ε4 or GBA mutations as the reference group, respectively.
GBA carriers include carriers of any GBA mutation, including variants of unknown significance.
Abbreviations: N, number; CI, confidence interval; Ref., reference; GBA, glucocerebrosidase gene; APOE, apolipoprotein E gene; MAPT, microtubule-associated protein tau gene; SNCA, alpha synuclein gene.
FIG. 1.
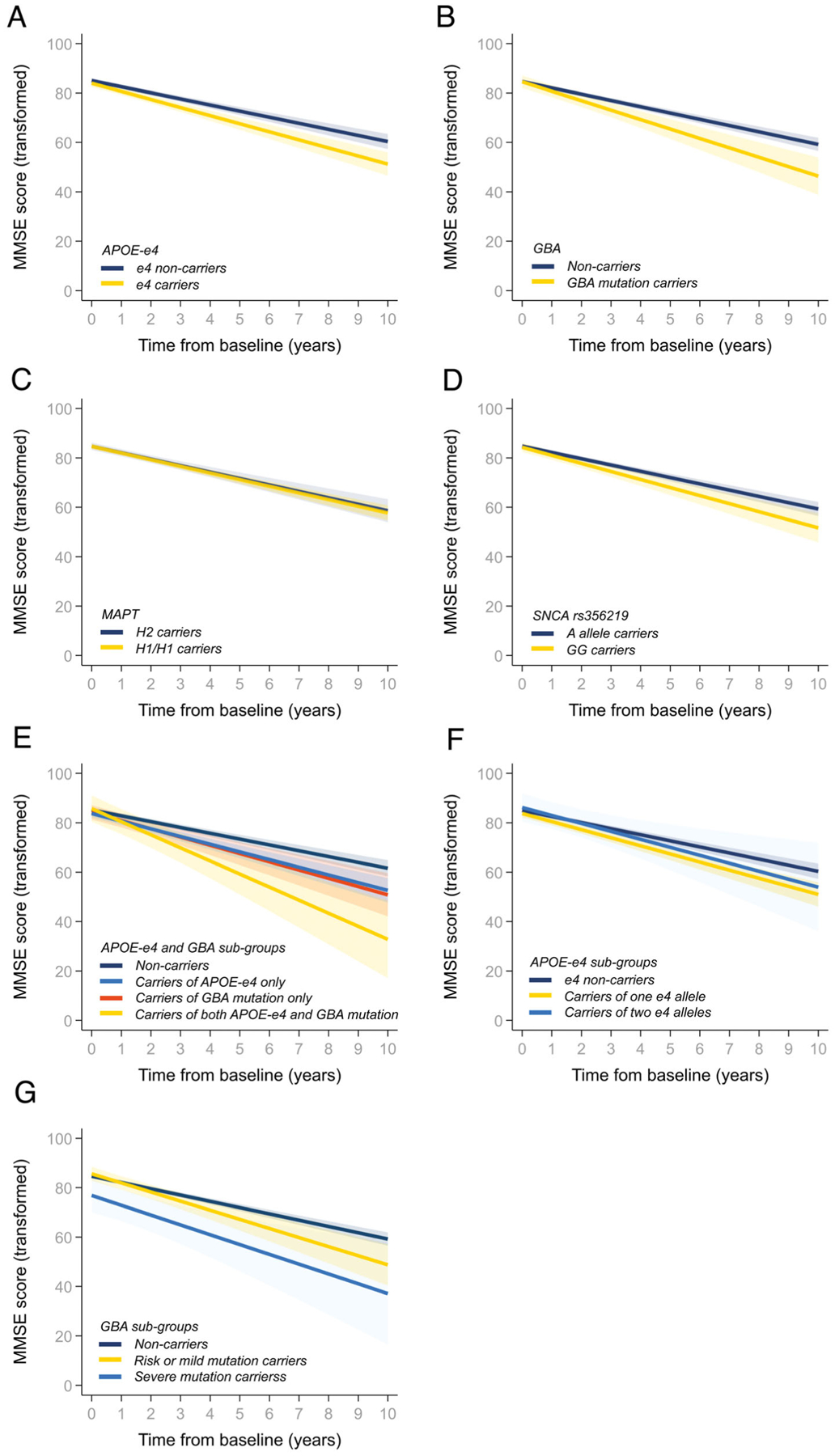
Prediction of Mini–Mental State Examination (MMSE) scores over time. Patients grouped by APOE, GBA, MAPT, and/or SNCA genotypes as outlined in the figure keys. MMSE scores were transformed before analysis as described in the Methods.
Based on the significant findings in the primary analysis, we explored the combined effect of harboring both an APOE-ε4 allele and GBA mutation and found that carriers of both declined faster than carriers of either APOE-ε4 or GBA, compared to non-carriers (Table 2, Fig. 1E). Over the 10 years, non-carriers were predicted to decline from approximately 29 to 26 MMSE points, while carriers of both APOE-ε4 and GBA mutations declined to 18 points.
Finally, in secondary analysis, carriers of one APOE-ε4 allele were predicted to have a faster annual change in MMSE score, whilst progression in ε4/ε4 carriers was not significantly different compared to non-carriers, though confidence intervals were wide as only 20 participants harbored ε4/ε4 (Table 2, Fig. 1F). Further, both carriers of risk or mild and carriers of severe GBA mutations were predicted to experience faster decline in MMSE than non-carriers, although these differences were only significant for those with the risk or mild mutations (Table 2, Fig. 1G).
APOE-ε4 and GBA Mutations Affect the Progression to PDD
At the study end, 290 of 1002 patients had developed PDD and the cumulative proportion of dementia accounting for deaths and losses to follow-up was 46.7%. APOE-ε4 and GBA had a significant impact on the rate of progression to PDD, while no effect was observed for either MAPT or SNCA rs356219 (Table 3, Fig. 2A–D). The model for APOE included a significant time-varying effect of the genetic variable (Table 3), which was also found in a competing risk model with death as a competing outcome (Supplementary Table 5) and indicated that the risk of PDD associated with the ε4 allele decreased over time. Patients carrying any GBA mutation were at 1.8 times higher risk of dementia (P = 0.001), and this remained significant after we excluded the 6 patients with GBA variants of unknown severity (HR 1.68; 95% CI, 1.19 to 2.37; P = 0.003) or the 278 patients screened for selected GBA variants using TaqMan and RFLP assays (HR 1.78; 95% CI, 1.10 to 2.86, P = 0.019). Further, carriers of both APOE-ε4 and GBA mutations were at a 5.2 times higher risk of progressing to PDD than non-carriers (Table 3, Fig. 2E).
TABLE 3.
Cox regression analysis evaluating the effect of genotype on the development of dementia
| Group | Total PD, Na | PDD, N (%) | Adjusted HR (95% CI) | P |
|---|---|---|---|---|
| APOE-ε4 | ||||
| ε4 Non-carriers | 665 | 169 (25.4) | Ref. | |
| ε4 Carriers | 281 | 109 (38.8) | 3.57 (2.15–5.93)b | <0.001 |
| APOE ⋆ time | 0.88 (0.79–0.99)b | 0.028 | ||
| APOE-ε4 subgroupsc | ||||
| Carriers of one ε4 allele | 261 | 99 (37.9) | 3.14 (1.96–5.02)b | <0.001 |
| Carriers of two ε4 alleles | 20 | 10 (50.0) | 6.39 (2.53–16.12)b | <0.001 |
| APOE ⋆ time | 0.91 (0.82–0.99)b | 0.047 | ||
| GBA | ||||
| Non-carriers | 867 | 239 (27.6) | Ref. | |
| GBA mutation carriersd | 100 | 42 (42.0) | 1.76 (1.26–2.46) | 0.001 |
| GBA subgroupsc | ||||
| Risk or mild mutation carriers | 81 | 32 (39.5) | 1.56 (1.07–2.26) | 0.020 |
| Severe mutation carriers | 13 | 7 (53.8) | 2.71 (1.26–5.82) | 0.011 |
| MAPT | ||||
| H2 carriers | 294 | 81 (27.6) | Ref. | |
| H1/H1 carriers | 676 | 200 (29.6) | 1.17 (0.90–1.52) | 0.248 |
| SNCA rs356219 | ||||
| A-allele carriers | 775 | 217 (28.0) | Ref. | |
| GG carriers | 181 | 62 (34.3) | 1.21 (0.91–1.60) | 0.199 |
| APOE-ε4 and GBA subgroups | ||||
| Non-carriers of APOE-ε4 and GBA mutations | 577 | 139 (24.1) | Ref. | |
| Carriers of both APOE-ε4 and GBA mutations | 23 | 14 (60.9) | 5.19 (2.88–9.38) | <0.001 |
Models adjusted for sex, age at baseline, education and stratified for study cohort.
Numbers include participants who had information available for education and time to event or censoring after the baseline visit.
Model includes interaction between the APOE variable and time. The presented HRAPOE for the APOE-ε4 variable refers to the hazard ratio for the respective carriers at time = 0 years (baseline). HR at any point of disease duration can be calculated: HRAPOE ⋆ (HRAPOE ⋆ time)t, where t defines the time point of interest in years.
The models of APOE-ε4 or GBA subgroups include non-carriers of APOE-ε4 or GBA mutations as the reference group, respectively.
GBA carriers include carriers of any GBA mutation, including variants of unknown significance.
Abbreviations: PD, Parkinson’s disease; PDD, Parkinson’s disease dementia; N, number; HR, hazard ratio; CI, confidence interval; Ref., reference; APOE, apolipoprotein E gene; GBA, glucocerebrosidase gene; MAPT, microtubule-associated protein tau gene; SNCA, alpha synuclein gene.
FIG. 2.
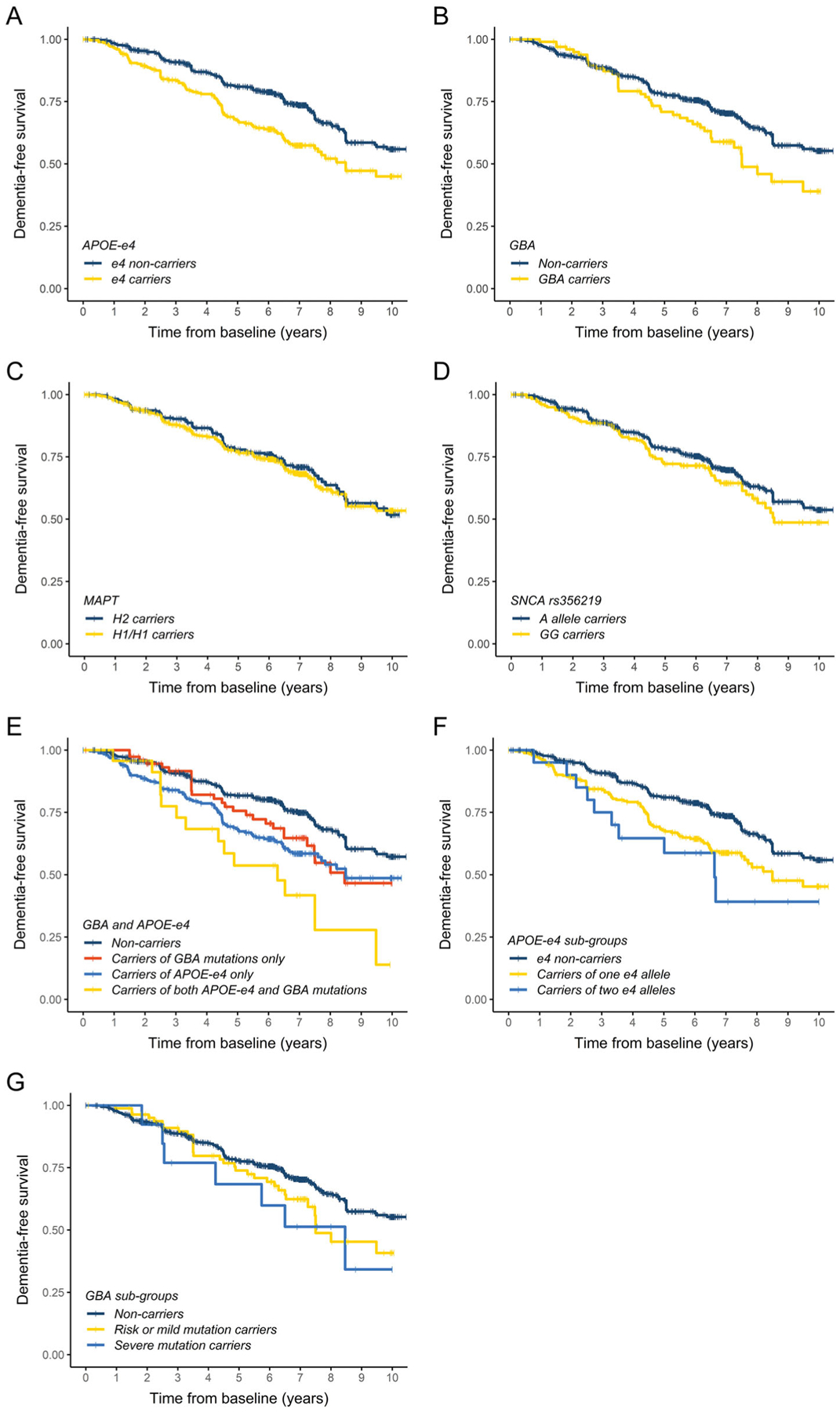
Kaplan–Meier plots of Parkinson’s disease (PD) dementia-free survival. Patients grouped by APOE, GBA, MAPT, and/or SNCA genotypes as outlined in the figure keys.
Secondary analysis showed a dose-dependent risk of developing PDD associated with APOE-ε4: when compared to non-carriers, carriers of one ε4 allele were at 3.1 times higher risk of progressing to dementia, while those who carried two ε4 alleles were at 6.4 times higher risk (Table 3, Fig. 2F). Among the GBA subgroups, the risk of developing PDD was 1.6 times higher in carriers of risk or mild mutations, and 2.7 times higher in carriers of severe mutations, when compared to non-carriers (Table 3, Fig. 2G).
Discussion
Using the largest pooled study of population-based cohorts to date, we have provided data on the impact of genetic variants in APOE, GBA, MAPT and SNCA on cognitive decline over the first 10 years of PD. We have shown that both APOE-ε4 and GBA mutations have an independent and additive effect on cognitive outcomes, adding to mounting evidence that these variants are key drivers of cognitive decline in PD, whilst common variants in SNCA and MAPT showed no significant impact. Understanding the role of common variants on the pattern of cognitive decline over the natural course of PD will support more accurate disease prognosis and should be considered when designing clinical trials.
Carriers of APOE-ε4 were predicted to experience faster cognitive decline, measured using both the annual change in MMSE and the time to a clinical diagnosis of dementia, reinforcing earlier evidence that APOE is a major risk factor for cognitive decline in PD6–8 as well as other dementias.4,5 Further, APOE-ε4 had a dose-dependent effect, with about a threefold increased risk of dementia per ε4 allele, comparable to results from a small cohort of neuropathologically confirmed PD cases (HR per ε4 allele 1.82, 95% CI 1.16 to 2.83).14 Interestingly, we found that the risk of PDD associated with APOE-ε4 was not constant over the course of the disease: carrier status had a large impact on the development of dementia in the early stages of the disease, while no effect of APOE-ε4 was seen by 10 years from diagnosis. This may explain why a recent report failed to show an association of APOE-ε4 and the development of dementia in a cohort that the authors note was underrepresented for early dementia cases.33 Similar observations have also been made in AD, showing the APOE genotype–related risk for AD decreases significantly with age,34 and would support the importance of early initiation of neuroprotective treatment (when available) in APOE-ε4 carriers.
As previously reported,6 also by cohorts included in this study,35,36 GBA carriers were at twofold increased risk of progressing to dementia. This was also reflected in the faster rate of global cognitive decline in patients carrying GBA mutations. Further, we show that patients with GBA mutations enriched in neuropathic GD progress to dementia faster than patients with GBA mutations linked to non-neuropathic GD or risk of PD, compared to non-carriers. Previous studies have used different classification schemes for GBA variants, but consistently show that the risk of cognitive impairment increases with the severity of GBA mutation.35,37,38 Contrary to our findings, several longitudinal studies showed no association with the GBA PD-risk mutations, although these were either small studies39 or grouped E326K and T369M together with synonymous or intronic variants,7,40 potentially diluting their effect. Our study reinforces previous findings35,41,42 that not only severe GBA mutations, but also the PD-risk variants are important players in modifying the cognitive progression in PD and contribute to the clinical heterogeneity among GBA carriers. Given that E326K/T369M are the commonest GBA mutations, this knowledge is relevant for a large GBA-PD subpopulation. We also show that the severe (neuropathic GD) GBA mutations were associated with a greater degree of cognitive impairment at baseline, which indicates that the clinical continuum linked to GBA mutations is apparent already at diagnosis.
Lastly, we observed that the risk of cognitive impairment was further increased in individuals harboring both a GBA mutation and the APOE-ε4 allele. This small subgroup was at fivefold increased risk of progressing to PDD compared to non-carriers. A similar trend was previously observed in a study of 298 patients with PD, where 3 of 6 carriers of both APOE-ε4 and GBA severe mutations progressed to dementia (HR 2.95; 95% CI 0.80 to 10.90).7 A faster decline in MMSE in GBA carriers with the APOE-ε4 allele was also recently shown in 100 Ashkenazi Jewish patients with DLB.43 The increased risk of cognitive impairment observed in carriers of both APOE-ε4 and GBA mutations is possibly due to the combination of neurodegenerative mechanisms mediated by these genotypes. The compromised activity of mutated β-glucocerebrosidase facilitates the accumulation and aggregation of α-synuclein and APOE-ε4 exacerbates the brain accumulation and subsequent deposition of amyloid-β.1 Interestingly, two recent studies indicated a novel role for APOE-ε4 in enhancing the α-synucleinopathy, and particularly the spread of Lewy body pathology,44,45 reinforcing the importance of APOE-ε4 as a potential therapeutic target in PDD.
Besides its potential impact on cognitive decline in PD, the direct influence of APOE-ε4 on α-synuclein pathology could contribute to the earlier age of PD onset in the ε4 carriers observed in our study, and reported previously.46,47 Both of these studies used time of self-reported onset of cardinal PD symptoms, and our study reaffirms this finding of a younger age at clinical PD diagnosis in APOE-ε4 carriers in a population-based cohort.
Previous reports on MAPT and SNCA and cognition in PD are mixed. We show a small effect of rs356219 on global cognitive decline before adjustment for multiple comparisons, replicating our previous finding using a subset of 443 patients included in this current work.30 A larger, independent cohort will be required to validate this result, but given the small effect size, the impact of this variant is unlikely to be clinically meaningful. Few studies have reported the impact of MAPT or rs356219 on the progression to dementia. In keeping with our findings, a longitudinal study of 298 Spanish patients followed retrospectively found no association between MAPT or rs356219 and the time to dementia,7 and a study of 514 patients showed no association between MAPT haplotype and the years from PD motor onset until PDD.33 Contrary to these and our results, an association between MAPT H1/H1 genotype and dementia onset was found in two previous survival analyses.14,17 Both of these were high-quality studies but were notably smaller (129 patients from the CamPaIGN population-based study, included in this work, and an independent sample of 152 neuropathologically confirmed cases). Many others have shown no association between cognitive test performance, cognitive diagnosis, or rate of cognitive decline and the H1 haplotype15,16,48–51 or rs356219,15,52 and our study provides additional evidence that the MAPT H1 haplotype and SNCA rs356219 do not play a key role in cognitive decline in PD.
Longitudinal studies represent a gold standard for tracking disease progression but are traditionally hampered by small sample size, short follow-up time, and losses to follow-up. Considering this, our study has notable strengths, presenting data from the largest, population-based sample of mostly incident PD patients to date, with regular follow-up over up to 10 years. Further, the cohorts had uniform design, used standardized diagnostic criteria for PD and PDD, and attrition (18%) was very low. An additional strength is the applied time-to-event analyses that were also performed allowing for competing risk of death. Potential limitations include incomplete screening for GBA mutations, although most participants were genotyped for the common variants and the number of rare mutations or complex GBA variants, such as RecNCiI, undetected is expected to be modest. Further, we chose MMSE scores to assess global cognition, and alternatives such as the Montreal Cognitive Assessment (MoCA) are more sensitive to mild and domain-specific changes53 and may have revealed subtle effects for some loci. Lastly, it will be important to expand this work to consider promising new candidates, such as the RIMS2 locus from the first genome-wide survival analysis.6
Evidence for the impact of common genetic variants on dementia in the general PD population is important for predicting the prognosis of newly diagnosed patients. In our study, 36% of patients were carriers of either APOE-ε4 or GBA, which places many individuals at risk of a more severe disease course, and the importance of these results is augmented by the additional effect of GBA and APOE carrier status on reducing the age at disease onset. Use in clinical practice will necessitate more precise estimates of when patients develop dementia and it will be important to establish the success of combining GBA and APOE mutations with other predictors. More immediately, the impact of genetic loci on the rate of cognitive decline can be useful in improving clinical trials of putative cognitive neuroprotective agents in PD populations. MMSE is a popular outcome measure in clinical trials due to its short application time and sensitivity to the effect of treatment,54 although trials have largely been ineffective, in part attributed to heterogeneity in patient selection and variability in disease progression. Our models estimate that carriers of either GBA or APOE variants will decline on average 0.5 MMSE points per year, whilst carriers of both variants would average 1.1 points decline per year. Although modest, this is substantially higher than estimates of 0.1–0.2 points per year in other longitudinal studies of unselected cohorts,55–57 and suggests that inclusion of genetic variants in trial inclusion criteria could improve homogeneity and trial power.
In conclusion, our findings provide evidence for the role of both GBA and APOE in the rate of cognitive decline in the general PD population. We show that both APOE-ε4 and GBA mutations are risk factors for cognitive impairment, and the effect of APOE-ε4 on PDD risk is greater in early disease, which should be considered when interpreting the current literature and designing future trials. This knowledge may further improve the accuracy of disease prognosis, especially for those with a younger age at onset who are not traditionally identified as of high risk of rapid cognitive decline.
Supplementary Material
Acknowledgments:
We would like to thank all participants, study personnel from each study, and funders of individual studies and of PICC. We would like to thank Artur Wozniak and Adrian Martin from the University of Aberdeen, Data Management Department, for help in developing the PICC database.
We acknowledge the contributions of members of the individual study groups as detailed below.
Members of PICC Steering Group: Dr. Angus D. Macleod, Dr. Carl E. Counsell (Chair), University of Aberdeen, UK; Prof. Ole-Bjørn Tysnes, University of Bergen, Norway; Marta Camacho, Dr. Caroline Williams-Gray, University of Cambridge, UK; Dr. Rachael A. Lawson, Newcastle University, UK; Dr. Jodi Maple-Grødem, Prof. Guido Alves, Stavanger University Hospital, Norway; Prof. Lars Forgren, Umeå University, Sweden.
CamPaIGN study: Roger A. Barker, Thomas Foltynie, Sarah L. Mason, Caroline H. Williams-Gray.
ICICLE-PD Study: David Burn, Lynn Rochester, Alison J. Yarnall, Rachael A. Lawson, Gordon W. Duncan, Tien K. Khoo.
NYPUM Study: Lars Forsgren, Jan Linder, Mona Edström, Jörgen Andersson, Linda Eriksson, David Bäckström, Gun-Marie Hariz, Magdalena Domellöf.
ParkWest Study: ParkWest Principal investigators: Guido Alves (Norwegian Centre for Movement Disorders, Stavanger University Hospital) and Ole-Bjørn Tysnes (Haukeland University Hospital). Study personnel: Michaela Dreetz Gjerstad, Kenn Freddy Pedersen, Elin Bjelland Forsaa, Veslemøy Hamre Frantzen, Anita Laugaland, Jodi Maple-Grødem, Johannes Lange, Karen Simonsen, Eldbjørg Fiske and Ingvild Dalen (Stavanger University Hospital); Bernd Müller, Geir Olve Skeie and Marit Renså (Haukeland University Hospital); Wenche Telstad, Aliaksei Labusau and Jane Kastet (Førde Hospital); Ineke HogenEsch, Marianne Kjerandsen and Liv Kari Håland (Haugesund Hospital); Karen Herlofson, Solgunn Ongre, and Siri Bruun (Sørlandet Hospital Arendal).
PICNICS study: Roger A. Barker, Marta Camacho, Gemma Cummins, Jonathan R. Evans, David P. Breen, Ruwani S. Wijeyekoon, Caroline H. Williams-Gray.
PINE Study: Medical: Carl E. Counsell, Kate S. M. Taylor, Robert Caslake, Angus D. Macleod, David J. M. McGhee, Diane Swallow; Research nurse/assistant: Joanne Gordon, Clare Harris, Ann Hayman, Nicola Johannesson, Hazel Forbes; Data management: Valerie Angus, Alasdair Finlayson, David Dawson, Katie Wilde, David Ritchie, Artur Wozniak; Statisticians: Neil Scott, Shona Fielding; Radiology: Prof. Alison Murray; Pathology: Ishbel Gall, Dr. James MacKenzie, Prof. Colin Smith; Secretarial: Aileen Sylvester, Susan Mitchell, Pam Rebecca, Ann Christie, and Diane McCosh.
Funding agencies:
This work was supported by the Research Council of Norway (287842). The CamPaIGN study has received funding from the Wellcome Trust, the Medical Research Council, the Patrick Berthoud Trust, and the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). The ICICLE-PD study was funded by Parkinson’s UK (J-0802, G-1301, G-1507) and supported by the Lockhart Parkinson’s Disease Research Fund, National Institute for Health Research (NIHR) Newcastle Biomedical Research Unit and Centre based at Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University. The NYPUM study was supported by grants from the Swedish Medical Research Council, Erling-Persson Foundation, the Swedish Brain Foundation (Hjärnfonden), Umeå University, Västerbotten County Council, King Gustaf V and Queen Victoria Freemason Foundation, Swedish Parkinson Foundation, Swedish Parkinson Research Foundation, Kempe Foundation, Swedish PD Association, the European Research Council, and the Knut and Alice Wallenberg Foundation. The Norwegian ParkWest study has received funding from the Research Council of Norway (177966), the Western Norway Regional Health Authority (911218), the Norwegian Parkinson’s Research Foundation, and Rebergs Legacy. The PICNICS study was funded by the Cure Parkinson’s Trust, the Van Geest Foundation, the Medical Research Council, Parkinson’s UK, and the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). The PINE study was funded by Parkinson’s UK (grant numbers G0502, G0914, and G1302), the Scottish Chief Scientist Office (CAF/12/05, PCL/17/10), Academy of Medical Sciences, NHS Grampian endowments, the BMA Doris Hillier award, RS Macdonald Trust, the BUPA Foundation, and SPRING. The PICC collaboration has been supported by The Chief Scientist Office of the Scottish Government (PCL/17/10), the Academy of Medical Sciences, Parkinson’s UK (initial collaborator meeting) and the Norwegian Association for Public Health. C.R.S.’s work was supported by NIH grants NINDS/NIA R01NS115144, U01NS095736, U01NS100603, and the American Parkinson Disease Association Center for Advanced Parkinson Research. This research was funded in whole, or in part by the UKRI Medical Research Council [MR/R007446/1]. For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Relevant conflicts of interest/financial disclosures:
A.A.S., I.D., K.F.P., M.C., D.B., L.F., C.T., G.H., G.L., R.A.L., A.J.Y., C.H.W.G., C.E.C., O.B.T., G.A., J.M.G.: None. S.W.R.: PhD supported by a Merck, Sharpe and Dohme studentship awarded though the University of Cambridge. C.R.S.: Brigham and Women’s Hospital holds a US provisional patent application on the polygenic hazard score for predicting PD progression, on which C.R.S. is named as inventor. C.R.S. has served as consultant, scientific collaborator and on scientific advisory boards for Sanofi; ADM: Grant support from the Chief Scientist Office of the Scottish Government (PCL/17/10), Academy of Medical Sciences, NHS Grampian Endowments.
Financial Disclosures
A.A.S.: Supported by Research Council of Norway (287842).
I.D.: None.
K.F.P.: None.
M.C.: Received grant support from the Evelyn Trust and the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014).
D.B.: Personal research grant awarded by the Swedish Brain Foundation (Hjärnfonden), and grant support from Umeå University, Västerbotten County Council, and the Swedish Parkinson Research Foundation.
L.F.: Received grant support from the Swedish Medical Research Council.
C.T.: None
SWR: NHS employment.
G.H.: Receives funding from Parkinson’s UK (G-2003), The Michael J. Fox Foundation (ID-15643 and ID-15749), and the Wellcome Trust (203,105/Z/16/Z).
G.L.: Supported by the National Natural Science Foundation of China (31900475), Young Talent Recruitment Project of Guangdong (2019QN01Y139), Shenzhen Basic Research Project (JCYJ20190807161601692), and the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (2021qntd43).
C.R.S.: Brigham and Women’s Hospital holds a US provisional patent application on the polygenic hazard score for predicting PD progression, on which C.R.S. is named as inventor. Outside this work, C.R.S. has served as consultant, scientific collaborator, or on scientific advisory boards for Sanofi, Berg Health, Pfizer, Biogen, and has received grants from NIH, US Department of Defense, American Parkinson Disease Association, and The Michael J. Fox Foundation (MJFF).
R.A.L.: Supported by Parkinson’s UK Senior Research Fellowship (F-1801), received grants from the Lewy Body Society (LBS/010/2020), the Medical Research Council Discovery Medicine North Doctoral Training Partnership, and the National Institute for Health Research (NIHR) Newcastle Biomedical Research Unit and Centre based at Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University.
A.J.Y.: Received funding from NIHR BRC, EU IMI, NIHR, Lewy Body Society, Parkinson’s UK, Dunhill Medical Trust, Cure Parkinson’s Trust, The Michael J. Fox Foundation, and Weston Brain Institute. Received funding and/or honoraria from Bial, Britannia, UCB, Abbvie, GSK, Teva-Lundbeck, GE Healthcare, and Genus for attending educational events.
C.H.W.G.: RCUK/UKRI Research Innovation Fellowship awarded by the Medical Research Council; grant support from the Cambridge Centre for Parkinson-Plus, the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014), The Michael J. Fox Foundation, the Evelyn Trust, Cure Parkinson’s, and Parkinson’s UK; consultancy payments from Evidera, Inc./GlaxoSmithKline.
A.D.M.: Grant support from the Chief Scientist Office of the Scottish Government (PCL/17/10), NHS Grampian Endowments.
C.E.C.: Grant support last 12 months NHS Grampian Endowments, RS Macdonald Trust.
O.B.T.: Research grant from the Norwegian ClinBeForsk as support for a clinical research trial on ALS.
G.A.: The Research Council of Norway (287842), University of Stavanger, Norwegian Parkinson Research Foundation, and Reberg’s Legacy.
J.M.G.: Received funding from the Norwegian Parkinson’s Disease Association and is supported by The Research Council of Norway (287842).
Footnotes
Supporting Data
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
Data Availability Statement
Anonymized data that support the findings of this study are available on request and with the correct permissions from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1.Aarsland D, Batzu L, Halliday GM, Geurtsen GJ, Ballard C, Chaudhuri KR, Weintraub D. Parkinson disease-associated cognitive impairment. Nat Rev Dis Primers 2021;7(1):47. [DOI] [PubMed] [Google Scholar]
- 2.Leroi I, McDonald K, Pantula H, Harbishettar V. Cognitive impairment in Parkinson disease: impact on quality of life, disability, and caregiver burden. J Geriatr Psychiatry Neurol 2012;25(4):208–214. [DOI] [PubMed] [Google Scholar]
- 3.Fagan ES, Pihlstrøm L. Genetic risk factors for cognitive decline in Parkinson’s disease: a review of the literature. Eur J Neurol 2017; 24(4):e561–e520. [DOI] [PubMed] [Google Scholar]
- 4.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013;45(12):1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rongve A, Witoelar A, Ruiz A, et al. GBA and APOE ε4 associate with sporadic dementia with Lewy bodies in European genome wide association study. Sci Rep 2019;9(1):7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu G, Peng J, Liao Z, et al. Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson’s disease. Nat Genet 2021;53:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huertas I, Jesús S, García-Gomez FJ, et al. Genetic factors influencing frontostriatal dysfunction and the development of dementia in Parkinson’s disease. PloS One 2017;12(4):e0175560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan MMX, Lawton MA, Jabbari E, et al. Genome-wide association studies of cognitive and motor progression in Parkinson’s disease. Mov Disord 2021;36(2):424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neumann J, Bras J, Deas E, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009; 132(Pt 7):1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Chen J, Xu C, Feng J, Li J. Effects of glucocerebrosidase gene polymorphisms and mutations on the risk of Parkinson’s disease dementia: a meta-analysis. Neurosci Lett 2020;714:134544. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Shu L, Sun Q, Pan H, Guo J, Tang B. A comprehensive analysis of the association between SNCA polymorphisms and the risk of Parkinson’s disease. Front Mol Neurosci 2018;11:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang D, Nalls MA, Hallgrímsdóttir IB, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 2017;49(10):1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campêlo CLC, Cagni FC, de Siqueira FD, et al. Variants in SNCA gene are associated with Parkinson’s disease risk and cognitive symptoms in a Brazilian sample. Front Aging Neurosci 2017;9:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tunold J-A, Geut H, Rozemuller JMA, et al. APOE and MAPT are associated with dementia in neuropathologically confirmed Parkinson’s disease. Front Neurol 2021;12:631145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mata IF, Leverenz JB, Weintraub D, et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol 2014;71(11):1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paul KC, Rausch R, Creek MM, et al. APOE, MAPT, and COMT and Parkinson’s disease susceptibility and cognitive symptom progression. J Parkinsons Dis 2016;6(2):349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams-Gray CH, Mason SL, Evans JR, Foltynie T, Brayne C, Robbins TW, Barker RA. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J Neurol Neurosurg Psychiatry 2013;84(11):1258–1264. [DOI] [PubMed] [Google Scholar]
- 18.Yarnall AJ, Breen DP, Duncan GW, et al. Characterizing mild cognitive impairment in incident Parkinson disease: the ICICLE-PD study. Neurology 2014;82(4):308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linder J, Stenlund H, Forsgren L. Incidence of Parkinson’s disease and parkinsonism in northern Sweden: a population-based study. Mov Disord 2010;25(3):341–348. [DOI] [PubMed] [Google Scholar]
- 20.Alves G, Müller B, Herlofson K, et al. Incidence of Parkinson’s disease in Norway: the Norwegian ParkWest study. J Neurol Neurosurg Psychiatry 2009;80(8):851–857. [DOI] [PubMed] [Google Scholar]
- 21.Breen DP, Evans JR, Farrell K, Brayne C, Barker RA. Determinants of delayed diagnosis in Parkinson’s disease. J Neurol 2013;260(8):1978–1981. [DOI] [PubMed] [Google Scholar]
- 22.Caslake R, Taylor K, Scott N, et al. Age-, gender-, and socioeconomic status-specific incidence of Parkinson’s disease and parkinsonism in Northeast Scotland: the PINE study. Parkinsonism Relat Disord 2013;19(5):515–521. [DOI] [PubMed] [Google Scholar]
- 23.Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17(5):427–442. [DOI] [PubMed] [Google Scholar]
- 24.Fahn S, Elton RL. The Unified Parkinson’s Disease Rating Scale. In: Fahn S, Marsden CD, Calne DM, et al. , eds. Recent Development in Parkinson’s Disease. Florham Park: MacMillan Healthcare Information; 1987:153–163. [Google Scholar]
- 25.Goetz CG, Tilley BC, Shaftman SR, et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008;23(15):2129–2170. [DOI] [PubMed] [Google Scholar]
- 26.Goetz CG, Stebbins GT, Tilley BC. Calibration of unified Parkinson’s disease rating scale scores to Movement Disorder Society-unified Parkinson’s disease rating scale scores. Mov Disord 2012;27(10):1239–1242. [DOI] [PubMed] [Google Scholar]
- 27.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12(3):189–198. [DOI] [PubMed] [Google Scholar]
- 28.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th edition. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- 29.Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 2007;22(12):1689–1707. quiz 1837 [DOI] [PubMed] [Google Scholar]
- 30.Szwedo AA, Pedersen CC, Ushakova A, et al. Association of SNCA Parkinson’s disease risk polymorphisms with disease progression in newly diagnosed patients. Front Neurol 2020;11:620585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beutler E, Gelbart T, Scott CR. Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis 2005;35(3):355–364. [DOI] [PubMed] [Google Scholar]
- 32.Philipps V, Amieva H, Andrieu S, et al. Normalized Mini-Mental State Examination for assessing cognitive change in population-based brain aging studies. Neuroepidemiology 2014;43(1):15–25. [DOI] [PubMed] [Google Scholar]
- 33.Phongpreecha T, Cholerton B, Mata IF, et al. Multivariate prediction of dementia in Parkinson’s disease. NPJ Parkinson’s Dis 2020; 6(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu L, Caselli RJ. Age stratification corrects bias in estimated hazard of APOE genotype for Alzheimer’s disease. Alzheimers Dement (N Y) 2018;4:602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lunde KA, Chung J, Dalen I, et al. Association of glucocerebrosidase polymorphisms and mutations with dementia in incident Parkinson’s disease. Alzheimers Dement 2018;14(10): 1293–1301. [DOI] [PubMed] [Google Scholar]
- 36.Stoker TB, Camacho M, Winder-Rhodes S, et al. Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J Neurol Neurosurg Psychiatry 2020;91(7):695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cilia R, Tunesi S, Marotta G, et al. Survival and dementia in GBA-associated Parkinson’s disease: the mutation matters. Ann Neurol 2016;80(5):662–673. [DOI] [PubMed] [Google Scholar]
- 38.Liu G, Boot B, Locascio JJ, et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol 2016;80(5):674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winder-Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013;136(Pt 2):392–399. [DOI] [PubMed] [Google Scholar]
- 40.Jesús S, Huertas I, Bernal-Bernal I, et al. GBA variants influence motor and non-motor features of Parkinson’s disease. PLoS One 2016;11(12):e0167749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis MY, Johnson CO, Leverenz JB, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 2016;73(10):1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Straniero L, Asselta R, Bonvegna S, et al. The SPID-GBA study: sex distribution, penetrance, incidence, and dementia in GBA-PD. Neurol Genet 2020;6(6):e523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shiner T, Mirelman A, Rosenblum Y, et al. The effect of GBA mutations and APOE polymorphisms on dementia with Lewy bodies in Ashkenazi Jews. J Alzheimers Dis 2021;80(3):1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dickson DW, Heckman MG, Murray ME, et al. APOE ε4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 2018;91(12):e1182–e1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davis AA, Inman CE, Wargel ZM, et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med 2020;12(529):eaay3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pankratz N, Byder L, Halter C, et al. Presence of an APOE4 allele results in significantly earlier onset of Parkinson’s disease and a higher risk with dementia. Mov Disord 2006;21(1):45–49. [DOI] [PubMed] [Google Scholar]
- 47.Li YJ, Hauser MA, Scott WK, et al. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology 2004;62(11):2005–2009. [DOI] [PubMed] [Google Scholar]
- 48.Morley JF, Xie SX, Hurtig HI, et al. Genetic influences on cognitive decline in Parkinson’s disease. Mov Disord 2012;27(4):512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 2012;72(4):587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mata IF, Johnson CO, Leverenz JB, et al. Large-scale exploratory genetic analysis of cognitive impairment in Parkinson’s disease. Neurobiol Aging 2017;56:e211–e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ezquerra M, Campdelacreu J, Gaig C, et al. Lack of association of APOE and tau polymorphisms with dementia in Parkinson’s disease. Neurosci Lett 2008;448(1):20–23. [DOI] [PubMed] [Google Scholar]
- 52.Goris A, Williams-Gray CH, Clark GR, et al. Tau and alpha-synuclein in susceptibility to, and dementia in, Parkinson’s disease. Ann Neurol 2007;62(2):145–153. [DOI] [PubMed] [Google Scholar]
- 53.Skorvanek M, Goldman JG, Jahanshahi M, et al. Global scales for cognitive screening in Parkinson’s disease: critique and recommendations. Mov Disord 2018;33(2):208–218. [DOI] [PubMed] [Google Scholar]
- 54.Chou KL, Amick MM, Brandt J, et al. A recommended scale for cognitive screening in clinical trials of Parkinson’s disease. Mov Disord 2010;25(15):2501–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burn DJ, Rowan EN, Allan LM, Molloy S, O’Brien JT, McKeith IG. Motor subtype and cognitive decline in Parkinson’s disease, Parkinson’s disease with dementia, and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2006;77(5):585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mollenhauer B, Zimmermann J, Sixel-Doring F, et al. Baseline predictors for progression 4 years after Parkinson’s disease diagnosis in the De Novo Parkinson Cohort (DeNoPa). Mov Disord 2019;34(1):67–77. [DOI] [PubMed] [Google Scholar]
- 57.Jo S, Kim SO, Park KW, Lee SH, Hwang YS, Chung SJ. The role of APOE in cognitive trajectories and motor decline in Parkinson’s disease. Sci Rep 2021;11(1):7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized data that support the findings of this study are available on request and with the correct permissions from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.