

Keywords: angiotensin, cardiomyopathy, epinephrine, mitofusin, neuropathy
Abstract
Neurohormonal signaling and mitochondrial dynamism are seemingly distinct processes that are almost ubiquitous among multicellular organisms. Both of these processes are regulated by GTPases, and disturbances in either can provoke disease. Here, inconspicuous pathophysiological connectivity between neurohormonal signaling and mitochondrial dynamism is reviewed in the context of cardiac and neurological syndromes. For both processes, greater understanding of basic mechanisms has evoked a reversal of conventional pathophysiological concepts. Thus, neurohormonal systems induced in, and previously thought to be critical for, cardiac functioning in heart failure are now pharmaceutically interrupted as modern standard of care. And, mitochondrial abnormalities in neuropathies that were originally attributed to an imbalance between mitochondrial fusion and fission are increasingly recognized as an interruption of axonal mitochondrial transport. The data are presented in a historical context to provide insight into how scientific thought has evolved and to foster an appreciation for how seemingly different areas of investigation can converge. Finally, some theoretical notions are presented to explain how different molecular and functional defects can evoke tissue-specific disease.
INTRODUCTION
The physiological effects of a neurohormone were first recognized at University College of London by the English physician George Oliver and physiologist Edward Schafer, who described increased heart rate and blood pressure with arteriole constriction in animals receiving extracts of adrenal medulla (1). The active principal, epinephrine, was purified in 1899 by John Jacob Abel at Johns Hopkins University, and independently in 1900 by Japanese chemist Jokichi Takamine (2). The chemical form, derived by Thomas Aldrich of Parke-Davis in 1901, was patented and ushered in a century of research and therapeutic development based on modulating neurohormone signaling.
Central to understanding neurohormonal effects was the notion, advanced by William Bayliss and Ernest Starling from their studies of secretin, that epinephrine acted as a chemical messenger secreted by one organ to modulate the function of others. The concept that adrenaline effects were specifically mediated by “adrenotropic” receptors emerged from multiple studies linking sympathetic nerve activity to epinephrine-like responses in uterine, bronchial, and arteriolar smooth muscle. However, the observation that sympathetic stimulation evoked distinct end-organ effects under different experimental conditions, such as in the presence of sympatholytic ergot, engendered a controversy over whether there were multiple factors released from sympathetic nerve endings versus multiple different receptor/detectors for a single factor. The controversy was resolved in favor of multiple receptors by H.H. Dale who described both a “receptive mechanism for adrenaline” at the neuromuscular junction, and a “mixed response” consisting of either smooth muscle contraction or relaxation in response to the single hormone (3, 4). Thirty years later, the hypothetical adrenotropic receptors were pharmacologically defined in groundbreaking studies by Raymond P. Ahlquist who discovered differences in the rank-order potency of a series of structurally distinct sympathomimetic amines to provoke vasoconstriction, pupillary dilation and uterine contraction. Ahlquist designated the two types of receptors “α-” and “β-adrenotrophic receptors” (5). This classic study laid the foundation for subsequent identification of hundreds of individual members of the superfamily of 7-transmembrane (7-TM) spanning receptors (6) and the rational therapeutic application of receptor subtype-specific agonists (as in β-AR activating isoprenaline and its successors for asthma) and antagonists (as in β1-AR blocking metoprolol for cardiac insufficiency).
Different functional effects of the nine modern adrenergic receptor (AR) subtypes (α1A, α1B, α1D, α2A, α2B, α2C, β1, β2, and β3) and indeed for all 7-TM receptors, derive from their coupling to specific heterotrimeric (i.e., having 3 different protein subunits, α, β, and γ) G-protein effectors. This mix-and-match receptor-effector design confers rich functional diversity through tissue-specific expression of 7-TM receptors (providing organ-selective responses to hormonal stimulation) and coupling of distinct receptor subtypes to different G-protein signal effectors. Whereas many hundreds of 7-TM receptors have been identified, they are coupled to only a few heterotrimeric G-proteins, encoded by 16 α subunit genes, 6 β subunit genes, and 12 γ subunit genes. Using prototypical adrenergic receptors as exemplars (Fig. 1), β1-AR coupling to Gs stimulates adenylyl cyclase and mediates positive cardiac inotropy (the vigor of myocardial contraction) and chronotropy (the rate of cardiac contraction), α2-AR coupling to Gi inhibits adenylyl cyclase and suppresses synaptic neurotransmitter release, and α1-AR coupling to Gq/11 activates phospholipase C and provokes vasoconstriction. Thus, a century-long investigative path beginning with the observation that crude extracts of adrenal medulla provoke acute tachycardia and hypertension led to our modern understanding of one of the most widespread stimulus-response systems in biology, responsible for such diverse physiology as the fight-or-flight adrenergic response and the senses of vision and smell. The structural determinants of G-protein coupled 7-TM receptor signaling and functional details beyond the scope of this manuscript have been elegantly reviewed by Nobel Laureate Brian Kobilka et al. (7).
Figure 1.

Mix-and-match of neurohormone receptors and effectors for functional diversity and tissue-specificity of response. Ribbon models of receptors were produced using swissmodel.expasy.org. Blue is amino terminus; red is carboxy terminus. Heterotrimeric G-proteins subunit color: Gα, green; Gβ, red; Gγ, purple.
NEUROHORMONES, BIOMECHANICS, AND CARDIAC STRESS ADAPTATION
Clinical therapeutics based on mimicking, interrupting, or otherwise modulating neurohormone synthesis or signaling are a cornerstone of modern medical therapy. Indeed, therapeutic interdiction of the adrenergic and renin-angiotensin systems has become the foundation for current pharmacological care of cardiac hypertrophy and heart failure. However, for decades after Powell and Slater (8) observed that dichloroisoprenaline (a chemical analog of norepinephrine) inhibited adrenergic receptors, and Nobel Laureate Sir James Black subsequently developed prototype β-adrenergic receptor antagonists (i.e., β-blockers) including propranolol (9–11), adrenergic blockade was proscribed in heart failure. Propanolol, the first β-blocker to be widely employed, found immediate therapeutic use in acute ischemic heart disease because it suppressed myocardial work (approximated by the product of heart rate and systolic blood pressure), thereby diminishing myocardial oxygen requirements to levels that could be accommodated by a diseased coronary arterial tree. The negative inotropic and chronotropic effects of cardiac β-adrenergic receptor blockade (12) were, by unassailable mechanistic logic and reports of adverse patient experiences to sympatholytic therapy (13, 14), viewed as contraindications to β-AR blockade in heart failure. Even advocates of β-blocker therapy cautioned against their use in heart failure: “Patients with left ventricular insufficiency should not be given a β-receptor blocking drug without prior administration of digitalis and diuretics” (15).
It is worth briefly considering how β-blocker therapy went from being contraindicated to a first-line, life-prolonging therapy in heart failure. When β-blockers were first developed and introduced into clinical practice, conventional wisdom regarding the regulation of integrated cardiovascular function was deeply rooted in biophysics, within the field of “cardiac mechanics.” Both acute cardiac pump function and chronic structural adaptation of the heart were understood in terms of two 19th century mathematical formulae: “Ohm’s law” was used to describe how the heart pump and its vascular conduits interact to maintain tissue perfusion, and the “Laplace relationship” described how ventricular remodeling (i.e., changes in wall thickness and/or chamber dimension) affected overall ventricular pump function (Fig. 2).
Figure 2.

How Ohm’s law and the Laplace equation can explain cardiac adaptation.
Ohm’s law is most recognized for its application to electrical circuits, but as applied to the cardiovascular system it describes how blood flow (I, or cardiac output) is determined by the pressure differential across the vascular bed (E, which for the systemic circulation is the difference between systemic arterial pressure and left atrial/pulmonary capillary wedge pressure) and vascular resistance (R, which increases as the integrated cross sectional area of the arteriolar bed decreases, i.e., during vasoconstriction) (Fig. 2). When tissue perfusion pressure (E) is constant/normal, Ohm’s law predicts that pump function/cardiac output (I) will decrease as vascular resistance (R) increases. This relationship can be used to understand the norepinephrine-mediated adrenergic fight or flight response: In the context of great emotional excitation, intense physical stress or acute massive blood loss, the imperatives are to maintain tissue perfusion pressure (E) by increasing systemic vascular resistance (R) via vasoconstricting α1-AR, and to enhance cardiac output (I) via the inotropic effects of myocardial β1-AR (see Fig. 1). According to Ohm’s law, an isolated decrease in cardiac output, as with primary cardiac insufficiency or acute massive blood loss, lowers tissue perfusion (Fig. 2). The integrated cardiovascular system reacts by invoking the adrenergic fight-or-flight response, including elaboration of epinephrine and norepinephrine. Thus, increased β1-AR mediated myocardial inotropy and chronotropy enhances pump function while intense α1-AR mediated vasoconstriction reduces blood flow to skin and periphery, thereby preferentially shunting the cardiac output to major organs and brain. Peripheral vasoconstriction and increased heart rate resulting from adrenergic stimulation underlie some of the classic physical signs of systolic heart failure, such as cool and pale extremities with a rapid and thready pulse. However, the Laplace relationship shows that increased systolic blood pressure from peripheral vasoconstriction further stresses the heart by increasing afterload (Fig. 2). Although a transient increase in cardiac afterload would likely be tolerated by a normal heart during fight-or-flight, increased afterload will further compromise cardiac pump function in primary cardiac insufficiency caused by, for example, acute myocardial infarction and viral or toxic myocarditis. Taken together, these concepts provided support for the notion that heart failure patients are “critically dependent on sympathetic activity … to maintain cardiac output” (15) and therefore should avoid β-blockers.
The Laplace relationship (Fig. 2) as applied to the left ventricle (LV) describes how chamber geometry and arterial pressure help determine LV ejection performance, i.e., the efficiency with which the heart pumps blood into its arterial conduits. In the context of LV systolic function, wall stress (σ) represents afterload, i.e., the intrinsic and extrinsic forces that oppose ventricular ejection; increased wall stress/afterload impedes ventricular ejection. The Laplace relationship predicts that afterload/systolic wall stress will increase as LV (systolic) pressure (p) increases (i.e., hypertension) and as LV ventricular size (r) increases (i.e., dilated cardiomyopathy), to the detriment of pump function. Conversely, wall stress will favorably decrease as LV wall thickness (h) increases, as in compensated cardiac hypertrophy (16, 17). Importantly, pressure is a variable in both the Laplace and Ohm relationships and is regulated in part via neurohormone-mediated vasoconstriction. The phenomenon wherein hearts modify their chamber geometry in response to different pathophysiological conditions is called cardiac remodeling and is driven in large part by hypertrophic growth or programmed death of constituent cardiac myocytes. The next section reviews the role of neurohormone signaling in these cellular events.
THE NEUROHORMONAL HYPOTHESIS OF CARDIAC HYPERTROPHY AND HEART FAILURE
As introduced, early observations that circulating epinephrine and norepinephrine levels were increased in heart failure and that β-blocker administration to heart failure patients caused acute functional deterioration, led to the widely accepted notion that failing hearts rely upon the positive inotropic, chronotropic, and vasoconstrictor effects of catecholaminergic stimulation to sustain cardiac pump output and peripheral blood pressure (18–22). The counter-hypothesis was that neurohormonal activity contributed to heart failure, which suggested that interrupting neurohormone signaling could be therapeutic. Various contrarian mechanisms were proposed in support of therapeutic trials of neurohormonal blockade in heart failure, including the idea that myocardial β-adrenergic receptor downregulation from heart failure catecholamine excess could be reversed by gentle β-AR therapy, i.e., “resensitization” (23–25). Another putative mechanism held that catecholamines and other vasoconstricting neurohormones could directly stimulate cardiac myocyte hypertrophy, independent of mechanical stress produce by increased hemodynamic load. This “neurohormonal hypothesis” was formalized by Milton Packer (26, 27), but the concept is founded upon a series of paradigm-changing studies enabled by Paul Simpson’s development of an experimental platform in which the response of tissue cultured cardiac myocytes to neurohormonal activation could be studied in the absence of altered mechanical loading (28–30). Simpson (28) initially observed that epinephrine provoked hypertrophy of unloaded neonatal rat cardiac myocytes in culture. Others subsequently adapted neonatal rat or embryonic chick cardiac myocyte systems to demonstrate analogous prohypertrophic effects of angiotensin II (31, 32) and endothelin I (33, 34). Later, Sadoshima and Izumo (35) applied neonatal rat cardiac myocytes to deformable substrates, revealing the existence of a stretch-stimulated endogenous renin-angiotensin system within cardiac myocytes, and an autocrine/paracrine system that linked mechanical stress to local, endogenous cardiomyocyte neurohormone release. Taken together, these studies showed the sufficiency of neurohormonal activation through multiple independent pathways to induce cardiac myocyte hypertrophy in vitro.
Neonatal rat cardiac myocytes in tissue culture are unlikely to react to epinephrine, angiotensin II or endothelin I in a manner that exactly recapitulates the in vivo responses of functioning hearts. Thus, it remained possible that hemodynamic stress was the dominant direct in vivo stimulus for cardiac remodeling, and that neurohormonal effects were required to sustain in vivo cardiac pump function and blood pressure as originally envisioned. Alternately, neurohormones might play a subordinate prohypertrophic role compared with the dominant effects of increased hemodynamic load. These possibilities required in vivo testing, but the straightforward study of administering the neurohormones continuously over a period of time (as with an osmotic mini-pump) and determining if that provoked cardiac remodeling could not provide an answer because the vasoconstricting effects of prohypertrophic neurohormones increases blood pressure (p), leading us back to increased biomechanical stress and the Laplace and Ohm equations.
A new experimental approach was needed to bypass the confounding vasoconstrictor effects of systemic neurohormonal stimulation and validate the hypothesis that in vivo neurohormonal stimulation of cardiac myocytes directly stimulates cardiomyocyte hypertrophy and ventricular remodeling. The development of cardiac-targeted transgenesis in mice, using promoter constructs employing cardiomyocyte-specific genes to drive cardiomyocyte-directed transgene expression (36–38), provided experimental alternatives (Fig. 3). One idea was to overexpress neurohormone receptors in mouse hearts, which was anticipated to constitutively increase signaling through that neurohormone signaling pathway in the absence of the neurohormones themselves. Indeed, transgenic receptor overexpression proved useful for exploring the qualitative and quantitative consequences of myocardial β-adrenergic receptor signaling (39–42). However, receptor expression met with variable success for prohypertrophic neurohormones (43–46), possibly because G-protein coupled receptor (GPCR) signaling is tightly regulated: chronic receptor activity induces functional receptor uncoupling and physical receptor downregulation in a negative feedback loop that ultimately suppressed the signaling pathways being experimentally activated (reviewed in Ref. 47). The solution to uncovering direct neurohormonal effects on the heart without experimental confounders was to circumvent both the prohypertrophic neurohormones and their individual GPCR receptors through transgenic cardiac myocyte-targeted expression of the common effector for prohypertrophic neurohormones and their respective receptors [i.e., epinephrine (α1-AR), angiotensin II (AT1R) and endothelin (ET) receptors], the Gq signaling protein (Fig. 3). The expectation was that overexpression of the alpha subunit of heterotrimeric Gq (Gαq), which is the component of the heterotrimer that binds both GPCRs and their common immediate downstream effector phospholipase C (48), would tonically activate signaling downstream of all three receptors without evoking ligand-mediated receptor desensitization or downregulation. The Dorn laboratory was the first to do this, and the result was what has been described as “pressure overload hypertrophy without pressure overload” (49, 50). Despite normal left ventricular systolic pressure, cardiomyocyte Gq overexpression recapitulated left ventricular wall thickening, the increase in cardiac myocyte cross-sectional area, β-AR desensitization, and reexpression of fetal cardiac genes that are hallmarks of pressure overload hypertrophy (49, 51–53). Together, these findings provided strong support for the notion that Gq signaling through myocardial neurohormone receptors is sufficient to stimulate in vivo cardiac hypertrophy in the absence of any abnormal hemodynamic load or stress. Subsequent studies employing a Gq inhibitory peptide (54) or cardiac-directed ablation of the genes encoding Gαq and functionally overlapping Gα11 (55) revealed suppression of pressure overload hypertrophy when cardiomyocyte Gq signaling was interrupted. Thus, Gq signaling was shown to be both necessary and sufficient for pressure-overload cardiac hypertrophy in mice.
Figure 3.

Experimental approaches used for in vivo testing of the neurohormonal theory of cardiac hypertrophy/heart failure.
The natural history of cardiac hypertrophy when the inciting hemodynamic stress is not relieved is progression to dilated cardiomyopathy with overt heart failure, called “decompensation.” In vivo Gq manipulation mechanistically linked “adaptive” cardiac hypertrophy to Gq-mediated neurohormone signaling, but did not reveal how or why this mechanism for functional compensation ultimately fails. In biomechanical terms of the Laplace equation: Why does the increase in ventricular wall thickness (h) that normalizes wall stress (σ) in the face of pressure overload (p) ultimately decompensate? Because development of transgenic Gq-mediated cardiac hypertrophy was a cellular event wherein enlargement and transcriptional reprogramming of individual cardiomyocytes collectively produced hypertrophic ventricular remodeling, we asked whether maladaptive ventricular dilation might likewise have its mechanistic foundations within individual cardiomyocytes. The idea that adaptive ventricular hypertrophy predisposes to maladaptive dilated cardiomyopathy was strengthened by the observation that a modest left ventricular pressure stimulus that would not normally cause heart failure provoked dilated cardiomyopathy in Gq transgenic mice (56). Moreover, a human GNAQ gene polymorphism that increases expression of Gαq was linked to accelerated mortality among African American patients with heart failure (57). These correlative studies implied detrimental synergy between hemodynamic stress and Gq-mediated neurohormone signaling.
The mechanism for maladaptive ventricular dilation in Gq mice was uncovered in Gq transgenic mice with greater Gq signaling activity than the original “compensated hypertrophy” Gq mouse line (58). These dual heterozygous Gq mice (carrying two independent Gq transgenes on different chromosomal loci) developed spontaneous adverse cardiac remodeling, i.e., left ventricular chamber dilation and wall thinning in the absence of any abnormal extracardiac hemodynamic stress. The underlying cellular mechanism mediating the transition to decompensated dilated cardiomyopathy (and also for a unique peripartum cardiomyopathy that afflicted Gq transgenic dames) was identified as apoptotic cardiac myocyte drop-out linked to markedly increased expression of proapoptotic factors (59). Likewise, a constitutively active Gαq mutant in unloaded cultured neonatal rat cardiac myocytes provoked apoptosis (58). Induction of proapoptotic genes was not a nonspecific feature of all forms of hypertrophy, but seemed to be specifically evoked by pathological hypertrophy signaling through the neurohormone/Gq signaling pathway (59, 60). Thus, the very process of neurohormone-stimulated cardiac myocyte hypertrophy, which can initially compensate for increased ventricular loading via the Laplace mechanism, carries within it changes in gene expression that culminates in programmed cardiac myocyte death, maladaptive ventricular remodeling and overt heart failure.
Collectively, these results provided mechanistic support for clinical trials of neurohormone antagonists in heart failure. Ironically, the major clinical trials demonstrating therapeutic efficacy of neurohormonal blockade were largely complete before neurohormonal mechanisms leading to cardiac hypertropy and failure were understood (Fig. 4). The first proof of concept study of neurohormonal blockade in heart failure used the α1-AR blocker prazocin (a vasodilator) to unload the heart. As reported in 1980 by Colucci et al. (61), prazocin-treated patients with severe heart failure showed initial improvement in objective metrics for left ventricular performance as well as symptoms. However, the benefits diminished over time, presumably because of increased activity of the renin-angiotensin system (RAS). Indeed, interrupting the RAS pathway with angiotensin converting enzyme (ACE) inhibitors captopril or enalapril was subsequently shown to be both symptom-relieving and life-prolonging as stand-alone therapy for heart failure of various causes (62–65). Perhaps more surprising were observations that β-AR antagonists (“β-blockers”) used in combination with ACE inhibitors further reduced hospitalizations and prolonged survival in chronic heart failure (66–68). By contrast, endothelin A receptor blockade with either darusentan or bosentan added to conventional therapy with ACE inhibitors and β-blockers showed no benefit for cardiac remodeling, heart failure symptoms or outcomes (69, 70). Thus, interrupting AT1 and β-AR signaling were proven to be strikingly beneficial in heart failure and their administration is the standard of modern medical care.
Figure 4.

Approximate timeline of basic and clinical studies testing neurohormone effects in cardiomyocyte hypertrophy and clinical heart failure. Key references are provided, color-coded by neurohormone system. Struck-through references indicate a negative study.
CARDIAC MALADAPTATION, NEUROHORMONES, AND MITOCHONDRIA
The sections above describe the convoluted conceptual path from early recognition that neurohormones regulate acute myocardial function via the Gs-mediated fight-or-flight response to a scientific understanding that neurohormonal responses determine chronic cardiac adaptation via Gq-mediated transcriptional programs for adaptive cardiomyocyte hypertrophy and maladaptive programmed cardiomyocyte death. The current section delves more deeply into the role of mitochondria as central regulators of cardiomyocyte fate in myocardial hypertrophy and the transition to dilated cardiomyopathy.
Hearts have the highest metabolic rate and greatest mitochondrial density of any mammalian organ. It has been calculated that an average human heart, which is ∼40% mitochondria by mass, will generate and consume ∼5kg of ATP in a single minute (71, 72). The reliance of myocardium upon mitochondrial ATP suggests that mitochondrial fitness can be an important determinant of cardiac health and integrated cardiovascular function, both under normal conditions and when the heart is damaged, diseased or chronically overloaded. Thus, it is not surprising that metabolic remodeling, wherein the normal adult myocardial preference for fatty acids as mitochondrial substrates reverts to a fetal myocardium-like preference for carbohydrates, is recognized in many myocardial disease (73–76).
Mitochondria are essential to cardiac pump functioning and stress-adaptation, but are unique in that they can either sustain or terminate the life of cells within which they reside. Although mitochondrial metabolism creates life-sustaining ATP via oxidative phosphorylation, mitochondria are also the “gatekeepers” for programmed cell death via apoptotic and nonapoptotic pathways (77–80). The roles played by mitochondria in apoptosis and programmed necrosis, and the respective roles of caspase signaling and activation of the mitochondrial permeability transition pore (MPTP) in these programmed cell death pathways, have been extensively reviewed elsewhere (81–83) and so will not be reproduced here. Rather, we address the question of how the transformation of cardiac myocyte mitochondria from cell-sustaining ATP generators into cell-killing engines of programmed death contributes to cardiac maladaptation.
Transcriptional and epitranscriptional reprogramming during pathological (as opposed to exercise-induced or physiological) (17) cardiac hypertrophy was initially recognized primarily for reexpression of embryonic heart genes, the so-called “fetal gene expression program” (84–90). However, microarray profiling of mRNAs differentially expressed in hypertrophied/compensated (i.e., nonfailing) Gq hearts revealed abnormally high levels of multiple transcripts linked to apoptosis (60). Of particular interest was Bnip3l or Nix (91), a Bcl2 family protein originally identified by Greenberg as proapoptotic (92). Contextually, Nix was upregulated in Gq and pressure-overload hypertrophies, whereas closely related proapoptotic Bnip3 was upregulated after ischemic myocardial injury (93). Cardiomyocyte-targeted transgenic Nix overexpression proved sufficient to induce cardiomyocyte apoptosis, provoking adverse ventricular remodeling and dilated cardiomyopathy (91, 94). Moreover, germ-line or cardiomyocyte-specific genetic Nix ablation suppressed cardiomyocyte apoptosis and attenuated ventricular dilation in Gq-mediated and pressure overload hypertrophy (95); Bnip3 plays an analogous role mediating cardiomyocyte apoptosis after postischemic cardiac injury (96). Together, these results showed that mitochondrial death protein Nix is transcriptionally induced during compensated hypertrophy, and revealed that Nix is both necessary and sufficient for maladaptive hypertrophy remodeling leading to dilated cardiomyopathy.
Conventional apoptosis is mediated by a well-characterized sequence of events: proapoptotic Bcl2-family proteins form macromolecular pores in mitochondrial outer membranes, permitting release of sequestered cytochrome C (mw 12,000 Da) into the cytosol. Extramitochondrial cytochrome C interacts with cytosolic Apaf-1 to form the multimeric apoptosome, which binds and activates caspase 9 in a ternary complex (97–100). Both Nix and Bnip3 localize to mitochondria and interact with proapoptotic Bax to initiate “intrinsic” pathway mitochondrial apoptosis mediated by the caspase cascade (92, 101) (Fig. 5).
Figure 5.

Hypertrophy-associated programmed death pathways induced by Gq-coupled neurohormone stimulation. Signaling events connecting a prohypertrophic, Gq-coupled receptor to apoptotic and necrotic programmed cell death are shown as described in the text. Transcriptional upregulation of Nix is not specifically shown, but is part of the hypertrophy-associated change in gene expression profile. cyt C, cytochrome C; IP3R, IP3 receptor; MCU, mitochondrial calcium uniporter; MPTP, mitochondrial permeability transition pore; PKC, protein kinase C; PLC, phospholipase C. Nix ribbon diagram was produces using swissmodel.expasy.org; red is Nix carboxy terminal transmembrane domain. Red box is expanded in Fig. 7D.
It is worth considering why potent proapoptotic factors such as Nix and Bnip3 are expressed in terminally differentiated cardiac myocytes that, if lost, cannot readily be replaced. One possibility is that apoptosis, which plays a central role in determining cardiac chamber and valve structure in developing embryonic hearts (102), is simply part of a fetal transcriptional program that is reexpressed in myocardial diseases. Alternately, Nix may have a necessary physiological function, independent of apoptosis. Indeed, although the majority of Nix localizes to mitochondrial outer membranes where it is positioned to activate caspase-dependent apoptosis, a substantial fraction of Nix localizes instead to endo/sarcoplasmic (ER/SR) reticulum where it can, through poorly understood mechanisms, activate calcium uptake and increase ER/SR calcium levels (103, 104) (Fig. 5). Mitochondria are physically associated with ER/SR and can sense ER/SR calcium release, which in the case of cardiomyocyte SR is cyclic, regulated by Gs-coupled β1-AR (105, 106), and is a primary determinant of cardiomyocyte contraction and cardiac failure (107). An increase in SR calcium stores such as that induced by Nix can therefore sensitize mitochondria to SR calcium crosstalk (108, 109). SR calcium release increases with the frequency of cardiomyocyte contraction (increased heart rate) provokes enhanced mitochondrial calcium uptake that accelerates calcium-sensitive mitochondrial metabolic pathways, resulting in accelerated ATP production (110, 111). In this way, Nix expression might help functionally compensate for greater demands on the cardiac pump. However, this same mitochondria-SR calcium crosstalk mechanism can, if of sufficient magnitude, activate the mitochondrial permeability transition pore (MPTP) leading to permeabilization of outer mitochondrial membranes and a catastrophic loss of mitochondrial structural integrity that halts or reverses ATP production (112) (Fig. 5). Nix is unusual in that it can activate both intrinsic caspase-dependent apoptosis and MPTP-dependent “programmed necrosis” pathways in mouse hearts (104, 113). Consequently, therapeutic cardiomyocyte salvage in neurohormone-dependent pressure overload hypertrophy aimed at preventing the transition to dilated cardiomyopathy and heart failure will likely require interrupting pathways leading to both mitochondria-mediated apoptosis and MPTP-mediated programmed cell death.
MITOCHONDRIAL DYNAMICS PROTEINS AND CARDIAC MALADAPTATION
Accumulated data support an important role for modulated mitochondrial metabolism and mitochondria-mediated programmed cell death in neurohormone-stimulated responses to cardiac injury and chronic hemodynamic stress. In addition to their unique dual roles in cell-sustaining ATP production and cell-terminating apoptosis/MPTP-mediated necrosis, mitochondria are exceptional in their ability to structurally remodel and relocate, referred to as “mitochondrial dynamism.” Dynamic behavior of mitochondria has been appreciated for over a century (114), but is highly contextual. For example, cultured fibroblasts are a preferred experimental system for studying mitochondrial fusion and fission because fibroblast mitochondria exist as an interconnected collective that undergoes almost continuous structural remodeling via these two dynamic processes (115, 116). On the other hand, fibroblasts are a poor platform to study mitochondrial motility because only a few fibroblast mitochondria undergo directed subcellular transport at any given time. Thus, mitochondrial motility is more readily assessed in neuronal processes wherein, under basal conditions, up to 30% of mitochondria are undergoing active transport (116–119).
Compared to fibroblasts and neurons, cardiomyocyte mitochondria appear relatively hypodynamic (120, 121). Moreover, cardiomyocyte mitochondria exist as many physically distinct organelles interspersed between myofilaments rather than as a filamentous network. While mitochondrial fission and fusion can certainly play roles in heart disease, as reviewed elsewhere (122–124), physically interconnected mitochondrial networks that undergo fusion/fission-mediated physical remodeling are not present in cardiac myocytes. Rather, cardiac myocyte mitochondria exhibit transient physical connectivity referred to as “kiss and run,” and interorganelle communications which, instead of being mediated by complete organelle fusion, involve reversable formation of tunneling nanotubes (125–127) and possibly other mechanisms (128, 129). Furthermore, mitochondrial turnover via fusion and fission is slower in cardiac myocytes than in other cell types (130). Mitochondrial motility is even less important to myocardial homeostasis than fusion and fission because the tightly organized intracellular architecture of cardiac myocytes restrains subcellular organelle transport. Nevertheless, the principal effectors of mitochondrial dynamics, including fusion proteins mitofusins (MFN) 1 and 2 and optic atrophy 1 (OPA1), and the fission protein dynamin-related protein 1 (DRP1), are expressed at high levels in mammalian myocardium (131–134). Moreover, cardiomyocyte-specific ablation of the genes encoding each of these mitochondrial fusion/fission proteins induces striking pathological cardiac phenotypes, revealing the necessity for mitochondrial fusion and fission proteins during both cardiac development and normal adult heart functioning (130, 135–140) (Fig. 6). The inference was that mitochondrial dynamics factors might be functioning in a noncanonical manner in the heart, i.e., that they are functioning in additional ways than just mediating mitochondrial dynamism (141).
Figure 6.

Schematic depiction of cardiac phenotypes provoked by cardiomyocyte-targeted genetic mitofusin manipulation. Arrow represents developmental stage.
The prototypical noncanonical function for a mitochondrial dynamics protein is mitofusin (MFN) 2 tethering of mitochondria to endo/sarcoplasmic reticulum. As introduced, physical approximation of mitochondria to ER/SR facilitates calcium transport from reticular structures to neighboring mitochondria, thereby modulating both mitochondrial metabolism and MPTP-mediated cell death (142, 143) (reviewed in Refs. 108, 109, 144). Indeed, mitochondrial calcium uptake through the uniporter (MCU), which requires at least micromolar concentrations of calcium that are not normally achieved in cytoplasm (145, 146), is thought to require close physical connections between mitochondria and ER/SR that form privileged mitochondrial-reticular microdomains through which calcium can transfer between organelles without diffusing into the cytoplasm (147, 148). Multimeric MFN2 is embedded within outer mitochondrial membranes from which it extends out into cytosolic space to dimerize in trans with mitofusins (1 or 2) on adjacent mitochondria (Fig. 7); this is its canonical function in mitochondrial-mitochondrial tethering and is required for subsequent outer membrane fusion (149–151). However, a minor fraction of MFN2 localizes to ER/SR, enabling mitochondrial-ER/SR tethering (152) that helps form calcium microdomains for trans-organelle calcium signaling (153–155). An evaluation of the consequences of cardiac-targeted MFN2 gene ablation on cardiomyocyte SR-mitochondria physical approximation and trans-organelle calcium signaling supported the concept that MFN2 promotes functional microdomains which aid mitochondrial sensing of SR calcium release (143). However, in this study MFN2 ablation produced only a transient delay in the mitochondrial reaction to SR calcium release prompted by increased work. The conclusion was that MFN2 tethering of mitochondria to SR contributes to the bioenergetic feedback response during physiological stress, but this aspect of MFN2 biology does not readily explain the development of severe cardiac phenotypes in mitofusin-deficient embryonic and adult hearts.
Figure 7.

Mitofusin 2 hypothetical active structure and putative multimeric structures. A: human MFN2 monomer in extended conformation with labelled structural domains. TM is transmembrane domain. B: comparison with Gαq shows a similar GTPase domain but absence of dynamin-like core and TM structures present in MFN2. C, left: putative MFN2 cis-homodimer, envisioned to assemble into dynamin-like rosettes on organelle membranes. D: cross section of how two MFN2 rosettes might interact in trans across cytoplasmic space tethering outer mitochondrial membranes (OMM) to sarcoplasmic reticulum (SR). This panel is greatly expanded view of red box in Fig. 5.
An important clue to alternate roles played by mitochondrial dynamics factors in vertebrate and invertebrate myocardium was the discovery that, under defined conditions, MFN2 is a central effector of the PINK1-Parkin pathway of mitophagic mitochondrial quality control (156). PINK1 is a kinase imported into, and then immediately degraded by, healthy mitochondria. This kinase is stabilized only in damaged mitochondria, wherein it can initiate mitophagy signaling through the PINK-Parkin pathway. Parkin is an E3 ubiquitin ligase activated by PINK1 in damaged mitochondria (157). When phosphorylated by mitochondrial PINK1, MFN2 exchanges an ability to interact with MFN1 and MFN2 on other mitochondria (e.g., during mitochondrial fusion) for an ability to bind cytosolic Parkin, thus initiating mitophagy (158–160). The consequence of Parkin recruitment to mitochondria by MFN2 and other “Parkin receptors” is polyubiquitination of a hundred or more outer mitochondrial membrane proteins, which attracts autophagosomes that engulf the damaged organelle and transport it to lysosomes for decomposition (157). Thus, unphosphorylated MFN2 actively promotes mitochondrial fusion, but is inactive for mitophagy (and vice versa).
The discovery of MFN2 functioning in mitophagy and the delineation of a mechanism by which it regulates fusion versus Parkin binding suggested an experimental means for distinguishing the functional impact of MFN2-mediated mitochondrial fusion versus mitophagy in mouse hearts in vivo: Cardiomyocyte-directed expression of MFN2 mutants engineered either to mimic or prevent PINK1-mediated phosphorylation of fusion/mitophagy switch sites. Conventional wisdom was that PINK1-Parkin mitophagy was the dominant mechanism for mitochondrial quality control, wherein individual damaged mitochondria initiate a pathway culminating in their selective suicide (157). If MFN2 was an essential intermediate between PINK1 and Parkin in mitophagy, then suppression of MFN2-mediated mitochondrial Parkin recruitment should interrupt mitophagy, resulting in accumulation of abnormal and damaged mitochondria due to defective quality control at the organelle level. However, this is not what was observed. Rather, suppression of MFN2-Parkin binding elicited a perinatal developmental defect characterized by cardiomyocyte retention of fetal-type mitochondria and failure to replace them with adult-type mitochondria, as is normal within a few days to weeks after birth (161). Failure to transition from fetal heart carbohydrate-based mitochondrial metabolism to postnatal fatty-acid based mitochondrial metabolism was incompatible with life. These studies uncovered a previously unappreciated function for PINK1-Parkin mediated mitophagy as the process mediating the normal developmentally regulated generalized turnover of mitochondria that enables the transition from fetal to adult myocardial metabolism. Indeed, at least for adult mouse myocardium, the PINK1-Parkin mitophagy pathway does not appear to play a major role in homeostatic mitochondrial quality control, but instead is a stress-induced pathway activated after cardiac injury (156, 162–164).
NONCANONICAL FUNCTIONING OF MITOCHONDRIAL FUSION PROTEINS IN EXTRACARDIAC DISEASES
The results described in the previous section reveal unexpected mechanisms by which mitochondria contribute to myocardial disease. It seems intuitively obvious that mitochondrial metabolic dysfunction would compromise pumping efficiency of this most mitochondrial-rich organ. Yet, what has been observed are mitochondrial pathways to cardiac disease, including programmed cardiomyocyte death and interrupted mitophagy, in which altered ATP production is a secondary, collateral or unrelated event. Although investigating context-driven noncanonical functioning by mitochondrial dynamics factors in the heart is still in its early stages, the notion that hypodynamic myocardial mitochondria may be uniquely suited to uncovering functioning of mitochondrial dynamics proteins beyond fusion and fission gives rise to the concept that organ-specific mitochondrial dynamic physiology can exist beyond the heart.
It seems noteworthy that no clinical cardiac-specific diseases are yet known to be caused by mutations of MFN1, MFN2, OPA1, or DRP1. Rather, the cardiac-disease associations are almost entirely in animal models using gene knockout techniques. Clinically, MFN1- linked human disease has not been described. OPA1 mutations are most recognized as causing autosomal dominant optic atrophy (165–168) (although a homozygous missense OPA1 mutation is reported to cause mitochondrial DNA depletion syndrome with lethal infantile encephalopathy and hypertrophic cardiomyopathy in two siblings) (169). De novo heterozygous mutations in the DNM1 gene that encodes the mitochondrial fission protein DRP1 were reported as a very rare cause of Developmental and Epileptic Encephalopathy type 31 (170), but are not an identified cause of common genetic diseases. Indeed, the only relatively common genetic condition linked to mutations of a mitochondrial dynamics protein is the predominantly pediatric peripheral neuropathy, Charcot-Marie-Tooth disease type 2 A (CMT2A). CMT2A is caused by ∼100 different mutations of MFN2 (171, 172). MFN2 mutations have also been identified as a cause of rare genetic lipodystrophy and variant forms of dominant optic atrophy (173, 174). Excellent reviews of MFN2 and CMT2A are available for the interested reader (175–178). Here CMT2A is discussed as the prototype disease incontrovertibly caused by a disturbance of mitochondrial dynamism.
Because CMT2A is caused by mutations in a mitofusin, it is widely considered to be a disease of interrupted or incomplete mitochondrial fusion (179–181). However, CMT2A characteristically has the greatest pathological impact on the distal portions of nerves innervating the upper and lower extremities, i.e., the sections of the longest nerves in the body that are farthest from the neuronal cell bodies, or soma. Because mitochondrial fusion is a ubiquitous process that is seemingly essential to every cell type and tissue, it is curious that a mutational fusion defect would have such a specific pathophysiological impact. Indeed, systemic interruption of mitochondrial fusion in mice by ablating either the Mfn1 or Mfn2 genes was embryonic lethal (182) and conditional ablation of Mfn2 in mouse hearts, livers and brains evokes dysfunction of the respective organs (183–187). Exclusivity of clinical disease with mutations of MFN2 (vs. MFN1) and specificity of disease within distal long peripheral neurons of CMT2A patients with MFN2 mutations suggested to some that neurons might rely more than other cell types on MFN2 versus MFN1 for mitochondrial fusion (188, 189). Support for the idea that MFN1 cannot functionally compensate for mutant MFN2 specifically in neurons was provided by reports that cultured dermal fibroblasts of patients with CMT2A did not exhibit the expected fragmented mitochondria (190, 191). However, absence of mitochondrial pathology in cultured fibroblasts of patients with CMT2A was recently discovered to be a function of the fibroblast culture conditions, and not attributable to any distinct resistance to MFN2 CMT2A mutations in nonneuronal cells (116). An alternate hypothesis that could explain unique neuronal sensitivity of long peripheral neurons to MFN2 mutations is that, as in the heart, MFN2 has a noncanonical function with specific biological relevance to long neurons. Accumulating data suggest that this extra function is regulation of mitochondria motility.
Synaptic release and reuptake of neurohormones is an energy intensive process requiring mitochondrial ATP that, because ATP is chemically unstable at physiological temperature and pH, must be generated locally by mitochondria located near neuronal synapses (192, 193). Since mitochondrial biogenesis must occur near the neuronal nucleus, wherein over 99% of genes encoding mitochondrial proteins are encoded and transcribed, delivery of fresh mitochondria to distal neuro-muscular or neuroneuronal junctions requires that mitochondria be transported from neuronal soma and through axons. For this reason, abnormal mitochondrial motility compromises neuronal signaling, repair and regeneration and is a commonly described feature of multiple neurodegenerative diseases (180, 194).
The best understood mechanism of mitochondrial motility involves calcium-regulated binding of outer mitochondrial membrane MIRO proteins to Trafficking kinesin-binding (TRAK/Milton) 1 and 2 adaptor proteins that couple mitochondria and other cellular cargo to dynein and kinesin-1 molecular motors for directed transport along microtubules (195–200). Severe mitochondrial dysmotility is a hallmark of neuronal die-back in CMT2A (201, 202). Baloh first suggested a mechanistic link between CMT2A MFN2 mutations and mitochondrial transport by showing that 1) MFN2 normally binds MIRO1 and MIRO2; and 2) CMT2A MFN2 mutants exhibit defective mitochondrial transport (183, 203, 204). Collectively, the observational data support a regulatory role for MFN2 in mitochondrial motility, perhaps by priming the critical MIRO-TRAK interaction (205). Confirming this hypothesis will require more fully delineating the key determinants and biological consequences of MFN2-MIRO interactions.
The most compelling evidence supporting both a central pathological role for mitochondrial dysmotility in CMT2A caused by MFN2 mutations and a key role for MFN2 as a regulator of axonal mitochondrial trafficking may be the ability of small molecular activators of mitofusins to reverse mitochondrial dysmotility and neuromuscular degeneration in preclinical CMT2A models (202). Reprogrammed CMT2A patient motor neurons, cultured CMT2A mouse neurons, and neuronal axons of CMT2A mouse sciatic nerves all exhibit nearly complete loss of mitochondrial trafficking through neuronal processes (206). To the extent studied, mitochondrial dysmotility in CMT2A appears agnostic to different MFN2 CMT2A mutations (206). Importantly, pharmacological mitofusin activation with either fusogenic peptides (207) or structurally dissimilar small molecules (206, 208–210) has normalized CMT2A mitochondrial dysmotility. In vivo, mitofusin activation reversed functional and neuroelectrophysiological metrics of neuromuscular degeneration and promoted neuronal regrowth by restoring mitochondrial occupancy at axonal growth buds and neuromuscular junctions (206). Taken together, these results implicate MFN2 in neuronal mitochondrial transport/delivery, thereby further expanding its recognized biological functions.
MFN2 multifunctioning seems attributable to its ability to pair with protein partners depending upon cellular and pathophysiological context (Fig. 8). Given the central importance of PINK1-MFN2-Parkin mitophagy to cardiac myocytes, versus mitochondrial transport for neurons, this concept suggests that a hypothetical MFN2 mutation impairing mitophagy and sparing mitochondrial motility would evoke cardiomyopathy instead of neuropathy. Likewise, a hypothetical MFN2 mutation that suppresses mitochondrial fusion without impairing motility might produce either milder neuropathy than is typical for CMT2A or a different spectrum of organ involvement. Because the focus on MFN2 mutations has been almost entirely on their suppression of mitochondrial fusion, significant effort will be required to test these notions.
Figure 8.

Schematic depiction of the relationship between MFN2 protein partnering, function, and disease manifestation when disrupted.
THE “BIG PICTURE”
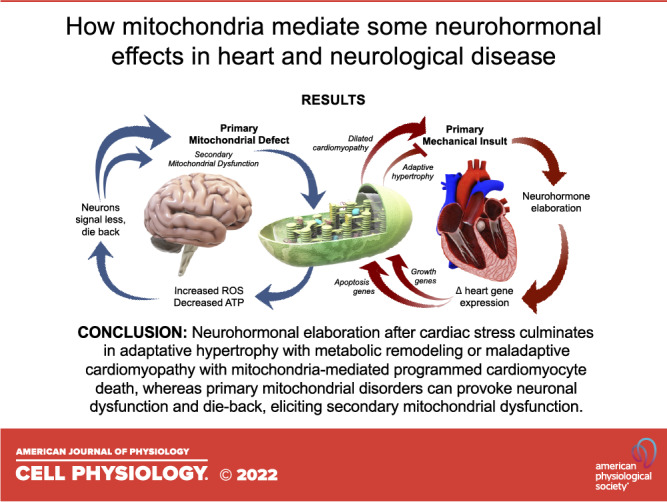
In this overview, I have employed two seemingly different cellular processes, neurohormonal signaling and mitochondrial dynamics, as examples of how biological pathways can be inextricably but differentially linked in cardiac and neurological disease. In heart disease, neurohormonal signaling is an important proximal effector of adaptation to hemodynamic stress, mediated in large part by induction of transcriptional programs leading to cardiomyocyte hypertrophy and apoptosis. Furthermore, perinatal cardiac development seems uniquely dependent upon mitochondrial replacement mediated by MFN2-Parkin for metabolic adaptation to the extrauterine milieu. Thus, increased signaling through neurohormone receptor-effectors in response to mechanical injury or stress is a proximate cause of pathological changes to biological pathways regulating replacement of cardiomyocyte mitochondria by mitophagy and inducing programmed cardiomyocyte death. In neurological diseases caused by primary damaging mutations or secondary dysregulation of mitochondrial dynamics factors the same connections exist, but the pathological cycle is reversed. As in CMT2A caused by MFN2 mutations, suppression of mitochondrial transport along neuronal processes interrupts neuronal signaling because cytotoxic senescent or damaged mitochondria accumulate at neuronal synapses, thereby impairing neurohormone release/reuptake and ultimately provoking neuronal die-back. A more complete understanding of how cardiac myocytes and neurons rely in different ways upon ubiquitous protein signaling factors, such as heterotrimeric G-proteins and MFN2 dynamin GTPases reviewed here, may provide insights into organ system selectivity in genetic and nongenetic disease.
GRANTS
This work was supported by National Institutes of Heart/National Heart, Lung, and Blood Institute (NIH/NHLBI) R35 HL135736 and NIH/National Institute of Neurological Disorders and Stroke (NINDS) R42 NS115184 and R42NS113642.
DISCLOSURES
G.W.D. is an inventor on patents that cover the use of small molecule mitofusin activators to treat neurodegenerative diseases and is the founder of Mitochondria in Motion, Inc., a Saint Louis based biotech R&D company that aims to enhance mitochondrial trafficking and fitness in neurodegenerative diseases.
AUTHOR CONTRIBUTIONS
G.W.D. prepared figures; drafted manuscript; edited and revised manuscript; approved final version of manuscript.
ACKNOWLEDGMENTS
G.W.D. is the Philip and Sima K. Needleman Professor at the Washington University in St. Louis School of Medicine.
This article is part of the special collection “Advances in GPCRs: Structure, Mechanisms, Disease, and Pharmacology.” Wei Kong, MD, PhD, and Jinpeng Sun, PhD, served as Guest Editors of this collection.
REFERENCES
- 1.Oliver G, Schäfer EA. The physiological effects of extracts of the suprarenal capsules. J Physiol 18: 230–276, 1895. doi: 10.1113/jphysiol.1895.sp000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rao Y. The first hormone: adrenaline. Trends Endocrinol Metab 30: 331–334, 2019. doi: 10.1016/j.tem.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Dale HH. On some physiological actions of ergot. J Physiol 34: 163–206, 1906. doi: 10.1113/jphysiol.1906.sp001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dale HH. On the action of ergotoxine; with special reference to the existence of sympathetic vasodilators. J Physiol 46: 291–300, 1913. doi: 10.1113/jphysiol.1913.sp001592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahlquist RP. A study of the adrenotropic receptors. Am J Physiol 153: 586–600, 1948. doi: 10.1152/ajplegacy.1948.153.3.586. [DOI] [PubMed] [Google Scholar]
- 6.Lefkowitz RJ. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci 25: 413–422, 2004. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature 459: 356–363, 2009. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powell CE, Slater IH. Blocking of inhibitory adrenergic receptors by a dichloro analog of isoproterenol. J Pharmacol Exp Ther 122: 480–488, 1958. [PubMed] [Google Scholar]
- 9.Black JW, Crowther AF, Shanks RG, Smith LH, Dornhorst AC. A new adrenergic beta-receptor antagonist. Lancet 1: 1080–1081, 1964. doi: 10.1016/S0140-6736(64)91275-9. [DOI] [PubMed] [Google Scholar]
- 10.Black J. A life in new drug research. Br J Pharmacol 160, Suppl 1: S15–S25, 2010. doi: 10.1111/j.1476-5381.2010.00848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyall J. James Black. BMJ 340: c1817, 2010. doi: 10.1136/bmj.c1817. [DOI] [Google Scholar]
- 12.Prichard BN, Gillam PM. Use of propranolol (inderal) in treatment of hypertension. Br Med J 2: 725–727, 1964. doi: 10.1136/bmj.2.5411.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillam PM, Prichard BN. Propranolol in the therapy of angina pectoris. Am J Cardiol 18: 366–369, 1966. doi: 10.1016/0002-9149(66)90055-5. [DOI] [PubMed] [Google Scholar]
- 14.Wolfson S, Gorlin R. Cardiovascular pharmacology of propranolol in man. Circulation 40: 501–511, 1969. doi: 10.1161/01.cir.40.4.501. [DOI] [PubMed] [Google Scholar]
- 15.Prichard BN. The second Lilly Prize Lecture, University of Newcastle, July 1977. beta-Adrenergic receptor blockade in hypertension, past, present and future. Br J Clin Pharmacol 5: 379–399, 1978. doi: 10.1111/j.1365-2125.1978.tb01644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spike BT, Daria D, Jegga AG, Geiger H, Aronow BJ, Molkentin JD, Macleod KF, Kalfa TA, Dorn GW 2nd.. Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci USA 104: 6794–6799, 2007. doi: 10.1073/pnas.0610666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dorn GW 2nd. The fuzzy logic of physiological cardiac hypertrophy. Hypertension 49: 962–970, 2007. doi: 10.1161/HYPERTENSIONAHA.106.079426. [DOI] [PubMed] [Google Scholar]
- 18.Chidsey CA, Harrison DC, Braunwald E. Augmentation of the plasma nor-epinephrine response to exercise in patients with congestive heart failure. N Engl J Med 267: 650–654, 1962. doi: 10.1056/NEJM196209272671305. [DOI] [PubMed] [Google Scholar]
- 19.Chidsey CA, Braunwald E, Morrow AG, Mason DT. Myocardial norepinephrine concentration in man. Effects of reserpine and of congestive heart failure. N Engl J Med 269: 653–658, 1963. doi: 10.1056/NEJM196309262691302. [DOI] [PubMed] [Google Scholar]
- 20.Spann JF Jr, Chidsey CA, Pool PE, Braunwald E. Mechanism of norepinephrine depletion in experimental heart failure produced by aortic constriction in the guinea pig. Circ Res 17: 312–321, 1965. doi: 10.1161/01.res.17.4.312. [DOI] [PubMed] [Google Scholar]
- 21.Chidsey CA, Sonnenblick EH, Morrow AG, Braunwald E. Norepinephrine stores and contractile force of papillary muscle from the failing human heart. Circulation 33: 43–51, 1966. doi: 10.1161/01.cir.33.1.43. [DOI] [PubMed] [Google Scholar]
- 22.Braunwald E, Chidsey CA, Pool PE, Sonnenblick EH, Ross J Jr, Mason DT, Spann JF, Covell JW. Congestive heart failure. Biochemical and physiological considerations. Combined clinical staff conference at the National Institutes of Health. Ann Intern Med 64: 904–941, 1966. doi: 10.7326/0003-4819-64-4-904. [DOI] [PubMed] [Google Scholar]
- 23.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N Engl J Med 307: 205–211, 1982. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 24.Lowes BD, Gilbert EM, Abraham WT, Minobe WA, Larrabee P, Ferguson D, Wolfel EE, Lindenfeld J, Tsvetkova T, Robertson AD, Quaife RA, Bristow MR. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med 346: 1357–1365, 2002. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- 25.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of β-adrenergic signaling in heart failure? Circ Res 93: 896–906, 2003. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- 26.Packer M. The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J Am Coll Cardiol 20: 248–254, 1992. doi: 10.1016/0735-1097(92)90167-l. [DOI] [PubMed] [Google Scholar]
- 27.Packer M. Evolution of the neurohormonal hypothesis to explain the progression of chronic heart failure. Eur Heart J 16, Suppl F: 4–6, 1995. doi: 10.1093/eurheartj/16.suppl_f.4. [DOI] [PubMed] [Google Scholar]
- 28.Simpson P. Norepinephrine-stimulated hypertrophy of cultured rat myocardial cells is an alpha 1 adrenergic response. J Clin Invest 72: 732–738, 1983. doi: 10.1172/JCI111023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long CS, Ordahl CP, Simpson PC. Alpha 1-adrenergic receptor stimulation of sarcomeric actin isogene transcription in hypertrophy of cultured rat heart muscle cells. J Clin Invest 83: 1078–1082, 1989. doi: 10.1172/JCI113951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waspe LE, Ordahl CP, Simpson PC. The cardiac beta-myosin heavy chain isogene is induced selectively in alpha 1-adrenergic receptor-stimulated hypertrophy of cultured rat heart myocytes. J Clin Invest 85: 1206–1214, 1990. doi: 10.1172/JCI114554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker KM, Aceto JF. Angiotensin II stimulation of protein synthesis and cell growth in chick heart cells. Am J Physiol Heart Circ Physiol 259: H610–H618, 1990. doi: 10.1152/ajpheart.1990.259.2.H610. [DOI] [PubMed] [Google Scholar]
- 32.Miyata S, Haneda T. Hypertrophic growth of cultured neonatal rat heart cells mediated by type 1 angiotensin II receptor. Am J Physiol Heart Circ Physiol 266: H2443–H2451, 1994. doi: 10.1152/ajpheart.1994.266.6.H2443. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki T, Hoshi H, Mitsui Y. Endothelin stimulates hypertrophy and contractility of neonatal rat cardiac myocytes in a serum-free medium. FEBS Lett 268: 149–151, 1990. doi: 10.1016/0014-5793(90)80995-U. [DOI] [PubMed] [Google Scholar]
- 34.Bogoyevitch MA, Clerk A, Sugden PH. Activation of the mitogen-activated protein kinase cascade by pertussis toxin-sensitive and -insensitive pathways in cultured ventricular cardiomyocytes. Biochem J 309: 437–443, 1995. doi: 10.1042/bj3090437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sadoshima J, Izumo S. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: potential involvement of an autocrine/paracrine mechanism. EMBO J 12: 1681–1692, 1993. doi: 10.1002/j.1460-2075.1993.tb05813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the α-myosin heavy chain gene promoter in transgenic mice. J Biol Chem 266: 24613–24620, 1991. [PubMed] [Google Scholar]
- 37.Rindt H, Gulick J, Knotts S, Neumann J, Robbins J. In vivo analysis of the murine beta-myosin heavy chain gene promoter. J Biol Chem 268: 5332–5338, 1993. [PubMed] [Google Scholar]
- 38.Lee KJ, Hickey R, Zhu H, Chien KR. Positive regulatory elements (HF-1a and HF-1b) and a novel negative regulatory element (HF-3) mediate ventricular muscle-specific expression of myosin light-chain 2-luciferase fusion genes in transgenic mice. Mol Cell Biol 14: 1220–1229, 1994. doi: 10.1128/MCB.14.2.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, Johnson TD, Bond RA, Lefkowitz RJ. Enhanced myocardial function in transgenic mice overexpressing the β2-adrenergic receptor. Science 264: 582–586, 1994. doi: 10.1126/science.8160017. [DOI] [PubMed] [Google Scholar]
- 40.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in β1-adrenergic receptor transgenic mice. Proc Natl Acad Sci USA 96: 7059–7064, 1999. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dorn GW 2nd, Tepe NM, Lorenz JN, Koch WJ, Liggett SB. Low- and high-level transgenic expression of β2-adrenergic receptors differentially affect cardiac hypertrophy and function in Gαq-overexpressing mice. Proc Natl Acad Sci USA 96: 6400–6405, 1999. doi: 10.1073/pnas.96.11.6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A, Dorn GW 2nd.. Early and delayed consequences of β2-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation 101: 1707–1714, 2000. doi: 10.1161/01.cir.101.14.1707. [DOI] [PubMed] [Google Scholar]
- 43.Milano CA, Dolber PC, Rockman HA, Bond RA, Venable ME, Allen LF, Lefkowitz RJ. Myocardial expression of a constitutively active alpha 1B-adrenergic receptor in transgenic mice induces cardiac hypertrophy. Proc Natl Acad Sci USA 91: 10109–10113, 1994. doi: 10.1073/pnas.91.21.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hein L, Stevens ME, Barsh GS, Pratt RE, Kobilka BK, Dzau VJ. Overexpression of angiotensin AT1 receptor transgene in the mouse myocardium produces a lethal phenotype associated with myocyte hyperplasia and heart block. Proc Natl Acad Sci USA 94: 6391–6396, 1997. doi: 10.1073/pnas.94.12.6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhai P, Yamamoto M, Galeotti J, Liu J, Masurekar M, Thaisz J, Irie K, Holle E, Yu X, Kupershmidt S, Roden DM, Wagner T, Yatani A, Vatner DE, Vatner SF, Sadoshima J. Cardiac-specific overexpression of AT1 receptor mutant lacking Gαq/Gαi coupling causes hypertrophy and bradycardia in transgenic mice. J Clin Invest 115: 3045–3056, 2005. doi: 10.1172/JCI25330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ainscough JF, Drinkhill MJ, Sedo A, Turner NA, Brooke DA, Balmforth AJ, Ball SG. Angiotensin II type-1 receptor activation in the adult heart causes blood pressure-independent hypertrophy and cardiac dysfunction. Cardiovasc Res 81: 592–600, 2009. doi: 10.1093/cvr/cvn230. [DOI] [PubMed] [Google Scholar]
- 47.Magalhaes AC, Dunn H, Ferguson SS. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br J Pharmacol 165: 1717–1736, 2012. doi: 10.1111/j.1476-5381.2011.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mizuno N, Itoh H. Functions and regulatory mechanisms of Gq-signaling pathways. Neurosignals 17: 42–54, 2009. doi: 10.1159/000186689. [DOI] [PubMed] [Google Scholar]
- 49.D’Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW 2nd.. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA 94: 8121–8126, 1997. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dorn GW 2nd, Brown JH. Gq signaling in cardiac adaptation and maladaptation. Trends Cardiovasc Med 9: 26–34, 1999. doi: 10.1016/S1050-1738(99)00004-3. [DOI] [PubMed] [Google Scholar]
- 51.Dorn GW 2nd, Tepe NM, Wu G, Yatani A, Liggett SB. Mechanisms of impaired β-adrenergic receptor signaling in G(αq)-mediated cardiac hypertrophy and ventricular dysfunction. Mol Pharmacol 57: 278–287, 2000. [PubMed] [Google Scholar]
- 52.Hahn HS, Marreez Y, Odley A, Sterbling A, Yussman MG, Hilty KC, Bodi I, Liggett SB, Schwartz A, Dorn GW 2nd.. Protein kinase Cα negatively regulates systolic and diastolic function in pathological hypertrophy. Circ Res 93: 1111–1119, 2003. doi: 10.1161/01.RES.0000105087.79373.17. [DOI] [PubMed] [Google Scholar]
- 53.Matkovich SJ, Zhang Y, Van Booven DJ, Dorn GW 2nd.. Deep mRNA sequencing for in vivo functional analysis of cardiac transcriptional regulators: application to Gαq. Circ Res 106: 1459–1467, 2010. doi: 10.1161/CIRCRESAHA.110.217513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science 280: 574–577, 1998. doi: 10.1126/science.280.5363.574. [DOI] [PubMed] [Google Scholar]
- 55.Wettschureck N, Rütten H, Zywietz A, Gehring D, Wilkie TM, Chen J, Chien KR, Offermanns S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Gαq/Gα11 in cardiomyocytes. Nat Med 7: 1236–1240, 2001. doi: 10.1038/nm1101-1236. [DOI] [PubMed] [Google Scholar]
- 56.Sakata Y, Hoit BD, Liggett SB, Walsh RA, Dorn GW 2nd.. Decompensation of pressure-overload hypertrophy in Gαq-overexpressing mice. Circulation 97: 1488–1495, 1998. doi: 10.1161/01.cir.97.15.1488. [DOI] [PubMed] [Google Scholar]
- 57.Liggett SB, Kelly RJ, Parekh RR, Matkovich SJ, Benner BJ, Hahn HS, Syed FM, Galvez AS, Case KL, McGuire N, Odley AM, Sparks L, Kardia SL, Dorn GW 2nd.. A functional polymorphism of the Gαq (GNAQ) gene is associated with accelerated mortality in African-American heart failure. Hum Mol Genet 16: 2740–2750, 2007. doi: 10.1093/hmg/ddm229. [DOI] [PubMed] [Google Scholar]
- 58.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW 2nd.. Enhanced Gαq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci USA 95: 10140–10145, 1998. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayakawa Y, Chandra M, Miao W, Shirani J, Brown JH, Dorn GW 2nd, Armstrong RC, Kitsis RN. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Gαq transgenic mice. Circulation 108: 3036–3041, 2003. doi: 10.1161/01.CIR.0000101920.72665.58. [DOI] [PubMed] [Google Scholar]
- 60.Aronow BJ, Toyokawa T, Canning A, Haghighi K, Delling U, Kranias E, Molkentin JD, Dorn GW 2nd.. Divergent transcriptional responses to independent genetic causes of cardiac hypertrophy. Physiol Genomics 6: 19–28, 2001. doi: 10.1152/physiolgenomics.2001.6.1.19. [DOI] [PubMed] [Google Scholar]
- 61.Colucci WS, Wynne J, Holman BL, Braunwald E. Long-term therapy of heart failure with prazosin: a randomized double blind trial. Am J Cardiol 45: 337–344, 1980. doi: 10.1016/0002-9149(80)90656-6. [DOI] [PubMed] [Google Scholar]
- 62.Pfeffer MA, Lamas GA, Vaughan DE, Parisi AF, Braunwald E. Effect of captopril on progressive ventricular dilatation after anterior myocardial infarction. N Engl J Med 319: 80–86, 1988. doi: 10.1056/NEJM198807143190204. [DOI] [PubMed] [Google Scholar]
- 63.Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN; SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 325: 293–302, 1991. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- 64.Cohn JN, Johnson G, Ziesche S, Cobb F, Francis G, Tristani F, Smith R, Dunkman WB, Loeb H, Wong M, Bhat G, Goldman S, Fletcher RD, Doherty J, Hughes CV, Carson P, Cintron G, Shabetai R, Haakenson C. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 325: 303–310, 1991. doi: 10.1056/NEJM199108013250502. [DOI] [PubMed] [Google Scholar]
- 65.Pfeffer MA, Braunwald E, Moyé LA, Basta L, Brown EJ Jr, Cuddy TE, Davis BR, Geltman EM, Goldman S, Flaker GC, Klein M, Lamas GA, Packer M, Rouleau J, Rouleau JL, Rutherford J, Wertheimer JH, Hawkins CM. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction—Results of the survival and ventricular enlargement trial. N Engl J Med 327: 669–677, 1992. doi: 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- 66.Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med 334: 1349–1355, 1996. doi: 10.1056/NEJM199605233342101. [DOI] [PubMed] [Google Scholar]
- 67. MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 353: 2001–2007, 1999. [PubMed] [Google Scholar]
- 68.Packer M, Fowler MB, Roecker EB, Coats AJ, Katus HA, Krum H, Mohacsi P, Rouleau JL, Tendera M, Staiger C, Holcslaw TL, Amann-Zalan I, DeMets DL; Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) Study Group. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 106: 2194–2199, 2002. doi: 10.1161/01.cir.0000035653.72855.bf. [DOI] [PubMed] [Google Scholar]
- 69.Anand I, McMurray J, Cohn JN, Konstam MA, Notter T, Quitzau K, Ruschitzka F, Lüscher TF; EARTH investigators. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet 364: 347–354, 2004. doi: 10.1016/S0140-6736(04)16723-8. [DOI] [PubMed] [Google Scholar]
- 70.Packer M, McMurray JJV, Krum H, Kiowski W, Massie BM, Caspi A, Pratt CM, Petrie MC, DeMets D, Kobrin I, Roux S, Swedberg K, Packer M, Caspi A, Kiowski W, Krum H, Pratt C, Swedberg K; ENABLE Investigators and Committees. Long-term effect of endothelin receptor antagonism with bosentan on the morbidity and mortality of patients with severe chronic heart failure: primary results of the ENABLE Trials. JACC Heart Fail 5: 317–326, 2017. doi: 10.1016/j.jchf.2017.02.021. [DOI] [PubMed] [Google Scholar]
- 71.Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res 113: 709–724, 2013. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tian R, Colucci WS, Arany Z, Bachschmid MM, Ballinger SW, Boudina S, Bruce JE, Busija DW, Dikalov S, Dorn GW 2nd, Galis ZS, Gottlieb RA, Kelly DP, Kitsis RN, Kohr MJ, Levy D, Lewandowski ED, McClung JM, Mochly-Rosen D, O’Brien KD, O’Rourke B, Park JY, Ping P, Sack MN, Sheu SS, Shi Y, Shiva S, Wallace DC, Weiss RG, Vernon HJ, Wong R, Longacre LS. Unlocking the secrets of mitochondria in the cardiovascular system: path to a cure in heart failure—a report from the 2018 National Heart, Lung, and Blood Institute Workshop. Circulation 140: 1205–1216, 2019. doi: 10.1161/CIRCULATIONAHA.119.040551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci 1188: 191–198, 2010. doi: 10.1111/j.1749-6632.2009.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dorn GW 2nd, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev 29: 1981–1991, 2015. doi: 10.1101/gad.269894.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Piquereau J, Ventura-Clapier R. Maturation of cardiac energy metabolism during perinatal development. Front Physiol 9: 959, 2018. doi: 10.3389/fphys.2018.00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol 15: 457–470, 2018. doi: 10.1038/s41569-018-0044-6. [DOI] [PubMed] [Google Scholar]
- 77.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730, 2001. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139, 2003. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 79.Halestrap AP, Doran E, Gillespie JP, O’Toole A. Mitochondria and cell death. Biochem Soc Trans 28: 170–177, 2000. doi: 10.1042/bst0280170. [DOI] [PubMed] [Google Scholar]
- 80.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 46: 821–831, 2009. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 81.Hengartner MO. The biochemistry of apoptosis. Nature 407: 770–776, 2000. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 82.Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol 192: 131–137, 2002. doi: 10.1002/jcp.10111. [DOI] [PubMed] [Google Scholar]
- 83.Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab 21: 206–214, 2015. doi: 10.1016/j.cmet.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chien KR, Knowlton KU, Zhu H, Chien S. Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. FASEB J 5: 3037–3046, 1991. doi: 10.1096/fasebj.5.15.1835945. [DOI] [PubMed] [Google Scholar]
- 85.Hu Y, Matkovich SJ, Hecker PA, Zhang Y, Edwards JR, Dorn GW 2nd.. Epitranscriptional orchestration of genetic reprogramming is an emergent property of stress-regulated cardiac microRNAs. Proc Natl Acad Sci USA 109: 19864–19869, 2012. doi: 10.1073/pnas.1214996109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microRNA expression in human heart disease. Physiol Genomics 31: 367–373, 2007. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 87.Latronico MV, Catalucci D, Condorelli G. MicroRNA and cardiac pathologies. Physiol Genomics 34: 239–242, 2008. doi: 10.1152/physiolgenomics.90254.2008. [DOI] [PubMed] [Google Scholar]
- 88.Matkovich SJ, Hu Y, Eschenbacher WH, Dorn LE, Dorn GW 2nd.. Direct and indirect involvement of microRNA-499 in clinical and experimental cardiomyopathy. Circ Res 111: 521–531, 2012. doi: 10.1161/CIRCRESAHA.112.265736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Matkovich SJ, Hu Y, Dorn GW 2nd.. Regulation of cardiac microRNAs by cardiac microRNAs. Circ Res 113: 62–71, 2013. doi: 10.1161/CIRCRESAHA.113.300975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matkovich SJ, Edwards JR, Grossenheider TC, de Guzman Strong C, Dorn GW 2nd.. Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc Natl Acad Sci USA 111: 12264–12269, 2014. doi: 10.1073/pnas.1410622111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, Aronow BJ, Lorenz JN, Dorn GW 2nd.. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med 8: 725–730, 2002. doi: 10.1038/nm719. [DOI] [PubMed] [Google Scholar]
- 92.Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, Shi L, Dubik D, Greenberg A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem 274: 7–10, 1999. doi: 10.1074/jbc.274.1.7. [DOI] [PubMed] [Google Scholar]
- 93.Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirschenbaum LA, Dorn GW 2nd.. Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J Biol Chem 281: 1442–1448, 2006. doi: 10.1074/jbc.M509056200. [DOI] [PubMed] [Google Scholar]
- 94.Syed F, Odley A, Hahn HS, Brunskill EW, Lynch RA, Marreez Y, Sanbe A, Robbins J, Dorn GW 2nd.. Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circ Res 95: 1200–1206, 2004. doi: 10.1161/01.RES.0000150366.08972.7f. [DOI] [PubMed] [Google Scholar]
- 95.Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW 2nd.. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation 117: 396–404, 2008. doi: 10.1161/CIRCULATIONAHA.107.727073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest 117: 2825–2833, 2007. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pan G, O’Rourke K, Dixit VM. Caspase-9, Bcl-XL, and Apaf-1 form a ternary complex. J Biol Chem 273: 5841–5845, 1998. doi: 10.1074/jbc.273.10.5841. [DOI] [PubMed] [Google Scholar]
- 98.Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274: 11549–11556, 1999. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 99.Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell 9: 423–432, 2002. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- 100.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495–516, 2007. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene 27, Suppl 1: S114–S127, 2008. doi: 10.1038/onc.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fisher SA, Langille BL, Srivastava D. Apoptosis during cardiovascular development. Circ Res 87: 856–864, 2000. doi: 10.1161/01.res.87.10.856. [DOI] [PubMed] [Google Scholar]
- 103.Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, Molkentin JD, Kranias EG, Dorn GW 2nd.. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest 119: 203–212, 2009. doi: 10.1172/JCI36445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW 2nd.. Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci USA 107: 9035–9042, 2010. doi: 10.1073/pnas.0914013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hata JA, Williams ML, Koch WJ. Genetic manipulation of myocardial beta-adrenergic receptor activation and desensitization. J Mol Cell Cardiol 37: 11–21, 2004. doi: 10.1016/j.yjmcc.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 106.Pleger ST, Boucher M, Most P, Koch WJ. Targeting myocardial beta-adrenergic receptor signaling and calcium cycling for heart failure gene therapy. J Card Fail 13: 401–414, 2007. doi: 10.1016/j.cardfail.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 107.Chacon E, Ohata H, Harper IS, Trollinger DR, Herman B, Lemasters JJ. Mitochondrial free calcium transients during excitation-contraction coupling in rabbit cardiac myocytes. FEBS Lett 382: 31–36, 1996. doi: 10.1016/0014-5793(96)00138-x. [DOI] [PubMed] [Google Scholar]
- 108.Dorn GW 2nd, Scorrano L. Two close, too close: sarcoplasmic reticulum-mitochondrial crosstalk and cardiomyocyte fate. Circ Res 107: 689–699, 2010. doi: 10.1161/CIRCRESAHA.110.225714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dorn GW 2nd, Maack C. SR and mitochondria: calcium cross-talk between kissing cousins. J Mol Cell Cardiol 55: 42–49, 2013. doi: 10.1016/j.yjmcc.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 110.Cortassa S, Aon MA, Marbán E, Winslow RL, O’Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J 84: 2734–2755, 2003. doi: 10.1016/S0006-3495(03)75079-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu T, O’Rourke B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J Bioenerg Biomembr 41: 127–132, 2009. doi: 10.1007/s10863-009-9216-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 1787: 1395–1401, 2009. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun 304: 463–470, 2003. doi: 10.1016/s0006-291x(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 114.Lewis MR, Lewis WH. Mitochondria in tissue culture. Science 39: 330–333, 1914. doi: 10.1126/science.39.1000.330. [DOI] [PubMed] [Google Scholar]
- 115.Detmer SA, Chan DC. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol 176: 405–414, 2007. doi: 10.1083/jcb.200611080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dang X, Walton EK, Zablocka B, Baloh RH, Shy ME, Dorn GW 2nd.. Mitochondrial phenotypes in genetically diverse neurodegenerative diseases and their response to mitofusin activation. Cells 11: 1053, 2022. doi: 10.3390/cells11061053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist 14: 12–18, 2008. doi: 10.1177/1073858407307354. [DOI] [PubMed] [Google Scholar]
- 118.Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13: 77–93, 2012. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mandal A, Drerup CM. Axonal transport and mitochondrial function in neurons. Front Cell Neurosci 13: 373, 2019. doi: 10.3389/fncel.2019.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dorn GW 2nd. Mitochondrial dynamism and heart disease: changing shape and shaping change. EMBO Mol Med 7: 865–877, 2015. doi: 10.15252/emmm.201404575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dorn GW 2nd. Cardiac-specific research platforms engender novel insights into mitochondrial dynamism. Curr Opin Physiol 3: 110–115, 2018. doi: 10.1016/j.cophys.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Knowlton AA, Liu TT. Mitochondrial dynamics and heart failure. Compr Physiol 6: 507–526, 2015. doi: 10.1002/cphy.c150022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ong SB, Kalkhoran SB, Hernández-Reséndiz S, Samangouei P, Ong SG, Hausenloy DJ. Mitochondrial-shaping proteins in cardiac health and disease - the long and the short of it! Cardiovasc Drugs Ther 31: 87–107, 2017. doi: 10.1007/s10557-016-6710-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nan J, Zhu W, Rahman MS, Liu M, Li D, Su S, Zhang N, Hu X, Yu H, Gupta MP, Wang J. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta Mol Cell Res 1864: 1260–1273, 2017. doi: 10.1016/j.bbamcr.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 125.Liu X, Weaver D, Shirihai O, Hajnóczky G. Mitochondrial 'kiss-and-run': interplay between mitochondrial motility and fusion-fission dynamics. EMBO J 28: 3074–3089, 2009. doi: 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Huang X, Sun L, Ji S, Zhao T, Zhang W, Xu J, Zhang J, Wang Y, Wang X, Franzini-Armstrong C, Zheng M, Cheng H. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc Natl Acad Sci USA 110: 2846–2851, 2013. doi: 10.1073/pnas.1300741110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cao Y, Xu C, Ye J, He Q, Zhang X, Jia S, Qiao X, Zhang C, Liu R, Weng L, Liu Y, Liu L, Zheng M. Miro2 regulates inter-mitochondrial communication in the heart and protects against tac-induced cardiac dysfunction. Circ Res 125: 728–743, 2019. doi: 10.1161/CIRCRESAHA.119.315432. [DOI] [PubMed] [Google Scholar]
- 128.Picard M, McManus MJ, Csordás G, Várnai P, Dorn GW 2nd, Williams D, Hajnóczky G, Wallace DC. Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat Commun 6: 6259, 2015. doi: 10.1038/ncomms7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Glancy B, Hartnell LM, Combs CA, Femnou A, Sun J, Murphy E, Subramaniam S, Balaban RS. Power grid protection of the muscle mitochondrial reticulum. Cell Rep 19: 487–496, 2017. [Erratum in Cell Rep 23: 2832, 2018]. doi: 10.1016/j.celrep.2017.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chen Y, Liu Y, Dorn GW 2nd.. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 109: 1327–1331, 2011. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res 84: 91–99, 2009. doi: 10.1093/cvr/cvp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tong M, Zablocki D, Sadoshima J. The role of Drp1 in mitophagy and cell death in the heart. J Mol Cell Cardiol 142: 138–145, 2020. doi: 10.1016/j.yjmcc.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hernandez-Resendiz S, Prunier F, Girao H, Dorn G, Hausenloy DJ; EU-CARDIOPROTECTION COST Action (CA16225). Targeting mitochondrial fusion and fission proteins for cardioprotection. J Cell Mol Med 24: 6571–6585, 2020. doi: 10.1111/jcmm.15384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen L, Liu B, Qin Y, Li A, Gao M, Liu H, Gong G. Mitochondrial fusion protein Mfn2 and its role in heart failure. Front Mol Biosci 8: 681237, 2021. doi: 10.3389/fmolb.2021.681237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res 111: 1012–1026, 2012. doi: 10.1161/CIRCRESAHA.112.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kasahara A, Cipolat S, Chen Y, Dorn GW 2nd, Scorrano L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 342: 734–737, 2013. doi: 10.1126/science.1241359. [DOI] [PubMed] [Google Scholar]
- 137.Wai T, García-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Rupérez FJ, Barbas C, Ibañez B, Langer T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350: aad0116, 2015. doi: 10.1126/science.aad0116. [DOI] [PubMed] [Google Scholar]
- 138.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd.. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21: 273–285, 2015. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116: 264–278, 2015. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 140.Song M, Franco A, Fleischer JA, Zhang L, Dorn GW 2nd.. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab 26: 872–883.e5, 2017. doi: 10.1016/j.cmet.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang W, Fernandez-Sanz C, Sheu SS. Regulation of mitochondrial bioenergetics by the non-canonical roles of mitochondrial dynamics proteins in the heart. Biochim Biophys Acta Mol Basis Dis 1864: 1991–2001, 2018. doi: 10.1016/j.bbadis.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662, 2005. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 143.Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, Nerbonne JM, Dorn GW 2nd, Maack C. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res 111: 863–875, 2012. doi: 10.1161/CIRCRESAHA.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.De la Fuente S, Sheu SS. SR-mitochondria communication in adult cardiomyocytes: a close relationship where the Ca2+ has a lot to say. Arch Biochem Biophys 663: 259–268, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Harrington JL, Murphy E. The mitochondrial calcium uniporter: mice can live and die without it. J Mol Cell Cardiol 78: 46–53, 2015. doi: 10.1016/j.yjmcc.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Murphy E, Steenbergen C. Regulation of mitochondrial Ca2+ uptake. Annu Rev Physiol 83: 107–126, 2021. doi: 10.1146/annurev-physiol-031920-092419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86: 369–408, 2006. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 148.Kohlhaas M, Maack C. Calcium release microdomains and mitochondria. Cardiovasc Res 98: 259–268, 2013. doi: 10.1093/cvr/cvt032. [DOI] [PubMed] [Google Scholar]
- 149.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46: 265–287, 2012. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 150.Dorn GW 2nd. Evolving concepts of mitochondrial dynamics. Annu Rev Physiol 81: 1–17, 2019. doi: 10.1146/annurev-physiol-020518-114358. [DOI] [PubMed] [Google Scholar]
- 151.Dorn GW 2nd. Mitofusins as mitochondrial anchors and tethers. J Mol Cell Cardiol 142: 146–153, 2020. doi: 10.1016/j.yjmcc.2020.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.de Brito OM, Scorrano L. Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid Redox Signal 10: 621–633, 2008. doi: 10.1089/ars.2007.1934. [DOI] [PubMed] [Google Scholar]
- 153.Dorn GW 2nd, Song M, Walsh K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J Mol Cell Cardiol 78: 123–128, 2015. doi: 10.1016/j.yjmcc.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernández-Alvarez MI, Zorzano A, De Stefani D, Dorn GW 2nd, Scorrano L. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci USA 113: 11249–11254, 2016. doi: 10.1073/pnas.1606786113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Han S, Zhao F, Hsia J, Ma X, Liu Y, Torres S, Fujioka H, Zhu X. The role of Mfn2 in the structure and function of endoplasmic reticulum-mitochondrial tethering in vivo. J Cell Sci 134: jcs253443, 2021. doi: 10.1242/jcs.253443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gustafsson AB, Dorn GW 2nd.. Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol Rev 99: 853–892, 2019. doi: 10.1152/physrev.00005.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065, 2012. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen Y, Dorn GW 2nd.. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340: 471–475, 2013. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW 2nd.. Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ Res 114: 257–265, 2014. doi: 10.1161/CIRCRESAHA.114.302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li J, Dang X, Franco A, Dorn GW 2nd. Reciprocal regulation of mitofusin 2-mediated mitophagy and mitochondrial fusion by different PINK1 phosphorylation events. Front Cell Dev Biol 10: 868465, 2022. doi: 10.3389/fcell.2022.868465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW 2nd.. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350: aad2459, 2015. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 288: 915–926, 2013. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Song M, Gong G, Burelle Y, Gustafsson AB, Kitsis RN, Matkovich SJ, Dorn GW 2nd.. Interdependence of Parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ Res 117: 346–351, 2015. doi: 10.1161/CIRCRESAHA.117.306859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Woodall BP, Orogo AM, Najor RH, Cortez MQ, Moreno ER, Wang H, Divakaruni AS, Murphy AN, Gustafsson ÅB. Parkin does not prevent accelerated cardiac aging in mitochondrial DNA mutator mice. JCI Insight 4: e127713, 2019. doi: 10.1172/jci.insight.127713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26: 207–210, 2000. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 166.Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26: 211–215, 2000. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 167.Ferré M, Bonneau D, Milea D, Chevrollier A, Verny C, Dollfus H, Ayuso C, Defoort S, Vignal C, Zanlonghi X, Charlin JF, Kaplan J, Odent S, Hamel CP, Procaccio V, Reynier P, Amati-Bonneau P. Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel OPA1 mutations. Hum Mutat 30: E692–E705, 2009. doi: 10.1002/humu.21025. [DOI] [PubMed] [Google Scholar]
- 168.Weisschuh N, Schimpf-Linzenbold S, Mazzola P, Kieninger S, Xiao T, Kellner U, Neuhann T, Kelbsch C, Tonagel F, Wilhelm H, Kohl S, Wissinger B. Mutation spectrum of the OPA1 gene in a large cohort of patients with suspected dominant optic atrophy: identification and classification of 48 novel variants. PLoS One 16: e0253987, 2021. doi: 10.1371/journal.pone.0253987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Spiegel R, Saada A, Flannery PJ, Burté F, Soiferman D, Khayat M, Eisner V, Vladovski E, Taylor RW, Bindoff LA, Shaag A, Mandel H, Schuler-Furman O, Shalev SA, Elpeleg O, Yu-Wai-Man P. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J Med Genet 53: 127–131, 2016. doi: 10.1136/jmedgenet-2015-103361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 95: 360–370, 2014. [Erratum in Am J Hum Genet 100: 179, 2017]. doi: 10.1016/j.ajhg.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schröder JM, Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36: 449–451, 2004. [Erratum in Nat Genet 36: 660, 2004]. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 172.Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, Day J, Feely S, Finkel RS, Grider T, Kirk CA, Herrmann DN, Laura M, Li J, Lloyd T, Sumner CJ, Muntoni F, Piscosquito G, Ramchandren S, Shy R, Siskind CE, Yum SW, Moroni I, Pagliano E, Zuchner S, Scherer SS, Shy ME; Inherited Neuropathies Consortium. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry 86: 873–878, 2015. doi: 10.1136/jnnp-2014-308826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Züchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, Hamilton SR, Van Stavern G, Krajewski KM, Stajich J, Tournev I, Verhoeven K, Langerhorst CT, de Visser M, Baas F, Bird T, Timmerman V, Shy M, Vance JM. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol 59: 276–281, 2006. doi: 10.1002/ana.20797. [DOI] [PubMed] [Google Scholar]
- 174.Rocha N, Bulger DA, Frontini A, Titheradge H, Gribsholt SB, Knox R, Page M, Harris J, Payne F, Adams C, Sleigh A, Crawford J, Gjesing AP, Bork-Jensen J, Pedersen O, Barroso I, Hansen T, Cox H, Reilly M, Rossor A, Brown RJ, Taylor SI, McHale D, Armstrong M, Oral EA, Saudek V, O’Rahilly S, Maher ER, Richelsen B, Savage DB, Semple RK. Human biallelic MFN2 mutations induce mitochondrial dysfunction, upper body adipose hyperplasia, and suppression of leptin expression. eLife 6: 28414270, 2017. doi: 10.7554/eLife.23813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cartoni R, Martinou JC. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol 218: 268–273, 2009. doi: 10.1016/j.expneurol.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 176.Bombelli F, Stojkovic T, Dubourg O, Echaniz-Laguna A, Tardieu S, Larcher K, Amati-Bonneau P, Latour P, Vignal O, Cazeneuve C, Brice A, Leguern E. Charcot-Marie-Tooth disease type 2A: from typical to rare phenotypic and genotypic features. JAMA Neurol 71: 1036–1042, 2014. doi: 10.1001/jamaneurol.2014.629. [DOI] [PubMed] [Google Scholar]
- 177.Stuppia G, Rizzo F, Riboldi G, Del Bo R, Nizzardo M, Simone C, Comi GP, Bresolin N, Corti S. MFN2-related neuropathies: clinical features, molecular pathogenesis and therapeutic perspectives. J Neurol Sci 356: 7–18, 2015. doi: 10.1016/j.jns.2015.05.033. [DOI] [PubMed] [Google Scholar]
- 178.Dorn GW 2nd. Mitofusin 2 dysfunction and disease in mice and men. Front Physiol 11: 782, 2020. doi: 10.3389/fphys.2020.00782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci 1201: 21–25, 2010. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- 180.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9: 505–518, 2008. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol 15: 235–259, 2020. doi: 10.1146/annurev-pathmechdis-012419-032711. [DOI] [PubMed] [Google Scholar]
- 182.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200, 2003. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 30: 4232–4240, 2010. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pham AH, Meng S, Chu QN, Chan DC. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum Mol Genet 21: 4817–4826, 2012. doi: 10.1093/hmg/dds311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sebastián D, Hernández-Alvarez MI, Segalés J, Sorianello E, Segalés JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Orešič M, Pich S, Burcelin R, Palacín M, Zorzano A. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA 109: 5523–5528, 2012. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet 21: 4827–4835, 2012. doi: 10.1093/hmg/dds352. [DOI] [PubMed] [Google Scholar]
- 187.Han S, Nandy P, Austria Q, Siedlak SL, Torres S, Fujioka H, Wang W, Zhu X. Mfn2 ablation in the adult mouse hippocampus and cortex causes neuronal death. Cells 9: 116–118, 2020. doi: 10.3390/cells9010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Filadi R, Pendin D, Pizzo P. Mitofusin 2: from functions to disease. Cell Death Dis 9: 330, 2018. doi: 10.1038/s41419-017-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhou Y, Carmona S, Muhammad A, Bell S, Landeros J, Vazquez M, Ho R, Franco A, Lu B, Dorn GW 2nd, Wang S, Lutz CM, Baloh RH. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J Clin Invest 129: 1756–1771, 2019. [Erratum in J Clin Invest 131: e147307, 2021]. doi: 10.1172/JCI124194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Loiseau D, Chevrollier A, Verny C, Guillet V, Gueguen N, Pou de Crescenzo MA, Ferré M, Malinge MC, Guichet A, Nicolas G, Amati-Bonneau P, Malthièry Y, Bonneau D, Reynier P. Mitochondrial coupling defect in Charcot-Marie-Tooth type 2A disease. Ann Neurol 61: 315–323, 2007. doi: 10.1002/ana.21086. [DOI] [PubMed] [Google Scholar]
- 191.Amiott EA, Lott P, Soto J, Kang PB, McCaffery JM, DiMauro S, Abel ED, Flanigan KM, Lawson VH, Shaw JM. Mitochondrial fusion and function in Charcot-Marie-Tooth type 2A patient fibroblasts with mitofusin 2 mutations. Exp Neurol 211: 115–127, 2008. doi: 10.1016/j.expneurol.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47: 365–378, 2005. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 193.Lin MY, Sheng ZH. Regulation of mitochondrial transport in neurons. Exp Cell Res 334: 35–44, 2015. doi: 10.1016/j.yexcr.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther 342: 619–630, 2012. doi: 10.1124/jpet.112.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol 131: 1315–1326, 1995. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schwartz T. Functional insights from studies on the structure of the nuclear pore and coat protein complexes. Cold Spring Harb Perspect Biol 5: a013375, 2013. doi: 10.1101/cshperspect.a013375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.López-Doménech G, Covill-Cooke C, Ivankovic D, Halff EF, Sheehan DF, Norkett R, Birsa N, Kittler JT. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J 37: 321–336, 2018. doi: 10.15252/embj.201696380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Henrichs V, Grycova L, Barinka C, Nahacka Z, Neuzil J, Diez S, Rohlena J, Braun M, Lansky Z. Mitochondria-adaptor TRAK1 promotes kinesin-1 driven transport in crowded environments. Nat Commun 11: 3123, 2020. doi: 10.1038/s41467-020-16972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Fenton AR, Jongens TA, Holzbaur ELF. Mitochondrial adaptor TRAK2 activates and functionally links opposing kinesin and dynein motors. Nat Commun 12: 4578, 2021. doi: 10.1038/s41467-021-24862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci USA 105: 20728–20733, 2008. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lovas JR, Wang X. The meaning of mitochondrial movement to a neuron's life. Biochim Biophys Acta 1833: 184–194, 2013. doi: 10.1016/j.bbamcr.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dorn GW. Mitofusin activation enhances mitochondrial motility and promotes neuroregeneration in CMT2A. Neural Regen Res 16: 2201–2203, 2021. doi: 10.4103/1673-5374.310684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci 27: 422–430, 2007. doi: 10.1523/JNEUROSCI.4798-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Misko AL, Sasaki Y, Tuck E, Milbrandt J, Baloh RH. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci 32: 4145–4155, 2012. doi: 10.1523/JNEUROSCI.6338-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tang BL. MIRO GTPases in mitochondrial transport, homeostasis and pathology. Cells 5: 1, 2015. doi: 10.3390/cells5010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Franco A, Dang X, Walton EK, Ho JN, Zablocka B, Ly C, Miller TM, Baloh RH, Shy ME, Yoo AS, Dorn GW 2nd.. Burst mitofusin activation reverses neuromuscular dysfunction in murine CMT2A. eLife 9: e61119, 2020. doi: 10.7554/eLife.61119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, Chen Y, Mennerick S, Hodgson L, Mochly-Rosen D, Dorn GW 2nd.. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540: 74–79, 2016. doi: 10.1038/nature20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rocha AG, Franco A, Krezel AM, Rumsey JM, Alberti JM, Knight WC, Biris N, Zacharioudakis E, Janetka JW, Baloh RH, Kitsis RN, Mochly-Rosen D, Townsend RR, Gavathiotis E, Dorn GW 2nd.. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 360: 336–341, 2018. doi: 10.1126/science.aao1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dang X, Zhang L, Franco A, Li J, Rocha AG, Devanathan S, Dolle RE, Bernstein PR, Dorn GW 2nd.. Discovery of 6-phenylhexanamide derivatives as potent stereoselective mitofusin activators for the treatment of mitochondrial diseases. J Med Chem 63: 7033–7051, 2020. [Erratum in J Med Chem 63: 10086, 2020]. doi: 10.1021/acs.jmedchem.0c00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhang LD, Franco A, Zhao H, Dorn GW 2nd.. Piperine derivatives enhance fusion and axonal transport of mitochondria by activating mitofusins. Chemistry (Easton) 4: 655–668, 2022. doi: 10.3390/chemistry4030047. [DOI] [Google Scholar]