

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) dysregulates antiviral signaling, immune response, and cell metabolism in human body. Viral genome and proteins hijack host metabolic network to support viral biogenesis and propagation. However, the regulatory mechanism of SARS‐CoV‐2‐induced metabolic dysfunction has not been elucidated until recently. Multiomic studies of coronavirus disease 2019 (COVID‐19) revealed an intensive interaction between host metabolic regulators and viral proteins. SARS‐CoV‐2 deregulated cellular metabolism in blood, intestine, liver, pancreas, fat, and immune cells. Host metabolism supported almost every stage of viral lifecycle. Strikingly, viral proteins were found to interact with metabolic enzymes in different cellular compartments. Biochemical and genetic assays also identified key regulatory nodes and metabolic dependencies of viral replication. Of note, cholesterol metabolism, lipid metabolism, and glucose metabolism are broadly involved in viral lifecycle. Here, we summarized the current understanding of the hallmarks of COVID‐19 metabolism. SARS‐CoV‐2 infection remodels host cell metabolism, which in turn modulates viral biogenesis and replication. Remodeling of host metabolism creates metabolic vulnerability of SARS‐CoV‐2 replication, which could be explored to uncover new therapeutic targets. The efficacy of metabolic inhibitors against COVID‐19 is under investigation in several clinical trials. Ultimately, the knowledge of SARS‐CoV‐2‐induced metabolic reprogramming would accelerate drug repurposing or screening to combat the COVID‐19 pandemic.
Keywords: antiviral response, metabolic reprogramming, mitochondria metabolism, posttranslational modification, SARS‐CoV‐2
SARS‐CoV‐2 deregulates cell metabolism in multiple organs. The interaction between SARS‐CoV‐2 and human hosts remodels metabolic network to regulate viral infection and replication. Metabolism‐based therapy holds promise in the combat against SARS‐CoV‐2.
1. INTRODUCTION
The coronavirus disease 2019 (COVID‐19) pandemic has become the biggest global health crisis since the beginning of the 21st century. 1 To date, this villainous virus has swiped over 110 countries and caused millions of deaths. 2 , 3 Public health interventions such as immunization and social distancing remain the most effective countermeasure to severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) transmission. 4 Progress in the clinical treatment of COVID‐19 patients remains dismal. Compounds that suppressed viral infection in vitro turned out to be mostly ineffective in clinical treatment of COVID‐19. 5 , 6 Monoclonal antibodies were selectively used to treat high‐risk patients due to a high cost. 7 , 8 Of note, nucleotide analogs such as remdesivir displayed broad activity against the circulating variants. 9 Nirmatrelvir, a compound targeting viral protease to restrict viral replication, was clinically used to treat mild‐to‐moderate COVID‐19 patients. 10 Nirmatrelvir remains active against Omicron variant. 11 Additionally, engineered natural killer cells are emerging as immunotherapeutic tools against COVID‐19. 12 Elucidation of COVID‐19 pathogenesis is essential for discovering new therapeutic opportunities to combat SARS‐CoV‐2.
SARS‐CoV‐2 is a single‐stranded RNA virus that targets multiple tissues in human body. Lung epithelium infection by SARS‐CoV‐2 causes life‐threatening pneumonia. SARS‐CoV‐2 virions are encapsulated in lipid bilayer. The viral genome encodes at least 14 open reading frames (ORFs), which give rise to 29 different proteins. 13 , 14 The products encoded by viral genome allow SARS‐CoV‐2 to dysregulate antiviral signaling, immune response, and cell metabolism in human body.
Of note, the SARS‐CoV‐2 genome does not encode metabolic enzymes required for viral genomic replication, protein synthesis, and lipogenesis. To replicate itself, SARS‐CoV‐2 hijacks the metabolic network and biogenesis programs in host cells. For example, SARS‐CoV‐2 boosted lipid biosynthesis to support the generation of lipid bilayer‐enveloped virions in a fashion similar to other coronaviruses. 15 , 16 Accordingly, SARS‐CoV‐2 infection causes systemic metabolic changes in the human body. In recent years, multiomics profiling of clinical samples from COVID‐19 patients has revealed key metabolic features of SARS‐CoV‐2 biology. The metabolic network of host cells interacted with the viral genome and its products to regulate viral replication and propagation. Viral hijacking of host cells also generated metabolic vulnerabilities of SARS‐CoV‐2 replication, which could be explored to develop new therapies for SARS‐CoV‐2 infection. Here, we reviewed recent advances in the metabolic interaction between SARS‐CoV‐2 and human body.
2. METABOLIC FEATURES OF COVID‐19 PATIENTS
SARS‐CoV‐2 infection causes systemic changes at transcriptomic, proteomic, and metabolomic levels. Multiomic profiling becomes a powerful tool in elucidating the metabolic impact of SARS‐CoV‐2 at different stages of infection. 17 To date, SARS‐CoV‐2 has been shown to dysregulate host metabolism in multiple tissues and organs (Figure 1).
FIGURE 1.
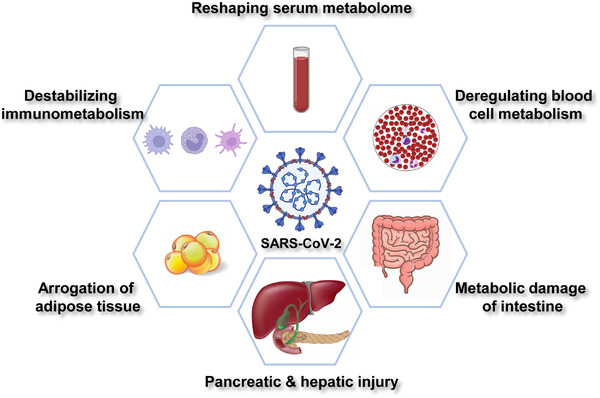
Metabolic features of COVID‐19. SARS‐CoV‐2 infection mediates systemic alterations in host metabolism. COVID‐19 is featured by dysregulated metabolism in serum, blood cells, intestine, pancreas, liver, adipose tissue, and immune system.
2.1. Reshaping serum metabolome
Serum samples are easily accessible from COVID‐19 patients. Alterations of serum metabolites provide mechanistic insights into viral infection‐induced metabolic reprogramming. Recent metabolomic profiling has revealed dramatic changes in glucose, amino acids, and lipid metabolism in COVID‐19 serum (Figure 1).
Dysregulation of glucose metabolism is a key feature as determined by plasma metabolome. COVID‐19 patients generally exhibited an elevation of serum glucose levels, which was in line with an upregulation of glycolytic intermediates. 18 Glucose metabolism supports the tricarboxylic acid cycle in the mitochondria and produces malate, an indicator of mitochondria activity. Interestingly, plasma malate was decreased after SARS‐CoV‐2 infection. 18 This observation suggests that mitochondria respiration is potentially suppressed due to deficient oxygenation in the pneumonia state. 19 However, lactate, the glycolytic product produced under hypoxic conditions, did not show significant changes in SARS‐CoV‐2‐positive patients. Reduced oxygenation may have a minimal effect on glycolysis at least in serum metabolome. 18 Notably, metabolites in the pentose phosphate pathway (PPP) showed a modest increase in COVID‐19 serum. 18 PPP intermediate xylulose 5‐phosphate (X5P) was decreased in a different cohort of COVD19 patients, which could be explained by cohort‐specific variations. 20
Alterations in amino acid metabolism are closely linked to the inflammatory and immune response in human body. 21 , 22 Kynurenine is a tryptophan‐derived amino acid and a key metabolite regulating inflammation and immunity. Circulating kynurenine was strongly increased in COVID‐19 patients and correlated with interleukin‐6 (IL6) levels. 18 Circulating amino acids also serve as physiological indicators of organ functions. For instance, amino acids generated from the urea cycle, such as arginine, ornithine, and citrulline, were dysregulated in patient serum. 18 , 23 The nitrogen acceptor carbamoyl phosphate was also decreased. 20 Both observations point to renal dysfunction in COVID‐19 patients. 18 Gluconeogenic amino acids including alanine, glycine, and serine were increased, suggesting a systemic increase in energy requirement. 18 Further, the decrease in sulfur‐containing amino acids such as cysteine and taurine potentially indicated SARS‐CoV‐2‐mediated oxidative stress. 18 Noteworthy, specific amino acids or their derivatives correlated with COVID‐19 development. 24 γ‐aminobutyric acid (GABA) is a neurotransmitter and immune‐modulatory metabolite. 25 Downregulation of GABA correlated with disease severity and could be used for stratification of COVID‐19 patients. 26
Serum lipid levels are intimately linked to COVID‐19 development. Both acylcarnitines and free fatty acids were increased in COVID‐19 patient serum. 18 A remarkable disturbance of sphingolipid and arachidonic acid metabolism pathways was also observed. 26 Notably, a percentage of COVID‐19 patients are asymptomatic and become hidden drivers of the pandemic. Serum metabolome profiling in asymptomatic patients revealed dysregulation of arachidonic acid metabolism, fatty acid β‐oxidation, and bile acid synthesis. 27 As an immune‐modulatory metabolite, arachidonic acid may be utilized as a marker for the antiviral response during asymptomatic infection.
2.2. SARS‐CoV‐2 infection dysregulates blood cell metabolism
Circulating blood cells are implicated in SARS‐CoV‐2‐induced metabolic dysfunction (Figure 1). C‐reactive protein (CRP) is widely used as a serological inflammation marker. 28 CRP levels were positively linked to glucose transporter 3 and monocarboxylate transporter SLC16A3 (solute carrier family 16 member 3) levels in blood cells, 29 which indicated that glucose uptake was enhanced to maintain energy supply in COVID‐19 patients. Surprisingly, patients with high CRP showed decreased mitochondrial respiration and TCA cycle activity. 29 These findings collectively suggest a reliance on glycolysis but not mitochondrial respiration for energy production in circulating blood cells.
Although red blood cells (RBCs) cannot support viral replication due to the lack of nucleus, angiotensin‐converting enzyme 2 (ACE2)‐interacting proteins were expressed on RBC membrane that potentially allows viral entry. 30 Proteomic analysis of RBCs from COVID‐19 patients revealed an elevation of glycolytic enzyme phosphofructokinase, which was in accordance with an increase in glycolytic activity. Besides, short‐ and medium‐chain saturated fatty acids, acyl‐carnitines, and sphingolipids were decreased, indicating remodeling of lipid metabolism and cell membrane composition in RBCs. Importantly, antioxidant enzymes such as peroxiredoxin 1 (PRDX1), superoxide dismutase 1 (SOD1), and glucose‐6‐phosphate dehydrogenase (G6PD) were decreased. This is a possible outcome of oxidative damage‐induced protein degradation. 31 , 32
2.3. Metabolic disorder of intestinal function and microecology
Intestinal epithelium expresses viral receptor ACE2 and is therefore vulnerable to SARS‐CoV‐2 infection. 33 Both intestinal function and microbiota metabolism were altered in COVID‐19 patients (Figure 1). Intestinal epithelium downregulated ACE2 expression after SARS‐CoV‐2 infection, leading to upregulation of sodium‐glucose cotransporter (SGLT). 34 Because SGLT is responsible for glucose absorption from the blood, intestinal SARS‐CoV‐2 infection contributes to the onset of metabolic disorders in COVID‐19 patients. Additionally, SARS‐CoV‐2 evaded amino acid transportation by suppressing solute carrier family 6 member 19 (SLC6A19), the major luminal Na+‐dependent neutral amino acids transporter in the intestine. 35 Spike protein recognized the ACE2–SLC6A19 complex and promoted its internalization. Consequently, neutral amino acid transportation was impaired. 36 Notably, tryptophan and glutamine are major substrates of SLC6A19. Both amino acids play important role in cytokine release, tight junction formation and microbial defense. 37 Disturbance of SLC6A19 activity by SARS‐CoV‐2 infection contributed to leaky gut and microbial dysbiosis, further exacerbating cytokine storm in COVID‐19 patients. 38
SARS‐CoV‐2 targets intestinal microecology to induce inflammation. 39 The diversity of fecal microbiota was significantly decreased in COVID‐19 patients. 40 Genomic and metabolomic profiling of fecal samples revealed broad metabolic alterations between COVID‐19 patients and healthy controls. 41 Strikingly, short‐chain fatty acid and isoleucine biosynthesis were suppressed ever after COVID‐19 recovery. 41 SARS‐CoV‐2 infection possibly resulted in sustained metabolic damage to gut microbiome and intestine function. Consistently, fecal microbiomes of COVID‐19 patients were enriched with opportunistic pathogens. 42 , 43 Specific bacterial species, such as Coprobacillus, Clostridium ramosum, and Clostridium hathewayi, were found in positive correlation with the severity of SARS‐CoV‐2 infection. Meanwhile, Faecalibacterium prausnitzii, Eubacterium rectale, and bifidobacteria were downregulated or even absent in COVID‐19 patients. 44 Fecal microbial markers might serve as a noninvasive diagnostic tool for COVID‐19. 39
Imbalanced microecology promoted intestinal disease development, such as diarrhea and abdominal pain. 45 , 46 After clearance of SARS‐CoV‐2, more than half of patients suffered from post‐acute COVID‐19 syndrome. 47 , 48 , 49 , 50 Impaired microecology potentially deregulated intestinal metabolism and host immune response. Therefore, restoration of intestinal microecology may alleviate post‐acute COVID‐19 syndrome.
2.4. Metabolic effects of pancreatic and hepatic injury
Meta‐analysis of COVID‐19 serum showed that amylase and lipase were increased in a part of SARS‐CoV‐2‐positive patients, which correlated with worse clinical outcomes. 51 This finding points to viral injury of the pancreas and liver 52 , 53 (Figure 1). Pancreatic and hepatic injuries may further cause metabolic complications that deteriorate clinical outcomes, as diabetic patients suffered more from lung damage in COVID‐19 cases. 54
SARS‐CoV‐2 infection resulted in long‐term hyperglycemia in nearly a third of individuals who are normoglycemic before infection. 55 Accordingly, clinic follow‐up showed that SARS‐CoV‐2 infection caused new‐onset diabetes or aggravation of pre‐existing metabolic dysfunction including obesity, hypertension, ketoacidosis, nonalcoholic fatty liver diseases, and diabetes. 34 The increase in glucose levels in COVID‐19 patients is at least in part due to impaired pancreatic function. Pancreatic beta cells express ACE2 receptor and its associating proteins that allow SARS‐CoV‐2 infection. 56 Coronavirus‐like particles were recently found in autophagolysosomes of pancreatic acinar cells of SARS‐CoV‐2 patients. These particles potentially impaired pancreatic islet function by altering the expression and distribution of host proteins. 57 In addition, an abnormal serum cytokine profile was reported to cause insulin resistance and β cell hyperstimulation in patients without diabetic history. 55
Liver injury represents another feature of SARS‐CoV‐2 infection. Approximately 15–53% of SARS‐CoV‐2 patients developed clinical features and pathological changes resembling hepatic injury. 58 , 59 ACE2 is overexpressed in both parenchyma hepatic cells and nonparenchyma hepatic cells. 60 SARS‐CoV‐2 could bind to ACE2 protein expressed at hepatocytes, bile duct cells, and hepatic endothelial cells to induce metabolic reprogramming or even apoptosis. However, which cell types serve as the major targets of SARS‐CoV‐2 under physiological conditions remains to be elucidated. 61 Studies using cultured liver cancer cell lines suggested that viral spike (S) protein could bind to cholesterol. 62 Further, high‐density lipoprotein (HDL) scavenger receptor B type 1 (SR‐B1) mediated SARS‐CoV‐2 attachment and entry. 63 , 64 During this process, HDL enhanced cell surface binding of SARS‐CoV‐2 S protein. 63 This observation explains why high cholesterol presented as a risk factor for severe COVID‐19 diseases. 65 Importantly, COVID‐19 autopsy studies revealed dysregulated glucose and fatty acid metabolism in the liver. 66 Specifically, glycogenolysis, galactose degradation, and glycolysis were suppressed while fatty acid oxidation and oxidative phosphorylation were activated in COVID‐19 cases. 66 These data indicate that SARS‐CoV‐2 proteins may rewire specific metabolic pathways in human liver during its lifecycle.
Although the exact role of liver injury in COVID‐19 development remained under discussion, 67 viral infection‐induced elevation of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) was positively relevant to disease severity. 68 Patients with hepatocellular carcinoma or those who underwent liver transplantation exhibited worse outcomes after SARS‐CoV‐2 infection. 69 Dysregulation of liver metabolism potentially contributed to the complications observed in severe COVID‐19 patients. 66 , 68 , 70 , 71 , 72
2.5. Arrogating adipose tissue
Obesity has been recognized as a high‐risk factor in COVID‐19 patients. Patients with metabolic syndrome are prone to develop more severe diseases upon exposure to SARS‐CoV‐2. 73 Mechanistic studies suggest that both cell state and nutrition status are key determinants of ACE2 levels in host adipocytes. 34 ACE2 expression was induced during the in vitro differentiation of adipocytes, implying that SARS‐CoV‐2 only targeted adipocytes in their mature state. 74 Additionally, the lipid‐rich microenvironment where adipocytes lived promoted ACE2 expression. 75 Rich lipid storage within adipocytes not only facilitated lipid raft formation on the cell membrane that perquisites viral entry, but also provided building blocks for viral capsules 54 (Figure 1). Consequently, SARS‐CoV‐2 arrogated adipocytes for rapid replication and expansion, leading to adipose tissue dysfunction and insulin resistance. 74 Inhibition of lipase‐mediated lipid breakdown strongly suppressed viral propagation in adipocytes. 76 Interestingly, SARS‐CoV‐2 was found in the adipose tissue of overweight males but not in females. 76
2.6. Destabilizing immunometabolism
SARS‐CoV‐2‐induced destabilization of immunometabolism is reflected in serum metabolome (Figure 1). Kynurenine metabolism belongs to one of the most prominent pathways that regulate host inflammatory events. 76 Plasmic kynurenic acid (KA) and its derivatives were proposed to be markers for viral infection. 77 , 78 In addition to serological changes, SARS‐CoV‐2 mediated specific immunometabolic responses in host T cells. Effector T cells and memory T cells are vital for immune response and viral clearance. While effector T cells have enhanced glycolysis, memory T cells rely on oxidative phosphorylation and fatty acid oxidation for energy production. 79 In COVID‐19 patients, a unique population of T cells was characterized by overexpression of voltage‐dependent anion channel 1 and translocase of outer mitochondrial membrane. Both proteins reside in mitochondrial membrane and sustain cellular respiration. Yet, mitochondria in these lymphocytes have misshaped cristae and deficient respiration. 80 The aberrant mitochondrial morphology explained a preapoptotic phenotype of this population and dysfunctional immune response in COVID‐19 patients.
Aged patients usually went through rapid and progressive deterioration of COVID‐19. 34 Coronavirus infection models established with aged mice phenocopied human SARS‐CoV‐2 infection and revealed immunometabolic alterations. A glycolysis‐to‐ketolysis switch protected host animals against inflammatory damage after murine beta coronavirus (mCoV) infection. Interestingly, a ketogenic diet promoted the expansion of protective γδ T cells and alleviated inflammation in mCoV‐infected aged mice. 81 Ketone metabolism may be beneficial in the immune defense against SARS‐CoV‐2 infection.
3. HOST METABOLISM MODULATES SARS‐COV‐2 LIFECYCLE
SARS‐CoV‐2 lifecycle comprises at least four stages: entry, replication, assembly, and release 82 (Figure 2). Multiple types of viruses, including SARS‐CoV‐2, human immunodeficiency virus, and influenza virus, use lipid rafts on host cell membrane as an entry point of the infection cycle. Receptor proteins and endocytosis machinery usually coexist in lipid rafts. SARS‐CoV‐2 infection starts from the interaction between viral S protein and ACE2 (Figure 2). Further, transmembrane serine protease 2 (TMPRSS2), which locates in proximity to ACE2, primes viral infection through enzymatic cleavage of S protein. 83 The ACE2‐containing lipid raft mediates the internalization of virions to form endosomes. 84 Subsequent proteolytic cleavage of S protein by cysteine proteases in late endosome or lysosome allows the release of viral nucleocapsid into host cytoplasm. 85 After that, the virus replicates in double‐membrane vesicles (DMVs). DMVs are structurally similar to autophagosomes. Nonstructural proteins induce the formation of DMVs, of which cholesterol‐rich sites support viral replication. 86 Therefore, cholesterol metabolism supports both viral entry and replication. 87 Viral genome‐encoded structural and accessory proteins are synthesized by the host ribosomes at the endoplasmic reticulum (ER). These proteins are subsequently transferred to the Golgi apparatus where they will be assembled with viral genome, encapsulated into vesicles, and released into extracellular space via exocytosis. 88 , 89 Noteworthy, host metabolic network intertwines with both antiviral signaling and viral biogenesis to modulate SARS‐CoV‐2 lifecycle. Host cells provide essential building blocks for the biogenesis of viral genomes, proteins, and lipid membranes. Recent studies have identified key metabolic events that support viral amplification.
FIGURE 2.
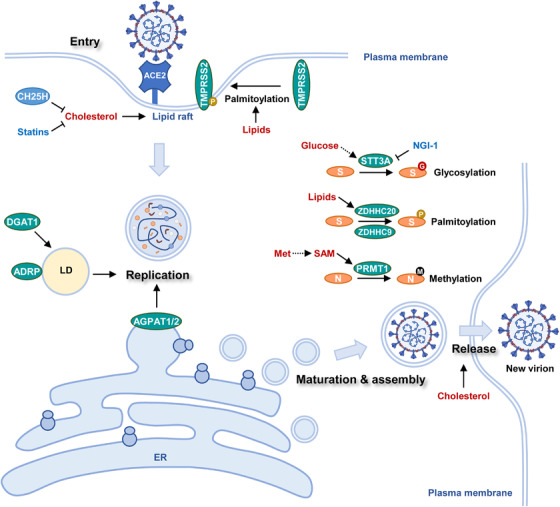
Host metabolism modulates SARS‐CoV‐2 lifecycle. SARS‐CoV‐2 lifecycle intensively interacts with host metabolism. Viral entry, replication, assembly, and release are modulated by host metabolism. Host cells provide metabolic support for SARS‐CoV‐2 biogenesis and maturation of viral proteins. ACE2 (angiotensin‐converting enzyme 2), TMPRSS2 (transmembrane serine protease 2), CH25H (cholesterol 25‐hydroxylase), DGAT1 (diacylglycerol O‐acyltransferase 1), ADRP (adipose differentiation‐related protein), LD (lipid droplet), AGPAT1/2 (1‐acylglycerol‐3‐phosphate O‐acyltransferase 1/3), ER (endoplasmic reticulum), S (spike protein), STT3A (STT3 oligosaccharyltransferase complex catalytic subunit A), ZDHHC20 (zinc finger DHHC‐type palmitoyltransferase 20), ZDHHC9 (zinc finger DHHC‐type palmitoyltransferase 9), Met (methionine), SAM (S‐adenosylmethionine), PRMT1 (protein arginine methyltransferase 1), N (Nucleocapsid protein),
3.1. Lipid and cholesterol metabolism regulates SARS‐COV‐2 entry
Cholesterol is a key component of lipid raft and provides structural support for the ACE2 receptors. Upon SARS‐CoV‐2 infection, interferon (IFN) induces cholesterol 25‐hydroxylase (CH25H), an ER membrane‐bound protein. CH25H converts cholesterol into 25‐hydrocholesterol (25HC) and suppresses viral infection through two different mechanisms: (1) CH25H activation depletes cholesterol on the plasma membrane to destabilize ACE2 receptor‐containing lipid raft; (2) 25HC generated by CH25H activates acyl‐CoA:cholesterol acyltransferase on the ER, leading to internalization of cholesterol on the plasma membrane, further decreasing its cholesterol content (Figure 2). Both actions impeded viral entry and conferred a broad antiviral activity to 25HC. 90
Interestingly, the importance of cholesterol in COVID‐19 development was firstly noticed in clinical records of hospitalization. Patients pretreated with statins, a blood cholesterol‐lowering drug, before hospital admission generally had reduced severity and morbidity. 91 Hence, cholesterol‐reducing agents hold great potential to suppress viral infection (Figure 2). At least three different strategies were proposed to inhibit cholesterol metabolism 92 : (1) Sequestrants‐induced removal of cholesterol. Sequestrants such as β‐cyclodextrin could absorb cholesterol‐containing bile acids and sterols to deplete plasma cholesterol 93 ; (2) Activation of cholesterol efflux. Compounds such as LXR agonists upregulate the expression of ATP‐binding cassette transporters A1 and G1, further promoting cholesterol secretion. Besides, cholesterol carriers or acceptors such as HDL or apolipoprotein A‐I could be employed to reduce cholesterol levels in circulation; (3) Inhibition of cholesterol synthesis. Chemical inhibitors of cholesterol synthesis other than statins are potently effective against viral infection.
Viral entry is also modulated by lipid metabolism. Lipid species not only support lipid raft formation as structural components but also serve as precursors for post‐translational modification (PTM). Fatty acylation is a PTM process during which fatty acids, such as palmitate, are attached to cysteine, serine, or threonine residues of proteins. 94 Due to its hydrophobic nature, the palmitoyl chain enhances the affinity between modified proteins and lipid membranes. 95 TMPRSS2, the viral entry priming protease, is at least in part regulated by palmitoylation 96 (Figure 2). Increased lipid levels in obese patients may result in TMPRSS2 hyper‐palmitoylation and enhance its membrane localization, explaining the SARS‐CoV‐2 vulnerability in overweight individuals.
3.2. Lipid droplets support viral replication
After entering into cells, SARS‐CoV‐2 genome and proteins exist in DMVs and further hijack host metabolism for the biogenesis of new viral nucleotides and proteins. 97 DMVs are centers for viral replication where the replication and transcription complexes anchor. The biogenesis of DMVs depends on host lipid phosphatidic acid (PA). Acylglycerolphosphate acyltransferase (AGPAT) 1 and 2 in the ER accelerated viral replication by increasing PA synthesis. 98 Unexpectedly, lipid droplets (LDs) were recently found to be uncanonical sites for viral expansion. LDs are lipid‐rich organelles with monolayered membranes. Monocytes from COVID‐19 patients accumulated LDs compared with their counterparts from healthy individuals. Strikingly, SARS‐CoV‐2 proteins and double‐stranded RNA were located in close proximity to LDs. Electron microscopy of infected cells uncovered the colocalization of viral particles and LDs. 99 LDs potentially served as a replicating center and assembly platform for SARS‐CoV‐2. Diacylglycerol O‐acyltransferase 1 (DGAT1) is the key enzyme responsible for triacylglycerol synthesis and LD formation. Besides, adipocyte differentiation‐related protein (ADRP) associates with LDs and maintains their structure. Depletion of either gene effectively downregulated SARS‐CoV‐2 amplification 100 (Figure 2).
3.3. Assembly and maturation of SARS‐CoV‐2 rely on host metabolism
Viral proteins undergo a series of PTMs before they are assembled into new virions. Host cells provide metabolic precursors for PTM of viral proteins. The membrane‐bound S protein of SARS‐CoV‐2 was sequentially modified by lipid species with the help of zinc finger DHHC‐type palmitoyltransferase 20 and 9 (ZDHHC20 and ZDHHC9) 101 (Figure 2). Notably, S protein acylation was indispensable for its maturation and viral budding at the Golgi apparatus. Besides, S protein acylation increased the fusion capacity of SARS‐CoV‐2 and thus enhanced its infectivity. Inhibition of protein acylation is a promising strategy to restrict viral infection. 101
Sugar groups contribute to viral protein maturation in the form of glycosylation. Mass spectrometry studies uncovered that the majority of SARS‐CoV‐2 encoded proteins were glycosylated. 102 SARS‐CoV‐2 employed host glycosylation enzymes, in particular STT3 oligosaccharyltransferase complex catalytic subunit A (STT3A), to modify S proteins (Figure 2). Once conjugated on spike protein, the sugar groups shielded the viral particle from immunosurveillance. 102 , 103 Chemical inhibition of spike protein glycosylation by NGI‐1 greatly weakened viral infectivity. 104 Animal studies also supported the important role of lipids and sugars in COVID‐19 development. After a continuous high‐fat high‐sugar diet, SARS‐CoV‐2‐infected Syrian hamsters showed increased weight loss and lung dysfunction, delayed viral clearance, and prolonged viral shedding. 105 Therefore, restriction of dietary lipid and sugar intake is potentially beneficial for COVID‐19 patients who have obesity or metabolic syndrome.
PTMs are also found on the nucleocapsid (N) protein. The N protein complexes with both SARS‐CoV‐2 genome and viral membrane proteins during virion assembly. Arginine residues (R95 and R177) in the N protein were methylated by protein arginine methyltransferase 1 (PRMT1) in the host cells. Nucleocapsid protein methylation promoted viral packaging 106 (Figure 2). Chemical inhibition of PRMT1 downregulated N protein methylation and impaired SARS‐CoV‐2 replication. 106 Arginine methylation level is regulated by the availability of S‐adenosyl methionine (SAM), the methyl donor produced from methionine. 107 Therefore, methionine metabolism potentially modulates viral assembly and packaging (Figure 2).
3.4. Lipid raft modulates SARS‐CoV‐2 transmission
Mature SARS‐CoV‐2 virions are released into extracellular space through membrane rearrangements. Interestingly, both SARS‐CoV and SARS‐CoV‐2 can avoid exposure to extracellular environment through direct cell‐to‐cell transmission. Intercellular channels or syncytia facilitated the uncanonical transfer of the virus. 108 Notably, lipid raft was involved in the formation of intercellular channeling tubes and syncytia. Thus, targeting lipid rafts suppressed both viral infection and transmission 109 (Figure 2).
4. MECHANISMS OF REMODELING HOST METABOLISM BY SARS‐COV‐2
The metabolic changes in host cells could be outcomes of either host antiviral response or SARS‐CoV‐2‐induced metabolic remodeling. SARS‐CoV‐2 genome has a compact size and consists of ∼30,000 nucleotides. Proteins encoded by the viral genome fall into three categories: (1) four structural proteins including spike protein (S), envelope protein (E), membrane protein (M), and nucleocapsid protein (N); (2) 16 nonstructural proteins (nsp1‐16); and (3) accessory proteins including ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF9c, and ORF10 110 (Figure 3A). Viral factors remodel host metabolism, which at least partially phenocopies reprogrammed cancer metabolism. In cancer cells, oncogenic mutations rewire metabolic networks to sustain malignant proliferation. 111 Viral genome and its products likewise manipulate host cell metabolism to ensure rapid amplification of SARS‐CoV‐2. In agreement with enhanced biosynthetic needs, SARS‐CoV‐2 replication is dependent on anabolic signaling pathways such as phosphatidylinositol 3‐kinase/protein kinase B/mammalian target of rapamycin signaling. 112 Although our knowledge remains in its infancy, multiomic profiling of virus–host interaction has shed light on how SARS‐CoV‐2 remodels host metabolism.
FIGURE 3.
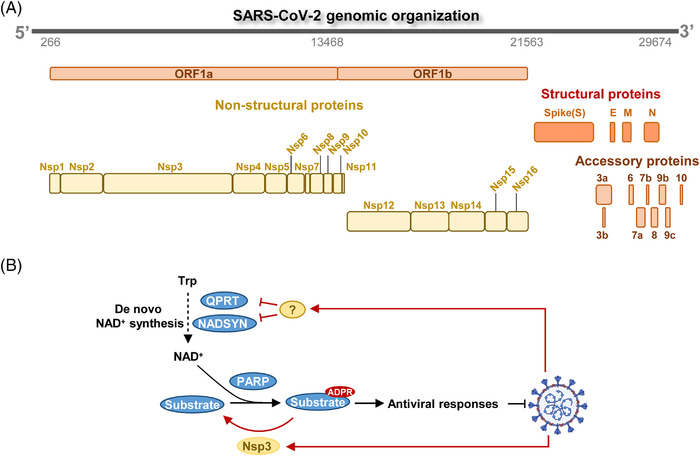
Mechanisms of remodeling host metabolism by SARS‐CoV‐2. (A) Organization of SARS‐CoV‐2 genome. SARS‐CoV‐2 genome encodes nonstructural proteins (Nsps), structural proteins, and accessory proteins. Structural proteins include Spike protein (S), Envelope protein (E), Membrane protein (M), and Nucleocapsid protein (N). (B) SARS‐CoV‐2 reshapes NAD+ metabolism to suppress antiviral response. SARS‐CoV‐2 protein Nsp3 counteracts with PARP to suppress ADPR modification and antiviral responses. Unknown viral factor(s) suppresses de novo NAD+ biosynthesis, which also inhibits PARP‐induced ADP‐ribosylation (ADPR) modification of substrate protein.
4.1. SARS‐CoV‐2 proteins disturb NAD+ metabolism to suppress antiviral response
NAD+ metabolism is perhaps one of the best studied metabolic pathways during SARS‐CoV‐2 infection (Figure 3B). Intracellular NAD+ pool is maintained by tryptophan‐dependent de novo NAD+ synthesis and salvage NAD+ biosynthesis. NAD+ serves as a donor for ADP‐ribose units and is required by poly (ADP‐ribose) polymerase (PARP) to mediate ADP‐ribosylation (ADPR) of antiviral proteins (Figure 3B). Host cells overexpress PARP and activate antiviral proteins by upregulating their ADP‐ribosylation. 113 , 114 Activation of PARP quickly exhausts intracellular NAD+. Notably, SARS‐CoV‐2 adopts at least two different strategies to suppress antiviral responses. First, viral protein Nsp3 is an ADPR hydrolase that removes ADPR modification to counteract host PARPs 115 (Figure 3B). Second, SARS‐CoV‐2 restricts NAD+ supply with unknown factors to suppress antiviral signaling. Specifically, quinolinate phosphoribosyl transferase (QPRT) and NAD+ synthetase (NADSYN), the enzymes responsible for de novo NAD+ synthesis, were decreased by CoV infection (Figure 3B). Host cells could partially compensate for NAD+ shortage by enhancing salvage NAD+ biosynthesis. Importantly, chemical activation of NAD+ biosynthesis repressed viral infection and replication. 116
4.2. Virus–host interactome reveals metabolic targets of SARA‐CoV‐2
SARS‐CoV‐2 genome and its products potentially modulate host metabolism at multiple levels such as transcription, translation, and PTM of metabolic regulators 14 , 63 , 117 , 118 , 119 , 120 , 121 , 122 , 123 (Figure 4A). Mechanisms underlying how viral proteins hijack host cell metabolism remain largely unclear. Protein–protein interactions provide a molecular basis for SARS‐CoV‐2‐mediated metabolic remodeling. To comprehensively illustrate the metabolic interaction between SARS‐CoV‐2 and human body, we extracted the interactions between viral proteins and host metabolic enzymes from published proteomic studies 14 , 123 and grouped them by metabolic pathways and subcellular compartments (Figure 4B). Unexpectedly, viral proteins interacted with multiple central pathways in host cell metabolism, such as sugar metabolism, amino acid metabolism, lipid metabolism, nucleotide metabolism, and redox homeostasis. Another surprising observation from protein interaction studies is that viral proteins interacted with metabolic enzymes from different subcellular locations. Viral proteins might enter into different organelles to rewire metabolic pathways in a compartmentalized manner. Of note, phosphoglycerate kinase 2 (PGK2), fructose‐bisphosphate aldolase b (ALDOB), and hexokinase 3 (HK3) are cytosolic enzymes involved in glycolysis. 124 , 125 , 126 Interaction with viral proteins may impact the enzymatic activity or stability of these enzymes, resulting in dysregulation of glucose metabolism in host cells upon SARS‐CoV‐2 infection (Figure 4C). G6PD catalyzes the rate‐limiting step of the PPP, which provides NADPH and pentose phosphates for fatty acid and nucleic acid synthesis. 127 Individuals with G6PD deficiency suffer a hemolytic crisis when exposed to oxidants or microbes. They are supposed to be more susceptible to SARA‐CoV‐2 infection. 128 , 129 , 130
FIGURE 4.
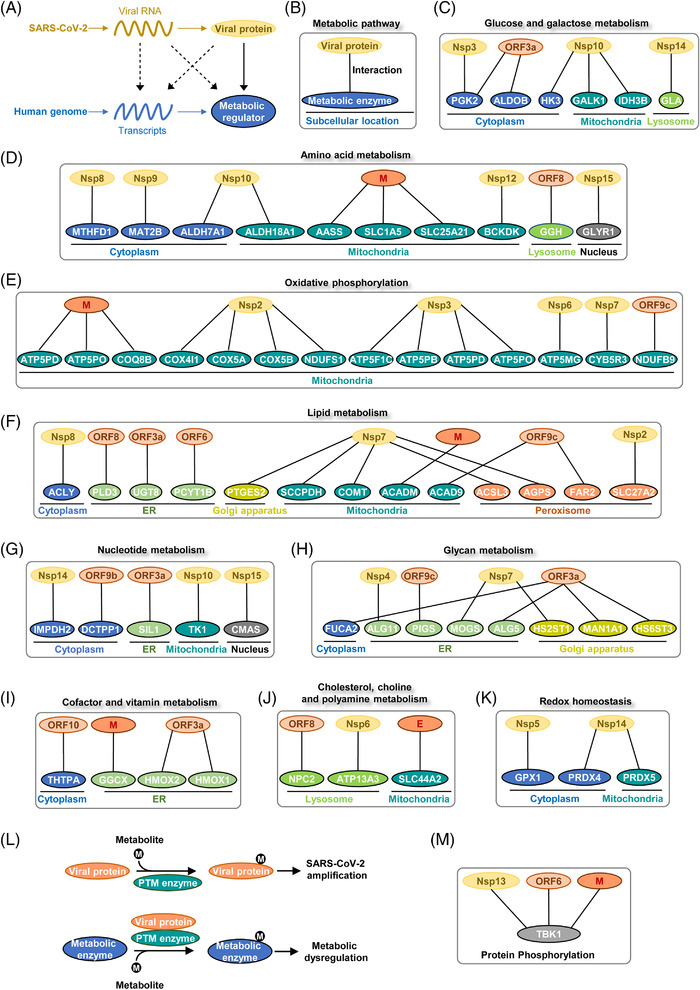
Protein–protein interactions between human and SARS‐CoV‐2. (A) Models showing how viral genome products (RNAs, proteins) interact with host metabolic networks. Protein–protein interactions between SARS‐CoV2 and humans have been elucidated in proteomic profiling studies. (B) Proteomic profiling of SARS‐CoV‐2 interactome revealed the physical association between viral proteins and host metabolic enzymes. The protein–protein interactions are categorized by metabolic pathways and subcellular location. (C) Viral proteins interacted with host enzymes involved in glucose and galactose metabolism. (D) Physical interactions between viral proteins and host enzymes in amino acid metabolism. (E) Viral proteins interacted with host proteins involved in oxidative phosphorylation. (F) Physical associations between viral proteins and host enzymes in lipid metabolism. (G) Viral proteins interacted with host proteins involved in nucleotide metabolism. (H) Interactions between viral proteins and host enzymes involved in glycan metabolism. (I) Viral proteins interacted with host enzymes involved in cofactor and vitamin metabolism. (J) Viral proteins were associated with host enzymes involved in cholesterol, choline and polyamine metabolism. (K) Viral proteins interacted with host enzymes involved in redox homeostasis. (L) Different modes of the interaction between host metabolism, PTM enzyme, and viral infection. Viral proteins may serve as either substrates or modulators of PTM enzymes to interact with host metabolic networks. (M) The physical association of host TBK1 with viral proteins was used as an example to explain virus–host metabolism interaction.
SARS‐CoV‐2 proteins have broad interactions with transporters and enzymes from amino acid metabolism. For instance, M protein was associated with transporter solute carrier family 1 member 5 (SLC1A5), transporter solute carrier family 25 member 21 (SLC25A21), and lysine degradation enzyme aminoadipate‐semialdehyde synthase (AASS) 131 , 132 , 133 (Figure 4D). Notably, SARS‐CoV‐2 protein may be able to sense the metabolic status of host cells. Viral protein Nsp12 belongs to RNA‐dependent RNA polymerases. Nsp12 contains a nucleotidyltransferase domain distant from the catalytic center. This domain functions as a nucleotide sensor and activates RNA synthesis in the presence of nucleotides. 134 , 135 Hence, Nsp12 may sense host nucleotide availability and correspondingly modulates replicative efficacy of viral genome. Interestingly, Nsp12 interacted with branched‐chain keto acid dehydrogenase kinase, a mitochondrial enzyme responsible for branched‐chain amino acid (BCAA) catabolism 136 (Figure 4D). Two cofactors of Nsp12, Nsp7, and Nsp8, interacted with the electron transport chain (ETC) and ribosome proteins in mitochondria 137 (Figure 4E). These observations indicate that BCAA metabolism and mitochondrial respiration are potentially under the control of viral proteins.
Lipid metabolism and nucleotide biosynthesis are reprogrammed during SARS‐CoV‐2 infection. 100 Enzymes from both fatty acid beta‐oxidation (acyl‐CoA dehydrogenases, ACADM and ACAD9) and lipogenesis (ATP citrate lyase, ACLY) were found to interact with viral proteins. 138 , 139 , 140 Strikingly, Nsp7 seems to be a potent regulator of lipid metabolism as it interacted with four different enzymes from this pathway. Among binding partners of Nsp7, acyl‐CoA synthetase long‐chain family member 3 (ACSL3) converts free long‐chain fatty acids into fatty acyl‐CoA esters to support lipid biosynthesis 141 (Figure 4F). Intriguingly, ACSL3 has been found to maintain the replication of different viruses and serves as a promising therapeutic target for COVID‐19. 142 In accordance, SARS‐CoV‐2 infection reduced ATP production and deregulated fatty acid metabolism in a cell‐based infection model. 143 Unknown virulent factors from SARS‐CoV‐2 upregulated lipid uptake and lipogenesis genes in monocytes. 99 DGAT and ADRP were also upregulated to promote the formation of LDs. Moreover, viral nucleocapsid protein interacted with ADRP on the LD surface to facilitate viral replication. 144
Viral genome replication and transcription consume large amounts of nucleotides. Inosine monophosphate dehydrogenase 2 (IMPDH2) and thymidine kinase 1 (TK1) are rate‐limiting enzymes that control guanine and thymidine nucleotide biosynthesis, respectively. 145 , 146 Viral proteins may boost nucleotide biosynthesis by activating these enzymes (Figure 4G). Noteworthy, folate metabolism provides one‐carbon units to maintain nucleotide synthesis. 147 SARS‐CoV‐2 infection resulted in folate metabolism remodeling to enhance de novo purine synthesis and viral genome replication. 148
Glycans play vital roles in cellular structure maintenance, protein or lipid modification and cell‐to‐cell communications. 149 , 150 Importantly, ORF3a was associated with fucosidase 2 (FUCA2) which catalyzes the hydrolysis of carbohydrate moieties from glycoproteins. FUCA2 has been revealed as an oncogenic factor due to its immunosuppressive activity. 151 Whether ORF3a induces host immunosuppression through interacting with FUCA2 still needs further validation (Figure 4H).
Cofactor and vitamin metabolism are candidates for viral targets as well. Heme metabolism end‐products, such as ferrous iron, biliverdin, and bilirubin, have antiviral effects depending on their antioxidant, anti‐inflammatory, and anti‐apoptotic functions. 152 , 153 , 154 , 155 Heme oxygenase (HMOX), an essential enzyme in heme catabolism, might be a therapeutic target for COVID‐19. 156 , 157 Viral ORF3a interacted with both isozymes, HMOX1 and HMOX2, implying a significant role of heme metabolism during SARS‐CoV‐2 infection (Figure 4I).
As mentioned earlier, cholesterol is essential for SARS‐CoV‐2 entry and replication. Once entered into cells, SARS‐CoV‐2 in turn upregulated host cholesterol metabolism by modulating SREBP2‐mediated gene expression. 158 Viral proteins ORF8, Nsp6, and E interacted with enzymes in cholesterol, choline, and polyamine metabolism. The biological function of these interactions remains unclear (Figure 4J).
Redox imbalance represented a key metabolic outcome of COVID‐19. 36 , 83 , 159 , 160 , 161 Redox homeostasis is involved in the whole lifecycle of SARS‐CoV‐2 and host inflammatory responses. 162 , 163 , 164 Viral protein Nsp5 and Nsp14 interacted with redox‐regulatory enzymes in both cytoplasm and mitochondria (Figure 4K). SARS‐CoV‐2 may coordinate redox balance and biosynthetic processes in different cellular compartments to maximize its amplification efficacy. Metabolic interaction between SARS‐CoV‐2 and human hosts will provide more clues for therapeutic opportunities.
4.3. The effect of SARS‐CoV‐2 on mitochondrial function and metabolism
A remarkable number of metabolic enzymes from SARS‐CoV‐2 interactome reside in mitochondria, highlighting a vital role of mitochondria in viral lifecycle. Mitochondria generate ATP and metabolic intermediates to support the biosynthesis of macromolecules. 165 Moreover, they function as hubs in cell metabolism, stress signaling and immune response. 166 , 167 Importantly, viral proteins bind to multiple enzymes residing in the mitochondria matrix (Figures 4C–4K). Although it remains unclear whether or not mitochondria serve as the replicating platform for SARS‐CoV‐2, accumulating evidence has shown that mitochondria function is broadly affected by SARS‐CoV‐2 infection.
SARS‐CoV‐2 remodels mitochondria metabolism after entering into host cells. In cancer cells, suppression of mitochondria activity drives the redistribution of carbon flux into glycolysis and PPP to satisfy anabolic needs. 32 , 135 Similarly, mitochondrial activity is suppressed in multiple models of viral infection. 168 The interaction between viral proteins and oxidative phosphorylation implies that viral factors may directly modulate cellular respiration and carbon fluxes (Figure 4E). In support of this speculation, the transcriptional activity of nuclear factor erythroid 2‐related factor 2 (NRF2), a nuclear factor that drives mitochondrial biogenesis, was broadly suppressed in COVID‐19 patients' biopsies. 167 , 169 , 170 , 171 Chemical activation of NRF2 exerted antiviral effects toward SARS‐CoV‐2 infection. 168 Of note, mouse hepatitis virus infection model suggested that endogenous metabolites could alter mitochondrial metabolism and correspondingly modulate viral replication efficacy. 172 Thus, metabolic interventions may be employed to restrain mitochondria metabolism and SARS‐CoV‐2 replication. 172 , 173 It is worthy to mention that previous proteomic studies utilized overexpressed viral proteins as baits to track virus–host interaction. 63 , 123 Further efforts are required to exclude potential experimental artifacts in physical association with mitochondrial enzymes. Which viral factors function as drivers of metabolic remodeling remains an open biological question.
Mitochondria‐derived reactive oxygen species (ROS) act as metabolic signals that modulate SARS‐CoV‐2 amplification. Mitochondria utilize ETC to fulfill oxidative phosphorylation and energy production (Figure 4E). Leakage of electrons from the ETC results in the production of ROS, making mitochondria a major source of oxidative stress. SARS‐CoV‐2 infection triggered mitochondrial ROS accumulation to interrupt host antiviral responses. In COVID‐19 patients, elevated mitochondrial ROS associated with mitochondrial membrane depolarization. 174 Specifically, ORF3a caused mitochondrial damage and elevated mitochondrial ROS to upregulate hypoxia‐inducible factor 1α (HIF‐1α). HIF‐1α further induced proinflammatory responses to enhance viral infection. 175 In monocytes, elevated mitochondrial ROS stabilized HIF‐1α and promoted glycolysis. Dysregulation of monocyte metabolism inhibited T cell activity and reduced lung epithelial cell viability. 176 In a complementary study, cluster of differentiation 147 (CD147, also known as basigin) was found to strongly bind to S protein of SARS‐CoV‐2. The consequent ROS accumulation contributed to metabolic dysfunction of T cells and impaired virus clearance. Nutrients such as fatty acid and glucose were proposed to alleviate mitochondrial ROS production in T cells to fight against viral infection. 177 , 178
SARS‐CoV‐2 also affects mitochondrial mass and organelle dynamics. Mitochondria maintain organelle‐specific genomes (mtDNA). Both bulk and single‐cell RNA sequencing of COVID‐19 blood samples showed that mtDNA‐encoded gene expression levels were decreased. This phenomenon is possibly resulted from reduced expression of mitochondrial RNA polymerase. 179 , 180 Unexpectedly, T cells, monocytes, and granulocytes were reported to have increased mitochondrial mass. However, only T cells exhibited higher oxidative phosphorylation capacity and architecture disruption. 178 Besides, hepatocytes from the liver samples of SARS‐CoV‐2‐positive patients were also characterized by cristae disruption. 181 Mechanistically, ORF9b promoted mitochondrial elongation by proteasomal degradation of dynamin‐related protein 1 (DRP1), a protein responsible for membrane remodeling during mitochondrial division. 182 Viral proteins ORF9c, M, Nsp6, and ORF3a were shown to localize to mitochondrial membrane and induce morphological changes. 183 Of note, viral proteins encoded by SARS‐CoV‐2 genome were reported to interact with multiple proteins located in mitochondrial inner membrane. Mitochondrial permeability transition pore (mPTP) is a protein complex formed in the inner membrane under specific pathological conditions. 184 The opening of mPTP increases mitochondria permeability and leads to mitochondrial swelling. 185 Viral proteins may hamper mitochondrial function through binding to mPTP and destructing mitochondrial inner membrane integrity. Depolarization of mitochondria could further impair mitophagy and cause apoptosis. 174 , 183 , 186 , 187 Notably, mPTP blocker cyclosporin A was demonstrated to protect cardiomyocytes from SARS‐CoV‐2‐induced autophagy and cell death. 183
The cytokines released during SARS‐CoV‐2 infection are critical determinants of mitochondrial function and metabolism. 188 IFN‐γ induced by viral infection acted on Toll‐like receptor signaling in macrophages and initiated mitochondria‐driven cell death. 189 , 190 In this process, nitric oxide synthase was induced to promote caspase‐8 cleavage and activate mitochondrial apoptotic effectors. Importantly, the mitochondria‐driven apoptosis potentially increased disease severity in COVID‐19 patients. 189 In critically ill COVID‐19 cases, both pro‐inflammatory (IL‐1, IL‐2, IL‐6, TNF‐α) and anti‐inflammatory (IL‐4, IL‐10) cytokines were presented at high levels. 191 The metabolic influences of these cytokines on mitochondrial function remains vague. Previous studies have shown that IL‐1 suppressed mitochondrial fatty acid oxidation to drive tissue‐specific inflammation. 192 , 193 IL‐4 was reported to promote mitochondrial glutaminolysis and α‐ketoglutarate production in macrophages. 194
4.4. The interaction between PTM enzymes and host metabolism during SARS‐CoV‐2 infection
PTMs of viral proteins represent a key regulatory mechanism of SARS‐CoV‐2 lifecycle. 195 In addition to known modifications on the spike and nucleocapsid proteins (Figure 2), a variety of PTMs presumably exists on SARS‐CoV‐2 proteome. Previous studies of human and animal coronavirus have revealed numerous PTM events on viral proteins, such as glycosylation, phosphorylation, ADP‐ribosylation, SUMOylation, and ubiquitination. 195 , 196 Noteworthy, SARS‐CoV‐2 proteins interacted with posttranslational modifying enzymes involved in protein methylation, phosphorylation, palmitoylation, and glycosylation. 14 Because metabolic intermediates serve as substrates in the majority of PTM reactions, 197 , 198 cell metabolism has an intimate relationship with PTM of viral proteins. 199 , 200 , 201 For example, protein kinase transfers phosphate group from ATP to modify its substrate protein in the form of phosphorylation. From this perspective, PTM enzymes potentially connect metabolic status to their downstream proteins. Given the fact that PTMs on spike and nucleocapsid proteins contribute to viral protein maturation and assembly, it would not be surprising that host metabolism is possibly involved in the regulation of other steps in SARS‐CoV‐2 lifecycle. The PTMs of viral proteins potentially interconnect host metabolic network with viral lifecycle to coordinate the replication and dissemination of viral particles. Therefore, PTMs may act as metabolic checkpoints of viral lifecycle and pave the way for metabolic intervention of COVID‐19. Additionally, metabolites are emerging as signaling molecules that regulate cellular biogenesis. 202 Whether SARS‐CoV‐2 senses specific metabolic signals to prime viral replication remains an unsettled question. Proteomic profiling of SARS‐CoV‐2 proteins at different time points of infection would help to elucidate the dynamics of viral protein modification.
Viral proteins could act as either substrates or regulators of host PTM enzymes (Figure 4L). During SARS‐CoV‐2 infection, PTM enzymes are potential regulatory nodes that connect host metabolism and viral proteins (Figure 4L). Of note, TANK‐binding kinase 1 (TBK1) is a vital regulator of innate immunity and antiviral response. Previously, viral proteins Nsp13, ORF6, and M were shown to associate with TBK1. 14 Through phosphorylating target proteins, TBK1 not only facilitated the activation of NF‐κB pathway but also promoted the expression of IFN‐stimulated genes. 203 , 204 , 205 , 206 Notably, Nsp13 and M protein promoted TBK1 degradation via autophagic process or ubiquitination, resulting in inhibition of host innate immune response 207 (Figure 4M). Besides, TBK1 was previously reported to modulate energy metabolism by suppressing adenosine 5‘‐monophosphate‐activated protein kinase. 208 , 209 It is reasonable to speculate that other PTM enzymes may be involved in viral infection through interconnecting cell metabolism and SARS‐CoV‐2.
5. DISCOVERY OF METABOLISM‐BASED THERAPY TO COMBAT SARS‐COV‐2
The deepening knowledge of COVID‐19 metabolism enables the discovery of new therapeutic targets. Clinical evidence combined with genetic or chemical screening has revealed several metabolic vulnerabilities of SARS‐CoV‐2. Metabolism‐based therapy would provide powerful expansion to the arsenal against SARS‐CoV‐2.
5.1. Targeting cholesterol metabolism: lessons from human genetics
Human genetic diversity provides insights into disease progression and treatment. 210 In the case of COVID‐19, genetic evidence highlights the role of Niemann‐Pick disease type C1 (NPC1) gene in viral infection. NPC1 locates at the lysosomal membrane and mediates cholesterol efflux 211 (Figure 5A). Inherent mutations of this gene sequester cholesterol in the lysosome and lead to dysregulation of lysosomal proteolytic activity. As proteolysis of spike proteins is an essential step for viral infection, NPC1 mutations conferred a broad resistance against filovirus, retrovirus, and togavirus. 212 Homozygous deletion of Npc1 also protected mice from viral infection, 212 making NPC1 an exploitable target for COVID‐19 treatment. 213 Small molecule inhibitors of NPC1, such as U18666A, showed a broad antiviral activity including coronavirus. 214 , 215 Of note, imipramine and cepharanthine are FDA‐approved drugs targeting the NPC1 pathway. Both compounds reduced the infectivity of several single‐stranded RNA viruses 216 , 217 (Figure 5A).
FIGURE 5.
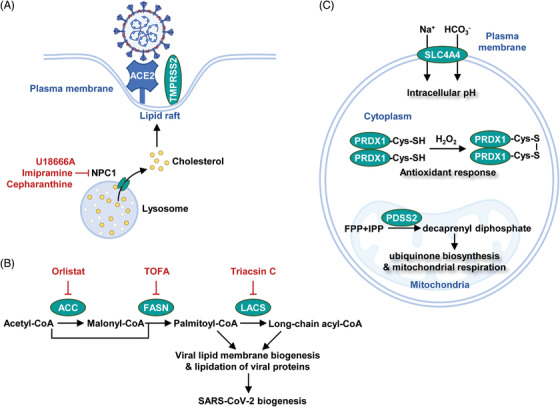
Discovery of metabolism‐based therapy to combat SARS‐CoV‐2. (A) NPC1, a lysosomal protein, mediated cholesterol efflux to promote the formation of lipid rafts on the plasma membrane. Chemical inhibition of NPC1 suppressed lipid raft‐induced viral entry. (B) Lipid synthesis supported SARS‐CoV‐2 biogenesis. Chemical inhibition of lipogenic enzymes, such as ACC (acetyl‐CoA carboxylase), FASN (fatty acid synthase), and LACS (long‐chain acyl‐CoA synthetases), suppressed viral replication. (C) Metabolic dependencies identified by high‐throughput screening. SLC4A4 (solute carrier family 4 member 4), PRDX1 (peroxiredoxin 1), and PDSS2 (decaprenyl diphosphate synthase subunit 2) were identified to be necessary metabolic factors for SARS‐CoV‐2 infection
In addition, human genome‐wide association studies identified a link between solute carrier family transporter 6A20 (SLC6A20) with interstitial pneumonia in COVID‐19 patients. 218 SLC6A20 locates on the plasma membrane and mediates proline/glycine uptake in a calcium‐dependent manner. Further investigations are required to pinpoint the therapeutic opportunities behind SLC6A20.
5.2. Lipid and glucose metabolism as feasible therapeutic targets
Clinical observations in hospitalized COVID‐19 patients revealed a link between statin use with improved outcomes. 219 , 220 Statin potentially slowed COVD‐19 pathogenesis by suppressing lipid metabolism. However, concerns were also raised because statin increased the expression of ACE2 receptor. Alternative inhibitors of lipid biosynthesis showed promising activity against SARS‐CoV‐2. For example, inhibitors of fatty acid synthase (orlistat), long‐chain acyl‐CoA synthetases (triacsin C), and acetyl‐CoA carboxylase (TOFA) effectively suppressed virus infection (Figure 5B). In contrast, lipid β‐oxidation seems to play a minor role in virus replication. 221 Interestingly, polyunsaturated fatty acids weakened viral infection. Linolenic acid and eicosapentaenoic acid (EPA) suppressed TMPRSS2 protease to block SARS‐CoV‐2 entry. 222
Diabetic COVID‐19 patients exhibited lower mortality if prescribed glucose‐lowering drugs. 223 , 224 The virulence of SARS‐CoV‐2 may be determined by environmental factors such as glucose concentration in the tissue microenvironment. The efficacy of glucose‐lowering drugs in nondiabetic patients remains unclear.
5.3. Genome‐wide screening of essential metabolic genes for viral infection
Genome‐wide CRISPR screening serves as a powerful tool for identifying key factors for viral infection. 225 In monkey kidney cells, metabolic genes including solute carrier family transporter 4A4 (SLC4A4, a bicarbonate cotransporter and pH modulator), PRDX1 (a peroxide‐detoxifying enzyme), and decaprenyl diphosphate synthase subunit 2 (PDSS2, an enzyme in ubiquinone biosynthesis), were found to be essential for viral entry and replication 226 (Figure 5C). Hence, cellular acid‐base balance, redox signaling, and cellular respiration potently govern SARS‐CoV‐2 infectivity. Compounds targeting these metabolic processes may be translated into COVID‐19 therapies.
5.4. Clinical trials of metabolic intervention of COVID‐19
After years of drug discovery, remdesivir is still one of the most effective drugs recommended for SARS‐CoV‐2 treatment. 227 , 228 , 229 Remdesivir inhibits the RNA‐dependent RNA polymerase encoded by the viral genome to impair RNA synthesis and SARS‐CoV‐2 replication. 230 Compared to compound screening from scratch, repurposing clinically approved drugs is more economic. 231 Chemical screening in the field of cancer metabolism has generated a rich number of drugs that suppress various metabolic pathways. 232 , 233 Because SARS‐CoV‐2 reprograms host cell metabolism in a fashion similar to cancer cells. The preclinical drugs targeting cancer metabolism may be directly tested for antiviral activity. Indeed, bioactive molecules targeting metabolic enzymes in lipid metabolism and protein glycosylation were found to inhibit SARS‐CoV‐2 infection. 142 Several metabolites from commensal microbiota were predicted to bind to SARS‐CoV‐2 spike, awaiting experimental validation. 234
Importantly, several antiviral compounds suppress different metabolic pathways and are under clinical evaluation (Table 1). Palmitoylethanolamide (PEA) is a lipid‐derived peroxisome proliferator‐activated receptor‐α (PPAR‐α) agonist that prevents SARS‐CoV‐2 entry and replication. 235 PEA was found to promote β‐oxidation and LD degradation. 235 Baricitinib, an inhibitor of the Janus‐associated kinases 1 and 2 (JAK1 and JAK2), was demonstrated to modulate fatty acid oxidation in SARS‐CoV‐2 infected patients. 236 Fenofibrate is a cholesterol‐lowering drug that holds potential to treat COVID‐19. 237
TABLE 1.
Clinical trials of metabolic intervention of COVID‐19
| Drug | Metabolic target | Clinical trial number | Phase | Status | Drug type |
|---|---|---|---|---|---|
| PEA 235 |
Lipid metabolism (PPAR‐α) |
NCT04619706 | 2 | Terminated | Chemical |
| NCT04568876 | 4 | Completed | |||
| Pioglitazone 249 |
Lipid metabolism (PPAR‐γ) |
NCT04535700 | 4 | Recruiting | Chemical |
| Baricitinib 236 |
Fatty acid metabolism (JAK1/JAK2) |
NCT04421027 | 3 | Completed | Chemical |
| Fenofibrate 237 | Cholesterol metabolism | NCT04517396 | 2 | Completed | Chemical |
| VPA 250 |
Glucose metabolism (HDAC) |
NCT04513314 | 4 | Not yet recruiting | Chemical |
| Masitinib 240 |
Potentially glucose metabolism (Tyrosine kinase) |
NCT05047783 | 2 | Recruiting | Chemical |
| Methotrexate 251 | Folate metabolism | NCT04610567 | 1 | Recruiting | Chemical |
| Cholecalciferol 252 | Steroid metabolism | NCT04952857 | 4 | Completed | Chemical |
| Nicotinamide 253 | Nicotinamide metabolism | NCT04751604 | NA | Active, not recruiting | Chemical |
| L‐citrulline 254 | Amino acid metabolism | NCT04404426 | NA | Completed | Chemical |
| Coenzyme Q10 255 | Mitochondrial metabolism | NCT04960215 | 2 | Completed | Chemical |
In addition to lipid metabolism, glucose metabolism is emerging as one of the major targets for antiviral drugs (Table 1). Valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, was reported to lower glucose levels and inhibit cytokine response. Both actions may contribute to its inhibitory effect on SARS‐CoV‐2 entry. 238 Masitinib, a tyrosine kinase inhibitor, inactivates viral protease to restrict viral replication. Although Masitinib has a significant effect on body glucose metabolism, its exact metabolic target is still unknown. 239 , 240 Of note, folate metabolism provides one‐carbon units to fuel nucleotide biosynthesis. 241 Methotrexate suppresses folate metabolism and has been used for the treatment of multiple cancers. The efficacy of methotrexate in COVID‐19 development is under investigation in a clinical trial. 242
6. DISCUSSION
SARS‐CoV‐2‐induced metabolic reprogramming plays a key role in COVID‐19 progression. Due to the limitation of animal models that recapitulate human COVID‐19 development, the mechanism of how SARS‐CoV‐2 rewires host metabolism remains poorly understood. Besides, SARS‐CoV‐2 is rapidly evolving and accumulating mutations worldwide. Newly occurring mutations confer new traits to SARS‐CoV‐2 and breach immune protection. Whether newly evolved SARS‐CoV‐2 variants remodel host metabolic in specific manners requires further investigation. Metabolic vulnerabilities shared by major SARS‐CoV‐2 strains will open the window to the discovery of broad‐spectrum antiviral compounds. Meanwhile, strain‐specific remodeling of host metabolic pathway creates the opportunity for precise and personalized treatment of COVID‐19.
6.1. Systemic profiling of metabolic interactions between host and SARS‐CoV‐2
SARS‐CoV‐2 was reported to cause long‐term metabolic complications in infected individuals. 243 The impact of viral genome and its products on host cell metabolism remains largely unclear. It is necessary to evaluate the influence of each viral protein and RNA on host metabolic networks, such as mRNA expression, protein level, and catalytic activity of rate‐limiting enzymes. Besides, SARS‐CoV‐2‐mediated metabolic alterations vary among different tissues. In‐depth profiling of tissue‐specific metabolic reprogramming is essential to resolving COVID‐19 pathology. Of note, SARS‐CoV‐2 replication produces viral subgenome. 13 Genomic and subgenomic RNAs are potentially neglected regulators of host metabolism.
6.2. Humanized animal model for SARS‐CoV‐2 infection
The lack of humanized SARS‐CoV‐2 infection model remains a bottleneck in COVID‐19 studies. Recently, a lung organoid model indicated that SARS‐CoV‐2 infection downregulated lipid metabolism, urea cycle, folate metabolism, and glutamine metabolism. 244 Mouse models have been widely used to study coronavirus infection, but they could not faithfully reproduce the antiviral responses in humans. 245 The development of humanized animal models for COVID‐19 would undoubtedly accelerate vaccine development and drug screening. 246
6.3. Emerging mutations and new challenges
The new SARS‐CoV‐2 variant Omicron has exacerbated the global COVID‐19 pandemic. Novel COVID‐19 mutations mainly occur in the receptor‐binding domain of the spike protein. 247 Whether these mutations rewire unique metabolic pathways in host cells remains unknown. It is thus important to track the metabolic changes caused by different variants, and correspondingly identify metabolic determinants their infectivity, virulence, and transmissibility. 248 These efforts would give rise to predesigned therapies and enable our chase to be faster than SARS‐CoV‐2 mutations. 248
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
Not applicable.
AUTHOR CONTRIBUTIONS
T. W., Q. Z., and Y. P. W. conceptualized the work. T. W., Y. C., H. Z., Z. W., C. H. M., Y. Y., L. C., S. X., X. Y., Q. Z., and Y. P. W. wrote the paper. T. W., Q. Z., and Y. P. W prepared figures and table; T. W., Q. Z., and Y. P. W. supervised the work and revised the manuscript. T. W., Y. C., and H. Z. contributed equally to this work.
ACKNOWLEDGMENTS
We thank Dr. Ruikang Liu (National Institute of Allergy and Infectious Diseases, Bethesda, USA) for her critical reading of the manuscript. This work was supported by the National Natural Science Foundation of China (Nos. 81790251, 81790250, 81772946, and 81502379 to Y. P. W.; Nos. 92049113 and 32070759 to T. W.; No. 81402053 to L. C.; No. 81902673 to Q. Z.; No. 82102923 to Y. C.), the Shanghai Rising‐Star Program (No. 20QA1401700 to Y. P. W.), the Natural Science Foundation of Shanghai (No. 22ZR1414200 to Y. P. W.), the Young Elite Scientist Sponsorship Program by China Association for Science and Technology (No. 2018QNRC001 to Y. P. W.), the “Chenguang Program” of Shanghai Education Development Foundation and Shanghai Municipal Education Commission (No. 14CG15 to Y. P. W.), Shanghai Committee of Science and Technology (No. 15ZR1424500 to T. W.), and Shanghai Jiao Tong University School of Medicine (Nos. 18zxy004 and YCTSQN2021005 to T. W.; No. YG2021QN39 to Y. C.). Part of the figures was prepared with BioRender.
Wang T, Cao Y, Zhang H, et al. COVID‐19 metabolism: Mechanisms and therapeutic targets. MedComm. 2022;3:e157. 10.1002/mco2.157
Contributor Information
Tianshi Wang, Email: tianshi777@shsmu.edu.cn.
Quan Zheng, Email: 184277@shsmu.edu.cn.
Yi‐Ping Wang, Email: yiping_wang@fudan.edu.cn.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Mallah SI, Ghorab OK, Al‐Salmi S, et al. COVID‐19: breaking down a global health crisis. Ann Clin Microbiol Antimicrob. 2021;20(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou H, Yang J, Zhou C, et al. A Review of SARS‐CoV2: compared With SARS‐CoV and MERS‐CoV. Front Med (Lausanne). 2021;8:628370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Acuti Martellucci C, Flacco ME, Cappadona R, Bravi F, Mantovani L, Manzoli L. SARS‐CoV‐2 pandemic: an overview. Adv Biol Regul. 2020;77:100736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guerstein S, Romeo‐Aznar V, Dekel M, et al. The interplay between vaccination and social distancing strategies affects COVID19 population‐level outcomes. PLoS Comput Biol. 2021;17(8): e1009319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peng H, Ding C, Jiang L, et al. Discovery of potential anti‐SARS‐CoV‐2 drugs based on large‐scale screening in vitro and effect evaluation in vivo. Sci China Life Sci. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sahebnasagh A, Avan R, Saghafi F, et al. Pharmacological treatments of COVID‐19. Pharmacol Rep. 2020;72(6):1446‐1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miguez‐Rey E, Choi D, Kim S, Yoon S, Sandulescu O. Monoclonal antibody therapies in the management of SARS‐CoV‐2 infection. Expert Opin Investig Drugs. 2022;31(1):41‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brobst B, Borger J. Benefits and risks of administering monoclonal antibody therapy for coronavirus (COVID‐19). StatPearls. 2022. [PubMed] [Google Scholar]
- 9. Kokic G, Hillen HS, Tegunov D, et al. Mechanism of SARS‐CoV‐2 polymerase stalling by remdesivir. Nat Commun. 2021;12(1):279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hammond J, Leister‐Tebbe H, Gardner A, et al. Oral nirmatrelvir for high‐risk, nonhospitalized adults with Covid‐19. N Engl J Med. 2022;386(15):1397‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sacco MD, Hu Y, Gongora MV, et al. The P132H mutation in the main protease of Omicron SARS‐CoV‐2 decreases thermal stability without compromising catalysis or small‐molecule drug inhibition. Cell Res. 2022;32(5):498‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu T, Ma R, Dong W, et al. Off‐the‐shelf CAR natural killer cells secreting IL‐15 target spike in treating COVID‐19. Nat Commun. 2022;13(1):2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang D, Jiang A, Feng J, et al. The SARS‐CoV‐2 subgenome landscape and its novel regulatory features. Mol Cell. 2021;81(10):2135‐2147. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li J, Guo M, Tian X, et al. Virus‐host interactome and proteomic survey reveal potential virulence factors influencing SARS‐CoV‐2 pathogenesis. Med (N Y). 2021;2(1):99‐112. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nardacci R, Colavita F, Castilletti C, et al. Evidences for lipid involvement in SARS‐CoV‐2 cytopathogenesis. Cell Death Dis. 2021;12(3):263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ebrahimi KH, McCullagh JSO. A lipidomic view of SARS‐CoV‐2. Biosci Rep. 2021;41(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Du M, Cai G, Chen F, Christiani DC, Zhang Z, Wang M. Multiomics evaluation of gastrointestinal and other clinical characteristics of COVID‐19. Gastroenterology. 2020;158(8):2298‐2301. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas T, Stefanoni D, Reisz JA, et al. COVID‐19 infection alters kynurenine and fatty acid metabolism, correlating with IL‐6 levels and renal status. JCI Insight. 2020;5(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu EPK, Reinhold J, Yu H, et al. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arterioscler Thromb Vasc Biol. 2017;37(12):2322‐2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu D, Shu T, Yang X, et al. Plasma metabolomic and lipidomic alterations associated with COVID‐19. Natl Sci Rev. 2020;7(7):1157‐1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li JT, Li KY, Su Y, et al. Diet high in branched‐chain amino acid promotes PDAC development by USP1‐mediated BCAT2 stabilization. Natl Sci Rev. 2022;9(5):nwab212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma QX, Zhu WY, Lu XC, et al. BCAA‐BCKA axis regulates WAT browning through acetylation of PRDM16. Nat Metab. 2022;4(1):106‐122. [DOI] [PubMed] [Google Scholar]
- 23. Matsuyama T, Yoshinaga SK, Shibue K, Mak TW. Comorbidity‐associated glutamine deficiency is a predisposition to severe COVID‐19. Cell Death Differ. 2021;28(12):3199‐3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cuperlovic‐Culf M, Cunningham EL, Teimoorinia H, et al. Metabolomics and computational analysis of the role of monoamine oxidase activity in delirium and SARS‐COV‐2 infection. Sci Rep. 2021;11(1):10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Q, Zhang L, Shan H, et al. The immuno‐behavioural covariation associated with the treatment response to bumetanide in young children with autism spectrum disorder. Transl Psychiatry. 2022;12(1):228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Masoodi M, Peschka M, Schmiedel S, et al. Disturbed lipid and amino acid metabolisms in COVID‐19 patients. J Mol Med (Berl). 2022;100(4):555‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu J, Yuan Y, Chen YY, Xiong CF, Zhang Z, Feng YQ. Carboxylic submetabolome‐driven signature characterization of COVID‐19 asymptomatic infection. Talanta. 2022;239:123086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sproston NR, Ashworth JJ. Role of C‐reactive protein at sites of inflammation and infection. Front Immunol. 2018;9:754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sullivan KD, Galbraith MD, Kinning KT, et al. The COVIDome explorer researcher portal. Cell Rep. 2021;36(7):109527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. D'Alessandro A, Dzieciatkowska M, Nemkov T, Hansen KC. Red blood cell proteomics update: is there more to discover?. Blood Transfus. 2017;15(2):182‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thomas T, Stefanoni D, Dzieciatkowska M, et al. Evidence of structural protein damage and membrane lipid remodeling in red blood cells from COVID‐19 patients. J Proteome Res. 2020;19(11):4455‐4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang YP, Zhou LS, Zhao YZ, et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 2014;33(12):1304‐1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lamers MM, Beumer J, van der Vaart J, et al. SARS‐CoV‐2 productively infects human gut enterocytes. Science. 2020;369(6499):50‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steenblock C, Schwarz PEH, Ludwig B, et al. COVID‐19 and metabolic disease: mechanisms and clinical management. Lancet Diabetes Endocrinol. 2021;9(11):786‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Camargo SM, Singer D, Makrides V, et al. Tissue‐specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology. 2009;136(3):872‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science. 2020;367(6485):1444‐1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Obukhov AG, Stevens BR, Prasad R, et al. SARS‐CoV‐2 infections and ACE2: clinical outcomes linked with increased morbidity and mortality in individuals with diabetes. Diabetes. 2020;69(9):1875‐1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Penninger JM, Grant MB, Sung JJY. The role of angiotensin converting enzyme 2 in modulating gut microbiota, intestinal inflammation, and coronavirus infection. Gastroenterology. 2021;160(1):39‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu Z, Yang Z, Wang Y, et al. Recent advance of ACE2 and microbiota dysfunction in COVID‐19 pathogenesis. Heliyon. 2021;7(7):e07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ren Z, Wang H, Cui G, et al. Alterations in the human oral and gut microbiomes and lipidomics in COVID‐19. Gut. 2021;70(7):1253‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang F, Wan Y, Zuo T, et al. Prolonged impairment of short‐chain fatty acid and L‐isoleucine biosynthesis in gut microbiome in patients with COVID‐19. Gastroenterology. 2022;162(2):548‐561. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gu S, Chen Y, Wu Z, et al. Alterations of the gut microbiota in patients with coronavirus disease 2019 or H1N1 influenza. Clin Infect Dis. 2020;71(10):2669‐2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zuo T, Zhang F, Lui GCY, et al. Alterations in gut microbiota of patients with COVID‐19 during time of hospitalization. Gastroenterology. 2020;159(3):944‐955. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yeoh YK, Zuo T, Lui GC, et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID‐19. Gut. 2021;70(4):698‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xiao F, Tang M, Zheng X, Liu Y, Li X, Shan H. Evidence for gastrointestinal infection of SARS‐CoV‐2. Gastroenterology. 2020;158(6):1831‐1833. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Han C, Duan C, Zhang S, et al. Digestive symptoms in COVID‐19 patients with mild disease severity: clinical presentation, stool viral RNA testing, and outcomes. Am J Gastroenterol. 2020;115(6):916‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen Y, Gu S, Chen Y, et al. Six‐month follow‐up of gut microbiota richness in patients with COVID‐19. Gut. 2022;71(1):222‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu Q, Mak JWY, Su Q, et al. Gut microbiota dynamics in a prospective cohort of patients with post‐acute COVID‐19 syndrome. Gut. 2022;71(3):544‐552. [DOI] [PubMed] [Google Scholar]
- 49. Nalbandian A, Sehgal K, Gupta A, et al. Post‐acute COVID‐19 syndrome. Nat Med. 2021;27(4):601‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang C, Huang L, Wang Y, et al. 6‐month consequences of COVID‐19 in patients discharged from hospital: a cohort study. Lancet. 2021;397(10270):220‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barlass U, Wiliams B, Dhana K, et al. Marked elevation of lipase in COVID‐19 disease: a cohort study. Clin Transl Gastroenterol. 2020;11(7):e00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. de‐Madaria E, Siau K, Cardenas‐Jaen K. Increased amylase and lipase in patients with COVID‐19 pneumonia: don't blame the pancreas just yet!. Gastroenterology. 2021;160(5):1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang F, Wang H, Fan J, Zhang Y, Wang H, Zhao Q. Pancreatic injury patterns in patients with coronavirus disease 19 pneumonia. Gastroenterology. 2020;159(1):367‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guo W, Li M, Dong Y, et al. Diabetes is a risk factor for the progression and prognosis of COVID‐19. Diabetes Metab Res Rev. 2020:e3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Montefusco L, Ben Nasr M, D'Addio F, et al. Acute and long‐term disruption of glycometabolic control after SARS‐CoV‐2 infection. Nat Metab. 2021;3(6):774‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wu CT, Lidsky PV, Xiao Y, et al. SARS‐CoV‐2 infects human pancreatic beta cells and elicits beta cell impairment. Cell Metab. 2021;33(8):1565‐1576. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ji N, Zhang M, Ren L, et al. SARS‐CoV‐2 in the pancreas and the impaired islet function in COVID‐19 patients. Emerg Microbes Infect. 2022;11(1):1115‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xu L, Liu J, Lu M, Yang D, Zheng X. Liver injury during highly pathogenic human coronavirus infections. Liver Int. 2020;40(5):998‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Morgan K, Samuel K, Vandeputte M, Hayes PC, Plevris JN. SARS‐CoV‐2 infection and the liver. Pathogens. 2020;9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Delorey TM, Ziegler CGK, Heimberg G, et al. COVID‐19 tissue atlases reveal SARS‐CoV‐2 pathology and cellular targets. Nature. 2021;595(7865):107‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shoemark DK, Colenso CK, Toelzer C, et al. Molecular simulations suggest vitamins, retinoids and steroids as ligands of the free fatty acid pocket of the SARS‐CoV‐2 Spike protein*. Angew Chem Int Ed Engl. 2021;60(13):7098‐7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gordon DE, Hiatt J, Bouhaddou M, et al. Comparative host‐coronavirus protein interaction networks reveal pan‐viral disease mechanisms. Science. 2020;370(6521). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wei C, Wan L, Yan Q, et al. HDL‐scavenger receptor B type 1 facilitates SARS‐CoV‐2 entry. Nat Metab. 2020;2(12):1391‐1400. [DOI] [PubMed] [Google Scholar]
- 65. Diaz LA, Idalsoaga F, Cannistra M, et al. High prevalence of hepatic steatosis and vascular thrombosis in COVID‐19: a systematic review and meta‐analysis of autopsy data. World journal of gastroenterology. 2020;26(48):7693‐7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nie X, Qian L, Sun R, et al. Multi‐organ proteomic landscape of COVID‐19 autopsies. Cell. 2021;184(3):775‐791. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bangash MN, Patel JM, Parekh D, et al. SARS‐CoV‐2: is the liver merely a bystander to severe disease?. J Hepatol. 2020;73(4):995‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wanner N, Andrieux G, Badia IMP, et al. Molecular consequences of SARS‐CoV‐2 liver tropism. Nat Metab. 2022;4(3):310‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cornberg M, Buti M, Eberhardt CS, Grossi PA, Shouval D. EASL position paper on the use of COVID‐19 vaccines in patients with chronic liver diseases, hepatobiliary cancer and liver transplant recipients. J Hepatol. 2021;74(4):944‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bai L, Li H. Innate immune regulatory networks in hepatic lipid metabolism. J Mol Med (Berl). 2019;97(5):593‐604. [DOI] [PubMed] [Google Scholar]
- 71. Tilg H, Adolph TE, Dudek M, Knolle P. Non‐alcoholic fatty liver disease: the interplay between metabolism, microbes and immunity. Nat Metab. 2021;3(12):1596‐1607. [DOI] [PubMed] [Google Scholar]
- 72. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus‐infected pneumonia in Wuhan, China. Jama. 2020;323(11):1061‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Smith M, Honce R, Schultz‐Cherry S. Metabolic syndrome and viral pathogenesis: lessons from influenza and coronaviruses. J Virol. 2020;94(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Reiterer M, Rajan M, Gomez‐Banoy N, et al. Hyperglycemia in acute COVID‐19 is characterized by insulin resistance and adipose tissue infectivity by SARS‐CoV‐2. Cell Metab. 2021;33(11):2174‐2188. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gupte M, Boustany‐Kari CM, Bharadwaj K, et al. ACE2 is expressed in mouse adipocytes and regulated by a high‐fat diet. Am J Physiol Regul Integr Comp Physiol. 2008;295(3):R781‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zickler M, Stanelle‐Bertram S, Ehret S, et al. Replication of SARS‐CoV‐2 in adipose tissue determines organ and systemic lipid metabolism in hamsters and humans. Cell Metab. 2022;34(1):1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cihan M, Dogan O, Ceran Serdar C, Altuncekic Yildirim A, Kurt C, Serdar MA. Kynurenine pathway in Coronavirus disease (COVID‐19): potential role in prognosis. J Clin Lab Anal. 2022;36(3):e24257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cai Y, Kim DJ, Takahashi T, et al. Kynurenic acid may underlie sex‐specific immune responses to COVID‐19. Sci Signal. 2021;14(690). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Thompson EA, Cascino K, Ordonez AA, et al. Metabolic programs define dysfunctional immune responses in severe COVID‐19 patients. Cell Rep. 2021;34(11):108863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ryu S, Shchukina I, Youm YH, et al. Ketogenic diet restrains aging‐induced exacerbation of coronavirus infection in mice. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sturley SL, Rajakumar T, Hammond N, et al. Potential COVID‐19 therapeutics from a rare disease: weaponizing lipid dysregulation to combat viral infectivity. J Lipid Res. 2020;61(7):972‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Inoue Y, Tanaka N, Tanaka Y, et al. Clathrin‐dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J Virol. 2007;81(16):8722‐8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wang H, Yang P, Liu K, et al. SARS coronavirus entry into host cells through a novel clathrin‐ and caveolae‐independent endocytic pathway. Cell Res. 2008;18(2):290‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double‐membrane vesicles. mBio. 2013;4(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Drucker DJ. Diabetes, obesity, metabolism, and SARS‐CoV‐2 infection: the end of the beginning. Cell Metab. 2021;33(3):479‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92(4):418‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ujike M, Taguchi F. Incorporation of spike and membrane glycoproteins into coronavirus virions. Viruses. 2015;7(4):1700‐1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang S, Li W, Hui H, et al. Cholesterol 25‐Hydroxylase inhibits SARS‐CoV‐2 and other coronaviruses by depleting membrane cholesterol. EMBO J. 2020;39(21):e106057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Diaz‐Arocutipa C, Melgar‐Talavera B, Alvarado‐Yarasca A, et al. Statins reduce mortality in patients with COVID‐19: an updated meta‐analysis of 147 824 patients. Int J Infect Dis. 2021;110:374‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sviridov D, Miller YI, Ballout RA, Remaley AT, Bukrinsky M. Targeting lipid rafts‐a potential therapy for COVID‐19. Front Immunol. 2020;11:574508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Carter GC, Bernstone L, Sangani D, Bee JW, Harder T, James W. HIV entry in macrophages is dependent on intact lipid rafts. Virology. 2009;386(1):192‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yang Y, Hsu JM, Sun L, et al. Palmitoylation stabilizes PD‐L1 to promote breast tumor growth. Cell Res. 2019;29(1):83‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pei X, Li KY, Shen Y, et al. Palmitoylation of MDH2 by ZDHHC18 activates mitochondrial respiration and accelerates ovarian cancer growth. Sci China Life Sci. 2022. [DOI] [PubMed] [Google Scholar]
- 96. Shulla A, Heald‐Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. 2011;85(2):873‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kim D, Kim S, Park J, et al. A high‐resolution temporal atlas of the SARS‐CoV‐2 translatome and transcriptome. Nat Commun. 2021;12(1):5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Tabata K, Prasad V, Paul D, et al. Convergent use of phosphatidic acid for hepatitis C virus and SARS‐CoV‐2 replication organelle formation. Nat Commun. 2021;12(1):7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dias SSG, Soares VC, Ferreira AC, et al. Lipid droplets fuel SARS‐CoV‐2 replication and production of inflammatory mediators. PLoS Pathog. 2020;16(12):e1009127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Casari I, Manfredi M, Metharom P, Falasca M. Dissecting lipid metabolism alterations in SARS‐CoV‐2. Prog Lipid Res. 2021;82:101092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Mesquita FS, Abrami L, Sergeeva O, et al. S‐acylation controls SARS‐CoV‐2 membrane lipid organization and enhances infectivity. Dev Cell. 2021;56(20):2790‐2807. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gong Y, Qin S, Dai L, Tian Z. The glycosylation in SARS‐CoV‐2 and its receptor ACE2. Signal Transduct Targeted Ther. 2021;6(1):396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sartore G, Bassani D, Ragazzi E, Traldi P, Lapolla A, Moro S. In silico evaluation of the interaction between ACE2 and SARS‐CoV‐2 Spike protein in a hyperglycemic environment. Sci Rep. 2021;11(1):22860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Huang HC, Lai YJ, Liao CC, et al. Targeting conserved N‐glycosylation blocks SARS‐CoV‐2 variant infection in vitro. EBioMedicine. 2021;74:103712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Port JR, Adney DR, Schwarz B, et al. High‐fat high‐sugar diet‐induced changes in the lipid metabolism are associated with mildly increased COVID‐19 severity and delayed recovery in the Syrian Hamster. Viruses. 2021;13(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cai T, Yu Z, Wang Z, Liang C, Richard S. Arginine methylation of SARS‐Cov‐2 nucleocapsid protein regulates RNA binding, its ability to suppress stress granule formation, and viral replication. J Biol Chem. 2021;297(1):100821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Huang W, Li N, Zhang Y, Wang X, Yin M, Lei QY. AHCYL1 senses SAH to inhibit autophagy through interaction with PIK3C3 in an MTORC1‐independent manner. Autophagy. 2022;18(2):309‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Niyogi K, Hildreth JE. Characterization of new syncytium‐inhibiting monoclonal antibodies implicates lipid rafts in human T‐cell leukemia virus type 1 syncytium formation. J Virol. 2001;75(16):7351‐7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Li GM, Li YG, Yamate M, Li SM, Ikuta K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome‐coronavirus life cycle. Microbes Infect. 2007;9(1):96‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Srinivasan S, Cui H, Gao Z, et al. Structural genomics of SARS‐CoV‐2 indicates evolutionary conserved functional regions of viral proteins. Viruses. 2020;12(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang YP, Zhou W, Wang J, et al. Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol Cell. 2016;64(4):673‐687. [DOI] [PubMed] [Google Scholar]
- 112. Abu‐Eid R, Ward FJ. Targeting the PI3K/Akt/mTOR pathway: a therapeutic strategy in COVID‐19 patients. Immunol Lett. 2021;240:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Shang J, Smith MR, Anmangandla A, Lin H. NAD+‐consuming enzymes in immune defense against viral infection. Biochem J. 2021;478(23):4071‐4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Fehr AR, Singh SA, Kerr CM, Mukai S, Higashi H, Aikawa M. The impact of PARPs and ADP‐ribosylation on inflammation and host‐pathogen interactions. Genes Dev. 2020;34(5‐6):341‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Grunewald ME, Chen Y, Kuny C, et al. The coronavirus macrodomain is required to prevent PARP‐mediated inhibition of virus replication and enhancement of IFN expression. PLoS Pathog. 2019;15(5):e1007756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Heer CD, Sanderson DJ, Voth LS, et al. Coronavirus infection and PARP expression dysregulate the NAD metabolome: an actionable component of innate immunity. J Biol Chem. 2020;295(52):17986‐17996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ziv O, Price J, Shalamova L, et al. The short‐ and long‐range RNA‐RNA interactome of SARS‐CoV‐2. Mol Cell. 2020;80(6):1067‐1077. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yang SL, DeFalco L, Anderson DE, et al. Comprehensive mapping of SARS‐CoV‐2 interactions in vivo reveals functional virus‐host interactions. Nat Commun. 2021;12(1):5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Schmidt N, Lareau CA, Keshishian H, et al. The SARS‐CoV‐2 RNA‐protein interactome in infected human cells. Nat Microbiol. 2021;6(3):339‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kamel W, Noerenberg M, Cerikan B, et al. Global analysis of protein‐RNA interactions in SARS‐CoV‐2‐infected cells reveals key regulators of infection. Mol Cell. 2021;81(13):2851‐2867. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Banerjee AK, Blanco MR, Bruce EA, et al. SARS‐CoV‐2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell. 2020;183(5):1325‐1339. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chen Z, Wang C, Feng X, et al. Interactomes of SARS‐CoV‐2 and human coronaviruses reveal host factors potentially affecting pathogenesis. EMBO J. 2021;40(17):e107776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wyatt E, Wu R, Rabeh W, Park HW, Ghanefar M, Ardehali H. Regulation and cytoprotective role of hexokinase III. PLoS One. 2010;5(11):e13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bu P, Chen KY, Xiang K, et al. Aldolase B‐mediated fructose metabolism drives metabolic reprogramming of colon cancer liver metastasis. Cell Metab. 2018;27(6):1249‐1262. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wu ST, Liu B, Ai ZZ, et al. Esculetin inhibits cancer cell glycolysis by binding tumor PGK2, GPD2, and GPI. Front Pharmacol. 2020;11:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Luzzatto L, Ally M, Notaro R. Glucose‐6‐phosphate dehydrogenase deficiency. Blood. 2020;136(11):1225‐1240. [DOI] [PubMed] [Google Scholar]
- 128. Yang HC, Ma TH, Tjong WY, Stern A, Chiu DT. G6PD deficiency, redox homeostasis, and viral infections: implications for SARS‐CoV‐2 (COVID‐19). Free Radic Res. 2021;55(4):364‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Jamerson BD, Haryadi TH, Bohannon A. Glucose‐6‐Phosphate dehydrogenase deficiency: an actionable risk factor for patients with COVID‐19?. Arch Med Res. 2020;51(7):743‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Garcia AA, Koperniku A, Ferreira JCB, Mochly‐Rosen D. Treatment strategies for glucose‐6‐phosphate dehydrogenase deficiency: past and future perspectives. Trends Pharmacol Sci. 2021;42(10):829‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Guo Y, Wu J, Wang M, et al. The metabolite saccharopine impairs neuronal development by inhibiting the neurotrophic function of glucose‐6‐phosphate isomerase. J Neurosci. 2022;42(13):2631‐2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang Y, Gao J, Hu S, et al. SLC25A21 suppresses cell growth in bladder cancer via an oxidative stress‐mediated mechanism. Front Oncol. 2021;11:682710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Yoo HC, Park SJ, Nam M, et al. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 2020;31(2):267‐283. e12. [DOI] [PubMed] [Google Scholar]
- 134. Wang B, Svetlov V, Wolf YI, Koonin EV, Nudler E, Artsimovitch I. Allosteric activation of SARS‐CoV‐2 RNA‐dependent RNA polymerase by remdesivir triphosphate and other phosphorylated nucleotides. mBio. 2021;12(3):e0142321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Xu SN, Wang TS, Li X, Wang YP. SIRT2 activates G6PD to enhance NADPH production and promote leukaemia cell proliferation. Sci Rep. 2016;6:32734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sivanand S, Vander Heiden MG. Emerging roles for branched‐chain amino acid metabolism in cancer. Cancer Cell. 2020;37(2):147‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Bera SC, Seifert M, Kirchdoerfer RN, et al. The nucleotide addition cycle of the SARS‐CoV‐2 polymerase. Cell Rep. 2021;36(9):109650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ma APY, Yeung CLS, Tey SK, et al. Suppression of ACADM‐mediated fatty acid oxidation promotes hepatocellular carcinoma via aberrant CAV1/SREBP1 signaling. Cancer Res. 2021;81(13):3679‐3692. [DOI] [PubMed] [Google Scholar]
- 139. Batchuluun B, Pinkosky SL, Steinberg GR. Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat Rev Drug Discov. 2022;21(4):283‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Jiang N, Xie B, Xiao W, et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47‐mediated immune evasion. Nat Commun. 2022;13(1):1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Quan J, Bode AM, Luo X. ACSL family: the regulatory mechanisms and therapeutic implications in cancer. Eur J Pharmacol. 2021;909:174397. [DOI] [PubMed] [Google Scholar]
- 142. Santos‐Beneit F, Raskevicius V, Skeberdis VA, Bordel S. A metabolic modeling approach reveals promising therapeutic targets and antiviral drugs to combat COVID‐19. Sci Rep. 2021;11(1):11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Yaneske E, Zampieri G, Bertoldi L, Benvenuto G, Angione C. Genome‐scale metabolic modelling of SARS‐CoV‐2 in cancer cells reveals an increased shift to glycolytic energy production. FEBS Lett. 2021;595(18):2350‐2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Yuan S, Yan B, Cao J, et al. SARS‐CoV‐2 exploits host DGAT and ADRP for efficient replication. Cell Discov. 2021;7(1):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Bitter EE, Townsend MH, Erickson R, Allen C, O'Neill KL. Thymidine kinase 1 through the ages: a comprehensive review. Cell Biosci. 2020;10(1):138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Burrell AL, Kollman JM. IMPDH dysregulation in disease: a mini review. Biochem Soc Trans. 2022;50(1):71‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wang J, Zhu ZH, Yang HB, et al. Cullin 3 targets methionine adenosyltransferase IIalpha for ubiquitylation‐mediated degradation and regulates colorectal cancer cell proliferation. FEBS J. 2016;283(13):2390‐2402. [DOI] [PubMed] [Google Scholar]
- 148. Zhang Y, Guo R, Kim SH, et al. SARS‐CoV‐2 hijacks folate and one‐carbon metabolism for viral replication. Nat Commun. 2021;12(1):1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Rabu C, McIntosh R, Jurasova Z, Durrant L. Glycans as targets for therapeutic antitumor antibodies. Future Oncol. 2012;8(8):943‐960. [DOI] [PubMed] [Google Scholar]
- 150. Guindolet D, Woodward AM, Gabison EE, Argueso P. Glycogene expression profile of human limbal epithelial cells with distinct clonogenic potential. Cells. 2022;11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhong A, Chen T, Xing Y, Pan X, Shi M. FUCA2 is a prognostic biomarker and correlated with an immunosuppressive microenvironment in pan‐cancer. Front Immunol. 2021;12:758648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Espinoza JA, Gonzalez PA, Kalergis AM. Modulation of antiviral immunity by heme oxygenase‐1. Am J Pathol. 2017;187(3):487‐493. [DOI] [PubMed] [Google Scholar]
- 153. Huang H, Falgout B, Takeda K, Yamada KM, Dhawan S. Nrf2‐dependent induction of innate host defense via heme oxygenase‐1 inhibits Zika virus replication. Virology. 2017;503:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. El Kalamouni C, Frumence E, Bos S, et al. Subversion of the heme oxygenase‐1 antiviral activity by Zika virus. Viruses. 2018;11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Espinoza JA, Leon MA, Cespedes PF, et al. Heme oxygenase‐1 modulates human respiratory syncytial virus replication and lung pathogenesis during infection. J Immunol. 2017;199(1):212‐223. [DOI] [PubMed] [Google Scholar]
- 156. Hooper PL. COVID‐19 and heme oxygenase: novel insight into the disease and potential therapies. Cell Stress Chaperones. 2020;25(5):707‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Singh D, Wasan H, Reeta KH. Heme oxygenase‐1 modulation: a potential therapeutic target for COVID‐19 and associated complications. Free Radic Biol Med. 2020;161:263‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Sohrabi Y, Reinecke H, Godfrey R. Altered cholesterol and lipid synthesis mediates hyperinflammation in COVID‐19. Trends Endocrinol Metab. 2021;32(3):132‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Silvagno F, Vernone A, Pescarmona GP. The role of glutathione in protecting against the severe inflammatory response triggered by COVID‐19. Antioxidants (Basel). 2020;9(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II‐mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97(8):1916‐1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lan J, Ge J, Yu J, et al. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215‐220. [DOI] [PubMed] [Google Scholar]
- 162. Suhail S, Zajac J, Fossum C, et al. Role of oxidative stress on SARS‐CoV (SARS) and SARS‐CoV‐2 (COVID‐19) infection: a review. Protein J. 2020;39(6):644‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Paul BD, Lemle MD, Komaroff AL, Snyder SH. Redox imbalance links COVID‐19 and myalgic encephalomyelitis/chronic fatigue syndrome. Proc Natl Acad Sci USA. 2021;118(34). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kaundal RK, Kalvala AK, Kumar A. Neurological implications of COVID‐19: role of redox imbalance and mitochondrial dysfunction. Mol Neurobiol. 2021;58(9):4575‐4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Wang ZH, Chen L, Li W, Chen L, Wang YP. Mitochondria transfer and transplantation in human health and diseases. Mitochondrion. 2022;65:80‐87. [DOI] [PubMed] [Google Scholar]
- 166. Wang Y, Xiao M, Chen X, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. 2015;57(4):662‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Wang YP, Sharda A, Xu SN, et al. Malic enzyme 2 connects the Krebs cycle intermediate fumarate to mitochondrial biogenesis. Cell Metab. 2021;33(5):1027‐1041. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Olagnier D, Farahani E, Thyrsted J, et al. SARS‐CoV2‐mediated suppression of NRF2‐signaling reveals potent antiviral and anti‐inflammatory activity of 4‐octyl‐itaconate and dimethyl fumarate. Nat Commun. 2020;11(1):4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Yan WW, Liang YL, Zhang QX, et al. Arginine methylation of SIRT7 couples glucose sensing with mitochondria biogenesis. EMBO Rep. 2018;19(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Xu J, Guo H, Xing Z, et al. Mild oxidative stress reduces NRF2 SUMOylation to promote Kras/Lkb1/Keap1 mutant lung adenocarcinoma cell migration and invasion. Oxid Med Cell Longev. 2020;2020:6240125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Guo H, Xu J, Zheng Q, et al. NRF2 SUMOylation promotes de novo serine synthesis and maintains HCC tumorigenesis. Cancer Lett. 2019;466:39‐48. [DOI] [PubMed] [Google Scholar]
- 172. Lee SR, Roh JY, Ryu J, Shin HJ, Hong EJ. Activation of TCA cycle restrains virus‐metabolic hijacking and viral replication in mouse hepatitis virus‐infected cells. Cell & bioscience. 2022;12(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Qian L, Zhang F, Yin M, Lei Q. Cancer metabolism and dietary interventions. Cancer Biol Med. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Shang C, Liu Z, Zhu Y, et al. SARS‐CoV‐2 causes mitochondrial dysfunction and mitophagy impairment. Frontiers in microbiology. 2021;12:780768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Duan X, Tang X, Nair MS, et al. An airway organoid‐based screen identifies a role for the HIF1alpha‐glycolysis axis in SARS‐CoV‐2 infection. Cell Rep. 2021;37(6):109920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Codo AC, Davanzo GG, Monteiro LB, et al. Elevated glucose levels favor SARS‐CoV‐2 infection and monocyte response through a HIF‐1alpha/glycolysis‐dependent axis. Cell Metab. 2020;32(3):498‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. He J, Shangguan X, Zhou W, et al. Glucose limitation activates AMPK coupled SENP1‐Sirt3 signalling in mitochondria for T cell memory development. Nat Commun. 2021;12(1):4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Siska PJ, Decking SM, Babl N, et al. Metabolic imbalance of T cells in COVID‐19 is hallmarked by basigin and mitigated by dexamethasone. The Journal of clinical investigation. 2021;131(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Medini H, Zirman A, Mishmar D. Immune system cells from COVID‐19 patients display compromised mitochondrial‐nuclear expression co‐regulation and rewiring toward glycolysis. iScience. 2021;24(12):103471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Miller B, Silverstein A, Flores M, et al. Host mitochondrial transcriptome response to SARS‐CoV‐2 in multiple cell models and clinical samples. Sci Rep. 2021;11(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Chu H, Peng L, Hu L, et al. Liver histopathological analysis of 24 postmortem findings of patients with COVID‐19 in China. Front Med (Lausanne). 2021;8:749318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Gatti P, Ilamathi HS, Todkar K, Germain M. Mitochondria targeted viral replication and survival strategies‐prospective on SARS‐CoV‐2. Frontiers in pharmacology. 2020;11:578599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ramachandran K, Maity S, Muthukumar AR, et al. SARS‐CoV‐2 infection enhances mitochondrial PTP complex activity to perturb cardiac energetics. iScience. 2022;25(1):103722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Guo L. Mitochondrial ATP synthase inhibitory factor 1 interacts with the p53‐cyclophilin D complex and promotes opening of the permeability transition pore. J Biol Chem. 2022;298(5):101858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Wo L, Zhang X, Ma C, et al. LncRNA HABON promoted liver cancer cells survival under hypoxia by inhibiting mPTP opening. Cell Death Discov. 2022;8(1):171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nature reviews Molecular cell biology. 2012;13(12):780‐788. [DOI] [PubMed] [Google Scholar]
- 187. He J, Cheng J, Wang T. SUMOylation‐mediated response to mitochondrial stress. International journal of molecular sciences. 2020;21(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Tan Y, Tang F. SARS‐CoV‐2‐mediated immune system activation and potential application in immunotherapy. Medicinal research reviews. 2021;41(2):1167‐1194. [DOI] [PubMed] [Google Scholar]
- 189. Simpson DS, Pang J, Weir A, et al. Interferon‐gamma primes macrophages for pathogen ligand‐induced killing via a caspase‐8 and mitochondrial cell death pathway. Immunity. 2022;55(3):423‐441. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Xiao M, Bian Q, Lao Y, et al. SENP3 loss promotes M2 macrophage polarization and breast cancer progression. Mol Oncol. 2022;16(4):1026‐1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Peman J, Ruiz‐Gaitan A, Garcia‐Vidal C, et al. Fungal co‐infection in COVID‐19 patients: should we be concerned?. Revista iberoamericana de micologia. 2020;37(2):41‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Chi Z, Chen S, Xu T, et al. Histone deacetylase 3 couples mitochondria to drive IL‐1beta‐dependent inflammation by configuring fatty acid oxidation. Mol Cell. 2020;80(1):43‐58. e7. [DOI] [PubMed] [Google Scholar]
- 193. Zhou H, Wang H, Yu M, et al. IL‐1 induces mitochondrial translocation of IRAK2 to suppress oxidative metabolism in adipocytes. Nature immunology. 2020;21(10):1219‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Zhou W, Hu G, He J, et al. SENP1‐Sirt3 signaling promotes alpha‐ketoglutarate production during M2 macrophage polarization. Cell Rep. 2022;39(2):110660. [DOI] [PubMed] [Google Scholar]
- 195. Fung TS, Liu DX. Post‐translational modifications of coronavirus proteins: roles and function. Future Virol. 2018;13(6):405‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Chen F, Yan H, Guo C, et al. Assessment of SENP3‐interacting proteins in hepatocytes treated with diethylnitrosamine by BioID assay. Acta Biochim Biophys Sin (Shanghai). 2021;53(9):1237‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Qian X, Li X, Shi Z, et al. PTEN suppresses glycolysis by dephosphorylating and inhibiting autophosphorylated PGK1. Mol Cell. 2019;76(3):516‐527. e7. [DOI] [PubMed] [Google Scholar]
- 198. Tarazona OA, Pourquie O. Exploring the influence of cell metabolism on cell fate through protein post‐translational modifications. Dev Cell. 2020;54(2):282‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Sanchez EL, Lagunoff M. Viral activation of cellular metabolism. Virology. 2015;479‐480:609‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Thaker SK, Ch'ng J, Christofk HR. Viral hijacking of cellular metabolism. BMC Biol. 2019;17(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Mullen PJ, Garcia G Jr, Purkayastha A, et al. SARS‐CoV‐2 infection rewires host cell metabolism and is potentially susceptible to mTORC1 inhibition. Nat Commun. 2021;12(1):1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Wang YP, Li JT, Qu J, Yin M, Lei QY. Metabolite sensing and signaling in cancer. J Biol Chem. 2020;295(33):11938‐11946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Fitzgerald KA, McWhirter SM, Faia KL, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4(5):491‐496. [DOI] [PubMed] [Google Scholar]
- 204. Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential requirement for TANK‐binding kinase‐1 in type I interferon responses to toll‐like receptor activation and viral infection. J Exp Med. 2004;199(12):1651‐1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Pomerantz JL, Baltimore D. NF‐kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK‐related kinase. EMBO J. 1999;18(23):6694‐6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Kawai T, Akira S. Signaling to NF‐kappaB by Toll‐like receptors. Trends Mol Med. 2007;13(11):460‐469. [DOI] [PubMed] [Google Scholar]
- 207. Sui C, Xiao T, Zhang S, et al. SARS‐CoV‐2 NSP13 inhibits type I IFN production by degradation of TBK1 via p62‐dependent selective autophagy. J Immunol. 2022;208(3):753‐761. [DOI] [PubMed] [Google Scholar]
- 208. Reilly SM, Chiang SH, Decker SJ, et al. An inhibitor of the protein kinases TBK1 and IKK‐varepsilon improves obesity‐related metabolic dysfunctions in mice. Nat Med. 2013;19(3):313‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Zhao P, Wong KI, Sun X, et al. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. 2018;172(4):731‐743. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Claussnitzer M, Cho JH, Collins R, et al. A brief history of human disease genetics. Nature. 2020;577(7789):179‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Ballout RA, Sviridov D, Bukrinsky MI, Remaley AT. The lysosome: a potential juncture between SARS‐CoV‐2 infectivity and Niemann‐Pick disease type C, with therapeutic implications. FASEB J. 2020;34(6):7253‐7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Herbert AS, Davidson C, Kuehne AI, et al. Niemann‐pick C1 is essential for ebolavirus replication and pathogenesis in vivo. mBio. 2015;6(3):e00565‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Prabhakara C, Godbole R, Sil P, et al. Strategies to target SARS‐CoV‐2 entry and infection using dual mechanisms of inhibition by acidification inhibitors. PLoS Pathog. 2021;17(7):e1009706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Takano T, Endoh M, Fukatsu H, Sakurada H, Doki T, Hohdatsu T. The cholesterol transport inhibitor U18666A inhibits type I feline coronavirus infection. Antiviral Res. 2017;145:96‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Lu F, Liang Q, Abi‐Mosleh L, et al. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nat Commun. 2020;11(1):1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Zhang X, Chen W, Li P, et al. Agonist‐specific voltage‐dependent gating of lysosomal two‐pore Na(+) channels. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Yao Y, Ye F, Li K, et al. Genome and epigenome editing identify CCR9 and SLC6A20 as target genes at the 3p21.31 locus associated with severe COVID‐19. Signal Transduct Target Ther. 2021;6(1):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Wu KS, Lin PC, Chen YS, Pan TC, Tang PL. The use of statins was associated with reduced COVID‐19 mortality: a systematic review and meta‐analysis. Ann Med. 2021;53(1):874‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Scheen AJ. Statins and clinical outcomes with COVID‐19: meta‐analyses of observational studies. Diabetes Metab. 2021;47(6):101220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Williams CG, Jureka AS, Silvas JA, et al. Inhibitors of VPS34 and fatty‐acid metabolism suppress SARS‐CoV‐2 replication. Cell Rep. 2021;36(5):109479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Goc A, Niedzwiecki A, Rath M. Polyunsaturated omega‐3 fatty acids inhibit ACE2‐controlled SARS‐CoV‐2 binding and cellular entry. Sci Rep. 2021;11(1):5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Khunti K, Knighton P, Zaccardi F, et al. Prescription of glucose‐lowering therapies and risk of COVID‐19 mortality in people with type 2 diabetes: a nationwide observational study in England. Lancet Diabetes Endocrinol. 2021;9(5):293‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Scheen AJ. Metformin and COVID‐19: from cellular mechanisms to reduced mortality. Diabetes Metab. 2020;46(6):423‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Li B, Clohisey SM, Chia BS, et al. Genome‐wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat Commun. 2020;11(1):164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Wei J, Alfajaro MM, DeWeirdt PC, et al. Genome‐wide CRISPR screens reveal host factors critical for SARS‐CoV‐2 infection. Cell. 2021;184(1):76‐91. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Williamson BN, Feldmann F, Schwarz B, et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS‐CoV‐2. Nature. 2020;585(7824):273‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Cusinato J, Cau Y, Calvani AM, Mori M. Repurposing drugs for the management of COVID‐19. Expert opinion on therapeutic patents. 2021;31(4):295‐307. [DOI] [PubMed] [Google Scholar]
- 229. Rodrigues L, Bento Cunha R, Vassilevskaia T, Viveiros M, Cunha C. Drug repurposing for COVID‐19: a review and a novel strategy to identify new targets and potential drug candidates. Molecules. 2022;27(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Yin W, Mao C, Luan X, et al. Structural basis for inhibition of the RNA‐dependent RNA polymerase from SARS‐CoV‐2 by remdesivir. Science. 2020;368(6498):1499‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Fajgenbaum DC, Rader DJ. Teaching old drugs new tricks: statins for COVID‐19?. Cell Metab. 2020;32(2):145‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Martinez‐Outschoorn UE, Peiris‐Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14(1):11‐31. [DOI] [PubMed] [Google Scholar]
- 233. Mullard A. FDA approves first‐in‐class cancer metabolism drug. Nat Rev Drug Discov. 2017;16(9):593. [DOI] [PubMed] [Google Scholar]
- 234. Dragelj J, Mroginski MA, Ebrahimi KH. Hidden in plain sight: natural products of commensal microbiota as an environmental selection pressure for the rise of new variants of SARS‐CoV‐2. Chembiochem. 2021;22(20):2946‐2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Fonnesu R, Thunuguntla V, Veeramachaneni GK, et al. Palmitoylethanolamide (PEA) inhibits SARS‐CoV‐2 entry by interacting with S protein and ACE‐2 receptor. Viruses. 2022;14(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Kalil AC, Patterson TF, Mehta AK, et al. Baricitinib plus remdesivir for hospitalized adults with Covid‐19. N Engl J Med. 2021;384(9):795‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Feher M, Joy M, Munro N, Hinton W, Williams J, de Lusignan S. Fenofibrate as a COVID‐19 modifying drug: laboratory success versus real‐world reality. Atherosclerosis. 2021;339:55‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Stakisaitis D, Kapocius L, Valanciute A, et al. SARS‐CoV‐2 infection, sex‐related differences, and a possible personalized treatment approach with valproic acid: a review. Biomedicines. 2022;10(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti‐SARS drugs. Science. 2003;300(5626):1763‐1767. [DOI] [PubMed] [Google Scholar]
- 240. Drayman N, DeMarco JK, Jones KA, et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS‐CoV‐2. Science. 2021;373(6557):931‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Zhang F, Wang D, Li J, et al. Deacetylation of MTHFD2 by SIRT4 senses stress signal to inhibit cancer cell growth by remodeling folate metabolism. J Mol Cell Biol. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Gowda P, Patrick S, Joshi SD, Kumawat RK, Sen E. Repurposing methotrexate in dampening SARS‐CoV2‐S1‐mediated IL6 expression: lessons learnt from lung cancer. Inflammation. 2022;45(1):172‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Ayres JS. A metabolic handbook for the COVID‐19 pandemic. Nat Metab. 2020;2(7):572‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Pei R, Feng J, Zhang Y, et al. Host metabolism dysregulation and cell tropism identification in human airway and alveolar organoids upon SARS‐CoV‐2 infection. Protein Cell. 2021;12(9):717‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Takaki H, Oshiumi H, Shingai M, Matsumoto M, Seya T. Development of mouse models for analysis of human virus infections. Microbiol Immunol. 2017;61(3‐4):107‐113. [DOI] [PubMed] [Google Scholar]
- 246. Munoz‐Fontela C, Dowling WE, Funnell SGP, et al. Animal models for COVID‐19. Nature. 2020;586(7830):509‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Ortega JT, Jastrzebska B, Rangel HR. Omicron SARS‐CoV‐2 variant spike protein shows an increased affinity to the human ACE2 receptor: an in silico analysis. Pathogens. 2021;11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Kim S, Nguyen TT, Taitt AS, et al. SARS‐CoV‐2 omicron mutation is faster than the chase: multiple mutations on spike/ACE2 interaction residues. Immune Netw. 2021;21(6):e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Carboni E, Carta AR, Carboni E. Can pioglitazone be potentially useful therapeutically in treating patients with COVID‐19?. Med Hypotheses. 2020;140:109776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Pitt B, Sutton NR, Wang Z, Goonewardena SN, Holinstat M. Potential repurposing of the HDAC inhibitor valproic acid for patients with COVID‐19. Eur J Pharmacol. 2021;898:173988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Ganjei Z, Faraji Dana H, Ebrahimi‐Dehkordi S, Alidoust F, Bahmani K. Methotrexate as a safe immunosuppressive agent during the COVID‐19 pandemic. Int Immunopharmacol. 2021;101(Pt B):108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Giannini S, Passeri G, Tripepi G, et al. Effectiveness of in‐hospital cholecalciferol use on clinical outcomes in comorbid COVID‐19 patients: a hypothesis‐generating study. Nutrients. 2021;13(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Esam Z, Akhavan M, Lotfi M, Bekhradnia A. Molecular docking and dynamics studies of nicotinamide riboside as a potential multi‐target nutraceutical against SARS‐CoV‐2 entry, replication, and transcription: a new insight. J Mol Struct. 2022;1247:131394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Fiorentino G, Coppola A, Izzo R, et al. Effects of adding L‐arginine orally to standard therapy in patients with COVID‐19: a randomized, double‐blind, placebo‐controlled, parallel‐group trial. Results of the first interim analysis. EClinicalMedicine. 2021;40:101125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Medvegy M, Simonyi G. Supplementary therapeutic possibilities to alleviate myocardial damage due to microvascular dysfunction in coronavirus disease 2019 (COVID‐19). Cardiol Ther. 2021;10(1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.