

Abstract
Interstitial lung diseases (ILDs) other than idiopathic pulmonary fibrosis (IPF) have an array of immunomodulatory treatment options compared with IPF, due to their inflammatory component. However, there is a relative paucity of guidance on the management of this heterogeneous group of diseases. In ILDs other than IPF, immunosuppression is the cornerstone of therapy, with varying levels of evidence for different immunomodulatory agents and for each specific ILD. Classification of ILDs is important for guiding treatment decisions. Immunomodulatory agents mainly include corticosteroids, mycophenolate mofetil (MMF), azathioprine, methotrexate, cyclophosphamide and rituximab. In this review, the available evidence for single agents in the most common ILDs is first discussed. We then reviewed practical therapeutic approaches in connective tissue disease–related ILD and interstitial pneumonia with autoimmune features, scleroderma-related ILD, vasculitis and dermatomyositis with hypoxemic respiratory failure, idiopathic non-specific interstitial pneumonia, hypersensitivity pneumonitis sarcoidosis, fibrosing organizing pneumonia and eosinophilic pneumonia. The treatment of acute exacerbations of ILD is also discussed. Therapy augmentation in ILD is dictated by the recognition of progression of disease. Criteria for the evaluation of progression of disease are then discussed. Finally, specific protocol and measures to increase patients’ safety are reviewed as well, including general monitoring and serologic surveillance, Pneumocystis jirovecii prophylaxis, patients’ education, genetic testing for azathioprine, MMF serum levels and cyclophosphamide administration protocols. Immunomodulatory therapies are largely successful in the management of ILDs and can be safely managed with the application of specific protocols, precautions and monitoring.
Keywords: immunomodulatory, immunosuppressive, interstitial lung disease, therapy, treatment
Introduction
Interstitial lung disease (ILD) encompasses a heterogeneous group of pulmonary diseases characterized by inflammation and fibrosis of the lung parenchyma. 1 The classification of these ILDs is important for informing treatment decisions. Idiopathic pulmonary fibrosis (IPF) is an idiopathic interstitial pneumonia (IIP) clearly distinguished from other subtypes.1,2 IPF is characterized by severe, progressive fibrosis and has poor prognosis, with few, but well-defined, treatment options. 2 Many of the other ILDs, however, have an inflammatory component in addition to a fibrotic one.
In non-IPF ILDs, the process usually starts with alveolitis, developing when CD4 T cells are activated by antigen-presenting cells. As a result, cytokines are released, and alveolar macrophages, T lymphocytes or neutrophils accumulate in the alveoli and interstitium. Persistent inflammation can result in organization into granuloma and often leads to tissue injury and eventual fibrosis.3,4 The inflammatory component allows for an array of therapeutic options, with immunosuppression being the mainstay of therapy, while the fibrotic component may or may not be progressive.1,5 As immunomodulatory therapies have increased risk of harm in IPF, particularly increased mortality, 6 diagnostic accuracy is crucial to determine the best treatment course. For simplicity, from now on, we will refer to any non-IPF fibrotic ILD with a fibrotic component (not necessarily progressive) as ‘fibrosing ILDs’.
In this article, in addition to reviewing the literature regarding immunomodulatory therapies, we discuss the evidence for specific treatment approaches, including precautions and monitoring.
Immunomodulatory therapies
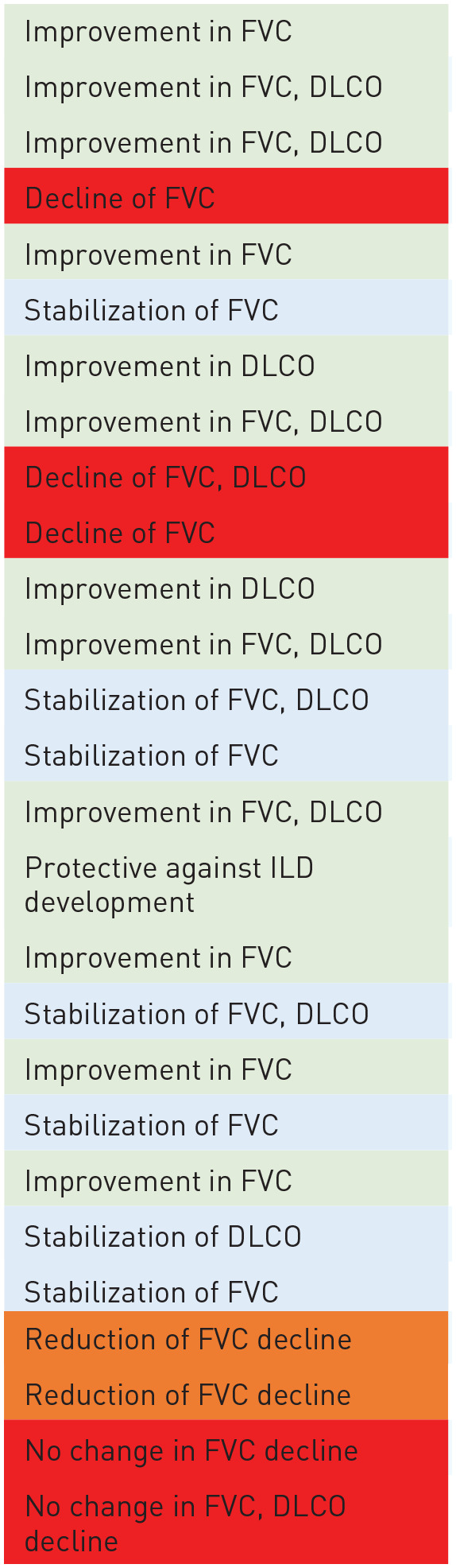
Currently, immunosuppression is still the mainstay of therapy in ILDs other than IPF. Therapies include corticosteroids, mycophenolate mofetil (MMF), azathioprine (AZA), methotrexate (MTX), cyclophosphamide (CYC) and rituximab (RTX) (Table 1).
Table 1.
Best published evidence with objective lung function data in ILDs for each drug.
| Drug | Condition studied | Efficacy observed a | Level of evidence b | References |
|---|---|---|---|---|
| Prednisone | Eosinophilic pneumonia |
|
B | Philit et al. 7 |
| Sarcoidosis | C | Paramothayan and Jones 8 | ||
| HP (non-fibrotic) | C | De Sadeleer et al. 9 | ||
| SSc | C | Steen et al. 10 | ||
| Mycophenolate mofetil | SSc | A | Tashkin et al. 11 | |
| HP | B | Morisset et al. 12 | ||
| CTD-ILD | C | Fischer et al. 13 | ||
| Sarcoidosis | C | Hamzeh et al. 14 | ||
| Azathioprine | HP | B | Morisset et al. 12 | |
| Sarcoidosis | B | Vorselaars et al. 15 | ||
| CTD-ILD | C | Boerner et al. 16 | ||
| SSc | A | Hoyles et al. 17 | ||
| Methotrexate | Sarcoidosis | B | Vorselaars et al. 15 | |
| RA-ILD | B | Juge et al. 18 | ||
| Cyclophosphamide | SSc | A | Tashkin et al. 11 | |
| NSIP | C | Corte et al. 19 | ||
| Rituximab | SSc | A | Sircar et al. 20 | |
| Sarcoidosis | B | Sweiss et al. 21 | ||
| CTD-ILD | B | Duarte et al. 22 | ||
| RA-ILD | C | Vadillo et al. 23 | ||
| Nintedanib | SSc | A | Distler et al. 24 | |
| PPF | A | Flaherty et al. 25 | ||
| Pirfenidone | Unclassifiable ILD | A | Maher et al. 26 | |
| PPF | A | Behr et al. 27 |
CTD-ILD, connective tissue disease–related interstitial lung disease; DLCO, diffusing capacity of the lungs for carbon monoxide; FVC, forced vital capacity; HP, hypersensitivity pneumonitis; ILD, interstitial lung disease; NSIP, nonspecific interstitial pneumonia; PPF-ILD, progressive pulmonary fibrosis; RA-ILD, rheumatoid arthritis interstitial lung disease; SSc, systemic sclerosis.
Colour Legend:
Green: improvement
Blue: stabilization
Orange: reduced/slowed rate of decline
Red: decline or no change in amount of decline
Level of Evidence Legend:
A: Randomized clinical trial
B: Multi-centre retrospective study or small (n < 20) clinical trial
C: Single-centre retrospective study or systematic review of single-centre studies, and case series
D: Case report/series
Corticosteroids
Corticosteroids are frequently used as the first-line therapy in ILD for their anti-inflammatory and immunosuppressive effects. Corticosteroids inhibit leukocyte movement and access to inflamed tissues, interfere with leukocyte, fibroblast and endothelial cell function, and suppress humoral factors. 28 Despite their wide use, there is a surprising lack of high-quality data in ILD.
The efficacy of corticosteroids is dependent, to some extent, on the stage of ILD. Patients with sarcoidosis, cryptogenic organizing pneumonia (COP), acute hypersensitivity pneumonitis (HP) and eosinophilic pneumonia generally respond rapidly and often to a full recovery.7,8,29 In a 2018 retrospective study on non-fibrotic HP, corticosteroids increased forced vital capacity (FVC) significantly, but did not have any impact on diffusing lung capacity for carbon monoxide (DLCO) decline. 9 In fibrosing ILD, although complete reversal is evidently not possible, short-term corticosteroids still have a role in stabilizing rapidly progressive disease. 30
In connective tissue disease (CTD)-ILD, available data are contrasting10,31,32 and corticosteroids are weaned off whenever possible, to avoid long-term side effects. A number of studies have shown benefits, including improved modified Rodnan skin score and improvement or stabilization in pulmonary function tests (PFTs), with steroid combined with other agents in systemic sclerosis–related ILD (SSc-ILD).33,34 However, high-dose steroids have been shown to increase the risk of scleroderma renal crisis and are thus often avoided. 35
Mycophenolate mofetil
MMF is an immunosuppressant that inhibits inosine monophosphate dehydrogenase and exerts a cytostatic effect on lymphocytes. 36 MMF is currently the most widely used first-line, steroid-sparing agent in fibrosing ILD as it is generally effective, well tolerated13,37 and less toxic than CYC. 11
In 2016, a randomized trial compared 2 years of MMF therapy with 1 year of oral CYC, followed by 1 year of placebo in SSc-ILD patients. A significant improvement in FVC and Rodney skin score over 2 years was observed with both treatments, with no significant differences between drugs, although MMF was associated with less toxicity. 11 Both regimens were associated with a significant improvement in the extent of high-resolution computed tomography (HRCT) ILD changes at 2 years 38 and improvements in health-related quality of life. 39
MMF was associated with an improvement in or stability of FVC and DLCO in a retrospective study of 125 CTD-ILD patients [including 19 with interstitial pneumonia with autoimmune features (IPAF)] over 2.5 years. 13 In a study by McCoy et al., 40 there was a non-significant improvement in FVC and DLCO slope after MMF in IPAF. Similarly, a 2022 study found an association between combination therapy with prednisone and MMF and decreased disease progression. 41 In myositis-related ILD, a retrospective study found that patients treated with MMF had a significant improvement in FVC and a decrease in mean prednisone dose requirement after 24 months of therapy. 42
In chronic HP, patients treated with MMF or AZA had a significant improvement in DLCO12,43 and reduced prednisone requirements. 43
Despite its effectiveness in other fibrosing ILDs, MMF has not been shown to be an effective therapy in sarcoidosis. A retrospective study of sarcoidosis patients reported no change in lung function in patients unresponsive to other steroid-sparing agents and treated with MMF for 1 year. 14
Azathioprine
AZA is an immunosuppressant agent that inhibits purine synthesis and DNA replication in lymphocytes, and is widely used as the second-line therapy in fibrosing ILD. 44 Data are unanimously positive, but largely limited to retrospective series.
In SSc-ILD, AZA therapy following intravenous CYC induction has been shown to stabilize or improve lung function in both a 2008 retrospective study and in the randomized Fibrosing Alveolitis in Scleroderma (FAST) trial.17,45 In CTD-ILD, AZA has been shown to stabilize or improve lung function during treatment. 16
In sarcoidosis, AZA is often used as the second-line therapy. A 2013 retrospective study comparing AZA and MTX effect found significant steroid-sparing effect and improvement in FVC and DLCO with both therapies. 15
In chronic HP, a retrospective study in patients treated with AZA showed significant improvement in FVC after 24 months of treatment. 46 Other studies showed that chronic HP patients treated with either MMF or AZA had significant improvement in DLCO12,43 and reduced prednisone requirements. 43
Methotrexate
MTX is a folate analogue that interferes with purine and pyrimidine synthesis and has anti-inflammatory and immunosuppressant effects. 47 Recent evidence has shown that pulmonary toxicity from MTX is much rarer than previously thought. 48
In rheumatoid arthritis (RA)-related ILD, MTX does not cause ILD and is actually protective. A 2021 study comparing the use of MTX in patients with RA-ILD to patients with RA without ILD found that ILD detection was significantly delayed in MTX users compared with never-users. 18 Other studies showed increased survival 49 and improved lung function 50 in MTX-treated RA-ILD patients.
In sarcoidosis, MTX is a highly effective second-line therapy after prednisone. In a 2013 retrospective study of 145 patients treated with MTX and 55 with AZA, daily prednisone requirements decreased with both treatments. 15 In addition, FVC and DLCO increased significantly. 15 Similar findings were reported by a small randomized trial in patients treated with MTX, compared with placebo. 49
Cyclophosphamide
CYC is regarded as the third-line treatment for fibrosing ILD, being more immunosuppressive and toxic, but also as an effective rescue therapy. CYC is a potent alkylating immunosuppressant that is used in numerous hematologic malignancies and autoimmune conditions.50,51
The best data were reported in SSc-ILD. The Scleroderma Lung Study I (SLS-I), a randomized, placebo-controlled trial investigating the effect of oral CYC on lung function and symptoms in 145 patients with SSc-ILD across 13 centres, reported a mean absolute difference in FVC of 2.53% between groups (p < 0.03) at 12 months, but no significant difference in DLCO. In addition, the CYC arm demonstrated improved dyspnoea and less disability. 52 Data from the same trial also demonstrated decreased cough frequency with 12 months of CYC, although this was not sustained after discontinuation of therapy. 53 HRCT changes observed after a year of oral CYC parallel these improvements, with fibrosis being significantly worse in placebo-treated patients.54,55 In the SLS II study, CYC and MMF were both effective, but comparatively, MMF was better tolerated. 11
The multicenter, randomized FAST trial explored the effect of low-dose prednisone and intravenous (iv) CYC for 6 months, followed by maintenance AZA in SSc-ILD. Compared with placebo, predicted FVC in the treatment group improved by 4.2%, but only with a trend towards significance (p = 0.08) after 12 months. 17
Due to potential bladder toxicity, continuing CYC long-term is challenging. In SLS-I, the beneficial effects of 1-year treatment with CYC on lung function and health status dissipated after 18 months, while favourable effects on dyspnea continued through 24 months. 56 Considering both SLS-I and SLS-II trials, significant improvement in FVC lasted for 12 months, but not beyond that. 57
In a retrospective study, CYC showed positive results in the treatment of severe progressive nonspecific interstitial pneumonia (NSIP) resistant to other treatments, with stabilization of lung function. 19 In a 2017 study of iv CYC in patients with steroid-refractory IPAF, an increase in FVC at 6 months was observed (p = 0.002). 58
Notably, CYC is the mainstay of therapy in vasculitis. In antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis, randomized trials have found that prednisone and iv-pulse CYC induce remission as frequently as prednisone and oral CYC. 59 A 1998 study found similar survival, time of remission and relapse rate between groups, 59 while a long-term follow-up of patients from the Cyclophosphamide Daily Oral versus Pulsed (CYCLOPS) study found higher risk of relapse with pulse CYC. 60 Importantly, the total CYC dose is reduced with iv administration. 59
Rituximab
RTX is a monoclonal antibody that targets CD20 on B-lymphocytes 61 and is the object of increasing interest as third- or even second-line option in the therapeutic algorithm of fibrosing ILDs. Although almost all data reported on RTX are positive, there unfortunately is a lack of high-quality trials.
In progressive CTD-ILD, a 2020 retrospective study reported significant improvement in FVC and DLCO after 1 year of treatment with RTX, with sustainable improvement in DLCO remaining at 2 years. 62 Another retrospective multicentre cohort study on 49 patients with CTD-ILD found stabilization of DLCO and significant improvement of FVC after 1 year of RTX. 22 In a further retrospective study on CTD-ILD patients, the addition of RTX to MMF reduced daily prednisone requirements, although no significant changes in lung function were seen. 63
In a 2018 open-label, randomized trial on 60 patients with early SSc-ILD treated with RTX versus CYC for 6 months, there was a significant improvement in FVC in the RTX group (from 61% to 68%) and a non-significant decline in the CYC group. 20 Other studies have had similarly positive findings. 64 A European prospective, observational, non-randomized study comparing SSc patients treated with RTX to matched, untreated patients, however, did not find any significant difference in FVC or DLCO in the two cohorts over 2 years, but no decline either. 65
Evidence supporting the use of RTX in RA-ILD has not been as strong. In a retrospective observational study of 44 patients treated with RTX for arthritis, 16% of patients improved and 52% of patients stabilized in terms of FVC, DLCO and radiographic extent on HRCT. 66 A 2020 study from the Spanish registry found that patients treated with RTX versus other therapies had a lower risk of functional decline (decline in FVC ⩾5%). 23 Another British registry study demonstrated improved survival compared with tumour necrosis factor α (TNFα) inhibitors. 67
There is a lack of trials exploring the effectiveness of RTX in IPAF; however, in a 2021 case series from two medical centres, 41 of 44 patients with PFTs had improvement or stability in FVC after treatment with RTX. 68
Idiopathic inflammatory myositis–related ILDs, particularly antisynthetase syndrome-ILD, have shown good response to RTX in observational studies, with stabilization or improvement in radiographic extent 69 and FVC.69,70
RTX has been shown to be an effective rescue therapy in patients with treatment-refractory fibrosing ILD.71–75 In a study of 50 severe, progressive ILD patients unresponsive to other immunosuppressants, RTX resulted in a median improvement in FVC of 6.7% (p < 0.01) and stabilized DLCO within 6–12 months. 73 A retrospective, observational study analysed SSc-ILD patients treated with RTX for worsening lung function, despite steroids and immunosuppression with CYC and MMF. Among the 15 patients who completed 2 years of RTX, there was significant improvement in FVC and DLCO. 74 However, in ‘refractory’ pulmonary sarcoidosis, an open-label, phase I/II trial found inconsistent response to RTX, with only 5 of 10 patients having >5% absolute improvement in FVC. 21
The Evaluation of Efficacy and Safety of Rituximab with Mycophenolate Mofetil in Patients with Interstitial Lung Diseases (EVER-ILD) trial is a double-blind, placebo-controlled randomized trial currently underway, comparing RTX induction followed by MMF with placebo and MMF in patients with severe and progressive NSIP, refractory to other immunosuppressants. 76
Treatment approaches
Aside from cases in which the risk–benefit analysis favours careful observation, 77 the general approach to immunosuppressive therapy in ILD is based on a dynamic, stepwise process, where treatment is augmented when progression of disease or lack of expected improvement is observed, and is stepped down when lung function has reached a steady plateau. 78 This approach implies a regular reassessment of treatments and doses in each individual patient (Table 2).
Table 2.
Suggested approach to treatment by condition. a
| Condition | Treatments | Approach | |
|---|---|---|---|
| CTD-ILD, IPAF | Prednisone | First line10,31,32,41 | |
| MMF | First line with prednisone or second line13,40,41,79 | ||
| AZA | First line with prednisone or second line 16 | ||
| RTX | Third line22,62,63,68 | ||
| CYC | Third line58,80 | ||
| RA-ILD | MTX | Second line if required for joint disease81,82 | |
| Tocilizumab | Fourth line 83 | ||
| SSc-ILD | MMF | First line 11 | |
| CYC | Second line11,52 | ||
| RTX | Third line20,64 | ||
| Tocilizumab | Third line 84 | ||
| Vasculitis or Dermatomyositis with hypoxemic respiratory failure | Methylprednisolone pulse | First line 85 | |
| CYC | First line 59 | ||
| RTX | Second line75,86 | ||
| AZA | Third line (maintenance only) 87 | ||
| MMF | Third line (maintenance only)13,88 | ||
| NSIP | Prednisone | First line 89 | |
| MMF | Second line 79 | ||
| AZA | Second line 89 | ||
| CYC | Third line19,89 | ||
| HP | Prednisone | First line9,29 | |
| Chronic HP | MMF | Second line12,43 | |
| AZA | Second line12,43,46 | ||
| Sarcoidosis | Prednisone | First line8,89 | |
| MTX | Second line15,49 | ||
| AZA | Second line 15 | ||
| RTX | Third line 21 | ||
| Infliximab | Third line 90 | ||
| Fibrosing organizing pneumonia | Prednisone | First line 89 | |
| MMF | Second line 91 | ||
| CYC | Second line 92 | ||
| Eosinophilic pneumonia | Prednisone | First line 7 |
AZA, azathioprine; CTD-ILD, connective tissue disease related interstitial lung disease; CYC, cyclophosphamide; HP, hypersensitivity pneumonitis; IPAF, interstitial pneumonia with autoimmune features; NSIP, nonspecific interstitial pneumonia; MTX, methotrexate; MMF, mycophenolate mofetil; RA-ILD, rheumatoid arthritis interstitial lung disease; RTX, rituximab; SSc-ILD, systemic sclerosis related interstitial lung disease.
Nintedanib can be considered for progressive pulmonary fibrosis regardless of the subtype.
A common approach is to start oral prednisone 0.5–1 mg/kg for a limited period of time, to achieve improvement or at least stabilization of disease, 89 and then to introduce 8–10 weeks later a steroid-sparing agent, to avoid long-term side effects. The steroid is then tapered to a smaller dose and eventually completely stopped, if stabilization of ILD is achieved.
Increasingly, however, steroid-sparing agents such as MMF and AZA are started upfront, especially when the disease at presentation is severe, with supplemental oxygen requirements. In SSc-ILD, where the efficacy of MMF and CYC monotherapy is established,11,52 and where corticosteroids may cause a renal crisis, 35 prednisone may in fact not be used at all. Extrapolating this evidence to other ILDs, MMF or AZA may be used upfront without prednisone, when the absence of rapid progression of disease has been ascertained or when significant contraindications to the use of steroids are present. In the study by Morisset et al., 12 for example, 77% of patients with HP were treated with either MMF or AZA, without prednisone in advance.
When ILD is clinically significant, with physiologic compromise, an early, complete cessation of immunomodulatory therapy may trigger an acute exacerbation (AE) or rapid progression of disease, with potentially fatal outcome. A cautious, gradual decrease is instead adopted, with the aim of minimizing immunosuppression whenever possible. A complete discontinuance of therapy is possible in sarcoidosis and COP, but not always achieved in other types of ILD.
Since immunosuppressive therapy is often a long-term commitment in ILD, when 2 or more agents are used for a period longer than 2 months, Pneumocystis jirovecii pneumonia (PJP) prophylaxis is generally provided. PJP prophylaxis is also usually recommended in the literature with a dose of prednisone ⩾25 mg/day, 93 although there is no full consensus on this. 94 The risk of PJP is particularly high in patients receiving an initial dose of ⩾60 mg/day prednisone or equivalent. 93 In practice, it is therefore generally accepted that high-dose, prolonged courses of prednisone merit PJP prophylaxis, whether as monotherapy or in combination with another agent. In contrast, when a single non-steroid agent is used, PJP prophylaxis is not required, with the notable exception of CYC, which is considered significantly immunosuppressive by itself. 95 The occurrence of PJP can create major diagnostic difficulties in ILD, as it can be confused with progression of disease or AE. Considering this, and the extremely high morbidity of PJP pneumonia in patients with underlying ILD, 96 whenever two or more agents are used (either ⩾2 steroid-sparing agents or a steroid-sparing agent in combination with low-dose corticosteroid), PJP prophylaxis is likely indicated.
It is well known that prolonged steroid use accelerates bone loss and increases risk of osteoporosis. Bisphosphonates are therefore recommended in the British Thoracic Society ILD guidelines for ILD patients treated with steroids. 89 The American College of Rheumatology recommends that all adults taking ⩾2.5 mg/day of prednisone for ⩾3 months optimize their calcium and vitamin D intake. Addition of osteoporosis pharmacotherapy (such as bisphosphonates and denosumab) is based on age and fracture risk. 97
In aggressive presentations of ILD with severe hypoxemic respiratory failure, presenting with diffuse, bilateral ground glass opacities, such as vasculitis, 98 dermatomyositis/polymyositis, 99 or AEs of any fibrosing ILD, 100 a methylprednisolone iv pulse may stop rapid progression of disease and stabilize the patient. The dose of 10 mg/kg of methylprednisolone iv is usually administered for 3 consecutive days. In rapidly progressive ILD, the institution of very high-dose immunosuppression for a limited period is preferred over a low or average level of therapy with a prolonged treatment course, where adverse events are inevitable.
Surveillance and patient education are both fundamental aspects of the immunomodulatory treatment of ILD to avoid and reduce significant adverse events, as well as increase patient adherence. 78 While the education of patients and caregivers in clinic is always helpful, it is recommended to also provide written information in lay language about the specific drug(s) used.
Finally, drug-specific protocols of therapy, discussed below, allow further reduction of toxicity.
MMF serum levels
MMF is a pro-drug of mycophenolic acid (MPA). MMF is rapidly absorbed from the gastrointestinal tract and undergoes extensive pre-systemic de-esterification to become MPA, the active moiety. After an oral dose, MMF in systemic circulation quickly disappears and the plasma concentration of MPA rises rapidly, reaching its maximum concentration within 1 h. 101 Food intake can delay the rate of MMF absorption, but does not affect the extent of it. Co-administration of antacids or cholestyramine decreases the extent of absorption by approximately 20% and 40%, respectively. 102
Although not routinely adopted in clinical practice, monitoring of serum levels of MPA can be helpful to ensure therapeutic levels in patients who cannot tolerate the full dose 103 Unfortunately, there is no published experience on the use of MMF guided by serum levels, but extrapolating the evidence from transplant experience, a level of 1.0–3.5 µg/ml should be targeted. 104 This approach may potentially allow a reduction in the dose of MMF with improved tolerability, while still achieving a therapeutic level of MPA.
Genetic testing for AZA
Thiopurine methyltransferase (TPMT) genetic profiling can be used to identify intermediate and slow metabolizers of AZA who are at higher risk of developing bone marrow suppression. 105 In addition, HLA-DQA1-HLA-DRB genetic profiling can predict the risk of pancreatitis with AZA. 106 A recent study demonstrated that genetic testing was associated with a significantly reduced incidence of major adverse events and a lower rate of AZA discontinuation, but the total number of adverse events did not change, as available genetic testing does not predict the risk of liver dysfunction or other side effects. 107 While the cost-effectiveness of systematic genetic testing for AZA has not yet been demonstrated, it is very likely to increase patient safety.
CYC treatment protocol to reduce toxicity
CYC is the most potent immunosuppressive drug in the pulmonologist’s armamentarium for ILD. The British Thoracic Society recommended the use of iv rather than oral CYC, 89 given preferable side effect profile. 107 The rate of leukopenia, severe infections, and gonadal toxicity were reduced in the iv administration route, compared with oral, without differences in patient outcomes. 59 The recommended iv dose is 500–750 mg/m2 monthly, 78 but frequency can be increased in severe cases with hypoxemic respiratory failure. However, the total dose should not exceed 20 g, as the risk of bladder cancer increases above that level. 108 It is unusual to exceed 12 g in a treatment course of ILD.
To reduce the risk of hemorrhagic cystitis and bladder cancer, the administration of 250–500 ml of normal saline before and after infusion and good hydration for the following 72 h is recommended. 109 The concomitant administration of ondansetron reduces the frequency of emesis. 110
The white blood cell nadir usually occurs 10–14 days after an iv pulse, and bi-weekly surveillance is strongly recommended. These precautions, together with dose adjustments dictated by regular surveillance, should ensure a safe administration of CYC in most cases.
Recognizing progression of disease
Therapy augmentation in ILD is undoubtedly dictated by the recognition of progression of disease. However, there is currently no consensus as to how disease progression should be defined in ILD patients.24,25,111,112 A number of end-points have been proposed in clinical trials exploring fibrosing ILDs. 5 In IPF and other ILDs, most studies have defined disease progression as a decline in FVC, measured as the change from baseline or as a categorical change (typically ⩾10% predicted).24,25,111,112 A decline in FVC is a well-defined predictor of mortality in IPF.113,114 Nevertheless, a recent study showed remarkable heterogeneity in FVC trajectories, depending on the ILD subtype. 115 Patient-reported outcomes (PRO), imaging features, acute worsening events, mortality, exercise capacity and quality of life measures are often used as secondary end-points. 5
In daily clinical practice, progression of ILD is highlighted by the integration of multiple domains, including deterioration in lung function tests, worsening of fibrosis on chest HRCT, worsening of symptoms and exercise capacity. Measurement of FVC and DLCO is considered the best tool in monitoring disease progression. However, the main limitations are represented by test variability and confounding pulmonary comorbidities, such as emphysema or pulmonary hypertension. The recent 2022 American Thoracic Society guideline proposes a definition of progressive pulmonary fibrosis (PPF) as ⩾2/3 of (1) worsening respiratory symptoms, (2) physiologic (absolute fall ⩾5% in FVC and ⩾10% in DLCO within 1 year) and (3) radiographic evidence of progression. 116
Respiratory symptoms are meaningful in detecting disease progression. Although there are no data on fibrosing ILD, chronic cough in IPF is not only often refractory but is also considered an independent predictor of disease progression. 117 Similarly, changes in dyspnea score, for example, have been demonstrated to be independently predictive of survival in ILD patients. 118
PRO and experiences are key to understanding needs and facilitating patient-centred care. Symptoms should be measured across the disease course. In fact, in IPF, PRO measures are considered secondary outcomes in clinical trials. 119
Reduced exercise capacity is an essential characteristic of progressive fibrosing ILDs, and a decline in 6-min walk distance (6MWD), at least in IPF, is a strong, independent predictor of mortality. 120 6MWD can be affected by numerous factors, including age, body size, comorbidities and the use of supplemental oxygen during the test, and these issues need to be considered in result interpretation of both individual and serial tests. 121
HRCT has a role in staging and quantifying the extent of diffuse lung diseases. However, there currently is a need to create a reproducible HRCT staging system for the evaluation of clinically significant changes. Many studies recognize the extent of fibrosis as a strong predictor of outcome in patients with IPF.122,123 However, studies using a visual, semiquantitative score of parenchymal abnormalities to predict the mortality rate are considered poorly reproducible. 124 Computer-based quantification of disease on CT has been used in a variety of ILDs and have significantly improved human-based CT evaluation.125–129 Quantitative CT also has several limitations, mainly related to the fact that it is heavily influenced by CT dose, slice thickness and reconstruction kernel. 126
When progression of fibrosing ILD occurs, a role for anti-fibrotic therapy may be considered.24–27,130 Although this is not the object of this review, given the recent published evidence, we included anti-fibrotic agents in the suggested approaches to therapy (Tables 1 and 2). Combination therapy of MMF or other immunomodulatory agents with either nintedanib or pirfenidone is considered tolerable and safe. The decision as to whether the best management for a patient with progressive phenotypes of ILD is to intensify immunosuppression, introduce second-line therapy with anti-fibrotic therapy, or combine these two approaches is challenging and will require future studies specifically designed to address combination therapy and with well-defined criteria for truly progressive ILD. 131
Conclusion
Immunomodulatory therapy is largely successful in the treatment of ILD and can be safely managed with the application of specific protocols, precautions, monitoring and patient education. This is reflected by consistently better outcomes reported for fibrosing ILD other than IPF compared with IPF, despite a remarkable scarcity of clinical trials on immunosuppressive agents. There is currently a key need to clarify the optimal timing and sequence of treatments.
Acknowledgments
None.
Footnotes
ORCID iD: Marco Mura  https://orcid.org/0000-0002-2159-7083
https://orcid.org/0000-0002-2159-7083
Contributor Information
Laura van den Bosch, Division of Pulmonary Medicine, University of Alberta, Edmonton, AB, Canada.
Fabrizio Luppi, Department of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy; Respiratory Unit, San Gerardo Hospital, ASST Monza, Monza, Italy.
Giovanni Ferrara, Division of Pulmonary Medicine, University of Alberta, Edmonton, AB, Canada.
Marco Mura, London Health Sciences Centre, Victoria Hospital, 800 Commissioners Road East, Room E6-203, London, ON N6A 5W9, Canada.
Declarations
Ethics approval and consent to participate: This is a review manuscript and no ethics approval was required.
Consent for publication: All authors have read and approved the submitted manuscript. No other consent for publication was required.
Author contributions: Laura van den Bosch: Conceptualization; Data curation; Resources; Writing – original draft; Writing – review & editing.
Fabrizio Luppi: Conceptualization; Data curation; Writing – original draft; Writing – review & editing.
Giovanni Ferrara: Conceptualization; Data curation; Resources; Writing – original draft; Writing – review & editing.
Marco Mura: Conceptualization; Data curation; Resources; Supervision; Writing – original draft; Writing – review & editing.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
Competing interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Availability of data and material: This is a review paper.
References
- 1. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188: 733–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Travis WD, King TE, Bateman ED, et al. American thoracic society/European respiratory society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304. [DOI] [PubMed] [Google Scholar]
- 3. Semenzato G, Adami F, Maschio N, et al. Immune mechanisms in interstitial lung diseases. Allergy 2000; 55: 1103–1120. [DOI] [PubMed] [Google Scholar]
- 4. Spagnolo P, Distler O, Ryerson CJ, et al. Mechanisms of progressive fibrosis in connective tissue disease (CTD)-associated interstitial lung diseases (ILDs). Ann Rheum Dis 2021; 80: 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cottin V, Hirani NA, Hotchkin DL, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev 2018; 27: 180076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Network TIPFCR. Prednisone, azathioprine, and N -acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366: 1968–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Philit F, Etienne-Mastroïanni B, Parrot A, et al. Idiopathic acute eosinophilic pneumonia: a study of 22 patients. Am J Respir Crit Care Med 2002; 166: 1235–1239. [DOI] [PubMed] [Google Scholar]
- 8. Paramothayan S, Jones PW. Corticosteroid therapy in pulmonary sarcoidosis. JAMA 2002; 287: 1301–1307. [DOI] [PubMed] [Google Scholar]
- 9. De Sadeleer L, Hermans F, De Dycker E, et al. Effects of corticosteroid treatment and antigen avoidance in a large hypersensitivity pneumonitis cohort: a single-centre cohort study. J Clin Med 2018; 8: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steen VD, Lanz JK, Jr, Conte C, et al. Therapy for severe interstitial lung disease in systemic sclerosis. a retrospective study. Arthritis Rheum 1994; 37: 1290–1296. [DOI] [PubMed] [Google Scholar]
- 11. Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med 2016; 4: 708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morisset J, Johannson KA, Vittinghoff E, et al. Use of mycophenolate mofetil or azathioprine for the management of chronic hypersensitivity pneumonitis. Chest 2017; 151: 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fischer A, Brown KK, Du Bois RM, et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J Rheumatol 2013; 40: 640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hamzeh N, Voelker A, Forssén A, et al. Efficacy of mycophenolate mofetil in sarcoidosis. Respir Med 2014; 108: 1663–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vorselaars ADM, Wuyts WA, Vorselaars VMM, et al. Methotrexate vs azathioprine in second-line therapy of sarcoidosis. Chest 2013; 144: 805–812. [DOI] [PubMed] [Google Scholar]
- 16. Boerner EB, Cuyas M, Theegarten D, et al. Azathioprine for connective tissue disease-associated interstitial lung disease. Respiration 2020; 99: 628–636. [DOI] [PubMed] [Google Scholar]
- 17. Hoyles RK, Ellis RW, Wellsbury J, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum 2006; 54: 3962–3970. [DOI] [PubMed] [Google Scholar]
- 18. Juge PA, Lee JS, Lau J, et al. Methotrexate and rheumatoid arthritis associated interstitial lung disease. Eur Respir J 2021; 57: 2000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Corte TJ, Ellis R, Renzoni EA, et al. Use of intravenous cyclophosphamide in known or suspected, advanced non-specific interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis 2009; 26: 132–138. [PubMed] [Google Scholar]
- 20. Sircar G, Goswami RP, Sircar D, et al. Intravenous cyclophosphamide vs rituximab for the treatment of early diffuse scleroderma lung disease: open label, randomized, controlled trial. Rheumatology 2018; 57: 2106–2113. [DOI] [PubMed] [Google Scholar]
- 21. Sweiss NJ, Lower EE, Mirsaeidi M, et al. Rituximab in the treatment of refractory pulmonary sarcoidosis. Eur Respir J 2014; 43: 1525–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duarte AC, Cordeiro A, Fernandes BM, et al. Rituximab in connective tissue disease–associated interstitial lung disease. Clin Rheumatol 2019; 38: 2001–2009. [DOI] [PubMed] [Google Scholar]
- 23. Vadillo C, Nieto MA, Romero-Bueno F, et al. Efficacy of rituximab in slowing down progression of rheumatoid arthritis–related interstitial lung disease: data from the NEREA Registry. Rheumatology 2020; 59: 2099–2108. [DOI] [PubMed] [Google Scholar]
- 24. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis–associated interstitial lung disease. N Engl J Med 2019; 380: 2518–2528. [DOI] [PubMed] [Google Scholar]
- 25. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 2019; 381: 1718–1727. [DOI] [PubMed] [Google Scholar]
- 26. Maher TM, Corte TJ, Fischer A, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med 2020; 8: 147–157. [DOI] [PubMed] [Google Scholar]
- 27. Behr J, Prasse A, Kreuter M, et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med 2021; 9: 476–486. [DOI] [PubMed] [Google Scholar]
- 28. Strehl C, Spies CM, Buttgereit F. Pharmacodynamics of glucocorticoids. Clin Exp Rheumatol 2011; 29: S13–S18. [PubMed] [Google Scholar]
- 29. Kokkarinen JI, Tukiainen HO, Terho EO. Effect of corticosteroid treatment on the recovery of pulmonary function in farmer’s lung. Am Rev Respir Dis 1992; 145: 3–5. [DOI] [PubMed] [Google Scholar]
- 30. Ejima M, Okamoto T, Suzuki T, et al. Efficacy of treatment with corticosteroids for fibrotic hypersensitivity pneumonitis: a propensity score-matched cohort analysis. BMC Pulm Med 2021; 21: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iudici M, Valentini G, Van der Goes MC, et al. Glucocorticoids in systemic sclerosis: weighing up the benefits and risks – a systematic review. Clin Exp Rheumatol 2013; 31: 157–165. [PubMed] [Google Scholar]
- 32. Yamano Y, Taniguchi H, Kondoh Y, et al. Multidimensional improvement in connective tissue disease-associated interstitial lung disease: two courses of pulse dose methylprednisolone followed by low-dose prednisone and tacrolimus. Respirology 2018; 23: 1041–1048. [DOI] [PubMed] [Google Scholar]
- 33. Griffiths B, Miles S, Moss H, et al. Systemic sclerosis and interstitial lung disease: a pilot study using pulse intravenous methylprednisolone and cyclophosphamide to assess the effect on high resolution computed tomography scan and lung function. J Rheumatol 2002; 29: 2371–2378. [PubMed] [Google Scholar]
- 34. Vanthuyne M, Blockmans D, Westhovens R, et al. A pilot study of mycophenolate mofetil combined to intravenous methylprednisolone pulses and oral low-dose glucocorticoids in severe early systemic sclerosis. Clin Exp Rheumatol 2007; 25: 287–292. [PubMed] [Google Scholar]
- 35. Trang G, Steele R, Baron M, et al. Corticosteroids and the risk of scleroderma renal crisis: a systematic review. Rheumatol Int 2012; 32: 645–653. [DOI] [PubMed] [Google Scholar]
- 36. Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000; 47: 85–118. [DOI] [PubMed] [Google Scholar]
- 37. Swigris JJ, Olson AL, Fischer A, et al. Mycophenolate mofetil is safe, well tolerated, and preserves lung function in patients with connective tissue disease-related interstitial lung disease. Chest 2006; 130: 30–36. [DOI] [PubMed] [Google Scholar]
- 38. Goldin JG, Kim GHJ, Tseng CH, et al. Longitudinal changes in quantitative interstitial lung disease on computed tomography after immunosuppression in the Scleroderma Lung Study II. Ann Am Thorac Soc 2018; 15: 1286–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Volkmann ER, Tashkin DP, LeClair H, et al. Treatment with mycophenolate and cyclophosphamide leads to clinically meaningful improvements in patient-reported outcomes in scleroderma lung disease: results of scleroderma lung study II. ACR Open Rheumatol 2020; 2: 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McCoy SS, Mukadam Z, Meyer KC, et al. Mycophenolate therapy in interstitial pneumonia with autoimmune features: a cohort study. Ther Clin Risk Manag 2018; 14: 2171–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joerns EK, Adams TN, Newton CA, et al. Variables associated with response to therapy in patients with interstitial pneumonia with autoimmune features. J Clin Rheumatol 2022; 28: 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huapaya JA, Silhan L, Pinal-Fernandez I, et al. Long-term treatment with azathioprine and mycophenolate mofetil for myositis-related interstitial lung disease. Chest 2019; 156: 896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fiddler CA, Simler N, Thillai M, et al. Use of mycophenolate mofetil and azathioprine for the treatment of chronic hypersensitivity pneumonitis–a single-centre experience. Clin Respir J 2019; 13: 791–794. [DOI] [PubMed] [Google Scholar]
- 44. Maltzman JS, Koretzky GA. Azathioprine: old drug, new actions. J Clin Invest 2003; 111: 1122–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bérezné A, Ranque B, Valeyre D, et al. Therapeutic strategy combining intravenous cyclophosphamide followed by oral azathioprine to treat worsening interstitial lung disease associated with systemic sclerosis: a retrospective multicenter open-label study. J Rheumatol 2008; 35: 1064–1072. [PubMed] [Google Scholar]
- 46. Terras Alexandre A, Martins N, Raimundo S, et al. Impact of azathioprine use in chronic hypersensitivity pneumonitis patients. Pulm Pharmacol Ther 2020; 60: 101878. [DOI] [PubMed] [Google Scholar]
- 47. Genestier L, Paillot R, Quemeneur L, et al. Mechanisms of action of methotrexate. Immunopharmacology 2000; 47: 247–257. [DOI] [PubMed] [Google Scholar]
- 48. Sathi N, Chikura B, Kaushik VV, et al. How common is methotrexate pneumonitis? A large prospective study investigates. Clin Rheumatol 2012; 31: 79–83. [DOI] [PubMed] [Google Scholar]
- 49. Baughman RP, Winget DB, Lower EE. Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. Sarcoidosis Vasc Diffuse Lung Dis 2000; 17: 60–66, https://europepmc.org/article/med/10746262 (accessed 15 February 2021). [PubMed] [Google Scholar]
- 50. Hall AG, Tilby MJ. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev 1992; 6: 163–173. [DOI] [PubMed] [Google Scholar]
- 51. Teles KA, Medeiros-Souza P, Lima FAC, et al. Cyclophosphamide administration routine in autoimmune rheumatic diseases: a review. Rev Bras Reumatol Engl Ed 2017; 57: 596–604. [DOI] [PubMed] [Google Scholar]
- 52. Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med 2006; 354: 2655–2666. [DOI] [PubMed] [Google Scholar]
- 53. Theodore AC, Tseng CH, Li N, et al. Correlation of cough with disease activity and treatment with cyclophosphamide in scleroderma interstitial lung disease: findings from the scleroderma lung study. Chest 2012; 142: 614–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goldin J, Elashoff R, Kim HJ, et al. Treatment of scleroderma-interstitial lung disease with cyclophosphamide is associated with less progressive fibrosis on serial thoracic high-resolution CT scan than placebo: findings from the scleroderma lung study. Chest 2009; 136: 1333–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim HJ, Brown MS, Elashoff R, et al. Quantitative texture-based assessment of one-year changes in fibrotic reticular patterns on HRCT in scleroderma lung disease treated with oral cyclophosphamide. Eur Radiol 2011; 21: 2455–2465. [DOI] [PubMed] [Google Scholar]
- 56. Tashkin DP, Elashoff R, Clements PJ, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med 2007; 176: 1026–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Volkmann ER, Tashkin DP, Sim M, et al. Cyclophosphamide for systemic sclerosis-related interstitial lung disease: a comparison of scleroderma lung study I and II. J Rheumatol 2019; 46: 1316–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wiertz IA, van Moorsel CHM, Vorselaars ADM, et al. Cyclophosphamide in steroid refractory unclassifiable idiopathic interstitial pneumonia and interstitial pneumonia with autoimmune features (IPAF). Eur Respir J 2018; 51: 1702519. [DOI] [PubMed] [Google Scholar]
- 59. Haubitz M, Schellong S, Göbel U, et al. Intravenous pulse administration of cyclophosphamide versus daily oral treatment in patients with antineutrophil cytoplasmic antibody-associated vasculitis and renal involvement: a prospective, randomized study. Arthritis Rheum 1998; 41: 1835–1844. [DOI] [PubMed] [Google Scholar]
- 60. Harper L, Morgan MD, Walsh M, et al. Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Ann Rheum Dis 2012; 71: 955–960. [DOI] [PubMed] [Google Scholar]
- 61. Weiner GJ. Rituximab: mechanism of action. Semin Hematol 2010; 47: 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Robles-Perez A, Dorca J, Castellví I, et al. Rituximab effect in severe progressive connective tissue disease-related lung disease: preliminary data. Rheumatol Int 2020; 40: 719–726. [DOI] [PubMed] [Google Scholar]
- 63. Zhu L, Chung MP, Gagne L, et al. Rituximab versus mycophenolate in the treatment of recalcitrant connective tissue disease–associated interstitial lung disease. ACR Open Rheumatol 2021; 3: 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jordan S, Distler JH, Maurer B, et al. Effects and safety of rituximab in systemic sclerosis: an analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis 2015; 74: 1188–1194. [DOI] [PubMed] [Google Scholar]
- 65. Elhai M, Boubaya M, Distler O, et al. Outcomes of patients with systemic sclerosis treated with rituximab in contemporary practice: a prospective cohort study. Ann Rheum Dis 2019; 78: 979–987. [DOI] [PubMed] [Google Scholar]
- 66. Md Yusof MY, Kabia A, Darby M, et al. Effect of rituximab on the progression of rheumatoid arthritis–related interstitial lung disease: 10 years’ experience at a single centre. Rheumatology 2017; 56: 1348–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Druce KL, Iqbal K, Watson KD, et al. Mortality in patients with interstitial lung disease treated with rituximab or TNFi as a first biologic. RMD Open 2017; 3: e000473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. D’Silva KM, Ventura IB, Bolster MB, et al. Rituximab for interstitial pneumonia with autoimmune features at two medical centres. Rheumatol Adv Pract 2021; 5: ii1–ii9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Doyle TJ, Dhillon N, Madan R, et al. Rituximab in the treatment of interstitial lung disease associated with antisynthetase syndrome: a multicenter retrospective case review. J Rheumatol 2018; 45: 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Andersson H, Sem M, Lund MB, et al. Long-term experience with rituximab in anti-synthetase syndrome-related interstitial lung disease. Rheumatology 2015; 54: 1420–1428. [DOI] [PubMed] [Google Scholar]
- 71. Keir GJ, Maher TM, Hansell DM, et al. Severe interstitial lung disease in connective tissue disease: rituximab as rescue therapy. Eur Respir J 2012; 40: 641–648. [DOI] [PubMed] [Google Scholar]
- 72. Marie I, Dominique S, Janvresse A, et al. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012; 106: 581–587. [DOI] [PubMed] [Google Scholar]
- 73. Keir GJ, Maher TM, Ming D, et al. Rituximab in severe, treatment-refractory interstitial lung disease. Respirology 2014; 19: 353–359. [DOI] [PubMed] [Google Scholar]
- 74. Narváez J, LLuch J, Molina-Molina M, et al. Rituximab as a rescue treatment added on mycophenolate mofetil background therapy in progressive systemic sclerosis associated interstitial lung disease unresponsive to conventional immunosuppression. Semin Arthritis Rheum 2020; 50: 977–987. [DOI] [PubMed] [Google Scholar]
- 75. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol 2018; 37: 1983–1989. [DOI] [PubMed] [Google Scholar]
- 76. Bejan-Angoulvant T, Naccache JM, Caille A, et al. Evaluation of efficacy and safety of rituximab in combination with mycophenolate mofetil in patients with nonspecific interstitial pneumonia non-responding to a first-line immunosuppressive treatment (EVER-ILD): a double-blind placebo-controlled randomized trial. Respir Med Res 2020; 78: 100770. [DOI] [PubMed] [Google Scholar]
- 77. Wells AU, Kouranos V. An IPF-like disease course in disorders other than IPF: how can this be anticipated, recognized, and managed? Expert Rev Clin Immunol 2021; 17: 1091–1101. [DOI] [PubMed] [Google Scholar]
- 78. Baughman RP, Meyer KC, Nathanson I, et al. Monitoring of nonsteroidal immunosuppressive drugs in patients with lung disease and lung transplant recipients: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2012; 142: e1S–e111S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chartrand S, Swigris JJ, Stanchev L, et al. Clinical features and natural history of interstitial pneumonia with autoimmune features: a single center experience. Respir Med 2016; 119: 150–154. [DOI] [PubMed] [Google Scholar]
- 80. Barnes H, Holland AE, Westall GP, et al. Cyclophosphamide for connective tissue disease-associated interstitial lung disease. Cochrane Database Syst Rev 2018; 1: CD010908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rojas-Serrano J, Herrera-Bringas D, Pérez-Román DI, et al. Rheumatoid arthritis-related interstitial lung disease (RA-ILD): methotrexate and the severity of lung disease are associated to prognosis. Clin Rheumatol 2017; 36: 1493–1500. [DOI] [PubMed] [Google Scholar]
- 82. Fraenkel L, Bathon JM, England BR, et al. 2021 American College of Rheumatology Guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol 2021; 73: 1108–1123. [DOI] [PubMed] [Google Scholar]
- 83. Manfredi A, Cassone G, Furini F, et al. Tocilizumab therapy in rheumatoid arthritis with interstitial lung disease: a multicentre retrospective study. Intern Med J 2020; 50: 1085–1090. [DOI] [PubMed] [Google Scholar]
- 84. Khanna D, Lin CJF, Furst DE, et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med 2020; 8: 963–974. [DOI] [PubMed] [Google Scholar]
- 85. Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116: 488–498. [DOI] [PubMed] [Google Scholar]
- 86. Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010; 363: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349: 36–44. [DOI] [PubMed] [Google Scholar]
- 88. Koukoulaki M, Jayne DR. Mycophenolate mofetil in anti-neutrophil cytoplasm antibodies-associated systemic vasculitis. Nephron Clin Pract 2006; 102: c100–7. [DOI] [PubMed] [Google Scholar]
- 89. Bradley B, Branley HM, Egan JJ, et al. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax 2008; 63: v1–58. [DOI] [PubMed] [Google Scholar]
- 90. Baughman RP, Drent M, Kavuru M, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med 2006; 174: 795–802. [DOI] [PubMed] [Google Scholar]
- 91. Paul C, Lin-Shaw A, Joseph M, et al. Successful treatment of fibrosing organising pneumonia causing respiratory failure with mycophenolic acid. Respiration 2016; 92: 279–282. [DOI] [PubMed] [Google Scholar]
- 92. Davison AG, Heard BE, Mcallister WAC, et al. Cryptogenic organizing pneumonitis. Q J Ofmedtdnt, New Ser LII 1983; 207: 382–394, https://academic.oup.com/qjmed/article/52/3/382/1585108 [PubMed] [Google Scholar]
- 93. Park JW, Curtis JR, Moon J, et al. Prophylactic effect of trimethoprim-sulfamethoxazole for pneumocystis pneumonia in patients with rheumatic diseases exposed to prolonged high-dose glucocorticoids. Ann Rheum Dis 2018; 77: 644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Liebling M, Rubio E, Ie S. Prophylaxis for pneumocystis jiroveci pneumonia: is it a necessity in pulmonary patients on high-dose, chronic corticosteroid therapy without AIDS? Expert Rev Respir Med 2015; 9: 171–181. [DOI] [PubMed] [Google Scholar]
- 95. Mecoli CA, Saylor D, Gelber AC, et al. Pneumocystis jiroveci pneumonia in rheumatic disease: a 20-year single-centre experience. Clin Exp Rheumatol 2017; 35: 671–673. [PubMed] [Google Scholar]
- 96. Hamada S, Ichiyasu H, Inaba M, et al. Prognostic impact of pre-existing interstitial lung disease in non-HIV patients with pneumocystis pneumonia. ERJ Open Res 2020; 6: 00306–02019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Buckley L, Guyatt G, Fink HA, et al. 2017 American College of Rheumatology Guideline for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res 2017; 69: 1095–1110. [DOI] [PubMed] [Google Scholar]
- 98. Miller A, Chan M, Wiik A, et al. An approach to the diagnosis and management of systemic vasculitis revised version with tracked changes removed. Clin Exp Immunol 2010; 160: 143–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schiopu E, Phillips K, MacDonald PM, et al. Predictors of survival in a cohort of patients with polymyositis and dermatomyositis: effect of corticosteroids, methotrexate and azathioprine. Arthritis Res Ther 2012; 14: R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Suzuki A, Kondoh Y, Brown KK, et al. Acute exacerbations of fibrotic interstitial lung diseases. Respirology 2020; 25: 525–534. [DOI] [PubMed] [Google Scholar]
- 101. Jeong H, Kaplan B. Therapeutic monitoring of mycophenolate mofetil. Clin J Am Soc Nephrol 2007; 2: 184–191. [DOI] [PubMed] [Google Scholar]
- 102. Bullingham RES, Nicholls AJ, Kamm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet 1998; 34: 429–455. [DOI] [PubMed] [Google Scholar]
- 103. Yabuki H, Matsuda Y, Watanabe T, et al. Plasma mycophenolic acid concentration and the clinical outcome after lung transplantation. Clin Transplant 2020; 34: e14088. [DOI] [PubMed] [Google Scholar]
- 104. Knoop C, Haverich A, Fischer S. Immunosuppressive therapy after human lung transplantation. Eur Respir J 2004; 23: 159–171. [DOI] [PubMed] [Google Scholar]
- 105. Coenen MJ, de Jong DJ, van Marrewijk CJ, et al. Identification of patients with variants in TPMT and dose reduction reduces hematologic events during thiopurine treatment of inflammatory bowel disease. Gastroenterology 2015; 149: 907–917. [DOI] [PubMed] [Google Scholar]
- 106. Heap GA, Weedon MN, Bewshea CM, et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet 2014; 46: 1131–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Taha N, Hosein K, Grant-Orser A, et al. TPMT and HLA-DQA1-HLA-DRB genetic profiling to guide the use of azathioprine in the treatment of interstitial lung disease: first experience. Pulm Pharmacol Ther 2021; 66: 101988. [DOI] [PubMed] [Google Scholar]
- 108. Vlaovic P, Jewett MAS. Cyclophosphamide-induced bladder cancer. Can J Urol 1999; 6: 745–748, http://www.ncbi.nlm.nih.gov/pubmed/11178599 (accessed 17 April 2021). [PubMed] [Google Scholar]
- 109. West NJ. Prevention and treatment of hemorrhagic cystitis. Pharmacotherapy 1997; 17: 696–706. [PubMed] [Google Scholar]
- 110. Carden PA, Mitchell SL, Waters KD, et al. Prevention of cyclophosphamide/cytarabine-induced emesis with ondansetron in children with leukemia. J Clin Oncol 1990; 8: 1531–1535. [DOI] [PubMed] [Google Scholar]
- 111. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–2082. [DOI] [PubMed] [Google Scholar]
- 112. King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2083–2092. [DOI] [PubMed] [Google Scholar]
- 113. Du Bois RM, Weycker D, Albera C, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med 2011; 184: 1382–1389. [DOI] [PubMed] [Google Scholar]
- 114. Paterniti MO, Bi Y, Rekić D, et al. Acute exacerbation and decline in forced vital capacity are associated with increased mortality in idiopathic pulmonary fibrosis. Ann Am Thorac Soc 2017; 14: 1395–1402. [DOI] [PubMed] [Google Scholar]
- 115. Oldham JM, Lee CT, Wu Z, et al. Lung function trajectory in progressive fibrosing interstitial lung disease. Eur Respir J 2021: 2101396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2022; 205: E18–E47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ryerson CJ, Abbritti M, Ley B, et al. Cough predicts prognosis in idiopathic pulmonary fibrosis. Respirology 2011; 16: 969–975. [DOI] [PubMed] [Google Scholar]
- 118. Khadawardi H, Mura M. A simple dyspnoea scale as part of the assessment to predict outcome across chronic interstitial lung disease. Respirology 2017; 22: 501–507. [DOI] [PubMed] [Google Scholar]
- 119. Kalluri M, Luppi F, Ferrara G. What patients with idiopathic pulmonary fibrosis and caregivers want: filling the gaps with patient reported outcomes and experience measures. Am J Med 2020; 133: 281–289. [DOI] [PubMed] [Google Scholar]
- 120. du Bois RM, Albera C, Bradford WZ, et al. 6-minute walk distance is an independent predictor of mortality in patients with idiopathic pulmonary fibrosis. Eur Respir J 2014; 43: 1421–1429. [DOI] [PubMed] [Google Scholar]
- 121. Lancaster LH. Utility of the six-minute walk test in patients with idiopathic pulmonary fibrosis. Multidiscip Respir Med 2018; 13: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Best AC, Meng J, Lynch AM, et al. Idiopathic pulmonary fibrosis: physiologic tests, quantitative CT indexes, and CT visual scores as predictors of mortality. Radiology 2008; 246: 935–940. [DOI] [PubMed] [Google Scholar]
- 123. Sumikawa H, Johkoh T, Colby TV, et al. Computed tomography findings in pathological usual interstitial pneumonia: relationship to survival. Am J Respir Crit Care Med 2008; 177: 433–439. [DOI] [PubMed] [Google Scholar]
- 124. Walsh SL, Calandriello L, Sverzellati N, et al. Interobserver agreement for the ATS/ERS/JRS/ALAT criteria for a UIP pattern on CT. Thorax 2016; 71: 45–51. [DOI] [PubMed] [Google Scholar]
- 125. Walsh SLF, Humphries SM, Wells AU, et al. Imaging research in fibrotic lung disease; applying deep learning to unsolved problems. Lancet Respir Med 2020; 8: 1144–1153. [DOI] [PubMed] [Google Scholar]
- 126. Wu X, Kim GH, Salisbury ML, et al. Computed tomographic biomarkers in idiopathic pulmonary fibrosis: the future of quantitative analysis. Am J Respir Crit Care Med 2019; 199: 12–21. [DOI] [PubMed] [Google Scholar]
- 127. Jacob J, Bartholmai BJ, Rajagopalan S, et al. Evaluation of computer-based computer tomography stratification against outcome models in connective tissue disease-related interstitial lung disease: a patient outcome study. BMC Med 2016; 14: 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jacob J, Bartholmai BJ, Rajagopalan S, et al. Mortality prediction in idiopathic pulmonary fibrosis: evaluation of computer-based CT analysis with conventional severity measures. Eur Respir J 2017; 49: 1601011. [DOI] [PubMed] [Google Scholar]
- 129. Jacob J, Bartholmai BJ, Egashira R, et al. Chronic hypersensitivity pneumonitis: identification of key prognostic determinants using automated CT analysis. BMC Pulm Med 2017; 17: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wells AU, Flaherty KR, Brown KK, et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases–subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med 2020; 8: 453–460. [DOI] [PubMed] [Google Scholar]
- 131. Mura M. Use of nintedanib in interstitial lung disease other than idiopathic pulmonary fibrosis: much caution is warranted. Pulm Pharmacol Ther 2021; 66: 101987. [DOI] [PubMed] [Google Scholar]