

Abstract
The purpose of the immune system is simple—protect the human organism from foreign (antigenic) invasion and resultant disease. The innate immune response (“our best friend”) does a great job of accomplishing that defense under most circumstances. But sometimes, innate immunity confronts an adversary (a pathogen) that overwhelms it and produces a “dysregulated,” adaptive immune response. The first clinical effect is acute inflammation with an array of familiar signs and symptoms (pain, redness, swelling, sometimes fever). If not reversed within days to weeks, this negative pathological condition progresses from the acute state to its more devastating successor, chronic inflammation (“our worst enemy”), and the progenitor of all human disease. This chapter presents the clinical, histological, and pharmacological stages and basic immunotherapeutic efforts to arrest and reverse the “inflammatory cascade.” Unsuccessful efforts allow the adaptive immune system and chronic inflammation to begin an inexorable, pathological course toward autoimmune disease, cancers, and the ravages of infectious pandemics like COVID19.
Keywords: Acute inflammation, Adaptive immunity, Antigenic, Chronic inflammation, Dysregulate, Fever, Pain, Redness, Swelling
It's not whether you get knocked down. It's whether you get up again.
Vince Lombardi.
1. The path to “dysregulation”
Wouldn't it be nice if our body won all its battles against antigens? Needless to say, life doesn't work that way. Sometimes, the innate (natural) immune system can't quite handle the load. Maybe, for some genetic, pathological (disease) or environmental reason (e.g., smoking—I always love to include that) your immune system is compromised (“immunocompromised”) or weakened or suppressed (“immunosuppression”). Perhaps the antigen is not being removed effectively (persistent cause—smoking, pollution, allergic to something), or it keeps reoccurring (reexposure) as the innate system tries to eliminate it. Or maybe the antigen is over abundant or too pernicious (virulent) for the innate immune response to overcome it. In such conditions, after a few days to a week of feeling “not so great,” the strength of the human immune system begins to demonstrate more aggressive activity called the “adaptive immune response.” All in all, this adaptive immune system serves as a powerful defender and protector … to a point.
Adaptive immunity is more vigorous than the innate form. Its intensity and duration are controlled by the patient's genetic makeup (notice how I move from “person” to “patient” about here?). As adaptive immunity advances, it begins to disrupt the homeostasis (maintenance of a stable condition) of your body and your overall immune system. This disruption is also referred to as “dysregulation” of the immune system.
But notwithstanding such disruption, four specific mechanisms that produce positive effects in adaptive immunity are believed to still be at work. First is something called “feedback inhibition” where removal of the antigen reduces its innate immune stimulus and thus decreases production of antibodies and cytokines, effectively reducing and reversing the response. The second is the neuroendocrine and neurogenic pathways that modulate cytokine production and reduction (“downregulation”) to control the immune response. This complex neurological control mechanism is the ultimate regulatory hierarchy for chronic inflammation and will be discussed in Chapter 4, page XXX. The third mechanism is when T-suppressor cells (remember TS, also referred to as T regulatory or Treg cells) reduce T-helper cells (TH) and thus produce a commensurate reduction in B-cell activity (which is controlled by T-helper cells, as you'll recall from the innate immune discussion in Chapter 1). It's a bit of “connecting the dots,” but the result is worth the trip, that is, a reduction in a dysregulated immune response.
The fourth mechanism for reestablishing a regulated immune system is a very complex genetic mechanism creating a system of idiotype antigen-specific B cells called the “Idiotype-Anti-idiotype Regulatory Circuit. This is a process that selfgenerates, through genetic cloning, creating its own immunogenic stimuli that induces anti-idiotype-specific antibodies (“antibodies 1, 2, etc.”) that establish the antibody idiotype-specific regulatory circuit. Confused? Don't worry, so are many scientists who have been studying this extraordinary complex process for years. The process remains the center of much immunology research that has far-reaching implications, especially in vaccines and cancer research. Because of its importance and value in infectious disease (i.e., vaccines for pandemics) and its value in cancer research, let's postpone its description here and revisit this important aspect of adaptive immunity with a fuller discussion (with illustrations) in Chapter 6 on cancer and Chapters 7 (on infectious pandemics).
2. Acute inflammation
Failure to remove an offending antigen in a timely manner, or malfunction of any one of the four mechanisms described above (feedback inhibition, neurogenic modulation, TS cells, and genetic cloning) can lead to a pathophysiological response (i.e., an abnormal bodily process) resulting in a clinical effect you have heard about and undoubtedly experienced yourself, called acute inflammation. The word “acute” refers to symptoms and conditions of rapid onset and usually of short duration. This “dysregulation” of the immune system could lead to a more prolonged, destructive process referred to as chronic inflammation (chronic simply meaning a prolonged condition). This later level of adaptive immunity, that is, chronic inflammation has potentially devastating consequences and is the process I mentioned in the Preface as the basis of all disease. We will be taking this up in detail in Section 2, Chapter 4.
These advancing clinical (inflammatory) effects of adaptive immunity and their various potential endpoints are the next level of active immunity (remember that was the generic term introduced in Chapter 1 for innate and adaptive immunity). So, adaptive immunity represents the second half of our earlier stated paradox about the immune system (“best friend and worst foe”). Adaptive immunity could indeed be considered “our most dangerous foe.” (NB: All inflammation, acute and chronic is characterized in medical terminology by the suffix “ … i t i s.” Thus, any condition mentioned, henceforth, under any disease category with the suffix “ … itis” [and there are a lot more of them coming up] should be considered an inflammation.)
2.1. Immunopathophysiology (“dysregulation” and hypersensitivity)
We can look at adaptive immunity as a race to eliminate the bad guy (antigens), a competition which, in most cases (given an otherwise healthy person), the combined innate and adaptive immune systems will win. If, however, the underlying health of the patient is not adequate enough to sustain the activity of the adaptive immune response, things could begin to deteriorate or “dysregulate.”
Among the cellular reactions associated with the adaptive immune response (all those T's and B's and so on), there are four classic “types” of “hypersensitivity reactions” (Coomb's Gell classification referred to as Type I to Type IV—remember Fig. 1.6 and Satchel Paige?). These hypersensitivity reactions (also referred to as “overreactions” because the immune system is now beginning to go beyond its basic “protective” functions) precipitate the next phase of immunity or the “inflammatory cascade” (diagrammed and discussed below in Fig. 2.1 ). Each overreaction is induced by different types of antigen categories and each characterized by specific cellular responses and “types” of immunoglobulin antibodies [1]. The four types (always designated by Roman numerals—why? I have no idea) include
Type I: The immediate, allergic (or anaphylactic) hypersensitivity response;
Type II: The cytotoxic hypersensitivity reaction;
Type III: The immune-complex hypersensitivity reaction; and
Type IV: The cell-mediated, delayed reaction
Figure 2.1.
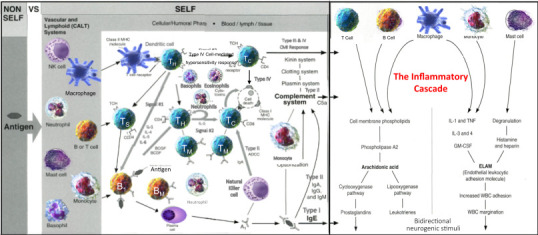
The inflammatory cascade—diagram #5.
The inflammatory cascade is a pharmacological array of cellular and humoral elements producing a pharmacodynamic process leading to the clinical manifestations of acute inflammation.
Source: Louis J Catania © 2022.
All four of these hypersensitivity reactions (I like the term “overreaction” better because it more accurately describes these reactions, but “hypersensitivity reaction” is the scientifically preferred term, so I'll stick with it … but cheat occasionally with a parenthetical “overreaction” if I think it helps the discussion). These hypersensitivity reactions usually commence during the acute inflammatory stage of the adaptive immune process (which we pretty much described at the end of Chapter 1 (page 20) under the section “From regulated to dysregulated—from innate to adaptive immunity”). Back there we described the more challenging molecular biology of the process which I'm hoping you understood. Now we'll get to the interesting clinical process which you'll be able to relate to far more than the cellular-chemical stuff. This is the manifestation (the pathophysiology) of all that molecular activity. It's the acute clinical phase or the reactions people see (and feel) as their immune system becomes more real to them (and us).
But allow me to forewarn you at this point. The more advanced and serious pathophysiological characteristics for each of the four types of hypersensitivity reactions will manifest far more distinctly during the chronic or continuing inflammatory process. I will describe each of the hypersensitivity types here, but we will be revisiting them in more depth in the discussion on chronic inflammation in Chapter 4. And trust me. Given the magnitude of the chronic inflammatory process on human health, you'll want to understand inflammation at its earlier (simpler) acute phases to better appreciate its profound effects on human health if and when it progresses. So now, given your Chapter 1 basis in the anatomy, physiology, immunology, and pathophysiology of active immunity (remember that generic term for combined innate and adaptive immunity?), let's discuss the nexus between all that immunobiology and the first classic, clinical level of immunology. First some quick descriptions of the hypersensitivity reactions themselves [2].
2.1.1. Type I: the immediate, allergic (or anaphylactic) hypersensitivity response
This is the most common reaction, produced by an antigen referred to as an “allergen.” Examples of this response include hay fever, eczema, hives, asthma, food allergy, insect bites and stings, dust, pollen, and on and on. Like its antigen cousin, the allergen can be inhaled, ingested, or enter through the skin. After a susceptible person is exposed to an allergen, the body starts producing a large quantity of IgE antibodies. This results in the reoccurrence of the allergic (anamnestic) response, sometimes with increasing intensity with each reexposure to the allergen. Included among its cytokines, are histamine, eosinophil complement, and heparin, which along with other inflammatory symptoms, produce itching. With the allergic response, symptoms can also include sneezing, and congestion from the release of histamine that is caused by IgE degranulating mast cells (cells that have histamine granules on their surface). In its most severe form, allergic hypersensitivity can produce a life-threatening condition called anaphylaxis (massive accumulation of Type I cytokines) that can cause blood pressure to drop suddenly, and airways narrowing, thereby blocking breathing (anaphylactic shock) [3].
2.1.2. Type II: the cytotoxic hypersensitivity reaction
Type II cytotoxic hypersensitivity reaction (sometimes referred to as antibody-dependent cell-mediated cytotoxicity [ADCC]) involves mainly IgM or IgG antibodies, natural killer (NK) cells and macrophages directed against antigens that cause cell destruction by complement activation (back in Chapter 1, Fig. 1.6). Type II reactions occur within hours of exposure and usually last for a day or so, but could be prolonged. Common forms include blood transfusion reactions and drug sensitivities such as penicillin.
2.1.3. Type III: the immune-complex hypersensitivity reaction
Type III, immune-complex hypersensitivity, is a reaction mediated by the formation of antigen-antibody aggregates called “immune complexes.” These reactions are not mediated by antibodies but rather involve the interaction of T cells, monocytes, and macrophages, sometimes (just to confuse us) also referred to as cell-mediated reactions (Type IV). The reaction can take hours, days, or even weeks to develop (into chronic inflammation), depending on whether or not there is anamnestic memory from previous antigen.
The smaller immune complexes that might become antigen bound to an antibody at the antigen binding site (called an epitope) are not cleared by macrophages and APCs and tend to insert themselves into small blood vessels, joints, and kidneys. These immune complexes can cause an array of symptoms (localized arthus [local vasculitis] inflammatory reactions or urticaria [histamine reactions from degranulated mast cells]. Type III diffuse inflammatory reactions are associated with autoimmune diseases, for example, systemic lupus erythematosus [SLE], rheumatoid arthritis, etc.—see Chapter 5).
2.1.4. Type IV: the cell-mediated, delayed reaction
Type IV, cell-mediated, delayed reaction takes several days to develop and does not involve antibodies. Rather, the reaction involves the activation of phagocytes (pathogen specific), NK cells, antigen-specific cytotoxic T-lymphocytes, and the release of various cytokines in response to an antigen (signal #2 in Fig. 1.5 displays this effect). Developing over a two-to-three-day course, in Type IV hypersensitivity reactions CD4 plus TH cells recognize antigen in a complex with Class 2 MHC (see Chapter 1, page 11) APCs.
This reaction provides immune protection through either humoral (body fluids, serum, and chemicals, e.g., interferons, interleukins—signal #2) or through cell-mediated immunity where the protective immune function is associated with cells. These cells include CD4 cells, NK cells, and helper TH cells that provide protection against pathogens, and cytotoxic TC cells that cause death by apoptosis (programmed cell death [4]) without the use of cytokines. Together the reactions are most effective in removing virus-infected cells, but also participate in defending against fungi, protozoans, cancers, and intracellular bacteria. Type IV hypersensitivity also plays a role in temporal arteritis, Hashimoto's thyroiditis, symptoms of tuberculosis, celiac disease, graft-rejection, and chronic transplant rejection.
2.2. The inflammatory cascade
OK, now back to the straight clinical stuff regarding presentations in acute inflammation. Consistent with the definition of its name, acute inflammation and its associated clinical manifestations usually occur within hours to days (minutes in Type I immediate, allergic IgE response) with ulceration occurring shortly thereafter if untreated. Somewhat ironic in this sequence of the clinical inflammatory process is that all of the effects up to (but not including) ulceration, are actually part of the immune system's healing process (an added paradox of a “best friend—worst enemy” combination!). The adaptive immune response is using many tools, like the cellular components (T and B cells), antibodies, chemicals (cytokines), and more in amounts not normal (“pathophysiological”) to your body. Here, the regular (“physiological”) activity is working diligently to resolve the “pathological” (disease) process in your body. But those valiant efforts are also producing abnormal byproducts like excessive cellular debris and added chemicals called “pro-inflammatory cytokines.” Accumulation of these byproducts is itself a basis for continuation of the acute inflammatory pathophysiological process.
Acute inflammation is a fairly efficient immunopathological defense mechanism of the adaptive immune system. (You'll notice how the “immuno” prefix is beginning to show up in more and more descriptive prefixes, e.g., “immunopathological.” This will be increasing throughout the rest of this book with many more “immuno …” labels. But, I digress—yet again.) This broadly defined, nonspecific, acute inflammatory, immunopathological process produces an observable clinical response referred to as the “inflammatory cascade.”
Fig. 2.1 (Diagram #5) is the continuum or the flow diagram [5] (started with Fig. 1.3 in Chapter 1) that I warned you about (with more to come). The left side of the inflammatory cascade in Fig. 2.1 represents the biochemistry and “pharmacodynamics” (how drugs work and their effects) most associated with acute inflammation and its medical treatments. You'll notice how the drug therapies (corticosteroids and NSAIDs [nonsteroidal antiinflammatory drugs]) produce their effects at particular sites within the pharmacological tree, blocking chemical pathways (phospholipids in the case of corticosteroids and NSAIDs for the cyclooxygenase and lipoxygenase pathways), the chemicals that lead to the dilation of blood vessels (vasodilation or the first level of acute inflammation—see Fig. 2.2 ) and pain (produced by chemical prostaglandins). Also, the chemicals, histamine and heparin, and mast cell molecule degranulation associated with the Type I allergic hypersensitivity reaction are immunomodulated within this pharmacological tree as well (right side). The balance of the biochemistry and molecular biology on the right side of this pharmacological diagram relate more to chronic inflammation and their relevance will be describe in greater detail in Chapter 4. Also, bidirectional neural stimuli (from innate immunity) continue its downregulating activity which may mitigate or even reverse the inflammatory, adaptive immune activity. But its label, “bidirectional,” also indicates its progressive, neurogenic inflammatory stimuli (discussed further in Chapter 4, page 87).
Figure 2.2.
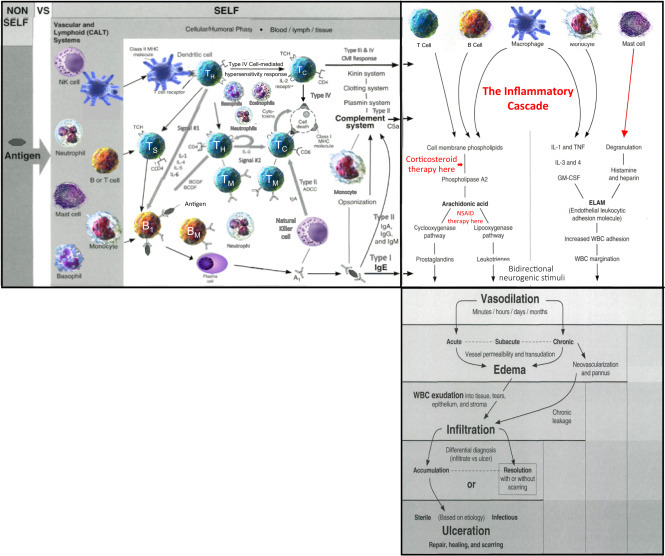
Clinical manifestations of acute inflammation—diagram #6.
Acute inflammation includes the clinical manifestations of vasodilation (rubor), edema (tumor and dolor), infiltration (tumor and dolor) and, if not timely controlled, ulceration (functio laesa).
Source: Louis J Catania © 2022.
Acute inflammation can occur anywhere, internally (e.g., acute gastritis, hepatitis, etc.) or externally (e.g., contact dermatitis, keratitis, etc.) producing the inflammatory cascade. The “cascade” includes the pharmacological components producing inflammation and their subsequent clinical manifestations. The pathophysiology and histopathology (tissue reactions) of acute inflammation as defined in the inflammatory cascade are being produced by the cells and mediators of the adaptive immune response (so, believe it or not, all of Chapter 1 does have some relevance) resulting in a dynamic unfolding of clinical events (Fig. 2.2—Diagram #6: the continuum or flow from Fig. 2.1—I think by now you can begin to see a flow diagram of active immunity emerging from the earliest APC (Fig. 1.3), through innate immunity (Figs. 1.4 and 1.5), then adaptive immunity, and now inflammation (Fig. 2.2). And believe it or not, there's yet still more to come).
3. Clinical considerations in acute inflammation
The observable signs and symptoms that flow from the inflammatory cascade (Fig. 2.2) are classic and familiar to all of us. We certainly were not the first generation nor even the first (or second) century to recognize or describe them. They are so classic that they were first observed as far back as 450 BCE and then documented and named by a Greek forefather of medicine, Celsius, in 38 AD (I'll give you the specific names, one by one in the upcoming description. Pretty excited, I'll bet!). The classic signs and symptoms of acute inflammation are quite predictable with almost any degree of early inflammation. With proper treatment approaches (described below), unless they reach the level of ulceration, acute inflammation usually diminishes and resolves relatively rapidly with no consequences. When unresolved, due to a lack of timely treatment, no response to treatment or no treatment at all, the condition can spiral into a destructive organ and/or tissue ulcerative process with tissue changes and scarring and more so, into an insidious and destructive chronic inflammatory stage (Chapter 4). But let's assume we're diligent patients. We understand the implications of acute inflammation and mostly, we just want to feel better.
3.1. Signs and symptoms
Acute inflammation is the immune system's attempt to help rather than hurt (as I mentioned previously, a bit of a paradox in and of itself) by reestablishing a homeostatic (stable, normal, balanced) state in the tissue(s) or organ(s) involved. Thus, each of the classic clinical acute inflammatory features has a physiological goal to achieve. Notwithstanding these worthy objectives, these physiological effects produce a disequilibrium (a “dysregulated”) subjective (pain and discomfort) and objective (physiologic) response. So, while acute inflammation is trying to help us, we're not all too happy with the help.
We know from our earlier discussion in Chapter 1 (page 13) about active immunity that we need a lot of WBCs (lymphocytes) and their accompanying antibodies to attack and remove antigens. Well, as you recall, most of those WBCs, particularly the neutrophils (remember, “the first responders”?) are in the blood (inside the circulatory system's blood vessels). Upon getting the immune signals from activated macrophages, TH cells (APCs), inflammatory mediators, cytokines, etc., the main phalanx of cells, countless neutrophils, lymphocytes, and NK cells must now get out of the blood and into the tissue where they are needed. This is accomplished by a process of vasodilation (increased caliber of the blood vessels) that opens the pores in the blood vessel walls. This allows the WBCs to exit the blood vessels (diapedesis—described below) and migrate to the needed area, where the antigen is attacking. So far, so good.
Together, the dilated blood vessels produce redness (aka “rubor” from the red blood cells [RBCs]—our first Celsius term) and the increased blood flow creates heat (aka “calor”—our second Celsius term). Though poorly understood, it is believed that the inflammatory mediators associated with this adaptive immune response send signals to the brain center (hypothalamus) that controls body temperature. The nervous system sends chemical signals, “dysregulators” that generate the heat to the involved tissue (or organ). Interleukin is a big player in this process (it's called the “leukocytic pyrogen”). These signals or pyrogenic mediators contribute to the direct calor at the inflammatory site as well as a fever, often associated with inflammation. This is a great illustration of how the immune mediators, especially interleukins and interferons, act as the “wireless” (Wi-Fi, if you will) communication system for immune responses (i.e., nervous system talking to the immune system—see Chapter 4, page 84).
All of this also suggests, as mentioned above, that an overabundance of these mediators and thus, their cumulative stealthy signals, could become overly aggressive and produce an “attack” on the body rather than beneficial, communicating signals. This could generate an antigenic response, or more appropriately termed an “autoantigenic” (signals coming from within the body) response (another immune paradox—seems like they're beginning to pile up since adaptive immunity kicked in). I'll bet you can see where I'm going with that evocative comment. Autoimmunity—“the mother of all immune system paradoxes!” Right on and more on the serious risk of autoimmunity in Chapter 5.
Meanwhile, back at those dilated blood vessels, protein-rich plasma is escaping. While aiding in the tissue defense, that plasma (fluid serum) is also producing edema or tissue swelling (aka “tumor”—third Celsius term). There actually is a reason and some good and some bad in this edema effect (usual, when you let nature call the shots). The plasma fluid is loosening the tissue and allowing the WBC cellular elements to migrate more effectively to the antigenic site (that's good) while continually accumulating (aka “infiltration”—not so good). Certainly, these plasma and cellular effects are beneficial, but as they accumulate, they also are becoming increasingly more apparent physically (e.g., pus), more uncomfortable to the patient, and more “foreign” (remember, those WBCs don't belong in that tissue, let alone in large quantities). This increasing loss of tissue or organ homeostasis is generating other chemical mediators as well (e.g., prostaglandins) that are increasing pain symptoms (aka “dolor”—and that's Celsius' last contribution to inflammation … I think. You never know with these Romans. He probably figured out some way to measure temperature. Naa. That was Fahrenheit, wasn't it?).
Hopefully, all of these activities in the tissue(s) or organ structure(s), especially the potent cellular elements like neutrophil phagocytosis (pathogen ingestion and externally appearing clinically as “pus”) and NK cells along with the T and B cells, are dispatching their immunologic duties of neutralizing the antigen in a timely manner (the mixed leukocytic reaction or MLR). The longer it takes them, however, the more at risk adjacent, healthy tissue becomes as cellular debris and neutrophils begin to interpret otherwise healthy (“self”) surrounding tissue as “foreign.” This possible, idiosyncratic, paradoxical effect can lead to that normal, otherwise healthy tissue destruction and loss, and eventual scarring (aka “ulceration”). Along with this tissue or organ destruction (including potential DNA disruptions), permanent loss of tissue or organ function (aka “functio laesa”) can occur. So ultimately, what started out as a positive, physiological, mitigating, rectifying immune process, has become “an enemy within us.”
3.2. Treatment approaches
The variety of treatment considerations (manipulations of the pharmacology in Fig. 2.3 —Diagram #7) related to the inflammatory cascade is extensive, but as stated numerous times up to this point, the first treatment is always the removal of the cause (the antigen). Such removal can range from simple hygiene; to antibiotics or antivirals for a bacterial or viral infectious antigen respectively (external or internal) and let's not forget, especially in this age of pandemics, vaccines as anti-virals. Type I reactions (allergy) includes removal of the allergen, antihistamine, decongestants, and mast cell stabilizing drugs. In more severe reactions (including anaphylaxis), topical and oral corticosteroids and injectable epinephrine may be required. In all forms of acute inflammation, cold (wet or ice) compresses are enormously valuable in reducing inflammatory edema by producing vasoconstriction and commensurate reduction in vasodilation of associated blood vessels (i.e., rubor with tumor or edema) that are supplying inflammatory WBCs through the blood vessel wall (diapedesis) to the affected site.
Figure 2.3.
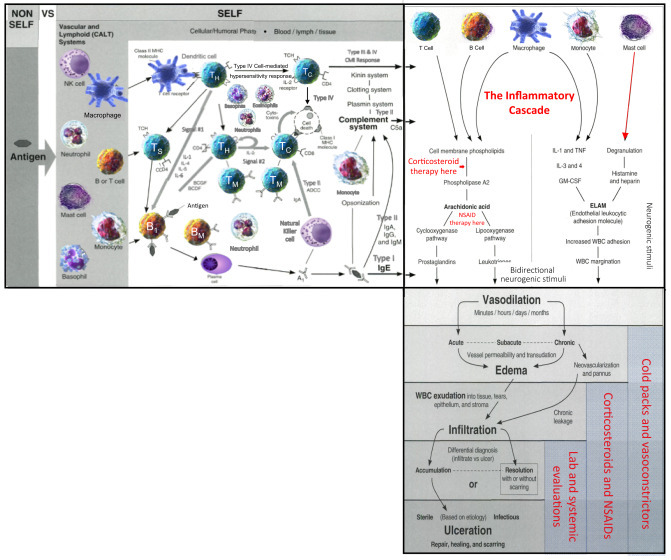
Treatment considerations in acute inflammation—diagram #7.
The clinical manifestations of the inflammatory cascade (vasodilation, edema, infiltration, ulceration) are treated generically with removal of the cause (antigen); cold compresses (for vasodilation and edema); corticosteroids for infiltration; and aggressive treatments for ulceration).
Source: Louis J Catania © 2022.
This critically important diapedesis process is instigated by the endothelial leukocytic adhesion molecule (ELAM on the right column of Fig. 2.3). A combination of cytokines induces the ELAMs to stimulate leukocytic (neutrophils in particular) adhesion to the endothelial vessel wall. Then cellular migration associated with surface proteins and chemokines produce blood vessel wall permeability (diapedesis) and extravasation (cell escape from the vessel) of the neutrophils. Fig. 2.4 illustrates this dynamic process. Once outside the blood vessel, as we've been describing, the circulating leukocytes migrate to the disease site(s), where they are activated by various cytokines and chemokines secreted by the macrophages and dendritic cells to help neutralize the antigen. But, without the assistance of removal of the cause by physical or antiinfective therapies, those inflammatory WBCs and chemical mediators become potentially destructive to normal tissue.
Figure 2.4.
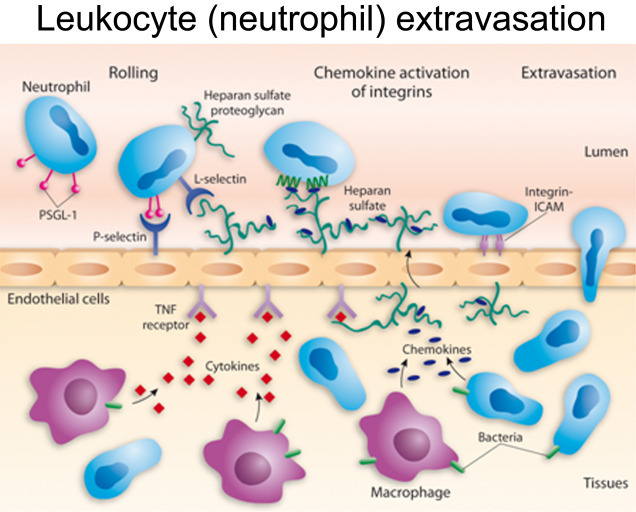
Leukocyte (neutrophil) extravasation.
Inflammatory vasodilation (redness or rubor) opens the pores of the blood vessels to allow WBCs (neutrophils) to “infiltrate” into the tissue (diapedesis) and attack the invading antigen.
Source: National Institute of Health, U.S. Dept. of Health and Human Services.
Topical and systemic corticosteroids are used to mitigate signs and symptoms in more acute inflammatory reactions (from Fig. 2.2). These drugs produce “masking effects” (reduction of signs and symptoms) but they are palliative rather than curative as is sometimes thought. NSAIDs (nonsteroidal antiinflammatories such as aspirin, ibuprofen, or naproxen) provide pain relief (as antiprostaglandins—from Fig. 2.3). It's also important to remember that as immunosuppressive agents, corticosteroids are reducing the defense mechanisms of the inflammatory reaction and thus its beneficial effects (WBCs, phagocytosis, etc.) against the invading antigen (a therapeutic paradox). When this antigen is an infectious agent, the steroid is reducing its immune mitigation and allowing it to survive, if not proliferate. This also increases the potential for additional opportunistic infectious agents to attack. Thus, corticosteroids have an attendant risk of secondary and/or increasing infection. Need I say, astute clinical diagnosis and management is essential in all steroid usage. This same risk will be mentioned again in Chapter 4 when we discuss immunosuppressive drugs.
Injury repair and stress reduction (physical, physiological, psychological) therapies are also valuable. Beyond these therapies, treatment is palliative and directed to the involved site (joint, muscle, internal organ, skin, etc.) to reduce the inflammatory process and mitigate the pain (assuming the antigen has been removed). Additional therapeutic measures include nutritional and vitamin supplements, omega-3 sources, compression, stress reduction, and exercise. We can do all sorts of additional chapters (and books) on these “additional therapeutic” measures and their benefits which are voluminous. But I'll have to leave that to other more knowledgeable experts and resources on those subjects. But you really should investigate such measures for improved health and wellness, especially exercise where the literature is exploding on its advantages in sickness and in health.
4. Using the adaptive immune response to help and prevent disease
We have been talking about “our friends,” the active, innate, and adaptive immune systems being our defense against foreign, nonself antigens and 99+% of the time doing a great job. In such approaches, we call upon the immune system as we incite, or better still “inspire” it to rally to a preemptive protection against antigens, mostly infectious. The COVID-19 pandemic painfully awakened us to the critical importance of immunology and genetics, especially as we race to find an antiviral drug and vaccines to save humanity. In such cases, we are attempting to modulate, manipulate, and enhance the adaptive immune system to use its powerful cellular and humoral properties to provide a defense against the invading antigen (specifically, the SARS-CoV-2 virus). We might clone neutralizing antibodies (monoclonal antibodies) to bolster the immune attack on the antigen. We might extract programmed antibodies from previously infected patients (convalescent plasma) as preventive therapy. We might try to trick the antigen with modified doppelgangers (copies of itself—in this case, the infectious agent) or introduce an altered form of another viral substance (e.g., an attenuated infectious agent or a recombinant viral vector) to stimulate a controlled production of antibodies to effect an immunity. And thus, we must consider vaccines and vaccination or immunization, (see Chapter 7) a process by which, preemptively (or thereafter) we can beneficially “manipulate” the immune system in multiple ways.
We've gotten substantially better at all these approaches, thanks to dedicated researchers, improved genetic technologies (next-gen sequencing or NGS—see Chapter 3 on genetics), and certainly the help of artificial intelligence (AI) that uses big data analytics (immunoinformatics—see page 210) to search billions of databases for virtually instant answers and expedited solutions. All of these applications of the adaptive immune system to help and prevent disease will be thoroughly addressed in Chapter 5 regarding therapeutic considerations for autoimmune diseases, in Chapter 6 regarding monoclonal antibodies in the treatment of cancers, and in Chapter 7 regarding immunotherapies for COVID-19. But I felt it would be worthwhile during this basic science discussion on adaptive immunity to accentuate its importance, regarding its value in clinical healthcare. It's kind of a “love–hate” scenario and “rubber meets the road” analogy regarding the benefits we enjoy from adaptive immunity and receive as patients from the work the basic scientists provide.
5. The path to chronic inflammation
Through proper diagnosis and removal of the cause, adaptive immunity paradoxically can remain a human biological “friend.” Such immunity's clinical manifestation of acute inflammation, in spite of a little discomfort, will usually result in health and wellness. But, if not managed, controlled, and resolved within a reasonable period of time (weeks to months at most), the adaptive immune system can advance to a condition called “chronic inflammation” or truly, “Enemy #1.” This form of inflammation differs from acute inflammation as we mentioned in the beginning of this chapter, in its cellular pathology and clinical symptoms ranging from nothing at all to those of acute inflammation and much worse. To further confuse the issue, it should be noted that, though poorly understood, chronic inflammation can also develop spontaneously, that is without an apparent antigen and acute inflammatory precursor episode.
The development of chronic inflammation could reasonably be considered an advanced form of acute inflammation. But its pathogenesis, histopathology, immunochemistry, and most of all, its clinical course contradicts such a clinical evolution. Rather, chronic inflammation provides irrefutable clinical confirmation that it is a distinct and unique clinical entity, notwithstanding the name it shares with acute inflammation. At the point of clinical diagnosis of chronic inflammation, we can no longer consider the immune system “a friend.” We are now faced with the dark side of our immune system, or “the enemy within us.” That discussion begins with chronic inflammation (Chapter 4) as a distinct and separate disease entity, while often evolving from its “distant cousin,” acute inflammation. Chronic inflammation now devolves into an array of serious abnormalities from autoimmune diseases, cancers and beyond which we will address in Section 2, “The enemy within us.”
But first, as promised, in the Introduction to this Section 1, I want to set the tone and provide information needed for a full understanding of Section 2 discussions. This last chapter in Section 1 (Chapter 3) will introduce, in rather abbreviated fashion, the basic science and concepts of genetics, specifically immunogenetics and immunogenomics. This will lay the groundwork for the substantial amount of genetic science that will appear throughout Section 2. My hope is that the Chapter 3 information on genetics will provide comfort for you in its Section 2 clinical applications. I know we can get through this together, so let's keep going.
6. Brief research summaries on the innate and adaptive immune system
(Reference citations for each research study presented below can be found in the corresponding footnote. Also, a listing of available scientific reference sources and databases used by the author are included in the book's Acknowledgments.)
As described in Chapter 1 on innate immunity, research in the field of immunology (and genetics), especially with the help of AI, is rapidly advancing our understanding of the immune system and its clinical effects on our bodies. Here again, research regarding adaptive immunity will give more practical examples of the benefits to humanity being realized through immunology research. The following are three examples of such immunological research.
-
1.
The diagnosis of acute appendicitis is challenging, especially due to the frequently unspecific clinical picture it presents as. Inflammatory blood markers and imaging methods like ultrasound are limited because they have to be interpreted by experts, yet still do not offer sufficient diagnostic certainty. A recent study presents a method for automatic diagnosis of appendicitis as well as the differentiation between complicated and uncomplicated inflammation using values and parameters that are routinely and unbiasedly obtained for each patient with suspected appendicitis. A total of 590 patients (473 patients with histologically confirmed appendicitis and 117 with negative histopathological findings) were analyzed retrospectively with modern AI algorithms. Results revealed the capability to prevent two out of three patients without appendicitis from receiving “useless” surgery as well as one out of three patients with uncomplicated appendicitis. This clinical study and outcome has the potential to change the current therapeutic approach for appendicitis and it demonstrates the capability of AI algorithms to significantly improve inflammatory disease diagnostics even based on routine diagnostic parameters [6].
-
2.
The inflammatory response runs through all stages of acne. It involves both innate and adaptive immunity. A study aimed to explore the candidate genes and their relative signaling pathways in inflammatory acne used AI data-mining analysis. Aberrant, differentially expressed genes (DEGs) and pathophysiological pathways involved in acne were identified using bioinformatic (AI) analysis. There were 12 DEGs identified and the pathways included chemokine signaling pathway, cytokine-cytokine receptor interaction, and cell-mediated phagocytosis. Discovery of these pathways will serve as a basis for further understanding the pathogenesis and potential therapeutic targets of inflammatory acne [7].
-
3.
Innate and adaptive immune memory is defining features of the adaptive immune system, although their induction is distinctly different. Innate immune memory, sometimes referred to as “trained immunity” is a primitive form of adaptation and provides an increased but nonspecific response to reinfection. Adaptive immune memory is more advanced, with an increased magnitude of response mediated through epigenetic (see Chapter 3, page 62) changes, as well as specific mediation through gene recombination. An integrated model of innate and adaptive immune memory is important for a better understanding of host defense, and for identifying the most effective approaches to modulate it for the benefit of patients with infections and immune-mediated diseases [8].
Chapter highlights (key point and paradoxical-related information)
-
1.
As innate immunity advances to adaptive immunity, “our worst enemy,” immunity moves from “friend to foe.”
-
2.
As adaptive immunity advances, it begins to disrupt the homeostasis (maintenance of a stable condition) of your body and your overall immune system. This disruption is referred to as “dysregulation” of the immune system.
-
3.
Failure to remove an offending antigen in a timely manner or malfunction of any one of four mechanisms (feedback inhibition, neurogenic modulation, TS cells, and genetic cloning) leads to adaptive immunity.
-
4.
This “dysregulation” of the immune system produces the clinical condition of acute inflammation.
-
5.
In dysregulation, there are four classic hypersensitivity reactions (Types I to IV) which induce the ”inflammatory cascade” of biochemical reactions representing acute inflammation.
-
6.
Acute inflammation can be considered a “healing” process through vasodilation, edema, WBC infiltration all of which also produce negative clinical signs (fever, swelling) and symptoms (fever, pain). Furthermore, acute inflammation can become destructive in the form of ulceration.
-
7.
Clinical features of the inflammatory cascade lead to physiological effects producing a disequilibrium and a “dysregulated” immune state.
-
8.
Disequilibrium produces physiological stress and an “autoantigenic” response further disrupting tissue homeostasis.
-
9.
Principal treatment of acute inflammation is removal of the antigenic cause with supportive and palliative (cold compresses, steroids, etc.) measures.
-
10.
Prolonged acute inflammation can lead to the destructive process referred to as chronic inflammation.
References
- 1.Dispenza M.C. Classification of hypersensitivity reactions. Allergy Asthma Proc. November 2019;40(6):p470–473. doi: 10.2500/aap.2019.40.4274. 4pp. [DOI] [PubMed] [Google Scholar]
- 2.Allarakha S., Suyog Uttekar P. What are the four types of allergic reactions? Med Net. August 13, 2020 [Google Scholar]
- 3.Allergy and the immune system. Johns Hopkins Health; 2019. [Google Scholar]
- 4.Jorgensen I., Rayamajhi M., Miao E. Programmed cell death as a defense against infection. Nat Rev Immunol. 2017;17:151–164. doi: 10.1038/nri.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catania L.J. McGraw Hill; 1996. Primary care of the anterior segment 2nd edition (textbook) pp. 22–82. [Chapter 2]. Clinical Considerations on Anterior Segment Pathology and Immunology. [Google Scholar]
- 6.Reismann J., Romualdi A., Kiss N., et al. Diagnosis and classification of pediatric acute appendicitis by artificial intelligence methods: an investigator-independent approach. PLoS One. September 25, 2019;14(9) doi: 10.1371/journal.pone.0222030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faria B., Vistulo de Abreu F. Cellular frustration algorithms for anomaly detection applications. PLoS One. July 8, 2019;14(7):e0218930. doi: 10.1371/journal.pone.0218930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Netea M.G., Schlitzer A., Placek K., Joosten L.A.B., Schultze J.L. Innate and adaptive immune memory: an evolutionary continuum in the host's response to pathogens. Cell Host Microbe. January 9, 2019;25(1):13–26. doi: 10.1016/j.chom.2018.12.006. [DOI] [PubMed] [Google Scholar]