

Abstract
A rapidly expanding and clinically distinct group of CNS diseases are caused by pathogenic autoantibodies that target neuroglial surface proteins. Despite immunotherapy, patients with these neuroglial surface autoantibody (NSAb)-mediated diseases often experience clinical relapse, high rates of long-term morbidity and adverse effects from the available medications. Fundamentally, the autoantigen-specific B cell lineage leads to production of the pathogenic autoantibodies. These autoantigen-specific B cells have been consistently identified in the circulation of patients with NSAb-mediated diseases, accompanied by high serum levels of autoantigen-specific antibodies. Early evidence suggests that these cells evade well-characterized B cell tolerance checkpoints. Nearer to the site of pathology, cerebrospinal fluid from patients with NSAb-mediated diseases contains high levels of autoantigen-specific B cells that are likely to account for the intrathecal synthesis of these autoantibodies. The characteristics of their immunoglobulin genes offer insights into the underlying immunobiology. In this Review, we summarize the emerging knowledge of B cells across the NSAb-mediated diseases. We review the evidence for the relative contributions of germinal centres and long-lived plasma cells as sources of autoantibodies, discuss data that indicate migration of B cells into the CNS and summarize insights into the underlying B cell pathogenesis that are provided by therapeutic effects.
An increasing number of CNS disorders are known to be associated with autoantibodies that target the extra-cellular domains of neuronal or glial proteins1,2. In these neuroglial surface autoantibody (NSAb)-mediated diseases, early immunotherapy can substantially reduce morbidity and mortality3,4 so they are collectively considered to be ‘not-to-miss’ conditions. Typically, these diseases are characterized by autoantibodies that target a single autoantigen in any individual patient. The most common autoantigens involved include aquaporin 4 (AQP4) in neuromyelitis optica spectrum disorders (NMOSD), and the NMDA receptor, leucine-rich glioma inactivated protein 1 (LGI1) and contactin-associated protein-like 2 (CASPR2) in clinically distinct forms of encephalitis (TABLE 1). On the basis that these antigens are expressed on the surface of live cells, the autoantibodies that target them are considered to be directly causative. Indeed, increasingly robust evidence from multimodal in vitro and in vivo techniques confirms that the pathogenic mechanisms of diseases mediated by autoantigen-reactive immunoglobulins have overlapping and distinct characteristics1,5–13 (TABLE 1). In this context, reductionist study of the disease-related autoantigen-specific B cells is a logical approach to the comprehensive characterization of these archetypal autoantibody-mediated diseases of the CNS14–16.
Table 1 |.
Clinical characteristics of CNS neuroglial surface antibody-mediated diseases
| CNS target | Median age, years (range) | Gender ratio (M:F) | Clinical presentation | CSF and CNS characteristics | Main IgG subclassa | Putative mechanisms |
|---|---|---|---|---|---|---|
| Neuronal targets b | ||||||
| NMDA receptor3,14,55,151 | ~24 (0–90) | 1:4 | Flu-like prodrome; psychiatric features, which cross traditional categories; multimorphic movement disorder (dystonia, stereotypies and chorea, especially orofacial involvement); dysautonomia and coma are common; seizures typically a minor feature | CSF lymphocytic pleocytosis ± oligoclonal bands; normal in ~20%; MRI often normal (~ 80%) despite severe illness | IgG1 | Internalization via antigen crosslinking152–154 |
| LGI14,15,93 | ~60 (30–80) | 3:2 | Very frequent focal seizures (including pathognomonic faciobrachial dystonic seizures); amnesia; behavioural change; insomnia | CSF normal in ~ 75%; MRI shows medial temporal lobe swelling in ~40% | IgG4 | Functional block7,11,155, internalization4,11 |
| CASPR216,101,156 | ~60 (30–80) | 9:1 | Neuropsychiatric changes; cognitive impairment; dysautonomia; sleep disturbance; neuromyotonia and neuropathic pain | CSF normal in ~70%; imaging normal in ~70% | IgG4 | Functional modulation of antigen or neighbouring proteins6 |
| GABAA receptor157–159 | ~40 (0–90) | 1:1 | Seizures (including status epilepticus, epilepsia partialis continua); cognitive impairment; movement disorders | CSF pleocytosis in 25–50% ± oligoclonal bands and elevated protein; MRI abnormal in ~80% | IgG1 | Selective decrease in receptor surface density157 |
| GABAB receptor160,161 | ~60 (16–80) | 1.5:1 | Seizures (including status epilepticus); amnesia and cognitive impairment, often with underlying small-cell lung cancer | CSF pleocytosis in ~50%; MRI abnormal in ~70% | IgG1 | Functional modulation of antigen162 |
| Glial targets b | ||||||
| AQP469,163,164 | ~28 (2–90) | 1:10 | Optic neuritis; longitudinally extensive myelitis; area-postrema syndrome; other rarer presentations | CSF pleocytosis in ~50%; swelling on MRI | IgG1 | Complement activation; internalization69,104 |
| MOG99,165,166 | ~31 (0–80) | 1:1 | Myelitis and optic neuritis, plus forms of encephalitis, in particular ADEM; recovery often better than with AQP4 autoantibodies | CSF pleocytosis in ~44–80%; MRI abnormal in ~45% | IgG1 | Complement activation167–169 |
ADEM, acute disseminated encephalomyelitis; AQP4, aquaporin 4; CASPR2, contactin-associated protein-like 2; CSF, cerebrospinal fluid; LGI1, leucine-rich glioma inactivated protein 1; MOG, myelin oligodendrocyte glycoprotein.
IgG1 subclasses can activate the complement system.
Some of the specified proteins are expressed in glia and neurons; categorization is based on the cell type that has received more study.
B cells are a crucial component of the adaptive immune system, and some ultimately differentiate into plasmablasts or plasma cells, collectively referred to as antibody-secreting cells (ASCs). ASCs are responsible for all antibody production in vivo. In health, antibodies protect against infection, and their self-reactivity is limited by functional immune checkpoints that reduce the proportion of B cells that are autoreactive and minimize the responsiveness of these cells — a phenomenon known as immune tolerance17. In autoantibody-mediated diseases, autoreactive B cells evade tolerance checkpoints and aberrantly break tolerance to autoantigens. These cells can target self-proteins or molecules and, upon differentiation into ASCs, secrete autoantibodies with pathogenic potential18.
In this Review, we examine the immunobiology of pathogenic B cells in NSAb-mediated diseases of the CNS. Much of our current knowledge has arisen from the study of AQP4 autoantibodies, and some from studies of NMDA receptor and LGI1 autoantibodies. We use knowledge of these NSAb-mediated diseases — particularly NMOSD — to illustrate concepts of B cell biology that may be informative in other NSAb-mediated diseases. We discuss evidence for the involvement of defective immune tolerance mechanisms, for the generation and propagation of autoreactive B cells and ASCs across anatomically distinct compartments, and for the potential to model these diseases on the basis of traditional paradigms of long-lived immune memory. Where applicable, we also propose testable hypotheses for future study.
Failure of B cell tolerance
General principles.
The B cell receptor (BCR) is the membrane-tethered antigen receptor on a B cell that is secreted as an antibody by ASCs. During early B cell development, pseudo-random recombination of heavy-chain and light-chain gene segments results in vast diversity of the BCR repertoire within an individual. This diversity enables B cells to recognize a wide range of foreign antigens, particularly those on invading pathogens. However, the diversity also creates an intrinsic risk of generating autoreactive pathogenic BCRs19,20.
B cell receptor.
(BCR). An immunoglobulin molecule consisting of two paired identical heavy and light chains that forms a transmembrane receptor protein on the surface of B cells and signals to the B cell, largely via its interaction with its antigen or antigens.
In health, autoreactive BCRs are minimized by at least three established checkpoints along the developmental pathway20–23 (FIG. 1). In the bone marrow, a substantial proportion of polyreactive and autoreactive early immature B cells (CD34−CD19+CD10+CD38+IgM+) are removed or modified via mechanisms of central tolerance — apoptosis, induction of anergy and receptor editing17 — and only then pass into the periphery. This counter-selection is believed to depend on exposure of BCRs to autoantigens, with BCR signalling strength key to B cell fate24.
Fig. 1 |. B cell tolerance checkpoints in humans.
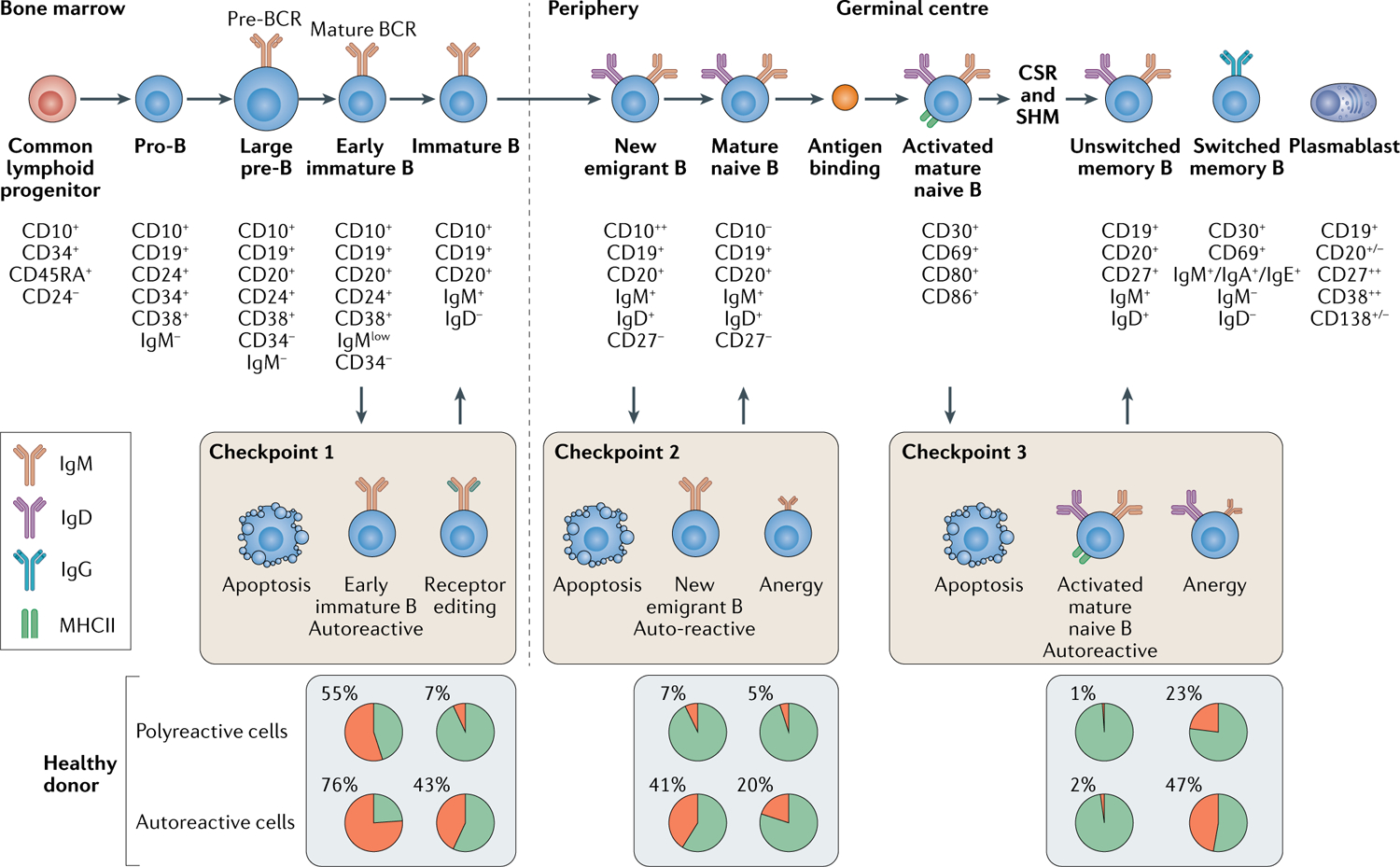
Pro-B cells in the bone marrow, which derive from common lymphoid progenitor cells, undergo B cell receptor heavy chain gene rearrangement. Newly rearranged heavy chains on pre-B cells acquire a surrogate invariant light chain to form a pre-B cell receptor (BCR). The suurogate invariant light chain is exchanged for a bona fide, paired light chain in the early immature B cell so that immunoglobulin M (IgM) is expressed on the B cell membrane. At this point (Checkpoint 1), autoreactive BCRs are negatively selected through apoptosis or rescued via receptor editing of paired functional light chains170. Positively selected or rescued B cells then exit the bone marrow as new emigrant B cells. Further negative selection occurs at the transition between new emigrant and mature naive B cells (Checkpoint 2) in the periphery20. Upon encountering their cognate antigen, mature naive B cells can enter germinal centre reactions, where T cell-dependent immunoglobulin class switch recombination (CSR) and somatic hypermutation (SHM) occur, and they upregulate MHC class II. Within germinal centres, SHM can lead IgG+ switched memory B cells to inadvertently acquire autoreactive BCRs with pathogenic potential21. IgM+ unswitched memory B cells can be further negatively selected (Checkpoint 3)22. The relative proportions of polyreactive and autoreactive B cells at each checkpoint are shown as the pie charts. MHCII, major histocompatibility complex class II.
Anergy.
A cell state in which B cells persist in the periphery but have limited responses to antigen; anergy is a mechanism that silences many self-reactive B cells.
Receptor editing.
A process in which autoreactive heavy or light chains of B cell receptors are exchanged, thereby changing the specificity of the antigen receptor and rescuing an autoreactive B cell receptor.
Upon entry into the periphery, B cells are exposed to a peripheral tolerance checkpoint (FIG. 1) that further reduces the numbers of autoreactive B cells and is thought to depend not only on intrinsic properties of BCR signalling strength but also extrinsic factors, such as T regulatory (Treg) cells25 and cytokines, including B cell-activating factor (BAFF)26,27. Subsequently, autoreactivity can be acquired or increased as a result of somatic hypermutation21. This process diversifies BCRs and increases affinity towards a target antigen28. Indeed, up to 20% of post-germinal centre immunoglobulin class-switched IgG+ memory B cells have been found to express autoreactive BCRs21. However, if the target autoantigen is expressed within the germinal centre microenvironment (FIG. 1), these autoreactive B cells fail to survive29. Therefore, germinal centre B cells that recognize rare or tissue-specific autoantigens are less likely to be eliminated. Indeed, though most established neuroglial targets of pathogenic autoantibodies are detectable outside the nervous system, they are dominantly expressed in the CNS and their extra-neural expression varies greatly. An alternative paradigm proposes that these autoantigen-specific B cells can undergo positive selection through the recognition of cross-reactive foreign antigens, a process often referred to as molecular mimicry, but evidence for this mechanism is limited30. Overall, a breakdown of the tolerance processes can result in a higher frequency of CNS-reactive B cells in disease.
B cell-activating factor.
(BAFF). A cytokine that belongs to the tumour necrosis factor ligand family and is expressed in B cell lineage cells, acting as a potent B cell activator.
Somatic hypermutation.
The introduction of point mutations within the immunoglobulin variable regions in the presence of an antigen and T helper cells.
Germinal centre.
A site within a secondary lymphoid organ where the genes that encode immunoglobulins in B cells undergo somatic hypermutation, thereby sequentially increasing their affinity for the antigen; a process that typically requires b cells to undergo rounds of proliferation, differentiation and interactions with antigen and T cells.
Evidence from NSAb-mediated diseases.
Insights into the aetiology and propagation of NSAb-mediated diseases can be gained by determining the earliest point at which autoantigen-specific B cells are generated. Circulating autoantigen-specific B cells have been detected in patients with NMOSD31, LGI1 antibody encephalitis11, NMDA receptor antibody encephalitis32 and myelin oligodendrocyte glycoprotein antibody disease33. This consistent observation across diseases can be combined with knowledge of how B cell tolerance breaks down in NMOSD to illustrate concepts that could be translatable to other NSAb-mediated conditions.
In cultures of B cells from patients with NMOSD, antigen-naive and memory B cells secreted AQP4-reactive autoantibodies, which were absent from cells from healthy controls31. This observation confirmed the existence of AQP4-reactive B cells before and after germinal centre maturation. An alternative approach to studying unmutated, naive B cells is the prediction of their BCR sequences on the basis of their counter-part autoantigen-specific memory B cells. These predicted sequences are reconstructed from the most closely aligned germline immunoglobulin genes and are referred to as unmutated common ancestors (UCAs). Use of this approach has shown that, upon reversion to UCAs, AQP4-specific autoantibodies that were cloned from BCRs of intrathecal B cells from patients with NMOSD lose their ability to bind to AQP4. However, some of these UCAs exhibit characteristics of autoantigen reactivities beyond AQP4, termed polyreactivity and/or autoreactivity34. Taken together, these observations suggest that some AQP4-specific B cells evade early tolerance checkpoints in NMOSD and that their AQP4 reactivity derives from an intrinsic BCR affinity, whereas others are subsequently increased via somatic hypermutation in germinal centres. Also, in patients with NMOSD, higher frequencies of polyreactive and autoreactive B cells suggest the existence of more global disturbances of central and peripheral tolerance checkpoints34. This finding mirrors those in NSAb-mediated diseases of the neuromuscular junction, autoimmune myasthenia gravis35 and in non-neurological autoantibody-associated diseases, such as systemic lupus erythematosus (SLE)36 and rheumatoid arthritis37. By contrast, in multiple sclerosis — another autoimmune disease of the CNS in which B cells are strongly implicated in the pathogenesis — only the peripheral checkpoints seem to be dysfunctional in a subgroup of patients38. These early observations of impaired B cell tolerance mechanisms must be investigated across several NSAb-mediated diseases to highlight shared or distinctive aspects of the immunobiology and to offer insights into the relative efficacies of divergent therapeutic approaches.
Unmutated common ancestors.
(UCAs). Antibodies that are derived computationally and represent the most accurately matched germline B cell receptors from which the mutated antibodies were derived.
Sources of autoantibody production
Two conventional models have been proposed to describe the way in which B cells that evade tolerance checkpoints contribute to the pool of secreted autoantigen-specific antibodies (FIG. 2). These two models are often discussed as dichotomous concepts, but are likely to often coexist so could both contribute to the perpetuation of circulating autoantibodies. For our purposes, however, we consider the evidence for each of these models discretely.
Fig. 2 |. Sources of autoantibodies.
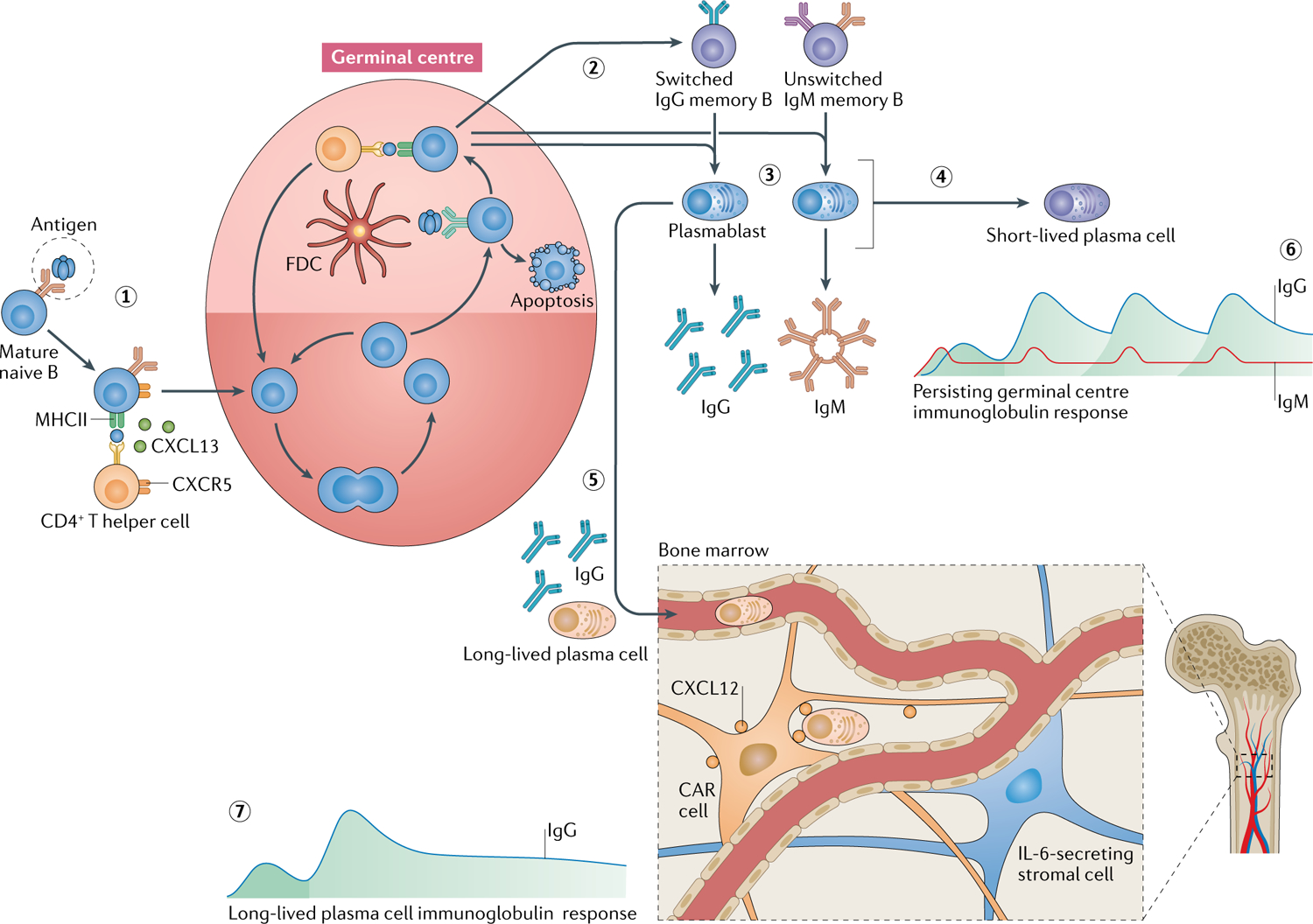
Two major sources of circulating autoantibodies have been proposed: plasmablasts that emerge from perpetually active germinal centre reactions in secondary lymphoid organs and long-lived plasma cells that reside in the bone marrow. Mature, naive B cells that encounter an autoantigen enter germinal centre reactions (1) and, in the presence of antigen and cognate CD4+ T helper cells, undergo somatic hypermutation to increase the affinity of their B cell receptors171. After successful maturation, unswitched or switched memory B cells exit the germinal centre (2) and can give rise to circulating plasmablasts that secrete IgM, IgG or IgA (not shown) (3). Plasmablasts can differentiate into short-lived plasma cells with a limited lifespan; sometimes this can happen rapidly via an extrafollicular response with limited dependence on typical germinal centre reactions (4)172. Alternatively, long-lived plasma cells can migrate into the bone marrow survival niche (5), supported by IL-6-secreting stromal cells and C–X–C motif chemokine ligand 12 (CXCL12)-abundant reticular (CAR) cells, where they can survive for many years. In the active germinal centre model, IgM and IgG are continually produced (6). By contrast, with a long-lived plasma cell response, IgG production is ongoing after a remote autoimmunization (7). CXCL13, C–X–C motif chemokine ligand 12; CXCR5, C–X–C motif chemokine receptor 5; FDC, follicular dendritic cell; MHCII, major histocompatibility complex class II.
Proposed models.
In the first proposed model, autoimmunized B cells continually enter germinal centre reactions and generate short-lived, circulating ASCs that secrete the disease-relevant autoantibodies39. This model assumes ongoing autoantigen re-challenge. IgMs are typically the first antibodies to be produced in a humoral response so, in this scenario, autoantigen-specific IgMs should be regularly generated and detectable in the circulation39. The possible sources of B cells that enter such germinal centre reactions are new autoantigen-specific naive B cells that continually evade compromised tolerance checkpoints, and autoantigen-specific memory B cells that were generated by earlier autoimmunizing events.
In the second model, autoantibody-secreting long-lived plasma cells (LLPCs) become niched in the bone marrow after an autoimmunizing event40. This model assumes that germinal centre-based activation generates a set of plasma cells that successfully migrate to the bone marrow and can thereafter secrete the bulk of the autoantibodies, largely autonomously. If the autoimmunization was transient, only a limited number of circulating B cells with autoantigen reactivity would persist41.
Overall, experimental and clinical evidence indicates contributions of both active germinal centres and LLPCs to the circulating autoantibody repertoire in patients with NSAb-mediated diseases. This evidence is discussed below.
Germinal centre reactions.
Several lines of evidence indicate an ongoing contribution of germinal centre reactions to NSAb production over the course of disease. First, circulating autoantigen-specific B cells have been robustly detected at various time points of disease11,31–33,42. The cells in this peripheral population have the potential to differentiate into ASCs and participate in autoantibody secretion. One study has identified these circulating ASCs43 and another infers their existence44, but their detection has not been reproducible — perhaps because ex vivo survival of ASCs is limited — so their existence remains unconfirmed31.
Second, although the existence and pathogenic relevance of IgM autoantibodies has been challenged45, autoantigen-specific IgMs can be detected in some patients with NMOSD46 and some with NMDA receptor antibody encephalitis32,47, albeit often at low levels. Observed levels of IgM are highest at disease onset but these antibodies often persist for several months after immunomodulatory therapy32, far longer than the half-life of circulating IgM, which is ~5 days48. Their persistent presence can be taken as evidence of recurrent germinal centre reactions triggered by repeated autoimmunization or of the establishment of IgM-secreting plasma cells49.
Third, ectopic germinal centres have been identified in the ovarian teratomas that are present in a subgroup of patients with NMDA receptor antibody encephalitis. These teratomas are enriched for identifiable tertiary lymphoid structures when compared with teratomas from people without NMDA receptor antibody encephalitis. Histologically, the tertiary lymphoid structures appear as dense aggregates of closely apposed B cell and T cell infiltrates with surrounding plasma cells32,50–53, abnormal neuroglial tissue51,54 and mature dendritic cells50,51, and consistent expression of the NR1 subunit, the autoantigen within the NMDA receptor32. Furthermore, cultured B cells from teratomas of patients with NMDA receptor antibody encephalitis can secrete NR1-specific IgG in vitro, demonstrating that tumour-resident B cells are a likely source of NMDA receptor autoantibody production in these patients32. Consequently, the clinical observation that expedited tumour removal is associated with improved outcomes might be explained by the elimination of a source of active germinal centres that generate NMDA receptor antibodies3,14,55.
Tertiary lymphoid structures.
Ectopic lymphoid-like tissues with features of secondary lymphoid organs, such as segregated T and B cell zones, mutational activity, follicular dendritic cell networks and high endothelial venules.
Fourth, studies of chemokines indicate involvement of germinal centres. C–X–C motif chemokine ligand 13 (CXCL13), which is secreted by T follicular helper cells in particular, is a proposed marker of germinal centre reactions, as it acts via the CXCR5 receptor to recruit B cells and T cells into germinal centre reactions56. Evidence suggests that CXLC13 levels are markedly elevated in the cerebrospinal fluid (CSF) of patients with NMDA receptor antibody encephalitis57–59, and in the serum and CSF of patients with LGI1 antibody encephalitis60.
T follicular helper cells.
Germinal centre resident CD4+ T follicular helper cells that provide potent survival and proliferative signals for B cells.
Fifth, CD20+ B cell depletion with rituximab has clinical benefits in NMOSD and NMDA receptor antibody encephalitis, and smaller studies suggest it has similar efficacy in other NSAb-mediated diseases3,61–63. However, this effect is not mediated by a reduction in autoantibody production, demonstrated by the fact that autoantibody levels are not markedly lowered by rituximab as expected owing to the lack of CD20 expression on plasmablasts and bone marrow resident LLPCs64–69. Rather, depletion of naive and memory B cells is the major observed effect of rituximab and repopulation of circulating memory B cells (CD19+CD27+) predicts clinical relapse70.
Finally, evidence suggests that B cell activation by T cells, which is classically dependent upon human leukocyte antigen class ii (HLA class II) molecules, is involved in NSAb-mediated diseases, and germinal centres are major sites of these B cell–T cell interactions. For example, HLA class II alleles are strongly associated with LGI1 antibody encephalitis (HLA-DRB1*07:01)70–73 and CASPR2 antibody encephalitis (HLA-DRB1*11:01)72, suggesting an HLA-restricted, T cell-dependent immune response. Associations between HLA class II alleles and NMOSD have been observed far less consistently, but the presence of immunoglobulin class-switched autoantibodies with somatic mutations is also consistent with a T cell-dependent immune response74. T cell antigens are presented to CD4+ T cells in complex with HLA class II molecules on antigen-presenting cells, including B cells. The amount of antigen that is captured and presented on HLA class II molecules helps govern B cell division, somatic mutation of genes that encode immunoglobulins and engagement of T follicular helper cells in germinal centres75,76. Therefore, strong associations between HLA class II alleles and NSAb-mediated diseases suggest preferential presentation of HLA-associated autoantigen-derived peptides to autoreactive CD4+ T cells in some of these diseases. By contrast, NMDA receptor autoantibodies are minimally mutated, which suggests a more limited role for T cells in NMDA receptor encephalitis (discussed further in Autoantigen-specific B cells in the CNS).
Human leukocyte antigen class II.
(HLA class ii). Proteins expressed on the surface of antigen-presenting cells and, if loaded with a cognate peptide, can activate antigen-specific CD4+ T cells. Also known as major histocompatibility complex class ii.
Bone marrow plasma cells.
Direct evidence for the presence of bone marrow-resident LLPCs in NSAb-mediated diseases is very limited, largely reflecting the inaccessibility of the bone marrow compartment. In one study of neurological disease with glutamic acid decarboxylase (GAD) autoantibodies, bone marrow mononuclear cells from a patient were found to secrete GAD autoantibodies in vitro. Although this condition is not widely accepted as an NSAb-mediated disease, the study demonstrated the existence of autoantigen-specific LLPCs77. Other indirect evidence for involvement of LLPCs in NSAb-mediated diseases comes from therapeutic observations. Not only do CD20− LLPCs evade rituximab-mediated depletion, but they are also resistant to drugs such as cyclophosphamide, as they do not divide78. However, they are thought to be deleted by immune ablation that precedes bone marrow transplantation79. Use of autologous haematopoietic stem cell transplantation for NMOSD led to 9 of 11 patients becoming AQP4 autoantibody-seronegative and relapse-free80, whereas conversion to seronegative status is rare with other treatments, including rituximab64–66,81. These clinical observations suggest that LLPCs contribute to AQP4 autoantibody production50.
Glutamic acid decarboxylase.
(GAD). A cytoplasmic antigen, autoantibodies to which can be found at high levels in patients with neurological syndromes such as stiff-person syndrome, cerebellar ataxia, limbic encephalitis and forms of epilepsy and diabetes.
Further indirect evidence that LLPCs are involved in autoantibody production in disease comes from targeting IL-6, a cytokine that is associated with survival of ASCs82. IL-6 levels in the CSF and serum are high in patients with NMOSD83,84 and in the CSF of patients with NMDA receptor antibody encephalitis58, and these high levels might support the autoantigen-specific plasma cells observed in the CSF of patients (see Autoantigen-specific B cells in the CNS). Blockade of the IL-6 receptor with tocilizumab and depletion of plasma cells with the proteasome inhibitor bortezomib had clinical benefits in patients with NMOSD and NMDA receptor antibody encephalitis85–90, implicating LLPCs in autoantibody production. However, both drugs can affect several other immune cell types, as their target molecules are not exclusive to ASCs or LLPCs90,91. In addition, high autoantibody levels persist in most patients even after these treatments, so further studies are needed to investigate the source of these residual antibodies.
Autoantibody access to CNS targets
Historically, the CNS has been considered an immune-privileged organ, in part owing to the presence of the tight blood–brain barrier (BBB) and a lack of draining lymphatics. However, discoveries in the past 10 years have led to the questioning of this concept, and we now know that peripheral immune cells can cross the intact BBB, a functional lymphatic system drains cellular and soluble components from the CSF into the cervical lymph nodes, immune responses or tolerance can be induced by CNS-derived antigens in the cervical lymph nodes, and ectopic tertiary lymphoid structures in the meninges can be a site of affinity maturation92. Nevertheless, crossing the BBB is a highly regulated, multistep process, and most available passive transfer models of CNS autoantibody-mediated diseases have necessitated an artificial active breakdown of the BBB or direct injection of the autoantibodies into the CNS compartment13. However, these studies did not address a major outstanding question: whether autoantibody entry into the CNS occurs via diffusion of IgG or via B cell trafficking from the periphery (FIG. 3).
Fig. 3 |. Autoantibody access to CNS targets.
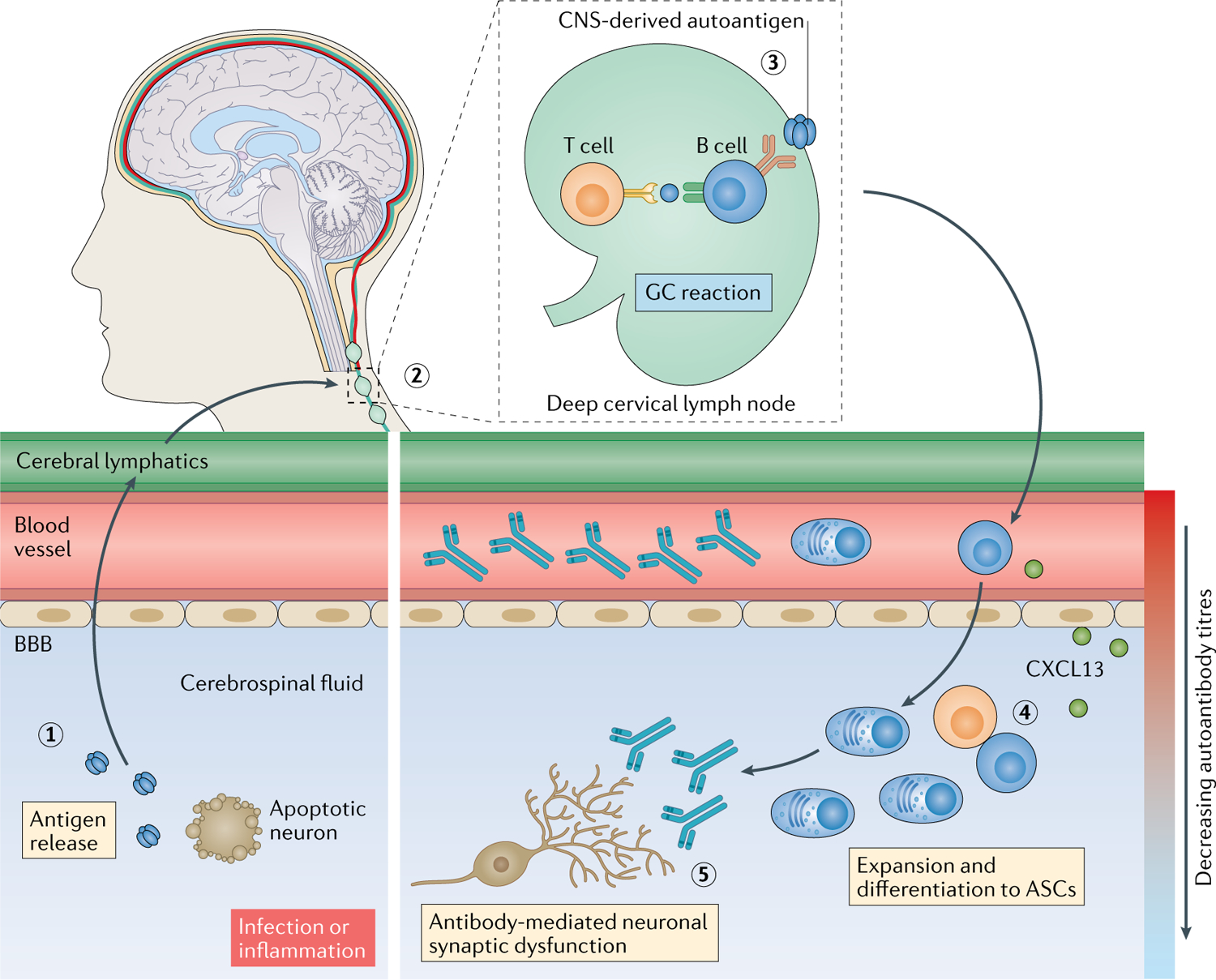
Tissue destruction, either as a result of infection112 or a pro-inflammatory milieu, can release soluble neuronal autoantigens into the cerebrospinal fluid (1), from where they can drain via the recently (re)discovered cerebral lymphatic drainage system173,174 into cervical lymph nodes (2). Here, B cell responses can be induced against CNS-derived autoantigens, generating antigen-specific B cells, plasma cells and high concentrations of autoantibodies in circulation (3). Activated B cells can then migrate into the intrathecal compartment, driven, for example, by a gradient of the C–X–C motif chemokine ligand 13 (CXCL13). In the intrathecal compartment, clonal expansion, class switching, affinity maturation and differentiation into antibody-secreting cells (ASCs) that secrete pathogenic autoantibodies can occur (4) and can be associated with ectopic tertiary lymphoid structures. These intrathecal antibodies can directly lead to neuronal dysfunction (5). BBB, blood–brain barrier; GC, germinal centre.
Serum and CSF autoantibody levels.
A consistent finding across all NSAb-mediated diseases is that median concentrations of the autoantibodies in the serum are 4-fold to 640-fold higher than those in the CSF (FIG. 4a). In some diseases, such as LGI1 antibody encephalitis and NMOSD, the antigen-specific autoantibodies cannot even be detected in CSF samples from ~10% of patients93,94. Further work is needed to determine whether they are absent or just not detectable with current methods. By contrast, in other diseases, such as NMDA receptor antibody encephalitis, antibodies are not detectable in the serum of ~15% of patients95. However, this finding is not consistent with the observed overall ratio of antibodies in the serum to those in the CSF across the field of NSAb-mediated diseases (FIG. 4a). The difficulties in detecting some serum NMDA receptor antibodies might be influenced by variability in technical parameters, including the use of fixed versus live cell-based assays, the use of neuronal tissue-based assays and the way in which absolute levels are normalized. Where live cell-based assays are used, serum NMDA receptor antibodies are detected in almost all cases (Al-Diwani and Irani, unpublished)55,96.
Fig. 4 |. Studies of intrathecal B cells.
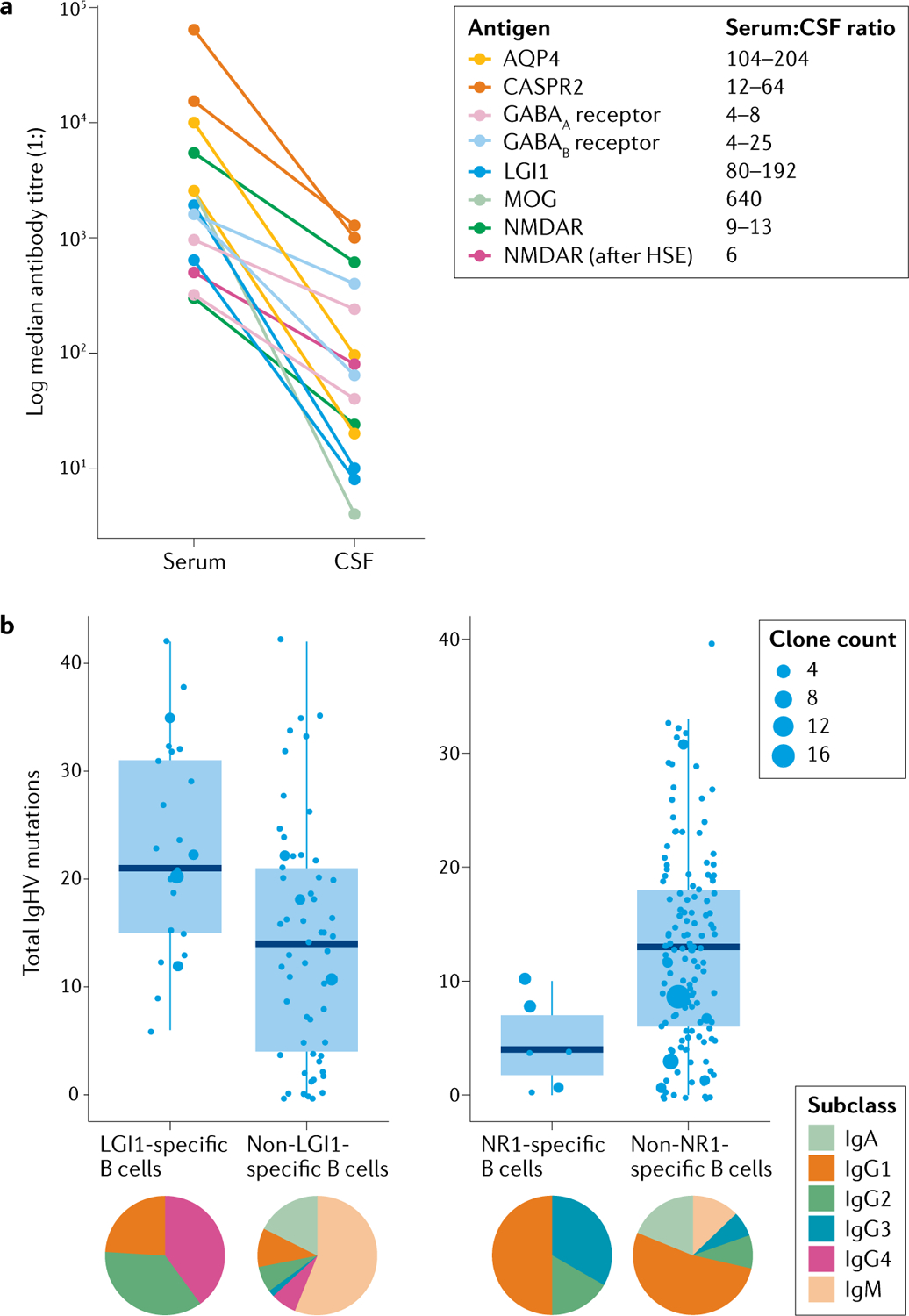
a | Median serum and cerebrospinal fluid (CSF) antibody titres in individuals demonstrate that autoantibodies are consistently present at higher levels in the serum than in the CSF, implying an initial peripheral generation of the autoantibodies. Data were adapted from multiple studies of different autoantibodies11,55,83,94,95,97,99–101,107,112,158,175,176. b | Immunoglobulin heavy chain (IgHV) somatic hypermutation of leucine-rich glioma inactivated protein 1 (LGI1)-specific (left) and NR1-specific (right) intrathecal B cells differs from that of non-target antigen-reactive B cells and among each other. This observation implies that different mechanisms operate to generate these two autoantibody populations and the non-antigen-specific cells. The proportions of immunoglobulin subclasses also differ between B cell populations (pie charts), again implicating different immunological pathways in the generation of each autoantibody population. Data adapted from previous studies5,12. AQP4, aquaporin 4; CASPR2, contactin-associated protein-like 2; NMDAR, N-methyl-d-aspartate receptor; MOG, myelin oligodendrocyte glycoprotein; HSE; herpes simplex virus encephalitis.
Intrathecal synthesis of autoantibodies is a measure taken to infer the presence of autoantigen-specific B cells in the CSF. If the ratio of autoantibody levels in the serum to those in the CSF falls below the 400:1 ratio of total IgG concentrations, this is taken to denote intrathecal synthesis. Such reduced ratios are consistently observed across NSAb-mediated diseases (FIG. 4a). However, only in a few studies are autoantibody levels strictly normalized to the total level of IgGs to determine the more formal biochemical measure of antibody index94,97–101. Nonetheless, tissue-specific and compartment-specific cellular studies provide more definitive evidence of intrathecal autoantibody production in the NSAb-mediated diseases and are discussed in the following section.
Antibody index.
A calculation used to assess intrathecal IgG synthesis by comparing the ratio of specific autoantibody levels to total levels of IgG in serum and CSF (antibody index = Qspec/QIgG, where Qspec = CSF/serum quotient for specific IgG, and QIgG = CSF/serum quotient for total IgG; values >4 are taken as evidence for intrathecal autoantigen-specific IgG synthesis).
Autoantigen-specific B cells in the CNS.
Lymphocyte infiltrates within the CNS parenchyma have been identified in patients with NSAb-mediated autoimmune diseases. In NMOSD, CD20+ B cells and CD138+ plasma cells are present within lesions, consistent with autoantibody production within the CNS102–104. Similarly, in patients with NMDA receptor antibody encephalitis or LGI1 antibody encephalitis, occasional CD20+ B cells and CD138+ plasma cells have been observed in the CNS, within the meninges and perivascular cuffs53,105. However, antigenic specificities were not determined in any of these studies.
Direct evidence that autoantigen-specific B cells exist within the CNS in patients with NSAb-mediated diseases comes from successful cloning of human recombinant monoclonal antibodies from CSF ASCs or memory B cells from patients with NMOSD106, NMDA receptor antibody encephalitis5 and LGI1 antibody encephalitis12 (FIG. 4b). These studies have shown that autoantigen-specific B cells account for up to 83% of all CSF B cells, suggesting marked intrathecal enrichment. The autoantigen-specific B cells in the CSF of patients tend to represent their autoantigen-specific IgG subclasses in serum and CSF — typically IgG4 for LGI1 antibody encephalitis and IgG1 for NMDA receptor antibody encephalitis (FIG. 4b). Somatic hypermutation also differs between diseases: LGI1-specific BCRs have all undergone somatic hypermutation, whereas IgG-expressing NMDA receptor-specific ASCs are entirely or almost unmutated5,12 (FIG. 4b). Indirectly, this limited number of somatic mutations supports the idea that T cells have a limited role in NR1-reactive B cell development, and indicates the existence of unmutated, naive NMDA receptor-reactive precursors that evade central tolerance checkpoint mechanisms, similar to findings in NMOSD31.
The presence of mutated and clonally expanded autoantigen-specific B cell populations within the CSF indicates that they undergo affinity maturation and selective intrathecal expansion. Indeed, two BCR sequencing studies have revealed clonally related B cell clusters in the circulation and CNS in patients with LGI1 antibody encephalitis and NMOSD44,107. In NMOSD, the intrathecal AQP4-specific B cells comprise peripherally derived clones and clones exclusive to the CSF. These observations can be interpreted as evidence that some autoantigen-specific B cells migrate into the CSF. The observed marked clonal expansion of the CSF B cells could represent the products of germinal centre-like structures within the CNS, as have been identified frequently in multiple sclerosis108–110 and once in NMOSD111. The presence of potential ectopic germinal centre-like structures is consistent with the high CSF levels of CXCL13 observed in patients with NMDA receptor antibody encephalitis57–59 and in a few patients with LGI1 antibody encephalitis60. Taken together, the evidence suggests that B cells can access the CNS and can undergo further intrathecal maturation and expansion.
A human paradigm in which a proportion of patients with herpes simplex virus encephalitis (HSE) develop de novo secondary autoimmunity — usually associated with NMDA receptor antibodies in the serum and CSF112 — provides a key model of autoimmunity. In this model, the primary insult is identifiable within the CNS, and the lowered serum to CSF ratios of autoantibody levels discussed above are reproduced. One possible explanation for this phenomenon is that the destructive process of HSE leads to a sequence of events that begins with the release of CNS antigens into the draining cerebral lymphatics and, subsequently, cervical lymph nodes where these antigens provoke a germinal centre reaction. This reaction could be initiated by peripheral B cells that are previously naive to CNS-restricted antigens. Subsequently, these activated CNS antigen-matured B cells probably migrate back into the CNS and account for the observed intrathecal synthesis of the autoantibodies. However, the mechanisms by which autoantigen-specific B cells gain access to the CNS compartment remain unknown and should be addressed in future studies.
Immunotherapy and B cell biology
The acute treatment of NSAb-mediated diseases has traditionally followed a hierarchical immunotherapeutic approach with the aim of maximizing therapeutic efficacy while limiting adverse effects. Class 1 data on the use of immunotherapy in NSAb diseases is lacking despite a pressing clinical need for such data — well-powered, randomized controlled trials have only been conducted in NMOSD. Nevertheless, the mechanisms by which immunotherapies modulate the adaptive immune system are of great interest, and early inferences can be made from the available retrospective and prospective clinical studies. In this section, we focus on how B cells are modulated by the most commonly used immunotherapies and the implications for understanding the underlying disease biology.
First-line therapies.
Corticosteroids are often the first pharmacological interventions used in NSAb-mediated diseases and they broadly attenuate the immune response via genomic and non-genomic effects113. Upon corticosteroid treatment, the immediate landscape of peripheral B cell subsets remains relatively unchanged114. However, corticosteroid-induced inhibition of B cell proliferation, BCR-triggered signalling, plasma cell differentiation and immunoglobulin gene expression have all been reported in vitro and in vivo115–117. Furthermore, transcriptomic inhibition of some genes, such as those that encode IL-2 and the transcription factor GATA3 (the key driver of naive T cell differentiation into type 2 T helper cells), might also reduce T cell-dependent activation of autoreactive B cells118. Intriguingly, corticosteroids have faster effects in LGI1 antibody encephalitis4,93 than in either NMDA receptor antibody encephalitis or NMOSD. The immunobiology that underlies this observation is uncharacterized, although corticosteroid therapy has marked clinical efficacy in IgG4-related diseases119,120 and LGI1 autoantibodies are predominantly of the IgG4 subclass. Nonetheless, differences in the therapeutic effects of corticosteroids across NSAb-mediated diseases require further study.
Intravenous immunoglobulin (IVIg) is another first-line therapy that is effective in several autoimmune conditions, including autoantibody-mediated diseases. In the first randomized controlled trial of IVIg in LGI1 antibody encephalitis and CASPR2 antibody encephalitis, the effect of IVIg was slightly superior to that of placebo121. Proposed mechanisms by which IVIg exerts its therapeutic effect include inhibition of B cell activation by interference with IL-4, CD40, Toll-like receptors and BCR pathways, and the blockade of the variable domain of causative antibodies by anti-idiotype antibodies122–124. However, the precise mechanisms by which IVIg exerts its effect are yet to be elucidated.
Another first-line therapy for NSAb-mediated diseases is plasma exchange, the predominant therapeutic mechanism of which is thought to be the removal of circulating pathogenic autoantibodies125,126. A concomitant reduction in CSF levels of autoantibodies has been observed in patients who receive plasma exchange, which could explain the efficacy of the treatment in diseases of the CNS127,128. The mechanisms that underlie this phenomenon remain uncharacterized but could involve permissive removal of intrathecal immunoglobulins owing to BBB dysfunction, as observed in paraneoplastic autoantibody syndromes128. In addition, the bulk removal of immunoglobulins by plasma exchange has been shown to increase lymphocyte proliferation and antibody production129. This rebound state could increase the susceptibility of circulating ASCs and their precursors to concomitant cytotoxic immunotherapies. Indeed, synchronized plasma exchange and cyclophosphamide therapy in patients with SLE reduced in vitro antibody production from cultured B cells from these patients more than did cyclophosphamide alone129.
Later line therapies.
Rituximab is a chimeric monoclonal antibody that depletes only CD20+ B cells. This mechanism spares ASCs and does not generally affect autoantibody levels, yet rituximab is clinically effective in NSAb-mediated diseases66,82,130. These observations suggest that the efficacy of rituximab is mediated by removal of the antigen-presenting capacity of B cells or by B cell-independent factors. NMOSD relapses are temporally associated with the repopulation of memory B cells, suggesting that their restoration is directly involved in treatment failure64,66,131. However, rituximab therapy does not consistently reset defective early B cell tolerance checkpoints132, so newly formed autoreactive B cell populations could contribute to treatment failure. In addition, the dynamics of tissue-specific depletion seem to vary — B cells within secondary lymphoid organs and in the CSF are depleted to a lesser extent than B cells in circulation133,134. The latter finding is consistent with the observation that only a small fraction of peripherally administered rituximab reaches the CNS135. Consequently, the relative resistance of these compartments to rituximab might mean that tissue-resident autoantigen-specific B cells are not depleted. Finally, after rituximab therapy, the expansion of enduring B cells can result in a clonally restricted B cell repertoire136, so ongoing disease could be driven by the persistence of these autoreactive clones137.
Cyclophosphamide induces cell apoptosis through the DNA crosslinking action of its metabolite phosphoramide mustard. This mechanism prevents the proliferation of activated B cells. However, the non-proliferative LLPCs remain resistant to cyclophosphamide138,139. In NMDA receptor antibody encephalitis, cyclophosphamide has comparable efficacy to rituximab, and small series indicate that it has efficacy in NMOSD140,141. However, the use of the drug is often avoided for young women owing to concerns about infertility142,143.
Bortezomib, a small-molecule proteasome inhibitor, has also been used in patients with severe autoimmune encephalitis and NMOSD87–89,144. Theoretically, bortezomib promotes relatively selective apoptosis of plasma cells because the rate of immunoglobulin synthesis in these cells is very high. In vivo mouse studies have shown that bortezimib depletes plasma cells and protects against nephritis with lupus-like disease145,146. In humans, the use of bortezimib can ameliorate clinical manifestations in patients with refractory SLE147, thrombotic thrombocytopenia purpura148 and Sjögren syndrome149, making it a promising option for the management of autoantibody-mediated conditions. In multiple myeloma, use of cyclophosphamide in combination with bortezomib — a combination that targets both the LLPCs and their emerging precursors — is considerably more effective than bortezomib alone150. Such observations support the notion that isolated depletion of LLPCs is unlikely to terminate disease activity in the presence of peripheral B cell tolerance dysfunction. Accurate follow-up of patients who have been treated with bortezomib will better determine the long-term efficacy of this drug.
Conclusions
The study of B cells in NSAb-mediated diseases has revealed fundamental observations that implicate the breakdown of immune tolerance mechanisms in the pathogenesis of these diseases. Future studies of NSAb-associated diseases should aim to identify B cell-specific biomarkers to enable better monitoring of disease activity and to inform the rational administration of medications. Furthermore, elucidation of the mechanisms by which autoreactive B cells evade immune tolerance could uncover further mechanistic and aetiological insights.
In some NSAb-mediated diseases, it seems likely that conventional germinal centre reactions generate somatically mutated autoantibodies with pathogenic potential. In others, disease propagation seems to arise independent of, or in addition to, canonical pathways of long-lived autoantigen-specific immune memory. The pathogenic step of B cell access to the CNS means that pathways of their migration, differentiation into ASCs and survival within the CSF milieu could provide further opportunities for therapeutic targeting to eliminate intrathecal autoantibody production. Further studies should address the mechanisms by which these processes are affected by therapies, and whether this approach can offer opportunities to personalize therapy in the NSAb-mediated diseases.
Key points.
Autoantigen-specific B cells that target neuroglial surface proteins are responsible for the production of pathogenic neuroglial surface autoantibodies (NSAbs) in an increasing variety of antibody-mediated CNS diseases.
Pathogenic NSAbs are typically found at higher concentrations in the serum than in the cerebrospinal fluid.
Current evidence suggests that autoreactive B cells evade early B cell tolerance checkpoints in NSAb-mediated CNS diseases.
Enrichment of autoantigen-specific B cells in the cerebrospinal fluid in patients with NSAb-mediated diseases suggests that they migrate into this compartment and secrete pathogenic IgGs intrathecally.
Experimental and clinical evidence implicates both ongoing germinal centre reactions and long-lived plasma cells in the propagation of autoantibody production in NSAb-mediated diseases.
Acknowledgements
B.S. is supported by the Association of British Neurologists via the Patrick Berthoud Charitable Trust. M.R. is supported by the Austrian Science Fund (FWF J4157-B30). S.R.I. is supported by the Wellcome Trust (104079/Z/14/Z), BMA Research Grants (Vera Down grant (2013) and Margaret Temple grant (2017)), Epilepsy Research UK (P1201), the Fulbright UK–US Commission (MS Society research award) and the National Institute for Health Research (NIHR), Oxford Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the National Health Service, the NIHR or the Department of Health. K.C.O’C. is supported by the National Institute of Allergy and Infectious Diseases (NIAID) under award numbers R01-AI114780 and R21-AI142198, by a Neuromuscular Disease Research program award from the Muscular Dystrophy Association (MDA) under award number MDA575198 and by the Guthy-Jackson Charitable Foundation.
Competing interests
R.J.M.B.-R. is a co-founder and consultant for Alchemab Therapeutics Ltd and has consulted for Imperial College London and VHSquared. S.R.I. and M.R. are co-inventors on a patent to improve the specificity of autoantibody detection. S.R.I. is a co-applicant and receives royalties on patent application WO/2010/046716 entitled ‘Neurological Autoimmune Disorders’. The patent has been licensed for the development of assays for LGI1 and other voltage-gated potassium channel (VGKC)-complex antibodies. S.R.I. has received research support from CSL Behring, ONO Pharma and UCB. B.S. declares no competing interests.
References
- 1.Varley J, Vincent A & Irani SR Clinical and experimental studies of potentially pathogenic brain-directed autoantibodies: current knowledge and future directions. J. Neurol 262, 1081–1095 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramanathan S, Al-Diwani A, Waters P & Irani SR The autoantibody-mediated encephalitides: from clinical observations to molecular pathogenesis. J. Neurol 10.1007/s00415-019-09590-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Titulaer MJ et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 12, 157–165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson J et al. The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain 141, 348–356 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kreye J et al. Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 139, 2641–2652 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Patterson KR, Dalmau J & Lancaster E Mechanisms of Caspr2 antibodies in autoimmune encephalitis and neuromyotonia. Ann. Neurol 83, 40–51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petit-Pedrol M et al. LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain 141, 3144–3159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huijbers MG et al. MuSK myasthenia gravis monoclonal antibodies: valency dictates pathogenicity. Neurol. Neuroimmunol. Neuroinflamm 6, e547 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takata K et al. Characterization of pathogenic monoclonal autoantibodies derived from muscle-specific kinase myasthenia gravis patients. JCI insight 4, e127167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones BE et al. Autoimmune receptor encephalitis in mice induced by active immunization with conformationally stabilized holoreceptors. Sci. Transl Med 11, eaaw0044 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramberger M et al. Distinctive binding properties of human monoclonal LGI1 autoantibodies determine pathogenic mechanisms. Brain 143, 1731–1745 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kornau H-C et al. Human cerebrospinal fluid monoclonal LGI1 autoantibodies increase neuronal excitability. Ann. Neurol 87, 405–418 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Duan T & Verkman AS Experimental animal models of aquaporin-4-IgG-seropositive neuromyelitis optica spectrum disorders: progress and shortcomings. Brain Pathol. 30, 13–25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalmau J et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 7, 1091–1098 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irani SR et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 133, 2734–2748 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Sonderen A et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology 87, 521–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nemazee D Mechanisms of central tolerance for B cells. Nat. Rev. Immunol 17, 281–294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meffre E & O’Connor KC Impaired B-cell tolerance checkpoints promote the development of autoimmune diseases and pathogenic autoantibodies. Immunol. Rev 292, 90–101 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schatz DG & Ji Y Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol 11, 251–263 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Wardemann H et al. Predominant autoantibody production by early human B cell precursors. Science 301, 1374–1377 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Tiller T et al. Autoreactivity in human IgG+ memory B cells. Immunity 26, 205–213 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuiji M et al. A checkpoint for autoreactivity in human IgM+ memory B cell development. J. Exp. Med 203, 393–400 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meffre E The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann. NY Acad. Sci 1246, 1–10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benschop RJ, Brandl E, Chan AC & Cambier JC Unique signaling properties of B cell antigen receptor in mature and immature B cells: implications for tolerance and activation. J. Immunol 167, 4172–4179 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Wing JB & Sakaguchi S Foxp3(+) T(reg) cells in humoral immunity. Int. Immunol 26, 61–69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thien M et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity 20, 785–798 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Lesley R et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 20, 441–453 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Baumgarth N How specific is too specific? B-cell responses to viral infections reveal the importance of breadth over depth. Immunol. Rev 255, 82–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan TD et al. Elimination of germinal-center-derived self-reactive B cells is governed by the location and concentration of self-antigen. Immunity 37, 893–904 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Burnett DL et al. Germinal center antibody mutation trajectories are determined by rapid self/foreign discrimination. Science 360, 223–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson R et al. Condition-dependent generation of aquaporin-4 antibodies from circulating B cells in neuromyelitis optica. Brain 141, 1063–1074 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makuch M et al. N-methyl-D-aspartate receptor antibody production from germinal center reactions: therapeutic implications. Ann. Neurol 83, 553–561 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winklmeier S et al. Identification of circulating MOG-specific B cells in patients with MOG antibodies. Neurol. Neuroimmunol. Neuroinflamm 6, 625 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cotzomi E et al. Early B cell tolerance defects in neuromyelitis optica favour anti-AQP4 autoantibody production. Brain 142, 1598–1615 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J-Y et al. Compromised fidelity of B-cell tolerance checkpoints in AChR and MuSK myasthenia gravis. Ann. Clin. Transl Neurol 3, 443–454 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yurasov S et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J. Exp. Med 201, 703–711 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samuels J, Ng Y-S, Coupillaud C, Paget D & Meffre E Impaired early B cell tolerance in patients with rheumatoid arthritis. J. Exp. Med 201, 1659–1667 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kinnunen T et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J. Clin. Invest 123, 2737–2741 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boes M Role of natural and immune IgM antibodies in immune responses. Mol. Immunol 37, 1141–1149 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Leyendeckers H et al. Correlation analysis between frequencies of circulating antigen-specific IgG-bearing memory B cells and serum titers of antigen-specific IgG. Eur. J. Immunol 29, 1406–1417 (1999). [DOI] [PubMed] [Google Scholar]
- 41.Radbruch A et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol 6, 741–750 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Sharma R et al. Monoclonal antibodies from a patient with anti-NMDA receptor encephalitis. Ann. Clin. Transl Neurol 5, 935–951 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chihara N et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl Acad. Sci. USA 108, 3701–3706 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kowarik MC et al. CNS aquaporin-4-specific B cells connect with multiple B-cell compartments in neuromyelitis optica spectrum disorder. Ann. Clin. Transl Neurol 4, 369–380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hara M et al. Clinical and pathogenic significance of IgG, IgA, and IgM antibodies against the NMDA receptor. Neurology 90, e1386–e1394 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jarius S, Franciotta D, Bergamaschi R, Wildemann B & Wandinger K-P Immunoglobulin M antibodies to aquaporin-4 in neuromyelitis optica and related disorders. Clin. Chem. Lab. Med 48, 659–663 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Castillo-Gomez E et al. All naturally occurring autoantibodies against the NMDA receptor subunit NR1 have pathogenic potential irrespective of epitope and immunoglobulin class. Mol. Psychiatry 22, 1776–1784 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Barth WF, Wochner RD, Waldmann TA & Fahey JL Metabolism of human gamma macroglobulins. J. Clin. Invest 43, 1036–1048 (1964). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bohannon C et al. Long-lived antigen-induced IgM plasma cells demonstrate somatic mutations and contribute to long-term protection. Nat. Commun 7, 11826 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabata E et al. Immunopathological significance of ovarian teratoma in patients with anti-N-methyl-d-aspartate receptor encephalitis. Eur. Neurol 71, 42–48 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Chefdeville A et al. Immunopathological characterization of ovarian teratomas associated with anti-N-methyl-D-aspartate receptor encephalitis. Acta Neuropathol. Commun 7, 38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nolan A, Buza N, Margeta M & Rabban JT Ovarian teratomas in women with anti-N-methyl-D-aspartate receptor encephalitis: topography and composition of immune cell and neuroglial populations is compatible with an autoimmune mechanism of disease. Am. J. Surg. Pathol 43, 949–964 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Tuzun E et al. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol. 118, 737–743 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Day GS, Laiq S, Tang-Wai DF & Munoz DG Abnormal neurons in teratomas in NMDAR encephalitis. JAMA Neurol. 71, 717–724 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Irani SR et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 133, 1655–1667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Havenar-Daughton C et al. CXCL13 is a plasma biomarker of germinal center activity. Proc. Natl Acad. Sci. USA 113, 2702–2707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leypoldt F et al. Investigations on CXCL13 in anti-N-methyl-D-aspartate receptor encephalitis: a potential biomarker of treatment response. JAMA Neurol. 72, 180–186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Byun J-I et al. Distinct intrathecal interleukin-17/interleukin-6 activation in anti-N-methyl-d-aspartate receptor encephalitis. J. Neuroimmunol 297, 141–147 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Liba Z et al. Anti-N-methyl-D-aspartate receptor encephalitis: the clinical course in light of the chemokine and cytokine levels in cerebrospinal fluid. J. Neuroinflammation 13, 55 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin Y-T, Yang X, Lv J-W, Liu X-W & Wang S-J CXCL13 is a biomarker of anti-leucine-rich glioma-inactivated protein 1 encephalitis patients. Neuropsychiatr. Dis. Treat 15, 2909–2915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dale RC et al. Utility and safety of rituximab in pediatric autoimmune and inflammatory CNS disease. Neurology 83, 142–150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Damato V, Evoli A & Iorio R Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: a systematic review and meta-analysis. JAMA Neurol. 73, 1342–1348 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Cree BAC et al. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 64, 1270–1272 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Kim S-H, Kim W, Li XF, Jung I-J & Kim HJ Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch. Neurol 68, 1412–1420 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Valentino P, Marnetto F, Granieri L, Capobianco M & Bertolotto A Aquaporin-4 antibody titration in NMO patients treated with rituximab: a retrospective study. Neurol. Neuroimmunol. Neuroinflamm 4, e317 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pellkofer HL et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 76, 1310–1315 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Brown JWL et al. Long-term remission with rituximab in refractory leucine-rich glioma inactivated 1 antibody encephalitis. J. Neuroimmunol 271, 66–68 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Irani SR, Gelfand JM, Bettcher BM, Singhal NS & Geschwind MD Effect of rituximab in patients with leucine-rich, glioma-inactivated 1 antibody-associated encephalopathy. JAMA Neurol. 71, 896–900 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Waters P et al. Aquaporin-4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch. Neurol 65, 913–919 (2008). [DOI] [PubMed] [Google Scholar]
- 70.Kim T-J et al. Anti-LGI1 encephalitis is associated with unique HLA subtypes. Ann. Neurol 81, 183–192 (2017). [DOI] [PubMed] [Google Scholar]
- 71.van Sonderen A et al. Anti-LGI1 encephalitis is strongly associated with HLA-DR7 and HLA-DRB4. Ann. Neurol 81, 193–198 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Binks S et al. Distinct HLA associations of LGI1 and CASPR2-antibody diseases. Brain 141, 2263–2271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mueller SH et al. Genetic predisposition in anti-LGI1 and anti-NMDA receptor encephalitis. Ann. Neurol 83, 863–869 (2018). [DOI] [PubMed] [Google Scholar]
- 74.Gontika M & Anagnostouli M Human leukocyte antigens-immunogenetics of neuromyelitis optica or Devic′s disease and the impact on the immune-pathogenesis, diagnosis and treatment: a critical review. Neuroimmunol. Neuroinflamm 1, 44–50 (2014). [Google Scholar]
- 75.Gitlin AD, Shulman Z & Nussenzweig MC Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 509, 637–640 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shulman Z et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science 345, 1058–1062 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thaler FS et al. Abundant glutamic acid decarboxylase (GAD)-reactive B cells in gad-antibody-associated neurological disorders. Ann. Neurol 85, 448–454 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Mumtaz IM et al. Bone marrow of NZB/W mice is the major site for plasma cells resistant to dexamethasone and cyclophosphamide: implications for the treatment of autoimmunity. J. Autoimmun 39, 180–188 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Alexander T et al. Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long-term remission through de novo generation of a juvenile and tolerant immune system. Blood 113, 214–223 (2009). [DOI] [PubMed] [Google Scholar]
- 80.Burt RK et al. Autologous nonmyeloablative hematopoietic stem cell transplantation for neuromyelitis optica. Neurology 93, e1732–e1741 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jarius S et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 131, 3072–3080 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cassese G et al. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J. Immunol 171, 1684–1690 (2003). [DOI] [PubMed] [Google Scholar]
- 83.Sato DK et al. Cerebrospinal fluid aquaporin-4 antibody levels in neuromyelitis optica attacks. Ann. Neurol 76, 305–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Icoz S et al. Enhanced IL-6 production in aquaporin-4 antibody positive neuromyelitis optica patients. Int. J. Neurosci 120, 71–75 (2010). [DOI] [PubMed] [Google Scholar]
- 85.Araki M Blockade of IL-6 signaling in neuromyelitis optica. Neurochem. Int 130, 104315 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Lee W-J et al. Tocilizumab in autoimmune encephalitis refractory to rituximab: an institutional cohort study. Neurotherapeutics 13, 824–832 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scheibe F et al. Bortezomib for treatment of therapy-refractory anti-NMDA receptor encephalitis. Neurology 88, 366–370 (2017). [DOI] [PubMed] [Google Scholar]
- 88.Shin Y-W, Lee S-T, Kim T-J, Jun J-S & Chu K Bortezomib treatment for severe refractory anti-NMDA receptor encephalitis. Ann. Clin. Transl Neurol 5, 598–605 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Keddie S et al. Plasma cell depletion with bortezomib in the treatment of refractory N-methyl-d-aspartate (NMDA) receptor antibody encephalitis. Rational developments in neuroimmunological treatment. Eur. J. Neurol 25, 1384–1388 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Taylor J & Irani SR Bortezomib for neuromyelitis optica spectrum disorder: a new therapeutic option for the more severe forms? JAMA Neurol. 75, 129 (2018). [DOI] [PubMed] [Google Scholar]
- 91.Kishimoto T The biology of interleukin-6. Blood 74, 1–10 (1989). [PubMed] [Google Scholar]
- 92.Engelhardt B et al. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 132, 317–338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van Sonderen A et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology 87, 1449–1456 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Majed M, Fryer JP, McKeon A, Lennon VA & Pittock SJ Clinical utility of testing AQP4-IgG in CSF: guidance for physicians. Neurol. Neuroimmunol. Neuroinflamm 3, e231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gresa-Arribas N et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 13, 167–177 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jezequel J et al. Cell- and single molecule-based methods to detect anti-N-methyl-D-aspartate receptor autoantibodies in patients with first-episode psychosis from the OPTiMiSE project. Biol. Psychiatry 82, 766–772 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Dujmovic I et al. Temporal dynamics of cerebrospinal fluid anti-aquaporin-4 antibodies in patients with neuromyelitis optica spectrum disorders. J. Neuroimmunol 234, 124–130 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Jarius S et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J. Neuroinflammation 7, 52 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jarius S et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J. Neuroinflammation 13, 279 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Joubert B et al. Characterization of a subtype of autoimmune encephalitis with anti-contactin-associated protein-like 2 antibodies in the cerebrospinal fluid, prominent limbic symptoms, and seizures. JAMA Neurol. 73, 1115–1124 (2016). [DOI] [PubMed] [Google Scholar]
- 101.Bien CG et al. Anti-contactin-associated protein-2 encephalitis: relevance of antibody titres, presentation and outcome. Eur. J. Neurol 24, 175–186 (2017). [DOI] [PubMed] [Google Scholar]
- 102.Lucchinetti CF et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 125, 1450–1461 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Misu T et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 125, 815–827 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roemer SF et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 130, 1194–1205 (2007). [DOI] [PubMed] [Google Scholar]
- 105.Bien CG et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 135, 1622–1638 (2012). [DOI] [PubMed] [Google Scholar]
- 106.Bennett JL et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol 66, 617–629 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lehmann-Horn K et al. Intrathecal B-cell activation in LGI1 antibody encephalitis. Neurol. Neuroimmunol. Neuroinflamm 7, e669 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E & Aloisi F Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 14, 164–174 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lehmann-Horn K, Wang S-Z, Sagan SA, Zamvil SS & von Budingen H-C B cell repertoire expansion occurs in meningeal ectopic lymphoid tissue. JCI Insight 1, e87234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bell L, Lenhart A, Rosenwald A, Monoranu CM & Berberich-Siebelt F Lymphoid aggregates in the CNS of progressive multiple sclerosis patients lack regulatory T cells. Front. Immunol 10, 3090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chan KH, Lee R, Lau KK & Loong F Orbital ectopic lymphoid follicles with germinal centers in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. Front. Immunol 10.3389/fimmu.2017.01947 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Armangue T et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol. 17, 760–772 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liberman AC et al. Regulatory and mechanistic actions of glucocorticoids on T and inflammatory cells. Front. Endocrinol 9, 235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Olnes MJ et al. Effects of systemically administered hydrocortisone on the human immunome. Sci. Rep 6, 23002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee J et al. Corticosteroid modulation of immunoglobulin expression and B-cell function in COPD. FASEB J. 30, 2014–2026 (2016). [DOI] [PubMed] [Google Scholar]
- 116.Yan S, Deng X, Wang Q, Sun X & Wei W Prednisone treatment inhibits the differentiation of B lymphocytes into plasma cells in MRL/MpSlac-lpr mice. Acta Pharmacol. Sin 36, 1367–1376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Franco LM et al. Immune regulation by glucocorticoids can be linked to cell type-dependent transcriptional responses. J. Exp. Med 216, 384–406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cousins DJ, McDonald J & Lee TH Therapeutic approaches for control of transcription factors in allergic disease. J. Allergy Clin. Immunol 121, 801–803 (2008). [DOI] [PubMed] [Google Scholar]
- 119.Perugino CA & Stone JH Treatment of IgG4-related disease: current and future approaches. Z. Rheumatol 75, 681–686 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brito-Zeron P et al. Therapeutic approach to IgG4-related disease: a systematic review. Medicine 95, e4002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dubey D et al. Randomized placebo-controlled trial of intravenous immunoglobulin in autoimmune LGI1/CASPR2 epilepsy. Ann. Neurol 87, 313–323 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Konova E, Atanasova M, Stoykov S, Velkova A & Shoenfeld Y Idiotypic and anti-idiotypic elastin autoantibodies: implications for IVIg and pregnancy loss. J. Autoimmun 28, 46–54 (2007). [DOI] [PubMed] [Google Scholar]
- 123.Kazatchkine MD & Kaveri SV Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N. Engl. J. Med 345, 747–755 (2001). [DOI] [PubMed] [Google Scholar]
- 124.Sultan Y, Kazatchkine MD, Maisonneuve P & Nydegger UE Anti-idiotypic suppression of autoantibodies to factor VIII (antihaemophilic factor) by high-dose intravenous gammaglobulin. Lancet 2, 765–768 (1984). [DOI] [PubMed] [Google Scholar]
- 125.Guptill JT et al. Effect of therapeutic plasma exchange on immunoglobulins in myasthenia gravis. Autoimmunity 49, 472–479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Winters JL Plasma exchange: concepts, mechanisms, and an overview of the American Society for Apheresis guidelines. Hematol. Am. Soc. Hematol. Educ. Progr 2012, 7–12 (2012). [DOI] [PubMed] [Google Scholar]
- 127.Sun X et al. Clinical application of plasma exchange in pediatric anti-N-methyl-D-aspartate receptor encephalitis. Int. J. Clinial Exp. Med 10, 11945–11952 (2017). [Google Scholar]
- 128.Graus F, Abos J, Roquer J, Mazzara R & Pereira A Effect of plasmapheresis on serum and CSF autoantibody levels in CNS paraneoplastic syndromes. Neurology 40, 1621–1623 (1990). [DOI] [PubMed] [Google Scholar]
- 129.Hanly JG, Hong C, Zayed E, Jones JV & Jones E Immunomodulating effects of synchronised plasmapheresis and intravenous bolus cyclophosphamide in systemic lupus erythematosus. Lupus 4, 457–463 (1995). [DOI] [PubMed] [Google Scholar]
- 130.Hofmann K, Clauder A-K & Manz RA Targeting B cells and plasma cells in autoimmune diseases. Front. Immunol 9, 835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lebrun C et al. Only follow-up of memory B cells helps monitor rituximab administration to patients with neuromyelitis optica spectrum disorders. Neurol. Ther 7, 373–383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chamberlain N et al. Rituximab does not reset defective early B cell tolerance checkpoints. J. Clin. Invest 126, 282–287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Martin F & Chan AC B cell immunobiology in disease: evolving concepts from the clinic. Annu. Rev. Immunol 24, 467–496 (2006). [DOI] [PubMed] [Google Scholar]
- 134.Monson NL, Cravens PD, Frohman EM, Hawker K & Racke MK Effect of rituximab on the peripheral blood and cerebrospinal fluid B cells in patients with primary progressive multiple sclerosis. Arch. Neurol 62, 258–264 (2005). [DOI] [PubMed] [Google Scholar]
- 135.Petereit HF & Rubbert-Roth A Rituximab levels in cerebrospinal fluid of patients with neurological autoimmune disorders. Mult. Scler 15, 189–192 (2009). [DOI] [PubMed] [Google Scholar]
- 136.Bashford-Rogers RJM et al. Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature 574, 122–126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jiang AR, Fichtner ML, Hoehn KB & Stathopoulos P Single-cell immune repertoire tracing identifies rituximab refractory B cells that emerge during relapse. JCI Insight 10.1172/jci.insight.136471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cupps TR, Edgar LC & Fauci AS Suppression of human B lymphocyte function by cyclophosphamide. J. Immunol 128, 2453–2457 (1982). [PubMed] [Google Scholar]
- 139.Hoyer BF et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J. Exp. Med 199, 1577–1584 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kessler RA, Mealy MA & Levy M Treatment of neuromyelitis optica spectrum disorder: acute, preventive, and symptomatic. Curr. Treat. Options Neurol 18, 2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stellmann J-P et al. Immunotherapies in neuromyelitis optica spectrum disorder: efficacy and predictors of response. J. Neurol. Neurosurg. Psychiatry 88, 639–647 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Clowse MEB et al. Ovarian reserve diminished by oral cyclophosphamide therapy for granulomatosis with polyangiitis (Wegener’s). Arthritis Care Res. 63, 1777–1781 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nguyen QN et al. Cisplatin- and cyclophosphamide-induced primordial follicle depletion is caused by direct damage to oocytes. Mol. Hum. Reprod 25, 433–444 (2019). [DOI] [PubMed] [Google Scholar]
- 144.Zhang C et al. Safety and efficacy of bortezomib in patients with highly relapsing neuromyelitis optica spectrum disorder. JAMA Neurol. 74, 1010–1012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Neubert K et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat. Med 14, 748–755 (2008). [DOI] [PubMed] [Google Scholar]
- 146.Khodadadi L et al. Bortezomib plus continuous B cell depletion results in sustained plasma cell depletion and amelioration of lupus nephritis in NZB/W F1 mice. PLoS ONE 10, e0135081 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Alexander T et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythe matosus. Ann. Rheum. Dis 74, 1474–1478 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Patriquin CJ et al. Bortezomib in the treatment of refractory thrombotic thrombocytopenic purpura. Br. J. Haematol 173, 779–785 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Rosenberg AS et al. A role for plasma cell targeting agents in immune tolerance induction in autoimmune disease and antibody responses to therapeutic proteins. Clin. Immunol 165, 55–59 (2016). [DOI] [PubMed] [Google Scholar]
- 150.Kapoor P, Ramakrishnan V & Rajkumar SV Bortezomib combination therapy in multiple myeloma. Semin. Hematol 49, 228–242 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Al-Diwani A et al. The psychopathology of NMDAR-antibody encephalitis in adults: a systematic review and phenotypic analysis of individual patient data. Lancet Psychiatry 6, 235–246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hughes EG et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci 30, 5866–5875 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Moscato EH et al. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann. Neurol 76, 108–119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Planaguma J et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain 138, 94–109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ohkawa T et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J. Neurosci 33, 18161–18174 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sunwoo J-S et al. Clinical manifestations of patients with CASPR2 antibodies. J. Neuroimmunol 281, 17–22 (2015). [DOI] [PubMed] [Google Scholar]
- 157.Ohkawa T et al. Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis. J. Neurosci 34, 8151–8163 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Petit-Pedrol M et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 13, 276–286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pettingill P et al. Antibodies to GABAA receptor α1 and γ2 subunits: clinical and serologic characterization. Neurology 84, 1233–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lancaster E et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 9, 67–76 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dogan Onugoren M et al. Limbic encephalitis due to GABAB and AMPA receptor antibodies: a case series. J. Neurol. Neurosurg. Psychiatry 86, 965–972 (2015). [DOI] [PubMed] [Google Scholar]
- 162.Nibber A et al. Pathogenic potential of antibodies to the GABAB receptor. Epilepsia Open 2, 355–359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Collongues N et al. Neuromyelitis optica in France: a multicenter study of 125 patients. Neurology 74, 736–742 (2010). [DOI] [PubMed] [Google Scholar]
- 164.Pandit L et al. Demographic and clinical features of neuromyelitis optica: a review. Mult. Scler 21, 845–853 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hoftberger R et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult. Scler 21, 866–874 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Waters P et al. Serial anti-myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol. 77, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mader S et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J. Neuroinflammation 8, 184 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jarius S et al. Screening for MOG-IgG and 27 other anti-glial and anti-neuronal autoantibodies in ‘pattern II multiple sclerosis’ and brain biopsy findings in a MOG-IgG-positive case. Mult. Scler 22, 1541–1549 (2016). [DOI] [PubMed] [Google Scholar]
- 169.Hoftberger R et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein auto antibody. Acta Neuropathol. 139, 875–892 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wardemann H, Hammersen J & Nussenzweig MC Human autoantibody silencing by immunoglobulin light chains. J. Exp. Med 200, 191–199 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Silver J et al. Stochasticity enables BCR-independent germinal center initiation and antibody affinity maturation. J. Exp. Med 215, 77–90 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Victora GD & Nussenzweig MC Germinal centers. Annu. Rev. Immunol 30, 429–457 (2012). [DOI] [PubMed] [Google Scholar]
- 173.Louveau A et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Louveau A, Harris TH & Kipnis J Revisiting the mechanisms of CNS immune privilege. Trends Immunol. 36, 569–577 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.van Coevorden-Hameete MH et al. The expanded clinical spectrum of anti-GABABR encephalitis and added value of KCTD16 autoantibodies. Brain 142, 1631–1643 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Spatola M et al. Investigations in GABAA receptor antibody-associated encephalitis. Neurology 88, 1012–1020 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]