

Abstract
Alcohol consumption is a global healthcare problem with enormous social, economic, and clinical consequences. The liver sustains the earliest and the greatest degree of tissue injury due to chronic alcohol consumption and it has been estimated that alcoholic liver disease (ALD) accounts for almost 50% of all deaths from cirrhosis in the world. In this study, we used a modified Lieber-DeCarli (LD) diet to treat mice with alcohol and simulate chronic alcohol drinking. Using an untargeted metabolomics approach, our aim was to identify the various metabolites and pathways that are altered in the early stages of ALD. Histopathology showed minimal changes in the liver after 6 weeks of alcohol consumption. However, untargeted metabolomics analyses identified 304 metabolic features that were either up- or down-regulated in the livers of ethanol-consuming mice. Pathway analysis revealed significant alcohol-induced alterations, the most significant of which was in the FXR/RXR activation pathway. Targeted metabolomics focusing on bile acid biosynthesis showed elevated taurine-conjugated cholic acid compounds in ethanol-consuming mice. In summary, we showed that the changes in the liver metabolome manifest very early in the development of ALD, and when minimal changes in liver histopathology have occurred. Although alterations in biochemical pathways indicate a complex pathology in the very early stages of alcohol consumption, bile acid changes may serve as biomarkers of the early onset of ALD.
1. Introduction
Alcohol consumption is a global healthcare problem with enormous social, economic, and clinical consequences [1]. Chronic alcohol consumption causes multi-organ damage, with the liver sustaining the earliest and the greatest degree of tissue injury because it is the primary site of ethanol metabolism [2]. It has been estimated that alcoholic liver disease (ALD) accounts for almost 50% of all deaths from cirrhosis in the world [3]. The clinical spectrum of ALD is very broad, spanning from fatty liver to alcoholic hepatitis, fibrosis and cirrhosis, and hepatocellular carcinoma [4]. Although steatosis is usually asymptomatic and reversible with alcohol cessation in heavy drinkers who do not have other risk factors for steatosis (such as insulin resistance and/or obesity) [5], chronic alcohol consumption can lead to hepatocellular damage, inflammation and a variable degree of fibrosis and lobular distortion that may progress to cirrhosis [6].
Ethanol undergoes extensive metabolism in the liver, starting with oxidation to acetaldehyde, and subsequently to acetate [7]. The oxidation takes place in the main parenchymal cells of the liver (i.e., hepatocytes) and is catalyzed by alcohol dehydrogenase (ADH) and cytochrome P450 2E1 (CYP2E1). The product, acetaldehyde, is converted into acetate by aldehyde dehydrogenases (ALDH2, ALDH1B1 and ALDH1A1) [8]. Oxidative metabolism of alcohol generates an excess of reducing equivalents, primarily in the form of reduced nicotinamide adenine dinucleotide (NADH) which alters the cellular redox potential [2,6]. The accumulation of reactive oxygen species (ROS), mainly hydrogen peroxide (H2O2) and superoxide anion , exacerbates oxidative stress in the liver [9]. The produced radicals are highly reactive, and bind rapidly to ethanol or iron atoms to form reactive metabolites, promoting lipid peroxidation [2]. The cytotoxic effect of ethanol metabolism and ROS lead to cell death in ALD [10]. Ethanol also impairs fatty acid catabolism in the liver (in part by blocking peroxisome proliferator-activated receptor α-mediated responses) [11], inhibits AMP kinase [12], and stimulates the sterol regulatory element-binding protein 1, a membrane-bound transcription factor [13]. All of these effects lead to abnormal lipid metabolism in the liver and steatosis in ALD.
Early detection of initial forms of ALD in the primary care setting is critical. However, ALD is most commonly diagnosed at very advanced stages. Liver disease is symptomatically silent. While it is known that alcohol-induced hepatic steatosis may cause a rise in serum transaminases, elevation in the levels of these enzymes is non-specific and they may be normal in early liver disease. Changes in bilirubin and albumin indicate late liver disease (i.e., fibrosis) and are also poor screening tools [14]. Physicians therefore need a means by which patients with alcohol-related disease can be identified early, such as through the identification of abnormal liver biochemistry. Understanding the molecular mechanism(s) by which ethanol causes liver damage, especially pathways that mediate the development and progression of alcohol-induced organ pathology, may aid in early-stage prognosis and the development of therapeutic targets to prevent and treat ALD.
Our aim was to identify the various pathways that are altered in the early stages of ALD in the liver using an untargeted metabolomics approach in a mouse model simulating chronic alcohol consumption [15].
2. Materials and methods
2.1. Chemicals
Cholic acid, chenodeoxycholic acid, hyodeoxycholic acid, sodium glycochenodeoxycholate, sodium taurochenodeoxycholate glycocholic acid hydrate, sodium taurolithocholic acid, sodium taurodeoxycholate hydrate, sodium taurohyodeoxycholate hydrate, and taurocholic acid sodium salt hydrate were purchased from Sigma-Aldrich (Saint Louis, MI, USA). Glycodeoxycholic acid sodium salt (EMD, Millipore Corp., Burlington, MA, USA), ursodeoxycholic acid, deoxycholic acid (Chem-Cruz, Santa Cruz Biotechnology Inc., Dallas TX, USA), lithocholic acid (Cayman Chemical Company, Ann Arbor, MI, USA), and glycodeoxycholic acid, glycolithocholic acid, glycodeoxycholic acid, 7-ketodeoxycholate and 7-ketolithocholic acid were purchased from Toronto Research Chemicals (North York, ON, Canada). Formic acid (99+%) was purchased in 1 mL ampoules from Thermo Scientific (Rockford, IL). Ammonium formate and 2-propanol (both Optima® LC/MS grade), acetonitrile and water (both Optima® grade) were purchased from Fisher Chemical (Fair Lawn, NJ, USA).
2.2. Animal study
Ten wk old male C57BL/6J mice were fed a modified Lieber-DeCarli (LD) diet (F4473SP and F4474SP; Bio-Serv, Frenchtown, NJ) (ethanoltreated, n = 4) or an ethanol-free LD diet (control, n = 5). Briefly, the LD liquid diet is composed of 45% fat-derived calories, 15% protein-derived calories and 40% calories comprised varying concentrations of carbohydrate-derived or ethanol (EtOH)-derived calories (EDC). Ethanol-consuming mice began the study on a diet containing 2% EtOH (v/v) (10.8% EDC) and the amount of EtOH was increased 1% weekly until it reached 6% (v/v) (35% EDC) and the animals were maintained on 6% EtOH (v/v) for the remainder of the study. Control (CON) mice received a LD diet in which the EtOH content was substituted by carbohydrates. Mice were housed individually and had free access to drinking water. Food intake was recorded daily, and body weights were measured weekly. At the end of the 6-wk feeding regimen, mice were euthanized by CO2 asphyxiation, and the livers were promptly collected. A small piece was immediately fixed in 10% formalin for histologic evaluation. The remainder was snap frozen in liquid nitrogen for latter metabolomics analyses.
2.3. Histopathology
Formalin-fixed paraffin-embedded pieces of liver were sectioned at 5 μm, and stained with hematoxylin and eosin (H&E). Pathological changes in the liver were quantified used a procedure adapted for mice [16,17] from the validated histological scoring system established by Kleiner and Brunt [18]. Briefly, the extent of steatosis, the presence of inflammatory cells and foci, features of hepatocyte injury, and markers of tissue response, including increased cholangiocytes near the portal triad, accumulations of Mallory bodies, increased accumulation of highly glycosylated proteins in the nucleus, formation of hepatocyte giant cells, accumulation of inclusion bodies, hepatocyte polyploidization, hyalinized or thickened peri-ductal tissue and hyperplasic nodular regeneration [19,20] were determined by microscopic evaluation of H&E-stained tissue sections by a trained histopathologist. The entire section of liver from each mouse was examined and the extent of each of the above parameters was scored by the histopathologist who was blinded to the treatment of the mice.
2.4. Untargeted metabolomics
Sample preparation:
Liver samples (50.00 ± 2.50 mg for each sample) were subjected to three 20 s periods of 6,000 rpm homogenization (Precellys Evolution homogenizer, Bertin Technologies SAS, France) with ceramic beads in 500 μL of water and was adjusted according to the tissue weight. Dry ice was used to keep the temperature of homogenization low. The resultant tissue homogenates were decanted from the beads and aliquoted (100 μL/aliquot), one of which was subjected to analysis immediately while the others were stored in −80 °C for future use. 400 μL of methanol:acetonitrile (1:1, v/v) was added to the 100 μL of fresh homogenate sample and the resultant solution was vortexed for 30 s, and subjected to centrifugation at 15,000 rpm for 15 min at 4 °C. The supernatant was placed in a tube and incubated at −20 °C for 1 h (to promote further precipitation of proteins), and was subsequently subjected to centrifugation at 15,000 rpm for 15 min at 4 °C. The resultant supernatant was isolated and evaporated to dryness in a vacuum concentrator (Savant, SC210A, SpeedVac Concentrator, Thermo Scientific) coupled with a refrigerator vapor trap (Savant RVT5105, Thermo Scientific). Dry extracts were reconstituted in 150 μL of acetonitrile: water (1:1, v/v) and subjected to centrifugation at 15,000 rpm for 10 min at 4 °C to remove insoluble debris. The supernatant was transferred to LC-MS vials and stored at −80 °C for latter LC-MS analysis.
A quality control (QC) sample was prepared by pooling a small aliquot from each reconstituted tissue sample from all mice. The samples were analyzed using ultraperformance liquid chromatography (UPLC, Acquity I Class, Waters Corp., Milford, MA, USA) interfaced with a QTOF mass spectrometer (Xevo G2-XS, Waters Corp., Milford, MA, USA) operating in negative mode.
Data analysis:
ProteoWizard (version 3.06150) was used to convert raw MS data files to the.mzML format [19]. An optimized MZmine 2 (version 2.52) workflow was developed for feature list generation. The workflow included the exact mass detection algorithm, with a noise level set to 5 × 103. Chromatogram building was performed, including a minimum ion time span of 0.1 min, a minimum peak height set to 5 × 103, and the relative m/z tolerance set to 15 ppm. The next step was chromatogram deconvolution by applying the Wavelets (ADAP) algorithm [20]. The S/N threshold was set to 5, the minimum absolute intensity was set to 5 × 103, and a maximum peak duration range was set to 0.5 min. Isotopic features were removed by applying an isotopic peaks grouper algorithm, including a relative m/z tolerance of 15 ppm, a retention time tolerance of 0.1 min, the monotonic shape parameter was set to false, two was the maximum allowed charge, and the most intense isotope was kept as the final result.
The data was normalized by the median for each metabolite and log transformed and the final dataset was scaled by mean centering in Metaboanalyst [21,22]. The log-transformed data was used as an input to principal component analysis (PCA) for exploratory analysis. Because the PCA analysis revealed tight clustering of the quality control samples, no further data normalization, data imputation or transformation was performed. The log-transformed data were used as an input to univariate analysis where the Wilcoxon rank sum test in combination with FDR correction (q ≤ 0.05) was used to detect significant differences between the EtOH-consuming and CON mice. The threshold for the significantly up- or down-regulated metabolic features were defined as being a log2 fold-change ≥ |1|.
Annotation:
For metabolite annotation, the Metabolite annotation and Dysregulated Network Analysis (MetDNA) workflow were used. This implements a metabolic reaction network (MRN)-based recursive algorithm for metabolite annotation [23].
Pathway analysis:
Ingenuity Pathway Analysis software (IPA, QIAGEN Redwood City, USA, www.qiagen.com/ingenuity) was used to identify pathways, diseases and functions over-represented in the putatively annotated metabolic features. Significant (P < 0.05) pathway enrichment within a reference network was discerned using Fisher’s exact test.
2.5. Targeted metabolomic analysis of bile acids
UPLC-TOF-MRM method:
A previously published method by our group was used to quantify the bile acids in the liver tissue [24]. Briefly, a reversed phase liquid chromatographic (RPLC) separation and mass spectrometric detection were performed using UPLC (Acquity I Class, Waters Corp., Milford, MA, USA) interfaced with a QTOF mass spectrometer (Xevo G2-XS, Waters Corp., Milford, MA, USA). The chromatographic separation was performed on an Acquity UPLC BEH C18 column (50 × 2.1 mm; i.d. 1.7 μm) (Waters Corporation, Milford, MA, USA) equipped with a BEH C18 VanGuard pre-column (5 × 2.1 mm; i.d. 1.7 μm). The mobile phase consisted of a mixture of A (aqueous buffer containing 1 mM ammonium formate and formic acid (pH 4.39)) and B (acetonitrile/isopropanol (1:1 v/v)) at a total flow rate of 0.4 mL/min. The linear gradient elution started at a ramp of 20–30% B (0–3 min), 30–40% B (3–4 min), 50–70% B (4–5 min), 70–90% B (5–5.2 min), and continuing at 90% B up to 6 min, followed by 90–20% B in 0.1 min with 1.9 min equilibration time, for a total of 8 min. The injection volume for all samples and standard solutions was 2 μL. The column temperature was 55 °C, and the sample tray temperature was maintained at 8 °C. For mass spectrometry analysis, an electrospray ionization source was operated in negative mode (ESI—). The parameters were as follows: spray voltage 2 kV, cone voltage 30 V, source temperature 120 °C, desolvation temperature 500 °C, cone gas flow 50 L/h, and desolvation gas flow 900 L/h. The quadrupole-time-of-flight multiple reaction monitoring (TOF-MRM) was adopted for enhanced target quantitative analysis according to the transitions and collision energy levels described for each analyte listed in Table 1.
Table 1.
TOF-MRM transitions, retention time and collision energy level for the 19 bile acids that were quantified in liver samples.
| Analyte | Precursor ion (M–H)− | Production | Retention time (min) | Collision energy (eV) |
|---|---|---|---|---|
|
| ||||
| 7-ketodeoxycholate (7-keto-DCA) | 405.2646 | 405.2646 | 3.81 | 0 |
| 7-ketolithocholic acid (7-keto-LCA) | 389.2697 | 389.2697 | 4.45 | 0 |
| Chenodeoxycholic acid (CDCA) | 391.2853 | 391.2853 | 4.87 | 0 |
| Cholic acid (CA) | 407.2802 | 407.2802 | 4.4 | 0 |
| Deoxycholic acid (DCA) | 391.2853 | 391.2853 | 4.92 | 0 |
| Glycochenodeoxycholic acid (GCDCA) | 448.3068 | 74.0244 | 4.24 | −40 |
| Glycocholic acid (GCA) | 464.3017 | 74.0244 | 3.55 | −55 |
| Glycodeoxycholic acid (GDCA) | 448.3068 | 74.0244 | 4.33 | −40 |
| Glycohyodeoxycholic acid (GHDCA) | 448.3068 | 74.0244 | 3.43 | −40 |
| Glycolithocholic acid GLCA | 432.3119 | 74.0244 | 4.69 | −40 |
| Glycoursodeoxycholic acid (GUDCA) | 448.3068 | 74.0244 | 3.22 | −40 |
| Hyodeoxycholic acid (HDCA) | 391.2853 | 391.2853 | 4.46 | 0 |
| Lithocholic acid (LCA) | 375.2904 | 375.2904 | 5.31 | 0 |
| Taurochenodeoxycholic acid (TCDCA) | 498.2894 | 79.9568 | 4.13 | −70 |
| Taurocholic acid (TCA) | 514.2843 | 79.9568 | 3.3 | −65 |
| Taurodeoxycholic acid (TDCA) | 498.2894 | 79.9568 | 4.22 | −70 |
| Taurohyodeoxycholic acid (THDCA) | 498.2894 | 79.9568 | 3.29 | −65 |
| Taurolithocholate (TLCA) | 482.2946 | 79.9568 | 4.57 | −70 |
| Ursodeoxycholic acid (UDCA) | 391.2853 | 391.2853 | 4.37 | 0 |
Standard stock solutions preparation:
Standard stock solutions of bile acids were individually prepared by dissolving each bile acid in the appropriate amount of methanol:water (1:1 v/v) to achieve a final concentration of 1 mg/mL. All 19 bile acids were combined in a mixed intermediate solution at 5,000 ng/mL in acetonitrile:water (1:1). The intermediate solution was then serially diluted in acetonitrile:water (1:1) to obtain standard working solutions containing 1.6, 5, 16, 50, 160 and 500 ng/mL of cholic acid (CA), deoxycholic acid (DCA), hyodeoxycholic acid (HDCA), lithocholic acid (LCA), or taurolithocholate (TLCA), and 1.6, 5, 16, 50, 160, 500 and 1,600 ng/mL of 7-ketodeoxy-cholate (7-keto-DCA), 7-ketolithocholic acid (7-keto-LCA), chenodeoxycholic acid (CDCA), glycochenodeoxycholic acid (GCDCA), glycocholic acid (GCA), glycodeoxycholic acid (GDCA), glycohyodeoxycholic acid (GHDCA), glycolithocholic acid (GLCA), glycoursodeoxycholic acid (GUDCA), taurochenodeoxycholic acid (TCDCA), taurocholic acid (TCA), taurodeoxycholic acid (TDCA), taurohyodeoxycholic acid (THDCA), or ursodeoxycholic acid (UDCA). The limit of quantification (LOQ) for the bile acids was determined to be 4.8 pg/mg tissue.
Data acquisition and processing:
Data were acquired with MassLynx version 4.1 (Waters Corp., Milford, MA, USA). TargetLynx (Waters Corp., Milford, MA, USA) was used to integrate chromatograms of identified compounds. Statistical analyses were performed using GraphPad, Prism 7 (San Diego, CA).
2.6. Raw data availability
Both untargeted and targeted metabolomics datasets will be available at the NIH Metabolomics workbench (http://www.metabolomicsworkbench.org/).
3. Results
3.1. Histopathology
Hematoxylin and eosin-stained images from representative areas of liver from EtOH-consuming and CON mice are shown in Fig. 1A. Very few pathological changes occurred in the liver of mice treated with EtOH for 6 wk, i.e., no hepatocyte death, very few inflammatory cells or inflammatory cell foci, and no true markers of tissue response to injury. A low content of cytoplasmic lipid droplets (CLDs), predominantly in zone 2, were observed that were much more pronounced in the livers of EtOH-consuming mice. Since these CLDs were smaller than the hepatocyte nuclear diameter, they did not meet the criteria for macrosteatosis. Nor did they meet the criteria for microsteatosis because they did not completely fill the hepatocyte. Nevertheless, the CLDs indicate an increase in triacylglycerol storage in the zone 2 hepatocytes.
Fig. 1.
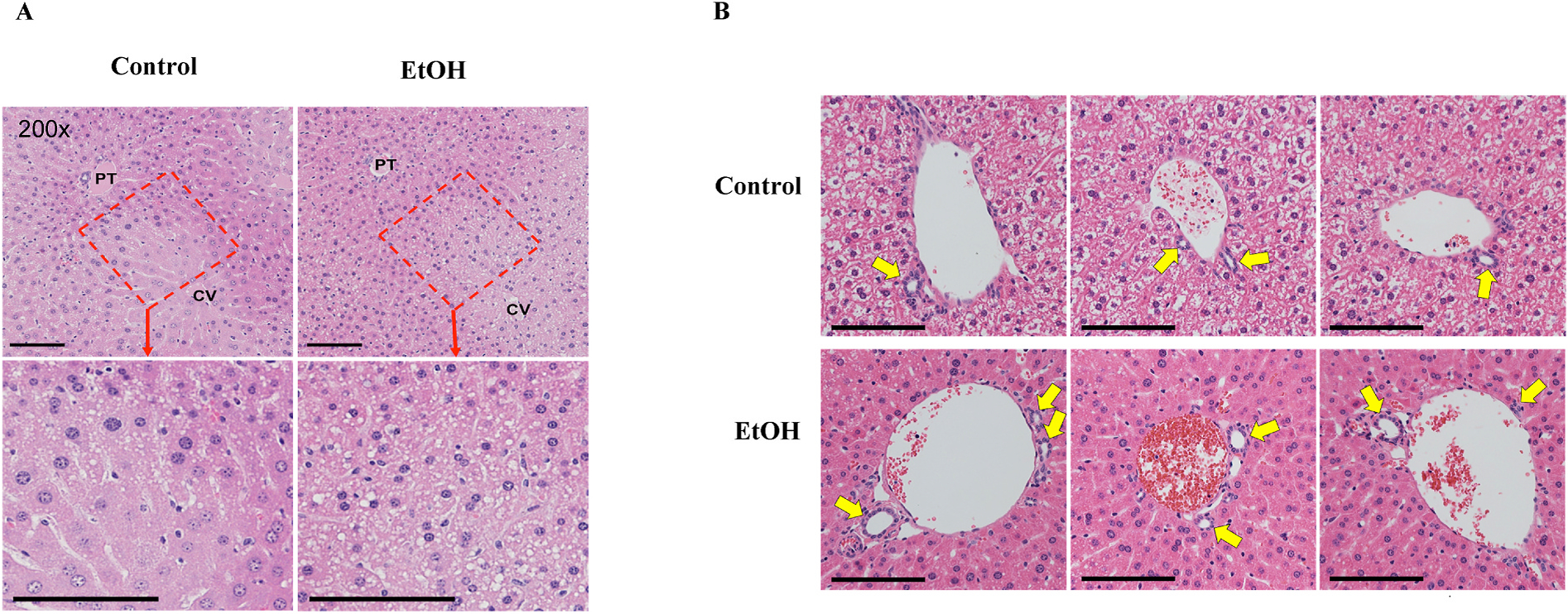
A) H&E images of liver sections from control and ethanol-consuming mice. Representative images of a liver lobule showing the full portal triad (PT) to central vein (CV) histology (200x magnification) (top panel). Cropped portions showing the zone 2 area from the 200x images, now at higher magnification are shown in the lower panels. B) Representative images of larger portal triads from 3 animals each from the control (top row) and ethanol-consuming group (bottom row), allowing comparison of the relative bile duct sizes. The bile ducts from the ethanol-consuming mice were slightly larger. Yellow arrows point to the bile ducts. Size bars represent 100 μ.
Curiously, the bile duct diameters of EtOH-consuming mice appeared to be slightly increased relative to the control mice (Fig. 1B). However, quantification of this difference was problematic due to the occurrence of multiple bile ducts near portal triads and variance in the size of the bile ducts in an approximate but not exact proportion with the size of the portal vein. Nevertheless, the phenotype of the bile duct in EtOH-consuming mice is one of lower, simple cuboidal epithelium, a stretched appearance, and a larger lumen.
3.2. Untargeted metabolomics and pathway analysis
Untargeted metabolomics was performed to determine the differences in metabolic profiles caused by ethanol treatment of mice. Comprehensive profiles were acquired from the liver extracts and a total of 4,729 features were detected. The principal component analysis showed separation along the first principal component (Fig. 2A), indicating relevant differences in the metabolome after ethanol consumption. The global metabolomics volcano plot analysis revealed 304 metabolic features to be significant (q ≤ 0.05) (Fig. 2B). Hierarchical clustering analysis (HCA) heatmap of the 100 most highly significant log-transformed metabolic features (Fig. 2C) showed a clear pattern of up- or down-regulated clusters of metabolic features in EtOH-consuming mice. Putative identification revealed 83 unique metabolic features that were used for pathway enrichment; the tentative metabolic pathways are shown in Supplemental Table S1. Of these, the farnesoid X receptor (FXR)/retinoid X receptor (RXR) activation pathway was the most significant (p = 3.2E-6), which is linked to bile acid homeostasis.
Fig. 2. Multivariate and univariate analysis of the control and EtOH-consuming liver samples:
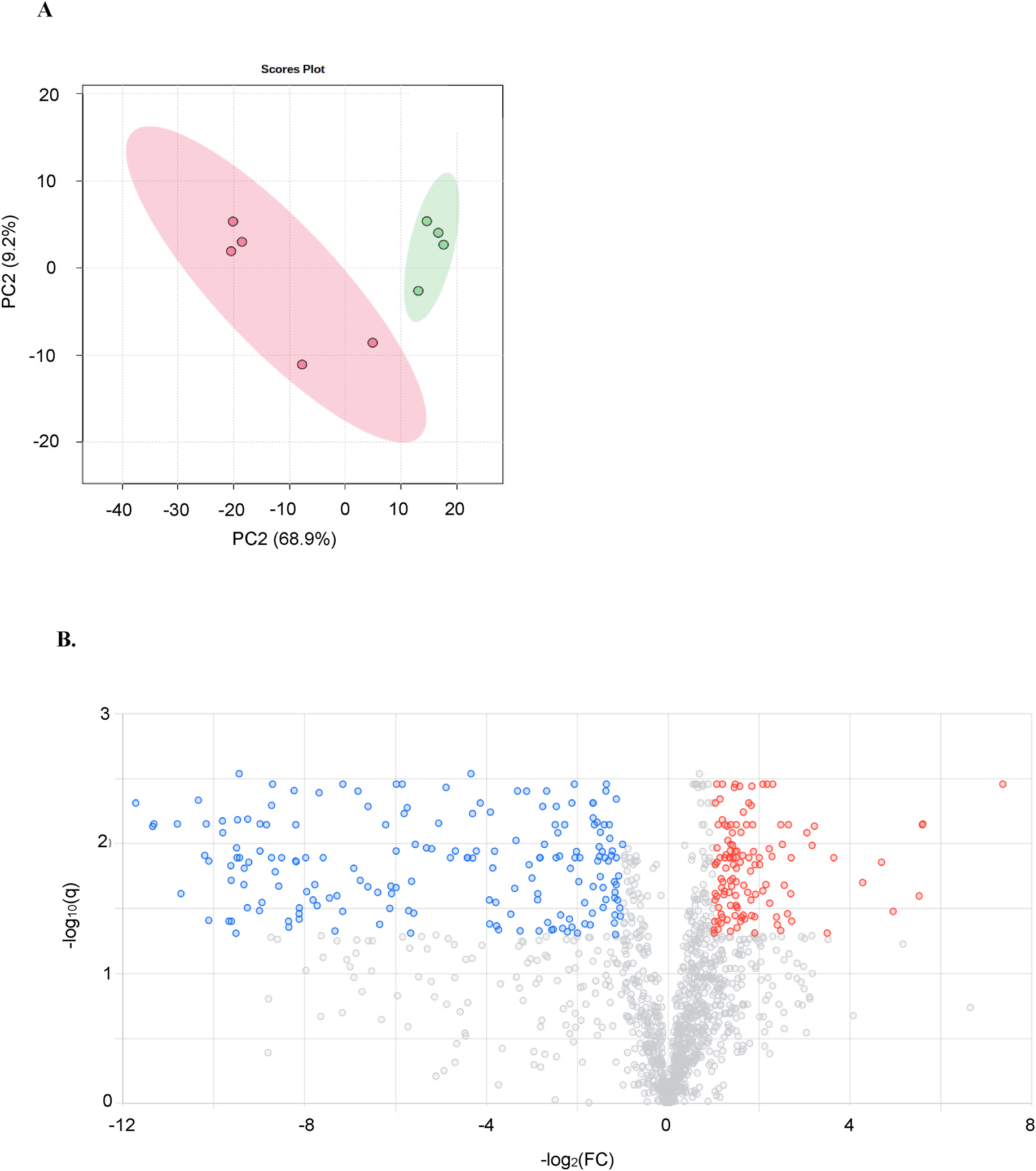
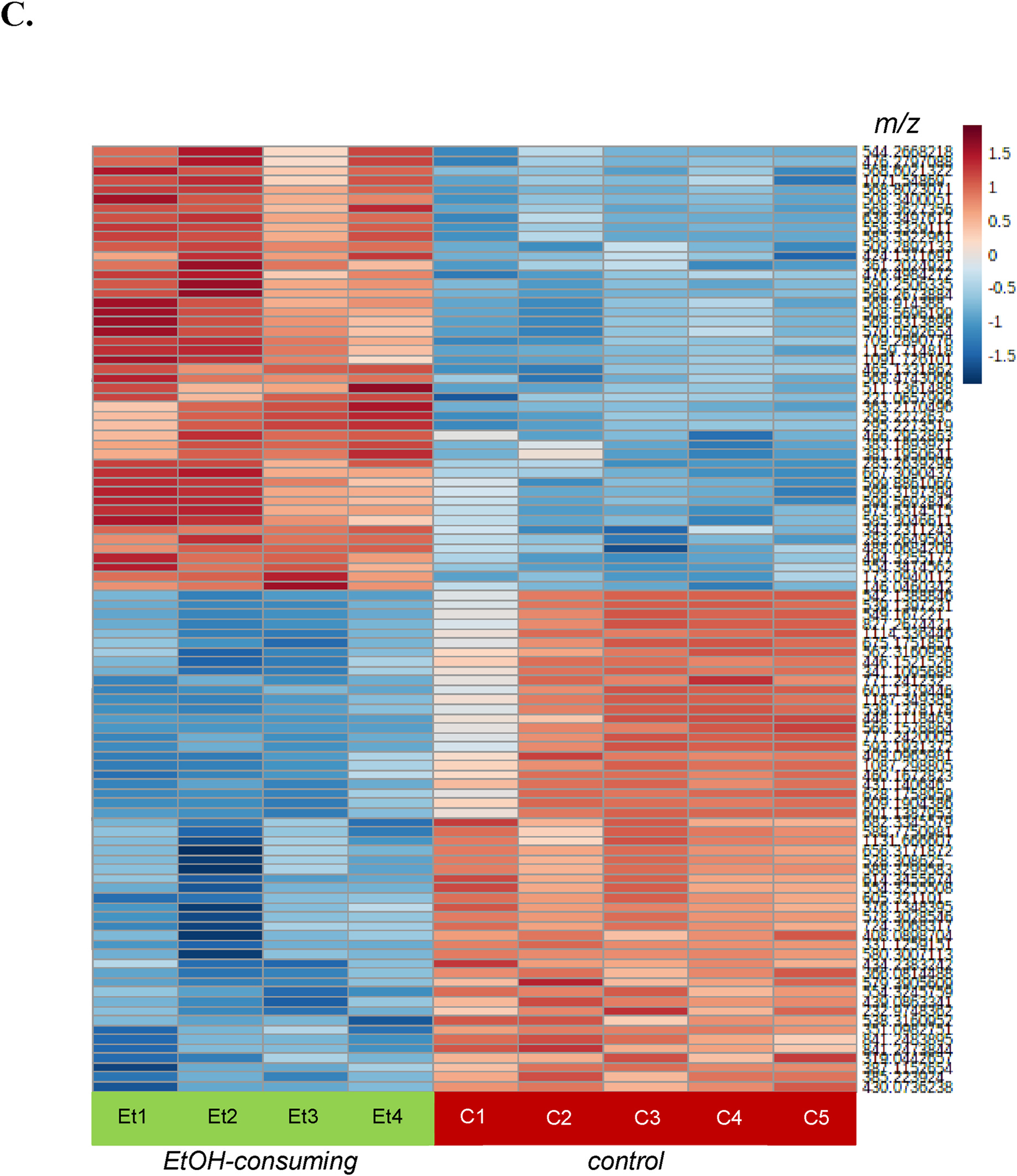
A) Principal component analysis scores plot of control (•) and EtOH-consuming (•) mice. B) Volcano plot highlighting 304 significant ions. Metabolic features that were significantly (q ≤ 0.05, univariant analysis vs. control) upregulated (red dots) or down-regulated (blue dots) in EtOH-consuming mice are shown. For the X-axis, the threshold was log2 fold-change ≥ |1| and for the Y-axis q-value ≤ 0.05. C) Heatmap of the 100 most highly significant metabolic features (i.e., m/z values) in individual control (C) and EtOH-consuming (Et) mice. Scale shows the log10 fold-change in each metabolic feature using a range from −1.5 (blue) to +1.5 (red).
3.3. Targeted metabolomics: bile acid biosynthesis pathway
Given the relationship of the FXR/RXR activation pathway to bile acids, targeted metabolomic analyses were conducted on bile acid synthesis in the liver (Fig. 3). While the targeted method was developed to measure 19 bile acids, only 12 were detected and quantified in the liver tissue of control and EtOH-consuming mice (Table 2). Of these, cholic acid (CA) showed an increasing trend (13.1 fold-change, p = 0.052) and its taurine conjugate, taurocholic acid (TCA) was increased 3.2-fold. Although the increase in deoxycholic acid (DCA) did not attain significance (8.9-fold change, p = 0.074), its taurine conjugates, taurodeoxycholic acid (TDCA) and taurohyodeoxycholic acid (THDCA), increased 2.0- and 2.7- fold, respectively. Levels of the glycine-conjugate of CA, glycocholic acid (GCA), could be measured in the liver tissue of EtOH-consuming mice (0.09 ± 0.03 ng/mg tissue), but they were below the limit of quantification (LOQ, 0.0048 ng/mg tissue) in the liver tissue of control mice. A similar pattern was observed for 7-ketodeoxycholate (7-keto-DCA), i.e., 0.58 ± 0.53 ng/mg tissue in EtOH-consuming mice and below the LOQ in control mice. In contrast, 7-ketolithocholic acid (7-keto-LCA) was detected in both groups with a 2-fold increase, even though it did not attain significance (p = 0.261), in EtOH-consuming mice. No differences between EtOH-consuming and control mice were found for liver levels of hyodeoxycholic acid (HDCA), chenodeoxycholic acid (CDCA), ursodeoxycholic acid (UDCA) or taurolithocholate (TLCA). The following glycine-conjugated bile acids, glycochenodeoxycholic acid (GCDCA), glycolithocholic acid (GLCA), glycoursodeoxycholic acid (GUDCA) and glycohyodeoxycholic acid (GHDCA), were not detected by our method and, as such, were not quantified. In the liver of EtOH-consuming mice, the ratio of CA:CDCA was increased (34.7) relative to control mice (0.95).
Fig. 3. Bile acid synthetic pathways.
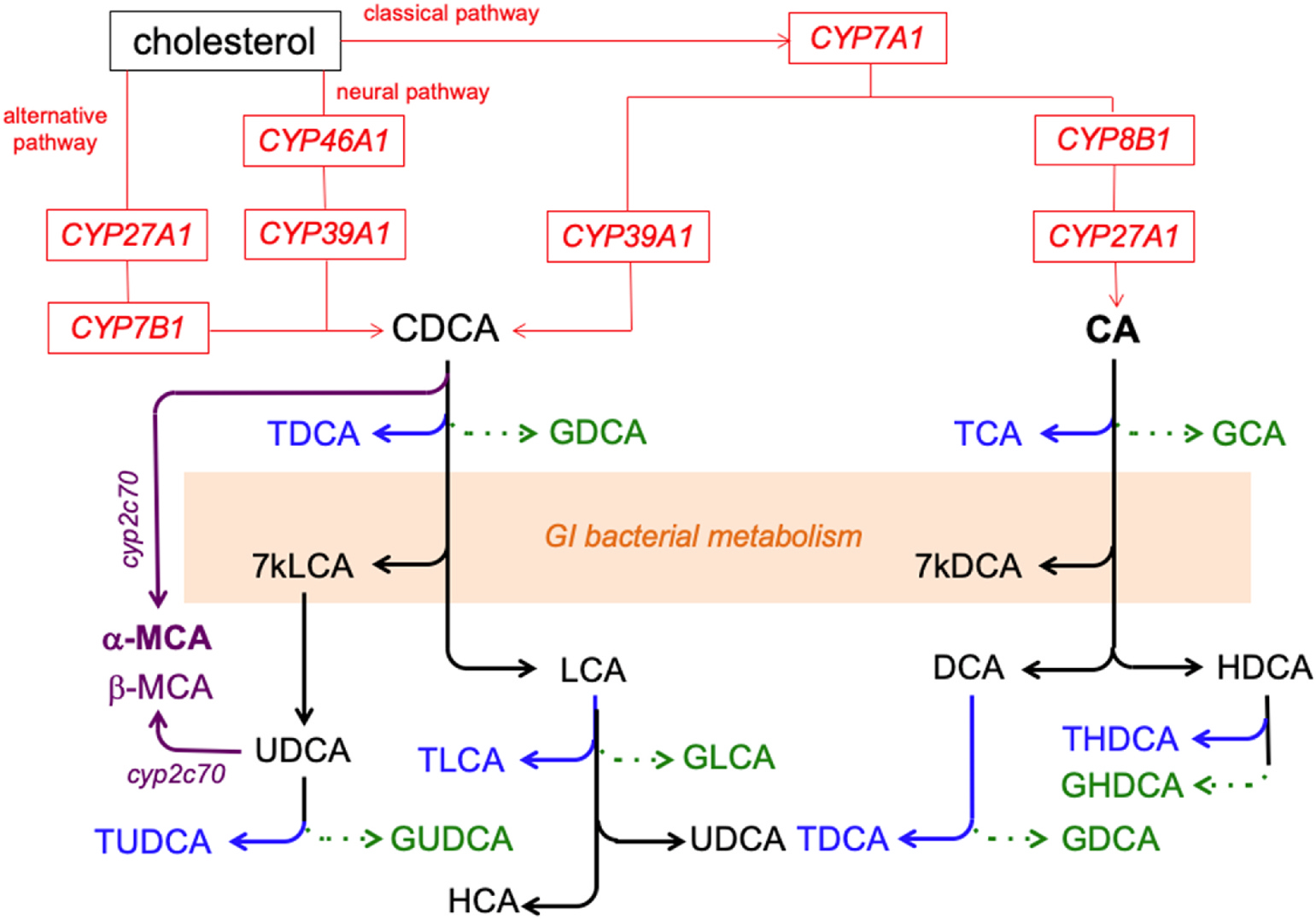
Cholesterol is enzymatically converted to the bile acids cholic acid (CA) or chenodeoxycholic acid (CDCA) by the sequential actions of specific CYP450 enzymes (CYP, red lines and text). These bile acids may be enzymatically converted to the secondary bile acids, deoxycholic acid (DCA), hyodeoxycholic acid (HDCA), lithocholic acid (LCA) or ursodeoxycholic acid (UDCA) by gastrointestinal bacteria. Alpha- and β-mur-icholic acid (MCA, primary bile acids in mice) can be formed from CDCA and UCDA (respectively) by Cyp2c70 (purple lines and text). The bile acids may undergo conjugation with glycine (green arrow and text) or taurine (blue arrow and text). Enzymes in the bacteria residing in the gastrointestinal tract can generate 7-keto-LCA (7 kLCA) and 7-keto-DCA (7kDCA) from CDCA and CA, respectively.
Table 2.
Quantification of bile acids in the liver of control and ethanol (EtOH)-consuming mice.
| Bile acid | Level of bile acid in liver samplea (ng/mg tissue) | Fold-Changeb | pc | |
|---|---|---|---|---|
| Contrbl cice | EtOH-consuming mice | |||
|
| ||||
| CA | 0.37 ± 0.21 | 4.86 ± 1.92 | 13.1 | 0.052 |
| CDCA | 0.39 ± 0.24 | 0.14 ± 0.07 | 0.4 | 0.089 |
| TCA | 9.00 ± 5.29 | 28.81 ± 15.62 | 3.2 | 0.031 |
| TLCA | 0.03 ± 0.001 | 0.03 ± 0.002 | 1.0 | 0.831 |
| DCA | 0.04 ± 0.005 | 0.38 ± 0.36 | 9.0 | 0.074 |
| UDCA | 0.19 ± 0.07 | 0.26 ± 0.13 | 1.37 | 0.325 |
| 7-keto-LCA | 0.03 ± 0.001 (N = 3) | 0.05 ± 0.003 | 2.0 | 0.261 |
| 7-keto-DCA | Below LOQ | 0.58 ± 0.53 (N = 3) | 240d | 0.036 |
| GCA | Below LOQ | 0.09 ± 0.03 (N = 3) | 37.5d | 0.001 |
| TDC d | 2.20 ± 0.73 | 4.47 ± 1.24 | 2.0 | 0.010 |
| THDCA | 0.45 ± 0.38 | 1.22 ± 0.41 | 2.7 | 0.022 |
| HDCA | 3.88 ± 2.86 | 1.14 ± 0.56 | 0.3 | 0.104 |
| GCDCA | ND | ND | ||
| GLCA | ||||
| GUDCA | ||||
| GDCA | ||||
| GHDCA | ||||
| LCA | ||||
ND Not detectable.
Data are presented as mean and associated SD from 4 to 5 mice (except where a different number (N) is specified).
Fold-chain= (average level of bile acid in ethanol-consuming mice)/(average level of bile acid in control mice).
Probability, Student’s unpaired t-test, levels in EtOH-consuming mice compared to those in control mice.
LOQ/2 (0.0024ng/mg tissue) was used to calculate the fold-change for levels that were below LOQ for 7-keto-DCA and GCA in control mice [1].
4. Discussion
The present study investigated the changes that occur in the metabolome in the early stages of ALD in a mouse model. The aim was to eludicate pathways affected by EtOH consumption and that mediate biochemical changes promoting damage to the liver.
Six weeks of EtOH consumption caused minor changes in the histology of the mouse liver, a result consistent with previous publications [25]. The only change identified in the present study was that the lumens of bile ducts in the EtOH-consuming mice were slightly wider than in control mice. In contrast to the histological results, the liver metabolome in EtOH-consuming mice was very different from the control mice, as reflected by the clear separation between the two groups as shown in the PCA plot. These results indicate that cellular activities in the liver are affected by alcohol consumption before the manifestation of histological indicators of tissue damage. To our knowledge, this is the first study reporting significant alcohol consumption-induced changes in the liver tissue metabolome during minimal overt histological signs of liver damage.
There are several studies using either rat or mouse models to explore the impact of EtOH on the metabolome [26–30] and of these, the Lieber DeCarli liquid method is amongst the most commonly used means of administering EtOH to the animals [26,29,30]. Our metabolomics data and multivariate analyses are consistent with the literature in showing distinct perturbations of the metabolome in EtOH-treated animals. In addition, a recent metabolomics study identified the FXR/RXR activation pathway as being highly significant in EtOH-treated mice [29]. This result is consistent with our analysis that revealed that EtOH consumption had the most significant impact on pathways associated with bile acid homeostasis. The farnesoid X receptor (FXR) has emerged as a key player in the control of numerous metabolic pathways [31,32]. It is a bile acid-binding transcription factor belonging to the superfamily of nuclear receptors [33]. The FXR can be activated by both free (e.g., CDCA, CA, DCA, LCA) and conjugated (e.g., GCDCA, TCA, TCDCA) bile acids [33,34]. The synthesis of bile acids is tightly regulated by negative feedback inhibition through FXR [35]. Multiple mechanisms of bile acid feedback regulation to ensure bile acid homeostasis is maintained and bile acid toxicity is controlled [36]. One such mechanism in the liver involves the induction of the negative nuclear receptor small heterodimer partner (SHP) by FXR, which inhibits the transcription of CYP7A1 and CYP8B1, genes responsible for the two rate-limiting enzymes in the classical pathway of bile acid synthesis that produce CA and CDCA from cholesterol [37]. Activation of the liver FXR/SHP pathway by high levels of bile acids is thought to be an adaptive response that protects the liver against cholestatic injury by inhibiting bile acid synthesis [38]. Bile acid pool size, bile acid composition and hydrophobicity play important roles in regulation of bile acid synthesis and lipid metabolism. The potency of the FXR activation in reporter gene assays is CDCA > LCA = DCA > CA, whereas hydrophilic bile acids, such as ursodeoxycholic acid (UDCA) and muricholic acid (MCA), do not activate FXR [39,40]. The conjugated ones only weakly activate FXR [41]. Upon bile acid activation, FXR plays a crucial role in linking bile acid regulation with lipoprotein, lipid and glucose metabolism. In addition, it regulates the inflammatory responses, barrier function and prevention of bacterial translocation in the intestinal tract [40,42]. There is emerging evidence that the activity of FXR is functionally impaired during chronic alcohol intake and that FXR activation could lead to attenuation of alcohol-related liver injury [43,44].
In the present study, levels of CA showed a trend to increase, and its taurine conjugate, TCA, was found to be increased in the liver of EtOH-consuming mice suggesting that the FXR pathway was affected by alcohol intake. If this were the case, one would have expected increased levels of CDCA and LCA to have occurred in EtOH-consuming mice as well. This was not observed. Specifically, CDCA levels were not altered, and LCA levels were not detected. This apparent discrepancy could be explained by the conversion of CDCA or CDCA products to α- or β-MCA in mice (Fig. 3) [45]. In the present study, only 7-keto-DCA was increased in the livers of EtOH-consuming mice. Our method was able to detect 7-keto-DCA in EtOH-consuming mice but not in control mice. Keto-bile acids are formed by the bacterial flora of the intestine through deconjugation, dehydroxylation and oxidoreduction [46]. A total of nine bacterial strains have been shown to synthesize 7-keto-DCA [47]. As such, the observed increase may reflect alterations in the gut microbiome by EtOH consumption. Several studies have shown that alcohol consumption increases the small intestinal bacteria and the intestinal permeability, leading to enhanced entry of endotoxin (which plays a major role in ALD) [48,49]. Interestingly, in our study, the CA bile acid synthesis pathway appeared to be the one primarily affected by EtOH consumption. In the CDCA pathway, hepatic levels of the bile acids were not altered by EtOH consumption except for a 2-fold increase in TDCA levels.
Changes in bile acid composition and synthesis can potentiate hepatotoxicity through pro-inflammatory mechanisms, membrane damage or cytotoxicity [50]. Bile acid synthesis in the liver occurs primarily through the ‘classical’ (neutral) pathway that involves cholesterol being converted to primary bile acids (i.e., CA and CDCA) by several enzymes, including CYP7A1, CYP8B1 and CYP27A1 [51]. A secondary bile acid synthesis pathway, the ‘alternative’ or oxysterol (acidic) pathway, is also present and is thought to assume a more important role during disease and liver insult wherein it may compensate for limitations in the classical pathway. For example, during the initiation of liver cirrhosis, the alternative pathway can become predominant [52,53]. This pathway utilizes extrahepatic cholesterol sources with the help of CYP27A1, CYP7B1 and CYP39A1 [54]. In humans, the ratio of CA:CDCA indicates whether a shift from the primary to secondary pathway has occurred, i.e., greater alternative pathway involvement would lead to higher levels of CDCA relative to CA [55]. In the present study, the ratio was increased in the liver of EtOH-consuming mice, suggesting a shift to the primary pathway. However, unlike humans, mice can produce α-MCA or β-MCA from products of the CDCA pathway. Since we have not measured the MCAs, which are the primary bile acids in mice, along with CA [31], the changes in the ratio may reflect increased MCA production from CDCA or UDCA. The accumulation of taurine-conjugated CA-derived bile acids (i.e., TCA, TDCA, THDCA) in EtOH-consuming mice were increased while other bile acids in the liver were not. Indeed, higher levels of taurine-conjugated bile acids were formed than glycine-conjugated bile acids in the present study, results consistent with previous studies in mice showing the predilection for taurine conjugation of bile acids in mice [56]. Ethanol consumption caused increases in the levels of multiple products of the CA pathway (e.g., TCA, CA, THDCA) but only of one (TDCA) in the CDCA pathway. Based on these results, it is tempting to speculate that EtOH consumption up-regulates the classical pathway and CYP8B1. However, mice can produce α-MCA or β-MCA from products of the CDCA pathway. Given that these metabolites were not measured in the present study, it is not possible to draw definitive conclusions about the effect of EtOH consumption on the CDCA pathway.
5. Conclusions
This is the first study to show striking alterations in the liver metabolome prior to the onset of histological signs of liver damage in a mouse model simulating chronic alcohol consumption. The untargeted metabolomics analysis of liver tissue revealed a clear impact of alcohol consumption, with 304 metabolic features being either up- or down- regulated. This result, along with the pathway analyses, indicate that significant alterations can be caused by alcohol at a stage of liver damage that only shows minor histological changes. Our data suggest that bile acids could serve as an early indicator of liver injury. It is anticipated that the results of the present study will stimulate further investigation of mechanisms involved in the early stages of ALD.
Supplementary Material
Acknowledgements
This work was funded in part by NIAAA grants AA022057 and AA028432.
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Vasilis Vasiliou reports financial support was provided by National Institutes of Health.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cbi.2022.109931.
References
- [1].Kirpich IA, Warner DR, Feng W, Joshi-Barve S, McClain CJ, Seth D, Zhong W, et al. , Mechanisms, biomarkers and targets for therapy in alcohol-associated liver injury: from genetics to nutrition: summary of the ISBRA 2018 symposium, Alcohol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Osna NA, Donohue TM Jr., Kharbanda KK, Alcoholic liver disease: pathogenesis and current management, Alcohol Res 38 (2017) 147–161. [PMC free article] [PubMed] [Google Scholar]
- [3].Liangpunsakul S, Haber P, McCaughan GW, Alcoholic liver disease in asia, europe, and north America, Gastroenterology 150 (2016) 1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Roth NC, Qin J, Histopathology of alcohol-related liver diseases, Clin. Liver Dis 23 (2019) 11–23. [DOI] [PubMed] [Google Scholar]
- [5].Lefkowitch JH, Morphology of alcoholic liver disease, Clin. Liver Dis 9 (2005) 37–53. [DOI] [PubMed] [Google Scholar]
- [6].Lucey MR, Mathurin P, Morgan TR, Alcoholic hepatitis, N. Engl. J. Med 360 (2009) 2758–2769. [DOI] [PubMed] [Google Scholar]
- [7].Marshall S, Chen Y, Singh S, Berrios-Carcamo P, Heit C, Apostolopoulos N, Golla JP, et al. , Engineered animal models designed for investigating ethanol metabolism, toxicity and cancer, Adv. Exp. Med. Biol 1032 (2018) 203–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Stagos D, Chen Y, Brocker C, Donald E, Jackson BC, Orlicky DJ, Thompson DC, et al. , Aldehyde dehydrogenase 1B1: molecular cloning and characterization of a novel mitochondrial acetaldehyde-metabolizing enzyme, Drug Metab. Dispos 38 (2010) 1679–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Farooq MO, Bataller R, Pathogenesis and management of alcoholic liver disease, Dig. Dis 34 (2016) 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Luedde T, Kaplowitz N, Schwabe RF, Cell death and cell death responses in liver disease: mechanisms and clinical relevance, Gastroenterology 147 (2014) 765–783 e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fischer M, You M, Matsumoto M, Crabb DW, Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice, J. Biol. Chem 278 (2003) 27997–28004. [DOI] [PubMed] [Google Scholar]
- [12].You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW, The role of AMP-activated protein kinase in the action of ethanol in the liver, Gastroenterology 127 (2004) 1798–1808. [DOI] [PubMed] [Google Scholar]
- [13].Ji C, Chan C, Kaplowitz N, Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model, J. Hepatol 45 (2006) 717–724. [DOI] [PubMed] [Google Scholar]
- [14].Hazeldine S, Hydes T, Sheron N, Alcoholic liver disease - the extent of the problem and what you can do about it, Clin. Med 15 (2015) 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen Y, Singh S, Matsumoto A, Manna SK, Abdelmegeed MA, Golla S, Murphy RC, et al. , Chronic glutathione depletion confers protection against alcohol-induced steatosis: implication for redox activation of AMP-activated protein kinase pathway, Sci. Rep 6 (2016) 29743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lanaspa MA, Andres-Hernando A, Orlicky DJ, Cicerchi C, Jang C, Li N, Milagres T, et al. , Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice, J. Clin. Invest 128 (2018) 2226–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Monks J, Orlicky DJ, Stefanski AL, Libby AE, Bales ES, Rudolph MC, Johnson GC, et al. , Maternal obesity during lactation may protect offspring from high fat diet-induced metabolic dysfunction, Nutr. Diabetes 8 (2018) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, et al. , Design and validation of a histological scoring system for nonalcoholic fatty liver disease, Hepatology 41 (2005) 1313–1321. [DOI] [PubMed] [Google Scholar]
- [19].Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, et al. , A cross-platform toolkit for mass spectrometry and proteomics, Nat. Biotechnol 30 (2012) 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Myers OD, Sumner SJ, Li S, Barnes S, Du X, One step forward for reducing false positive and false negative compound identifications from mass spectrometry metabolomics data: new algorithms for constructing extracted ion chromatograms and detecting chromatographic peaks, Anal. Chem 89 (2017) 8696–8703. [DOI] [PubMed] [Google Scholar]
- [21].Xia J, Psychogios N, Young N, Wishart DS, MetaboAnalyst: a web server for metabolomic data analysis and interpretation, Nucleic Acids Res 37 (2009) W652–W660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Southam AD, Weber RJ, Engel J, Jones MR, Viant MR, A complete workflow for high-resolution spectral-stitching nanoelectrospray direct-infusion massspectrometry-based metabolomics and lipidomics, Nat. Protoc 12 (2016) 310–328. [DOI] [PubMed] [Google Scholar]
- [23].Shen X, Wang R, Xiong X, Yin Y, Cai Y, Ma Z, Liu N, et al. , Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics, Nat. Commun 10 (2019) 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Charkoftaki G, Golla JP, Santos-Neto A, Orlicky DJ, Garcia-Milian R, Chen Y, Rattray NJW, et al. , Identification of dose-dependent DNA damage and repair responses from subchronic exposure to 1,4-dioxane in mice using a systems analysis approach, Toxicol. Sci (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Orlicky DJ, Roede JR, Bales E, Greenwood C, Greenberg A, Petersen D, McManaman JL, Chronic ethanol consumption in mice alters hepatocyte lipid droplet properties, Alcohol Clin. Exp. Res 35 (2011) 1020–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bradford BU, O’Connell TM, Han J, Kosyk O, Shymonyak S, Ross PK, Winnike J, et al. , Metabolomic profiling of a modified alcohol liquid diet model for liver injury in the mouse uncovers new markers of disease, Toxicol. Appl. Pharmacol 232 (2008) 236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ma T, Li Y, Zhu Y, Jiang S, Cheng C, Peng Z, Xu L, Differential metabolic pathways and metabolites in a C57bl/6J mouse model of alcoholic liver disease, Med. Sci. Mon. Int. Med. J. Exp. Clin. Res 26 (2020), e924602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gika HG, Wilson ID, Global metabolic profiling for the study of alcohol-related disorders, Bioanalysis 6 (2014) 59–77. [DOI] [PubMed] [Google Scholar]
- [29].Dong Y, Qiu P, Zhao L, Zhang P, Huang X, Li C, Chai K, et al. , Metabolomics study of the hepatoprotective effect of Phellinus igniarius in chronic ethanolinduced liver injury mice using UPLC-Q/TOF-MS combined with ingenuity pathway analysis, Phytomedicine 74 (2020), 152697. [DOI] [PubMed] [Google Scholar]
- [30].Deda O, Virgiliou C, Orfanidis A, Gika HG, Study of fecal and urinary metabolite perturbations induced by chronic ethanol treatment in mice by UHPLC-MS/MS targeted profiling, Metabolites 9 (2019) 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mertens KL, Kalsbeek A, Soeters MR, Eggink HM, Bile acid signaling pathways from the enterohepatic circulation to the central nervous system, Front. Neurosci 11 (2017) 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hou Y, Fan W, Yang W, Samdani AQ, Jackson AO, Qu S, Farnesoid X receptor: an important factor in blood glucose regulation, Clin. Chim. Acta 495 (2019) 29–34. [DOI] [PubMed] [Google Scholar]
- [33].Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, et al. , Identification of a nuclear receptor for bile acids, Science 284 (1999) 1362–1365. [DOI] [PubMed] [Google Scholar]
- [34].Wang H, Chen J, Hollister K, Sowers LC, Forman BM, Endogenous bile acids are ligands for the nuclear receptor FXR/BAR, Mol. Cell 3 (1999) 543–553. [DOI] [PubMed] [Google Scholar]
- [35].Wahlström A, Sayin SI, Marschall HU, Bäckhed F, Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism, Cell Metabol 24 (2016) 41–50. [DOI] [PubMed] [Google Scholar]
- [36].Chiang JY, Negative feedback regulation of bile acid metabolism: impact on liver metabolism and diseases, Hepatology 62 (2015) 1315–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chiang JY, Bile acids: regulation of synthesis, J. Lipid Res 50 (2009) 1955–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li T, Chiang JY, Bile acid signaling in metabolic disease and drug therapy, Pharmacol. Rev 66 (2014) 948–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Park JS, Seo JH, Youn HS, Gut microbiota and clinical disease: obesity and nonalcoholic Fatty liver disease, Pediatr. Gastroenterol. Hepatol. Nutr 16 (2013) 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ding L, Yang L, Wang Z, Huang W, Bile acid nuclear receptor FXR and digestive system diseases, Acta Pharm. Sin. B 5 (2015) 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vaquero J, Monte MJ, Dominguez M, Muntane J, Marin JJ, Differential activation of the human farnesoid X receptor depends on the pattern of expressed isoforms and the bile acid pool composition, Biochem. Pharmacol 86 (2013) 926–939. [DOI] [PubMed] [Google Scholar]
- [42].Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ, Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis, Cell 102 (2000) 731–744. [DOI] [PubMed] [Google Scholar]
- [43].Wu W, Zhu B, Peng X, Zhou M, Jia D, Gu J, Activation of farnesoid X receptor attenuates hepatic injury in a murine model of alcoholic liver disease, Biochem. Biophys. Res. Commun 443 (2014) 68–73. [DOI] [PubMed] [Google Scholar]
- [44].Manley S, Ding W, Role of farnesoid X receptor and bile acids in alcoholic liver disease, Acta Pharm. Sin. B 5 (2015) 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li T, Apte U, Bile acid metabolism and signaling in cholestasis, inflammation, and cancer, Adv. Pharmacol 74 (2015) 263–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB, Consequences of bile salt biotransformations by intestinal bacteria, Gut Microb 7 (2016) 22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Heinken A, Ravcheev DA, Baldini F, Heirendt L, Fleming RMT, Thiele I, Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease, Microbiome 7 (2019) 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Park BJ, Lee YJ, Lee HR, Chronic liver inflammation: clinical implications beyond alcoholic liver disease, World J. Gastroenterol 20 (2014) 2168–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Abhilash PA, Harikrishnan R, Indira M, Ascorbic acid suppresses endotoxemia and NF-kappaB signaling cascade in alcoholic liver fibrosis in Guinea pigs: a mechanistic approach, Toxicol. Appl. Pharmacol 274 (2014) 215–224. [DOI] [PubMed] [Google Scholar]
- [50].Zhang Y, Hong JY, Rockwell CE, Copple BL, Jaeschke H, Klaassen CD, Effect of bile duct ligation on bile acid composition in mouse serum and liver, Liver Int 32 (2012) 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K, Targeting bile-acid signalling for metabolic diseases, Nat. Rev. Drug Discov 7 (2008) 678–693. [DOI] [PubMed] [Google Scholar]
- [52].Crosignani A, Del Puppo M, Longo M, De Fabiani E, Caruso D, Zuin M, Podda M, et al. , Changes in classic and alternative pathways of bile acid synthesis in chronic liver disease, Clin. Chim. Acta 382 (2007) 82–88. [DOI] [PubMed] [Google Scholar]
- [53].Pandak WaK M, Genta, The acidic pathway of bile acid synthesis: not just an alternative pathway, Liver Res (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pandak WM, Kakiyama G, The acidic pathway of bile acid synthesis: not just an alternative pathway(*), Liver Res 3 (2019) 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].MahmoudianDehkordi S, Arnold M, Nho K, Ahmad S, Jia W, Xie G, Louie G, et al. , Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease-An emerging role for gut microbiome, Alzheimers Dement 15 (2019) 76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Li J, Dawson PA, Animal models to study bile acid metabolism, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis 1865 (2019) 895–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Both untargeted and targeted metabolomics datasets will be available at the NIH Metabolomics workbench (http://www.metabolomicsworkbench.org/).