

Abstract
The protein kinase family is characterized by substantial conservation of architectural elements that are required for both ATP binding and phosphotransferase activity. Many of these structural features have also been identified in homologous enzymes that phosphorylate a variety of alternative, non-protein substrates. A comparative structural analysis of these different kinase sub-classes is a portal to a greater understanding of reaction mechanisms, enzyme regulation, inhibitor-development strategies, and superfamily-level evolutionary relationships. To serve such advances, we review structural elements of the protein kinase fold that are conserved in the subfamily of inositol phosphate kinases (InsPKs) that share a PxxxDxKxG catalytic signature: inositol 1,4,5-trisphosphate kinase (IP3K), inositol hexakisphosphate kinase (IP6K), and inositol polyphosphate multikinase (IPMK). We describe conservation of the fundamental two-lobe kinase architecture: an N-lobe constructed upon an anti-parallel β-strand scaffold, which is coupled to a largely helical C-lobe by a single, adeninebinding hinge. This equivalency also includes a G-loop that embraces the β/γ-phosphates of ATP, a transition-state stabilizing residue (Lys/His), and a Mg-positioning aspartate residue within a catalytic triad. Furthermore, we expand this list of conserved structural features to include some not previously identified in InsPKs: a ‘gatekeeper’ residue in the N-lobe, and an ‘αF’-like helix in the C-lobe that anchors two structurally-stabilizing, hydrophobic spines, formed from non-consecutive residues that span the two lobes. We describe how this wide-ranging structural homology can be exploited to develop lead inhibitors of IP6K and IPMK, by using strategies similar to those that have generated ATP-competing inhibitors of protein-kinases. We provide several examples to illustrate how such an approach could benefit human health.
Introduction: a sub-family of inositol phosphate kinases defined by their PxxxDxKxG catalytic signature.
Each member of the multitudinous inositol phosphate (InsP) family consists of a unique, three-dimensional pattern of phosphates, sometimes including pyrophosphates, all of which are crammed around a six-carbon inositol ring (Abel et al., 2002; Hatch and York, 2010; Shears et al., 2017). As one might expect from such polar entities, they are water-soluble molecules that access the entire cytoplasm (it is presumed they can pass through nuclear pores, but there is no indication they can cross other membrane-barriers). Despite the freely-diffusible nature of the InsPs, they are occasionally misunderstood as being members of the phosphoinositide family, as noted previously (Schell, 2010). So, we will reiterate here that the term ‘phosphoinositides’ is reserved for those inositol-based signals that are lipid in nature (due to their diacylglycerol backbone); these are physicochemically- and functionally-distinct from the InsPs.
Due to the many important biological functions of the InsPs (Hatch and York, 2010; Shears, 2017), the kinases that synthesize them command considerable attention. Three of these kinases are grouped into a sub-family (see Fig. 1) that is defined by their PxxxDxKxG catalytic signature: inositol 1,4,5-trisphosphate kinase (IP3K; 2.7.1.127), inositol hexakisphosphate kinase (IP6K; E.C. 2.7.4.21), and inositol polyphosphate multikinase (IPMK, also known as ipk2 in plants and yeasts; E.C. 2.7.1.151)(Saiardi et al., 1999). We use the term ‘PDKG-InsPK’ (Nalaskowski and Mayr, 2004) to describe this group of enzymes.
Fig. 1. The kinase activities of IPMK, IP3K, and IP6K in the pathway of inositol phosphate synthesis in mammals.
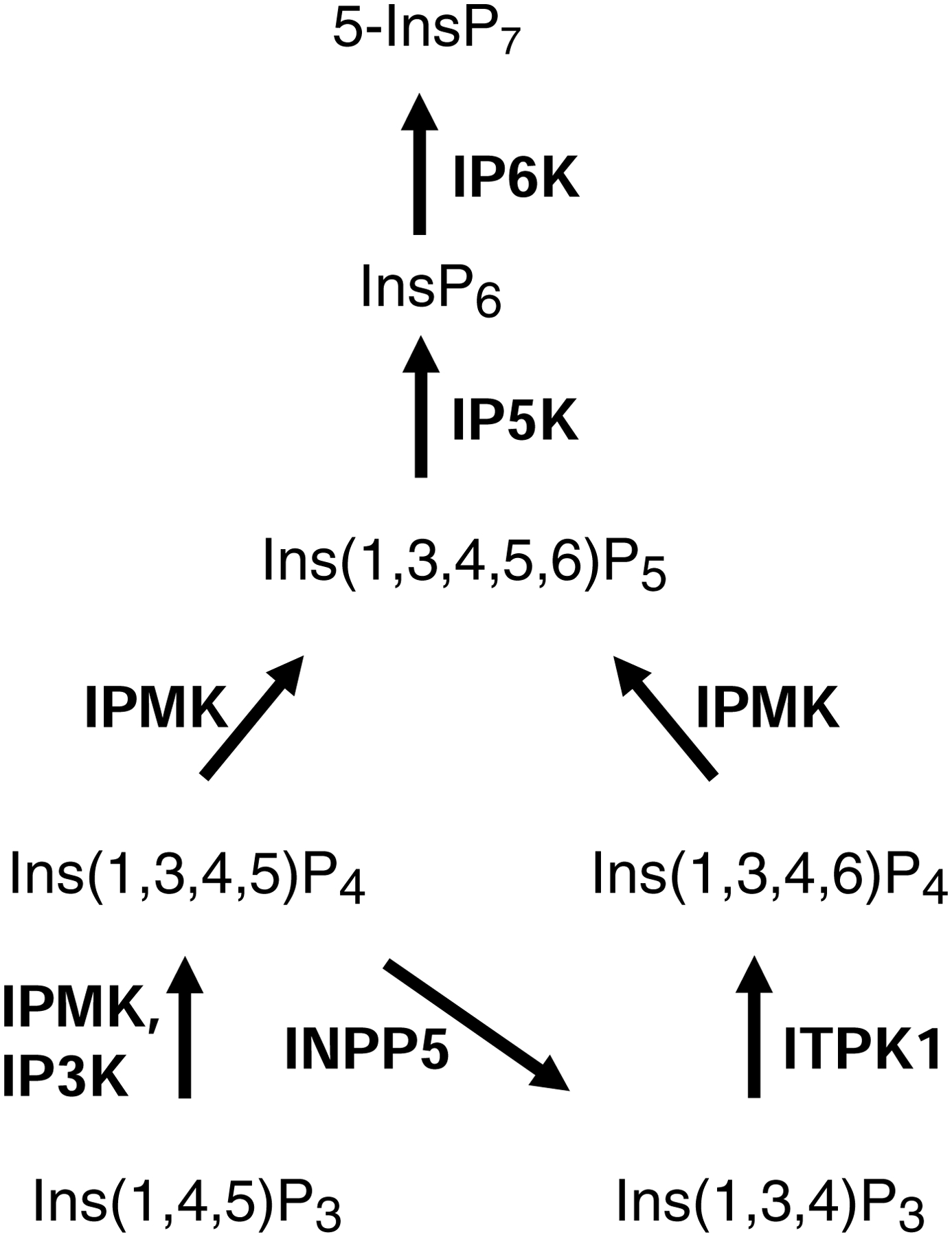
Each individual inositol phosphate is abbreviated as follows: ‘Ins’ refers to inositol; the subscripts denote the total number of phosphates (‘P’), and the numbers in parentheses describe the positions of the phosphate groups around the inositol ring. Note that ‘5-InsP7’ denotes an ‘inositol pyrophosphate’ that has a diphosphate group at the 5-position. Arrows depict metabolic steps, catalyzed by the following enzymes, with E.C. numbers in parentheses: 1, IPMK, inositol polyphosphate multikinase (2.7.1.151; also known as IPK2); IP3K, inositol trisphosphate 3-kinase (2.7.1.127); INPP5, inositol polyphosphate 5-phosphatase, (3.1.3.56); ITPK1, inositol trisphosphate 6-kinase/ inositol tetrakisphosphate 1-kinase (2.7.1.134); IP5K, inositol pentakisphosphate 2-kinase (2.7.1.158; also known as IPK1); IP6K, inositol hexakisphosphate kinase (2.7.4.21).
A major function for the IP3Ks is to terminate calcium-mobilization by Ins(1,4,5)P3. Such negative feedback is much more than just a signaling “off-switch”; it is an intrinsic component of the dynamics of calcium oscillations, which themselves influence countless aspects of cell biology (Gaspers et al., 2014; Politi et al., 2006). Additionally, the reaction product of IP3K activity – Ins(1,3,4,5)P4 – can serve as a precursor for synthesis of the ‘higher’ InsPs (Fig. 1). Ins(1,3,4,5)P4 has also been proposed to participate in the process of calcium signaling (Irvine and Schell, 2001), but that concept has never been conclusively validated (Schell, 2010; Shears et al., 2012). However, several other products of this kinase-dependent pathway, including in particular InsP6 and 5-InsP7 (Fig. 1), are widely recognized as being vital regulators of a diverse range of biological processes (Hatch and York, 2010; Shears, 2017).
IPMK (Fig. 1) is aptly named as a ‘multi-kinase’ (Saiardi et al., 1999). This enzyme utilizes both 3- and 6-kinase activities to phosphorylate Ins(1,4,5)P3 to InsP5 (Odom et al., 2000; Saiardi et al., 2000), it also uses a 5-kinase activity to convert Ins(1,3,4,6)P4 to InsP5 (Chang et al., 2002; Nalaskowski et al., 2002), while the yeast and mammalian IPMKs exhibit 3-kinase activity towards the phosphoinositide, PtdIns(4,5)P2, thereby producing PtdIns(3,4,5)P3 (Maag et al., 2011; Resnick et al., 2005). The latter lipid activates the transcriptional activity of nuclear receptor steroidogenic factor 1 (Blind et al., 2012). The enzyme’s InsP kinase activity is also advocated to regulate transcription in S. cerevisiae (Odom et al., 2000). The mechanisms by which the InsP4 and InsP5 products of IPMK regulate gene transcription are still being pursued (Hatch et al., 2018; Kim et al., 2017).
As for IP6Ks, these are of particular interest because they add a 5-β-phosphate to InsP6, yielding 5-InsP7, an ‘inositol pyrophosphate’ (Saiardi et al., 1999; Shears, 2017). The latter is an ‘energetic’ (Hand and Honek, 2007), multifunctional molecule that receives particular attention for its key roles in bioenergetic homeostasis, cancer, and aging (Chakraborty, 2017; Shears, 2017).
Bearing in mind the enormous significance of the PDKG-InsPK family, it is unsurprising that several groups have taken a structural approach to characterizing these enzymes (Bennett et al., 2006; Chamberlain et al., 2005; Endo-Streeter et al., 2012; Gonzalez et al., 2004; Holmes and Jogl, 2006; Miller and Hurley, 2004; Wang et al., 2014; Wang and Shears, 2017). Such information can derive insight into reaction mechanisms, enzyme regulation, and evolutionary relationships, and can also assist in the development of enzyme-specific inhibitors, to be used not only as research tools, but also as ‘lead’ compounds for the generation of drugs that can benefit human health. These are all areas that we believe will benefit from the comparative structural analysis described below.
Conservation in InsP kinases of the two-lobe architecture of protein kinases.
As first described for PKA (Knighton et al., 1991a), the conserved core structure of protein-kinases comprises a small N-terminal ‘N-lobe’ and a larger C-terminal ‘C-lobe’ (Fig. 2A). The architectural core of the N-lobe consists of five anti-parallel β-sheets which are coupled to a helical subdomain, the ‘C-helix’ (αC in Fig 2A); the latter spans the breadth of the N-domain (Taylor and Kornev, 2011). This C-helix lines a wedge-shaped cleft between the two lobes that accommodates the ATP.
Fig. 2. A cartoon describing conserved secondary and tertiary structures among PI3Kγ, PKA, IPMK and IP3KA.
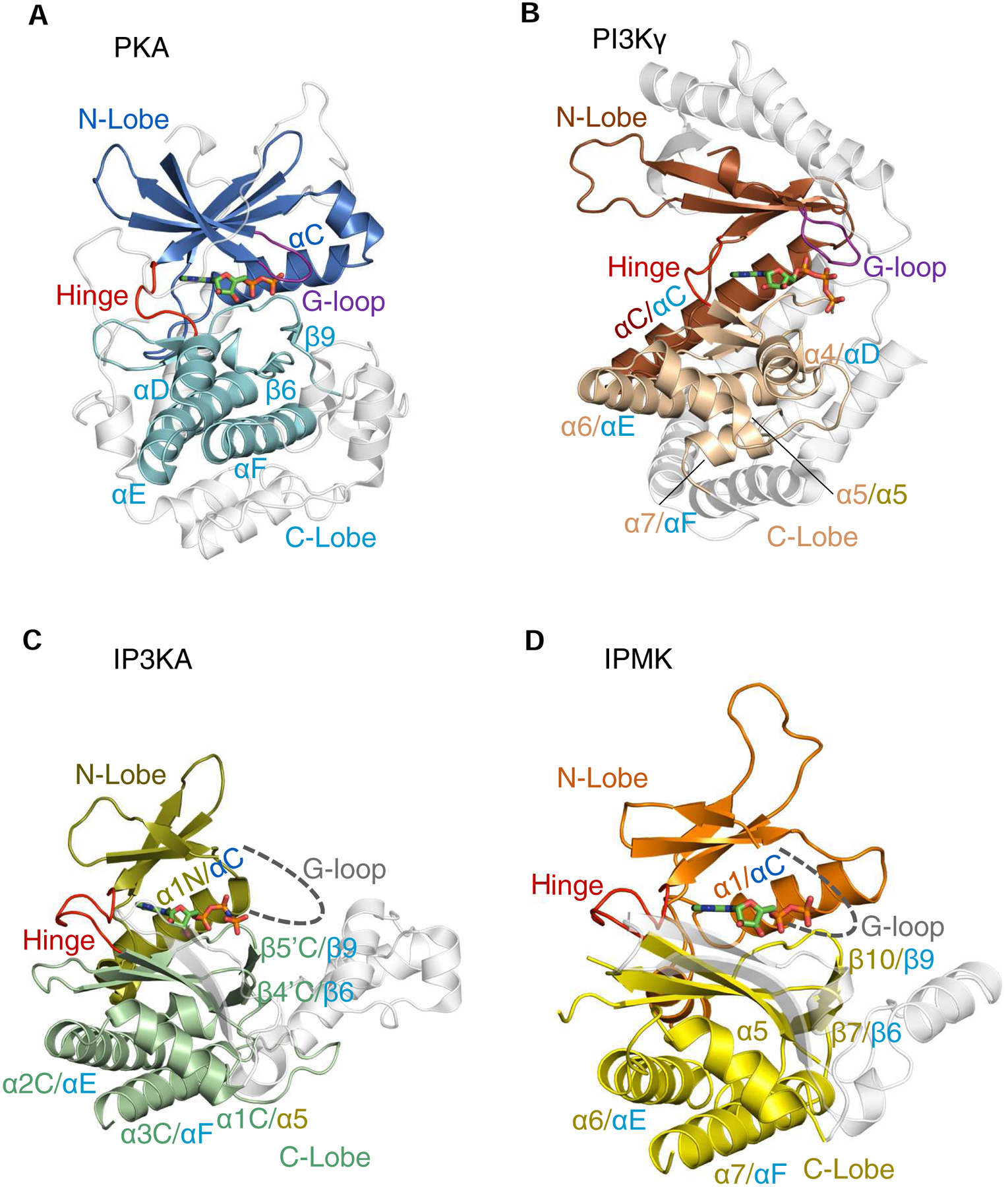
Ribbon plots of the following enzymes are shown (PDB access codes in parentheses): A, Murine protein kinase A (catalytic subunit)/ADP complex, E.C. 2.7.11.11 (1L3R); B, Sus scrofa PI3Kγ (catalytic subunit)/ATP complex (E.C. 2.7.11.1) (1E8X). C, Human IP3KA/AMP-PNP complex (1W2C) D, Human IPMK/ADP/IP3 complex (5W2H). The α-helices and β-strands that are colored are conserved in at least two of the structures shown; in cases where individual structural elements have two labels separated by a forward slash, the first is the label given in the original PDB entry, and the second label corresponds to the corresponding element in the homologous protein (color coding of labels matches that of the originating lobe). Non-conserved structural elements are depicted in light gray. Broken lines are used to depict the expected G-loops in IP3K and IPMK (their actual structures are not known). Bound nucleotides are shown as stick models. Note that the structural element in PI3Kγ that we describe as a ‘G-loop’ (panel B) was originally named ‘P-loop’ (Walker et al., 1999). For an explanation, see the text.
The realization that the protein kinase fold might occur in other cell-signaling contexts owes much to early demonstrations that it is present in phosphoinositide kinases such as PIP4K2B (E.C. 2.7.1.149; phosphatidylinositol-5-phosphate 4-kinase type 2β) (Rao et al., 1998) and the catalytic subunit of PI3Ks (E.C. 2.7.1.153; phosphoinositide 3-kinases; see Fig. 2B) (Walker et al., 2000; Walker et al., 1999). Subsequent publication of multiple sequence alignments that include InsP kinases suggested they, too, might possess a kinase-fold through a distant, evolutionary connection (Cheek et al., 2002). The latter study is particularly significant because protein superfamily relationships are not always clear from primary sequence alone, which is highly degenerate; structural information is much more reliable for predicting evolutionary links (Scheeff and Bourne, 2005). Nevertheless, the concept that protein kinases and InsP kinases have structural similarities was consolidated by a pair of 2004 studies that described the crystal structure of IP3KA (Gonzalez et al., 2004; Miller and Hurley, 2004). For example, the latter studies identified N- and C-lobes (Fig. 2C) with several structural elements corresponding to those previously characterized in protein kinases (Fig. 2A,C; Table 1). The protein kinases and PDKG-InsPKs also share the same functional distinctions between the two lobes: the N-lobes provide most of the structural features that are required for ATP-binding, while it is substrate recognition that is predominantly a feature of the C-lobe. The descriptions of the crystal structure of IPMK/Ipk2 from S. cerevisiae (Holmes and Jogl, 2006), and later the ortholog expressed in Arabidopsis (Endo-Streeter et al., 2012), have consolidated appreciation that PDKG-InsPKs possess the basic two-lobe architecture of protein-kinases. The human IPMK structure (Fig. 2D) was solved more recently, and is also the first of this series of studies to describe an enzyme/InsP crystal complex (Wang and Shears, 2017). The crystal structure of an IP6K expressed in Entamoeba histolytica (EhIP6K) was described in 2014 (Wang et al., 2014). These developments have prompted us to conduct a comparative analysis of the structures of PDKG-InsPKs, protein kinases and also PI3Ks (see below).
Table 1.
A list of conserved structural and catalytic elements in PKA, PI3Kγ, and PDKG-InsPKs.
| MmPKA | SsPI3Kγ | HsIP3KA | HsIPMK | HsIP6K2 | |
|---|---|---|---|---|---|
| PDB Code | 1L3R | 1E8X | 1W2C | 5W2H | Modeled |
| Conserved Secondary Structures | |||||
| N-lobe | β2–4 | kβ4–5 | β1N-2N | β0–1 | |
| αC | kα3 | α1N | α1 | ||
| β4–5 | kβ6–7 | β3N-4N | β2–3 | ||
| Hinge Region | β5-αD | kβ7- kβ8 | β4N-β1C | β3-β4 | |
| C-lobe | αD | α4 | - | - | |
| - | kα5 | α1C | α5 | ||
| αE | kα6 | α2C | α6 | ||
| β6 | - | β4’C | β7 | ||
| β7–8 | kβ9–10 | β4C-5C | β8–9 | ||
| β9 | - | β5’C | β10 | ||
| αF | kα7 | α3C | α7 | ||
| Catalytic Center | |||||
| G-loop | gtg 52 sfg | ask 807 kkp | lag 193 htg | vag 55 hmy | vgg 28 hsc |
| Transition State Stabilization | |||||
| Residue | K168 | H948 | K263 | K146 | K222 |
| Motif | Y/HRDxK | DRH | PxxDxKxG | PxxDxKxG | PxxDxKxG |
| Catalytic Triad | |||||
| DFG/A Motif | d 184 fg | d 964 fg | d 414 fg | d 385 fa | d 383 fa |
| Salt Bridge | K72---E91 | K833---D836 | K209---E215 | K75---E86 | K42---E48 |
| ATP Binding | |||||
| Gatekeeper | M120 | I879 | L248 | L130 | L216 |
| Hinge | e 121 yv | e 880 iv | q 249 dl | e 131 dv | e 207 nl |
| Ribose Binding | E127 | - | D261 | D144 | D220 |
| Structural Spines | |||||
| C- Spine (C-lobe) | |||||
| First Layer | L173 | M953 | L401 | L254 | L330 |
| Second Layer | M128, L172, I174 | I888, I952, I954 | L261, L400, F402, V412 | M143, L253, F255, V381 | L219, L229, V331, V379 |
| Third Layer (α-helix) | L227, M231 | L960, V986, F991 | L451, L455 | L406, L410 | L413, V417 |
| C- Spine (N-lobe) | |||||
| First Layer | V57, A70 | W812, I831 | F198, I207 | I65, V73 | V32, L40 |
| Second Layer | M71, I73 | I830, M878 | L208 | L66, L74 | L33, C41 |
| Third Layer | L40, F110, Y117 | L742, F832 | V238, Y245 | W117, P119, Y127 | V70, V72, F203 |
| R- Spine | |||||
| Residues | L95, L106, Y164, F185 | M842, I844, L845, I870, I944, F965 | L219, F234, V395, F417 | Y90, Y113, F248, F386 | Y52, Y66, F324, F384 |
| Anchoring contacts | D220---Y164 | G945---I944 | D442---V395 | D396---F248 | D403---F324 |
The Table summarizes the homologies between the indicated members of the kinase superfamily. The sources of this information are the structural data associated with the given PDB codes, with the exception of HsIP6K2, which is a homology model constructed as previously described, based on EhIP6K2 (Puhl-Rubio et al., 2018; Wang et al., 2014). Since HsIP6K2 is a model, we did not attempt to annotate “conserved secondary structures”. A single dash indicates no homologies entity has been identified. Note that the structural element in PI3Kγ that we describe as a ‘G-loop’ was originally named ‘P-loop’ (Walker et al., 1999). For an explanation, see the text.
Substrate recognition: protein kinases versus PDKG-InsPKs.
In the context that protein kinases, PI3Ks and PDKG-InsPKs may all be considered members of the same kinase superfamily, the ATP-binding N-lobe is evolutionarily robust. In contrast, the C-lobes exhibit more divergence in both sequence and structure, which has allowed substrate versatility.
Protein kinases accommodate a polypeptide chain is into a long, surface grove between the two lobes (Fig. 3A), but nevertheless, almost all of the ligand’s interactions with the protein involve the C-lobe (the exception being two hydrogen bonds with the G-loop) (Madhusudan et al., 2002). The more N-terminal section of the polypeptide substrate slots between αD and αF helices on one side, and the αG helix on the other (Taylor and Kornev, 2011). The residue that is placed immediately C-terminal to the phosphorylation site locks into a pocket formed by the so-called P + 1 loop (Taylor and Kornev, 2011). Both hydrophobic interactions and hydrogen bonds contribute to substrate positioning (Moore et al., 2003; Zhu et al., 2005).
Fig. 3. Electrostatic surface plots of substrate binding sites for PKA and IPMK.
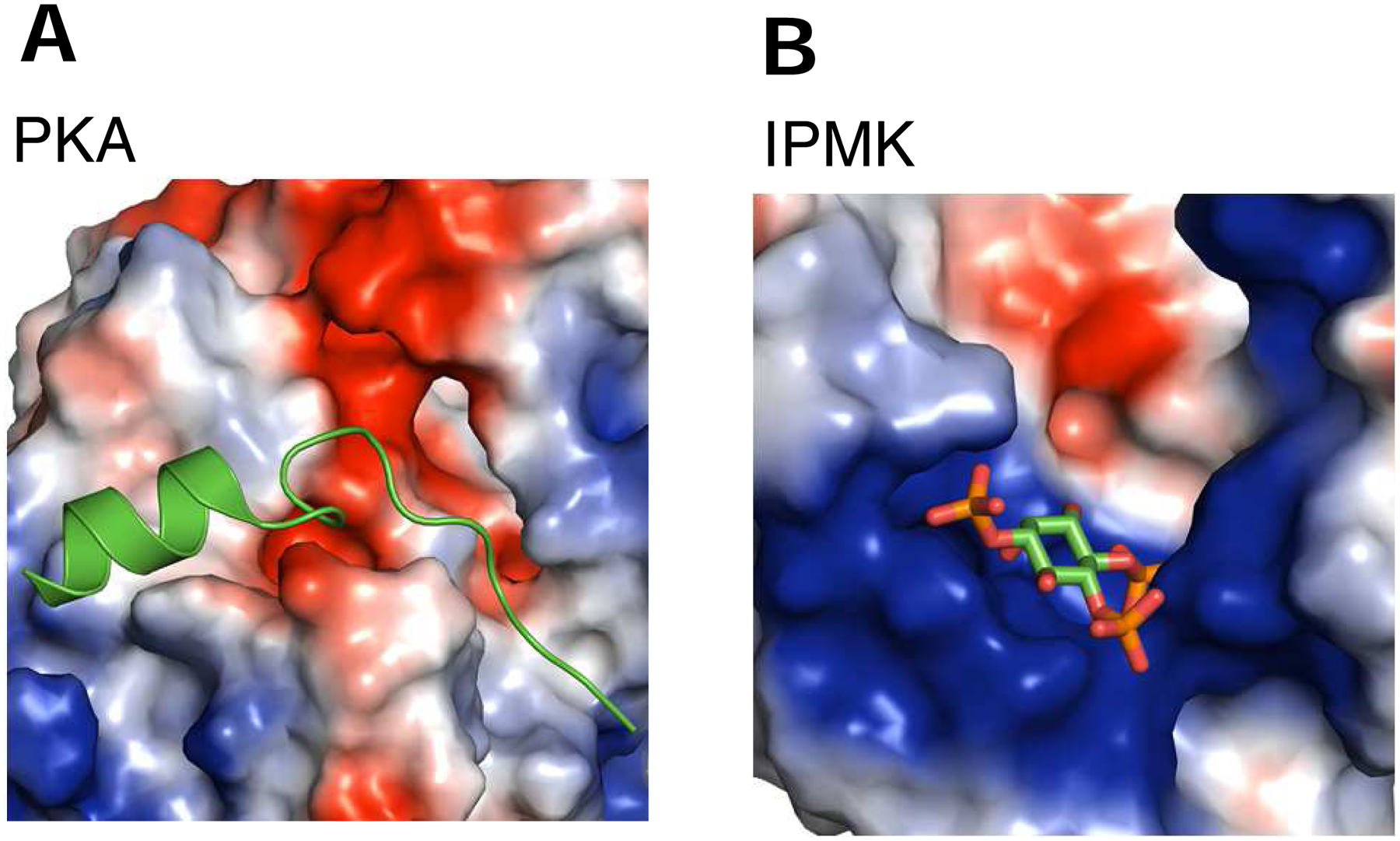
Shown are electrostatic surface plot with blue and red coloration to denote positive and negative electrostatic potentials, respectively, at physiological pH, for A, murine PKA in complex with a 20 residue peptide inhibitor (PDB, 1L3R; (Madhusudan et al., 2002)), B, human IPMK (PDB, 5W2H) in complex with Ins(1,4,5)P3; (Wang and Shears, 2017)). Substrates are colored green and red.
The substrates of the PDKG-InsPKs — small and highly negatively-charged inositol phosphates — have an inherently different physicochemical nature as compared to the polypeptides that are phosphorylated by protein kinases. So naturally, the two kinase subclasses possess distinctive ligand-binding elements, but they are still hosted by the C-lobes. For example, the aforementioned αD and αG helices that are so important for protein kinases are not conserved in PDKG-InsPKs (Fig. 2A,D; Table 1; (Miller and Hurley, 2004)). Instead, the ligand pocket of PDKG-InsPKs is considerably more electropositive (Fig. 3A,B). In the case of IPMKs and IP6Ks (Wang et al., 2014; Wang and Shears, 2017), substrate-binding involves rather short ‘IP helices’ that host arrays of positively-charged residues; nevertheless, they are spatially equivalent to the ‘activation segment’ of protein kinases (which corresponds to the ‘activation loop’ of PI3Ks) (Fig. 3B; 4A,B,C). Thus, we view the IP helices as part of the C-lobe, rather than their constituting a separate ‘domain’, as proposed by others (Endo-Streeter et al., 2012; Holmes and Jogl, 2006). For EhIP6K, two of these IP helices can be viewed as forming one jaw of an open clamshell (Wang et al., 2014); the other jaw is provided by another C-lobe element, an unusual, two-turn 310-helix. The two ‘jaws’ are not directly connected, but they approach within sufficient proximity to provide a metaphorical hinge (Wang et al., 2014).
Fig. 4. Cooperation between the lobes: R-Spines, gatekeepers, catalytic triads and other motifs.
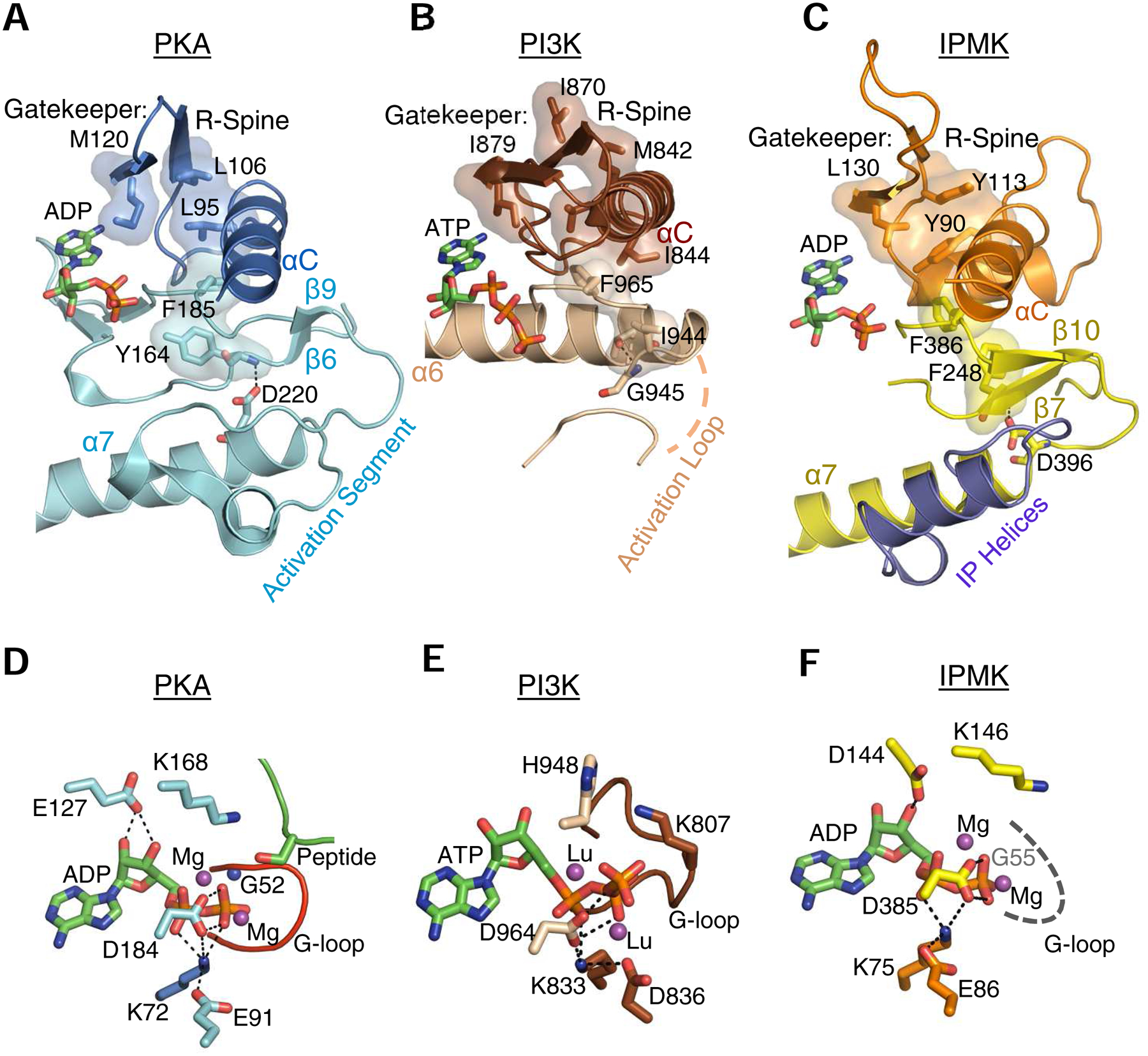
A, B and C highlight selected, conserved elements of murine PKA (PDB, 1L3R), human IPMK (PDB, 5W2H) and Sus scrofa PI3Kγ (PDB, 1E8X), respectively, shown as a composite of ribbon plots, with selected key residues as space-filling representations and stick-model depictions. For PKA and PI3Kγ, the identities of residues comprising the gatekeeper, R-spine and the anchoring α-helix are provided in reviews of this topic (Taylor and Kornev, 2011; Vadas et al., 2011); the corresponding features in IPMK were identified from its alignment with PKA (using Pymol) in which bound nucleotides were superimposed. Panels D, E, F are corresponding stick-model close-ups of the catalytic pockets. The dashed gray line in panel F denotes that the actual structure of the G-loop is disordered in the crystal structure. The residues that we denote as comprising a ‘catalytic triad’ (see text) are as follows: PKA, Lys72, Glu91, Asp184; PI3Kγ, Lys833, Asp836, Asp964; IPMK, Lys75, Glu86, Asp385 (also see Table 1). Thin, broken black lines depict interactions between residues in the catalytic triad, and also those involving the nucleotide’s ribose ring. Magenta balls depict the metal binding sites occupied by magnesium (or, in the case of PI3Kγ, lutetium (Walker et al., 1999)). All other color schemes match those in Fig. 2. Note that the structural element in PI3Kγ that we describe as a ‘G-loop’ (panel E) was originally named ‘P-loop’ (Walker et al., 1999). For an explanation, see the text.
For human IPMK, we have described the substrate-binding pocket as horseshoe-shaped, constructed from an α-helix, a classical (one turn) 310-helix, and also a proline loop in the N-lobe that positions its N-terminal Gln78 and C-terminal Arg82 for interactions with substrates. Human IPMK is therefore unique in being the only PDKG-InsPK family member shown to utilize residues in the N-lobe for substrate binding. It should be noted that a full description of the substrate-binding pocket is not available from published structures of IPMK orthologs from a yeast (Holmes and Jogl, 2006) and a plant (Endo-Streeter et al., 2012), as those crystal complexes do not contain substrates. However, we do know that those orthologs do not possess the proline-loop, which we therefore view as a relatively recent evolutionary development (Wang and Shears, 2017).
Finally, in IP3Ks, the IP helices are replaced by a distinct and more highly structured 63-residue insert into the C-lobe that is generally considered to be a separate domain of the protein (Gonzalez et al., 2004; Miller and Hurley, 2004) that evolved at a late stage of PDKG-InsPK evolution (Bennett et al., 2006).
To summarize, there are important differences between the C-lobes of protein kinases and PDKG-InsPKs that are necessary for the purposes of substrate specificity, while the ATP-binding N-lobes are much more alike. Nevertheless, in our new analysis (Table 1, and see below), we show that even the C-lobe has catalytic elements, and stabilizing scaffolds, that are conserved to a greater degree than has generally been appreciated.
Conservation in InsPKs of individual structural elements: human IPMK as an example.
In this section, we will focus on human IPMK as an exemplar for the degree of structural conservation between protein kinases and PDKG-InsPKs. Corresponding information for PI3Kγ and other PDKG-InsPKs are also provided in Table 1. Note that the data for human IP6K2 are less detailed (Table 1), because they are derived from a homology model that we previously created from the crystal structure of EhIP6K (Puhl-Rubio et al., 2018; Wang et al., 2014).
As mentioned above, IPMK contains two lobes that correspond to the N- and C-lobes of protein kinases. One of the most striking observations to emerge from a structural comparison of PKA with IPMK is the remarkable degree to which the N-lobes of these two proteins can be superimposed: the RMSD is 1.8 Å (Wang and Shears, 2017). For example, there is considerable overlap of the core scaffold of antiparallel β-sheets, even though there are just four of them in IPMK (Fig. 2D), while PKA has five, which are also longer (Fig. 2A). Additionally, the relative position of the αC-helix in PKA is very similar to that of the α1-helix in IPMK (Fig. 2A,D; Table 1).
An important conserved element in the N-lobe of protein kinases is the ‘Gly-loop’ between the β1 and β2 strands that makes contacts with nucleoside phosphates (Fig. 2A). This loop contains three Gly residues that are critical to the loop’s necessary structural characteristics and its appropriate degree of conformational flexibility (Knighton et al., 1991b). Mutagenic experiments have established that the most catalytically-critical of the three Gly residues is Gly52, which lies in the middle of the motif (Hemmer et al., 1997).
As for IPMK, multiple sequence alignments have suggested that there is a Gly-loop in the N-lobe that interacts with nucleotide phosphates (Gonzalez et al., 2004; Wang and Shears, 2017). Even though this loop only contains one Gly residue, it corresponds to the one that, in PKA (Gly52), is most critical to kinase activity (see above). Unfortunately, this idea does not yet have structural validation; putative Gly-loops are disordered in all published IPMK structures (Endo-Streeter et al., 2012; Holmes and Jogl, 2006; Wang and Shears, 2017); this situation presumably reflects loop flexibility. In fact, among all published structures of PDKG-InsPKs, there is just one (for IP3KB) which provides data supporting the possibility of a structural role for the Gly-loop in ATP-binding (Chamberlain et al., 2005). However, in the latter study, the reason that the loop can be observed in the structure is because it is stabilized by an intermolecular, antiparallel β-sheet. It is possible such a mechanism of protein dimerization may not be biologically relevant, and instead could arise as an artifact of the crystal packing.
Interestingly, the flexible loop in PI3K that corresponds to the Gly-loop does not actually contain any Gly residues, but it is still contacts with nucleotide phosphates through a lysine residue (Lys776 in PI3Kα; Lys807 in SsPI3Kγ; (Maheshwari et al., 2017; Walker et al., 2000)). As in protein kinases, the loop also links two antiparallel β-sheets that cover the adenine ring (Fig. 2B). In order to reflect this structural and functional conservation, we have taken the liberty of describing this PI3K structural element as a ‘G-loop’ (Fig. 2B, 4E; Table 1), although others have instead named it for its role in phosphate-binding (‘P-loop’; (Maheshwari et al., 2017; Walker et al., 2000)).
Another conserved element in IPMK, and PDKG-InsPKs in general, is the ‘hinge’ region (Fig. 2A,B,C,D; Table 1). The name of this element reflects it being the only contiguous connection between the N- and C-lobes. The backbone of the hinge makes hydrogen bonds with the nitrogen atoms in the adenine ring of ATP (Holmes and Jogl, 2006; Wang and Shears, 2017). Immediately C-terminal to the hinge in IPMK is the first secondary structural element in the C-lobe: a β-strand that hosts the catalytically-critical DxKxG sequence. There is a topologically equivalent DxK motif in protein kinases, although it occurs in a loop rather than a β-strand. In protein kinases, the Lys residue participates in charge neutralization in the transition state (Madhusudan et al., 2002). A similar role seems likely for the equivalent Lys residues in the PDKG-InsPKs, as proposed previously (Gonzalez et al., 2004). Mutation of this Lys residue in the PDKG-InsPKs strongly impairs InsP kinase activity (Saiardi et al., 2001; Togashi et al., 1997; Wang et al., 2014). The remarkable conservation of function for this particular residue is also seen in PPIP5K2 (E.C. 2.7.4.24; (Wang et al., 2012)) and IP5K (Franco-Echevarria et al., 2017), even though these are not PDKG-InsPKs.
In IPMK, Asp144 from within the DxKxG sequence contacts the ribose ring of the nucleotide (Wang and Shears, 2017). The catalytic importance of the corresponding residue in yeast IPMK/Ipk2 was first described by York and colleagues (Odom et al., 2000). The corresponding residue in the DxK motif of PKA (Asp166) is not spatially equivalent (it lies on the opposite side of the conserved Lys), but it is nevertheless functional: it serves a different purpose, optimizing the orientation of the target OH that is phosphorylated (Madhusudan et al., 2002). In place of Asp166, an alternative acidic residue (Glu127) assumes the function of binding the ribose ring (Madhusudan et al., 2002).
In contrast to the preponderance of β-strands in the N-lobe, the C-lobe of protein kinases is largely α-helical in structure, although it does also contain four very short β-strands. Between two of these, β8 and β9, there is a crucial component of the catalytic machinery: Asp184 within a ‘DFG’-motif. In protein kinases, this tripeptide is part of an “activation segment”, a flexible structure that switches the kinase between catalytically-active (“DFG-in”) and inactive (“DFG-out”) states, typically through phosphorylation/dephosphorylation of a serine/threonine or tyrosine residue that is present in the same segment (Taylor and Kornev, 2011). This DFG-Asp is particularly critical because it coordinates magnesium ions within the catalytic site (Fig. 4D). The same is true for PI3K (Fig. 4E; Table 1). In IPMK, Asp385 is the equivalent residue, although the context of the corresponding tripeptide is DFA rather than DFG (Fig. 4F; Table 1); again, a magnesium coordination function is envisaged for this Asp (Wang and Shears, 2017).
Additionally, in the human IPMK structure (Wang and Shears, 2017), Asp385 is only 3.5 Å from Lys75, which is close enough to form a salt bridge. This Lys75-Asp385 interaction has an equivalent in PKA: Lys72-Asp184, two residues that are also only 3.5 Å apart (Fig. 4D). Evidence supporting their formation of a salt bridge in PKA emerged from early cross-linking experiments (Buechler and Taylor, 1989). The corresponding salt bridge in PI3Kγ is Lys833-Asp964 (2.7 Å apart; Fig. 4E; Table 1). Of further interest is that, in IPMK, Lys75 itself forms another salt bridge with the α-phosphate of ATP and Glu86 (Wang and Shears, 2017). A corresponding Lys-Glu/Asp salt bridge is also a well-known, tightly-conserved property of protein kinases and PI3Ks (Taylor and Kornev, 2011). These intertwined salt bridges between the invariant Lys and two acidic residues leads us to consider them collectively as a ‘catalytic triad’ (Table 1) that activates magnesium and coordinates the kinase reaction.
Notwithstanding the importance of identifying the contributions of key polar and charged residues to the structure and function of kinases, it has been noted previously that this is insufficient for a full understanding of protein architecture and intramolecular connectivity (Taylor and Kornev, 2011). It was these knowledge-gaps that led to the development of bioinformatics techniques that uncovered dominating patterns of hydrophobic residues in all protein kinases. Consequently, it is now recognized that an unusually hydrophobic α-helix is a key architectural aspect of the C-lobe of protein kinases and PI3Ks, namely, αF (Fig. 2A) and kα7 (Fig. 2B), respectively. These helices anchor structurally stabilizing elements that are known as catalytic (C-) and regulatory (R-) spines (Taylor and Kornev, 2011; Vadas et al., 2011). For kinases that are in their active configuration, both the C- and R-spine are held together by hydrophobic interactions between a series of non-contiguous, hydrophobic residues that stretch from the C-lobe to the N-lobe (Fig. 4A,B; 5A,B).
Fig. 5. C-Spines in PKA, PI3K and IPMK.
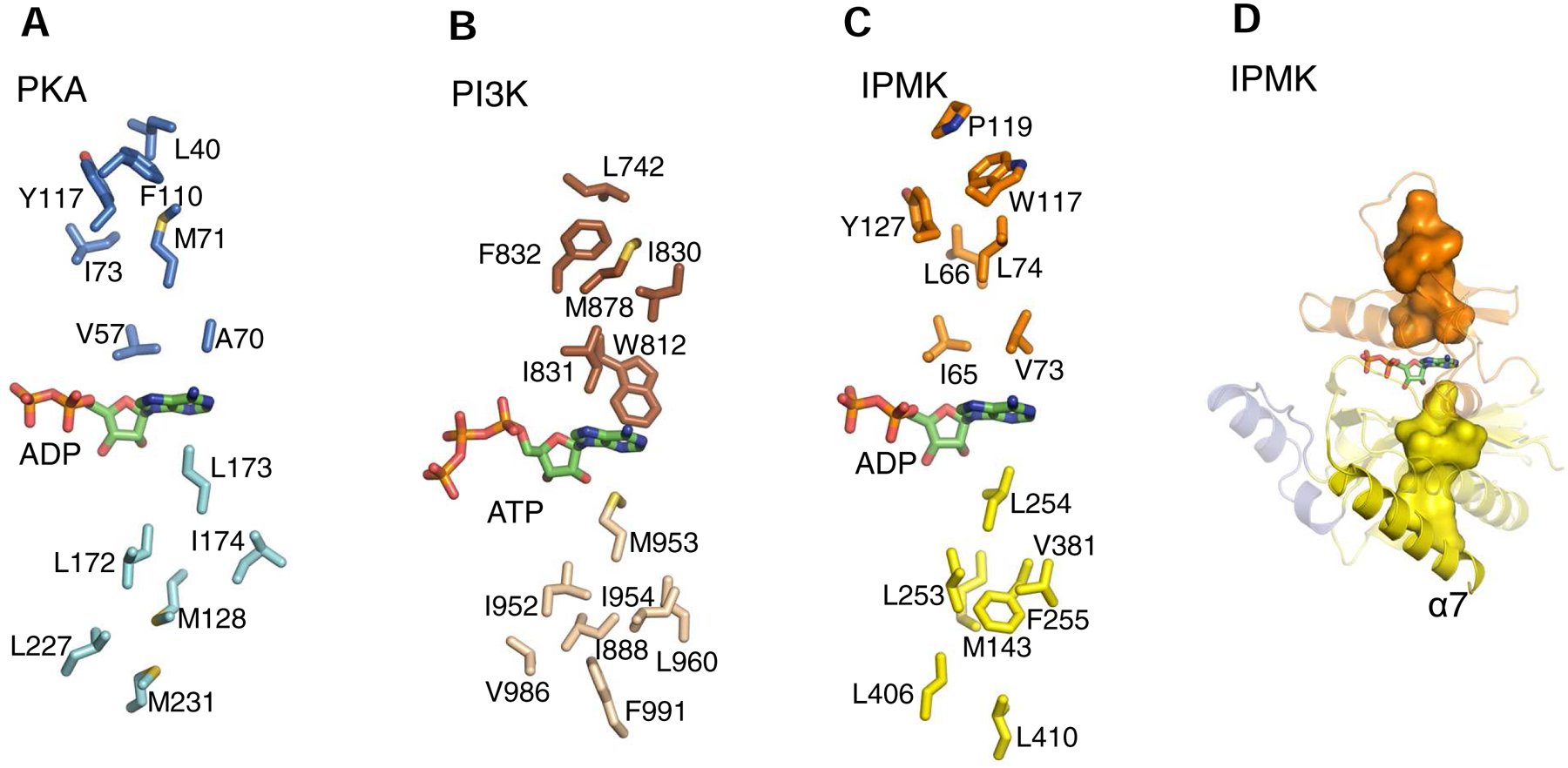
A, B and C describe the relative spatial positions of the individual residues (stick models) that comprise the C-spines of murine PKA (PDB, 1L3R), human IPMK (PDB, 5W2H) and Sus scrofa PI3Kγ (PDB, 1E8X), respectively. Individual residues were identified as described in the legend to Fig. 4. Color schemes match those in Fig. 2. Bound nucleotides are shown as green stick models (color schemes as in Fig. 2). D, shows the C-spine of IPMK as a space-filling model (yellow surface) anchored to the α7-helix.
In protein kinases (and PI3Ks), the C-spines stretch between the two lobes by linking through the adenine ring, as part of the process by which the two domains cooperate in positioning the nucleotide for catalysis (Taylor and Kornev, 2011; Vadas et al., 2011). Here, we provide the first description of equivalent structural features in IPMK and the other PDKG-InsPKs (Fig. 4C; Fig. 5C,D; Table 1). The C-spine in IPMK is completed by sandwiching the adenine ring of ATP between three layers of residues in the N-lobe and three layers in the C-lobe (Fig. 5C,D). Thus, Ile65 and Val73 can be depicted as the layer that stacks immediately above the nucleotide’s adenine ring, with a middle layer comprising Leu66 and Leu74, while Trp117, Pro119 and Phe127 form a top layer. In the C-lobe, Leu254 provides the first layer of the C-spine to stack below the adenine. Met143, Leu253, Phe255 and Val381 form the second layer. Leu406 and Leu410 form the third layer that anchors the C-spine to the α7 helix. We have also identified conserved C-spine residues in IP3K and IP6K (Table 1).
In protein kinases, the R-spine is so named because its dynamic assembly and disassembly is part of the structural reorganization that occurs as the enzyme toggles between its active and inactive states (Taylor and Kornev, 2011). The possibility has been raised that PI3Ks may utilize a similar structure for regulatory purposes (Maheshwari et al., 2017; Vadas et al., 2011). We have now determined that IPMK has an equivalent structure to the R-spine that is also comprised of bulky hydrophobic residues (Fig 4C). Also analogous to protein kinases, the backbone amide of Phe248 in the R-spine of IPMK hydrogen bonds with a conserved Asp (D396; Fig. 4C) in the anchoring C-lobe α-helix (α7).
We propose that in IPMK the R-spine imposes rigidity upon catalytically important elements from both the C- and N-lobes. In particular, we note that Phe386, a component of the R-spine, is also a highly-conserved residue from within the catalytically important DFG motif that is described above. Conservation of Phe386 underscores its structural contribution to the placement of the adjacent and catalytically critical Asp385.
There is currently no evidence that R-spines in PDKG-InsPKs undergo conformational changes or have regulatory properties. However, the fact that this architecturally-important spine is anchored to a remote distal α-helix opens up the possibility of an intramolecular connectivity network for conveyance of long-distance allosteric effects from either (a) distal mutations, (b) the binding of regulatory ligands or proteins, (c) covalent modifications.
There is an intriguing residue (Met210 in PKA) that is positioned between the C- and R-spines of PKA, and many other protein kinases, that is known as a ‘gatekeeper’. Its significance was originally considered to be only pharmacological; it sets the depth of the nucleotide pocket to which kinase inhibitors can have access (Liu et al., 1998). However, there also appear to be active states of certain protein kinases in which the gatekeeper is a component of the structurally-stabilizing R-spine (Azam et al., 2008). We believe that this architectural function is also applicable to the gatekeeper residue that we identified in IPMK, i.e., Leu130 (Fig. 4C). Irrespective of this structural role, in all PDKG-InsPKs (Table 1) the impact of the gatekeeper side-chain upon the space within the nucleotide-binding pocket is likely to be an important factor in the rational design of inhibitors of this kinase subclass. It is also worth noting that the architectural support provided by the αF-helix/hydrophobic spine structural complex is missing from IP5K, another InsP kinase that is not a member of the PDKG-InsPK family (Franco-Echevarria et al., 2017; Gosein and Miller, 2013). These and other structural differences between various types of InsP kinases can be exploited to develop target-specific inhibitors.
To help summarize this section, Fig. 6 provides a schematic of the high degree of conservation of important structural elements of protein kinases, PI3Ks and PDKG-InsPKs.
Fig. 6. Conserved aspects of nucleotide binding for PKA, PI3K and InsP kinases.
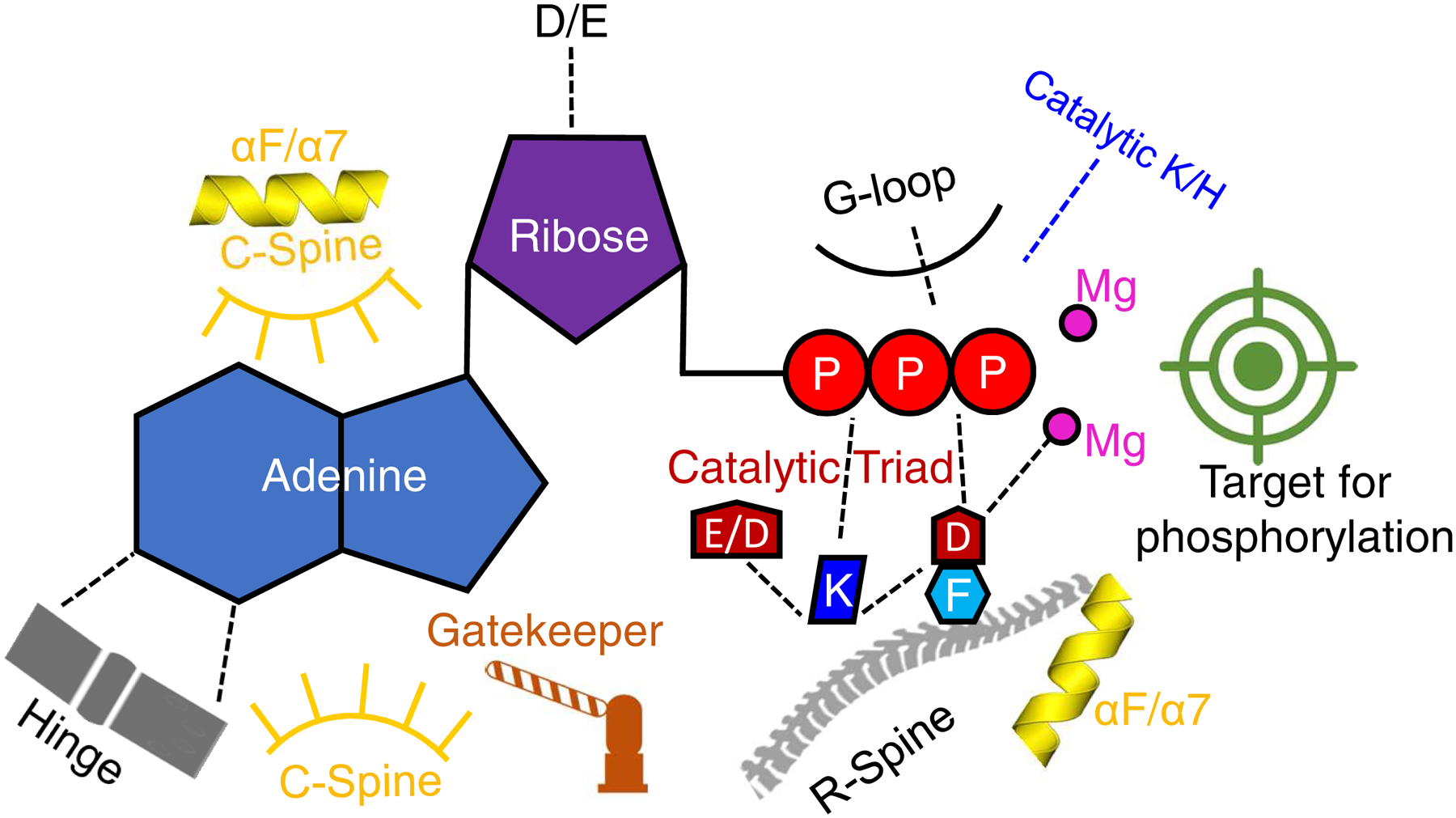
Shown is a graphical representation of highly conserved elements of protein kinases, PI3Ks and PDKG-InsPKs. Broken lines depict charged and polar contacts. For an explanation of catalytic triad, see Fig. 4 D,E,F and the text. ‘Catalytic K/H’ refers to the transition state stabilizing residue in the DxK/H motif (Table 1). See text and Table 1 for other details.
Human-health significance of the protein-kinase fold in InsP kinases.
Chemical probes that inhibit PDKG-InsPKs could be used as research tools for functional characterization of their kinase activities, and also to distinguish those activities from separate, non-catalytic roles mediated by protein-protein interactions. The only PDKG-InsPK inhibitor that is currently in common use is the pan-IP6K inhibitor N2-(m-(trifluoromethyl)benzyl) N6-(p-nitrobenzyl)purine (TNP) (Puhl-Rubio et al., 2018). However, this compound is compromised by weak (low micromolar) potency, inability to distinguish between different IP6K isoenzymes, and off-target side effects, (Ghoshal et al., 2016). Thus, there is increasing interest in developing new inhibitors that are more potent and specific. Moreover, drugs that inhibit these kinase activities could ultimately bring therapeutic benefits. For example, it has been argued that IP6Ks might be an appropriate target for the treatment of obesity, cancer, and aging (Chakraborty, 2017; Shears, 2017).
Lead molecules for inhibitor-development programs can be obtained by high throughput screening (HTS) of chemical libraries. There are HTS assays that are applicable to PDKG-InsPKs (Puhl-Rubio et al., 2018; Wormald et al., 2017). Furthermore, the identification of chemically tractable ‘hits’ can be facilitated by the curation and application of relatively small, focused libraries consisting of molecules that have appropriate functionally and/or chemically related properties (Barnash et al., 2017). To this end, we (Puhl-Rubio et al., 2018) have previously drawn attention to the nucleotide-binding elements in the protein kinase fold that are conserved in PDKG-InsPKs (as discussed above). Thus, we suggested that small-molecule inhibitors of protein kinases, that act by competing with nucleotide binding, might have the same action upon PDKG-InsPKs (Puhl-Rubio et al., 2018). We have screened human IP6K2 against a focused library of just over 5000 individual compounds that are all expected (or demonstrated) to occupy the ATP-binding site of protein kinases. From that library, 46 novel IP6K2 inhibitors were identified (Puhl-Rubio et al., 2018).
Naturally, we expected that our hits would also target protein kinases. This was found to be the case when we screened four of our preferred hits against a commercial kinase panel (Puhl-Rubio et al., 2018). However, we believe that a structure-based drug development strategy can exploit subtle differences in nucleotide-binding sites among the different members of the kinase superfamily, in order to develop specific inhibitors of individual PDKG-InsPKs. Conversely, compounds that are developed for inhibiting protein kinases should be tested against PDKG-InsPKs in order to guard against cross-pharmacological targeting.
Furthermore, there is growing interest in the substantial contributions that pathogenic fungal and protozoan PDKG-InsPKs make to the fitness and virulence of these organisms. It has become a viable proposition to develop drugs that can specifically target microbial PDKG-InsPKs, due to their limited homology with human orthologs (Cestari et al., 2016; Saiardi et al., 2018).
Finally, attention continues to be given to the potential therapeutic effects of natural products that are known to act as ATP-competitive inhibitors of protein kinases and PI3Ks (Liu et al., 2012; Navarro-Retamal and Caballero, 2016). In view of the work we describe in this review, we now propose that, in some cases at least, it may be inhibition of the PDKG-InsPKs by some natural products that brings therapeutic benefit.
Acknowledgement
Research in the authors’ laboratory is supported by the Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Statement
The authors declare there is no conflict of interest.
References
- Abel K, Anderson RA, Shears SB, 2002. Phosphatidylinositol and inositol phosphate metabolism. J. Cell Sci 114, 2207–2208. [DOI] [PubMed] [Google Scholar]
- Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ, 2008. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol 15(10), 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnash KD, James LI, Frye SV, 2017. Target class drug discovery. Nat Chem Biol 13(10), 1053–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett M, Onnebo SM, Azevedo C, Saiardi A, 2006. Inositol pyrophosphates: metabolism and signaling. Cell. Mol. Life Sci 63, 552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blind RD, Suzawa M, Ingraham HA, 2012. Direct Modification and Activation of a Nuclear Receptor-PIP2 Complex by the Inositol Lipid Kinase IPMK. Sci. Signal 5(229), ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buechler JA, Taylor SS, 1989. Dicyclohexylcarbodiimide cross-links two conserved residues, Asp-184 and Lys-72, at the active site of the catalytic subunit of cAMP-dependent protein kinase. Biochemistry 28(5), 2065–2070. [DOI] [PubMed] [Google Scholar]
- Cestari I, Haas P, Moretti NS, Schenkman S, Stuart K, 2016. Chemogenetic Characterization of Inositol Phosphate Metabolic Pathway Reveals Druggable Enzymes for Targeting Kinetoplastid Parasites. Cell Chem. Biol 23(5), 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, 2017. The inositol pyrophosphate pathway in health and diseases. Biol. Rev. Camb. Philos. Soc, doi: 10.1111/brv.12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain PP, Sandberg ML, Sauer K, Cooke MP, Lesley SA, Spraggon G, 2005. Structural insights into enzyme regulation for inositol 1,4,5-trisphosphate 3-kinase B. Biochemistry 44(44), 14486–14493. [DOI] [PubMed] [Google Scholar]
- Chang S-C, Miller AL, Feng Y, Wente SR, Majerus PW, 2002. The human homologue of the rat inositol phosphate multikinase is an inositol 1,3,4,6-tetrakisphosphate 5-kinase. jbc 277, 43836–43843. [DOI] [PubMed] [Google Scholar]
- Cheek S, Zhang H, Grishin NV, 2002. Sequence and structure classification of kinases. J. Mol. Biol 320, 855–881. [DOI] [PubMed] [Google Scholar]
- Endo-Streeter S, Tsui MK, Odom AR, Block J, York JD, 2012. Structural studies and protein engineering of inositol phosphate multikinase. J. Biol. Chem 287, 35360–35369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Echevarria E, Sanz-Aparicio J, Brearley CA, Gonzalez-Rubio JM, Gonzalez B, 2017. The Crystal Structure of Mammalian Inositol 1,3,4,5,6-Pentakisphosphate 2-Kinase Reveals a New Zinc Binding Site and Key Features for Protein Function. J. Biol. Chem 292, 10534–10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspers LD, Bartlett PJ, Politi A, Burnett P, Metzger W, Johnston J, Joseph SK, Hofer T, Thomas AP, 2014. Hormone-induced calcium oscillations depend on cross-coupling with inositol 1,4,5-trisphosphate oscillations. Cell Rep 9(4), 1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal S, Zhu Q, Asteian A, Lin H, Xu H, Ernst G, Barrow JC, Xu B, Cameron MD, Kamenecka TM, Chakraborty A, 2016. TNP [N2-(m-Trifluorobenzyl), N6-(p-nitrobenzyl)purine] ameliorates diet induced obesity and insulin resistance via inhibition of the IP6K1 pathway. Mol Metab 5(10), 903–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez B, Schell MJ, Letcher AJ, Veprintsev DB, Irvine RF, Williams RL, 2004. Structure of a human inositol 1,4,5-trisphosphate 3-kinase; substrate binding reveals why it is not a phosphoinositide 3-kinase. Mol Cell 15(5), 689–701. [DOI] [PubMed] [Google Scholar]
- Gosein V, Miller GJ, 2013. Roles of Phosphate Recognition in Inositol 1,3,4,5,6-Pentakisphosphate 2-Kinase Substrate Binding and Activation. J. Biol. Chem 288, 26908–26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand CE, Honek JF, 2007. Phosphate transfer from inositol pyrophosphates InsP5PP and InsP4(PP)2: a semi-empirical investigation. Bioorg. Med. Chem. Lett 17(1), 183–188. [DOI] [PubMed] [Google Scholar]
- Hatch AJ, Odom AR, York JD, 2018. Inositol phosphate multikinase dependent transcriptional control. Adv. Biol. Regul 64, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch AJ, York JD, 2010. SnapShot: Inositol phosphates. Cell 143(6), 1030–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmer W, McGlone M, Tsigelny I, Taylor SS, 1997. Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. jbc 272, 16946–16954. [DOI] [PubMed] [Google Scholar]
- Holmes W, Jogl G, 2006. Crystal structure of inositol phosphate multikinase 2 and implications for substrate specificity. J. Biol. Chem 281(49), 38109–38116. [DOI] [PubMed] [Google Scholar]
- Irvine RF, Schell M, 2001. Back in the water: the return of the inositol phosphates. Nature Reviews Molecular Cell Biology 2, 327–338. [DOI] [PubMed] [Google Scholar]
- Kim E, Ahn H, Kim MG, Lee H, Kim S, 2017. The Expanding Significance of Inositol Polyphosphate Multikinase as a Signaling Hub. Mol. Cells 40(5), 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM, 1991a. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253(5018), 407–414. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM, 1991b. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253(5018), 414–420. [DOI] [PubMed] [Google Scholar]
- Liu J, Hu Y, Waller DL, Wang J, Liu Q, 2012. Natural products as kinase inhibitors. Nat. Prod. Rep 29(3), 392–403. [DOI] [PubMed] [Google Scholar]
- Liu Y, Shah K, Yang F, Witucki L, Shokat KM, 1998. A molecular gate which controls unnatural ATP analogue recognition by the tyrosine kinase v-Src. Bioorg Med Chem 6(8), 1219–1226. [DOI] [PubMed] [Google Scholar]
- Maag D, Maxwell MJ, Hardesty DA, Boucher KL, Choudhari N, Hanno AG, Ma JF, Snowman AS, Pietropaoli JW, Xu R, Storm PB, Saiardi A, Snyder SH, Resnick AC, 2011. Inositol polyphosphate multikinase is a physiologic PI3-kinase that activates Akt/PKB. Proc. Natl. Acad. Sci. U. S. A 108(4), 1391–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhusudan PA, Xuong NH, Taylor SS, 2002. Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase. Nat. Struct. Biol 9(4), 273–277. [DOI] [PubMed] [Google Scholar]
- Maheshwari S, Miller MS, O’Meally R, Cole RN, Amzel LM, Gabelli SB, 2017. Kinetic and structural analyses reveal residues in phosphoinositide 3-kinase alpha that are critical for catalysis and substrate recognition. J Biol Chem 292(33), 13541–13550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GJ, Hurley JH, 2004. Crystal structure of the catalytic core of inositol 1,4,5-trisphosphate 3-kinase. Mol Cell 15(5), 703–711. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Adams JA, Taylor SS, 2003. Structural basis for peptide binding in protein kinase A. Role of glutamic acid 203 and tyrosine 204 in the peptide-positioning loop. J Biol Chem 278(12), 10613–10618. [DOI] [PubMed] [Google Scholar]
- Nalaskowski MM, Deschermeier C, Fanick W, Mayr GW, 2002. The human homologue of yeast ArgRIII protein is an inositol phosphate multikinase with predominantly nuclear localization. bj 366, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalaskowski MM, Mayr GW, 2004. The families of kinases removing the Ca2+ releasing second messenger Ins(1,4,5)P3. Curr Mol Med 4(3), 277–290. [DOI] [PubMed] [Google Scholar]
- Navarro-Retamal C, Caballero J, 2016. Flavonoids as CDK1 Inhibitors: Insights in Their Binding Orientations and Structure-Activity Relationship. PLoS One 11(8), e0161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom AR, Stahlberg A, Wente SR, York JD, 2000. A role for nuclear inositol 1,4,5-trisphosphate kinase in transcriptional control. Science 287, 2026–2029. [DOI] [PubMed] [Google Scholar]
- Politi A, Gaspers LD, Thomas AP, Hofer T, 2006. Models of IP3 and Ca2+ oscillations: frequency encoding and identification of underlying feedbacks. Biophys J 90(9), 3120–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhl-Rubio AC, Stashko MA, Wang H, Hardy PB, Tyagi V, Li B, Wang X, Kireev D, Jessen HJ, Frye SV, Shears SB, Pearce KH, 2018. Use of Protein Kinase-Focused Compound Libraries for the Discovery of New Inositol Phosphate Kinase Inhibitors. SLAS. Discov, 2472555218775323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VD, Misra S, Boronenkov IV, Anderson RA, Hurley JH, 1998. Structure of type IIbeta phosphatidylinositol phosphate kinase: a protein kinase fold flattened for interfacial phosphorylation. Cell 94(6), 829–839. [DOI] [PubMed] [Google Scholar]
- Resnick AC, Snowman AM, Kang BN, Hurt KJ, Snyder SH, Saiardi A, 2005. Inositol polyphosphate multikinase is a nuclear PI3-kinase with transcriptional regulatory activity. Proc. Natl. Acad. Sci. U. S. A 102(36), 12783–12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiardi A, Azevedo C, Desfougeres Y, Portela-Torres P, Wilson MSC, 2018. Microbial inositol polyphosphate metabolic pathway as drug development target. Adv. Biol. Regul 67, 74–83. [DOI] [PubMed] [Google Scholar]
- Saiardi A, Caffrey JJ, Snyder SH, Shears SB, 2000. Inositol Polyphosphate Multikinase (ArgRIII) Determines Nuclear mRNA Export in Saccharomyces cerevisiae. FEBS Lett 468, 28–32. [DOI] [PubMed] [Google Scholar]
- Saiardi A, Erdjument-Bromage H, Snowman A, Tempst P, Snyder SH, 1999. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr. Biol 9, 1323–1326. [DOI] [PubMed] [Google Scholar]
- Saiardi A, Nagata E, Luo HR, Snowman AM, Snyder SH, 2001. Identification and characterization of a novel inositol hexakisphosphate kinase. jbc 276, 39179–39185. [DOI] [PubMed] [Google Scholar]
- Scheeff ED, Bourne PE, 2005. Structural evolution of the protein kinase-like superfamily. PLoS. Comput. Biol 1(5), e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell MJ, 2010. Inositol trisphosphate 3-kinases: focus on immune and neuronal signaling. Cell. Mol. Life Sci 67, 1755–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shears SB, 2017. Intimate Connections: Inositol Pyrophosphates at the Interface of Metabolic Regulation and Cell-Signaling. J. Cell Physiol DOI: 10.1002/jcp.26017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shears SB, Baughman BM, Gu C, Nair VS, Wang H, 2017. The significance of the 1-kinase/1-phosphatase activities of the PPIP5K family. Adv Biol Regul 63, 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shears SB, Ganapathi SB, Gokhale NA, Schenk TM, Wang H, Weaver JD, Zaremba A, Zhou Y, 2012. Defining signal transduction by inositol phosphates. Subcell. Biochem 59, 389–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Kornev AP, 2011. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci 36(2), 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi S, Takazawa K, Endo T, Erneux C, Onaya T, 1997. Structural identification of the myoinositol 1,4,5-trisphosphate-binding domain in rat brain inositol 1,4,5-trisphosphate 3-kinase. Biochem J 326 (Pt 1), 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadas O, Burke JE, Zhang X, Berndt A, Williams RL, 2011. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal 4(195), re2. [DOI] [PubMed] [Google Scholar]
- Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL, 2000. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 6(4), 909–919. [DOI] [PubMed] [Google Scholar]
- Walker EH, Perisic O, Ried C, Stephens L, Williams RL, 1999. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 402(6759), 313–320. [DOI] [PubMed] [Google Scholar]
- Wang H, DeRose EF, London RE, Shears SB, 2014. IP6K structure and the molecular determinants of catalytic specificity in an inositol phosphate kinase family. Nature Communications 5:4178, doi: 10.1038/ncomms5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Falck JR, Hall TM, Shears SB, 2012. Structural basis for an inositol pyrophosphate kinase surmounting phosphate crowding. Nat. Chem. Biol 8(1), 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Shears SB, 2017. Structural features of human inositol phosphate multikinase rationalize its inositol phosphate kinase and phosphoinositide 3-kinase activities. J. Biol. Chem 292, 18192–18202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormald M, Liao G, Kimos M, Barrow J, Wei H, 2017. Development of a homogenous high-throughput assay for inositol hexakisphosphate kinase 1 activity. PLoS. ONE 12(11), e0188852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Fujii K, Liu Y, Codrea V, Herrero J, Shaw S, 2005. A single pair of acidic residues in the kinase major groove mediates strong substrate preference for P-2 or P-5 arginine in the AGC, CAMK, and STE kinase families. J Biol Chem 280(43), 36372–36379. [DOI] [PubMed] [Google Scholar]