

SUMMARY
Mammals differ more than 100-fold in maximum lifespan. Here, we conducted comparative transcriptomics on 26 species with diverse lifespans. We identified thousands of genes with expression levels negatively or positively correlated with species’ maximum lifespan (Neg- or Pos-MLS genes). Neg-MLS genes are primarily involved in energy metabolism and inflammation. Pos-MLS genes show enrichment in DNA repair, microtubule organization and RNA transport. Expression of Neg- and Pos-MLS genes is modulated by interventions including mTOR and PI3K inhibition. Regulatory networks analysis showed that Neg-MLS genes are under circadian regulation possibly to avoid persistent high expression, while Pos-MLS genes are targets of master pluripotency regulators OCT4 and NANOG, and are upregulated during somatic cell reprogramming. Pos-MLS genes are highly expressed during embryogenesis but significantly downregulated after birth. This work provides targets for antiaging interventions by defining pathways correlating with longevity across mammals and uncovering circadian and pluripotency networks as central regulators of longevity.
Keywords: Aging, longevity, comparative transcriptomics, epigenetic reprogramming, pluripotency, circadian clock, functional genomics
eTOC blurb
Lu et al. conducted comparative transcriptomics and identified genes with expression negatively or positively correlated with species’ maximum lifespan (Neg- or Pos-MLS genes). Neg-MLS genes are enriched in energy metabolism and inflammation, and controlled by circadian factors. Pos-MLS genes are enriched in DNA repair and controlled by the pluripotency network.
Graphical Abstract
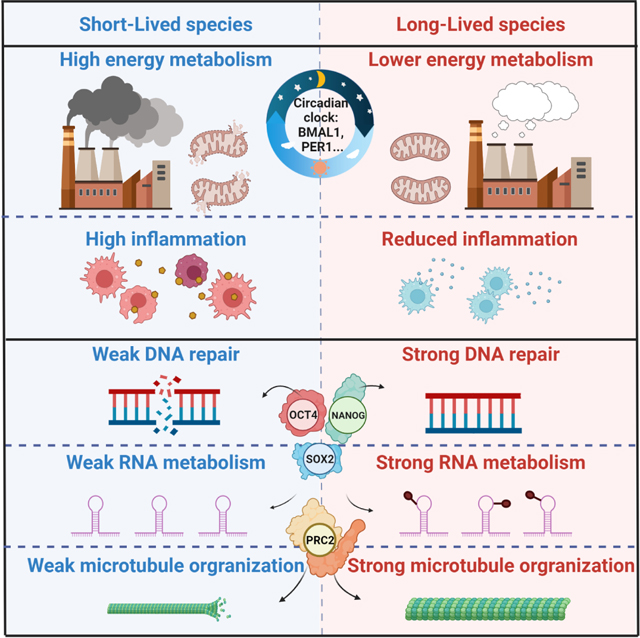
INTRODUCTION
Aging is characterized by a progressive loss of physiological capacity, making aging a major risk factor for multiple diseases (Lopez-Otin et al., 2016). Many aging theories have been proposed, and important longevity pathways have been discovered, with the help of model organisms including yeasts, nematodes, fruit flies, mice (Miller et al., 2020; Mitchell et al., 2015). However, lifespan extension experimentally achieved in model organisms using genetic or pharmacological manipulations pales in comparison with interspecies differences in lifespan. Natural selection has produced mammals with dramatically diverse aging rates that provide the opportunity to identify the underlying mechanisms of large changes in lifespan through comparative biology (Gorbunova et al., 2014; Zhao et al., 2021).
Previous studies have investigated the genomic features of mammals with extreme longevity such as naked mole-rats (Kim et al., 2011), beavers (Zhou et al., 2020), bowhead whales (Keane et al., 2015), elephants (Sulak et al., 2016) and bats (Foley et al., 2018). Long-lived rodents, like the naked mole-rats and beaver show enhanced resistance to DNA damage and cellular stress, which can be partially explained by enhanced activity of the Sirt6 gene (Tian et al., 2019). Hpgd, a tumor suppressor gene, is uniquely duplicated in the beaver among rodents (Zhang et al., 2021). Similarly, an increase in Tp53 copy number has been linked to increased cancer resistance enabling a longer lifespan in the elephants (Sulak et al., 2016). Sequencing of the bowhead whale genome revealed species-specific changes in DNA repair and cell cycle genes (Keane et al., 2015). Genomic analysis of 88 rockfish species identified a positive selection in DNA repair pathways in long-lived taxa (Kolora et al., 2021). In contrast, a comparative study of 45 killifish species revealed that genes involved in DNA repair and nutrient-sensing pathways are under relaxed selection in short-lived killifish (Cui et al., 2019).
Changes in gene expression and gene regulatory networks also play an important role in aging and the regulation of lifespan (Tabula Muris, 2020). African killifish tend to live longer when genes involved in cellular energy production are less active at a young age (Baumgart et al., 2016). Transcriptomic profiling of long- and short-lived mutant mice implicates a role of mitochondrial metabolism in aging (Fuentealba et al., 2021). Gene expression signatures of mammalian life histories have been examined in brain, kidney and liver (Fushan et al., 2015), but the species in this dataset were highly diverse phylogenetically. Hence, a large-scale comparative transcriptomic analysis of mammals that are closely related phylogenetically, but highly diverse in their MLS would have more power at identifying the signatures of longevity. Despite these initial findings of transcriptomic features correlating with longevity, the regulation of lifespan-associated genes remained to be determined.
Here, by performing extensive comparative transcriptomics analysis, we revealed common paths to longevity and uncovered molecular and transcriptional networks regulating the MLS-associated genes. Notably, we found that MLS-associated genes are controlled by circadian and pluripotency factors, suggesting that these two regulatory networks are tuned by evolutionary processes to give rise to striking differences in MLS between species.
RESULTS
Identification of genes associated with MLS by comparative transcriptomics
To systematically characterize transcriptomic signatures of longevity, we collected six tissues including brain, heart, kidney, liver, lung and skin from 141 individuals covering 26 species that belong to the orders Rodentia and Eutlipotyphyla. The MLS of these species ranged from 3 years to 37 years (Figure 1A; Figure S1A). We deep-sequenced ~13.1 trillion base pairs of RNA from 557 samples (78.5 ± 17.6 s.d. million reads per sample) using Illumina RNA-sequencing (RNA-Seq). Some tissues were not available for some of the individuals. Most of the raw reads (99.2%) showed high quality (Phred quality score > 20). On average, 92.6% of clean reads were successfully mapped to the corresponding reference genomes, 77.4% of which had unique mapping coordinates (Table S1).
Figure 1. Comparative transcriptomics of 26 mammalian species.

(A) Schematic workflow showing the comparative transcriptomics of 26 species with distinct MLS.
(B) Network plot showing the Neg-MLS and Pos-MLS genes overlapped with GenAge database. The node size (count) indicates the number of tissues in which the genes were significantly correlated with MLS. The color of connecting lines corresponds to the Spearman correlation coefficient. Orange circles showing representative Pos-MLS genes. Blue circles showing representative Neg-MLS genes.
(C-D) Barplots showing the number of genes for which expression levels positively (C) or negatively (D) correlate with MLS, body mass or both.
(E) Heatmap showing the Spearman correlation coefficient of Neg-MLS and Pos-MLS genes across tissues.
(F-G) Scatter plots showing the relative gene expression levels of Akt2 (F) and Msh2 (G) in six tissues across species. Spearman correlation coefficients (ρ) of gene expression and MLS are shown in the plots. Correlation coefficients for phylogenetically corrected (PIC) data are shown in parenthesis. P values were shown for the Spearman correlation coefficients. All the P values were adjusted for multiple tests with BH method.
See also Figures S1–2 and Table S1
Because 14 of the 26 genomes in this study lack gene annotations in the Refseq database (Pruitt et al., 2012), we developed a comprehensive pipeline (Figure S1B, also see Methods) to de novo assemble, annotate, and quantify the full-length transcripts from RNA-Seq samples. We used the mouse and guinea pig as examples to evaluate the performance of our pipeline. The gene expression levels calculated by our pipeline are highly consistent with the results from well-annotated Gencode annotation for the mouse (Figure S1C, R-square = 0.87) (Frankish et al., 2019). For guinea pig, whose genome is less completely assembled and annotated, high consistency of gene expression between our pipeline and Refseq annotation was also observed (Figure S1C, R-square = 0.92). To conduct inter-species comparisons of gene expression, different methods were used to normalize the expression levels of homologous genes across species. The results are highly consistent (Figure S1D and see Methods). As expected, the expression of tissue-specific marker genes was restricted to the corresponding tissues (Figure S2A). Hierarchical clustering of RNA-seq data revealed that samples were clustered by tissues instead of species (Figure S2B). These results suggest a high quality of sample integrity, RNA library preparation, transcriptome assembly and gene expression quantification in this study.
To reduce background variability, 16,021 homologous genes that have been annotated in at least 10 species were kept for the following analysis (Figure S2C). We computed Spearman’s rank correlation coefficient for every gene based on the expression and MLS with all the tissues merged and within each tissue across species. ~1,000–1,500 genes are negatively or positively correlated with MLS (Neg-MLS or Pos-MLS genes) in each tissue (Spearman |ρ| > 0.4, adj. p < 0.05) (Figures 1B–D and S2D; Table S2), suggesting a universal association between gene expression and lifespan of species across tissues. We next analyzed the overlap of MLS-associated genes between tissues. Compared to the expected distribution (Lex et al., 2014), MLS-associated genes tended to be tissue-specific or shared by multiple tissues (Figures S2E–F). Heatmap analysis revealed that most MLS-associated genes show a consistent positive or negative correlation with MLS, yet the strength of the correlation can differ by tissue (Figure 1E). These results suggest that both tissue-specific and global mechanisms are involved in longevity regulation. Previous studies showed that adult body mass (BM) correlates with MLS —with larger animals living, on average, longer than smaller ones (Magalhães et al., 2007). We confirmed this trend among the species used in this study (Figure S2G). As expected, genes that correlate with BM (Table S3) show significant overlap with Neg- and Pos-MLS genes (Figures 1C–D).
Interestingly, 46 of 136 mouse genes curated in the GenAge database (Tacutu et al., 2018) show a significant correlation with MLS (Figure 1B). Among these genes, AKT serine/threonine kinase 2 (Akt2) and Glutathione S-Transferase alpha 4 (Gsta4) were identified as Neg-MLS genes in most tissues, suggesting they may generally serve as negative regulators of longevity (Figures 1F and S2H). Indeed, disruption of Akt2 and Gsta4 in mice has been shown to extend the average lifespan by 9.1% and 13%, respectively (Ren et al., 2017; Singh et al., 2010). In contrast, the expression levels of DNA mismatch repair family gene MutS Homolog 2 (Msh2) and its paralog Msh6 tend to be positively correlated with MLS of species (Figures 1G and S2I). Intriguingly, previous studies revealed that about 50% of Msh2 knockout mice die by 8 months of age and all animals were dead by 12 months of age (Wei et al., 2003). Similarly, 40% of Msh6 knockout mice died at 9 months after birth (Edelmann et al., 1997). Depletion of Msh6 in Saccharomyces cerevisiae leads to a 50% decrease of the mean chronological lifespan (Laschober et al., 2010). Taken together, our results suggest that Neg-MLS and Pos-MLS genes are prevalent across tissues and may have functional roles in regulating lifespan.
Functional enrichment of genes negatively correlated with MLS
To identify a pan-tissue transcriptomic signature that correlates with MLS, we ranked all the genes by their Spearman correlation coefficient with MLS, with six tissues merged across species, and performed Gene Set Enrichment Analysis (GSEA). Neg-MLS genes showed strong enrichment in Gene Ontology (GO) and KEGG pathways including respirasome, oxidative phosphorylation, fatty acid catabolism, metabolism of ribonucleotide and glutathione, cytochrome complex and mitochondria (Figures 2A–B and S3A; Table S4A). These enriched terms are highly interconnected, supporting the notion that cellular energy metabolism is regulated in a coordinated manner. For example, the expression of Uqcrfs1, a respiratory chain gene, shows strong negative correlations with MLS across all six tissues (Figures 2C). Another module enriched with Neg-MLS genes contained inflammation-related functions, including leukocyte, lymphocyte, cytokine, chemokine, and cytosolic DNA sensing-related pathways. This is consistent with the causal role of chronic inflammation in aging-related diseases (Rea et al., 2018).
Figure 2. Functional enrichments of genes negatively correlated with MLS.

(A) The enrichment map networks visualization of genes that negatively correlate with MLS across six tissues. Each dot represents a Gene Ontology (GO) or KEGG functional term. Two functional terms are connected in the enrichment map network if they have a high overlap of genes. Groups of inter-related functional terms tend to cluster together. The dot sizes indicate the number of genes in a specific term.
(B) GSEA showing global enrichment of GO terms including Respirasome (upper panel) and Interferon-Alpha production (lower panel) in Neg-MLS genes. Normalized enrichment scores (NES) and false discovery rate (FDR) values are shown.
(C) Scatter plots showing the relative expression levels of a respiratory chain gene Uqcrfs1 in six tissues across species. Spearman correlation coefficients (ρ) of gene expression and MLS are shown in the plots. ρ calculated with gene expression after phylogenetically independent contrasts are shown in parenthesis.
(D) The heatmap showing the tissues-specific functional enrichments of Neg-MLS genes. Colors indicate NES values from GSEA in each tissue. Functional terms enriched across all tissues (A) are not shown here to avoid redundancy.
(E) GSEA showing specific enrichment of voltage-gated potassium channel in the Neg-MLS genes in the heart. NES and FDR values are shown.
(F) Scatter plots showing the relative expression level of potassium voltage-gated channel subfamily genes Kcna7 and Kcnb1 in the heart across species.
(G) GSEA showing specific enrichment of NF-Kappa B pathway in the Neg-MLS genes in the lung. NES and FDR values are shown.
(H) Scatter plots showing the relative expression levels of key regulators in NF-Kappa B pathway including Nfkb1and Kras in the lung across species.
Cellular respirasome is a major intracellular source of reactive oxygen species (ROS), which can trigger oxidative stress and inflammation (Mittal et al., 2014). We next asked whether the enrichment of Neg-MLS genes in cellular energy metabolism and inflammation are interdependent. The cellular respirasome-related metabolic pathways were further divided into oxidative phosphorylation, NADH dehydrogenase, citrate cycle and fatty acid metabolism. We found that the expression of energy metabolism genes is highly intercorrelated and shows negative correlations with the MLS of species (Figure S3B). Interestingly, the expression of energy metabolism genes showed a strong positive correlation with the expression of inflammation genes (Figure S3B). In addition, the expression of both energy metabolism and inflammation genes showed a weak positive correlation with the expression of genes that respond to ROS and oxidative stress. These results suggest that the enrichment of Neg-MLS genes in the inflammation pathway may be interdependent with energy metabolism pathways.
To identify genes associated with MLS, independent of BM, we built a linear model using BM to predict the MLS (Figure S2G) and used the residuals as BM-corrected MLS (Table S4). Similar to Neg-MLS genes, we found genes showing a negative correlation with BM-corrected MLS were overrepresented for respirasome and oxidative phosphorylation-related pathways, but the enrichment level decreased by ~50% compared to the Neg-MLS genes (Figure S3A). This result suggests that the metabolism enriched pathways of Neg-MLS genes can be partially explained by BM. In contrast, BM-corrected Neg-MLS genes showed even stronger enrichment in inflammation-related terms (Figure S3A), indicating that inflammation shows a stronger negative correlation with MLS than BM.
We next explored tissue-specific functional enrichments of Neg-MLS genes (Figure 2D). In the brain and heart, the expression of genes involved in voltage-gated potassium channel shows specific negative correlations with MLS (Figure 2E), which were exemplified by potassium voltage-gated channel subfamily genes Kcna7 and Kcnb1 in the heart (Figure 2F; ρ = −0.69 and −0.65, respectively). Interestingly, the potassium channel has a pivotal role in maintaining the rhythmicity of the heartbeat (Grandi et al., 2017), which is negatively correlated with MLS (Figure S3C) (Boudoulas et al., 2015). The inverse correlation of potassium channel genes with MLS might provide a molecular explanation of how heartbeat rate negatively correlates with MLS. In addition, the expression of genes related to cardiolipin metabolism (Figure 2D) also shows strong a negative correlation with MLS in the heart. Cardiolipin is a major membrane phospholipid in the mitochondria, which is essential for oxidative phosphorylation (Mejia et al., 2014). This result suggests that tissue-specific energy metabolism also contributes to lifespan regulation.
In the lung, we observed specific enrichments of Neg-MLS genes in immune-related functions including neutrophil activation, inflammatory response and NF-Kappa B pathway (Figure 2G). This may be due to the lung serving as a critical immune interface for airborne pathogens (Hartl et al., 2018). For instance, the lung expression of two master regulatory genes Nfkb1 and Kras in the NF-Kappa B pathway shows a strong negative correlation with MLS (Figure 2H, ρ = −0.73 and −0.67, respectively). The expression of genes involved in the collagen catabolic process also shows a specific negative correlation with MLS in the lung, which is consistent with the role collagen homeostasis plays in lung diseases like pulmonary fibrosis (Tsukui et al., 2020). In the kidney, gene expression of glucose transmembrane transporter shows specific negative correlation with MLS. In fact, the kidney is responsible for up to 20% of all glucose production and new anti-hyperglycemic drugs that inhibit glucose reabsorption protect the kidneys and heart from failing (Mather and Pollock, 2011). One of these drugs, canagliflozin, extends mouse lifespan, though only in males (Miller et al., 2020). In addition, the negative correlation of MLS and expression of genes involved in the long-chain fatty acid-CoA ligase activity, muscle cell apoptotic process, and myosin filament were specifically observed in the skin (Figure 2D), suggesting that proliferation and energy metabolism of skin cells may affect the lifespan of the organism.
We next tested whether the functional enrichments that negatively correlate with MLS result from the evolutionary history of species. We performed phylogenetically independent contrasts (PIC) of gene expression according to the phylogeny of the species in our study (Figure S1A). We found that respirasome and inflammation-related terms are still the most enriched terms (Figure S3D), suggesting that phylogenetic factors are not responsible for their negative correlation with MLS. Together, these results suggest higher expression of genes involved in energy metabolism and inflammation-related functions negatively correlates with species longevity.
Functional enrichments of genes positively correlated with MLS
The functions of Pos-MLS genes are dramatically different from those of Neg-MLS genes. Pos-MLS genes showed strong enrichment in three modules including DNA repair, organization of cilium and microtubule, RNA transport and localization (Figure 3A; Table S4B). In the DNA repair module, most DNA repair types are enriched, including base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR), and non-homologous end joining (NHEJ). Notably, enrichments of Pos-MLS gens in DNA repair were retained after correction with BM, albeit to a decreased level (Figure S3A). After correction for BM, pathways involved in cilium and microtubule also showed similarly decreased enrichment, but an increased enrichment was observed in the RNA export pathway (Figure S3A).
Figure 3. Functional enrichments of genes positively correlated with MLS.

(A) The enrichment map networks visualization of genes that are positively correlated with MLS across six tissues. Each dot represents a GO and KEGG functional term. Two functional terms are connected in the enrichment map network if they have a high overlap of genes. Groups of inter-related functional terms tend to cluster together. The dot size indicates the number of genes in a specific term.
(B) GSEA plots showing global enrichment of representative GO terms including mismatch repair and DNA DSB Repair. NES and FDR values are shown.
(C) Scatter plots showing the relative expression levels of a DNA mismatch repair gene Pms1 in six tissues across species. Spearman correlation coefficients (ρ) of gene expression and MLS are shown. Phylogenetically corrected values for ρ are shown in parenthesis.
(D) The heatmap showing NES of MLS-associated pathways based on the correlation of gene expression with MLS (left) and age (right). Negative NES values shown as Blue indicate expression of genes involved in this pathway tends to negatively correlate with MLS or/and age, i.e. downregulated during mouse aging. Positive NES values shown as Red indicate expression of genes involved in this pathway tends to positively correlate with MLS or/and age, i.e. upregulated during mouse aging.
Notably, Pos-MLS genes showed stronger enrichments in the DNA Double-Stranded Break (DSB) repair and MMR than in the BER and NER (Figures 3B and S4A; Normalized Enrichment Score [NES] = 2.4 (HR) versus 1.6 (NER)), which is consistent with our previous finding that more robust DNA DSB repair coevolves with longevity (Tian et al., 2019). For example, the expression of DNA mismatch repair family gene Pms1 shows a consistent positive correlation with MLS (average ρ = 0.68) in all six tissues (Figure 3C). Aprataxin and PNKP like factor (Aplf) is a component of the cellular response to DNA single- and double-strand breaks. Checkpoint kinase 1 (Chek1) is required for checkpoint mediated cell cycle arrest and activation of DNA repair. Protection of telomeres 1A (Pot1a) is involved in telomere maintenance (Lee et al., 2014). The expression of all these genes shows a strong positive correlation with MLS across multiple tissues (Table S2).
Besides canonical DNA repair genes, genes that encode the central component of centrioles spindle, assembly abnormal protein 6 (Sass6), Fanconi anemia (FA) core complex, and centromere protein S (Cenps) also show strong positive correlations with MLS across multiple tissues (Table S2). This might be partly due to the important role of centrioles and the FA complex in cell division, DNA damage response and prevention of chromosomal breakage (Duxin and Walter, 2015; Nigg and Holland, 2018; Ryu and Kim, 2019). The general enrichment of RNA transport and localization, exemplified by phosphorylated adaptor for RNA export (Phax, Table S2), across tissues suggests RNA homeostasis as an emerging regulator of longevity (Dominick et al., 2015; Park et al., 2017).
We also observed tissue-specific functional enrichment of Pos-MLS genes. The expression of genes involved L-Glutamate transmembrane transport, glial cell migration and extracellular matrix structure show a positive correlation with MLS in the brain (Figure S4B). As an example, extracellular matrix protein 2 (Ecm2) gene shows higher expression in the species with long lifespans (Figure S4C). Myelin basic protein (Mbp) shows a strong positive correlation with MLS in the liver (Figure S4D). Interestingly, the expression of myelin significantly decreases with age in the mouse brain (Ahn et al., 2017), while its role in the liver remains largely unknown. We also observed several functional terms including DNA replication, long-chain fatty acid biosynthesis, and keratinization that are specifically correlated with MLS in the skin (Figure S4B), indicating a role of normal skin function in longevity.
Similar to Neg-MLS genes, we also found that the positive correlation of these functions with MLS is sustained after PIC of gene expression (Figure S3D). Collectively, these data indicate that the high expression of genes involved in DNA repair and centriole assembly-related pathways constitute the transcriptomic signature of long MLS.
Expression of Neg- and Pos-MLS genes during aging
To determine how aging affects the expression of MLS-associated genes, we reanalyzed the published mouse gene expression data during aging (Schaum et al., 2020). Spearman correlation coefficients of gene expression with age were calculated. Interestingly, we found that Neg- and Pos-MLS genes can be both upregulated and downregulated during mouse aging, which is dependent on the enriched function (Figure 3D). Specifically, Neg-MLS genes involved in energy metabolism are downregulated in 14 out of 17 tissues during mouse aging (shown as negative correlations with age). In contrast, Neg-MLS genes involved in inflammation are upregulated in all17 tissues except the limb and subcutaneous adipose tissue (SCAT). As for Pos-MLS genes, we found that genes involved in DNA repair, RNA export and processing, spindle microtube and centriole assembly related pathways show moderate but consistent downregulation in most of the tissues except pancreas, marrow and skin, where there is a trend towards upregulation. Finally, Pos-MLS genes involved in the cilium pathway tend to be upregulated during mouse aging.
To test whether the observations in mice are conserved in other species, we reanalyzed the human gene expression data during aging (Ferraro et al., 2020). Human samples are more heterogeneous and biased towards older individuals (Figures S4E–F). Consistently with the mouse results, we found that in human Neg-MLS genes involved in energy metabolism show an overall downregulation in 40 out of 45 tissues or regions during human aging (Figure S4G). Furthermore, Neg-MLS involved in inflammation are globally upregulated in 31 of 45 tissues or regions during human aging. For Pos-MLS genes, we observed that genes involved in DNA repair, RNA export and processing, and spindle microtube tend to be downregulated in ~50% of all the tissues or regions. Pos-MLS genes involved in centriole assembly and cilium pathways tend to be upregulated during human aging.
These results suggest that MLS-associated genes are under dynamic regulation during human and mouse aging in a function- and tissue-specific manner. The different trajectories of MLS-associated gene expression during aging suggest that age-related expression changes of MLS-associated genes can be both harmful (inflammation, DNA repair), and adaptive (energy metabolism), though there is an alternative possibility that the downregulation of energy metabolism genes is a result of impaired mitochondrial function during aging (Sun et al., 2016).
Interventions regulating MLS-associated genes
We next identified potential interventions that regulate MLS-associated genes. We analyzed transcriptome profiling data after perturbations from Connectivity Map (CMap) (Subramanian et al., 2017) and other published datasets with aging-associated interventions.
CMap’s perturbations were classified as compound treatment (CP) and gene manipulation (Gene) (Table S5). We found that compounds activating protein kinase C, glucocorticoid receptor and progesterone receptor can downregulate the expression of Pos-MLS genes in all six tissues (Figure 4A. Similarly, compounds that inhibit ATPase, tubulin, heat shock protein, proteasome, purine synthesis and histone deacetylases (HDAC) can also downregulate Pos-MLS genes. Intriguingly, we found that inhibition of Beta-Amyloid (Aβ), a major risk factor for Alzheimer’s Disease (AD) and holding therapeutic promise (Wang et al., 2017), can upregulate the Pos-MLS genes. This striking link might provide novel insight into the neurotoxicity of Aβ. Manipulation of specific genes could also lead to the expression change of MLS-associated genes (Figure 4B). For example, overexpression of genes in mitogen-activated protein kinase (MAPK), mTOR pathways, and depletion of genes in the proteasome, mitotic regulation pathways including PTC1 and Polo-like kinase (PLK) can downregulate the overall expression of Pos-MLS genes (Table S5).
Figure 4. Interventions that regulate genes correlated with MLS.

(A-B) Heatmaps showing the perturbations including (A) compounds and (B) gene manipulations that regulate Pos-MLS genes. Compounds include both activators and inhibitors. Gene manipulation includes overexpression and loss of function induced by shRNA or CRISPR. The colors in the heatmap indicate the normalized connectivity score that was calculated by the CMap query. A positive or negative connectivity score indicates that the perturbation upregulates or downregulates the Pos-MLS genes, respectively. Only those terms with FDR < 0.01 in at least one tissue are shown in the heatmap. See Methods for details.
(C-D) Heatmaps showing the perturbations including compounds (C) and gene manipulations (D) that regulate Neg-MLS genes.
(E) GSEA of Neg-MLS and Pos-MLS genes in the liver RNA-seq data treated with different interventions. Barplot showing the NES values. * indicates FDR < 0.05. RNA-seq data is from (Tyshkovskiy et al., 2019)
(F) GSEA plots showing the Neg-MLS genes are downregulated in the liver treated with Rapamycin (upper panel) and 17-alpha-estradiol (lower panel) compared to the livers of control BL6 mice.
See also Table S5
Pos-MLS gene expression can be positively regulated by the inhibition of phosphoinositide 3-kinase (PI3K), cyclin dependent kinase (CDK), mammalian target of rapamycin (mTOR), RAF and protein synthesis (Figure 4A). Overexpression of genes involved in the Wnt signaling pathway, activator protein 1 (AP-1) and liver kinase B1 (LKB1), which directly phosphorylates and activates the AMP-activated protein kinase (AMPK) pathway, can upregulate the expression of Pos-MLS genes. Depletion of ribosomal 40S Subunit can also upregulate the expression of Pos-MLS genes (Figure 4B).
We also observed that several compounds and genes could regulate the expression of Neg-MLS genes. For example, inhibition of BCL, PI3K/AKT, EGFR, DNA polymerase and MAPK pathway can downregulate the expression of Neg-MLS in all six tissues (Figure 4C). Inhibitors of mTOR, RAF and phospholipase can downregulate the expression of Neg-MLS genes in all the tissues except the lung. Compounds that inhibit proteasome, topoisomerase, glucocorticoid receptor, ATPase and HDAC can lead to an upregulation of Neg-MLS genes. Overexpression of genes involved in ERBB1, HER2 and EGFR pathways and depletion of genes involved in the lysosome, cytokines, CD40 and RB1 pathways can downregulate Neg-MLS genes (Figure 4D). In contrast, depletion of genes that regulate proteasome, double-strand break repair, DNA replication and Notch pathways can upregulate the expression of Neg-MLS genes (Figure 4D).
Because gene profiling data from CMap were obtained from cell lines, we reanalyzed published datasets on mouse hepatic gene expression in response to 10 in vivo interventions including Rapamycin, 17-alpha-estradiol, Pit1 KO (Snell dwarf), growth hormone receptor (Ghr)-KO, Acarbose, Protandim, etc. (Figure 4E) (Tyshkovskiy et al., 2019). In agreement with CMap results, we found that mouse livers following 2 months treatment with rapamycin downregulated the expression of Neg-MLS genes compared to the WT B6 control mice. Similar to rapamycin, 17-alpha-estradiol treatment also showed a repressive role in the expression of Neg-MLS genes (Figure 4F). Accordingly, both rapamycin and 17-alpha-estradiol treatments tended to upregulate the expression of Pos-MLS genes. Other treatments like PIT1-KO, GHR-KO, Rilmenidine and Ascorbyl-palmitate also showed similar trends to those seen for rapamycin and 17-alpha-estradiol, but the p-values did not reach a statistical significance, which might be due to relatively small sample sizes (n=6) for the in vivo study. Interestingly, calorie restriction (CR) tended to upregulate both Neg-MLS and Pos-MLS genes, which is possibly related to the complex output of CR and its mimetics on healthy lifespan (Espada et al., 2020; Lee et al., 2021; Liao et al., 2010; Tang et al., 2016). Development of treatments that separate the “beneficial” effects from “deleterious” effects of CR may further improve the CR effect on lifespan.
Paradoxically, acarbose, methionine restriction and Protandim stimulated Neg-MLs genes and inhibited Pos-MLS genes, although the effects did not reach statistical significance. Acarbose and methionine restriction are believed to extend lifespan via a hormesis mechanism by inducing metabolic stress (Gems and Partridge, 2008). Protandim was proposed to activate the Nrf2 (Nfe2l2) antioxidant pathway (Abusarah et al., 2017; Reuland et al., 2013). While we observed a positive correlation of Nfe2l2 gene expression with MLS across species (average ρ = 0.45) (Table S2), the gene expression levels of Nfe2l2 were not upregulated in the mice treated with Protandim (Figure S4H). It is also possible, that while these interventions may affect an individual’s lifespan in mice, these pathways seem not to be involved in the evolution of longevity.
Together, these results provide new mechanistic and evolutionary insights into how longevity interventions affect the aging process at the molecular level. In addition, we also demonstrate that the transcriptomic signatures of species with long and short MLS could be applied to evaluate the performance of anti-aging interventions at the gene expression level.
Neg-MLS genes are regulated in a circadian manner
We next evaluated upstream transcriptional regulators (TRs) of MLS-associated genes through public data on chromatin accessibility and chromatin immunoprecipitation (ChIP)-seq (Qin et al., 2020). TR network analysis of Neg-MLS genes identified the core subunit of RNA Polymerase II (RNA Pol II) POLR2B and general transcriptional factors GTF2B and PRDM16 (Figure 5A and Table S6). One plausible explanation is that Neg-MLS genes are enriched in housekeeping functions like respirasome and oxidative phosphorylation (Figure 2A). Immune regulators including recombination-activating gene (RAG), RELA (NF-kB Subunit), signal transducer and activator of transcription (STAT) and interferon-regulatory factors (IRFs) were specifically observed in the TRs of lung Neg-MLS genes, which is consistent with the function of Neg-MLS genes in the lung (Figure 2D). Notably, we found that several hormone receptors including retinoid X receptor alpha (RXRA) and retinoic acid receptor alpha (RARA), as well as circadian regulator CLOCK were among the top-ranked TR list of Neg-MLS genes (Figure 5B). We next asked whether genes involved in energy metabolism and/or inflammation, are regulated by circadian regulators. We reanalyzed ChIP-seq data for CLOCK in the mouse liver and found that CLOCK shows significantly higher binding affinity to the promoter regions of Neg-MLS genes involved in energy metabolism compared to inflammation genes and Pos-MLS genes (Figures 5C–D and S5A). This is consistent with previous studies showing that circadian system orchestrates the expression of energy metabolism genes (Poggiogalle et al., 2018). To investigate the potential regulatory differences between energy metabolism genes, we divided them into two groups according to the correlations of gene expression with MLS. We found that metabolic genes that negatively correlate with MLS (ρ < −0.4) show ~2-fold higher binding signal for CLOCK compared to the metabolic genes that do not correlate with MLS (−0.1 < ρ < 0.1) (Figure S5A). These results confirm that Neg-MLS genes involved in energy metabolism are the specific targets of CLOCK.
Figure 5. Neg-MLS genes are regulated in a circadian manner.

(A) Regulatory network visualizing potential TRs of Neg-MLS genes. The connection between tissue and TR indicates that the TR shows a significantly higher binding preference to Neg-MLS genes identified in this tissue compared to random genes. The node size (count) indicates the number of tissues in which the genes were identified as top-ranked (30 proteins with lowest p values) regulators. TRs were classified into different functional categories highlighted with different colors. TRs positively or negatively regulating gene expression are shown in red and blue font, respectively. See Methods for details.
(B) Scatter plots showing the TR rank in the liver (left) and heart (right). Y-axis indicates the p values that were calculated using the Wilcoxon rank test comparison of the Neg-MLS genes and random gene background. The top regulators with the lowest p values are highlighted in red.
(C) Metagene analysis showing differential binding activities of circadian regulator CLOCK at Neg-MLS and Pos-MLS genes in mouse liver. Five kb around the TSS regions are shown. Liver ChIP-seq data is from (Annayev et al., 2014)
(D) Heatmaps showing differential binding activities of circadian regulator CLOCK at Neg-MLS, Pos-MLS and random genes in mouse liver.
(E) Metagene analysis showing dynamic binding activities of circadian regulators at Neg-MLS genes during the circadian cycle. Arrows indicate the direction of change in gene expression.
(F) Metagene analysis showing dynamic binding activities of RNA Pol II and histone modifications including H3K9ac and H3K27ac at Neg-MLS genes during the circadian cycle. Ser5P Pol II is an initiation form of Pol II. 8WG16 targets hypophosphorylated Pol II.
(G) Boxplots showing the dynamic transcriptional activity of Neg-MLS genes in two circadian cycles. CT, circadian time.
(H) Scatter plot showing the relative expression levels of Igf1 gene in the liver across species. Spearman correlation coefficients (ρ) of gene expression and MLS is shown.
(I) The barplot showing the dynamic transcription of the Igf1 gene in the liver.
Circadian ChIP-, RNA-seq data are from (Koike et al., 2012)
To further investigate how circadian TRs regulate the expression of Neg-MLS genes, we reanalyzed the ChIP-seq data of 7 master circadian TRs including BMAL1, CLOCK, NPAS2, PER1, PER2, CRY1, and CRY2 in the mouse liver at different circadian time courses (Koike et al., 2012). Metagene analysis revealed that the transcriptional activator BMAL1 binds to the transcription start site (TSS) region of Neg-MLS genes in a circadian manner between circadian time (CT) 0 and 12 with peak binding phases occurring at CT4 to 8 and then suffers a sharp decrease (Figure 5E). The heterodimeric partners of BMAL1, CLOCK and NPAS2, also bind in a time-dependent manner similar to BMAL1 (Figures 5E and S5B). In contrast, the transcriptional repressors PER1, PER2, and CRY2 then show rhythmic binding to Neg-MLS genes between CT12 and 20. CRY1 exhibits maximal binding at CT0 and 4, when BMAL1, CLOCK, and NPAS2 occupancy increases again at CT0. The co-occupation of activators and repressors indicates that the Neg-MLS genes are in a transcriptional poised phase at the beginning of the next cycle (Figure 5E).
We also analyzed the TSS occupancy of Neg-MLS genes by RNA Pol II as a function of the circadian cycle (Figure 5F). The initiation form of RNA Pol II phosphorylated on Ser5 peaks at CT0–4 (Figure 5F), which is consistent with the transcriptional poised phase in CT0–4 circadian window. The binding of unphosphorylated RNA Pol II (8WG16) peaks at CT12–16. Coordinately, active promoter histone modification markers H3K4me3 and H3K9ac also show peaks at CT12–16 and become decreased after CT16. As a control, we did not observe obvious changes in active histone modification marks H3K27ac and transcription elongation histone modification marks H3K36me3 and H3K79me2 (Figures 5F and S5B). Coinciding with TF binding, the transcription of Neg-MLS genes, but not genes with low binding of circadian regulators, shows a circadian cyclicity and peaks at CT12–16 in the mouse liver (Figure 5G and S5C). For example, insulin like growth factor 1 (Igf1), for which expression negatively correlates with MLS in the liver (ρ = −0.62) (Figure 5H), shows a circadian expression pattern and peaks at CT12 and CT36 in the 48h circadian time windows (Figure 5I). Other Neg-MLS genes that show similar oscillatory transcription include hypoxia inducible factor 1 subunit alpha (Hif1a), Ak2, which regulate cellular energy homeostasis and nucleotide metabolism, and Fbp1, which acts as a rate-limiting enzyme in gluconeogenesis. The expression of these genes in the liver shows consistently negative correlations with MLS (ρ = −0.47 ~ −0.64) (Figure S5D). Their expression levels usually peak at CT12 and become downregulated during the CT12–24 (Figures S5E and F). In conclusion, these results suggest that Neg-MLS genes in the liver are under circadian transcriptional control rather than maintaining consistently high expression.
Pos-MLS genes are upregulated by the master pluripotency factors
Transcriptional regulatory network analysis of Pos-MLS genes revealed TRs distinct from those of Neg-MLS genes (Figure 6A and Table S6). Epigenetic regulators including polycomb repressive complex 2 (PRC2) core components including SUZ12 and EZH2 and PRC1 component KDM2B were found to be TRs of Pos-MLS genes. The PRC2 contains methyltransferase activity generating the di-/trimethylated form of lysine 27 on histone H3 (H3K27me2/3) (van Mierlo et al., 2019), which might contribute to transcriptional repression of Pos-MLS genes. In support of this notion, the expression of PRC2 subunits Suz12, Eed and Ezh2 show consistently negative correlations with MLS (Figure S6A and Table S2). At the DNA level, key regulators of DNA demethylation, TET1 and TDG, were found to be TRs of Pos-MLS genes. We did not see a significant correlation between expression TET1 and TDG and MLS (Table S2). However, we found that gene expression of DNA methyltransferase 1 (Dnmt1) is negatively correlated with MLS (Figure S6B). These results suggest that the repression of Pos-MLS genes might be mediated by epigenetic mechanisms such as histone modification and DNA methylation.
Figure 6. Pos-MLS genes are targets of master pluripotency regulators and are upregulated during somatic cell reprogramming.

(A) Regulatory network visualizing potential TRs of Pos-MLS genes. The connection between tissue and TR indicates that TR shows a significantly higher binding preference to Pos-MLS genes identified in this tissue compared to random genes. The node size (count) indicates the number of tissues in which the genes were identified as top-ranked (30 with lowest p values) regulators. TRs were classified into different functional categories highlighted with different colors. TRs positively or negatively regulating gene expression are shown in red and blue font, respectively. See Methods for details.
(B) Scatter plots showing the TR rank in the heart (left) and liver (right). Y-axis indicates the p values that are calculated using the Wilcoxon rank test comparison of the pos-MLS genes and background. Top regulators with the lowest p values are labeled in red.
(C) Heatmaps showing differential binding activities of pluripotent regulator OCT4 at Pos-MLS genes during somatic reprogramming. Two clusters of genes, Pos-MLS-low (2125 genes) and Pos-MLS-high (1395 genes), were identified by kmeans method according to the low and high binding affinity of OCT4.
(D) The dot plot showing the GO enrichment of genes in the Pos-MLS-high cluster. The colors indicate the BH-adjusted p values. The dot sizes indicate the number of genes.
(E) Metagene analysis showing dynamic binding activities of pluripotent regulator OCT4 and RNA Pol II during the OSKM-induced somatic reprogramming. ChIP-seq data are from (Chen et al., 2016). Arrows indicate the direction of change in ChIP-seq signal.
(F) Boxplot showing the upregulation of Pos-MLS-high genes during the OSKM-induced somatic cell reprogramming. p values were calculated using two-tailed Student’s t-test. Microarray data are from (Chen et al., 2016).
(G) Heatmap showing the expression change of representative Pos- and Neg-MLS genes during the OSKM-induced somatic reprogramming. Each row represents a gene and its expression levels at different times during reprogramming were normalized by Z-score. Microarray data are from (Chen et al., 2016).
Notably, we also observed that master pluripotency regulators including OCT4 (encoded by Pou5f1), NANOG, SOX2 and KLF4 are among the top-ranked regulators for Pos-MLS genes (Figures 6A–B and S6C). OCT4, SOX2, KLF4 and MYC (OSKM) are known as Yamanaka factors for somatic cell reprogramming (Takahashi and Yamanaka, 2006). We next explored the binding pattern of pluripotency regulators during somatic cell reprogramming. We examined the binding pattern of OCT4 (Chen et al., 2016) to the genes that showed Pos-MLS association in at least one tissue. A heatmap revealed that exogenous OCT4 begins to bind to Pos-MLS genes as early as day 1 during the reprogramming process (Figure 6C). According to the binding strength of OCT4, the Pos-MLS genes were divided into two clusters. The first cluster containing 1,395 genes shows a high binding occupancy of OCT4. GO analysis of genes in the first cluster showed that they are mainly enriched in DNA repair, ribosome biogenesis and RNA methylation-related functions (Figure 6D). The other cluster shows relatively lower OCT4 binding during reprogramming, which is enriched in functional terms like organic hydroxy compound metabolism, protein secretion, and cilium organization (Figure S6D). During the reprogramming process, both exogenous OCT4 and total OCT4 begin to occupy the TSS regions of Pos-MLS genes at day 1 and peak at days 7–11. RNA Pol II also shows an increased biding to the TSS regions of Pos-MLS at day 3. Accordingly, we observed a significant increase in the expression of Pos-MLS genes with high OCT4 binding (Figures 6F–G). For example, FA complementation group I (Fanci) was upregulated ~3-fold at day 1 during reprogramming (Figure S6E) and expression of Fanci shows significant positive correlations with MLS (average ρ= 0.60) (Figure S6F). Suppressor of variegation 3–9 homolog 2 (Suv39h2), a histone methyltransferase for Lysine 9 of histone H3 (H3K9me3), showed ~4-fold upregulation at day 1, suggesting an involvement of heterochromatin (Figure S6E). In contrast, we observed an overall downregulation of Neg-MLS genes involved in inflammation but not cellular respirasome-based energy metabolism during the MEF reprogramming (Figure S6G). For example, Neg-MLS genes involved in inflammation including Nfkb1, Rela, Bmp2m, Tgfb1, Cxcl12, Il1rn, Stat1 and Ccl4 are downregulated during reprogramming (Figure 6G). In addition, Neg-MLS genes involved in nutrient sensing like insulin signaling pathway (Igf1, Irs1), Akt pathway (Akt1 and Akt2), and growth hormone receptor (Ghr) are also downregulated during reprogramming (Figure 6G). As controls, Pos-MLS genes with lower OCT4 binding and random genes did not show obvious changes in expression levels during reprogramming (Figure S6G).
We also examined the expression of Pou5f1 in adult tissues (Schaum et al., 2020). Low levels of Pou5f1 transcripts can be ubiquitously detected in all tissues used in this study (Figure S7A). The expression of Pou5f1 and other pluripotency factors shows no obvious downregulation or upregulation during aging in most tissues (Figure S7B). Based on our reanalysis of published gene expression data (Arjona et al., 2022; Booker et al., 2021; Ji et al., 2021; Luo et al., 2021; McCauley et al., 2021; Rinaldi et al., 2017; Sun et al., 2019), we note that the expression of Pou5f1 is low in adult stem cells, with the exception of female germline and spermatogonial stem cells (Figure S7C). In addition, stem cells comprise less than 1% of total cells in adult tissues (Bhartiya, 2021). These observations suggest that a signal from tissue stem cells did not contribute to the expression levels of pluripotency factors and their targets detected by bulk RNA-seq of the tissues. Rather the differences between short- and long-lived species originate from the bulk of differentiated cells.
We next explored the expression of Pos-MLS genes during embryonic stem cell (ESC) differentiation. In this process, we did not observe an overall expression change of Pos-MLS genes, suggesting that the expression of Pos-MLS genes can be sustained during the transition from naïve to primed pluripotency (Figure 7A). However, the expression of Pos-MLS genes was significantly downregulated in the trophectoderm (TE) (Figure 7A). TE formation and separation from inner cell mass represent the first lineage specification event during embryogenesis. Before the zygote formation, we observed a high expression of Pos-MLS genes during oocyte maturation (Figure 7A), which is consistent with a high expression of pluripotent gene Pou5f1 in female germline stem cells (Figure S7C). After fertilization, Pos-MLS genes can be detected in the zygote (Figure 7A), suggesting that Pos-MLS genes comprise a component of maternal RNAs. The abundance of Pos-MLS genes is further decreased in the early 2-cell stage and becomes upregulated at the late 2-cell stage embryos. Treatment of the zygotes with transcriptional inhibitor α-amanitin blocked the upregulation of Pos-MLS genes, suggesting that Pos-MLS genes are actively transcribed in zygotic genome activation. To make a direct comparison within tissues, we examined the expression of Pos-MLS genes in six tissues including brain, kidney, heart, lung, liver and limb (gene expression data for skin during embryogenesis is not available) at embryonic and postnatal stages (Davis et al., 2017; Dunham et al., 2012). Compared to the embryonic stages ranging from E10.5 to E15.5, expression of Pos-MLS genes shows dramatic downregulation in the tissues of 4-weeks old mice (Figure 7B and S7D–I). In contrast, there is no obvious change in overall expression levels for Neg-MLS genes between embryonic and postnatal stages (Figure S7D–I).
Figure 7. Conserved genes and pathways controlling lifespan across mammalian species.

(A) Boxplot showing the dynamic expression of Pos-MLS genes identified in all tissues during preimplantation development and gastrulation. Gene expression of Pos-MLS genes shows significant downregulation in TE compared to E3.5ICM during embryonic development. p values were calculated using a two-tailed Student’s t-test. RNA-seq data of gastrulation are from (Zhang et al., 2018). RNA-seq data of preimplantation development are from (Wu et al., 2016). ICM: inner cell mass. TE: trophectoderm. Epi: epiblasts. VE: visceral endoderm. Ect: ectoderm. PS: primitive streak. Mes: mesoderm. End: endoderm.
(B) Boxplot showing the expression of Pos-MLS genes identified in the heart. Gene expression of the fetal heart is shown as light blue. Gene expression of the postnatal heart (4-weeks old) is shown as orange. p values were calculated using a two-tailed Student’s t-test.
(C) Across species, gene expression is differentially regulated and highly correlated with MLS. Genes involved in respirasome, mitochondria, and fatty acid catabolism-related functions show higher expression in short-lived mammals. In comparison, genes involved in DNA damage response (DDR), DSB repair, mismatch repair, homologous recombination and centriole and microtubule-related functions tend to be upregulated in long-lived species. Neg-MLS and Pos-MLS also show distinct responses to interventions and are regulated by distinct transcriptional regulatory networks. Neg-MLS are regulated by circadian transcription factors while Pos-MLS genes are regulated by pluripotency factors.
See also Figure S7
Cumulatively, these results suggest that Pos-MLS genes, especially those genes involved in DNA repair, are upregulated by the master pluripotency factors during somatic reprogramming. Pos-MLS genes are also actively and dynamically transcribed during early embryonic development, when extensive reprogramming takes place.
DISCUSSION
Mammals differ over 100-fold in maximum lifespan (Tacutu et al., 2013), and this diversity makes comparative biology a fruitful avenue for exploring the mechanisms of aging and lifespan control (Gorbunova et al., 2014). Here we present comparative transcriptomics of 141 young adult animals covering 26 mammalian species with diverse MLSs. All samples were processed and sequenced in-house, reducing batch effects and sample processing variation.
We identified gene expression signatures both negatively and positively associated with MLS. We integrated our transcriptomic analysis with published transcriptome data from different aging-associated interventions and found interventions that have a negative or positive effect on the expression of MLS-associated genes. Furthermore, the analysis of gene regulatory networks revealed new connections between MLS-associated genes and emerging longevity and aging control pathways, such as circadian rhythms and somatic cell reprogramming (Figure 7C). Taken together, our study provides novel resources and insights relevant to the regulation of longevity across, and potentially within, species.
Neg-MLS genes were enriched for genes involved in apoptosis, mitochondria metabolism, fatty acid catabolism, oxidative phosphorylation and metabolism of ribonucleotides and glutathione. Indeed, excessive apoptosis is associated with accelerated aging and stem cell depletion (Tyner et al., 2002) and longevity may co-evolve with more stringent control of apoptosis. Although, functional mitochondria are required for viability, mild reduction in mitochondrial respiration has been associated with lifespan extension in model organisms including C. elegans (Dillin et al., 2002; Lee et al., 2003), Drosophila (Copeland et al., 2009) and mice (Dell’agnello et al., 2007; Liu et al., 2005). This is generally consistent with the “rate of living” theory proposing that a slower metabolic rate is associated with longevity. However, the relationship between mitochondrial activity and longevity is complex, as longer lifespans may be uncoupled from slow metabolism (Hwang et al., 2012).
The second group of Neg-MLS genes is related to immunity, inflammation, and cytosolic DNA sensing. Sterile, age-related “inflammaging” has emerged as a driving force underlying multiple age-related diseases (Franceschi et al., 2018). Hence, the evolution of longer lifespans may be associated with downregulation and more stringent control of inflammation. Consistent with our findings, comparative genomics analysis of 88 rockfish species showed expansion of negative regulators of inflammation in long-lived taxa (Kolora et al., 2021). Bats, a clade that is characterized by extreme longevity, especially when taking into account small body mass, evolved multiple mechanisms controlling inflammation. Strikingly, all bat species examined to date, have lost the entire PYHIN gene family responsible for cytoplasmic DNA sensing, which has been linked to sterile inflammation associated with aging (Ahn et al., 2016; Gorbunova et al., 2020). While our current study was limited to rodents and Eulipotyphla, we also observed downregulation of cytoplasmic DNA sensing, suggesting that downregulation of these pro-inflammatory pathways is a conserved signature of long-lived mammals. This finding may guide the future development of pharmacological inhibitors of cytoplasmic DNA sensors to improve human healthspan.
Pos-MLS genes were enriched for processes related to DNA repair. It has been proposed that DNA damage plays a causal role in aging via the induction of genomic instability, cellular senescence, and disruption of epigenetic landscapes (Korotkov et al., 2021; Schumacher et al., 2021; Yousefzadeh et al., 2021). Comparative genomics studies found that DNA repair genes are under positive selection or undergo duplications in the longest-lived mammal, the bowhead whale (Keane et al., 2015), in long-lived rockfishes (Kolora et al., 2021), and in bats (Huang et al., 2019; Koh et al., 2019). Our previous functional study demonstrated that DNA DSB repair positively correlates with MLS across rodent species (Tian et al., 2019). In this study, we reveal that genes involved in DNA repair coevolve with longevity at the transcriptional level.
DNA damage in circulating leukocytes measured with the comet assay was predictive of the risk of death in humans (Bonassi et al., 2021). Germline mutation rates in young adults were predictive of their longevity (Cawthon et al., 2020). Human genome-wide association studies (GWAS) also revealed that DNA damage response is one of the primary biological pathways regulating ovarian aging (Stolk et al., 2012). A study of 200,000 women reported that common alleles of DNA damage and repair genes were shaping the age of natural menopause. Higher expression of Chek1 was associated with a longer reproductive lifespan in women (Ruth et al., 2021). In our study, the expression of Chek1 shows a strong positive correlation with MLS, suggesting a conserved mechanism of longevity regulation in humans. Taken together, these studies indicate that interventions aimed to enhance DNA repair are a promising strategy to extend human healthspan.
Centrioles are structures that form the centrosome and the poles of the mitotic spindles (Breslow and Holland, 2019). Centrosome dysfunction contributes to chromosome instability and chromoanagenesis (Pihan, 2013). Moreover, DNA damage was found to induce centrosome amplification (Bourke et al., 2007), suggesting a functional role of the centrosome in DNA repair (Mullee and Morrison, 2016). Cilia are microtubule-based sensory organelles that extend from the surface of mammalian cells (Fliegauf et al., 2007). Changes in the primary cilium are frequently detected in hippocampal neurons of aged mice and in neurodegenerative disorders (Alvarez-Satta et al., 2019). It has been suggested that cilia safeguard cortical neurons in the mouse forebrain from environmental stress-induced dendritic degeneration (Ishii et al., 2021). The positive correlation in gene expression that was observed in this study further suggests that the cilium and microtubules are emerging regulators of longevity.
Nuclear transport of mRNAs, transfer RNAs (tRNAs), and ribosomal RNAs (rRNAs) is essential for transcription and translation. Whether RNA export plays a crucial role in regulating aging and longevity has remained largely elusive (Dominick et al., 2017; Park et al., 2017). Our study provides new links between RNA homeostasis and longevity regulation.
Circadian rhythms are part of the body’s internal clock and carry out essential functions to regulate physiological processes during a 24-hour period. A systematic decline of circadian control was reported during the aging process (Hood and Amir, 2017). Mice deficient in BMAL1 and CLOCK, two core components of the circadian clock, show a premature aging phenotype (Dubrovsky et al., 2010; Kondratov et al., 2006). The expression levels of Neg-MLS genes, are by definition, negatively correlated with MLS, suggesting a deleterious effect of their high-level expression on longevity. The Neg-MLS genes are enriched in housekeeping functions (Figure 2). Therefore, Neg-MLS genes seem to function as a double-edged sword where they are required for organismal function, but overactivation of these genes counteracts longevity. In this study, we found that core components of the circadian clock prevalently bind to the promoter regions of Neg-MLS gene, suggesting a direct regulatory role rather than a secondary effect. The activators and repressors in the circadian clock pathway show a dynamic yet coordinated time-course binding pattern to Neg-MLS genes, leading to their oscillated expression. We propose that circadian control sets time limits on the activation of many genes related to metabolic processes, limiting their lifespan shortening effects. Based on this hypothesis, long-lived species might have a more robust circadian regulation system than short-lived species like mice, which show increased mortality when subjected to jet-lag (Davidson et al., 2006).
Our study also suggests that a high level of nutrient metabolism, protein synthesis, DNA replication, and cell proliferation tend to downregulate the Pos-MLS genes and upregulate Neg-MLS genes. These observations are consistent with the aging life-history trade-off theory, which posits that investment in growth and reproduction at an early age reduces late-life fitness and shortens lifespan (Stearns, 1989). Interventions targeting PI3K, mTOR, tubulin, and HDAC related pathways could regulate the expression of both Neg-MLS and Pos-MLS genes, suggesting an intrinsic interconnection of Neg-MLS and Pos-MLS genes. In addition, the circadian control of Neg-MLS genes suggests that the design of pro-longevity lifestyle and pharmacological interventions must consider the circadian rhythm (Acosta-Rodriguez et al., 2021). In support of this notion, a recent study reported that time-restricted feeding extended fly lifespan and delayed the onset of aging markers (Ulgherait et al., 2021). Intermittent and time-controlled feeding regimens are being applied to mice and to humans offering a more tolerable alternative to calorie restriction (Chaix et al., 2014; Longo et al., 2021).
Recently, in vivo expression of somatic cell reprogramming factors (Yamanaka factors) was found to extend mouse lifespan and rescue certain age-related pathologies (Chen et al., 2021; Lapasset et al., 2011; Lu et al., 2020; Ocampo et al., 2016; Zhang et al., 2020). We found that Pos-MLS genes tend to be directly bound by pluripotency factors OCT4, NANOG, and are upregulated during cell reprogramming. We propose that the upregulation of Pos-MLS genes might, at least partially, contribute to the rejuvenation induced by reprogramming.
There are challenges on the path of employing somatic cell reprogramming for rejuvenation. During reprogramming, cells are losing identity and normal physiological function (Takahashi and Yamanaka, 2006). Expression of the Yamanaka factors in vivo can cause cancer or teratoma formation (Abad et al., 2013; Ohnishi et al., 2014). To avoid these caveats, new strategies including partial reprogramming by short-term expression of the Yamanaka factors or exclusion of the oncogenic MYC were proposed (Lu et al., 2020; Ocampo et al., 2016). To what degree these strategies can preserve somatic cell identities or repress tumor formation remains to be determined (Roux et al., 2021; Senis et al., 2018). Our study provides an alternative strategy to specifically target the downstream Pos-MLS genes instead of inducing somatic cell reprogramming.
Living organisms, experience DNA damage throughout their life, which results in the accumulation of mutations and epigenetic changes. After fertilization, the terminally differentiated gametes are converted into a totipotent zygote and a new life begins with a clean biological slate. Hence, the germ line is rejuvenated during development (Kerepesi et al., 2021). Interestingly, we observed that Pos-MLS genes are highly expressed in the oocyte, which might be related to the rejuvenation process as well as the reprogramming of a somatic nucleus during the somatic cell nuclear transfer (SCNT). Indeed, oocytes efficiently repair severe DNA double-strand breaks via homologous recombination to restore genetic integrity (Stringer et al., 2020) indicating that DNA repair is a key quality control mechanism in the female germ line. These results support the idea that direct modulation of Pos-MLS genes may be a promising strategy to induce efficient and safe rejuvenation.
In conclusion, our work identifies transcriptomic features that evolved in long-lived animal species and reveals their regulatory networks. These results further disentangle the molecular mechanism of aging and longevity regulation. Our study not only provides novel resources for identification of targets for rejuvenation therapies, but also builds broad connections to other emerging aspects in the aging field and provides evolutionary insights about the evolutionary strategies shaping lifespan control. Targeting the lifespan genes and their regulatory networks will facilitate the development of interventions to increase lifespan and healthspan, and may provide new insights into age-related diseases.
Limitations of Study
Since most of the species used in this study were caught in the wild, it is difficult to evaluate the precise age of these individuals. We presume that the samples used in this study were from young adults. We excluded juveniles based on size and appearance. Most individuals in wild rodent populations die before they reach advanced age due to predation and pathogens. Consistently, age structure analysis of wild rodents and shrews revealed that <5% of individuals survive for more than 1 year (Bishop and Hartley, 1976; Blair, 1953). Therefore, we believe, that the samples used in this study were from young adult individuals, at a comparable physiological stage across the species. We aimed to make a comprehensive investigation of compounds that can affect gene signatures (Cmap, published studies). However, it should be noted that the interventions predicted by Cmap were performed in vitro using tumor-derived and genetically manipulated immortalized cell lines, which may respond to interventions differently compared to normal tissues. The candidate interventions should be further validated using in vivo models.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vera Gorbunova (vera.gorbunova@rochester.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Raw sequencing reads are available in the NCBI Gene Expression Omnibus (GEO) and Sequence Read Archive (SRA) under accession numbers GSE190756.
Original data for creating all graphs in the paper are provided in Data S1.
This study does not generate any new codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian Species Sample Collection
All experiments were performed according to procedures approved by the University of Rochester Committee on Animal Resources (UCAR).
C57BL/6 mice were purchased from the Jackson Laboratory. Norway rats and golden hamsters were purchased from Charles River Laboratories. Capybaras were obtained from Bio Fau Assesoria e Comercio (São Paulo, Brazil). Pacas were from the animal facility at São Paulo State University. Outbred multicolored guinea pigs were purchased from Elm Hill Labs. Chinchillas were purchased from Moulton Chinchilla Ranch. Beavers, deer mice, muskrats, woodchucks, chipmunks, eastern mole, wild type mice, star-nosed mole, red, and gray squirrels were trapped in New York State by licensed hunters. Nutria samples were obtained through USDA Nutria Eradication Program, MD. Blind mole rats were caught in Upper Galilee Mountains in Israel. Bushy tail rats were purchased from Cascade Biological Supply, WA. Naked mole rats and Damaraland mole rats were from the colonies at University of Rochester. African spiny mice were from Texas Exotic animals. Octodon degus was from Dr. Nattan Insel, University of Montana. Chinese hamster was obtained from Dr. Sarelius Lab, University of Rochester. Ellobius lutescens were obtained from Dr. Goskun lab, Turkey; Short-tailed shrews were wild caught in Auburn, AL. Since most of the species used in this study were caught in the wild, it is difficult to evaluate the precise age of these individuals. We presume that the samples used in this study were from young adults since the age structure analysis of wild rodents and shrews revealed that <5% of individuals survive for more than 1 year (Bishop and Hartley, 1976; Blair, 1953). Care and maintenance were standard for the mice and rats. Naked mole rats were maintained as described previously (Tian et al., 2019). Damaraland mole rats are maintained in a similar way to naked mole rats but at 25 oC. The sex information of the subjects can be found in the Table S1.
METHOD DETAILS
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA library preparation
Frozen tissues were pulverized using a Cell Crusher piston chilled with liquid nitrogen to produce uniform powdered tissue. Approximately 30 mg of frozen tissue powder was used for Trizol RNA extraction. Contaminating DNA was removed from RNA samples using Purelink DNase kit along with Purelink RNA columns. Purified RNA quality was checked using agarose gel examination for rRNA integrity and Qubit analysis. The RNA samples were processed with the Illumina TruSeq stranded total RNA RiboZero Gold kit and then subjected to Illumina HiSeq 4000 paired-end 150-bp sequencing at the New York University Genome Technology Center.
RNA-seq based transcriptome assembly and annotation
The RNA-seq reads were first processed with Trim_Galore (version 0.6.6), which trimmed both adapter sequences and low-quality base calls (threshold: Phred quality score < 20). The clean reads were aligned to corresponding reference genome (see Table S1 for detail) by HISAT2 (v2.2.1) with the specific parameters “--dta --very-sensitive” (Kim et al., 2019). For those species without reference genomes, clean RNA-seq reads were aligned to the reference genome of neighboring species (see Table S1 for detail). For each RNA-seq sample, de novo transcriptome assembly was conducted by StringTie (v2.1) (Kovaka et al., 2019). The reference-based assemblies from all the samples in each species were merged by command “stringtie --merge” with the parameter “gap between transcripts to merge together” set to 50 bp. The transcripts with low expression levels were filtered by the parameter threshold of “TPM >0.5”. The redundant transcripts of the merged transcriptome assembly were removed using CD-HIT (v4.8.1) with a sequence identity threshold of 95% (-c 0.95) (Fu et al., 2012). To annotate the de novo transcriptome assembly, the transcript sequences of each species were blasted to the Gencode mouse gene annotation (Frankish et al., 2019) by Nucleotide BLAST (v2.10.1) (Camacho et al., 2009) with the parameters “- qcov_hsp_perc 10; -evalue 1e-30”. If a transcript sequence could be blasted to multiple transcript sequences in the Gencode database, only the one with the highest “bit score” was kept for the annotation of the transcript.
Gene expression analyses of tissues across species
The RNA-seq reads were first processed using Trim_Galore (version 0.6.6). The clean RNA-seq reads were used to quantify the gene expression with Salmon (version 1.4.0) (Patro et al., 2017). Transcript fasta files of species were got from the method section “RNA-seq based transcriptome assembly and annotation”. Transcript fasta files were indexed with the command “Salmon index” by default parameters. Clean RNA-seq reads were aligned to the indexed transcript by the specific parameters (--useVBOpt --seqBias --gcBias), which were set for sequence-specific bias correction and fragment GC bias correction. The reads counts of genes from Salmon were further normalized by trimmed mean of M-values (TMM) (Robinson et al., 2010) and median of ratios method (Love et al., 2014). The RNA-seq sample clustering was performed by the R package “pheatmap” (v1.0.12).
Phylogenetically independent contrasts
The normalized read counts obtained from Salmon were used as input for phylogenetically independent contrasts (PIC). The PIC was performed by the function “pic” in the R package APE (Analyses of Phylogenetics and Evolution) (Paradis and Schliep, 2019). The phylogenetic tree of species used in this study was generated by VertLife (Upham et al., 2019) and phyloT (https://phylot.biobyte.de/). The same phylogenetic trees were obtained from these two methods.
Gene Set Enrichment Analysis
Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) was performed to detect the functional enriched set (version 4.1.0). All genes were preranked by the values of Spearman’s rank correlation coefficient of gene expression and the maximum lifespan or body mass of corresponding species. Normalization mode in GSEA was set to “meandiv”. The gene sets of Kyoto Encyclopedia of Genes and Genomes (KEGG) were downloaded from https://www.genome.jp/kegg/ (2020.11). The gene sets of Gene Ontology (GO) were downloaded from http://geneontology.org/ (2021.02). Only those gene sets with a size of more than 15 genes and less than 800 genes were kept for further analysis. Network visualization and clustering groups with similar pathways were performed by the EnrichmentMap (Reimand et al., 2019). In each network, GO terms and pathways were shown as circles (nodes) that are connected with lines (edges) if the pathways share many genes. Only those GO terms or pathways with FDR < 0.1 were shown in the network plots.
Identification of interventions for Neg-MLS and Pos-MLS genes
Connectivity Map (CMap) database, containing the transcriptomic response of various human cells to thousands of chemical compounds and gene manipulations, was used to find the interventions that regulate Neg-MLS and Pos-MLS genes. Analysis was performed with the online “query” tool in https://clue.io/query. The query was run against the dataset released on (Dec 17, 2020). In each query, 150 genes with the lowest or highest Spearman’s rank correlation coefficient of gene expression and maximum lifespan of corresponding species were used as the input. FDR < 0.01 was used as the threshold to select statistically significant functions.
The in vivo (liver) RNA-seq data were obtained from gene expression omnibus (GEO) with accession number: GSE131901 (see key resource table). The adapter sequences and low-quality sequences of raw RNA-seq were trimmed by Trim_Galore (version 0.6.6). The clean RNA-seq reads were used to quantify the gene expression with Salmon (version 1.4.0). Gencode (Frankish et al., 2019) (version M25) was used for the genome-wide annotation of the gene in the mouse genome. The reads counts for gene levels were used as the input for differential expression analysis by DESeq2 (Anders and Huber, 2010). Low-expression genes with reads counts less than 10 were excluded from differential expression analysis. For each intervention, GSEA (Subramanian et al., 2005) was performed (version 4.1.0). All genes were preranked by the values of –log10 (adjusted p value)*(fold change)/abs(fold change), where abs(fold change) indicates the absolute value of fold change. Adjusted p value and fold change for each gene was obtained from DEseq2. 500 genes with lowest or highest Spearman’s rank correlation coefficient of gene expression and MLS in the liver were used as the gene sets for GSEA.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| RNA-seq of mice and Blind mole rats | Gene Expression Omnibus | GSE181413 |
| RNA-seq with different interventions in mouse liver | Gene Expression Omnibus | GSE131901 |
| Circadian RNA-seq data in mouse liver | Gene Expression Omnibus | GSE39860 |
| Circadian ChIP-seq data in mouse liver | Gene Expression Omnibus | GSE39977 |
| CLOCK ChIP-seq data in mouse liver | Gene Expression Omnibus | GSE53828 |
| OCT4 binding and Histone modification profiling during OSKM-mediated reprogramming | Gene Expression Omnibus | GSE67520 |
| ArrayExpress of OSKM-mediated reprogramming cells and the corresponding iPS cell line | Gene Expression Omnibus | GSE67462 |
| RNA-seq of mouse pre-implantation embryos | Gene Expression Omnibus | GSE66390 |
| RNA-seq of gastrulation in mouse | Gene Expression Omnibus | GSE76505 |
| RNA-seq of mouse tissues during aging | Gene Expression Omnibus | GSE132040 |
| RNA-seq of fetal mouse tissues | encodeproject.org | Heart, Limb, Liver, Lung and Kidney tissues: Barbara Wold, Caltech; Whole Brain: Bing Ren, UCSD |
| RNA-seq of human tissues during aging | The Genotype-Tissue Expression (GTEx) project | GTEx_Analysis_2017–06-05_v8_ |
| RNA-seq used for MLS gene identification | This study | GSE190756 |
| RNA-seq of Interfollicular Epidermal Stem Cells | Gene Expression Omnibus | GSE92423 |
| RNA-seq of Hair Follicle Stem Cells | Gene Expression Omnibus | GSE92423 |
| RNA-seq of mesenchymal stem cells | Gene Expression Omnibus | GSE156174 |
| RNA-seq of skeletal stem cells | Gene Expression Omnibus | GSE156174 |
| RNA-seq of neural stem cells | Gene Expression Omnibus | GSE156407 |
| RNA-seq of hematopoietic stem cells | Gene Expression Omnibus | GSE164284 |
| RNA-seq of adipose derived stem cells | Gene Expression Omnibus | GSE171946 |
| RNA-seq of skeletal muscle stem cells | Gene Expression Omnibus | GSE178070 |
| RNA-seq of melanocyte stem cells | Gene Expression Omnibus | GSE96966 |
| RNA-seq of Spermatogonial stem cells and Female germline stem cells | Gene Expression Omnibus | GSE134640 |
| Experimental models: Organisms/strains | ||
| Norway rats | Charles River Laboratories | N/A |
| Golden hamsters | Charles River Laboratories | N/A |
| Capybaras | Bio Fau Assesoria e Comercio | N/A |
| Pacas | São Paulo State University | N/A |
| Guinea pigs | Elm Hill Labs | |
| Beavers | Wild caught in New York State | N/A |
| Chinchillas | Moulton Chinchilla Ranch | N/A |
| Deer mice | Wild caught in New York State | N/A |
| Muskrats | Wild caught in New York State | N/A |
| Woodchucks | Wild caught in New York State | N/A |
| Chipmunks | Wild caught in New York State | N/A |
| Eastern mole | Wild caught in New York State | N/A |
| Wild mice | Wild caught in New York State | N/A |
| Star-nosed mole | Wild caught in New York State | N/A |
| Red squirrels | Wild caught in New York State | N/A |
| Gray squirrels | Wild caught in New York State | N/A |
| Nutria | USDA Nutria Eradication Program | N/A |
| Blind mole rats | Wild caught in Upper Galilee Mountains in Israel | N/A |
| Bushy tail rats | Cascade Biological Supply | N/A |
| Naked mole rats | University of Rochester | N/A |
| African spiny mice | Texas Exotic animals | N/A |
| Octodon degus | Gift from Dr. Nattan Insel, University of Montana | N/A |
| Chinese hamster | Gift from Dr. Sarelius Lab, University of Rochester | N/A |
| Damara mole-rat | University of Rochester | N/A |
| Ellobius lutescens | Dr. Goskun lab, Turkey | N/A |
| Short-tailed Shrew | Wild caught in Auburn, AL | N/A |
| Software and algorithms | ||
| Trim_Galore | Babraham Bioinformatics | version 0.6.6 |
| HISAT2 | Kim Lab at UT Southwestern Medical Center, (Kim et al., 2019) | v2.2.1 |
| StringTie | The Center for Computational Biology, Johns Hopkins University, (Kovaka et al., 2019) | v2.1 |
| CD-HIT | Godzik’s Lab at the Burnham Institute, (Fu et al., 2012) | v4.8.1 |
| Gencode | The GENCODE Project, (Frankish et al., 2019) | mV25 |
| Nucleotide BLAST | National Center for Biotechnology Information, (Camacho et al., 2009) | v2.10.1 |
| Salmon | Dr.Rob Patro, Stony Brook University, (Patro et al., 2017) | v1.4.0 |
| pheatmap | Raivo Kolde, University of Tartu | v1.0.12 |
| R | The R Project for Statistical Computing | V4.03 |
| APE | http://ape-package.ird.fr/ (Paradis and Schliep, 2019) | V5.5 |
| phyloT | https://phylot.biobyte.de/ | V2 |
| VertLife | https://vertlife.org/ (Upham et al., 2019) | N/A |
| GSEA | UC San Diego and Broad Institute | V4.1 |
| EnrichmentMap | https://www.baderlab.org/Software/EnrichmentMap (Reimand et al., 2019) | V3.3 |
| DESeq2 | (Anders and Huber, 2010) | V1.34.0 |
| edgeR | (Robinson et al., 2010) | V3.14 |
| Integrative Genomics Viewer | Broad Institute | V2.9.2 |
| UpSet2 | https://vdl.sci.utah.edu/upset2/ (Lex et al., 2014) | Last updated on December 22, 2021 |
| ClusterProfiler | (Wu et al., 2021) | V3.14 |
| LISA | (Qin et al., 2020) | LISA v2.2.5 |
| Cytoscape | https://cytoscape.org/ | V3.8.2 |
ChIP-seq analysis
The accession numbers of ChIP-seq data analyzed in this study are indicated in the KEY RESOURCES TABLE. The raw ChIP-seq reads were first processed using Trim_Galore (version 0.6.6). The clean reads were aligned to corresponding reference genome (mm10) by HISAT2 (v2.2.1) with the parameters “--no-spliced-alignment; --very-sensitive” (Kim et al., 2019). The heatmap and genome coverage signal of ChIP-seq were generated by deeptools (v 3.5.0) (Ramírez et al., 2014) and shown in the Integrative Genomics Viewer (IGV) (Robinson et al., 2011).
Gene expression analyses of circadian RNA-seq data
Circadian RNA-seq data were obtained from GEO with accession number: GSE39860.The adapter sequences and low-quality sequences of raw RNA-seq were trimmed by Trim_Galore (version 0.6.6). The clean RNA-seq reads were used to quantify the gene expression with Salmon (version 1.4.0). Gencode (Frankish et al., 2019) (version M25) was used for the genome-wide annotation of the gene in the mouse. To quantify the relative transcriptional activity of genes across the genome, we calculated the RNA-seq reads coverage in reads per kilobase per million reads (RPKM) of the intron regions for each gene. The intron signal serves as a representation of pre-mRNA expression or nascent transcription.
Gene Ontology analysis
GO analysis were performed by R package clusterProfiler (Release version 3.14) (Wu et al., 2021). GO comprises of three orthogonal ontologies, i.e. molecular function (MF), biological process (BP), and cellular component (CC). Fisher’s exact test was performed for each GO term and corresponding p value was calculated. All the p-values got from Fisher’s exact test were adjusted for multiple testing with Benjamini –Hochberg (BH) correction.
Transcriptional regulator (TR) prediction
TR prediction was performed with Landscape In Silico deletion Analysis (LISA v2.2.5) (Qin et al., 2020). 500 genes with lowest or highest Spearman’s rank correlation coefficient of gene expression and MLS in each tissue were used as the input for LISA. Corresponding network visualizations were conducted by Cytoscape (V3.8.2).
Upset plot
Upset plots shown in Figure S2 were generated with online tools: https://vdl.sci.utah.edu/upset2/ (Lex et al., 2014).
All of the statistical details of experiments can be found in the figure legends.
Supplementary Material
Table S1 Statistics of RNA-seq samples. Related to Figure 1.
Table S5 CMap interventions. Related to Figure 4.
Data S1. Unprocessed data underlying the display items in the manuscript, related to Figures 1–7 and S1–S7
Highlights.
Neg-MLS genes are involved in energy metabolism and inflammation
Pos-MLS genes are involved in DNA repair, microtubule organization and RNA transport
Neg-MLS genes are controlled by circadian network
Pos-MLS genes are controlled by pluripotency genes and upregulated during reprogramming
ACKNOWLEDGMENTS
The authors thank the Center for Integrated Research Computing (CIRC) at the University of Rochester for providing computational resources and technical support and the Genomics Research Center (GRC) at the University of Rochester for sequencing services. Work in the authors’ laboratories is supported by grants from the US National Institute of Aging.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abad M, Mosteiro L, Pantoja C, Canamero M, Rayon T, Ors I, Grana O, Megias D, Dominguez O, Martinez D, et al. (2013). Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 502, 340–345. [DOI] [PubMed] [Google Scholar]
- Abusarah J, Benabdoune H, Shi Q, Lussier B, Martel-Pelletier J, Malo M, Fernandes JC, de Souza FP, Fahmi H, and Benderdour M. (2017). Elucidating the Role of Protandim and 6-Gingerol in Protection Against Osteoarthritis. J Cell Biochem 118, 1003–1013. [DOI] [PubMed] [Google Scholar]
- Acosta-Rodriguez VA, Rijo-Ferreira F, Green CB, and Takahashi JS (2021). Importance of circadian timing for aging and longevity. Nat Commun 12, 2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn JH, Lee TK, Park JH, Cho JH, Kim IH, Lee JC, Hong S, Jeon YH, Kang IJ, Lee YJ, et al. (2017). Age-dependent differences in myelin basic protein expression in the hippocampus of young, adult and aged gerbils. Lab Anim Res 33, 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn M, Cui J, Irving AT, and Wang LF (2016). Unique Loss of the PYHIN Gene Family in Bats Amongst Mammals: Implications for Inflammasome Sensing. Sci Rep 6, 21722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Satta M, Moreno-Cugnon L, and Matheu A. (2019). Primary cilium and brain aging: role in neural stem cells, neurodegenerative diseases and glioblastoma. Ageing Res Rev 52, 53–63. [DOI] [PubMed] [Google Scholar]
- Anders S, and Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjona M, Goshayeshi A, Rodriguez-Mateo C, Brett JO, Both P, Ishak H, and Rando TA (2022). Tubastatin A maintains adult skeletal muscle stem cells in a quiescent state ex vivo and improves their engraftment ability in vivo. Stem Cell Reports 17, 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart M, Priebe S, Groth M, Hartmann N, Menzel U, Pandolfini L, Koch P, Felder M, Ristow M, Englert C, et al. (2016). Longitudinal RNA-Seq Analysis of Vertebrate Aging Identifies Mitochondrial Complex I as a Small-Molecule-Sensitive Modifier of Lifespan. Cell Syst 2, 122–132. [DOI] [PubMed] [Google Scholar]
- Bhartiya D. (2021). Adult tissue-resident stem cells-fact or fiction? Stem Cell Res Ther 12, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JA, and Hartley DJ (1976). The Size and Age Structure of Rural Populations of Rattus norvegicus Containing Individuals Resistant to the Anticoagulant Poison Warfarin. Journal of Animal Ecology 45, 623–646. [Google Scholar]
- Blair WF (1953). Population Dynamics of Rodents and Other Small Mammals. In Advances in Genetics, Demerec M, ed. (Academic Press; ), pp. 1–41. [DOI] [PubMed] [Google Scholar]
- Bonassi S, Ceppi M, Moller P, Azqueta A, Milic M, Neri M, Brunborg G, Godschalk R, Koppen G, Langie SAS, et al. (2021). DNA damage in circulating leukocytes measured with the comet assay may predict the risk of death. Sci Rep 11, 16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker CN, Haga CL, Boregowda SV, Strivelli J, and Phinney DG (2021). Transcriptional responses of skeletal stem/progenitor cells to hindlimb unloading and recovery correlate with localized but not systemic multi-systems impacts. NPJ Microgravity 7, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudoulas KD, Borer JS, and Boudoulas H. (2015). Heart Rate, Life Expectancy and the Cardiovascular System: Therapeutic Considerations. Cardiology 132, 199–212. [DOI] [PubMed] [Google Scholar]
- Bourke E, Dodson H, Merdes A, Cuffe L, Zachos G, Walker M, Gillespie D, and Morrison CG (2007). DNA damage induces Chk1-dependent centrosome amplification. EMBO Rep 8, 603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, and Holland AJ (2019). Mechanism and Regulation of Centriole and Cilium Biogenesis. Annu Rev Biochem 88, 691–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM, Meeks HD, Sasani TA, Smith KR, Kerber RA, O’Brien E, Baird L, Dixon MM, Peiffer AP, Leppert MF, et al. (2020). Germline mutation rates in young adults predict longevity and reproductive lifespan. Sci Rep 10, 10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaix A, Zarrinpar A, Miu P, and Panda S. (2014). Time-restricted feeding is a preventative and therapeutic intervention against diverse nutritional challenges. Cell Metab 20, 991–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Chen X, Li M, Liu X, Gao Y, Kou X, Zhao Y, Zheng W, Zhang X, Huo Y, et al. (2016). Hierarchical Oct4 Binding in Concert with Primed Epigenetic Rearrangements during Somatic Cell Reprogramming. Cell Rep 14, 1540–1554. [DOI] [PubMed] [Google Scholar]
- Chen Y, Luttmann FF, Schoger E, Scholer HR, Zelarayan LC, Kim KP, Haigh JJ, Kim J, and Braun T. (2021). Reversible reprogramming of cardiomyocytes to a fetal state drives heart regeneration in mice. Science 373, 1537–1540. [DOI] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T Jr., Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, and Walker DW (2009). Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol 19, 1591–1598. [DOI] [PubMed] [Google Scholar]
- Cui R, Medeiros T, Willemsen D, Iasi LNM, Collier GE, Graef M, Reichard M, and Valenzano DR (2019). Relaxed Selection Limits Lifespan by Increasing Mutation Load. Cell 178, 385–399 e320. [DOI] [PubMed] [Google Scholar]
- Davidson AJ, Sellix MT, Daniel J, Yamazaki S, Menaker M, and Block GD (2006). Chronic jet-lag increases mortality in aged mice. Current Biology 16, R914–R916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, et al. (2017). The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Research 46, D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, and Zeviani M. (2007). Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet 16, 431–444. [DOI] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, and Kenyon C. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401. [DOI] [PubMed] [Google Scholar]
- Dominick G, Berryman DE, List EO, Kopchick JJ, Li X, Miller RA, and Garcia GG (2015). Regulation of mTOR activity in Snell dwarf and GH receptor gene-disrupted mice. Endocrinology 156, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominick G, Bowman J, Li X, Miller RA, and Garcia GG (2017). mTOR regulates the expression of DNA damage response enzymes in long-lived Snell dwarf, GHRKO, and PAPPA-KO mice. Aging Cell 16, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrovsky YV, Samsa WE, and Kondratov RV (2010). Deficiency of circadian protein CLOCK reduces lifespan and increases age-related cataract development in mice. Aging (Albany NY) 2, 936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, and Walter JC (2015). What is the DNA repair defect underlying Fanconi anemia? Curr Opin Cell Biol 37, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann W, Yang K, Umar A, Heyer J, Lau K, Fan K, Liedtke W, Cohen PE, Kane MF, Lipford JR, et al. (1997). Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell 91, 467–477. [DOI] [PubMed] [Google Scholar]
- Espada L, Dakhovnik A, Chaudhari P, Martirosyan A, Miek L, Poliezhaieva T, Schaub Y, Nair A, Doring N, Rahnis N, et al. (2020). Loss of metabolic plasticity underlies metformin toxicity in aged Caenorhabditis elegans. Nat Metab 2, 1316–1331. [DOI] [PubMed] [Google Scholar]
- Ferraro NM, Strober BJ, Einson J, Abell NS, Aguet F, Barbeira AN, Brandt M, Bucan M, Castel SE, Davis JR, et al. (2020). Transcriptomic signatures across human tissues identify functional rare genetic variation. Science 369, eaaz5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, and Omran H. (2007). When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol 8, 880–893. [DOI] [PubMed] [Google Scholar]
- Foley NM, Hughes GM, Huang Z, Clarke M, Jebb D, Whelan CV, Petit EJ, Touzalin F, Farcy O, Jones G, et al. (2018). Growing old, yet staying young: The role of telomeres in bats’ exceptional longevity. Sci Adv 4, eaao0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, and Santoro A. (2018). Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 14, 576–590. [DOI] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, and Li W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba M, Fabian DK, Donertas HM, Thornton JM, and Partridge L. (2021). Transcriptomic profiling of long- and short-lived mutant mice implicates mitochondrial metabolism in ageing and shows signatures of normal ageing in progeroid mice. Mech Ageing Dev 194, 111437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gems D, and Partridge L. (2008). Stress-response hormesis and aging: “that which does not kill us makes us stronger”. Cell Metab 7, 200–203. [DOI] [PubMed] [Google Scholar]
- Gorbunova V, Seluanov A, and Kennedy BK (2020). The World Goes Bats: Living Longer and Tolerating Viruses. Cell Metab 32, 31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbunova V, Seluanov A, Zhang Z, Gladyshev VN, and Vijg J. (2014). Comparative genetics of longevity and cancer: insights from long-lived rodents. Nat Rev Genet 15, 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi E, Sanguinetti MC, Bartos DC, Bers DM, Chen-Izu Y, Chiamvimonvat N, Colecraft HM, Delisle BP, Heijman J, Navedo MF, et al. (2017). Potassium channels in the heart: structure, function and regulation. J Physiol 595, 2209–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl D, Tirouvanziam R, Laval J, Greene CM, Habiel D, Sharma L, Yildirim AO, Dela Cruz CS, and Hogaboam CM (2018). Innate Immunity of the Lung: From Basic Mechanisms to Translational Medicine. J Innate Immun 10, 487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood S, and Amir S. (2017). The aging clock: circadian rhythms and later life. J Clin Invest 127, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Whelan CV, Foley NM, Jebb D, Touzalin F, Petit EJ, Puechmaille SJ, and Teeling EC (2019). Longitudinal comparative transcriptomics reveals unique mechanisms underlying extended healthspan in bats. Nat Ecol Evol 3, 1110–1120. [DOI] [PubMed] [Google Scholar]
- Hwang AB, Jeong DE, and Lee SJ (2012). Mitochondria and organismal longevity. Curr Genomics 13, 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii S, Sasaki T, Mohammad S, Hwang H, Tomy E, Somaa F, Ishibashi N, Okano H, Rakic P, Hashimoto-Torii K, et al. (2021). Primary cilia safeguard cortical neurons in neonatal mouse forebrain from environmental stress-induced dendritic degeneration. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Ma Y, Shen J, Ni H, Lu Y, Zhang Y, Ma H, Liu C, Zhao Y, Ding S, et al. (2021). TBX20 Contributes to Balancing the Differentiation of Perivascular Adipose-Derived Stem Cells to Vascular Lineages and Neointimal Hyperplasia. Frontiers in Cell and Developmental Biology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane M, Semeiks J, Webb AE, Li YI, Quesada V, Craig T, Madsen LB, van Dam S, Brawand D, Marques PI, et al. (2015). Insights into the evolution of longevity from the bowhead whale genome. Cell Rep 10, 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerepesi C, Zhang B, Lee SG, Trapp A, and Gladyshev VN (2021). Epigenetic clocks reveal a rejuvenation event during embryogenesis followed by aging. Sci Adv 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EB, Fang X, Fushan AA, Huang Z, Lobanov AV, Han L, Marino SM, Sun X, Turanov AA, Yang P, et al. (2011). Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh J, Itahana Y, Mendenhall IH, Low D, Soh EXY, Guo AK, Chionh YT, Wang LF, and Itahana K. (2019). ABCB1 protects bat cells from DNA damage induced by genotoxic compounds. Nat Commun 10, 2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, and Takahashi JS (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolora SRR, Owens GL, Vazquez JM, Stubbs A, Chatla K, Jainese C, Seeto K, McCrea M, Sandel MW, Vianna JA, et al. (2021). Origins and evolution of extreme life span in Pacific Ocean rockfishes. Science 374, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, and Antoch MP (2006). Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev 20, 1868–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkov A, Seluanov A, and Gorbunova V. (2021). Sirtuin 6: linking longevity with genome and epigenome stability. Trends Cell Biol 31, 994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, and Pertea M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapasset L, Milhavet O, Prieur A, Besnard E, Babled A, Ait-Hamou N, Leschik J, Pellestor F, Ramirez JM, De Vos J, et al. (2011). Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev 25, 2248–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laschober GT, Ruli D, Hofer E, Muck C, Carmona-Gutierrez D, Ring J, Hutter E, Ruckenstuhl C, Micutkova L, Brunauer R, et al. (2010). Identification of evolutionarily conserved genetic regulators of cellular aging. Aging Cell 9, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MB, Hill CM, Bitto A, and Kaeberlein M. (2021). Antiaging diets: Separating fact from fiction. Science 374, eabe7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, and Ruvkun G. (2003). A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet 33, 40–48. [DOI] [PubMed] [Google Scholar]
- Lee Y, Brown EJ, Chang S, and McKinnon PJ (2014). Pot1a prevents telomere dysfunction and ATM-dependent neuronal loss. J Neurosci 34, 7836–7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lex A, Gehlenborg N, Strobelt H, Vuillemot R, and Pfister H. (2014). UpSet: Visualization of Intersecting Sets. IEEE Transactions on Visualization and Computer Graphics 20, 1983–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CY, Rikke BA, Johnson TE, Diaz V, and Nelson JF (2010). Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell 9, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, and Hekimi S. (2005). Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev 19, 2424–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Di Tano M, Mattson MP, and Guidi N. (2021). Intermittent and periodic fasting, longevity and disease. Nature Aging 1, 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Galluzzi L, Freije JMP, Madeo F, and Kroemer G. (2016). Metabolic Control of Longevity. Cell 166, 802–821. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Brommer B, Tian X, Krishnan A, Meer M, Wang C, Vera DL, Zeng Q, Yu D, Bonkowski MS, et al. (2020). Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Li X, Tian GG, Li D, Hou C, Ding X, Hou L, Lyu Q, Yang Y, Cooney AJ, et al. (2021). Offspring production of ovarian organoids derived from spermatogonial stem cells by defined factors with chromatin reorganization. J Adv Res 33, 81–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhães J.P.d., Costa J, and Church GM (2007). An Analysis of the Relationship Between Metabolism, Developmental Schedules, and Longevity Using Phylogenetic Independent Contrasts. The Journals of Gerontology: Series A 62, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather A, and Pollock C. (2011). Glucose handling by the kidney. Kidney Int Suppl, S1–6. [DOI] [PubMed] [Google Scholar]
- McCauley BS, Sun L, Yu R, Lee M, Liu H, Leeman DS, Huang Y, Webb AE, and Dang W. (2021). Altered Chromatin States Drive Cryptic Transcription in Aging Mammalian Stem Cells. Nat Aging 1, 684–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia EM, Cole LK, and Hatch GM (2014). Cardiolipin metabolism and the role it plays in heart failure and mitochondrial supercomplex formation. Cardiovasc Hematol Disord Drug Targets 14, 98–106. [DOI] [PubMed] [Google Scholar]
- Miller RA, Harrison DE, Allison DB, Bogue M, Debarba L, Diaz V, Fernandez E, Galecki A, Garvey WT, Jayarathne H, et al. (2020). Canagliflozin extends life span in genetically heterogeneous male but not female mice. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Scheibye-Knudsen M, Longo DL, and de Cabo R. (2015). Animal Models of Aging Research: Implications for Human Aging and Age-Related Diseases. Annual Review of Animal Biosciences 3, 283–303. [DOI] [PubMed] [Google Scholar]
- Mittal M, Siddiqui MR, Tran K, Reddy SP, and Malik AB (2014). Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20, 1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullee LI, and Morrison CG (2016). Centrosomes in the DNA damage response--the hub outside the centre. Chromosome Res 24, 35–51. [DOI] [PubMed] [Google Scholar]
- Nigg EA, and Holland AJ (2018). Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol 19, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo A, Reddy P, Martinez-Redondo P, Platero-Luengo A, Hatanaka F, Hishida T, Li M, Lam D, Kurita M, Beyret E, et al. (2016). In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 167, 1719–1733 e1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi K, Semi K, Yamamoto T, Shimizu M, Tanaka A, Mitsunaga K, Okita K, Osafune K, Arioka Y, Maeda T, et al. (2014). Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 156, 663–677. [DOI] [PubMed] [Google Scholar]
- Paradis E, and Schliep K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. [DOI] [PubMed] [Google Scholar]
- Park S, Park H-EH, Son HG, and Lee S-JV (2017). The role of RNA helicases in aging and lifespan regulation. Translational Medicine of Aging 1, 24–31. [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihan GA (2013). Centrosome dysfunction contributes to chromosome instability, chromoanagenesis, and genome reprograming in cancer. Front Oncol 3, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggiogalle E, Jamshed H, and Peterson CM (2018). Circadian regulation of glucose, lipid, and energy metabolism in humans. Metabolism 84, 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Brown GR, and Maglott DR (2012). NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res 40, D130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Fan J, Zheng R, Wan C, Mei S, Wu Q, Sun H, Brown M, Zhang J, Meyer CA, et al. (2020). Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol 21, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Research 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, and Ross OA (2018). Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front Immunol 9, 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, Wadi L, Meyer M, Wong J, Xu C, et al. (2019). Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat Protoc 14, 482–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Yang L, Zhu L, Xu X, Ceylan AF, Guo W, Yang J, and Zhang Y. (2017). Akt2 ablation prolongs life span and improves myocardial contractile function with adaptive cardiac remodeling: role of Sirt1-mediated autophagy regulation. Aging Cell 16, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuland DJ, Khademi S, Castle CJ, Irwin DC, McCord JM, Miller BF, and Hamilton KL (2013). Upregulation of phase II enzymes through phytochemical activation of Nrf2 protects cardiomyocytes against oxidant stress. Free Radic Biol Med 56, 102–111. [DOI] [PubMed] [Google Scholar]
- Rinaldi L, Avgustinova A, Martín M, Datta D, Solanas G, Prats N, and Benitah SA (2017). Loss of Dnmt3a and Dnmt3b does not affect epidermal homeostasis but promotes squamous transformation through PPAR-γ. eLife 6, e21697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux A, Zhang C, Paw J, Zavala-Solorio J, Vijay T, Kolumam G, Kenyon C, and Kimmel JC (2021). Partial reprogramming restores youthful gene expression through transient suppression of cell identity. bioRxiv, 2021.2005.2021.444556. [Google Scholar]
- Ruth KS, Day FR, Hussain J, Martínez-Marchal A, Aiken CE, Azad A, Thompson DJ, Knoblochova L, Abe H, Tarry-Adkins JL, et al. (2021). Genetic insights into biological mechanisms governing human ovarian ageing. Nature 596, 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu NM, and Kim JM (2019). Centrobin plays a role in the cellular response to DNA damage. Cell Cycle 18, 2660–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaum N, Lehallier B, Hahn O, Palovics R, Hosseinzadeh S, Lee SE, Sit R, Lee DP, Losada PM, Zardeneta ME, et al. (2020). Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583, 596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B, Pothof J, Vijg J, and Hoeijmakers JHJ (2021). The central role of DNA damage in the ageing process. Nature 592, 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senis E, Mosteiro L, Wilkening S, Wiedtke E, Nowrouzi A, Afzal S, Fronza R, Landerer H, Abad M, Niopek D, et al. (2018). AAVvector-mediated in vivo reprogramming into pluripotency. Nat Commun 9, 2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SP, Niemczyk M, Saini D, Sadovov V, Zimniak L, and Zimniak P. (2010). Disruption of the mGsta4 gene increases life span of C57BL mice. J Gerontol A Biol Sci Med Sci 65, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns SC (1989). Trade-Offs in Life-History Evolution. Functional Ecology 3, 259–268. [Google Scholar]
- Stolk L, Perry JR, Chasman DI, He C, Mangino M, Sulem P, Barbalic M, Broer L, Byrne EM, Ernst F, et al. (2012). Meta-analyses identify 13 loci associated with age at menopause and highlight DNA repair and immune pathways. Nat Genet 44, 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer JM, Winship A, Zerafa N, Wakefield M, and Hutt K. (2020). Oocytes can efficiently repair DNA double-strand breaks to restore genetic integrity and protect offspring health. Proc Natl Acad Sci U S A 117, 11513–11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437–1452 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulak M, Fong L, Mika K, Chigurupati S, Yon L, Mongan NP, Emes RD, and Lynch VJ (2016). TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Youle RJ, and Finkel T. (2016). The Mitochondrial Basis of Aging. Mol Cell 61, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Lee W, Mohri Y, Takeo M, Lim CH, Xu X, Myung P, Atit RP, Taketo MM, Moubarak RS, et al. (2019). A novel mouse model demonstrates that oncogenic melanocyte stem cells engender melanoma resembling human disease. Nature Communications 10, 5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabula Muris C. (2020). A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacutu R, Craig T, Budovsky A, Wuttke D, Lehmann G, Taranukha D, Costa J, Fraifeld VE, and de Magalhaes JP (2013). Human Ageing Genomic Resources: integrated databases and tools for the biology and genetics of ageing. Nucleic Acids Res 41, D1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacutu R, Thornton D, Johnson E, Budovsky A, Barardo D, Craig T, Diana E, Lehmann G, Toren D, Wang J, et al. (2018). Human Ageing Genomic Resources: new and updated databases. Nucleic Acids Res 46, D1083–D1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Tang D, Tao S, Chen Z, Koliesnik IO, Calmes PG, Hoerr V, Han B, Gebert N, Zornig M, Loffler B, et al. (2016). Dietary restriction improves repopulation but impairs lymphoid differentiation capacity of hematopoietic stem cells in early aging. J Exp Med 213, 535–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Firsanov D, Zhang Z, Cheng Y, Luo L, Tombline G, Tan R, Simon M, Henderson S, Steffan J, et al. (2019). SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell 177, 622–638 e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukui T, Sun K-H, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, Adams TS, Schupp JC, Poli SD, Rosas IO, et al. (2020). Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nature Communications 11, 1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. (2002). p53 mutant mice that display early ageing-associated phenotypes. Nature 415, 45–53. [DOI] [PubMed] [Google Scholar]
- Tyshkovskiy A, Bozaykut P, Borodinova AA, Gerashchenko MV, Ables GP, Garratt M, Khaitovich P, Clish CB, Miller RA, and Gladyshev VN (2019). Identification and Application of Gene Expression Signatures Associated with Lifespan Extension. Cell Metab 30, 573–593 e578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulgherait M, Midoun AM, Park SJ, Gatto JA, Tener SJ, Siewert J, Klickstein N, Canman JC, Ja WW, and Shirasu-Hiza M. (2021). Circadian autophagy drives iTRF-mediated longevity. Nature 598, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upham NS, Esselstyn JA, and Jetz W. (2019). Inferring the mammal tree: Species-level sets of phylogenies for questions in ecology, evolution, and conservation. PLoS Biol 17, e3000494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mierlo G, Veenstra GJC, Vermeulen M, and Marks H. (2019). The Complexity of PRC2 Subcomplexes. Trends Cell Biol 29, 660–671. [DOI] [PubMed] [Google Scholar]
- Wang J, Gu BJ, Masters CL, and Wang YJ (2017). A systemic view of Alzheimer disease - insights from amyloid-beta metabolism beyond the brain. Nat Rev Neurol 13, 703. [DOI] [PubMed] [Google Scholar]
- Wei K, Clark AB, Wong E, Kane MF, Mazur DJ, Parris T, Kolas NK, Russell R, Hou H Jr., Kneitz B, et al. (2003). Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev 17, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh M, Henpita C, Vyas R, Soto-Palma C, Robbins P, and Niedernhofer L. (2021). DNA damage—how and why we age? eLife 10, e62852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Tombline G, Ablaeva J, Zhang L, Zhou X, Smith Z, Zhao Y, Xiaoli AM, Wang Z, Lin J-R, et al. (2021). Genomic expansion of Aldh1a1 protects beavers against high metabolic aldehydes from lipid oxidation. Cell Reports 37, 109965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Qu J, Liu GH, and Belmonte JCI (2020). The ageing epigenome and its rejuvenation. Nat Rev Mol Cell Biol 21, 137–150. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Seluanov A, and Gorbunova V. (2021). Revelations About Aging and Disease from Unconventional Vertebrate Model Organisms. Annu Rev Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Dou Q, Fan G, Zhang Q, Sanderford M, Kaya A, Johnson J, Karlsson EK, Tian X, Mikhalchenko A, et al. (2020). Beaver and Naked Mole Rat Genomes Reveal Common Paths to Longevity. Cell Rep 32, 107949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Statistics of RNA-seq samples. Related to Figure 1.
Table S5 CMap interventions. Related to Figure 4.
Data S1. Unprocessed data underlying the display items in the manuscript, related to Figures 1–7 and S1–S7
Data Availability Statement
Raw sequencing reads are available in the NCBI Gene Expression Omnibus (GEO) and Sequence Read Archive (SRA) under accession numbers GSE190756.
Original data for creating all graphs in the paper are provided in Data S1.
This study does not generate any new codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.