

Abstract
Neuropilin-1 (NRP1) is a widely expressed cell surface receptor protein characterized by its pleiotropic function. Recent reports highlighted NRP1 as an additional entry point of the SARS-CoV-2 virus, enhancing viral infectivity by interacting with the S-protein of SARS-CoV-2. The ubiquitous distribution and mechanism of action of NRP1 enable the SARS-CoV-2 virus to attack multiple organs in the body simultaneously. Therefore, blocking NRP1 is a potential therapeutic approach against SARS-CoV-2 infection. The current study screened the South African natural compounds database (SANCDB) for molecules that can disrupt the SARS-CoV-2 S protein-NRP1 interaction as a potential antiviral target for SARS-CoV-2 cellular entry. Following excessive screening and validation analysis 3-O-Methylquercetin and Esculetin were identified as potential compounds to disrupt the S-protein-NRP1 interaction. Furthermore, to understand the conformational stability and dynamic features between NRP1 interaction with the selected natural products, we performed 200 ns molecular dynamics (MD) simulations. In addition, molecular mechanics-generalized Born surface area (MM/GBSA) was utilized to calculate the free binding energies of the natural products interacting with NRP1. 3-O-methylquercetin showed an inhibitory effect with binding energies ΔG of −25.52 ± 0.04 kcal/mol to NRP1, indicating the possible disruption of the NRP1-S-protein interaction. Our analysis demonstrated that 3-O-methylquercetin presents a potential antiviral compound against SARS-CoV-2 infectivity. These results set the path for future functional in-vitro and in-vivo studies in SARS-CoV-2 research.
Keywords: Virtual screening, SARS-CoV-2, S-Protein, Neuropilin-1, Natural product, Molecular dynamic simulation
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), responsible for the COVID-19 pandemic, is an RNA-enveloped virus highly transmissible by air droplets [1]. The SARS-CoV-2 genome encodes four structural proteins, including glycosylated spike (S), envelope (E) (required to infiltrate host cells), M (membrane), and N (nucleocapsid) proteins. Whereby the SARS-CoV-2 viral infection mainly affects the upper respiratory tract through binding of the spike (S) protein to the angiotensin-converting-enzyme 2 (ACE2) receptor [2,3]. However, SARS-CoV-2 has demonstrated a tropism characteristic of infection, where co-receptors or additional proteins can act as mediators for viral entry to multiple tissues/organs [[4], [5], [6], [7]]. The proteins that interact with SARS-CoV-2 S-protein acting as cellular entry mediators include the receptor for advanced glycation end products (RAGE), glucose-regulated protein 78 (GRP78), and angiotensin II receptor type 2 (AGTR2), CD147, heparan sulfate, and neuropilin-1 (NRP1) [6].
Neuropilin-1 (NRP1) is a highly conserved non-tyrosine kinase surface glycoprotein encoded by the NRP1 gene that functions as a cell surface receptor with a pleiotropic role in cellular signaling identified as a neuronal adhesion molecule involved in Semaphorin-mediated axonal guidance [8]. NRP1 acts as a receptor to Epstein-Barr virus (EBV) infection of nasopharyngeal epithelial cells [9] and the human T cell lymphotropic virus type 1 (HTLV-1) [10]. Recent studies reported the involvement of NRP1 in SARS-CoV-2 cell entry and infectivity [[11], [12], [13]]. NRP1 demonstrated higher expression levels than ACE2 in pulmonary, olfactory, and endothelial cells, contributing to the multisystem effect caused by SARS-CoV-2. Moreover, cells of patients infected with SARS-CoV-2 demonstrated elevated levels of RNA expression of NRP1 [11]. In comparison, cells isolated from bronchoalveolar lavage fluid (BALF) of patients with severe COVID-19 were highly enriched with NRP1 [14]. In a recent report involving Alzeimer's disease (AD) patients, ACE2 and NRP1 were preferentially expressed in the brain, and their expression levels determined the sensitivity to SARS-CoV-2 infection [15]. NRP1 gene expression was upregulated with increased AD severity [15], whereas the expression of the ACE2 gene showed a gradual increase linked to the severity of AD symptoms [16]. Indicating that NRP1 is a significant risk factor for SARS-CoV-2 infection in patients with AD [16]. In addition to being a cofactor that contributes to SARS-CoV-2 infecting host cells in the central nervous system (CNS), NRP1 may also stimulate intracellular signaling pathways that are associated with pathological complications related to CNS and glioblastoma [7,[17], [18], [19]]. Thus, the detected upregulation of NRP1 might contribute to the long-term neurological complications specifically in people who have contracted several SARS-CoV-2 infections [20].
NRP1 comprises a long N-terminal extracellular domain, a transmembrane domain, and a short cytosolic tail of 43–44 amino acids. The extracellular region consists of five domains that are critical for ligand binding, which involve a pair of calcium-binding (CUB) domains (a1/a2), two coagulation factor V/VIII-like discoidin domains (b1b2), and a Meprin/A5-Antigen/pto-Mu (MAM) domain (c) [21]. A critical binding site in NRP1 is a core conserved binding pocket formed by b1 interloop cleft. The binding pocket is specific for ligands with a C-terminal arginine residue [8,22], conforming to the C-end rule (CendR) [23]. As such, NRP1 can bind peptides/molecules with the [R/K]XX[R/K] motif, where X can be any amino acid at the carboxyl terminus of the peptide/protein.
SARS-CoV-2 infection is facilitated by cleaving the S protein at S1/S2 multi-arginine residue (RRAR) cleavage site targeted by furin [3,[24], [25], [26]]. Furin-mediated proteolytic activity exposes the conserved C-terminal RRAR motif in the S1 protein to the ligand-specific binding site in NRP1-expressing cells [8,23,27], where the ligand-specific NRP1-b1 interloop cleft interacts with the SARS-CoV-2 S-protein exposed RARR site. Daly et al. identified SARS-CoV-2 S-protein S1 residue R685 as the key residue for interacting with NRP1-b1, whereby they demonstrated that S1 CendR peptide (679NSPRRAR685) has a micromolar affinity (20.3 μM) to the NRP1-b1 domain. The NRP1-b1 binding to S1-679NSPRRAR685 was further corroborated by using monoclonal antibodies to block the interaction and consequently reducing the efficiency of SARS-CoV-2 infection [12]. Additionally, deleting the CendR motif reduced the S1-NRP1 association, and NRP1 depletion reduced SARS-CoV-2 uptake to half compared to control cells, indicating the role of NRP1 in facilitating SARS-CoV-2 entry into host cells [28,29]. SARS-CoV-2 infectivity was also partially blocked in an ex-vivo placental explant with anti-NRP1 and anti-ACE2 antibodies [30]. As a cofactor facilitating SARS-CoV-2 infection, NRP1 is becoming an attractive drug target for developing new therapeutic agents for COVID-19 treatment [31,32]. The role of NRP1 as a cofactor for SARS-CoV-2 infection was also shown in circulating plasmacytoid dendritic cells (pDCs). On pDCs the SARS-CoV-2 engages NRP1/CD304 to mitigate the antiviral interferon response, which hampers the immunological response after viral sensing. Additionally, removing the expression of surface NRP1/CD304 from pDCs reduced the inhibitory effect of SARS-CoV-2 on the antiviral response, permitting the induction of IFNα production [33]. Since SARS-CoV-2 infection forms an additional cellular entry position by S-protein interacting with NRP1, blocking the S1-679NSPRRAR685 interactions with NRP1-b1 with an antiviral drug would hinder virus infection of host cells.
In the present study, we screened the South African natural compounds database (SANCDB (https://sancdb.rubi.ru.ac.za/), [34] and employed molecular dynamic simulations to identify potential NRP1 antagonists. The SANCDB database enlists natural products with medicinal properties such as antiviral, anti-plasmodial, and anti-malarial effects. Following vigorous screening and validation analysis, we identified two natural products with the highest binding affinity to NRP1-b1 key residues, Y297, W301, T316, D320, S346, T349, and Y353. From the SANCDB database, two compounds Esculetin and 3-O-Methylquercetin were selected as compounds to block NRP1-b1 S-protein interaction. The conformational stability and dynamic features of NRP1-b1 bound to the two selected compounds were tested by subjecting each complex to 200 ns molecular dynamic (MD) simulations. In addition, molecular mechanics-generalized Born surface area (MM/GBSA) to extract free binding energies. Of the two compounds, 3-O-methylquercetin demonstrated the most stable complex with NRP1-b1, in addition to a tighter binding affinity (−25.52 ± 0.04 kcal/mol), indicating the ability to disrupt the NRP1-S-protein interaction. This study provides a basis for novel drug development targeting the NRP1-Spike interface to treat COVID-19.
2. Materials and methods
2.1. Structures retrieval, filtration, and preparation
The Protein Databank ((http://www.rcsb.org/) was used to retrieve the X-ray crystal structure of NRP1 (PDB ID:7JJC) [12,35]. The NRP1 structure was prepared and minimized using Chimera and AMBER simulation package using FF14SB force field [36,37]. To screen targets against ligand-specific NRP1 b1 interloop, we utilized compounds from the South African Natural Products database (SANCDB) (https://sancdb.rubi.ru.ac.za/) [34].
Prior to structure-based molecular search to identify potential hits against NRP1 from the SANCDB database. Using the FAFDrugs4 server, the SANCDB database compounds were screened for drug-like structures and toxicity [38]. The non-toxic compounds were subjected to further screening to assign appropriate stereochemistry, ring conformations, tautomer, and ionization state for NRP1 using PyRx software [39].
2.2. Virtual drugs screening and rescoring of the top hits
Identification of potential compounds from SANCDB database through virtual screening approach using AutoDock Vina. The docking site was defined based on the experimentally reported residues of NRP1 Y297, W301, T316, D320, S346, T349, and Y353 [40]. A grid box of 6.04 x −70.97 × 24.028 and grid dimensions of 56 Å × 58 Å × 50 Å were generated based on the residues above. The docking of the compounds underwent a multi-step screening process whereby in the first step, the exhaustiveness was set to 16. The compounds that passed the first step underwent the second screening with exhaustiveness set to 32. Finally, using induced-fit docking (IFD) protocol, the top hits were screened again using 64 exhaustiveness to validate the best hits and remove any false-positive results as performed in previous work [41]. AutoDockFR–AutoDock for Flexible Receptors (ADFR) [42] was employed for the IFD docking, which utilizes the scoring function of AutoDock4 to boost the success frequency of docking. A cross-validation test revealed that AutoDockFR has higher accuracy than AutoDock Vina and faster docking speed. The top hits from AutoDock Vina results were subjected to binding affinity analysis and MD simulations.
2.3. Bioactivity and dissociation constant (KD) determination
Molinspiration is a cheminformatic tool (https://www.molinspiration.com/cgi-bin/properties) predicting the bioactivity and IC50 of each compound. PRODIGY (PROtein binDIng enerGY prediction) was adopted to compute the KD values for the top hit compound to provide a knowledge of the inhibitory potential of each selected compound [43].
2.4. Molecular dynamics simulation of protein-ligand complexes
To explore the dynamic behavior of the top hits obtained from SANCDB database AMBER20 simulation package was used to run molecular dynamic (MD) simulations [44]. Drug topologies were generated through an antechamber using Amber general force field (GAFF). For the simulation, ff14SB forcefield simulated each solvated and neutralized complex using TIP3P water box and Na+ ions. The energy-minimization of systems was carried out in two stages with 12000 and 6000 conjugate gradient energy minimization cycles to relax the complexes and remove the bad clashes, followed by heating and equilibration. Long-range electrostatic interactions were quantified using the Particle Mesh Ewald (PME) method [45] while keeping 1.4 nm cut-off for van der Waals interactions, and Columbic interactions of short-range were fixed. Constant volume and temperature (NVT) ensemble and pressure and temperature (NPT) ensemble was equilibrated and set out at 1-bar pressure for 100 ps. Langevin thermostat at 300 K for constant temperature while Berendsen barostat was used for pressure control. A simulation time of 200 ns with time step 2fs for each protein-ligand complex was achieved. Structural-dynamics features such as stability, compactness, flexibility, and other features of these ligand-protein complexes were assessed by CPPTRAJ and PTRAJ [46].
2.5. The binding free energy calculations
Estimation of binding free energy determines the true inhibitory potential of any therapeutic agent. Hence, the binding free energy of protein-ligand complexes was computed using the script MMGBSA.PY by considering 10000 snapshots [[47], [48], [49], [50]]. This method of calculating free energy is extensively used to estimate the TBE of various ligands reported by various studies [51,52].
| (1) |
Here, ΔGbind denotes total free binding energy, while others denote the free energy of the protein, the ligand, and complex. The following equation was used to calculate specific energy term contribution to the total free energy:
| (2) |
In equation (2), Gbond, Gele, and GvdW denote bonded, electrostatic, and van der Waals interactions. G-pol and Gnpol are polar and nonpolar solvated free energies. The Gpol and Gnpol are calculated using the GB implicit solvent method with the SA term.
3. Results and discussion
Neuropilin-1 (NRP1) is a 130 kDa protein comprised of an 850 amino acid N-terminal domain, a 24-residue short membrane-spanning domain, and a cytoplasmic domain (∼40 residues). The NRP1 ectodomain includes five individual structural motifs (a1, a2, b1, b2, and c). Recently it was demonstrated that NRP1-b1 binds to the SARS-CoV-2-S-protein S1 domain at the furin S1-679NSPRRAR685 site (Fig. 1 ). Consequently, resulting in NRP1 being a co-receptor for SARS-CoV-2 cell entry and infectivity [11,12]. As such, disrupting the S-protein S1-679NSPRRAR685 interaction with NRP1 could protect against viral infection. In our study, we utilized the X-ray crystal structure of NRP1 (PDB ID:7JJC) (Fig. 1) to identify natural compounds from the SANCDB that can disrupt the S1-679NSPRRAR685 interactions with NRP1 [12]. In addition, the NRP1 residues positioned in the interaction interface with S1-679NSPRRAR685 were identified using a script InterfaceResidues.py and the protein structural analysis software PyMOL (Fig. 1B), which corroborated with the experimentally reported residues of NRP1-b1 Y297, W301, T316, D320, S346, T349, and Y353 by Daly et al. (Fig. 1B).
Fig. 1.

A) The interaction between NRP1-b1 (blue) to S-protein S1 domain (salmon). B) The NRP1-b1 interface residues Y297, W301, T316, D320, S346, T349, and Y353 are depicted in cyan and the S-protein S1 binding 679-NSPRRAR-685 site in salmon.
3.1. Virtual drug screening and molecular docking
The natural compounds database SANCDB screened 1012 compounds as potential inhibitors for the NRP1-b1 interactions interface, where 1004 compounds passed the ADMET analysis criteria [53]. The 1004 compounds underwent virtual screening with AutoDock Vina with docking scores ranging from −4.74 to −1.96 kcal/mol. Furthermore, the virtual screening cut-off criteria were set to −4.0 kcal/mol to shortlist the highly efficient binders, where 55 compounds produced docking scored higher than −4.5 kcal/mol when interacting with the NRP1 binding interface. The screening of the 55 compounds using the IFD method resulted in narrowing it down to six compounds with docking scores ranging from −5.91 to −4.17 kcal/mol (Table 1 ). Based on the defined criteria of docking score lower than −5.0 kcal/mol, only two compounds, 3-O-methylquercetin and Esculetin, fitting this criterion, were selected to analyze the interaction with the critical residues in the NRP1-b1 binding interface with S-protein S1-679NSPRRAR685. Furthermore, 3-O-methylquercetin and Esculetin complexes with NRP1-b1 underwent MD simulations to measure their conformational dynamics and stability.
Table 1.
Top hits identified through multi-steps screening and rescoring via IFD method. The table shows the 2D structure, the SANCDB ID, name, and docking scores of the top six hits.
| 2D structure | SANCDB ID | Compound Name | IFD Scores |
|---|---|---|---|
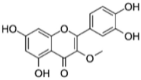 |
SANCDB00980 | 3-O-methylquercetin | −5.91 |
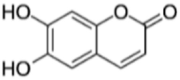 |
SANCDB00756 | Esculetin | −5.64 |
 |
SANCDB00361 | 4′-O-Demethyleucomol | −4.92 |
 |
SANCDB00130 | Makaluvic acid A | −4.89 |
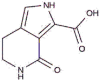 |
SANCDB00256 | Makaluvic acid C | −4.86 |
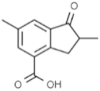 |
SANCDB00332 | 2,6- Dimethyl-1-oxo-4-indanecarboxylic acid | −4.17 |
3.2. Validation of molecular docking of selected ligands
In the case of NRP1, there is no pre-co-crystal data with a ligand. Therefore, to validate the docking procedure of the selected ligands to the NRP1-b1 druggable target. We targeted the interface by considering the previously reported residues [52] Y297, W301, T316, D320, S346, T349, and Y353, which are the RARR-furin binding site of SARS-CoV-2 S-protein reported in the X-ray crystal structure (PDB ID:7JJC) [12]. As a reference, the 2.36 Å-resolution X-ray crystal structure of the SARS-CoV-2 S-protein (PDB ID:7JJC) was superimposed with the docked structures of NRP1-b1 bound to 3-O-methylquercetin and Esculetin complexes (Fig. 2 A and B). From 10 conformers with the highest docking scores of NRP1-b1 bound to 3-O-methylquercetin and Esculetin demonstrated an RMSD of 1.05 and 1.03 Å, respectively, which is considered a valid docking score since the RMSD is lower than 2 Å [54,55].
Fig. 2.

The interaction between the docked structures of NRP1-b1 (blue) bound to (A) Esculetin and (B) 3-O-methylquercetin to form complexes with NRP1-b1.
3.3. Binding mode of 3-O-methylquercetin
3-O-methylquercetin is a flavonoid used as a natural product against numerous medical conditions. Which include anti-inflammatory, antioxidant, neuroprotective, bronchodilatory, vasodilatory, antinociceptive, immunomodulatory, antitumor, antiviral diseases. The protective effects of 3-O-Methylquercetin was demonstrated in human lungs and liver exposed to H2O2-induced cytotoxicity [56]. Pretreating lung and liver cells with 3-O-Methylquercetin prevented early symptoms of H2O2-induced anchorage dependency cell death, such as detachment and shrinkage of cells. This compound also attenuated the formation of reactive oxygen species (ROS) induced by H2O2, which protected cells from ROS-induced decrease in mitochondrial membrane potential and DNA fragmentation/damage, which are indicators of stress-induced apoptotic cell damage [56]. The protective effect of 3-O-Methylquercetin involved reducing caspase-3 activity by 48%. Additionally, exposing cells to this enhanced antioxidant expression of catalase and super oxide dismutase-2 in cells by 50%, suggesting its role as a cytoprotective agent mediating cell survival [56]. Furthermore, 3-O-methylquercetin has been tested against human influenza type A and multi-drug resistant strains of Mycobacterium tuberculosis [57]. The docking of 3-O-Methylquercetin against NRP-1 demonstrated a −5.91 kcal/mol score with four hydrogen bonds (H-bonds), one π-π interaction, and one salt-bridge. 3-O-methylquercetin formed H-bonds with S298, N300, D320, and Y353 are involved (Fig. 3 ). While residue W301 established p-p interaction and K351 formed the only salt bridge with 3-O-Methylquercetin. Moreover, 3-O-methylquercetin possesses a significant biological activity with a bioactivity score of 0.23 (Table 2 ), and a K d of 7.8 × 10−7 M, thus confirming this compound's strong inhibitory potential against the NRP1-b1 binding to S-protein S1 RARR site. In addition, 3-O-methylquercetin showed no violations to Lipinski rule [58], implying that it is a candidate compound that is more likely to be orally active and comply with the solubility and permeability requirements.
Fig. 3.

Binding mode of 3-O-methylquercetin. The left panel shows the surface representation of NRP1 and the binding conformation of 3-O-methylquercetin, while the right panel shows the interaction pattern of 3-O-methylquercetin with NRP1-b1. The red dashed lines represent hydrogen bonds in the left panel, and the blue lines represent salt bridges.
Table 2.
ADMET analysis, biological and toxicology properties.
| Compound Name | MW. | Source | Molecule Class | Biological Activity |
Lipinski Violation | AMESa Toxicity | Rat Oral LD50 (mol/kg) | Max. tolerated Dose (human) (log Mg/Kg/day) |
IC50 In vitro (μM) |
T.Pyriformis Toxicity ** (log μg/L) | HBD | HBA | Rotatable Bonds No. | TPSA | Bioactivity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3-O-Methylquercetin | 316.26 | Cheirolophus mauritanicus | Flavonoid | Antiviral | 0 | YES | 2.54 | 0.629 | 0.08 | 0.287 | 4 | 7 | 2 | 120.36 Å2 | 0.23 |
| Esculetin | 178.14 | Taeckholmia regis-jubae | Coumarin | Antioxidant | 0 | No | 2.13 | 0.034 | 25.18 | 0.467 | 2 | 4 | 0 | 70.67 Å2 | −0.22 |
AMES toxicity test, in-vitro testing to assess the potential carcinogenic effect of chemicals. ** Tetrahymena pyriformis, the most commonly ciliated model, used for toxicological studies.
3.4. Binding mode of esculetin
In traditional Chinese medicine, Esculetin is a derivative of coumarin isolated from Cortex Fraxini, possessing pharmacological activities against various diseases [59]. Esculetin has demonstrated anti-proliferative and anti-oxidative effects [60] and displayed an inhibitory activity on aldose reductase to deter galactose-induced cataractogenesis [61]. Esculetin was shown to alleviate H2O2-induced oxidative damage of lung fibroblasts in animals, and 0.1–10 μg/ml of esculetin displayed a protective effect against lipid peroxidation and DNA damage [62]. The compound demonstrated anti-inflammatory activity in human nasal epithelial cells, whereby using esculetin (10–40 μM) inhibited the production of inflammatory cytokines (IL-6 and IL-8) through the suppression of the NF-KB signaling pathway [63]. In another report, esculetin attenuated the histopathological change of lung, pulmonary wet-to-dy weight ratio, the infiltration of inflammatory cells and the generation of inflammatory cytokines in lung epithelial cells and mouse model studies [62]. In addition, computational studies using coumarin derivatives have blocked the interaction between the SARS-CoV-2 S-protein receptor-binding domain (RBD) and ACE2, revealing their inhibitory role against various SARS-CoV-2 targets [[64], [65], [66]]. The docking of Esculetin against NRP1-b1 (Fig. 4 ) demonstrated a score of −5.64 kcal/mol (Table 1) with three hydrogen bonds, including W301 and D320, present in the NRP1-b1 interaction interface with S-protein S1. On the other hand, with the bioactivity score of −0.22 and K d 5.3 × 10−7 M, Esculetin presented the potential of being a potent inhibitor that could abrogate the binding of S-protein to NRP1. Although ADMET analysis presented Esculetin with no AMES toxicity and no violations of Lipinski rule [58], the compound predicted a reduced bioactivity score (Table 2) compared to 3-O-methylquercetin.
Fig. 4.

Binding mode of Esculetin. The left panel shows the surface representation of NRP1 and the binding conformation of Esculetin, while the right panel shows the interaction pattern of Esculetin with NRP-b1.
3.5. Structural dynamic features of the NRP1-ligands complexes
The conformational stability and dynamic environment of NRP1-b1 bound to 3-O-methylquercetin and Esculetin were elucidated by running 200 ns MD simulations of the complexes. The root-mean-square deviation (RMSD) trajectories of the Cα-atoms demonstrated the dynamic stability and convergence of each system (Fig. 5 ), whereby the root mean square fluctuations (RMSF) of the Cα-atoms demonstrated the residual flexibility of each compound interacting with the NRP1-b1 interface (Fig. 7). The effect of 3-O-methylquercetin and Esculetin on binding with NRP1-b1 was confirmed by applying the MM/GBSA method to calculate the total binding free energy [66], as shown in Table 3 . The stability of the NRP1-b1 structure in complex with 3-O-methylquercetin and Esculetin extracted from the MD simulation trajectories at various simulation time scales shows that the potential hits remained stable during the simulation during the time intervals (Supplementary Fig. 1).
Fig. 5.

Dynamic stability of 3-O-methylquercetin-NRP1 and Esculetin-NRP1 complexes. (A) the structural stability of 3-O-methylquercetin-NRP1 complex, while (B) shows the Esculetin-NRP1 complex.
Fig. 7.

Root mean square fluctuations of 3-O-methylquercetin-NRP1 (orange) and Esculetin-NRP1 complexes (green). The x-axis shows the number of residues, and the y-axis shows the RMSF in Å.
Table 3.
The MM/GBSA binding free energy variables (Kcal/mol) of.
| Complexes | vdW | Electrostatic | GB | SA | ΔGbind |
|---|---|---|---|---|---|
| 3-O methylquercetin | −31.10 ± 0.046 | −2.91 ± 0.037 | 12.00 ± 0.036 | −3.50 ± 0.003 | −25.52 ± 0.04 |
| Esculetin | −22.49 ± 0.04 | −3.80 ± 0.06 | 9.32 ± 0.05 | −2.61 ± 0.05 | −19.59 ± 0.04 |
The 3-O-methylquercetin-NRP1 complex demonstrated a stable behavior during the 200ns simulation, with no drastic dynamic deviation. During the initial 12.5 ns, the 3-O-methylquercetin-NRP1 complex converged from 0.8 to 1.6 Å, after which the RMSD returned to 1.1 Å at 25 ns? The system stabilized at 1.1 Å from 25 to 75 ns, and then 3-O-methylquercetin-NRP1 complex experienced a dynamic fluctuation with an increase in RMSD from 1.1 to 1.6 Å from 75 to 85 ns? From 75 to 200 ns, the RMSD of the 3-O-methylquercetin-NRP1 complex averaged at 1.1 Å with a slight deviation at 140 ns? The overall stability of the 3-O-methylquercetin-NRP1 complex correlates with the higher IFD docking score of −5.91 kcal/mol and K d = 7.8 × 10−7 M. Furthermore, 3-O-methylquercetin forms a strong interaction with NRP1 due to the five H-bonds with the binding interface residues of NRP1. Furthermore, the conformational change of 3-O-methylquercetin interacting with NRP1 (Fig. 6A and S1) presented a stable complex with the RMSD fluctuating between 0.2 and 0.4 Å during the 200 ns simulation. 3-O-methylquercetin fluctuated to 0.4 Å the first 40 ns, after which the compound stabilized at 0.2 Å for the remainder of the simulation, further corroborating the stability of the 3-O-methylquercetin-NRP1 complex.
Fig. 6.

RMSD conformations 3-O-methylquercetin and Esculetin compounds interacting with NRP1. (A) the structural stability of 3-O-methylquercetin, while (B) shows the Esculetin complex.
On the other hand, the Esculetin-NRP1 complex demonstrated an unstable behavior compared to the 3-O-methylquercetin-NRP1 complex. The initial 20 ns of the Esculetin-NRP1 complex showed a very dynamic behavior whereby the RMSD decreased from 1.6 to 0.8 Å from 0 to 5 ns, following an increase to 2.4 Å from 5 to 10 ns? From 10 to 20 ns, the RMSD decreased to 0.8 Å for 5 ns? For the remainder of the 180 ns, the RMSD gradually fluctuated to 2.4 Å at 200 ns? The less stable Esculetin-NRP1 complex may be attributed to the weaker IFD docking and binding affinity. In addition, Esculetin only formed three H-bonds with NRP1 residues, and the interaction was not as stable as the 3-O-methylquercetin-NRP1 complex. The smaller molecular weight of Esculetin would result in a more significant conformational change to fit the binding pocket of NRP1-b1. The conformational change is further depicted in Fig. 6 B, with the RMSD of Esculetin interacting with NRP1, averaging an RMSD of 0.2 Å for the initial 40 ns, after which the compound fluctuated with an average RMSD of 0.4–0.5 Å for the remainder of the 200 ns simulation indicating a higher conformational change in comparison to3-O-methylquercetin.
3.6. Investigating residues fluctuations
To further understand the fluctuation of 3-O-methylquercetin-NRP1 and Esculetin-NRP1 complexes at the residue level, RMSF (root-mean-square fluctuation), these two systems were calculated (Fig. 7). The NRP1-b1- Esculetin complex demonstrates higher fluctuations in the interface residues (Fig. 7 green) than the 3-O-methylquercetin-NRP1-b1 complex. The higher fluctuations may result from the higher conformational sampling of Esculetin in the NRP1-b1 binding pocket, which corroborates with higher RMSD fluctuations. Overall, our findings have shown a very low mean RMSF, demonstrating the residues of NRP1-b1 in complex with 3-O-methylquercetin and Esculetin conforming to favorable energy minima [67]. The current findings are consistent with previous findings where low RMSF for the best compounds was reported when interacting with SARS-CoV-2 proteins [40].
3.7. Structural compactness evaluation
The radius of gyration (Rg) evaluates the structural compactness 3-O-methylquercetin and Esculetin-NRP1 complexes as a function of time (Fig. 8 ). The structural compactness of the interacting partners reveals essential information regarding the binding and unbinding events during the MD simulation [68]. Thus, our analysis revealed that the Rg results significantly correlate with the RMSD findings. The 3-O-methylquercetin-NRP1 complex compared to the Esculetin-NRP1 complex demonstrated more compact behavior during the simulation. The average Rg value for the 3-O-methylquercetin-NRP1 complex demonstrated an average RMSD of 15.1 Å (Fig. 5A), while the Esculetin-NRP1 complex showed an average Rg RMSD of 15.3 Å (Fig. 5B).
Fig. 8.

Structural compactness of 3-O-methylquercetin-NRP1 and Esculetin-NRP1 complexes. (A) the structural compactness of 3-O-methylquercetin-NRP1 complex, while (B) show the Esculetin-NRP1 complex.
Consequently, the 3-O-methylquercetin-NRP1-b1 complex compactness demonstrated by the Rg results corroborates the stability shown in the RMSD and RMSF data. These findings suggest that both complexes remained more compact, which ensured the stable binding and blockage of the critical residues of the interface. Although binding and unbinding events occurred during the 200 ns simulation, the ligands occupied the cavity robustly [69].
3.8. Binding free energy calculations
In a structure-based drug design, determining the binding free energies of a protein-ligand complex is a validating approach to re-evaluate the docking results [70]. In the current study, using MM/GBSA, the simulation trajectories consisting of 20000 frames were used to calculate the binding free energy. The calculation of binding free energies by MM/GBSA is widespread because it is more robust than the classical docking scores and less costly than the Alchemical free energy techniques. Our findings revealed vdW (−31.10 ± 0.046), electrostatic (−2.91 ± 0.037), GB (12.00 ± 0.036), SA (−3.50 ± 0.003), and the total binding energy DG (−25.52 ± 0.04) kcal/mol was reported for the 3-O-methylquercetin-NRP1 complex. On the hand, for Esculetin-NRP1 complex vdW (−22.49 ± 0.04), electrostatic (−3.80 ± 0.06), GB (9.32 ± 0.05), SA (−2.61 ± 0.05) and the total binding energy (−19.59 ± 0.04) kcal/mol was reported.
The higher vdW energies of 3-O-methylquercetin interactions with NRP1-b1 compared to Esculetin can result from a higher LogP for 3-O-methylquercetin (1.96) than Esculetin (1.02). The higher logP is attributed to higher lipophilicity, which supports by higher vdW forces. In addition to lipophilicity, the ligand geometries play an essential role in the vdW forces of protein-ligand interactions, affecting the total binding energy. On the other hand, the Esculetin-NRP1 complex demonstrated higher electrostatic energies due to the three electrostatic interactions formed with the NRP1-b1 binding interface (Fig. 4).
In general, both MM/GBSA results conclude that 3-O-methylquercetin and Esculetin interact robustly with interface residues of NRP1-b1 and may block the interactions with S-protein S1 domain. Furthermore, the 3-O-methylquercetin interactions with NRP1-b1 interface presented a ΔG -25.52 ± 0.04 kcal/mol (Table 3), which is 40 fold higher binding than Esculetin-NRP1 complex, since an increase in 1.4 kcal/mol equals a ten-fold increase in total binding energy [71]. The tighter 3-O-methylquercetin binding with NRP1 fits a more stable RMSD and RG results (Fig. 8). In addition, the higher number of H-bonds (five) present between 3-O-methylquercetin and the NRP1-b1 interface residues play a crucial role in protein-ligand stability demonstrated by the strong interactions and higher free binding energy.
4. Conclusions
The SARS-CoV-2 virus has demonstrated its ability to interact with multiple cellular receptors through the S-protein, including the receptor for advanced glycation end products (RAGE), glucose-regulated protein 78 (GRP78), angiotensin II receptor type 2 (AGTR2), CD147, heparan sulfate, and neuropilin-1 (NRP1). Subsequently, the multiple-entry sites of the SARS-CoV-2 have made it harder to design specific therapeutics to treat the disease. Here in this study, we describe two natural compounds from the SANCDB database that can target the interaction interface of the SARS-CoV-2 NRP1-S-protein complex. These natural products displayed high binding affinities to the interaction interface residues of NRP1, henceforth can disrupt the formation of the NRP1-S-protein complex in SARS-CoV-2.
Of the identified compounds, 3-O-methylquercetin presented a better IFD score of −5.91 kcal/mol and total binding energy of −25.52 ± 0.04 kcal/mol. Additionally, the 3-O-methylquercetin-NRP1 complex demonstrated more compact behavior during the simulation. Combined with its bioactive characteristics and no Lipinski rule violations, these results make 3-O-methylquercetin an attractive therapeutic candidate for SARS-CoV-2 infection. Although further functional in-vitro and in-vivo studies are required, we propose 3-O-methylquercetin as a natural product inhibiting NRP1-S-protein formation in SARS-CoV-2 infection.
Funding
Kuwait Foundation for the Advancement of Sciences
CRediT authorship contribution statement
Eman Alshawaf: Writing – original draft, Formal analysis, Conceptualization. Maha M. Hammad: Writing – original draft, Formal analysis, Conceptualization. Sulaiman K. Marafie: Writing – original draft. Hamad Ali: Writing – original draft. Fahd Al-Mulla: Writing – review & editing. Jehad Abubaker: Writing – review & editing, Conceptualization. Anwar Mohammad: Methodology, Formal analysis, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.micpath.2022.105701.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Leung N.H.L. Transmissibility and transmission of respiratory viruses. Nat. Rev. Microbiol. 2021;19(8):528–545. doi: 10.1038/s41579-021-00535-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shang J., et al. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA. 2020;117(21):11727–11734. doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walls A.C., et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zamorano Cuervo N., Grandvaux N. ACE2: evidence of role as entry receptor for SARS-CoV-2 and implications in comorbidities. Elife. 2020;9 doi: 10.7554/eLife.61390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies J., et al. Neuropilin-1 as a new potential SARS-CoV-2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID-19. Mol. Med. Rep. 2020;22(5):4221–4226. doi: 10.3892/mmr.2020.11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyrou I., et al. Not only ACE2—the quest for additional host cell mediators of SARS-CoV-2 infection: neuropilin-1 (NRP1) as a novel SARS-CoV-2 host cell entry mediator implicated in COVID-19. Signal Transduct. Targeted Ther. 2021;6(1):21. doi: 10.1038/s41392-020-00460-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veleri S. Neurotropism of SARS-CoV-2 and neurological diseases of the central nervous system in COVID-19 patients. Exp. Brain Res. 2022;240(1):9–25. doi: 10.1007/s00221-021-06244-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo H.-F., Vander Kooi C.W. Neuropilin functions as an essential cell surface receptor. J. Biol. Chem. 2015;290(49):29120–29126. doi: 10.1074/jbc.R115.687327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H.B., et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 2015;6:6240. doi: 10.1038/ncomms7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lambert S., et al. HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165. Blood. 2009;113(21):5176–5185. doi: 10.1182/blood-2008-04-150342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cantuti-Castelvetri L., et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518):856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daly J.L., et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370(6518):861–865. doi: 10.1126/science.abd3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayi B.S., et al. The role of Neuropilin-1 in COVID-19. PLoS Pathog. 2021;17(1):e1009153. doi: 10.1371/journal.ppat.1009153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao M., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26(6):842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 15.Lim K.-H., et al. Identifying new COVID-19 receptor neuropilin-1 in severe alzheimer's disease patients group brain using genome-wide association study approach. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.741175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim K.H., et al. Elevation of ACE2 as a SARS-CoV-2 entry receptor gene expression in Alzheimer's disease. J. Infect. 2020;81(3):e33–e34. doi: 10.1016/j.jinf.2020.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zalpoor H., et al. The roles of Eph receptors, neuropilin-1, P2X7, and CD147 in COVID-19-associated neurodegenerative diseases: inflammasome and JaK inhibitors as potential promising therapies. Cell. Mol. Biol. Lett. 2022;27(1):10. doi: 10.1186/s11658-022-00311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McFarland A.J., et al. Neurobiology of SARS-CoV-2 interactions with the peripheral nervous system: implications for COVID-19 and pain. PAIN Reports. 2021;6(1):e885. doi: 10.1097/PR9.0000000000000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chekol Abebe E., et al. Neuropilin 1: a novel entry factor for SARS-CoV-2 infection and a potential therapeutic target. Biologics. 2021;15:143–152. doi: 10.2147/BTT.S307352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zalpoor H., et al. Human Cell; 2022. Increased Neuropilin-1 Expression by COVID-19: a Possible Cause of Long-Term Neurological Complications and Progression of Primary Brain Tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parker M.W., et al. Function of members of the neuropilin family as essential pleiotropic cell surface receptors. Biochemistry. 2012;51(47):9437–9446. doi: 10.1021/bi3012143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parker M.W., et al. Structural basis for selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1. J. Biol. Chem. 2012;287(14):11082–11089. doi: 10.1074/jbc.M111.331140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teesalu T., et al. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. U. S. A. 2009;106(38):16157–16162. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmann M., Kleine-Weber H., Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell. 2020;78(4):779–784. doi: 10.1016/j.molcel.2020.04.022. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohammad A., et al. Structural analysis of ACE2 variant N720D demonstrates a higher binding affinity to TMPRSS2. Life Sci. 2020;259 doi: 10.1016/j.lfs.2020.118219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wrapp D., et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohammad A., et al. Higher binding affinity of furin for SARS-CoV-2 spike (S) protein D614G mutant could be associated with higher SARS-CoV-2 infectivity. Int. J. Infect. Dis. 2021;103:611–616. doi: 10.1016/j.ijid.2020.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z.-l, Buck M. Neuropilin-1 assists SARS-CoV-2 infection by stimulating the separation of Spike protein S1 and S2. Biophys. J. 2021;120(14):2828–2837. doi: 10.1016/j.bpj.2021.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moin A.S.M., et al. The relationship of soluble neuropilin-1 to severe COVID-19 risk factors in polycystic ovary syndrome. Metabolism Open. 2021;9 doi: 10.1016/j.metop.2021.100079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Argueta L.B., et al. Inflammatory responses in the placenta upon SARS-CoV-2 infection late in pregnancy. iScience. 2022;25(5) doi: 10.1016/j.isci.2022.104223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humayun F., et al. Abrogation of SARS-CoV-2 interaction with host (NRP1) neuropilin-1 receptor through high-affinity marine natural compounds to curtail the infectivity: a structural-dynamics data. Comput. Biol. Med. 2022;141 doi: 10.1016/j.compbiomed.2021.104714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Y., et al. Interfering effects on the bioactivities of several key proteins of COVID-19/variants in diabetes by compounds from Lianqiao leaves: in silico and in vitro analyses. Int. J. Biol. Macromol. 2022;207:715–729. doi: 10.1016/j.ijbiomac.2022.03.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Sluis R.M., et al. TLR2 and TLR7 mediate distinct immunopathological and antiviral plasmacytoid dendritic cell responses to SARS-CoV-2 infection. EMBO J. 2022;41(10):e109622. doi: 10.15252/embj.2021109622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ntie-Kang F., et al. NANPDB: a resource for natural products from northern african sources. J. Nat. Prod. 2017;80(7):2067–2076. doi: 10.1021/acs.jnatprod.7b00283. [DOI] [PubMed] [Google Scholar]
- 35.Rose P.W., et al. Nucleic acids research; 2016. The RCSB Protein Data Bank: Integrative View of Protein, Gene and 3D Structural Information; p. gkw1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pettersen E.F., et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Goddard T.D., Huang C.C., Ferrin T.E. Software extensions to UCSF chimera for interactive visualization of large molecular assemblies. Structure. 2005;13(3):473–482. doi: 10.1016/j.str.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 38.Lagorce D., et al. FAF-Drugs4: free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics. 2017;33(22):3658–3660. doi: 10.1093/bioinformatics/btx491. [DOI] [PubMed] [Google Scholar]
- 39.Dallakyan S., Olson A.J. Chemical Biology. Springer; 2015. Small-molecule library screening by docking with PyRx; pp. 243–250. [DOI] [PubMed] [Google Scholar]
- 40.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mohammad A., et al. Molecular simulation-based investigation of highly potent natural products to abrogate formation of the nsp10-nsp16 complex of SARS-CoV-2. Biomolecules. 2021;11(4) doi: 10.3390/biom11040573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ravindranath P.A., et al. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol. 2015;11(12):e1004586. doi: 10.1371/journal.pcbi.1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xue L.C., et al. PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics. 2016;32(23):3676–3678. doi: 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- 44.Case D.A., et al. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price D.J., Brooks C.L., III A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004;121(20):10096–10103. doi: 10.1063/1.1808117. [DOI] [PubMed] [Google Scholar]
- 46.Roe D.R., Cheatham T.E., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theor. Comput. 2013;9(7):3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 47.Sun H., et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014;16(31):16719–16729. doi: 10.1039/c4cp01388c. [DOI] [PubMed] [Google Scholar]
- 48.Hou T., et al. Characterization of domain–peptide interaction interface: prediction of SH3 domain-mediated protein–protein interaction network in yeast by generic structure-based models. J. Proteome Res. 2012;11(5):2982–2995. doi: 10.1021/pr3000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen F., et al. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Phys. Chem. Chem. Phys. 2016;18(32):22129–22139. doi: 10.1039/c6cp03670h. [DOI] [PubMed] [Google Scholar]
- 50.Miller B.R., III, et al. MMPBSA. py: an efficient program for end-state free energy calculations. J. Chem. Theor. Comput. 2012;8(9):3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y., et al. The systematic modeling studies and free energy calculations of the phenazine compounds as anti-tuberculosis agents. J. Biomol. Struct. Dyn. 2019;37(15):4051–4069. doi: 10.1080/07391102.2018.1537896. [DOI] [PubMed] [Google Scholar]
- 52.Khan A., et al. Deep-learning-based target screening and similarity search for the predicted inhibitors of the pathways in Parkinson's disease. RSC Adv. 2019;9(18):10326–10339. doi: 10.1039/c9ra01007f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7 doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eberhardt J., et al. AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021;61(8):3891–3898. doi: 10.1021/acs.jcim.1c00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zalevsky A.O., et al. PeptoGrid—rescoring function for AutoDock Vina to identify new bioactive molecules from short peptide libraries. Molecules. 2019;24(2):277. doi: 10.3390/molecules24020277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar A.D., et al. Protective effect of 3-O-methyl quercetin and kaempferol from Semecarpus anacardium against H(2)O(2) induced cytotoxicity in lung and liver cells. BMC Compl. Alternative Med. 2016;16(1):376. doi: 10.1186/s12906-016-1354-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lall N., Hussein A.A., Meyer J.J.M. Antiviral and antituberculous activity of Helichrysum melanacme constituents. Fitoterapia. 2006;77(3):230–232. doi: 10.1016/j.fitote.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 58.Lipinski C.A., et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 59.Liang C., et al. Pharmacological activities and synthesis of esculetin and its derivatives: a mini-review. Molecules. 2017;22(3):387. doi: 10.3390/molecules22030387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharifi-Rad J., et al. Natural coumarins: exploring the pharmacological complexity and underlying molecular mechanisms. Oxid. Med. Cell. Longev. 2021 doi: 10.1155/2021/6492346. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim C.S., et al. Esculetin, a coumarin derivative, inhibits aldose reductase activity in vitro and cataractogenesis in galactose-fed rats. Biomol Ther (Seoul) 2016;24(2):178–183. doi: 10.4062/biomolther.2015.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang L., Xie Q., Li X. Esculetin: a review of its pharmacology and pharmacokinetics. Phytother Res. 2022;36(1):279–298. doi: 10.1002/ptr.7311. [DOI] [PubMed] [Google Scholar]
- 63.Sun B., Wang B., Xu M. Esculetin inhibits histamine-induced expression of inflammatory cytokines and mucin in nasal epithelial cells. Clin. Exp. Pharmacol. Physiol. 2019;46(9):821–827. doi: 10.1111/1440-1681.13128. [DOI] [PubMed] [Google Scholar]
- 64.Junior A.G., et al. Natural agents modulating ACE-2: a review of compounds with potential against SARS-CoV-2 infections. Curr. Pharmaceut. Des. 2021;27(13):1588–1596. doi: 10.2174/1381612827666210114150607. [DOI] [PubMed] [Google Scholar]
- 65.Hamoda A.M., et al. Marine sponge is a promising natural source of anti-SARS-CoV-2 scaffold. Front. Pharmacol. 2021;12(1161) doi: 10.3389/fphar.2021.666664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chidambaram S.K., et al. In silico molecular docking: evaluation of coumarin based derivatives against SARS-CoV-2. J. Infect. Public Health. 2020;13(11):1671–1677. doi: 10.1016/j.jiph.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cournia Z., Allen B., Sherman W. Relative binding free energy calculations in drug discovery: recent advances and practical considerations. J. Chem. Inf. Model. 2017;57(12):2911–2937. doi: 10.1021/acs.jcim.7b00564. [DOI] [PubMed] [Google Scholar]
- 68.Torrens-Fontanals M., et al. How do molecular dynamics data complement static structural data of GPCRs. Int. J. Mol. Sci. 2020;21(16):5933. doi: 10.3390/ijms21165933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cuevas-Zuviría B., et al. Energy landscapes of ligand motion inside the tunnel-like cavity of lipid transfer proteins: the case of the pru p 3 allergen. Int. J. Mol. Sci. 2019;20(6):1432. doi: 10.3390/ijms20061432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nedyalkova M., et al. Inhibition ability of natural compounds on receptor-binding domain of SARS-CoV2: an in silico approach. Pharmaceuticals. 2021;14(12) doi: 10.3390/ph14121328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Loudon G.M. Roberts and Company; 2009. Organic Chemistry. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.