

Abstract
Objective.
Blood microbiome has been analyzed in cancer patients using machine learning. We recently published a methodological paper using microbial 16S rRNA sequencing to analyze human plasma microbiome with a substantial set of controls, including technical and biological replicates, blank extractions and amplifications and a thorough and combined bioinformatics analysis of the resulting control and plasma sequence data. Here, we aim to study whether the plasma microbiome represents the microbial community in the gut among systemic lupus erythematosus (SLE) patients and healthy controls.
Methods.
Paired plasma and stool samples from female SLE patients and female controls were assessed for microbiome composition by microbial 16S ribosomal RNA sequencing.
Results.
Decreased microbial α-diversity in stool compared to plasma and distinct plasma and gut β-diversity were found in both controls and patients. No difference of gut microbial diversity was found between patients and controls. However, plasma α-diversity was decreased in patients compared to controls. The top predominant bacteria differed between plasma and stool in any group. Although the predominant plasma and stool genus bacteria were similar in patients and controls, some were clearly different.
Conclusion.
Compared to the gut, the plasma microbiome contains distinct community and greater heterogeneity, indicating that the predominant circulating microbiome may originate from sites (e.g., oral or skin) other than the gastrointestinal tract. The decreased plasma but not gut alpha diversity in patients compared to controls implies an altered plasma microbiome in SLE, which may be important for systemic immune perturbations and lupus disease pathogenesis.
INTRODUCTION
Most studies have analyzed microbiome in stools or in samples from other mucosal sites that may not represent systemic microbiome and may not play a major role in systemic immunity. Recently, blood microbiome has been analyzed in cancer patients using a method of machine learning 1; notably, the method of machine learning requires a huge sample size (a total of 18,116 samples in this study). Rare studies have conducted blood microbiome in relatively small sample size studies 2,3. We recently published a methodological paper on human plasma microbiome analysis using a substantially expanded set of controls, including technical and biological replicates, blank extractions and amplifications and a thorough and combined bioinformatics analysis of the resulting control and plasma sequence data 4. The amplicon sequence variant (ASV)s from the water control was subtracted from the plasma microbiome analysis 4.
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by systemic inflammation and the production of autoantibodies against self-antigens leading to progressive organ damage. A previous study suggests that SLE patients had a distinct plasma microbiome composition with increased plasma levels of total microbial translocation that associated with disease pathogenesis 5. One hypothesis is that the translocation of bacterial products into the systemic circulation leads to systemic inflammation 6. The exact etiology of plasma microbial translocation remains unclear, but there is evidence to suggest that the source of blood bacterial translocation stems from increased intestinal permeability 6. Under normal, healthy conditions, the intestinal epithelium forms a protective barrier with assistance of protective factors such as mucus layers, and secretory IgA and defensins 6. In some disease states with a compromised barrier, increased plasma levels of microbial translocation may contribute to disease pathogenesis 5,6.
Once considered to be a sterile environment, it is now well-established that plasma contains bacterial products, both under normal conditions and disease states 1,5. While the gut microbiome has been extensively studied 7, rare studies have characterized the human plasma microbiome 1–3,8–10 date, no study has directly compared gut and plasma microbiome composition in healthy individuals and SLE patients. The role of dysbiosis and the circulating microbiome in the development of autoimmunity and SLE is being increasingly studied 11; however, the source of this microbial translocation has yet to be revealed, though there is evidence to suggest that the gastrointestinal tract is a major source 11. In this novel study, we compared the microbiome composition using stool and paired plasma samples among SLE patients and healthy controls to further understand the source of microbial translocation.
METHODS
Study participants
A single cohort was examined in this study and clinical information of patients are shown in Table 1. It consisted of 9 unrelated healthy controls and 11 patients with SLE. All patients and controls in this cohort were females. Samples were obtained from the Division of Rheumatology & Immunology at the Medical University of South Carolina. The criteria for inclusion in this study were age ≥ 18 years and ability to provide informed consent. The criteria for exclusion in this study were individuals who were pregnant or breastfeeding, had recent severe illness, contraindications to blood withdrawals, or had received antibiotics and probiotics within the previous 90 days. This study was approved by the Institutional Review Board of the Medical University of South Carolina, ethics approval number: Pro00082453. All participants have signed a consent form.
Table 1.
Demographic, clinical, and laboratory features of patients and controls
| Demographic features, controls (n=9) | Mean ± SD |
|---|---|
| Male/female, n | 9/0 |
| African-American race, n | 9 |
| Mean age ± SD (years) | 45 ± 15 |
| Demographic features, SLE patients (n=11) | Mean ± SD |
|
| |
| Male/female, n | 11/0 |
| African-American race, n | 11 |
| Age (years) | 51.4 ± 11.0 |
| Clinical manifestations of SLE patients | |
| SLEDAI | 1.72 ± 1.90 |
| Laboratory manifestations of SLE patients | |
| anti-dsDNA (IU/mL) | 57 ± 64 |
| C3 complement (mg/dL) | 117.2 ± 21.1 |
| C4 complement (mg/dL) | 23.7 ± 9.2 |
| Serum albumin (g/dL) | 3.6 ± 0.2 |
| BUN (mg/dL) | 14 ± 4 |
| Serum creatinine (mg/dL) | 0.8 ± 0.1 |
| Urine protein (mg/dL) | 26.3 ± 18.4 |
| Urine creatinine (mg/dL) | 143.1 ± 99.6 |
| Urine protein / creatinine ratio (mg/mg) | 0.210 ± 0.112 |
SLEDAI – Systemic Lupus Erythematosus Disease Activity Index, anti-dsDNA – anti-double stranded DNA antibodies, BUN – blood urea nitrogen
DNA extraction
The microbial DNA was extracted from 400 μl of plasma and 400 μl of water control using a QIAamp UCP Pathogen Mini Kit, according to the manufacturer’s protocol (Qiagen, Hilden, Germany). Bacterial DNA from 1 mg of stool was extracted using a QIAamp DNA Microbiome Kit, following the manufacturer’s protocol (Qiagen, Hilden, Germany). Polymerase chain reaction (PCR) primers 515/806 were used to amplify the V4 variable region of the 16S ribosomal RNA (16S rRNA) using a HotStarTaq Plus Master Mix (Qiagen) under the following conditions: denaturation at 94°C for 3 minutes, followed by 28 cycles at 94°C for 30 seconds, 53°C for 40 seconds, and 72°C for 1 minute, followed by a final elongation step at 72°C for 5 minutes. Sequencing was carried out using an Ion Torrent PGM according to the manufacturer’s guidelines (MR DNA, Shallowater, TX). A proprietary analysis pipeline was applied to process the sequencing data (MR DNA).
Sequence processing and taxonomic assignment
The microbiome analysis was described in our previous studies 5. In this study, we applied a quality-filtering strategy to plasma microbiome to efficiently exclude ASVs of contaminations and artifacts. ASVs distinguish sequence variation by a single nucleotide change and these single DNA sequences are created following the removal of erroneous sequences during high-throughput gene sequencing 5. Briefly, short sequences less than 200 bp, ambiguous base calls, and homopolymer runs exceeding 6 bp were removed. Next, singleton sequences and chimeras were removed and sequences were denoised and ASVs were clustered at 3% divergence (97% similarity). Final ASVs were taxonomically classified using BLASTn against a database derived from RDPII and NCBI (www.ncbi.nlm.nih.gov). The ASV table of raw counts was normalized to an ASV table of relative abundances, and taxa of the same type were grouped by phylum, class, order, family, genus, and species.
Statistical analysis
The microbiome diversity (α-diversity) was computed using the Simpson diversity index and the Shannon index in R (Version 1.2.5001), and significance was tested using the Wilcoxon rank sum test. The Wilcoxon rank sum test in R was applied to compare relative abundances, and P values were adjusted for multiple comparisons using the Benjamini-Hochberg false discovery rate (FDR) method. Statistical significance of beta-diversity was determined with permutational multivariate analysis of variance (R-vegan package with the function adonis).
RESULTS
Plasma has a distinct microbial community profile and increased species heterogeneity compared to the gut
To understand the potential source of the circulating microbiome, we examined differences in alpha and beta diversities of the gut and plasma microbiomes. The plasma microbiome of our cohort demonstrated significantly increased variance of species diversity compared to the gut microbiome (P < 0.0001) as measured by the Simpson diversity and Shannon-Wiener indices (Figure 1A). This difference was observed in either all samples or separated groups based on lupus disease condition. Consistently, beta diversity showed distinct plasma and gut microbial communities both in all samples and when separated into groups as reflected by PCoA plots, which were calculated using the weighted UniFrac metric (Figure 1B).
Figure 1. Distinct plasma microbiome from gut microbiome.
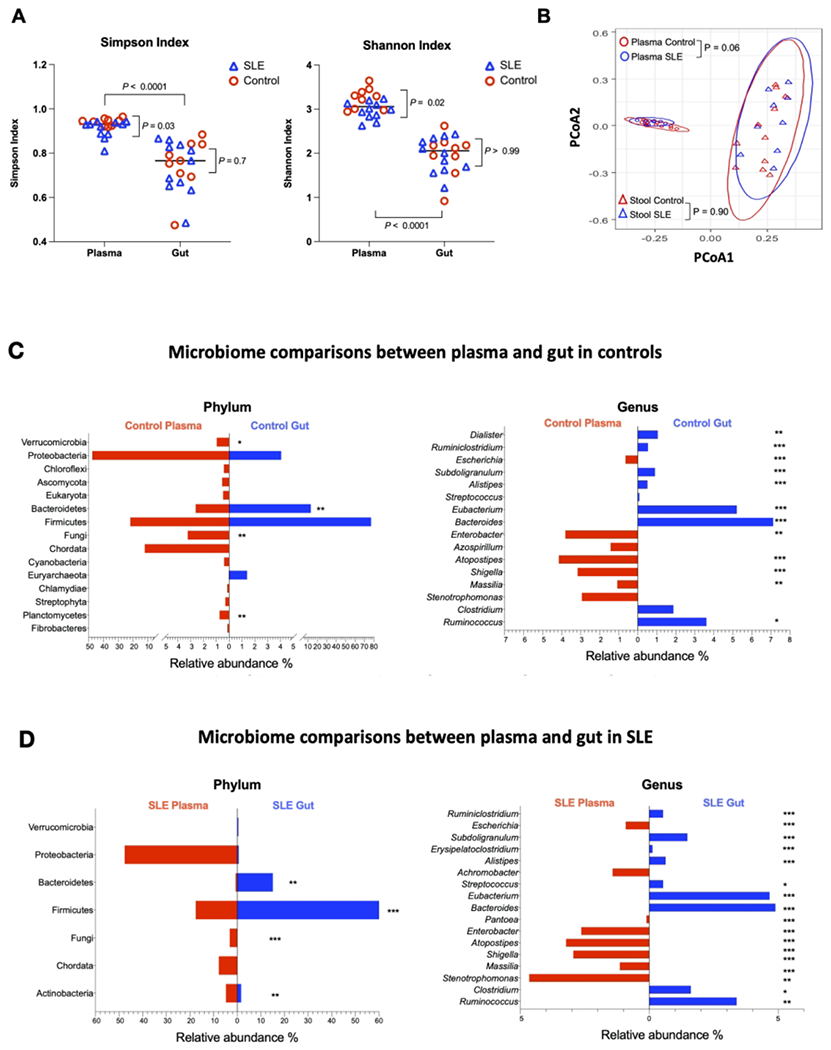
(A) Alpha diversity measured using Simpson and Shannon indices by site and patient status. (B) Principal coordinates analysis (PCoA) of beta diversity grouped by site and patient status. Median percentages of relative abundance at the phylum and genus levels in gut and plasma from healthy controls (C) and SLE patients (D). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Decreased plasma but not gut alpha diversity in SLE patients compared to controls
We found that the gut microbiome was similar in controls and patients. However, plasma from patients contained significantly decreased species diversity compared to healthy controls (Simpson diversity index, P = 0.03; Shannon-Wiener index, P = 0.02) (Figure 1A). Moreover, the beta diversity of plasma microbiome was marginally different in controls and patients (P = 0.06), but the beta diversity of gut microbiome was similar in controls and patients (P = 0.90) (Figure 1B).
Distinct microbiome in the plasma from the gut
The predominant microbiome in the plasma differed from that in the stool. There was a significant difference in the relative abundance of bacteria at each level of taxa between the gut and plasma microbiome. The largest difference was observed at the genus level. At the phylum level we found that the plasma microbiome was more enriched in Proteobacteria than the gut microbiome, and the gut microbiome was more enriched in Firmicutes and Bacteriodetes than that of the plasma (Figures 1C–1D). The ratio of gut enrichment of Firmicutes to Bacteriodetes is 4.58 versus 6.39 in patients and controls, respectively. The same was true when comparing the pooled samples from both groups as well as when comparing the two sites by patient status (Figures 1C–1D). The ratio of plasma enrichment of Firmicutes to Bacteriodetes is 25.87 versus 11.0 in patients and controls, respectively.
Microbiome composition between patients and controls
Next, the relative microbial abundance of both gut and plasma tended to be similar between patients and controls but with exceptions (Figures 2A–2D). After adjusting for FDR, the gut microbiome of SLE patients was significantly less enriched in Dialister and Azospirillum, and significantly more enriched in Verrucomicrobia phylum and Streptococcus genus than those in controls (Figures 2A–2B). The plasma microbiome of SLE patients was significantly more enriched in Gemmatimonadetes phylum only (Figure 2C).
Figure 2. Comparisons of plasma and gut microbiome between healthy controls and patients with SLE.
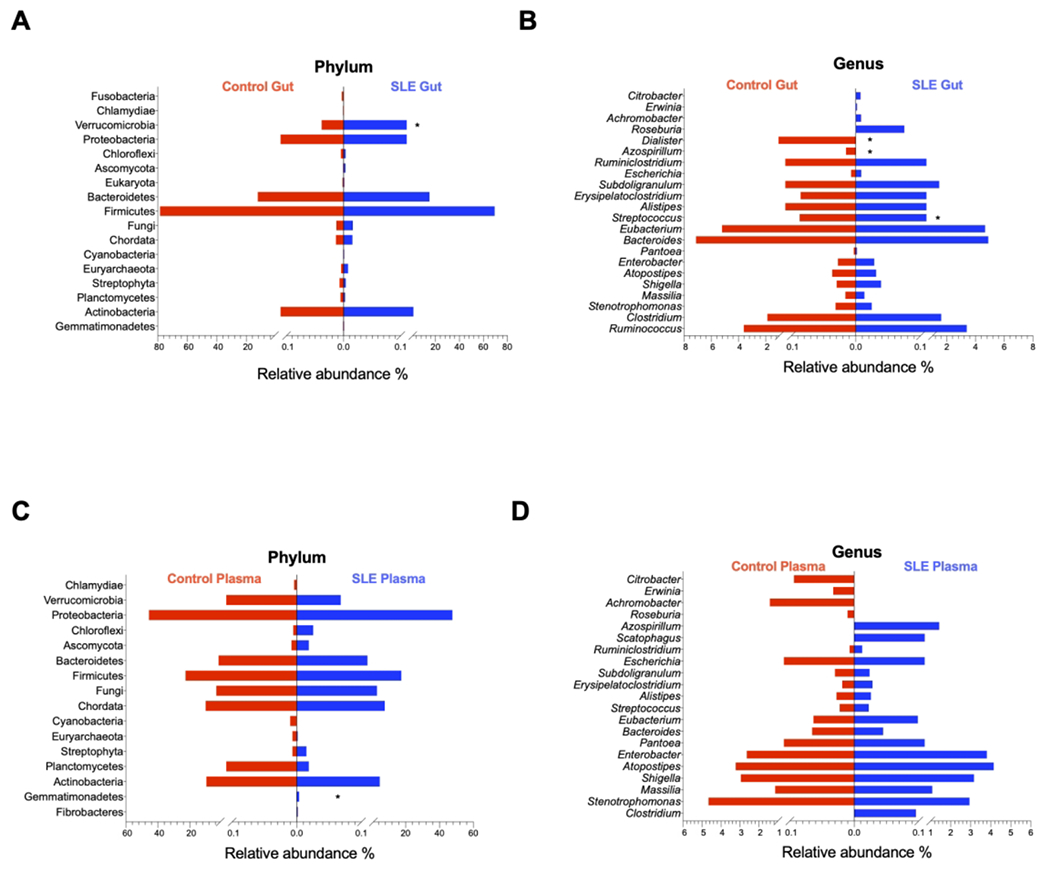
Median percentages of relative abundance of gut microbiome at phylum (A) and genus (B) levels, and plasma microbiome at phylum (C) and genus (D) levels in healthy controls versus SLE patients. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
In the current study, we directly examined the microbiome composition of the gut and plasma in SLE patients compared to healthy controls. We found that both microbial alpha and beta diversities of the gut microbiome were not significantly altered between healthy and diseased patients, which is in line with some previous studies 12,13. The fact that diversity was not significantly altered does not mean that the differences in microbiome is insignificant, and indeed, other studies show a difference in gut microbiome between SLE patients and controls 12. Nonetheless, we did find that plasma microbial alpha diversity was significantly decreased in SLE patients and beta diversity was marginally different in SLE patients compared to controls. Notably, the plasma microbial composition of both groups was significantly different from that of their own gut.
In a state of health, the gut microbiome consists primarily of Firmicutes and Bacteroidetes 14,15. The ratio of Firmicutes to Bacteroidetes in the gut has been shown to be decreased in SLE compared to healthy controls 12; consistently, we found a similar decreased ratio of Firmicutes and Bacteroidetes in the gut but not in the plasma of patients compared to controls (Figures 2A and 2C). Additionally, Lactobacillus spp. has been demonstrated to be enriched in the gut from SLE patients 16. A recent study on SLE and nephritis demonstrated that the abundance of Ruminococcus gnavus in the gut was inversely correlated with disease activity 17. In the current study, Lactobacillus spp. and Ruminococcus gnavus were not found to be enriched in either the plasma or gut microbiome in SLE, but Ruminococcus spp. was found to be increased in patient gut microbiome compared to those in controls.
Our results are consistent with previously characterized gut microbiome composition, and we additionally found a large discrepancy in the relative abundance of bacteria contained in the phyla Firmicutes, Proteobacteria, and Bacteroidetes between plasma and stool of both study groups. Although it was once hypothesized that transient or pathological increases in gut permeability was responsible for the circulating microbiome 18, our results may suggest an additional source for bacterial translocation. A possible source is the oral microbiome, which is distinct from the gut microbiome 19. There is evidence of plasma microbial translocation of oral bacteria following tooth extraction and even flossing in the general population 20. Furthermore, patients with SLE presented with decreased gut microbiota diversity similar to patients with Sjögren syndrome; in contrast, these two patient groups presented with an oral microbial composition that differed substantially 21. Another possible site is the skin, as its barrier function is disrupted not only in autoimmune diseases such as SLE and psoriasis, but also from cuts, scrapes, and burns 22. A study by Whittle et al characterizing the blood microbiome of healthy individuals and asthmatics found that blood microbiome was predominated by Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes, consistent with our results. They additionally compared their data with the Human Microbiome Project database and concluded that the blood microbiome is most similar to that of the skin and oropharynx 23. To our knowledge, ours is the first study directly comparing plasma and gut samples from the same individuals to show that there is little correlation between the human plasma and gut microbiomes.
A recent study shows that bacterial translocation from the small intestine to the liver in mice and SLE patients may drive autoimmunity 11. There is a paucity of data comparing stool and plasma microbiome composition in SLE and other autoimmune diseases. A study by Shukla et al revealed distinct plasma and gut microbiome in patients with myalgic encephalomyelitis/chronic fatigue syndrome and healthy controls between groups before and following exercise 24. Proteobacteria and Actinobacteria were relatively more abundant in plasma compared to stool in both groups 24. Consistently, we found increased relative abundance of Proteobacteria and Actinobacteria in plasma compared to stool in both study groups.
Enterobacteriaceae or Escherichia genus bacteria are believed to be prevalent in the gut microbiome in humans 25, however, our results show they were found in less than 1% in the gut microbiome but higher in abundance in the plasma microbiome regardless of patients or controls (Figures 2B and 2D). Notably, we did not find any Enterobacteriaceae or Escherichia genus bacteria in the water controls in the current study. Nonetheless, previous studies of the human gut microbiome often reported missing or similar low abundance (< 1%) of Enterobacteriaceae or Escherichia genus bacteria or related species bacteria using 16S sequencing 25 or metagenomic analysis 26. Thus, the low abundance of Enterobacteriaceae or Escherichia genus bacteria in the human gut microbiome is unlikely due to the limitation of microbial 16S sequencing. Notably, the previous concept of prevalent bacteria was mostly based on bacterial cultivation; however, bacteria differ largely in susceptibility to in vitro cultivation. Another possibility of the higher abundance of plasma Enterobacteriaceae or Escherichia genus bacteria compared to stool is that these bacterial antigens may have a greater ability to translocate to the systemic circulation. These hypotheses deserve further investigation.
This study is not without limitations. First, we are limited by the small sample size. Thus, this study could not control for potential confounders including but not limited to age, race, diet, and treatment regimens. Additionally, our cohort was all females, but there may be gender differences in microbiomes due to anatomical, genomic, biologic, and behavioral factors. Another limitation is the relatively low SLEDAI of our cohort. While ideally patients would have high disease activity at the time of sample collection, there are ethical limitations to altering patients’ treatment. Nonetheless, this is the first study to show that the microbiome in the plasma does not parallel that of the gut in patients with SLE and controls. This study also opens new directions for future studies linking the disease-associated plasma microbiome and systemic immune perturbations in SLE and other diseases, as well as comparisons of the plasma microbiome with those in other sites (e.g., oropharynx, skin, vaginal lavage) to determine which is the main source of plasma microbial translocation.
Acknowledgment.
We thank the funding agencies and all study participants.
This work was supported by grants from the National Institutes of Health: P60 AR062755 (Gilkeson, Kamen and Oates), P30AR072582 (Gilkeson, Kamen, and Oates), AR067459 (Kamen), AR068406 (Kamen), RR001070 (Kamen), and UL1 RR029882, UL1 TR001450, and the Medical Research Service at the Ralph H. Johnson VA Medical Center Merit grant VA CSRD MERIT (CX001211, Gilkeson).
Footnotes
Conflict of interest: none
Statement of ethics and consent: This study was approved by the Institutional Review Board of the Medical University of South Carolina (Pro00082453). All participants have signed the consent form.
Consent for publication. All authors approve the final version of this manuscript and consent for publication.
Competing interests. The authors declare that they have no competing interests.
Contributor Information
Warren James, College of Medicine, University of South Carolina.
Elizabeth Ogunrinde, Department of Microbiology and Immunology, Medical University of South Carolina.
Zhuang Wan, Department of Microbiology and Immunology, Medical University of South Carolina.
Diane L. Kamen, Division of Rheumatology and Immunology, Department of Medicine, Medical University of South Carolina
Jim Oates, Director and Endowed Chair, Division of Rheumatology and Immunology, Department of Medicine, Medical University of South Carllina.
Gary S. Gilkeson, Division of Infectious Diseases, Division of Rheumatology and Immunology, Department of Medicine, Medical University of South Carolina
Wei Jiang, Department of Microbiology and Immunology; Division of Infectious Diseases, Department of Medicine, Medical University of South Carolina.
Availability of data and material.
All data generated or analyzed during this study are included as supplementary files.
REFERENCES
- 1.Poore GD, Kopylova E, Zhu Q, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020;579:567–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puri P, Liangpunsakul S, Christensen JE, et al. The circulating microbiome signature and inferred functional metagenomics in alcoholic hepatitis. Hepatology 2018;67:1284–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schierwagen R, Alvarez-Silva C, Madsen MSA, et al. Circulating microbiome in blood of different circulatory compartments. Gut 2018. [DOI] [PubMed] [Google Scholar]
- 4.Luo Z, Alekseyenko AV, Ogunrinde E, et al. Rigorous Plasma Microbiome Analysis Method Enables Disease Association Discovery in Clinic. Front Microbiol 2020;11:613268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogunrinde E, Zhou Z, Luo Z, et al. A Link Between Plasma Microbial Translocation, Microbiome, and Autoantibody Development in First-Degree Relatives of Systemic Lupus Erythematosus Patients. Arthritis Rheumatol 2019;71:1858–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brenchley JM, Douek DC. Microbial translocation across the GI tract. Annu Rev Immunol 2012;30:149–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shreiner AB, Kao JY, Young VB. The gut microbiome in health and in disease. Curr Opin Gastroenterol 2015;31:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macherey-Nagel. Genomic DNA from blood. Genomic DNA from blood 2016. [Google Scholar]
- 9.clinical implications of circulating microbiome in cardiovascular disease patients. Journal of Cardiology & Current Research 2017;9:00317. [Google Scholar]
- 10.Nejman D, Livyatan I, Fuks G, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020;368:973–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manfredo Vieira S, Hiltensperger M, Kumar V, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 2018;359:1156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hevia A, Milani C, Lopez P, et al. Intestinal dysbiosis associated with systemic lupus erythematosus. MBio 2014;5:e01548–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Z, Shao T, Li H, Xie Z, Wen C. Alterations of the gut microbiome in Chinese patients with systemic lupus erythematosus. Gut Pathog 2016;8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science (New York, NY) 2005;308:1635–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature 2012;486:207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zegarra-Ruiz DF, El Beidaq A, Iñiguez AJ, et al. A Diet-Sensitive Commensal Lactobacillus Strain Mediates TLR7-Dependent Systemic Autoimmunity. Cell Host Microbe 2019;25:113–27.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Azzouz D, Omarbekova A, Heguy A, et al. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis 2019;78:947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 2006;12:1365–71. [DOI] [PubMed] [Google Scholar]
- 19.Gao L, Xu T, Huang G, Jiang S, Gu Y, Chen F. Oral microbiomes: more and more importance in oral cavity and whole body. Protein Cell 2018;9:488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crasta K, Daly CG, Mitchell D, Curtis B, Stewart D, Heitz-Mayfield LJ. Bacteraemia due to dental flossing. J Clin Periodontol 2009;36:323–32. [DOI] [PubMed] [Google Scholar]
- 21.van der Meulen TA, Harmsen HJM, Vila AV, et al. Shared gut, but distinct oral microbiota composition in primary Sjogren’s syndrome and systemic lupus erythematosus. Journal of autoimmunity 2019;97:77–87. [DOI] [PubMed] [Google Scholar]
- 22.Sirobhushanam S, Parsa N, Reed TJ, et al. Staphylococcus aureus Colonization Is Increased on Lupus Skin Lesions and Is Promoted by IFN-Mediated Barrier Disruption. J Invest Dermatol 2020;140:1066–74 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whittle E, Leonard MO, Harrison R, Gant TW, Tonge DP. Multi-Method Characterization of the Human Circulating Microbiome. Front Microbiol 2018;9:3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shukla SK, Cook D, Meyer J, et al. Changes in Gut and Plasma Microbiome following Exercise Challenge in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). PLoS One 2015;10:e0145453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinson JNV, Pinkham NV, Peters GW, et al. Rethinking gut microbiome residency and the Enterobacteriaceae in healthy human adults. ISME J 2019;13:2306–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pena-Gonzalez A, Soto-Giron MJ, Smith S, et al. Metagenomic Signatures of Gut Infections Caused by Different Escherichia coli Pathotypes. Appl Environ Microbiol 2019;85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included as supplementary files.