

Abstract
Myocardial infarction is a leading cause of death worldwide1. Although advances have been made in acute treatment, an incomplete understanding of remodelling processes has limited the effectiveness of therapies to reduce late-stage mortality2. Here we generate an integrative high-resolution map of human cardiac remodelling after myocardial infarction using single-cell gene expression, chromatin accessibility and spatial transcriptomic profiling of multiple physiological zones at distinct time points in myocardium from patients with myocardial infarction and controls. Multi-modal data integration enabled us to evaluate cardiac cell-type compositions at increased resolution, yielding insights into changes of the cardiac transcriptome and epigenome through the identification of distinct tissue structures of injury, repair and remodelling. We identified and validated disease-specific cardiac cell states of major cell types and analysed them in their spatial context, evaluating their dependency on other cell types. Our data elucidate the molecular principles of human myocardial tissue organization, recapitulating a gradual cardiomyocyte and myeloid continuum following ischaemic injury. In sum, our study provides an integrative molecular map of human myocardial infarction, represents an essential reference for the field and paves the way for advanced mechanistic and therapeutic studies of cardiac disease.
Subject terms: Transcriptomics, Data integration, Myocardial infarction
A time-resolved high-resolution map of human cardiac remodelling after myocardial infarction, integrating single-cell transcriptomic, chromatin accessibility and spatial transcriptomic data, provides a valuable resource for the field.
Main
Coronary heart disease driving acute myocardial infarction is the largest contributor to cardiovascular mortality, which in turn is the leading cause of death worldwide1. Substantial progress has been made in the acute therapy of myocardial infarction, focusing primarily on percutaneous coronary intervention resulting in decreased acute mortality. However, the morbidity and mortality caused by left ventricular cardiac remodelling after myocardial infarction remain unacceptably high2. Cardiac remodelling after myocardial infarction involves immune cell recruitment and demarcation of the infarcted area followed by resorption of necrotic tissue, phagocytosis, myofibroblast activation, scar formation and neovascularization3. Understanding the exact cellular and molecular mechanisms of cardiac remodelling processes from the acute ischaemic event to the chronic cardiac scar formation in their spatial context will be key to developing novel therapeutics.
Here we used a combination of single-cell gene expression, chromatin accessibility and spatially resolved transcriptomics to study the events of cardiac tissue reorganization and to characterize the cell-type-specific changes in gene regulation, providing an integrated spatial multi-omic map of cardiac remodelling after myocardial infarction. Our multi-omic data-driven approach, including spatial context, enables us to understand how a given cell state changes based on the cells’ neighbourhood and how this relates to transcriptional and regulatory variations. By deconvoluting spatial transcriptomics spots into cell-type abundances, we characterized cell niches occurring in different stages following acute myocardial infarction. We identified different cell states of cardiomyocytes, endothelial cells, myeloid cells and fibroblasts that are associated with disease progression on the basis of the integrated single-cell multi-omics data. Moreover, we inferred the gene-regulatory networks differentiating these cell states and projected this information onto specific tissue locations, thus mapping putative regulators controlling gene expression on specific myocardial tissue zones and disease stages. This enabled us to gain novel insights into the gene-regulatory programmes driving injury of cardiomyocytes, activated phagocytic macrophages and their relation to myofibroblast differentiation in cardiac tissue remodelling. Our results provide a comprehensive spatially resolved characterization of gene regulation of the human heart in homeostasis and after myocardial infarction. We have released our spatial multi-omics data through publicly available platforms to enable users to interactively explore the dataset. We anticipate that this data will be a reference map for future studies and ultimately for the development of novel therapeutics.
Multi-omic map of myocardial infarction
We applied an integrative single-cell genomics strategy with single-nucleus RNA sequencing (snRNA-seq) and single-nucleus assay for transposase-accessible chromatin sequencing (snATAC-seq) together with spatial transcriptomics from the same tissue mapping human cardiac cells in homeostasis and after myocardial infarction at unprecedented spatial and molecular resolution (Fig. 1a–c and Supplementary Table 1). We profiled a total of 31 samples from 23 individuals, including four non-transplanted donor hearts as controls, and samples from tissues with necrotic areas (ischaemic zone and border zone) and the unaffected left ventricular myocardium (remote zone) of patients with acute myocardial infarction (Fig. 1a). These acute myocardial infarction specimens were collected from heart tissues obtained at different time points after the onset of clinical symptoms (chest pain), before the patients received an artificial heart or a left-ventricular assist device because of cardiogenic shock and as a bridge to transplantation (Supplementary Fig. 1a–c). We also analysed nine human heart specimens at later stages after myocardial infarction (fibrotic zone; Fig. 1b) that exhibited ischaemic heart disease and were available from heart transplantation recipients at the time of orthotopic heart transplantation.
Fig. 1. Spatial multi-omic profiling of human myocardial infarction.

a, Study schematic. RZ, remote zone; BZ, border zone; IZ, ischaemic zone; FZ, fibrotic zone. b, Sampling time points. P indicates patient number. Asterisks indicate snRNA-seq samples that were used for validation only (P21–P23). c, Data modalities. GEX, gene expression. d, UMAP of snRNA-seq data from all samples (n = 191,795). vCMs, ventricular cardiomyocytes. vSMCs, vascular smooth muscle cells. e, Average marker gene expression after z-score transformation. Colours along the bottom correspond to the cell types in d. f, Uniform manifold approximation and projection (UMAP) of snATAC-seq data for all samples (n = 46,068). g, Chromatin accessibility of marker genes after z-score transformation. Colours along the bottom correspond to the cell types in d. h–j, Characterization of spatial transcriptomics data using cell-type deconvolution (h), pathway activity (i) and transcription factor (TF) binding activity (j) for control (Ctrl), border zone, ischaemic zone and fibrotic tissue samples. Max, maximum; min, minimum.
For each cardiac sample, we obtained 10-µm cryo-sections and isolated nuclei from the remaining tissue directly adjacent to the cryo-section with subsequent fluorescence-activated nuclei sorting (FANS) for snRNA-seq and snATAC-seq (Fig. 1c). After filtering out low-quality nuclei, we obtained a total of 191,795 nuclei from all samples for snRNA-seq, with an average of 2,020 genes per nucleus, together with chromatin accessibility data from 46,086 nuclei overall with an average of 28,066 fragments per nucleus (Supplementary Fig. 2a,b and Supplementary Tables 2–5). After controlling for data quality, the spatial transcriptomics datasets contained a total of 91,517 spots (average of 3,389 spots per specimen and 2,001 genes per spot) (Supplementary Figs. 2c,e–g and 3a,b). Quantification based on histology revealed an average of four nuclei per spatial transcriptomic spot from all slides (Supplementary Fig. 2c and Supplementary Table 6). Samples from the ischaemic zone had the lowest abundance of nuclei and an enriched expression of genes associated with cell death and the regulated necrosis pathway, suggesting increased necrotic cell death (Supplementary Fig. 2d). This integrated dataset represents, to our knowledge, the largest and most comprehensive multi-modal profiling of human myocardial infarction tissue including spatial information and samples at distinct disease progression stages. We devised an integrative data analysis approach spanning all three modalities of our single-cell experiments to study cardiac cell-specific information and cell-specific interactions in their spatial and disease progression context (Extended Data Fig. 1a).
Extended Data Fig. 1. Computational workflow and snRNA-Seq data analysis.

a, Schematic of the computational workflow for the main analyses of snRNA-seq, snATAC-seq, and spatial transcriptomics data. b, UMAP embedding of snRNA-seq data from all samples for patients, regions, and clusters. c, UMAP embedding of snRNA-seq data showing G2-M-phase cell-cycle score (left) and S-phase score (right). d, Barplots showing cell-type proportions of snRNA-seq data across all samples. Colours indicate different major cell types. e, Comparison of the generated snRNA-seq atlas to a previously published human heart cell atlas (HCA) for molecular profiles (left) and cell-type proportion (right). Left panel shows the adjusted P-value of the overlap of top gene markers of each cell-type between the different data sets (hypergeometric test). Right panel shows the Pearson correlation between the median proportion of each shared cell-type of the reference atlas and our control, border zone, and remote zone samples. f, UMAP embedding and annotation of an external dataset of three ischaemic zone samples following MI. g, Comparison of the generated snRNA-seq atlas to the external ischaemic data for molecular profiles (left) and cell-type proportion (right). Left panel shows the adjusted P-value of the overlap of top gene markers of each cell-type between the different data sets (hypergeometric test). Right panel shows the Pearson correlation between the median proportion of each shared cell-type of the external data set and our ischaemic samples.
We established a map of major human heart cell types using the snRNA-seq and snATAC-seq datasets independently. First, we clustered cells on the basis of the integrated snRNA-seq data from all samples after batch correction (Extended Data Fig. 1b). Clusters were annotated with curated marker genes from the literature4–6 and ten major cardiac cell types were identified (Fig. 1d,e). We also found an additional cluster with enriched expression of the cell-cycle marker gene MKI67, which showed a high score of cell-cycle G2/M and S phases and was mainly recovered in ischaemic zone samples (Extended Data Fig. 1c,d). To validate the annotations, we compared the data with a recent study on healthy human hearts4 and an independent novel dataset of ischaemic heart samples (n = 3, generated during this study) and observed a high agreement and correlation in terms of molecular profiles and cellular composition (Extended Data Fig. 1e–g). Of note, the cycling cells were also captured in the independent ischaemic dataset (Extended Data Fig. 1f).
We next integrated and clustered the snATAC-seq data from all samples (Extended Data Fig. 2a). These clusters were annotated on the basis of gene chromatin accessibility with the same markers as for snRNA-seq. This approach identified eight major cell types, matching all cell types from snRNA-seq data with the exception of two rare cell types (that is, mast cells and adipocytes) (Fig. 1f,g). Label transfer from snRNA-seq to snATAC-seq indicated that the annotations between these two modalities were consistent (Extended Data Fig. 2b,c). This was further supported by a high correlation of cellular composition between snRNA-seq and snATAC-seq and the presence of the same eight cell types in the majority of samples (Extended Data Fig. 2d,e). To explore regulatory information provided by the snATAC-seq, we performed transcription factor footprinting analysis using cell-type-specific pseudo-bulk ATAC-seq profiles. This revealed footprinting-based binding activity of known transcription factors such as MEF2C (ref. 7) in cardiomyocytes, CEBPD)8 in myeloid cells, FOS–JUNB9 in fibroblasts and SRF10 in vascular smooth muscle cells (vSMCs), which correlated with the expression of their predicted target genes in snRNA-seq data (Extended Data Fig. 2f). Together, our integrative analysis of transcriptomic and chromatin accessibility data defined a robust catalogue of cell types in the adult human heart across multiple modalities and samples.
Extended Data Fig. 2. snATAC-seq and spatial transcriptomics data analysis.

a, UMAP showing snATAC-seq data for all patients, regions, and clusters. b, Validation of snATAC-seq cell type annotation using snRNA-seq data. Left: UMAP showing the predicted labels for snATAC-seq data. Right: heatmap showing evaluation results by adjusted rand index (ARI). c, snATAC-seq cell-type proportions of all samples. d, Spearman correlations of cell-type compositions of samples estimated from snRNA-seq and and snATAC-Seq. Box-Whisker plots showing median and IQR (n = 3 for BZ, n = 4 for control, n = 4 for RZ, n = 5 for FZ, and n = 9 for IZ) e, Cell type detection for snRNA-seq and snATAC-seq data across samples. The colour represents in which modality was cell type detected: snRNA-seq and snATAC-seq, snRNA-seq only, or not recovered. f, Transcription factor (TF) binding and TF target expression per major cell type based on the snATAC-seq and snRNA-seq data. g, Overrepresented spatially variable biological processes (myocardium related, immune related and fibrosis related) across regions. Each cell contains the mean adjusted P-value of a hypergeometric test across all spatial transcriptomics samples belonging to each region. h, Spearman correlations of cell-type compositions of samples estimated from spatial transcriptomics and snRNA-seq (left) or snATAC-seq (right). Box-Whisker plots showing median and IQR (n = 3 for BZ, n = 4 for control, n = 4 for RZ, n = 5 for FZ, and n = 9 for IZ). i, GWAS SNP enrichment score across major cell types. j, Visualization of GWAS12 ll SNP enrichment (left ventricular ejection fraction) in spatial transcriptomics data. In d,h, each spot is a patient sample (n = 3 for borderzone (BZ), n = 4 for controls, n = 6 for fibrotic zone (FZ), n = 9 for ischaemic zone (IZ), n = 5 for remote zone (RZ)). Data are represented as boxplots where the middle line is the median, the lower and upper hinges correspond to the first and third quartiles, the upper whisker extends from the hinge to the largest value no further than 1.5 × IQR from the hinge (where IQR is the inter-quartile range) and the lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge, while data beyond the end of the whiskers are outlying points that are plotted individually. In a-c the number of spots of the bottom panels correspond to the barplots in the upper panel.
Molecular mapping of cell types in space
Using these data, we first identified overrepresented biological processes for each major histomorphological region (control, remote zone, border zone, ischaemic zone and fibrotic zone) using spatially variable genes (Supplementary Table 7). We identified cardiac muscle contraction in remote zones and controls, with adaptive immune system in the border and ischaemic zones and with matrisome processes in the fibrotic zones (Extended Data Fig. 2g). Overall, this analysis confirmed that the spatial data clearly reflect typical zones of biological processes following acute human myocardial infarction.
Since each spatial transcriptomics spot captured a group of cells, we increased its resolution by estimating the cell-type compositions of each spot. To this end, we deconvoluted each spot on the basis of the annotated snRNA-seq data from the same sample (Fig. 1h, Supplementary Figs. 2e–g and 3a,b, Supplementary Tables 8 and 9 and Methods). The estimated cell-type compositions from spatial transcriptomics of each patient generally agreed with their respective observed compositions in the snRNA-seq and snATAC-seq data (Extended Data Fig. 2h). We then estimated signalling pathway activities with PROGENy (Methods) for each spot from the spatial gene expression data. The comparison of spatially localized pathway activities with the estimated cellular abundance per spot enabled us to link the information on spatial cell composition to cellular function for each slide. For example, in areas with an abundance of fibroblasts, we detected increased TGFβ signalling activity, and in ischaemic regions, increased myeloid cell abundance occurred in areas of higher NFκB signalling activity (Fig. 1h,i).
Mapping the information obtained from the snATAC-seq data to space resulted in spatially resolved footprinting-based transcription factor binding activity, as exemplified by the previously described transcription factors associated with cardiomyocytes (for example, MEF2C; ref. 7), myeloid cells (for example, CEBPD8 and ATF111), fibroblasts (for example, FOS–JUNB9) and vSMCs (for example, SRF10) (Fig. 1j). To test the association of genetic variants with cell types, we performed enrichment analysis based on cell-type-specific pseudo-bulk ATAC-seq profiles and cardiomyopathy-related single nucleotide polymorphisms (SNPs) obtained from genome-wide association studies12 (GWAS). We focussed on SNPs relevant to left ventricular function, since we hypothesized that these might provide the most biologically relevant information for the cellular composition of myocardial tissue. This analysis revealed that SNPs associated with stroke volume and left ventricular end-diastolic volume were enriched in endothelial cells (Extended Data Fig. 2i), consistent with the role of the endothelial cells in cardiac relaxation and dilation13. SNPs associated with left ventricular end-systolic volume and left ventricular ejection fraction were enriched in cardiomyocytes, supporting the relationship between contraction and these left ventricular measures. We also visualized the spatial distribution of GWAS signals by mapping SNPs associated with left ventricular ejection fraction to each spot from spatial transcriptomics (Extended Data Fig. 2j). In summary, our integrated spatial atlas enabled us to map cell-type abundance, signalling pathway activities, transcription factor binding activity and GWAS signals across the complete spectrum of cardiac tissue zonations, providing an in-depth view at tissue remodelling processes following myocardial infarction in humans.
Spatial organization of myocardial tissue
To explore the spatial organization of the myocardial tissue, we leveraged the spatial transcriptomics data. Unsupervised clustering of spots from all samples on the basis of their cell-type compositions identified nine clusters, which we defined as major cell-type niches (Fig. 2a and Extended Data Fig. 3a–d). We hypothesized that these niches represent potential structural building blocks that are shared between different slides and could facilitate comparisons between subjects. Visualization of these niches in space revealed that some niches aligned closely with the underlying sample condition; for example, cell-type niche 8 was equally distributed across a control slide, whereas cell-type niche 5 localized to distinct regions on the ischaemic slide (Fig. 2b). We then tested the overrepresentation of the annotated cell types derived from snRNA-seq in the cell-type niches. We observed 4 myogenic cell-type niches (1, 7, 8 and 9), which were enriched with cardiomyocytes, endothelial cells, and pericytes (Fig. 2c); an inflammatory cell-type niche (niche 5); and a fibrotic cell-type niche (niche 4) containing fibroblasts, myeloid and lymphoid cells. The fibrotic cell-type niche (4) contained a higher proportion of fibroblasts, whereas the inflammatory cell-type niche (5) contained more myeloid and lymphoid cells (Fig. 2c). Finally, we observed niches associated with rare cell types of the myocardium, such as vSMCs (cell-type niches 3 and 6), adipocytes, lymphoid and cycling cells (cell-type niche 2) (Fig. 2c and Extended Data Fig. 3d). Our integrated results provide a comprehensive description of cellular colocalization events, enabling downstream molecular comparisons within this atlas across all tissue zonations. We next tested whether the abundances of major cell types within spots could be predicted by their spatial context described by the cell-type compositions of their neighbourhood. We evaluated three different neighbourhood area sizes using MISTy: (1) the importance of cell-type abundances within a spot (colocalization) (Fig. 2d), (2) in the local neighbourhood (radius of 1 spot), and (3) in an extended neighbourhood that expanded to a radius of 15 spots. We observed that endothelial cells were the most predictive of the abundance of vSMCs, pericytes, adipocytes and cardiomyocytes within all spots, probably reflecting dependencies between cell types of the vasculature (Fig. 2d). Lymphoid and myeloid cells showed strong dependencies with each other in line with zones of immune cell infiltration and inflammation—similarly captured by cell-type niche 5 (Fig. 2d). Notably, we observed strong dependencies between myeloid cells and fibroblasts, which were strongly co-enriched in niche 4 (Fig. 2d and Extended Data Fig. 3e), in line with a known key role of macrophages in fibroblast activation14 and fibroblasts in macrophage attraction15. Between immediate and extended neighbouring spots (Extended Data Fig. 3f–h), we observed stronger dependencies between cells associated with the cardiac vasculature (vSMCs, endothelial cells, pericytes and fibroblasts) indicating that the myocardial vascular network dominates cardiac tissue structural organization.
Fig. 2. Characterization of tissue organization using spatial transcriptomics data.
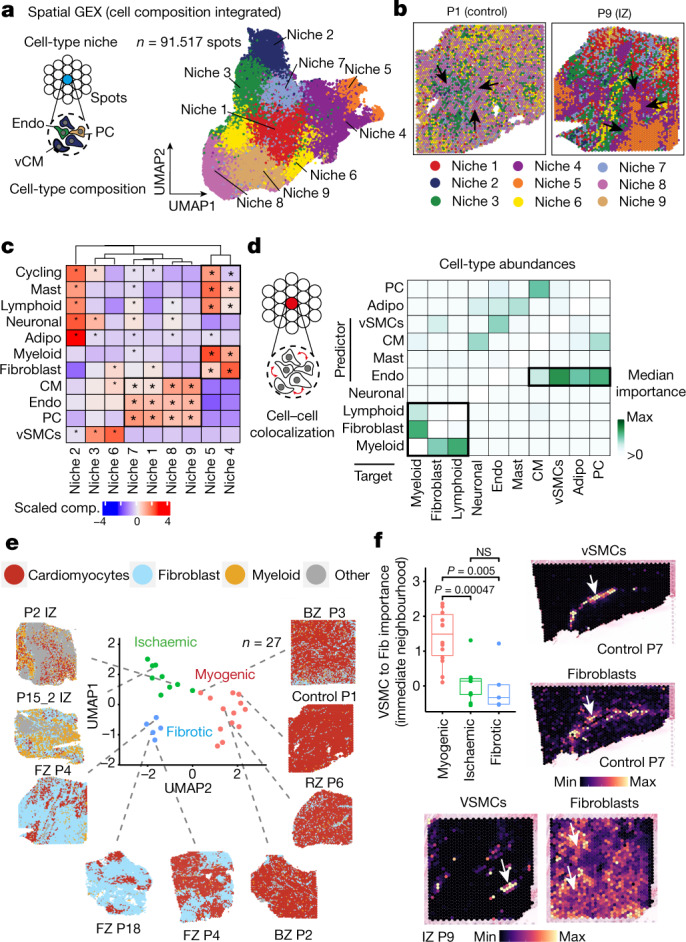
a, Schematic of cell-type niche definition and UMAP of spatial transcriptomics spots based on cell-type compositions. b, Mapping of cell-type niches in a control and an ischaemic zone sample. Arrows show niche 8 (left) and niche 4 (right). c, Scaled median cell-type compositions (comp.) within each niche. Asterisks indicate increased composition of a cell type in a niche compared with other niches (one-sided Wilcoxon rank sum test, adjusted (adj.) P < 0.05). Bold asterisks and outlines show the tissue modules discussed in the main text. d, Median importance of cell-type abundance in the prediction of abundances of other cell types within a spot. e, UMAP of all patient samples from spatial transcriptomics and visualization of abundance of the major cell types in myogenic (control, remote zone and border zone), ischaemic and fibrotic groups. f, Top left, importance of vSMC abundance in the immediate neighbourhood for prediction of fibroblast (Fib) abundance in myogenic, ischaemic and fibrotic groups (adj. P-value using a two-sided Wilcoxon rank-sum test is shown). In all box plots in this Article, the centre line corresponds to the median, the bottom and top hinges delineate the first and third quartiles, respectively, the top whisker extends from the hinge to the largest value no further than 1.5× the inter-quartile range (IQR) from the hinge and the bottom whisker extends from the hinge to the smallest value at most 1.5× IQR from the hinge; data beyond the end of the whiskers are outlying points and are plotted individually. Myogenic group: n = 14, ischaemic group: n = 9, fibrotic group: n = 5. Deconvoluted vSMCs and fibroblast abundance in a myogenic sample (top right) and in an ischaemic sample (bottom). For details on visualization, statistics and reproducibility, see Methods. NS, not significant. Adipo, adipocytes; CM, cardiomyocytes; PC, pericytes; Endo, endothelial cells.
Extended Data Fig. 3. Cell-type niches and spatially contextualized views analysis.

a-b, UMAP embedding of spatial transcriptomics (ST) spots based on major cell-type compositions coloured by the patient (a) or region (b). c, Cell-type niche compositions across patient sample. d, UMAP embedding of ST spots based on major cell-type compositions colored by the compositions of cycling cells, pericytes, adipocytes, endothelial cells and myeloid cells and their respective marker genes (RYR2 cardiomyocytes, PDGFRA fibroblasts, MKI67 cycling cells, ABCC9 pericytes, FASN adipocytes, VWF endothelial cells, IL7R myeloid cells, and MYH11 vSMCs). e, Standardized mean PROGENy pathway activities across different niches. f, H&E staining and cell2location cell-type abundance estimations of cardiomyocytes, fibroblasts and myeloid cells. Delineated areas represent myogenic or fibroblast/myeloid cell enriched tissue areas. g, Median standardized importances (> 0) of cell-type abundances in the prediction of other cell types within the immediate neighbourhood (upper part) and the extended neighbourhood (effective radius of 15 spots) (lower part) inferred from spatially contextualized models. Cell-type abundances of the immediate (upper panels) and extended neighbourhood of cardiomyocytes, fibroblasts and myeloid cells. h, Visualization examples of the dependencies between the abundance of endothelial cells and the abundance of pericytes in the immediate neighborhood, and vascular smooth muscle cells in the extended neighbourhood (effective radius of 15 spots) visualized on three tissues.
To link tissue organization to function, we analysed spatial dependencies between signalling pathways and cell types. Modelled importance of colocalized pathways captured relationships between PI3K and p53 signalling (Extended Data Fig. 4a–e), which showed a mutually exclusive spatial distribution (Extended Data Fig. 4c). Both pathways were related to the abundance of cardiomyocytes (Extended Data Fig. 4a). PI3K signalling in cardiomyocytes controls the hypertrophic response to preserve cardiac functions16, whereas p53 is known to act as a master regulator in cardiac homeostasis17. Spatial segregation of these cardiomyocyte-related pathways points towards functional cardiomyocyte heterogeneity. We observed colocalized and extended neighbourhood relationships of known key pathways in fibrosis including TGFβ and NFκB predicted by fibroblasts, and JAK–STAT and NFκB predicted by immune cells (Extended Data Fig. 4a–e). Overall, cardiomyocytes were the best predictor cell types of the activities of the estimated pathways. Hypoxia and WNT pathways showed a colocalization to cardiomyocytes in ischaemic specimens (Extended Data Fig. 4b–e), highly consistent with the cardiomyocyte differentiation events occurring after myocardial infarction18. Our results compiled principles of tissue organization of the human heart that relate to coordinated cellular processes and provide a basis for comparative analysis.
Extended Data Fig. 4. Spatial pathway and cell type dependency analysis.

a, Median standardized importances (> 0) of cell-type abundances within the spot and in the extended neighbourhood (effective radius = 15), and PROGENy pathway activities within a spot, and in the immediate or extended neighbourhood on the prediction of pathway activities inferred from spatially contextualized models. b, Standardized importances of cardiomyocyte abundances in the extended neighbourhood (radius of 15 spots) in predicting hypoxia pathway activity. Comparison was performed between two sample groups: control, border and remote zones samples (n = 10), and fibrotic and ischaemic samples (n = 15). Two-sided Wilcoxon rank sum test. c–e, Visualization examples of pathway to pathway and pathway to cell-type dependencies in spatial transcriptomics. (c) p53 and PI3K spatial pathway distribution. (d) JAK-STAT and NFkB activities’ dependencies to myeloid and lymphoid cells’ abundance. (e) Cardiomyocyte to WNT and hypoxia pathway dependency. f, Hierarchical clustering of the pseudo-bulk transcriptional profile of spatial transcriptomics slides of patient samples. Colour represents the three major sample groups. g, Comparisons of patient mean major cell-type proportions (inferred from snRNA, snATAC-seq data, and spatial transcriptomics) between myogenic, ischaemic and fibrotic groups. Two-sided Wilcoxon rank sum test. Each dot represents a patient sample (n=13 for myogenic group, n=9 for ischaemic group, n=5 for fibrotic group). h, Pairwise comparison between patient groups of the standardized importances of cell-type abundances within the same spot (intra), and in the immediate (juxta) or extended neighborhood (para) to predict other cell types’ abundances (two-sided Wilcoxon rank sum test). Myogenic vs ischaemic (left), myogenic vs fibrotic (middle), fibrotic vs ischaemic (right). * = adj. P-value <= 0.15. i, Comparison of standardized importances of lymphoid cells’ abundances in the immediate neighborhood (juxta) to predict myeloid cells between myogenic, ischaemic, and fibrotic groups (two-sided Wilcoxon rank sum test). Visualization of lymphoid and myeloid cells in an ischaemic (up) and control (down) sample. j, Comparison of standardized importances of pericytes’ abundances to predict cardiomyocytes’ abundances within the same spot (two-sided Wilcoxon rank sum test). Each dot represents a sample (n = 14 for myogenic group, n=9 for ischaemic group, n = 5 for fibrotic group). Spatial distributions of pericyte and cardiomyocytes abundances estimated from deconvolution in a myogenic sample (left) and a fibrotic sample (right). Arrows in the fibrotic enriched sample show a strong colocalization event. k, Comparison of the compositions of cell-type niches between myogenic, fibrotic, and ischaemic groups (Kruskal-Wallis test, line denotes an adj. P-value < 0.1). Comparison of the proportions of cell-type niches 4, 5, 7, and 8 between groups (adj. P-value estimated from two-sided Wilcoxon rank sum test) and visualization example. Each dot represents a patient sample (n for myogenic group = 13, n for ischaemic group = 7, n for fibrotic group = 5). l, Comparison of the proportions of cell-type niche 9 between groups (two-sided Wilcoxon rank sum test). Each dot represents a patient sample (n for myogenic group = 13, n for ischaemic group = 7, n for fibrotic group = 5). m, Pairwise comparison of the compositions of the cell-type niches between control, border and remote zone samples (two-sided Wilcoxon rank sum test). * = reflect changes with adj. P-value < 0.1, n for CTRL = 4, n for RZ = 5, n for BZ = 3. In b, g, i, j, k, l data are represented as boxplots where the middle line is the median, the lower and upper hinges correspond to the first and third quartiles, the upper whisker extends from the hinge to the largest value no further than 1.5 × IQR from the hinge (where IQR is the inter-quartile range) and the lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge, while data beyond the end of the whiskers are outlying points that are plotted individually.
Structural variation of cardiac tissue
To identify general tissue differences during remodelling after myocardial infarction, we compared the samples of distinct histomorphological regions, time points and individuals at the molecular and compositional level. We defined three major sample groups: myogenic-enriched (including control, border zone and remote zone), fibrotic-enriched (including all fibrotic zone samples, except one) and ischaemic-enriched (including all ischaemic zone samples) samples. Hierarchical clustering of their pseudo-bulk spatial transcriptomics supported this grouping and was displayed as a UMAP embedding (Fig. 2e and Extended Data Fig. 4f). Co-clustering of control, border zone and remote zone samples can be explained by the large abundance of functional myocardial tissue within these specimens (Fig. 2e). Since the pseudo-bulk profile of each spatial transcriptomic dataset combines information of multiple cell types, we next tested how differences in cellular composition determined by all modalities (that is, snRNA-seq, snATAC-seq and spatial transcriptomics) are associated with these three groups. Ischaemic-enriched samples showed a larger proportion of myeloid, lymphoid and cycling cells, with the lowest proportions of cardiomyocytes, representing cellular compositional changes expected after myocardial infarction. By contrast, fibroblasts and vSMCs were enriched in fibrotic-enriched samples (Extended Data Fig. 4g). These results indicate that the spatial transcriptomic data align with major histomorphological sample annotation and capture compositional hallmarks following myocardial infarction across our datasets.
We then analysed whether the cell-type compositional changes between sample groups were also reflected as changes in the spatial dependencies between the major cell types in spatial transcriptomics. To this end, we contrasted the importance, previously computed using MISTy, of each major cell type in predicting the others in the three different neighbourhood area sizes (colocalization, immediate and extended neighbourhood) between the three different sample groups (Extended Data Fig. 4h). We observed an increased spatial dependency in the immediate neighbourhood between lymphoid and myeloid cells in ischaemic samples compared with myogenic-enriched samples, reflecting the expected role that immune cell interactions have in cardiac repair following myocardial infarction (Extended Data Fig. 4i). Moreover, an increased colocalization of cardiomyocytes and pericytes in fibrotic-enriched samples revealed an exclusion of pericytes from scar tissue areas (Extended Data Fig. 4j). Similarly, the distribution of fibroblasts was better predicted by the presence of vSMCs in the immediate neighbourhood only in myogenic-enriched samples, where fibroblasts surrounded the vasculature, in contrast to ischaemic and fibrotic tissue specimens, where more extensive tissue scarring processes were captured (Fig. 2f).
We next compared compositions of cell-type niches between groups and observed differences in six out of nine cell-type niches (Extended Data Fig. 4k). Cell-type niches 8 and 9 (Extended Data Fig. 4k–l), mostly representing cardiac muscle structures, were more present in myogenic- and fibrotic-enriched samples compared with ischaemic-enriched samples, whereas cell-type niche 7, enriched in cardiomyocytes and pericytes (Extended Data Fig. 4k), was reduced in fibrotic-enriched samples. Niche 4, mainly associated with fibrotic structures (more fibroblasts than myeloid cells and thus termed fibrotic niche), was observed in higher proportions in fibrotic-enriched samples, whereas niche 5 (more myeloid cells than fibroblasts and thus termed inflammatory niche) was mainly present in ischaemic-enriched samples (Extended Data Fig. 4k). In summary, the major cell-type niches enabled us to categorize and compare interindividual spatial differences. Overall, this demonstrates the importance of cardiac vasculature in defining the overall myocardial architecture and the unique spatial dependencies of fibroblasts and myeloid cells, which facilitates gaining molecular insights of disease-specific spatial tissue remodelling.
Molecular variation following infarction
To study the molecular differences between similar tissue structures in an unbiased manner across samples, we generated a set of molecular niches by clustering of spots on the basis of their gene expression (Fig. 3a,b and Extended Data Fig. 5a–d). We identified molecular niches associated with inflammatory and fibrotic processes (molecular niches 3, 6 and 9), vSMCs (molecular niche 11) and myogenic-enriched regions (molecular niches 1, 2, 4, 5 and 12) (Fig. 3c). The molecular niches enriched in cardiomyocytes were depleted in ischaemic-enriched samples, whereas the fibrotic- and inflammatory-enriched molecular niches were depleted in myogenic-enriched samples (Fig. 3d and Extended Data Fig. 5e,f). The vSMC-enriched molecular niche 11 had a more distinct cell-type marker gene expression of vSMCs (MYH11) compared with the cell-type defined niche 6 (Fig. 3b versus Extended Data Fig. 3d).
Fig. 3. Characteristic tissue structures inferred from spatial transcriptomics data.
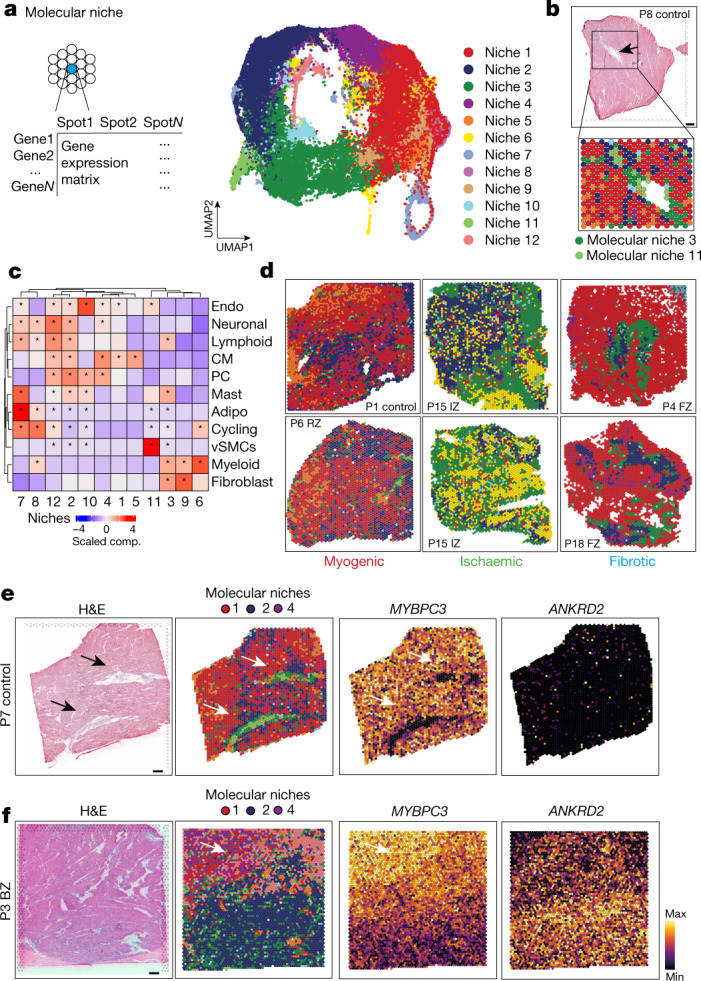
a, Schematic of molecular niche definition and UMAP of spatial transcriptomics spots based on gene expression. b, Spatial mapping of molecular niches. Arrows highlight molecular niche 11 (enriched in MYH11+ vSMCs) surrounded by molecular niche 3 (enriched in PDGFRA+ fibroblasts). c, Scaled median cell-type compositions within each molecular niche. Asterisks indicate increased composition of a cell type in a niche compared with other niches (one-sided Wilcoxon rank-sum test, adj. P < 0.05). d, Distribution of molecular niches in three different patient groups. Note the differential abundance of molecular niches 1 (red) and 6 (yellow). e,f, Haematoxylin and eosin (H&E) staining and visualization of molecular niches 1, 2 and 4 and gene expression (MYBPC3 and ANKRD2) of a control (e) and a border zone (f). Scale bars, 10 mm. For details on visualization, statistics and reproducibility, see Methods.
Extended Data Fig. 5. Characterization of molecular niches.

a, UMAP embedding of ST spots based on gene expression coloured by patients (left) and regions (right). b, Gene expression of RYR2, ABCC9, MYH11, PDGFRa, CD8A and IL7R. Colours refer to gene-weighted kernel density as estimated by using R package Nebulosa. c, Bar plots visualizing molecular niches proportion per patient. d, Standardized mean PROGENy pathway activities across different molecular niches. e, Visualization of molecular niches in control, IZ, and FZ samples. f, Comparison between patient groups of the proportions of molecular niches 5, 6, and 1 (adj. P-value from a two-sided Wilcoxon rank sum test). Each dot represents a patient sample (n for myogenic group = 13, n for ischaemic group = 9, n for fibrotic group = 5). g, Comparison between control (CTRL), border zone (BZ) and remote zone (RZ) of the proportions of molecular niches 1, 2, 3, and 4 (adj. P-value, two-sided Wilcoxon rank sum test, Box-Whisker plots showing median and IQR. Maximum and minimum values as described previously). Each dot represents a patient sample (n for BZ = 3, n for CTRL = 4, n for RZ group = 4). h, Top five differentially upregulated genes between spots belonging to molecular niches enriched with cardiomyocytes. Summary area under the curve (AUC) is used as a size effect of comparing the expression of one molecular niche against the rest. * = reflect FDR < 0.05 and summary AUC > 0.55 (n for mol. niche 0 = 30,058, n for mol. niche 1 = 19,958, n for mol. niche 3 = 7,360). Spatial distribution of the expression of MYLK3 in a control slide (upper) and a border zone (lower) i, Comparison between patient groups of the proportions of molecular niche 2 (adj. P-value, two-sided Wilcoxon rank sum test, Box-Whisker plots showing median and IQR. Maximum and minimum values as described previously). Each dot represents a patient sample (n for myogenic group = 13, n for ischaemic group = 9, n for fibrotic group = 5). j, Same as (i) for niche 3. In f, g, i, j data are represented as boxplots where the middle line is the median, the lower and upper hinges correspond to the first and third quartiles, the upper whisker extends from the hinge to the largest value no further than 1.5 × IQR from the hinge (where IQR is the inter-quartile range) and the lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge, while data beyond the end of the whiskers are outlying points that are plotted individually.
Of note, we observed molecular niches that enabled us to differentiate border zone, remote zone and control samples (Extended Data Fig. 5g), which were indistinguishable using the major cell-type niches (Extended Data Fig. 4m). Molecular niche 3, enriched in fibroblasts and immune cells, was more present in remote zones and border zones compared with control samples. Moreover, we observed differences in the proportions of the molecular niches 1, 2 and 4 among border zone, remote zone and controls (Extended Data Fig. 5g). These three molecular niches were enriched mainly in cardiomyocytes (Fig. 3c), but with a distinct molecular profile: among the top 5 upregulated genes of niche 2 was XIRP1, which encodes an intercalated-disc ion-channel-interacting protein and RRAD, which encodes a GTPase known to regulate L-type Ca2+ channels and contractile functions of the heart19; molecular niche 4 was enriched for SLC8A1 (also known as NCX1), which encodes the Na+/Ca2+ exchanger that is the major regulator of the Ca2+ efflux in cardiomyocytes and is critical to maintain Ca2+ homeostasis during excitation–contraction coupling20, and MPC1, which encodes mitochondrial pyruvate carrier, a known mitochondrial metabolic regulator of heart function21 (Extended Data Fig. 5h). Overall, molecular niche 1 was enriched in control and remote zone samples and niche 2 was enriched in the damaged tissue areas in border zone samples (Fig. 3e,f and Extended Data Fig. 5g). We observed slight changes in enrichment of molecular niches 2 and 4, and a depletion of niche 1 in border zones compared with controls (Extended Data Fig. 5g,i,j), suggesting that differences in cardiomyocyte phenotypes might also be present between these groups. In summary, the comparison of molecular niches pointed towards subtle changes between the remote myocardium and controls, and expected differences between border zone and both controls and remote zone that were not detectable in the cell-type niche comparison. Overall, this suggested the existence of functional differences between cardiomyocyte states in our data.
Disease-specific cardiomyocyte states
To further investigate distinct cardiomyocyte states, we aimed to understand the molecular heterogeneity of cardiomyocytes after myocardial infarction. We co-embedded the snRNA-seq and snATAC-seq data from cardiomyocytes into a common low-dimensional space and clustered the cells (Extended Data Fig. 6a). This uncovered five cell states of ventricular cardiomyocytes (vCM1–5), spanning multiple samples and modalities (Fig. 4a and Supplementary Table 10). Differential gene expression analysis revealed a significant upregulation of ANKRD1 in both vCM2 and vCM3, whereas NPPB showed a distinct upregulation and increased chromatin accessibility in vCM3 (Fig. 4b and Extended Data Fig. 6b,c). We validated this upregulation by single-molecule fluorescence in situ hybridization (smFISH) in an independent patient cohort (Fig. 4c and Extended Data Fig. 6d). Both NPPB and ANKRD1 have been reported to be upregulated in the border zone after myocardial infarction in mice22. vCM2 additionally showed enhanced expression of MYH7 (Extended Data Fig. 6b), a cardiomyocyte-associated stress gene that encodes the β-myosin heavy chain23. Thus, we annotated the vCM2-state as ‘pre-stressed’. In addition, we observed a higher correlation between ion-channel-related genes and vCM1 marker genes compared with ‘stressed’ vCM3 marker genes in spatial transcriptomics, which further highlights the functional differences between these two cardiomyocyte states (Extended Data Fig. 6e). Accordingly, we annotated the vCM3 state as stressed. Moreover, when comparing the differential expression of individual genes belonging to these ion-channel-related gene sets in snRNA-seq data, we observed mostly upregulations in vCM1 compared with vCM3 (Extended Data Fig. 6f,g). Cellular composition comparison between sample groups revealed that vCM1 was associated with myogenic-enriched samples and vCM3 was significantly associated with ischaemic-enriched samples. This was validated in an independent cohort using in situ hybridization, suggesting that these cardiomyocyte states represent distinct cellular stress states within the acute myocardial infarction phase (that is, vCM1, ‘non-stressed’; vCM2, ‘pre-stressed’; and vCM3, ‘stressed’) (Fig. 4c,d and Extended Data Fig. 6h,i).
Extended Data Fig. 6. Characterization of cardiomyocyte state clusters.

a, UMAP embedding of the integrated snRNA-seq and snATAC-seq data coloured by patients, regions, modalities, and clusters (resolution= 1). b, Dot plot showing the top 10 upregulated genes for each CM-state. c, State-specific pseudo bulk ATAC-seq profiles showing distinct chromatin accessibility for NPPB between different states. d, In situ RNA hybridization of NPPB, ANKRD1 and TNNT2 on human cardiac control tissue (upper panel). Note that only a single NPPB/ANKRD1 positive cardiomyocyte could be detected. (below) Representative image of the same in-situ staining of a human myocardial infarction sample. Note the NPPB and ANKRD1 expression in several cardiomyocytes (arrows). Scale bars: 25 µm (upper) + 50 µm (lower). e, Spearman correlation between the spatial expression of Ion-channel transport and transmural-ion channels related genes to the expression of the marker genes (state scores) of vCM1-3 (two-sided Wilcoxon rank sum test). Each dot represents a visium slide (n = 28). f, Differential gene expression of ion channel related genes in vCM1 contrasted to vCM3 cardiomyocytes (two-sided Wilcoxon rank sum test, area under the curve (AUC) is shown as size effect). Colours refer to the gene set membership of each gene. g, Visualization of vCM1 gene marker expression mapping (state scores) and the expression of RYR2 and ATP2B4 in a border zone sample. h, Comparison of vCM1 and vCM2 cell proportion between patient groups. P-values were calculated using Wilcoxon Rank Sum test (unpaired, two-sided). Each dot represents a sample (n = 13 for myogenic group, n = 7 for ischaemic group, and n = 4 for fibrotic group). i, Distribution of vCM3 marker gene expression (state-score) across molecular niche 1, 2, and 4. Each dot represents a spatial transcriptomics spot belonging to a molecular niche across samples (n = 30,058 for niche 1, n = 19,958 for niche 2, niche 4 = 7,360). Two-sided Wilcoxon rank sum test, adj. p-values (niche 1 vs niche 4 = 0, niche 2 vs niche 4 = 2e-09, niche 1 vs niche 2 = 0). j, Visualisation of the expression of ANKRD1 and NPPB, the spatial distribution of TGFβ, hypoxia, p53 and PI3K signaling activities, the spatial distribution of the expression of vCM-states marker genes (state score) for a border zone sample and the distribution of molecular niches 1,2, and 4. Box-Whisker plots showing median and IQR. Maximum and minimum values as described previously. In e, h, i data are represented as boxplots where the middle line is the median, the lower and upper hinges correspond to the first and third quartiles, the upper whisker extends from the hinge to the largest value no further than 1.5 × IQR from the hinge (where IQR is the inter-quartile range) and the lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge, while data beyond the end of the whiskers are outlying points that are plotted individually. In a-c the number of spots of the bottom panels correspond to the barplots in the upper panel.
Fig. 4. Sub-clustering of cardiomyocytes.

a, Sub-clustering of cardiomyocytes. b, Gene expression of ANKRD1 and NPPB. c, Left, smFISH staining of vCM3 marker genes. Scale bar, 50 µm. Right, quantification of NPPB and ANKRD1 signal relative to the TNNT2 signal. Two-sided Wilcoxon rank-sum test (control donors: n = 7, patients with myocardial infarction (MI): n = 10). d, Proportion of stressed vCM3 cells. Wilcoxon rank-sum test (unpaired, two-sided; myogenic: n = 13 myogenic, ischaemic: n = 7, fibrotic: n = 4). e–g, Expression of ANKRD1 and NPPB (e), TGFβ and hypoxia signalling activities (f) and expression of vCM-state marker genes (g) in a border zone sample. h, eGRN analysis including vCM1, vCM2 and vCM3. Each node represents a transcription factor (regulator) or a gene (target). i, Transcription factor activity and expression over pseudotime. Norm., normalized. j, Expression of transcription factor target genes in the border zone sample, as in e. k,l, Mean importance of the abundance of major cell types within a spot (k) and the local neighbourhood (within a 5-spot radius) (l) in the prediction of vCM3 in spatial transcriptomics. m, Importance of vSMC abundance predict vCM3 in myogenic, ischaemic and fibrotic groups within a spot (adj. P-value, two-sided Wilcoxon rank-sum test; myogenic: n = 9, ischaemic: n = 7, fibrotic: n = 4). n, Deconvoluted abundance of vSMCs or fibroblasts and vCM3 state scores in a border zone (left) and a control human heart (right). o, Importance of myeloid cell abundance in the local neighbourhood for predicting vCM3 in control and remote zone samples (two-sided t-test; controls: n = 3, remote zones: n = 3). For details on visualization, statistics and reproducibility, see Methods.
Next, we checked vCM marker genes in spatial transcriptomics in border zone samples, since spatial remodelling of this area is inextricably linked to the recovery of cardiac function. Interestingly, despite homogenous H&E staining and unique molecular identifier (UMI) distribution across spots (Supplementary Fig. 2g), we observed extensively heterogeneous spatial gene expression patterns of ANKRD1 and NPPB (Fig. 4e). Pathway analysis of the spatial gene expression data indicated an increased TGFβ signalling activity within the injured area (lower right), but a homogeneous distribution of hypoxia pathway activity (Fig. 4f). Mapping of cell states to space in a border zone sample revealed that vCM1 were solely located in the top left uninjured corner, vCM2 were located in the middle–top area, serving as a transition zone from injured towards remote myocardium, and vCM3 were primarily located below the transition zone within the injured area (Fig. 4g). Of note, such a spatially distributed pattern was also observed in another border zone sample, indicative of a similar remodelling process (Extended Data Fig. 6j).
Variability of cardiomyocyte states
To infer an enhancer-based gene-regulatory network (eGRN), we leveraged our multi-omics data to further investigate molecular mechanisms differentiating the relevant cardiomyocyte states (that is, vCM1–vCM3) (Methods and Supplementary Table 11). To this end, we paired the cells between snATAC-seq and snRNA-seq data and studied gene-regulatory changes along the cellular continuum from vCM1 to vCM3 (Extended Data Fig. 7a). Next, we estimated an enhancer-mediated transcription factor–target network by considering transcription factor activity (from snATAC-seq), expression of transcription factor and target genes (from snRNA-seq), and motif-supported peak-to-gene links (Extended Data Fig. 7b–d). Clustering of these transcription factors to the target network revealed three major modules, with each corresponding to a distinct cardiomyocyte state (Extended Data Fig. 7e).
Extended Data Fig. 7. Cardiomyocyte pseudotime and gene-regulatory network analysis.

a, Diffusion map embedding of pseudotime cardiomyocyte clusters vCM1, vCM2 and vCM3. b, Computational workflow for building gene regulatory network (upper) and schematic of linking TF to target genes through peak-to-gene links and predicted TF binding sites (lower). c, Heatmap showing TF binding activity and gene expression along the pseudotime trajectory. d, Heatmap of gene expression across pseudotime vCM1-vCM3. e, Heatmap showing the correlation between TF binding activity and gene expression along the pseudotime trajectory from vCM1 to vCM3. Colors on the top refer to pseudotime point where the TF showed the highest binding activity. Clustering analysis identified three modules. f, ANKRD1 and NPPB CM-stress marker gene expression along pseudotime vCM1-vCM3. g, Peak-to-gene links showing that JDP2 regulates TGFB2. Each loop represents a putative link between TGFB2 and a peak. Loop height represents the significance of the correlation and dash line represents threshold of significance (P = 0.05). ATAC-seq tracks were generated from pseudo-bulk chromatin profiles of vCM1, vCM2, and vCM3. Binding sites of JDP2 are highlighted. h, Line plots showing TF activity and expression after z-score normalization (y-axis) over pseudotime (x-axis) for JDP2 (left), its corresponding target gene expression after z-score normalization (y-axis) over pseudotime (x-axis) for TGFB2 (middle), and visualization of all target genes in the BZ sample (right). i, Comparison of standardized importances of myeloid abundances within the spot and fibroblast abundances in the local neighbourhood (radius of 5 spots) to predict vCM3 between patient groups samples. Each dot represents a sample (n = 9 for myogenic group, n = 7 for ischaemic group, n = 4 for fibrotic group). (lower panel) Spatial distribution of the state score of vCM3 and myeloid cell abundances in a RZ slide with histological evidence of a scar. (Two-sided Wilcoxon rank sum test, Box-Whisker plots showing median and IQR. Maximum and minimum values as described previously.) Data are represented as boxplots where the middle line is the median, the lower and upper hinges correspond to the first and third quartiles, the upper whisker extends from the hinge to the largest value no further than 1.5 × IQR from the hinge (where IQR is the inter-quartile range) and the lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge, while data beyond the end of the whiskers are outlying points that are plotted individually. In a-c the number of spots of the bottom panels correspond to the barplots in the upper panel.
We next used network analysis to visualize and detect major transcription factors (Fig. 4h). We identified the mineralocorticoid receptor (NR3C2), a major target of therapy for common heart failure, as a major regulator of the vCM1 state (Fig. 4h). Decreased NR3C2 expression has been associated with the development of severe heart failure and cardiac fibrosis24, and we observed decreased transcription factor binding activity and gene expression along the pseudotime of vCM1 to vCM3 differentiation (Fig 4i). Target genes of NR3C2 include several ion channel genes (such as SLC8A1), which also showed decreased gene expression along the pseudotime axis (Fig. 4i). Notably, these target genes were also differentially expressed in cardiomyocyte-enriched molecular niches (Fig. 3e,f and Extended Data Fig. 5h) and aligned spatially in the border zone with the vCM1 state (Fig. 4j). Notably, we also observed transcription factors (TBX3 and MEF2D) that were associated with pre-stressed stages of cardiomyocyte differentiation (Fig. 4h). Our analysis suggests that MEF2D, a cardiomyocyte factor controlling pacemaker function25, regulates the expression of the sarcomere protein MYBPC3 (Fig. 4i). MYBPC3, in turn, has been reported to regulate cardiomyocyte proliferation postnatally26. Of note, we identified MYBPC3 independently in our spatial data as being enriched in molecular niche 1 (Fig. 3e and Extended Data Fig. 5h).
We also identified ANKRD1, a mediator of cardiomyocyte response to stress27, as a target of MEF2D, suggesting a key regulatory role of MEF2D in the transition from vCM1 to vCM327 (Extended Data Fig. 7f). For vCM3 (stressed cardiomyocytes), we identified ATF3 as a regulator of the GTPase and Ca2+ regulator gene RRAD (Fig. 4h). We independently identified RRAD in molecular niche 2 (Extended Data Fig. 5h), which supports its relevance as a spatially differentially expressed gene of a distinct cardiomyocyte state, especially in border zone samples (Fig. 4i). We additionally identified the transcriptional regulator JDP2—which has a function in preventing cardiomyocyte hypertrophy and cell death28—as an important regulator of the vCM3 cardiomyocyte state, with TGFB2 as one of its target genes (Extended Data Fig. 7g,h). In summary, our cardiomyocyte states and major transcription factor regulators identified from the integrated snRNA-seq and snATAC-seq data reflect expression patterns associated with molecular niches supporting spatial changes of cardiomyocyte states during remodelling.
We next estimated the cell dependencies of the stressed cardiomyocyte state vCM3 with other cell types within each spatial spot and its local neighbourhood (radius of five spots) between sample groups (Fig. 4k–o). We observed that there was an increased importance of vSMCs in predicting vCM3 within a spot in myogenic and ischaemic samples (Fig. 4k), whereas fibroblasts and myeloid cells had a larger role in fibrotic samples (Fig. 4k). The local neighbourhood modelling of vCM3 revealed that the abundance of fibroblasts better explained vCM3 in myogenic-enriched samples compared with fibrotic samples (Fig. 4l and Extended Data Fig. 7i). To gain further insight, we visualized the dependencies of vSMCs and fibroblasts on vCM3 in myogenic-enriched samples and observed that their colocalization occurred in the perivascular niches (Fig. 4n). Overall, this demonstrates that the stressed cardiomyocyte state vCM3 occurs in the perivascular niche of larger blood vessels, highlighting the interaction of mesenchymal cells29 of the perivascular niche with stressed cardiomyocytes in this tissue area. Furthermore, we noticed that when comparing remote zone with control samples, stressed vCM3s are best predicted by myeloid cells (Fig. 4o). This underlines the importance of immune–cardiomyocyte interactions that could additionally explain the increased arrhythmia susceptibility in the remote regions of the post-infarct heart, since it has been shown that cardiac macrophages influence normal and aberrant cardiac conduction30. Our results showed that the stressed-cardiomyocyte vCM3 can be found in distinct spatial cell-type neighbourhoods enriched by different compositions of vSMCs, fibroblasts, adipocytes or myeloid cells.
Cardiac endothelial cell heterogeneity
Co-embedding of snRNA- and snATAC-seq data identified five subtypes of endothelial cells from all major vascular beds, namely capillary endothelial cells, arterial endothelial cells, venous endothelial cells, lymphatic and endocardial endothelial cells (Fig. 5a, Extended Data Fig. 8a–d, and Supplementary Table 12). Subtype-based pseudo-bulk ATAC-seq signals also revealed distinct chromatin accessibility of these marker genes (Extended Data Fig. 8c). Our analysis suggested POSTN as a characteristic marker for endocardial endothelial cells, which we validated using smFISH (Extended Data Fig. 8e). Analysis of cell proportions among the myogenic-enriched, ischaemic-enriched and fibrotic-enriched samples revealed a reduction of capillary endothelial cells in the ischaemic samples associated with a concordant increase in venous endothelial cells (Fig. 5b and Extended Data Fig. 8f,g). Furthermore, we observed that lymphatic endothelial cells were overall less abundant than the other populations, as expected, but were significantly increased in the ischaemic zone, suggesting an increased abundance of lymphatics modulating the immune response following cardiac injury31 (Fig. 5b).
Fig. 5. Sub-clustering of endothelial cells.
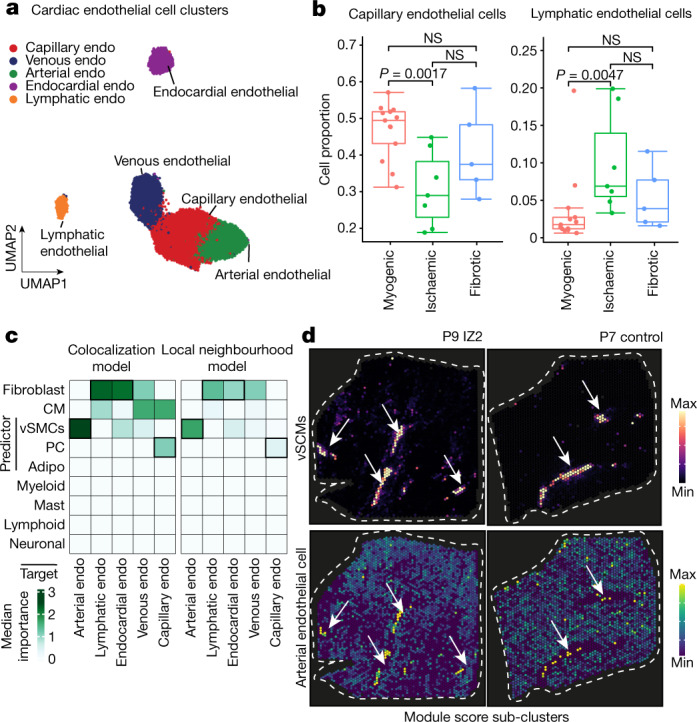
a, Sub-clusters of human endothelial (endo) cells using the integrated snRNA-seq and snATAC-seq data. b, Comparison of capillary endothelial cells and lymphatic endothelial cell proportion between donor and patient groups. Wilcoxon rank-sum test (unpaired, two-sided; myogenic: n = 13, ischaemic: n = 7, fibrotic: n = 4). c, Median importance of the abundance of major cell types within a spot (left) and the local neighbourhood (effective radius of 5 spots) (right) in the prediction of endothelial cell-state scores in spatial transcriptomics. d, Spatial distribution of the abundance of vSMCs and the state score of arterial endothelial cells in an ischaemic (left) and control (right) sample. Arrows point at colocalization events. For details on visualization, statistics and reproducibility, see Methods.
Extended Data Fig. 8. Endothelial cell heterogeneity.

a. UMAP embedding of integrated snRNA-seq and snATAC-seq data coloured by patients, regions, modalities and clusters (resolution= 0.9). b, Gene expression of NRG3, FLT4, SEMA3G and ACKR1. Colours refer to gene-weighted kernel density as estimated by using R package Nebulosa. c, Sub-cluster-specific pseudo bulk ATAC-seq tracks showing the chromatin accessibility of the genes from (b). d, Marker dot plot showing the DEGs for each endothelial cells state and subtype. e, Gene expression of SEM3G and POSTN. Colors refer to gene-weighted kernel density as estimated by using R package Nebulosa. Immunofluorescence of SEMA3G and ACTA2 (upper image). In situ mRNA (RNAscope) for POSTN and PECAM1 (magenta). Arrows highlight endocardial endothelial cells. Scale bars: 75 µm (upper) and 10 µm (lower). f, Endothelial cell state compositions across all patient samples. g, Comparison between patient groups of the venous endothelial cell proportion (two-sided Wilcoxon rank sum test). Each dot represents a patient sample (n for myogenic group = 13, n for ischaemic group = 9, n for fibrotic group = 5). h, Mean Pearson correlation between the abundance of major cell-types and endothelial cell-state scores across all spatial transcriptomics slides. i, Visualization of the spatial distribution of the abundances of pericytes and the state score of capillary endothelial cells sample. Arrows point at colocalization events. j, Visualization of the spatial distribution of molecular niches, the state score of capillary endothelial cells and the abundance of pericytes and cardiomyocytes. Arrows point at locations where molecular niche 10 is present together with high abundances of cardiomyocytes and pericytes. k, Endothelial cell state score distributions in all spots belonging to the molecular niche 10 across all slides (n = 1,874, P-value = 3.19e-298, obtained from a two sided Wilcoxon signed-rank test). l, Mean signalling pathway activities in the molecular niche 10 across different patient groups (adj. P-value of two-sided Wilcoxon Rank Sum test). Each dot represents a slide (n = 14 for myogenic group, n = 9 for ischaemic group, n = 5 for fibrotic group). m, Overrepresented hallmark pathways in the differentially upregulated genes of molecular niche 10 across different patient groups (hypergeometric tests, adj. P-values). In g, k, l data are represented as boxplots where the middle line is the median, the lower and upper hinges correspond to the first and third quartiles, the upper whisker extends from the hinge to the largest value no further than 1.5 × IQR from the hinge (where IQR is the inter-quartile range) and the lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge, while data beyond the end of the whiskers are outlying points that are plotted individually. In a–c the number of spots of the bottom panels correspond to the barplots in the upper panel.
We modelled the association of the different endothelial cell subtypes with the abundances of the other major cell types in spatial transcriptomics. We observed that the markers of arterial endothelial cells were best predicted by vSMCs within a spot and in the local neighbourhood (radius of five spots) reflecting the anatomy of arteries and arterioles in the heart (Fig. 5c,d and Extended Data Fig. 8h). Moreover, the expression of markers of capillary endothelial cells were best predicted by the presence of pericytes in the tissue, in line with the known presence and role of pericytes in direct contact with capillary endothelium32 (Extended Data Fig. 8i). The other endothelial subtypes were mainly predicted by the presence of fibroblasts within a spot and in the local neighbourhood (Extended Data Fig. 8h). Additionally, we observed that the abundance of myeloid cells correlated with the expression of markers of lymphatic endothelial cells (Extended Data Fig. 8h). Focusing on molecular niche 10, which contained the highest cell proportion of endothelial cells and additionally pericytes and mast cells (Extended Data Fig. 8j), we observed a significant enrichment of capillary endothelial cells (Extended Data Fig. 8k). Pathway analysis revealed a significantly higher hypoxia and TGFβ signalling activity in ischaemic and in fibrotic samples, underlining the importance of these processes in chronic fibrotic cardiac remodelling processes (Extended Data Fig. 8l). Pathways important for endothelial signalling in homeostasis such as PI3K and TRAIL showed a reduction in the fibrotic and ischaemic groups, respectively, highlighting further the differential endothelial cell signalling changes. Gene set enrichment analysis further revealed an altered metabolism (for example, fatty acid metabolism and oxidative phosphorylation) of this endothelial cell niche in diseased samples which was further associated with an increased inflammatory response via the TNF and NFκB pathways and increased apoptosis signalling33 (Extended Data Fig. 8m). In summary, we resolved all major endothelial cells states, localized them in space and described their spatial dependencies. Further, we identified a spatial niche enriched in capillary endothelial cells with complex metabolic and signalling changes.
Cardiac myofibroblast differentiation
To dissect molecular and cellular mechanisms of fibrogenesis in the human heart, we clustered all fibroblasts using the integrated snRNA-seq and snATAC-seq data and identified four sub-clusters (Fib1–4) (Fig. 6a, Extended Data Fig. 9a and Supplementary Table 13). Fib1 was marked by SCARA5, which we recently reported as a marker for myofibroblast progenitors in the human kidney34. Fib2 was marked by POSTN, COL1A1 and FN1, which, together with the fact that this population expresses most extracellular matrix (ECM)-related genes, suggests that Fib2 indeed comprises terminally differentiated myofibroblasts (Fig 6b and Extended Data Fig. 9a–c). Notably, Fib2 also exhibited an upregulation of RUNX1, which we recently reported as being involved in kidney myofibroblast differentiation35. Overexpression of RUNX1 in human heart PDGFRβ-expressing cells led to increased myofibroblast differentiation and matrix expression (Extended Data Fig. 9d). We validated the presence of high SCARA5 expression in fibroblasts by co-staining with the pan-fibroblast and myofibroblast marker COL15A1 as well as POSTN and COL1A1 in human heart tissues, and demonstrated that POSTN is significantly enriched in COL1A1+ cells compared with SCARA5+ cells (Extended Data Fig. 9e). Visualization of these markers in our spatial transcriptomics dataset suggested that Fib1 and Fib2 were enriched in mutually exclusive regions of the heart following injury (Fig. 6c and Extended Data Fig. 9f). Additionally, we observed that Fib1 comprised the highest proportion in myogenic-enriched samples, whereas Fib2 (myofibroblasts) were significantly enriched and Fib3 slightly reduced in ischaemic samples (Fig. 6d and Extended Data Fig. 9g,h).
Fig. 6. Characterization of mesenchymal–myeloid interaction.

a, UMAP of human cardiac fibroblasts (integrated snRNA-seq and snATAC-seq data). b, Expression of SCARA5, COL1A1, POSTN and FN1. c, Visualization of the markers in spatial transcriptomics data. d, Comparison of Fib1 and Fib2 compositions. Wilcoxon rank-sum test (unpaired, two-sided; myogenic: n = 13; ischaemic: n = 8, fibrotic: n = 5). e, Diffusion map of Fib1 and Fib2 populations. Colours refer to pseudotime points. f, Same as e, with colours referring to ECM score. g, eGRN analysis, including Fib1 and Fib2. Each node represents a transcription factor (regulator) or gene (target). Targets are coloured by clustering results and regulators are coloured by pseudotime with maximum transcription factor activity. The size of regulator nodes represents centrality. h, Transcription factor activity and expression over pseudotime and their corresponding target gene over pseudotime. i, Visualization of KLF4 and TEAD3 target genes and TGFβ pathway activity in a remote zone (left) and ischaemic zone (right) sample. j, UMAP of sub-clusters of human cardiac myeloid cells using the integrated snRNA-seq and snATAC-seq data. cDC, classical dendritic cell; MQ, macrophage. k, Gene expression of LYVE1, CCL18, ZBTB46 and SPP1. l, Median importance of myeloid cell states in the local neighbourhood in the prediction of fibroblast cell states. m, Cell-state scores of myofibroblasts (Fib2) and SPP1+ MQs in a remote zone sample. Arrows point to regions where there is an observed colocalization. n, In situ staining of CD163, POSTN and SPP1 on human cardiac myocardial infarction tissue. Arrows indicate CD163+SPP1+ macrophages near myofibroblasts. Scale: 10 µm. Quantification of SPP1+ macrophages relative to CD163+ macrophages from the in situ hybridization images (adj. P-value from a two-sided Wilcoxon rank-sum test, n = 8 control group, n = 6 fibrotic group, n = 12 ischaemic group). For details on visualization, statistics and reproducibility, see Methods.
Extended Data Fig. 9. Fibroblast heterogeneity and PDGFRb lineage tracing in murine MI.

a. UMAP embedding of integrated snRNA-seq and snATAC-seq data coloured by patients, regions, modalities and clusters (resolution = 0.9). b, Marker dot plot showing the DEGs for each fibroblast state. c, Box plots showing module score of NABA-collagen and NABA-core-matrisome score per fibroblast state. P-values were calculated using Wilcoxon rank sum (unpaired and two sided). d, Expression of COL1A1, ACTA2 and FN1 by RNA qPCR after RUNX1 overexpression with and without TGFβ compared to empty vector (EV) (n = 6). One-way ANOVA followed by Bonferroni correction. Error bars = S.D. e, In situ hybridization (RNAscope) on human myocardial tissue of SCARA5 and COL15a1 and quantification and comparison of SCARA5+/POSTN+ cells vs. POSTN+/COL1A1+ cells in human heart failure tissues (n = 7). Mann-Whitney test. Error bars = S.D. f, Visualization of SCARA5, POSTN, COL1A1 and FN1 on spatial transcriptomics slides from IZ and FZ human MI samples. g, Cell-type state compositions across patient sample. h, Comparison of Fib3 cell proportion between patient groups. P-values were calculated using Wilcoxon Rank Sum test (unpaired, two-sided). Each dot represents a sample (n = 13 for myogenic group, n = 7 for ischaemic group, and n = 4 for fibrotic group). i, Time course of lineage tracing experiment using PDGFRβCreER-tdTomato mice. j, Results of echocardiographic measurements EF (in %), and global longitudinal strain (GLS, in %), for each of the time points. One-way-ANOVA. n=4 day 0, n=3 day 4, n=4 day 7, n=4 day 14. Error bars = S.D. k, Quality measurements of single-cell RNA-Seq data from mouse MI experiment. l, UMAP representation of time points, cluster and gene expression (POSTN, SCARA5, COL1A1 and FN1) from mouse MI experiment. m, Confusion matrix comparing predicted fibroblasts states and obtained clusters for mouse dataset. n, UMAP representation of species cross-annotation of fibroblast clusters. o, Cell proportion of mouse fibroblast state Fib1 (SCARA5+) and Fib2 (POSTN+) per MI time-point. p, Box plots showing NABA collagens scores and NABA core matrisome score per fibroblast state (mouse Fib1 vs. Fib1) per time point. P-values were calculated using Wilcoxon rank sum (unpaired and two sided) (Sham: n = 578 for Fib1 and n = 685 for Fib2; Day 4: n = 1688 for Fib1 and n = 2498 for Fib2; Day 7: n = 203 for Fib1 and n = 330 for Fib2; Day 14: n = 2232 for Fib1 and n = 7684 for Fib2). q, Gene set enrichment analysis per human fibroblast-cell state. Scale bar: 10 µm. In c, d, h, k, p data are represented as boxplots where the middle line is the median, the lower and upper hinges correspond to the first and third quartiles, the upper whisker extends from the hinge to the largest value no further than 1.5 × IQR from the hinge (where IQR is the inter-quartile range) and the lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge, while data beyond the end of the whiskers are outlying points that are plotted individually.
To precisely understand differentiation trajectories of fibroblasts and transfer this knowledge to the human data, we performed inducible lineage tracing in mice using the pan-mesenchymal Cre driver Pdgfrb-CreER (crossed to a R26-tdTomato reporter) combined with scRNA-seq at different time points following myocardial infarction (Extended Data Fig. 9i–l). We integrated and annotated the cells by label transfer (Fib1–4) from human to mouse (Extended Data Fig. 9m,n). We observed an overall increase of the Fib2 population and collagens and ECM genes over time, whereas the Fib1 proportion was decreased, pointing towards a differentiation trajectory from SCARA5+ fibroblasts (Fib1) to myofibroblasts (Fib2) in mice (Extended Data Fig. 9o,p). Based on these observations, we inferred a pseudotime trajectory from Fib1 (SCARA5+) to Fib2 (myofibroblast) in the human samples, which was further supported by an increased enrichment of the ECM score (Fig. 6e,f) and of ECM biological gene ontology processes consistent with fibroblast-to-myofibroblast differentiation (Extended Data Fig. 9q).
To understand the regulatory mechanisms of these stromal cell differentiation processes we inferred a fibroblast eGRN (Fig. 6g, Extended Data Fig. 10a,b and Supplementary Table 14). Clustering resolved two eGRN modules that each corresponded to a distinct fibroblast state (Extended Data Fig. 10c) and identified potential regulators of myofibroblast differentiation (Fig. 6g). Among the transcription factors regulating the Fib1 module was KLF4, which regulates diverse cellular functions including cellular growth arrest, and is also one of the original reprogramming factors of induced pluripotent stem cells. Our network analysis highlighted the role of KLF4 in regulating SCARA5 and PCOLCE2 expression in Fib1, and it also targets MBLN1, an important regulator of cardiac wound healing36 and fibroblast-to-myofibroblast transition37. Concordantly, we observed reduced KLF4 binding activity and reduced SCARA5 expression in our pseudotime analysis (Fig. 6h), highlighting the role of KLF4 as a putative inhibitor of fibroblast activation. Among the transcription factors identified in the Fib2 module were TEAD3 (an effector of the Hippo pathway), GLI2 (in the hedgehog pathway) and RUNX2, which have been previously identified as regulators of myofibroblast differentiation38 (Fig. 6h and Extended Data Fig. 10d,e). Our network analysis revealed that both TEAD3 and GLI2 regulate bona fide myofibroblast target genes including COL1A1, TGFB1 and POSTN. Additionally, our network analysis identified the key anti-angiogenic regulator THBS139 as a direct target of TEAD3 and the recently identified cardiac fibrosis regulator MEOX1 in human cardiac myofibroblasts40. We next visualized the expression of the KLF4 and TEAD3 target genes in spatial transcriptomics slides and observed gradients and mutually exclusive spatial expression in defined cardiac regions of fibrotic responses, highlighting their differential spatial activity in the human heart (Fig. 6i and Extended Data Fig. 10d).
Extended Data Fig. 10. Gene regulatory network analysis of human cardiac fibroblasts.

a, Pseudotime heatmap showing gene expression (left) and TF binding activity (right) along the trajectory Fib1 to Fib2 (myofibroblasts). b, Pseudotime heatmap showing highly variable genes along the trajectory. c, Heatmap showing the correlation between TF binding activity and gene expression each TF from (a) and gene from (b). Each column represents a TF and each row represents a TF. Colours on the top refer to pseudotime labels for each TF estimated based on binding activity. d, Upper: line plots showing TF activity and expression after z-score normalization (y-axis) over pseudotime (x-axis) for GLI2 and their corresponding target gene expression after z-score normalization (y-axis) over pseudotime (x-axis) for TGFB1. Lower: visualization of target gene expression of KLF4 and GLI2, and TGFB pathway activity in the BZ sample. e, Line plots showing TF activity and expression after z-score normalization (y-axis) over pseudotime (x-axis) for RUNX2 and its corresponding target gene expression after z-score normalization (y-axis) over pseudotime (x-axis) for COL1A1. Visualization of RUNX2 target genes in the BZ sample (right).
Fibro-myeloid spatial relations
Myeloid-derived cells have been reported to have key roles in cardiac remodelling following myocardial infarction41. To understand their heterogeneity, we sub-clustered them using the multi-omic data and identified five sub-clusters across all myocardial infarction samples (Fig. 6j,k, Extended Data Fig. 11a–d and Supplementary Table 15). We observed that two clusters showed expression of resident myeloid cell markers42 (LYVE- and FOLR-expressing myeloid clusters), as well as a CCL18- and SPP1-expressing macrophage cluster and a monocyte and classical dendritic cell cluster (Fig. 6j and Extended Data Fig. 11b–d). We used an independent snRNA-seq dataset of three acute human myocardial infarction samples as reference for validation and found high concordance in terms of myeloid cell populations based on marker gene expression (Extended Data Fig. 11e). Cell proportion analysis revealed an increased abundance of a macrophage population defined by SPP1 expression in the ischaemic sample group, whereas CCL18+ macrophages were increased in fibrotic samples (Extended Data Fig. 11f). SPP1+ macrophages have been described in pulmonary fibrosis and COVID-1943,44, and recent work suggests a role of these cells in cardiac tissue remodelling in zebrafish45. We observed an upregulation of CD36 in the SPP1+ myeloid population; CD36 encodes a macrophage receptor known to be important for macrophage phagocytosis, binding to apoptotic and dead neutrophils and having a unique role in cardiac remodelling following myocardial infarction46 (Extended Data Fig. 11b). Indeed, smFISH staining of SPP1+ macrophages suggests increased phagocytic activity, since multiple intracellular vacuoles could be observed (Extended Data Fig. 11g,h). Quantification of multiplex in situ hybridization of SPP1, TREM2 and CCR2 in human myocardial infarction tissue specimens revealed that approximately half of all TREM2-expressing myeloid cells also express SPP1, whereas CCR2+ myeloid cells where less frequent (Extended Data Fig. 11i). Cell-dependency analyses of myeloid cell states revealed a close interaction for two identified LYVE+ resident macrophage populations, whereas the disease-enriched SPP1+ macrophages predicted the presence of CCL18+ macrophages (Extended Data Fig. 11j,k).
Extended Data Fig. 11. Myeloid cell heterogeneity and spatial mapping in human myocardial infarction.

a, UMAP embedding of integrated snRNA-seq and snATAC-seq data coloured by patient contribution, region, modality and clusters (resolution= 1). b, Marker dot plot showing the DEGs for each fibroblast state. c, Gene expression of MRC1, ITGAX, FLT3 and CD163. Colors refer to gene-weighted kernel density as estimated by using R package Nebulosa. d, Bar plots visualizing myeloid cell sub-population proportion per patient. e, Gene expression of MRC1, ITGAX, SPP1, CCL18, FLT3, ZBTB46 in an external dataset of ischaemic patients. Colors refer to gene-weighted kernel density as estimated by using R package Nebulosa. f, Comparison of myeloid cell proportion between patient groups. P-values were calculated using Wilcoxon Rank Sum test (unpaired, two-sided) (n = 13 for myogenic, n = 8 for ischaemic, and n = 5 for fibrotic group). g, In situ hybridization of CCR2, SPP1 and TREM2 on human myocardial infarction section IZ. Arrows point to phagocytotic vacuoles in the magnification. h, In situ hybridization of SPP1, POSTN and CD163 on human myocardial infarction section IZ. Note that SPP1+ macrophages colocalized in distinct tissue regions and appeared to have an enlarged cell-size. i, In situ hybridization quantification of cell type proportion per tissue section. n=7 patient tissues. Adjusted P-values were calculated using Wilcoxon signed-rank test. j, Median standardized importances (>0) of the cell-state scores of myeloid cells within the spot in the prediction of other myeloid cell-state scores in spatial transcriptomics. k, Mean pearson correlation between myeloid cell’s state scores across all spatial transcriptomics slides. Data in f, i are represented as boxplots where the middle line is the median, the lower and upper hinges correspond to the first and third quartiles, the upper whisker extends from the hinge to the largest value no further than 1.5 × IQR from the hinge (where IQR is the inter-quartile range) and the lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge, while data beyond the end of the whiskers are outlying points that are plotted individually.
Following acute myocardial infarction, an inflammatory response is triggered, resulting in tissue remodelling that can lead to heart failure47. It has been demonstrated that SPP1 itself can activate fibroblasts in vitro48, highlighting the fibro-myeloid signalling interaction as a crucial driver of the cardiac remodelling process. To further gain insights about the spatial dependencies of the myeloid and fibroblasts states, we modelled their marker expression using the spatial transcriptomics data. We observed that the presence of SPP1+ macrophages better predicted all fibroblasts states compared to other myeloid cell states, with a higher importance for myofibroblasts within a spot and in the local neighbourhood (Fig. 6l and Extended Data Fig. 12a). Myofibroblast marker expression aligned with a gradient of expression of the markers of SPP1+ macrophages (Fig. 6m). This pattern was also recovered by our cell-type niche definition, in which the inflammatory niche 5 was surrounded by the fibrotic-rich niche 4 (Extended Data Fig. 12b), which we could confirm by a higher expression of SPP1+ macrophages and myofibroblast marker genes in niche 5 compared with niche 4 (Extended Data Fig. 12c). As our data pointed towards a clear spatial association of myeloid cells and fibroblasts, and spatially associated cells are presumably more likely to communicate with each other, we next used receptor–ligand interaction analysis to study their cellular crosstalk. We observed an overall complex myeloid–fibroblast interaction (Extended Data Fig. 12d), and detected distinct changes in crosstalk between SPP1+ macrophages and Fib2. This included increased PDGF-C, PDGF-D and THBS1 signalling in ischaemic versus myogenic samples and increased ADAM17 and TGFB1 in fibrotic versus myogenic samples (Extended Data Fig. 12e). Of note, we observed enhanced TGFβ1 signalling in ischaemic versus myogenic samples towards Fib3 (Extended Data Fig. 12f). To validate the spatial interaction of SPP1+ macrophages and Fib2, we performed RNA in situ hybridization on human cardiac tissues following myocardial infarction and could confirm the spatial interaction and enrichment of SPP1+ macrophages in an independent tissue cohort (n = 26 patients) using an orthogonal method (Fig. 6n and Extended Data Fig. 12g).
Extended Data Fig. 12. Myeloid cell spatial modelling and cell-cell communication.

a, Median standardized importances (>0) of the cell-state scores of myeloid cells (colocalization) in the prediction of fibroblast cell-state scores in spatial transcriptomics. b, Spatial mapping of cell-type niches in a control and an ischaemic sample. Arrows in the IZ sample show niche 4 and how it surrounds niche 5. c, Distribution of myofibroblasts and SPP1+ macrophages marker gene expression (state-score) across cell-type niche 4 and 5. Each dot represents a spatial transcriptomics spot belonging to a molecular niche across samples (n = 12,427 for niche 4, n = 4,375 for niche 5) (two-sided Wilcoxon rank sum test, adj P-value = 6.02e-133 for myofibroblast, adj P-value = 0 for SPP1+ monocytes). Data are represented as boxplots where the middle line is the median, the lower and upper hinges correspond to the first and third quartiles, the upper whisker extends from the hinge to the largest value no further than 1.5 × IQR from the hinge (where IQR is the inter-quartile range) and the lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge, while data beyond the end of the whiskers are outlying points that are plotted individually. d, Cell-cell communication network representing number of ligand-receptor interactions (edge richness), expression of ligand-receptor pairs (LR scores; colour gradients) and cell centrality (Pagerank; node size) as estimated in snRNA-seq of myogenic, ischaemic and fibrotic samples. e, Sankey plots summarizing the top 50 ligand-receptor interactions for selected source and target cells and contrast. These ligand-receptor pairs are selected by absolute value of the difference in LRscore, as provided by CellPhoneDB method implemented in Liana. f. Sankey plot summarizing top 50 TGFbeta ligand-receptor interactions from Fib2 to SPP1+ Mac. cells. g, In situ hybridization mRNA (RNAscope) staining of CD163 (myeloid), POSTN (myofibroblast) and SPP1 on human cardiac MI tissue (crop-out in Fig. 6n). Arrows indicate CD163+SPP1+ macrophages near myofibroblasts. Scale: 25 µm.
In summary, we have decoded cellular fibroblast and myeloid heterogeneity and spatial modelling of the fibro-myeloid cell states, revealing a unique interaction of SPP1+ macrophages with myofibroblasts across the different stages of human cardiac tissue remodelling.
Discussion
In multicellular organs, such as the human heart, normal cellular function and tissue homeostasis depend on the interaction between neighbouring individual cell types. Single-cell technologies can profile the molecular heterogeneity of the different cell types and changes that occur during disease. However, without spatial context it is unclear how these different cell types interact in space to coordinate tissue functions. Here we provide a comprehensive map of the human heart at early and late stages after myocardial infarction compared to control hearts (non-transplanted donor hearts) by integrating spatial transcriptomics with single-nucleus gene expression and chromatin accessibility data.
Our computational analyses enabled an increased resolution of spatial transcriptomics by estimating cell-type compositions for each location and by estimating pathway activities, mapping transcription factor binding activities, and projecting GWAS SNPs. These different layers of biological information enabled us to link the organization in human heart tissue specimens of different histomorphological regions, different time points after myocardial infarction and different individuals to cellular functions. Here we characterized inflammatory and fibrotic remodelling events that differentiated functional myocardium from ischaemic and chronically remodelled tissue. We explored the effects that these remodelling events had on cardiac architecture, specifically on the vasculature and the dependencies between fibroblasts and myeloid cells. Furthermore, we identified spatial enrichment of different functional states of myogenic regions in control, remote and border zones that were not captured by looking at cell-type compositions or histology only.
Analysis of the integrated snRNA-seq and snATAC-seq data identified different cell states and subtypes for cardiomyocytes, endothelial cells, fibroblasts and myeloid cells. We observed distinct cardiomyocyte cell states associated with spatial distribution, pathway activity and disease condition. Leveraging our multi-omic data, we inferred an eGRN and identified potential regulators of cardiomyocytes and fibroblasts, which were also reflected in spatial transcriptomics data. Our data revealed a distinct niche of the border zone surrounding the injured myocardium, with a sharp border between injured and uninjured cell types and were marked by a gradient of ANKRD1 and NPPB expression. Late-stage remodelling after myocardial infarction was driven by fibrosis, with fibroblast-to-myofibroblast differentiation in distinct tissue areas. Our data provide novel insights into myofibroblast differentiation in human hearts after myocardial infarction, with distinct gene expression and gene-regulatory programmes driving this process. In addition, we decoded the fibroblast myeloid cellular heterogeneity after human myocardial infarction and identified a distinct cellular dependency between myofibroblasts and activated phagocytic macrophages (SPP1+CD36+). The combination of spatial technologies with single-cell data represented an opportunity to study how cardiac cell states are influenced by their tissue microenvironment. The identified interactions between cell types largely reflect the spatial organization of the tissue and, although many other factors are involved, these interactions provide hypotheses for further analysis. Of note, we observed high levels of cell death in the ischaemic samples, as expected, and thus also higher levels of ambient RNA, which could introduce a bias in the analyses. Furthermore, we cannot exclude an overestimation of cardiomyocytes in our cell-type proportion analysis, since about 25% of adult human cardiomyocytes are binucleated49, although multiple nuclei in a cell are reported to be transcriptionally homogenous50.
We envision that our publicly available atlas will serve as a reference for future studies integrating single-cell genomics and epigenomics with spatial gene expression data of the human heart. Furthermore, we believe that our data will facilitate the understanding of spatial gene expression and gene-regulatory networks within the human myocardium and will be a resource for future studies that aim to understand the function of distinct cardiac cell types in cardiac homeostasis and disease.
Methods
Ethics
The local ethics committee of the Ruhr University Bochum in Bad Oeynhausen, the RWTH Aachen University, Utrecht University and WUSTL approved all human tissue protocols (no. 220-640, EK151/09, 12/387 and no. 201104172 respectively). Human myocardial tissue was collected from non-transplanted donor hearts, patients after myocardial infarction undergoing heart transplantation, implantation of a total artificial heart or left ventricular assist device (LVAD) implantation. The study met all criteria of the code of conduct for responsible use of human tissue that is used in the Netherlands. The collection of the human heart tissue was approved by the scientific advisory board of the biobank of the University Medical Center Utrecht, The Netherlands (protocol no. 12/387). All patients provided informed consent and the study was performed in accordance with the Declaration of Helsinki. Written informed consent for collection and biobanking of tissue samples was obtained prior to LVAD implantation.
Human tissue processing and screening
Heart tissues were sampled by the surgeon and immediately frozen in liquid nitrogen. Tissues were homogenized in liquid nitrogen and 7–10 mm3 pieces were embedded in OCT compound (Tissue-Tek) and frozen on dry-ice. Ten-micrometre tissue cryosections were stained with H&E and the appropriate tissue regions were selected for further processing. In total 52 human tissue samples were screened this way and evaluated by a cardiac pathologist. For RNA quality control we minced a 3 × 3 mm3 heart tissue piece in liquid nitrogen and isolated the RNA using Qiagen RNeasy Mini kit (Qiagen) using a proteinase K digestion step as suggested in RNeasy Fibrous Tissue Mini Kit (Qiagen, 74704). RNA integrity number (RIN) analysis (Agilent) was performed using Bioanalyzer RNA 6000 Nano kits (Agilent, No. 5067). RIN ranged from >2 to a maximum of 8.8.
Spatial gene expression assay
Frozen heart samples were embedded in OCT (Tissue-Tek) and cryosectioned (Thermo Cryostar). The 10-µm section was placed on the pre-chilled Optimization slides (Visium, 10X Genomics, PN-1000193) and the optimal lysis time was determined. The tissues were treated as recommended by 10X Genomics and the optimization procedure showed an optimal permeabilization time of 12 or 18 min of digestion and release of RNA from the tissue slide. Spatial gene expression slides (Visium, 10X Genomics, PN-1000187) were used for spatial transcriptomics following the Visium User Guides. Brightfield histological images were taken using a 10X objective on the Nikon Eclipse TiE and a Leica Aperio Versa 200 scanner. Stitching of the raw images was performed using the NIS-Elements software. Next generation sequencing libraries were prepared according to the Visium user guide. Libraries were loaded at 300 pM and sequenced on a NovaSeq 6000 System (Illumina) as recommended by 10X Genomics.
Single-nuclei isolation of human hearts
Single-nuclei isolation was performed as previously described51. Briefly, heart tissue was cut into small pieces (0.5 mm3) in a sterile petri dish on ice and transferred to a tissue homogenizer. Nuclei isolation buffer 0.5 ml (EZ lysis buffer, NUC101, Sigma-Aldrich) plus RNase inhibitor (Protector RNase Inhibitor, Roche) were added to the tissue, and 10-15 strokes with pestle A were applied followed by 10–15 strokes of pestle B. The nuclei were stained with DAPI and FANS sorted using a Sony SH800 to enrich the nuclei. Nuclei isolation from three acute myocardial infarction samples from the WUSTL biobank was performed as described52.
scRNA-seq
Nuclei suspensions with a concentration ranging from 400–1000 nuclei per μl were loaded into the chromium controller (10X, Genomics, PN-120223′) on a Single Cell B chip (10X Genomics, PN-120262) and processed following the manufacturer’ original protocol to generate single-cell gel beads in the emulsion. The sequencing library was generated using the Chromium Single cell 3′ reagent Kit v3 (10X, PN-1000092) and Chromium i7 Multiplex Kit (10X Genomics, PN-120262). Quality control for the constructed library was performed by Tape Station. Libraries were sequenced on NovaSeq targeting 50,000 reads per cell 2 × 150 paired-end kits using the following read length: 28 bp Read1 for cell barcode and UMI, 8-bp I7 index for sample index, and 91-bp Read2 for transcript.
sc-ATAC-seq
The remaining nuclei after processing for 3′ scRNA-seq assay were centrifuged at 500g at 4 °C for 5 min and resuspended in 10 μl of nuclei suspension buffer. After tagmentation the nuclei suspension was loaded on the Chromium Chip E (10X Genomics, PN-1000082) in the Chromium controller according to the manufacturer’s protocol. The library was sequenced on an Illumina NovaSeq 6000 using the following read length: 50 bp Read1 for DNA fragments, 8 bp for i7 index for sample index, 16 bp i5 index for cell barcodes, and 50 bp Read2 for DNA fragments.
RNA in situ hybridization and image quantification
In situ hybridization was performed using formalin-fixed paraffin embedded tissue samples and the RNAscope Multiplex Detection KIT V2 (RNAscope, cat. no. 323100) and RNAscope 4-Plex Ancillary Kit (RNAscope, cat. no. 323120) following the manufacturer’s protocol with minor modifications. The antigen retrieval was performed for 30 min at 99 °C in a water bath (VWR). Tissue pretreatment and washing was performed as suggested by the RNAscope staining protocol. The following probes were used for the RNAscope assay: Hs-CD163 cat. no. 417061-C1, Hs-CCR2 cat. no. 438221-C1, Hs-ANKRD1 cat. no. 524241-C1, Hs-POSTN cat. no. 575941-C1, Hs-Col15a1 cat. no. 484001-C2, Hs-Col1a1 cat. no. 401891-C2, Hs-PECAM1-O2 cat. no. 487381-C2, Hs-NPPB cat. no. 448511-C2, Hs-TREM2 cat. no. 420491-C2, Hs-SPP1 cat. no. 420101-C2, Hs-NPR3 cat. no. 431241-C3, Hs-POSTN cat. no. 409181-C3, Hs-SCARA5 cat. no. 574781-C3, Hs-TNNT2 cat. no. 518991-C3, Hs-SPP1 cat. no. 420101-C4 and Hs-NFE2L1 cat. no. 53850.
Nuclei quantification of H&E stained Visium slides
To quantify nuclei from the H&E staining, we used VistoSeg53, an automated MATLAB pipeline for image analysis. Using this pipeline, the individual TIFF files were used for nuclei segmentation using k-means colour-based segmentation in the image processing toolbox. Next, the binary images were refined with the refineVNS() function for accurate detection of the nuclei. Then a CSV and JSON file was generated that contained the metrics to reconstruct the spot grid to allow for nuclei quantification per 10X Visium detection spot. Counting of nuclei was performed with the countNuclei() function. The images were checked individually with the spotspotcheck() function. Code is available at http://research.libd.org/VistoSeg.
Animal model of myocardial infarction
Myocardial infarction was performed as previously described54. In brief, 12-week-old male and female C57Bl/6J Pdgfrb-creER;tdTomato mice were subjected to chronic left anterior descending artery ligation. The mice were anaesthetized using isoflurane (2–2.5%). The mice were injected 30 min before surgery with metamizole (200 µg g−1 body weight) subcutaneously. Then they were intubated and ventilated with oxygen using a mouse respirator (Harvard Apparatus). Before incision, we injected bupivacaine (2.5 µg g−1 body weight) subcutaneously and intercostally for local analgesia. Then a left thoracotomy was performed, and myocardial infarction was induced by ligature of the left anterior descending artery with 0/7 silk (Seraflex, IO05171Z). The ribs, muscle layer and skin incision were closed. Metamizole was administered for three days via drinking water (1.25 mg ml−1, 1% sucrose) post-surgery. All mice were housed under standardized conditions in the Animal Facility of the University Hospital Aachen (Germany). The operating procedure was in accordance with European legislation and approved by local German authorities (LANUV, reference no. 81-02.04.2017.A410.). Mice were euthanized at different time points (sham, 4 days, 7 days and 14 days). As control, hearts from sham-operated, age-matched mice were taken (2 sham female and 2 sham male mice).
Inducible fate-tracing experiments
For inducible fate tracing, male and female Pdgfrb-creER;tdTomato mice (8 weeks of age) received tamoxifen (3 mg intraperitoneally) 4 times followed by a washout period of 21 days and were then subjected to myocardial infarction surgery or sham (12 weeks of age) as described above and euthanized at 4 days, 7 days and 14 days after surgery.
Echocardiography
Left ventricular heart function was determined by echocardiography performed on a small-animal ultrasound imager (Vevo 3100 and MX550D transducer, FUJIFILM Visualsonics). Recordings of short and long cardiac axis were taken in B mode (2D real-time) using a 40 MHz transducer (MX550D). During the procedure, mice were anaesthetized with 1–2% isoflurane. Ejection fraction (EF) and global longitudinal strain (GLS) were recorded and analysed with the VevoLab Software. The Simpson method was used to assess EF. The GLS was measured in the B-mode of the long axis.
smFISH spot quantification and nuclear segmentation
Images for smFISH were exported in native Nicon format (.nd2). Images were split by channel using bfconvert55 for further processing. RNA spots were quantified using the command line version of Radial Symmetry-FISH (RS-FISH)56. The sigma parameter from RS-FISH, determining spot size, was set to 2.9 for all images. Threshold settings in RS-FISH were manually determined for each channel and were set to the following values for cardiomyocyte state analysis: channel 1 (TNNT2) = 0.0107, channel 2 (ANKRD1) = 0.005, channel 3 (NPPB) = 0.0066. To remove spot counts resulting from low signal, high background images, we removed spots with an intensity lower than the 25th percentile of the channel intensity distribution across all images and applied a minimum intensity threshold of 600. For the quantification of CD163+SPP1+ macrophages, while we were not able to perform full cell segmentation, we performed nuclear segmentation using Mesmer57 with pre-trained nuclear segmentation models to identify all detectable nuclei in each image based on DAPI staining. We subsequently assigned spots to the closest nuclei based on euclidean distance and classified cells as positive or negative for the different markers (POSTN, CD163 and SPP1). Cells with more than 2 spots for a given marker were considered positive for that marker.
Masson trichrome staining
Masson’s trichrome staining was conducted using a ready-to-use kit (Trichrome Stain (Masson) Kit, HT15, Sigma-Aldrich) as described by the manufacturer.
Antibodies and immunofluorescence staining
Heart tissues were fixed in 4% formalin for 4 h at room temperature and then embedded in paraffin. For staining slides were blocked in 5% donkey serum followed by 1 h of incubation with the primary antibody, washing 3 times for 5 min in PBS, and subsequent incubation of the secondary antibodies for 45 min. Following DAPI (4′,6′-diamidino-2-phenylindole) staining (Roche, 1:10.000) the slides were mounted with ProLong Gold (Invitrogen, cat. no. P10144). The following antibodies were used: anti-ACTA2(aSMA)-Cy3 (C6198,1:250, Sigma-Aldrich), anti-SEMA3G (HPA001761, 1:100, Sigma-Aldrich), AF647 donkey anti-rabbit (1:200, Jackson Immuno Research).
Confocal imaging
Acquisition of images was performed using a Nikon A1R confocal microscope using 40× and 60× objectives (Nikon). Image processing was performed using the Nikon Software or ImageJ58.
Generation of a human PDGFRB+ cardiac cell line
PDGFRB+ cells were isolated from a 69-year-old male patient, undergoing left ventricular assist device surgery. To generate a single-cell suspension, the tissue was homogenized in a gentleMACS dissociator (Miltenyi) and digested with liberase (200 µg ml−1, Roche cat. no. 5401020001) and DNase (60 U ml−1), for 30 min at 37 °C. After filtering the cell suspension (70 µm mesh), cells were stained in two steps using a specific PDGFRB antibody (R&D cat. no. MAB1263 antibody, dilution 1:100) followed by Anti-Mouse IgG1-MicroBeads solution (Miltenyi, cat. no. 130-047-102). Following MACS isolation, cells were cultured in DMEM media (Thermo Fisher cat. no. 31885) for 20 days and immortalized using SV40-LT and HTERT. Retroviral particles were produced by transient transfection of HEK293T cells using TransIT-LT (Mirus). Two types of amphotropic particles were generated by co-transfection of plasmids pBABE-puro-SV40-LT (Addgene #13970) or xlox-dNGFR-TERT (Addgene #69805) in combination with a packaging plasmid pUMVC (Addgene #8449) and a pseudotyping plasmid pMD2.G (Addgene #12259). Retroviral particles were 100x concentrated using Retro-X concentrator (Clontech) 48 h post-transfection. Cell transduction was performed by incubating the target cells with serial dilutions of the retroviral supernatants (1:1 mix of concentrated particles containing SV40-LT or rather hTERT) for 48 h. Subsequently at 72 h after transduction, the transduced PDGFRb+ cells were selected with 2 μg ml−1 puromycin for 7 days.
Lentiviral overexpression of RUNX1
The human cDNA of RUNX1 was PCR amplified using the primer sequences 5′- atgcgtatccccgtagatgcc −3′ and 5′- tcagtagggcctccacacgg −3′. Restriction sites and N-terminal 1xHA-Tag were introduced into the PCR product using the primer 5′- cactcgaggccaccatgtacccatacgatgttccagattacgctcgtatccccgtagatgcc −3′ and 5′- acggaattctcagtagggcctccacac −3′. Subsequently, the PCR product was digested with XhoI and EcoRI and cloned into pMIG (pMIG was a gift from W. Hahn) (Addgene plasmid #9044 ;http://n2t.net/addgene:9044; RRID:Addgene_9044). Retroviral particles were produced by transient transfection in combination with packaging plasmid pUMVC (pUMVC was a gift from B. Weinberg (Addgene plasmid #8449)) and pseudotyping plasmid pMD2.G (pMD2.G was a gift from D. Trono (Addgene plasmid #12259 ; http://n2t.net/addgene:12259; RRID:Addgene_12259)) using TransIT-LT (Mirus). Viral supernatants were collected at 48–72 h post-transfection, clarified by centrifugation, supplemented with 10% FCS and Polybrene (Sigma-Aldrich, final concentration of 8 µg ml−1) and filtered with a 0.45-µm PES filter membrane (Millipore; SLHP033RS). Cell transduction was performed by incubating the PDGFB+ cells with viral supernatants for 48 h. eGFP-expressing single cells were sorted with a SH800 Cell Sorter.
Quantitative PCR with reverse transcription
Cell pellets were collected and washed with PBS followed by RNA extraction using the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Two-hundred nanograms total RNA was reverse transcribed with High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and quantitative PCR with reverse transcription was carried out as described29 Data were analysed using the 2Ct method. The primers used are listed in Supplementary Table 18.
Preprocessing of snRNA-seq, snATAC-seq and spatial transcriptome data
For snRNA-seq data, CellRanger software (v6.0.2) was used to perform the alignment with default options. Since the input consists of nuclei, we enabled the option ‘–include-introns’ to include intronic reads. For snATAC-seq data, the CellRanger ATAC pipeline (v2.2.0) was used with the default settings. For spatial transcriptome data, the SpaceRanger software (v1.3.2) was used to pre-process the sequencing data. The option ‘–reorient-images’ was enabled to allow for automatic image alignment. hg38 was used as the reference genome for human data alignment.
snRNA-seq data processing
To identify the major lineages representative of all of our specimens, we created a single-nuclei atlas analysing and integrating each snRNA-seq dataset using Seurat59 (v4.0.1).
Each dataset went through identical quality control processing. We discarded nuclei (1) in the top 1% in terms of the number of genes, (2) with less than 300 genes and less than 500 UMIs, (3) with more than 5% of mitochondrial gene expression, and (4) doublets as estimated using scDblFinder (v1.4.0)60 with default parameters. Count matrices were log-normalized for downstream analyses using a scaling factor of 10,000. We calculated a dissociation score for each cell using Seurat’s module score functions with a gene set provided by O’Flanagan et. al.61 and discarded the nuclei that belonged to the top 1%. To generate an integrated atlas of all samples, log-normalized expression matrices were merged and dimensionality reduction was performed on the collection of the top 3,000 most variable genes that were shared with most of the samples using principal component analysis (PCA). To select the collection of shared variable genes between samples, first we estimated the top 3,000 most variable genes per sample and then selected the top 3,000 most-recurrent genes from them across all samples. PCA correction was performed with harmony62 (v1.0) using as covariates the patient, sample, and batch labels. A shared nearest neighbour (SNN) graph was built with the first 30 principal components using Seurat’s FindNeighbors, and the cells were clustered with a Louvain algorithm with FindClusters. A high resolution (1) was selected to generate a large collection of nuclei clusters to capture representative major cell lineages, even if present in low proportions. Cluster markers were identified with Wilcoxon tests as implemented in Seurat’s FindAllMarkers function. Final assignment of cells to major cell lineages was based on literature marker genes. We filtered out small clusters (median number of nuclei across filtered clusters = 269) with low gene count distributions (median counts across filtered clusters = 756) or feature recovery (median number of genes across filtered clusters = 695), with marker genes that could not be assigned to known cell types of the heart. To visualize all nuclei in a two-dimensional embedding, a UMAP was created with Seurat’s RunUMAP function using the first 30 principal components of harmony’s PCA correction embedding. Major cell-type markers were estimated by performing differential expression analysis of cell-type and patient-specific pseudo-bulk profiles. Pseudo-bulk profiles were calculated by summing up the counts of all cells belonging to the same cell type and patient. Profiles coming from less than 10 cells or profiles from which the maximum gene expression was of less than 1,000 counts per library were discarded. Differentially expressed genes were calculated by fitting a quasi-likelihood negative binomial generalized log-linear model as implemented in edgeR (v3.32.1)63 (false discovery rate (FDR) < 0.15). Each cell type was compared against the rest.
Comparison with independent healthy and ischaemic human heart cell atlases
We compared our generated atlas with another reference human single-nuclei RNA-seq atlas4 at the molecular and compositional level. The counts matrix was downloaded directly from https://www.heartcellatlas.org and we selected the data coming from single-nuclei and left ventricle samples. Nuclei with fewer than 200 genes, and genes expressed in less than 3 nuclei were excluded. Log normalization with a scaling factor of 10,000 was performed with scanpy’s64 (v1.7.0) normalize_total function.
To evaluate our major cell-type annotation, we calculated the enrichment of the top 200 marker genes based on log fold change of each cell type defined in the reference atlas in the list of the top 200 marker genes of each of our defined cell types with hypergeometric tests. Marker genes of the reference atlas were calculated with Wilcoxon tests as implemented in scanpy’s 64 (v1.7.0) rank_genes_groups (adj. P < 0.01). Each cell type was compared against the rest. To evaluate the compositional stability of our control samples, we calculated the Pearson correlation between the median proportion of each shared cell type of the reference atlas and our control, border zone, and remote zone samples. Similarly, we compared our atlas to an independent collection of human heart nuclei derived from three ischaemic specimens. First, we analysed and integrated the smaller collection of samples using identical procedures as the ones used in our provided atlas. After nuclei clustering, we assigned each cluster to a cell type using literature markers. Cell-type markers were calculated with Wilcoxon tests (adj. P < 0.01) and the top 200 genes based on log fold change were selected. Marker overlap and compositional stability comparison with ischaemic specimens from our atlas were performed as described previously.
snATAC-seq data processing
To control the data quality, the fragment files were used as input for the package ArchR (v1.0.1)65, and low-quality cells were filtered out based on transcription start site (TSS) enrichment (> 4) and the number of unique fragments (> 3,000 and < 100,000). Doublets were identified and removed by using the functions addDoubletScores and filterDoublets from ArchR with default settings. Next, peaks were identified by using the function addReproduciblePeakSet for each sample. All peaks were merged to create a union peak set of which each peak was annotated as distal, promoter, exonic and intronic. A count matrix was constructed with the function addPeakMatrix. For dimensionality reduction, the method scOpen (v1.0.0)35 was used to generate a low-dimensional matrix of the cells. The algorithm Harmony62 was applied to correct the batch effects and integrate the data and UMAP was used to generate a 2D embedding for visualization. Cells were clustered using the Leiden algorithm with a resolution of 1. To annotate the clusters, a gene activity score matrix was created using the function addGeneScoreMatrix and marker genes were detected for each cluster using the function getMarkerFeatures. The same markers from snRNA-seq data were used to annotate the clusters.
Comparison between snRNA-seq and snATAC-seq data
The Seurat66 label-transferring approach was used to compare the annotation of snRNA-seq and snATAC-seq. To do so, the snRNA-seq data were used as reference and the function FindTransferAnchors was applied to identify a set of anchors using gene expression from snRNA-seq and gene activity score from snATAC-seq. Next, the cell labels from snRNA-seq were transferred to snATAC-seq by running the function TransferData. An adjusted rand index was calculated to evaluate the agreement between annotated and predicted cell labels for snATAC-seq data.
Cell-type-specific transcription factor binding and regulon activity
To estimate transcription factor binding activity for each major cell type identified from snATAC-seq data, we first aggregated the reads within each cell type and created a pseudo-bulk profile. Next, we used MACS2 (v2.2.7)67 to perform peak calling and removed the peaks from chrY, mitochondrial and unassembled ‘random’ contigs. We then predicted the transcription factor binding sites and estimated transcription factor binding activity using HINT-ATAC (v0.13.2)68 based on the JASPAR2020 database69. We linked the transcription factor binding sites to the nearest genes to create a cell-type-specific transcription factor–gene interaction. The number of ATAC-seq reads in the region with 100 bp up-stream and downstream of the the transcription factor binding site were used to indicate how strong the interaction was: each transcription factor–gene interaction was weighted as the ratio between the number of ATAC-seq reads around the transcription factor binding site associated with that gene and the maximum number of reads observed in any binding site of the transcription factor. All interactions with a weight larger than 0.3 were considered in downstream analysis. This generated weighted and filtered cell-type-specific regulons. To infer a transcription factor regulon activity score, we estimated the mean expression of the target genes in each cell-type-specific regulon. Cell-type pseudo-bulk profiles were filtered to contain only genes with at least 10 counts in 5% of the samples, before the estimation of normalized weighted means using decoupleR’s70 (v1.1.0) wmean function with 1,000 permutations. Regulon activities were standardized and correlated with transcription factor binding activities using Spearman correlations. The minimum correlation of 0.5 was used as threshold and the top 5 transcription factors per cell type were selected for visualization.
Cell-type-specific GWAS signal enrichment
GWAS summary statistics for 4 MRI based left ventricle function parameters12 were downloaded from the Cardiovascular Disease Knowledge Portal (https://cvd.hugeamp.org/). For each phenotype, GWAS summary statistics were clumped with Plink (v1.9)71 to identify index SNPs (clump-p1 = 0.0001, clump-kb = 250, clump-r2 = 0.5) using the European samples from 1000 Genomes as a reference population.
Next, we lifted over the coordinates of index SNPs from hg19 to hg38 using the LiftOver tools. For each major cell type, we generated an average chromatin accessibility profile by using snATAC-seq data from all cells. The cell-type-specific GWAS signal enrichment was performed using gchromVAR (v0.3.2)72 and enrichment scores were normalized to z-scores. P-values were calculated based on the z-scores and were corrected by the Benjamini–Hochberg method.
Cell-type-specific integration of snATAC-seq and snRNA-seq data and sub-clustering
For each major cell type that was recovered by both snATAC-seq and snRNA-seq, we aimed to identify sub-clusters spanning multiple samples and modalities. To do so, we devised a multi-step approach to integrate and cluster the data by controlling quality from sample-, cell-type- and modality-specific aspects.
To minimize the sample-specific effects, we only considered samples with a minimum number of cells in both snATAC-seq and snRNA-seq: for cardiomyocytes and endothelial cells (n_cells_ATAC > 300 and n_cells_RNA > 400); for fibroblasts (n_cells_ATAC > 100 and n_cells_RNA > 400); and for myeloid (n_cells_ATAC > 50 and n_cells_RNA > 200). This step controls for samples with low recovery of cells in a particular modality.
To further filter cell-type-specific low-quality cells from snRNA-seq and snATAC-seq data, we integrated the samples as selected in step 1 using Harmony to correct batch effects from patients and regions based on PCA space (30 dimensions) for snRNA-seq and LSI space (30 dimensions) for snATAC-seq data. We then clustered the cells using Seurat (resolution = 0.4) for each modality independently. We next excluded the clusters that were (i) enriched in a single sample; (ii) showed a lower data quality; (iii) showed a higher doublet score compared with others. Specifically, for cardiomyocytes, we removed 3 clusters from snATAC-seq data: 2 clusters (481 cells) were enriched in a single sample and another cluster (171 cells) showed a low number of unique fragments (average = 8,102). For fibroblasts, we removed 1 cluster (49 cells) from snATAC-seq (98% of cells from a single sample) and 1 cluster (1,172 cells) from snRNA-seq (average doublet score of 0.12). This step controls for cell type and modality-specific low-quality cells.
We next integrated the cells from snATAC-seq and snRNA-seq data. To this end, we used the gene activity score matrix of snATAC-seq estimated by ArchR and the gene expression data from snRNA-seq data as input for canonical component analysis by Seurat. The integrated data were projected into a PCA space (30 dimensions) and Harmony was used to correct the batch effects from samples and modalities. This step generated a co-embedded and batch-corrected dataset composed of cells from snRNA-seq and snATAC-seq samples.
For each major cell type, we defined the sub-clusters based on the co-embedded data using the Seurat (resolution = 0.9 or 1). Marker genes were identified by using the function FindAllMarkers. We next filtered clusters that were mainly driven by a single sample or modality. Finally, we merged and annotated the clusters based on the markers. The final statistics of the sub-clustering results for each major cell type were provided in Supplementary Table 16.
Analysis of snRNA-seq data from mouse fibroblasts
Cellranger mkfastq and count functions (version v6.0.2) with default parameters were used for demultiplexing and aligning the reads, respectively. Reads were aligned to the mouse reference transcriptome (mm10, Version=2020-A). Prior to alignment, reads for tdTomato were added to the reference. Quantified counts from each sample were aggregated and cells with counts <1,500 and >20,000 were filtered out. Further, cells with >5% reads mapped to mitochondrial genes, as well as cells with <500 genes were removed. Scrublet73 was used to detect potential doublets and only the resulting 40,495 cells with <0.2 scrublet score were kept for further analyses. The highly_variable_genes() function with seurat_v3 flavour implemented in Scanpy (version 1.8.1) was used to obtain the top 2,000 most highly variable genes. Count data was log-normalized using sc.pp.normalize_total(target_sum=1e4) followed by sc.pp.log1p(). The data was subset to the 2,000 genes, unwanted sources of variation from n_umi and mito_fraction were regressed out using sc.pp.regress_out(), and the top 30 principal components were estimated using sc.tl.pca(). Harmony was then used to account for large differences across samples using ‘sample’ as the batch indicator. Network neighbourhood graph was constructed using the function sc.pp.neighbors() with 30 adjusted principal components, cosine distance and n_neighbors = 10. Leiden clustering with resolution 1.0 was used to cluster the cells into 17 clusters. Marker genes were identified using the Wilcoxon test implemented in sc.tl.rank_genes_groups() function in Scanpy. Clusters were manually annotated using the marker genes. We next cleaned up the data by removing clusters with low data quality and re-clustered the data with resolution of 0.2. To annotate the cells, we used the label transfer approach from Seurat based on the sub-clustering results from human fibroblasts.
Gene-regulatory network inference for cardiomyocytes and fibroblasts
We inferred an eGRN for cardiomyocytes and fibroblasts using a multi-step approach including modality pairing, transcription factors and genes selection, and network construction.
We first paired the cells between snATAC-seq and snRNA-seq based on the previously described co-embedding space using an optimal matching approach74. This method returns a matching of a snATAC-seq cell to a unique cell in snRNA-seq. Next, we produced a diffusion map75 and created trajectories in this space using the function addTrajectory from ArchR (v1.0.1)65. For cardiomyocytes, we inferred a trajectory from clusters vCM1, vCM2 and vCM3, where vCM1 were considered as roots and vCM3 as the terminal state. For fibroblasts, we built a trajectory with SCARA5+ fibroblasts as root and myofibroblasts as terminal state.
Next, we predicted a single-cell-specific transcription factor binding activity score using the R package chromVAR (v1.16)76 from the snATAC-seq data based on motif from the JASPAR2020 database69. In contrast to HINT-ATAC, chromVAR provides transcription factor activity scores at single-cell level. We next selected transcription factors that display concordant binding activity (snATAC-seq) and its gene expression (snRNA-seq) (Pearson correlation > 0.1). This analysis identified 65 transcription factors for cardiomyocytes and 44 transcription factors for fibroblasts. We considered these transcription factors to be potential regulators. We sorted the transcription factors along the trajectory as defined in step 1 and assigned a pseudotime label to each transcription factor. Next, we selected highly variable genes using the snRNA-seq data along the trajectories as described65. We kept the top 10% variable genes and considered them as potential transcription factor targets.
To associate regulators with targets (that is, transcription factors with genes), we explored the correlation of peak accessibility and gene expression to identify peak-to-gene links. Specifically, for each gene, we consider peaks that are within 125 kb on either side of the transcription start sites, while excluding the promoter regions. This analysis generated a list of enhancer-to-promoter links. We only considered significantly correlated links (FDR < 0.0001) with a positive correlation as before65. Finally, we associated a transcription factor with a target gene if this gene was linked to an enhancer and this enhancer was predicted to be found by this transcription factor.
- To build a quantitative transcription factor–gene-regulatory network, we estimated the correlation of the transcription factor binding activity from snATAC-seq and target gene expression from snRNA-seq data and only considered those interactions with Pearson correlation >0.4. We visualized the network based on a force-layout, which places transcription factors (or target genes) with similar interactions close together. We coloured transcription factor nodes in the networks using the assigned pseudotime labels as inferred in step 2. To characterize the importance of transcription factors, we computed two measures: node betweenness (denoted by b)77 and pagerank (denoted by p)78. A final importance score for transcription factor i was calculated as:
Finally, to map the inferred GRN into spatial transcriptomics data, we used the target genes for each transcription factor and calculated a module score by using the function AddModuleScore from Seurat (v4.1.0).
Characterization of spatial transcriptomics datasets
Single-slide processing
Filtered feature-barcode expression matrices from SpaceRanger (v1.3.2) were used as initial input for the spatial transcriptomics analysis using Seurat (v4.0.1). Spots with less than 300 measured genes and less than 500 UMIs were filtered out. Ribosomal and mitochondrial genes were excluded from this analysis. Individual count matrices were normalized with sctransform79, and additional log-normalized (size factor = 10,000) and scaled matrices were calculated for comparative analyses using default settings.
Cell-type compositions were calculated for each spot using cell2location80 (v0.05). Reference expression signatures of major cell types were estimated using regularized negative binomial regressions and our integrated snRNA-seq atlas. We fitted a model in six downsampled iterations of our snRNA-seq atlas (30%) and generated a final reference matrix by taking the mean estimation. Each slide was later deconvoluted using hierarchical bayesian models as implemented in run_cell2location. We provided the following hyperparameters: 8 cells per spot, 4 factors per spot, and 2 combinations per spot. Additionally, for each spot we calculated cell-type proportions using the cell-type-specific abundance estimations. Cell-type compositions of the complete slide were calculated adding the estimated number of cells of each type across all spots. To compare the stability of estimated cell compositions between our different data modalities, we calculated Spearman correlations between the estimated cell type proportions of each slide and the observed cell type proportions in its corresponding snRNA-seq and snATAC-seq dataset.
Estimation of functional information from spatial data
For each spot, we estimated signalling pathway activities with PROGENy’s81,82 (v1.12.0) model matrix using the top 1,000 genes of each transcriptional footprint and the sctransform normalized data. Spatially variable genes were calculated with SPARKX83 (v1.1.1) using log-normalized data (FDR < 0.001). To obtain overrepresented biological processes from each list of spatially variable genes, we performed hypergeometric tests using the set of canonical pathways provided by MSigDB84 (FDR < 0.05).
Estimation of cell death molecular footprints from spatial data
To associate the differences in nuclei capture in snRNA-seq between the different samples to cell death processes, we leveraged the information from spatial transcriptomics to estimate the general expression of genes associated to cell death for each sample. For each unfiltered slide we estimated per spot the normalized gene expression of BioCarta’s84 ‘death pathway’ and Reactome’s85 ‘regulated necrosis pathway’ using the decoupleR (v1.1.0) wmean method and the sctransform normalized data. To have a final pathway score per slide, we calculated for each slide the mean ‘pathway expression’ across all spots.
Mapping transcription factor binding activity and GWAS enrichment to spatial data
To visualize the transcription factor binding activity estimated from snATAC-seq data in space, we used the estimated cell type proportion calculated from cell2location scores for mapping. Specifically, for each spot i and transcription factor j, we calculated the transcription factor binding activity as follows:
where Proportionik is the estimated proportion of cell type k, K is the number of cell types, and ACT′kj is the binding activity of transcription factor j in cell type k from snATAC-seq data. An equivalent approach was used to map GWAS scores into space.
Cell-state spatial mapping
To map the functional states of each cell type into spatial locations, we leveraged the deconvolution results of each slide and the set of differentially expressed genes of each recovered cell state. Given the continuous nature of cell states, we assumed that the collection of up and downregulated genes of a cell state represented its transcriptional fingerprint and could be summarized in a continuous score in locations where we could reliably identify the major cell type from which the state was derived. For a given major cell type of interest k, we identified spots where its inferred abundance was of at least 10%. To estimate state scores associated with cell type k, we used decoupleR’s (v1.1.0) normalized weighted mean method (wmean) and the set of the upregulated genes of each state defined with snRNA-seq and snATAC-seq (log fold change > 0; Wilcoxon tests, FDR < 0.05). The log fold change of each selected gene was used as the weight in the wmean function.
Analysis of ion channel-related genes
We related the expression of ion channel-related gene sets to the different cardiomyocyte cell states and their location in spatial transcriptomics. First we selected two different gene sets containing ion channel-related genes: (1) Reactome’s85 ‘ion channel transport’ and a curated list of transmural ion channels from Grant et al.86. Gene sets are provided in Supplementary Table 17. First, we calculated gene set scores for each spatial transcriptomics spot using decoupleR’s wmean function. Then we correlated these gene set scores to the spatial mapping of cardiomyocyte cell states in regions where we observed at least 10% of cardiomyocytes. Additionally, we evaluated if any of the genes belonging to these gene sets were differentially expressed between the vCM1 and stressed vCM3 population using Wilcoxon tests as implemented in scran’s (v1.18.5) findMarkers function (area under the curve (AUC) < 0.4, AUC > 0.6, FDR < 0.05).
Spatial map of cell dependencies
We used MISTy’s87 implementation in mistyR (v1.2.1) to estimate the importance of the abundance of each major cell type in explaining the abundance of the other major cell types. Cell-type cell2location estimations of all slides were modelled in a multi-view model using three different spatial contexts: (1) an intrinsic view that measures the relationships between the deconvolution estimations within a spot, (2) a juxta view that sums the observed deconvolution estimations of immediate neighbours (largest distance threshold = 5), and (3) a para view that weights the deconvolution estimations of more distant neighbours of each cell type (effective radius = 15 spots). The aggregated estimated standardized importances (median) of each view of all slides were interpreted as cell-type dependencies in different spatial contexts, such as colocalization or mutual exclusion. Nevertheless, the reported interactions did not imply any causal relation. Before aggregation, we excluded the importances of all predictors of target cell types whose R2 was less than 10% for each slide.
To associate tissue structures with tissue functions, we fitted a MISTy model to explain the distribution of PROGENy’s pathway activities standardized scores. The multi-view model consisted of the following predictors: (1) an intrinsic view to model pathway crosstalk within a spot, (2) a juxta view to model pathway crosstalk between neighbouring spots (largest distance threshold = 5), (3) a para view estimating pathway relations in larger tissue structures (effective radius = 15), (4) an intrinsic view and (5) a para view containing cell2location estimations (effective radius = 15). These last two views model explicitly the relations between cell-type compositions of spots and pathway activities. Cycling cells and TNF were not included in the described analyses. Before aggregation, we excluded the importances of all predictors of target pathway activities whose R2 was less than 10% for each slide.
Niche definitions from spatial transcriptomics data
To identify groups of spots in the different samples that shared similar cell-type compositions, we transformed the estimated cell-type proportions of each spatial transcriptomics spot and slide into isometric log ratios (ILR)88, and clustered spots into groups. These niches represent groups of spots that are similar in cell composition and represent potential shared structural building blocks of our different slides; we refer to these groups of spots as cell-type niches. Louvain clustering of spots was performed by first creating a shared nearest neighbour graph with k different number of neighbours (10, 20, 50) using scran’s89 (v1.18.5) buildSNNGraph function. Then, we estimated the clustering resolution that maximized the mean silhouette score of each cluster. We assigned overrepresented cell types in each structure by comparing the distribution of cell-type compositions within a cell-type niche versus the rest using Wilcoxon tests (FDR < 0.05). We tested if a given cell state was more representative of a cell-type niche by performing Wilcoxon tests between each niche and the rest (FDR < 0.05). Only positive state scores were considered in this analysis.
Additionally, to complement the repertoire of niches identified with cell-type compositions, we integrated and clustered the Visium spots of all slides using their log-normalized gene expression. We called these clusters molecular niches. Integration and clustering of spots was performed with the same methodology as the one used to create the snRNA-Seq atlas. A low resolution was used (0.2) to have a similar number of molecular niches as cell-type niches. Cell-type and cell-state enrichment was performed as mentioned before.
Differential expression analysis of molecular niches enriched with cardiomyocytes
Differential expression analysis between molecular niches enriched in cardiomyocytes (niche 0, niche 1, niche 3) was performed using the log-transformed expression of all spots belonging to a given niche. Wilcoxon tests were performed with scran’s89 (v1.18.5) findMarkers function. Genes with a summary AUC >0.55 and FDR <0.05 were considered upregulated genes.
Differential molecular profiles of the molecular niche 10 enriched with capillary endothelial cells
Differential expression analysis between ischaemic, fibrotic and myogenic-enriched spatial transcriptomic spots was performed with Wilcoxon tests as implemented in scran’s89 (v1.18.5) findMarkers function. To obtain overrepresented biological processes from upregulated genes, we performed hypergeometric tests using the set of hallmark pathways provided by MSigDB84. Normalized PROGENy’s pathway activities for each spot were calculated using decoupleR’s wsum method with 100 permutations on log-transformed data. Mean normalized pathway scores were calculated per slide and comparisons between groups were performed with Wilcoxon tests. Reported P-values were adjusted for multiple testing using the Benjamini–Hochberg procedure.
General differences in tissue organization
We annotated the different spatial transcriptomic slides into three groups based on histological differences with the help of pathologists: myogenic-enriched, fibrotic-enriched and ischaemic-enriched. A general comparison of the sampled patient specimens was performed at the compositional and molecular level.
Hierarchical clustering, with euclidean distances and Ward’s algorithm, was used to cluster the pseudo-bulk profiles of the spatial transcriptomics datasets (replicates where merged, n = 27). Genes with less than 100 counts in 85% of the sample size were excluded for this analysis. Log normalization (scale factor = 10,000) was performed. To visualize the general molecular differences between our samples, log-normalized pseudo-bulk profiles of the spatial transcriptomics datasets were projected in an UMAP embedding.
To identify compositional differences between our sample groups, we compared cell-type and niche compositions. To identify cell-type composition changes associated to the sample groups, mean cell-type compositions across single-cell and spatial datasets were compared with Kruskal–Wallis tests (FDR < 0.1). Pairwise comparisons of sample groups were performed with the Wilcoxon test. Additionally, to test which cell-type and molecular niches had different distributions between our group samples, we performed Kruskal–Wallis tests over the compositions of cell-type or molecular niches (FDR < 0.1). Additional pairwise comparisons were performed with Wilcoxon tests (P-values adjusted with Benjamini–Hochberg procedure). For this, we only consider slides where no single niche represents more than 80% of the spots. Also, we only consider niches representing more than 1% of the composition of at least 5 slides.
To identify differences between the structurally similar tissues captured in the myogenic-enriched group, we separated the samples into remote, border, and control zones and repeated the niche composition comparison described previously.
To identify patterns of tissue organization associated with a sample group, we tested if differential cell dependencies were captured by the MISTy models used to predict cell-type abundance (see ‘Spatial map of cell dependencies’). First, we filtered the standardized importance matrices of each sample’s MISTy model fitted to predict the abundance of major cell-types to contain only the values of target cell types predicted with an R2 greater than 0.05. Then, for each slide we created a spatial dependency vector where each element contains the importance of each possible pair of target and predicted cell types. Finally, we tested which cell interactions had higher importances in one of the sample groups compared to the rest using Wilcoxon tests (FDR < 0.25). To prioritize interactions, we only performed pairwise comparisons between sample groups for cell-type dependencies from which the maximum median importance across all groups was greater than 0.
Estimation of the effects of the spatial context on gene expression
We used mistyR (v1.2.1) to find the associations between the tissue organization and the spatial distribution of stressed cardiomyocytes and the different endothelial, myeloid and fibroblast cell states. We hypothesized that the distribution of specific cell states in the spatial transcriptomics slides could be modelled by the cell-type composition or cell-state presence of individual spots and their neighbourhood.
For a given collection of cell states of interest, we first defined regions of interest in every single slide as the collection of spots where the inferred abundance of the cell type from which the cell state was derived was at least 10%. These regions limit the target spots used in the MISTy model, however the whole slide is used to spatially contextualise the predictors. We used as predictors the abundances of cell types estimated with cell2location or cell states scores. To account only for the effects of the activation of a cell state, the state scores of predictor cell states were masked to 0 whenever their score was lower than 0. In all models we included two classes of spatially contextualized predictive views: an intrinsic (intra) and a local neighbourhood view (para, effective radius = 5).
Specifically we fitted the following models to answer four questions:
-
What are the main cell types whose abundance within a spot or in the local neighbourhood predict the stressed vCM3?
vCM3 ~ intra(cell-type abundance) + para(cell-type abundance)
-
What are the main cell types whose abundance within a spot or in the local neighbourhood predict the endothelial subtypes? How do the different subtypes relate to each other?
ECsubtypes ~ intra(ECsubtypes) + para(ECsubtypes) + intra(cell-type abundance) + para(cell-type abundance)
-
What are the myeloid cell states within a spot or in the local neighbourhood that better predict fibroblasts cell states? How do fibroblasts cell states relate to each other?
FibroblastStates ~ intra(FibroblastStates) + para(FibroblastStates) + intra(MyeloidStates) + para(MyeloidStates)
What are the main cell types whose abundance within a spot or in the local neighbourhood predict the myeloid cell states? How do the different states relate to each other?
MyeloidStates ~ intra(MyeloidStates) + para(MyeloidStates) + intra(cell-type abundance) + para(cell-type abundance)
Specific view importances were compared between patient groups as described previously with an R2 filter of 0.1.
Cell–cell communication analysis
To estimate ligand–receptor interactions between the sub-populations of fibroblasts and myeloid cells, we extracted gene expression matrix from the integrated snRNA-seq and snATAC-seq data for each sample group (that is, myogenic, ischaemic and fibrotic) and combined the matrices from all sub-populations. We next used LIANA (v0.0.9)90, a framework that compiles the results of state-of-the-art cell communication inference methods, to infer ligand–receptor interactions. We focused on the CellPhoneDB91 ligand–receptor method with Omnipath’s ligand–receptor database92 implemented in LIANA90. This was done by combining snRNA-seq samples of myogenic, ischaemic and fibrotic groups and subsetting only the fibroblasts and myeloid cells sub-states. Next, we used CrossTalker (v1.3.1)93 to find changes in cell–cell communication by contrasting ligand–receptor interactions predicted in myogenic vs. ischaemic samples and myogenic vs. fibrotic samples. The interactions considered by CrossTalkeR were obtained by filtering the output of LIANA90 (P > 0.01).
Visualization, statistics, and reproducibility
In data represented as box plots (Figs. 2f, 4c,d,m,o, 5b and 6d,n) the middle line corresponds to the median, the lower and upper hinges describe the first and third quartiles, the upper whisker extends from the hinge to the largest value no further than 1.5 × inter-quartile range (IQR) from the hinge and the lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge, and data beyond the end of the whiskers are outlying points that are plotted individually. In Figs. 4b and 5b,k, Colours refer to gene-weighted kernel density as estimated by using R package Nebulosa94. All reported P-values based on multi-comparison tests were corrected using the Benjamini–Hochberg method95. The depicted immunofluorescence micrographs are representative (Figs. 4c and 6n). The number of samples for each group was chosen on the basis of the expected levels of variation and consistency. The depicted RNAscope, immunofluorescence micrographs are representative and were performed at least twice, and all repeats were successful. Fig. 1a contains a panel from BioRender.com.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-022-05060-x.
Supplementary information
Sampling location, macroscopic and microscopic images. a, Schematic of human cardiac tissue sampling locations. LV-free wall n= 20. Apex n=11. b, Macroscopic images of myocardial tissue specimens. Arrows point to sampled infarcted tissue areas. c, Histological images (trichrome staining) of cardiac tissues included in this study. Arrows highlight the fibrotic tissue areas identified by trichrome staining. Scale bar sizes as indicated.
Quality measurements of different data modalities and samples. a, snRNA-Seq QC. Included are the number of nucleus per sample, number of genes (per nucleus), and number of unique molecular identifiers (UMIs) (per nucleus). Samples from different tissue regions are represented in different colors. b, snATAC-Sq QC. Included are the number of nucleus per sample, number of fragments per sample (per nucleus) and transcription start site (TSS) enrichment scores (per nucleus). c, Spatial transcriptomics QC. Included are the number of spots per sample, the number of UMIs (per spot), the number of quantified nucleus (per spot) and the number of genes (per spot). d, Mean “pathway expression” of BioCarta’s “Death Pathway” and Reactome’s “Regulated Necrosis Pathway” per unfiltered slide, and number of recovered nucleus per sample in snRNA-Seq data separated by sample regions. Right panels show for an ischaemic sample the normalized mean gene expression of the previously mentioned pathways per spot and the number of UMIs. n = 4 for control, n = 5 for RZ, n = 3 for BZ, n = 6 for FZ, and n = 9 for IZ. e-g, Spatial transcriptomic histology (hematoxylin and eosin stainings, H&Es) and UMI distribution across the slide for controls (e), remote zones (f) and border zone (g). Box-Whisker plots showing median and inter-quartile range (IQR). Raw data for (a)-(c) are provide in the Supplementary Tables 2-4. Scale bars 8mm.
Spatial quality controls. a-b, Spatial transcriptomic histology (hematoxylin and eosin stainings, H&Es) and UMI distribution across the slide for ischaemic zones (a) and fibrotic zone (b) samples. Scale bars 8mm.
FACS-gating strategy. a, Gating strategy on DAPI-positive nuclei isolated from frozen human heart tissue.
Clinical data for all samples used in this study.
Meta information and quality check for snRNA-seq samples.
Meta information and quality check for snATAC-seq samples.
Meta information for visium samples
Meta information for imaging samples
Number of nuclei per major cell type for snRNA-seq and snATAC-seq.
Spatially variable genes
Cell-type-specific gene expression estimated from snRNA-seq
Cell-type-specific gene chromatin accessibility estimated from snATAC-seq
Marker genes for cardiomyocytes states
Gene regulatory network for cardiomyocytes
Marker genes for endothelial cell subtypes
Marker genes for fibroblasts states
Gene regulatory network for fibroblasts
Marker genes for myeloid cell subtypes
Statistical results of the sub-clusters for cardiomyocytes, endothelial cells, fibroblasts, and myeloid cells.
Ion-channel related gene sets
qPCR Primer
Acknowledgements
This work was supported by grants of the German Research Foundation (DFG: SFBTRR219 to R.K.), CRU344-4288578857858 and CRU5011-445703531 to R.K., by two grants from the European Research Council (ERC-StG 677448, ERC-CoG 101043403), a grant from the Else Kroener Fresenius Foundation (EKFS), the Dutch Kidney Foundation (DKF), TASKFORCE EP1805, the NWO VIDI 09150172010072 and a grant from the Leducq Foundation, all to R.K., a grant from the Germany Society of Internal Medicine (DGIM) and a grant from the European Research Council (ERC-StG 101040726) to C.K., and a grant from the IZKF Faculty of Medicine at the RWTH Aachen University to I.G.C. This work was also supported by the BMBF eMed Consortia Fibromap (to I.G.C., R.K. and R.K.S.). J.S.-R., R.O.R.F. and R.T.L. are supported by Informatics for Life funded by the Klaus Tschira Foundation. D.Schapiro. and F.W. are supported by the German Federal Ministry of Education and Research (BMBF 01ZZ2004). P.B. is supported by the German Research Foundation (DFG, Project IDs 322900939, 454024652). 10X Genomics supported the project via the Visium challenge (to C.K.) and provided free data generation for Visium data of 8 specimens. We thank J. Cool and J. Hilton for supporting data submission to cellxgene Data Portal; E. Sapena Ventura and G. Yordanova for their support; J. Chew and S. Williams (10X Genomics) for their support; A. Valdeolivas, J. Lanzer, A. Gabor, P. Badia i Mompel and D. Dimitrov for fruitful discussions that shaped the computational analysis. We also acknowledge the support from E. Siera, U. Förster, V. Künstler, N. Graff, P. Cappelmann, L. Tenten, I.-K. Härthe and L. Palm. R.O.R.F. and J.S.-R. acknowledge support from the High Performance and Cloud Computing Group at the Zentrum für Datenverarbeitung of the University of Tübingen, the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 37/935-1 FUGG.
Extended data figures and tables
Source data
Author contributions
C.K. and R.K. planned and designed the study. R.K. obtained funding for the study. R.O.R.F. and Z.L. designed the data and computational analysis, advised by J.S.-R. and I.G.C. C.K., R.O.R.F., Z.L., S.H., R.T.L., M.Halder., M.T.H., D.S., S.M., G.S., K.H., J.T., I.G.C., F.P., N.K., T.S., R.J.A.V., J.A., P.B., J.S.-R. and RK analysed and interpreted the data. C.K., R.O.R.F., Z.L., M.H., I.G.C., J.S.-R. and R.K. wrote the manuscript and organized the figures. R.K.S., K.L., H.M., L.S. and S.S. edited the manuscript and advised on data interpretation. L.W.V.L., A.V., K.K., A.K., J.G., M.M., K.L. and H.M. organized patient tissue collection and biobanking and consent from the patients. N.K. established the PDGFRβ cell line from human hearts. S.Z., X.Z. and C.K. carried out the overexpression studies. C.K. together with X.Z., X.L. and Y.X. carried out all other single-cell and imaging experiments. Image analysis was performed by F.W. and D.Schapiro. R.M.H. and E.M.J.B. validated and sequenced the single-cell libraries. I.G.C., M.C., J.S.N. and Z.L. carried out snATAC-seq data analysis, its integration with snRNA-seq and spatial transcriptomics data, cell sub-state definitions and gene-regulatory networks. R.R., M.H., J.T. and J.S.-R. carried out the analysis and integration of spatial gene expression and snRNA-Seq data. R.O.R.F. designed the multi-omics patient comparison, advised by J.S.-R. C.K., H.M. and R.K. initiated the study. All authors read and approved the manuscript.
Peer review
Peer review information
Nature thanks Eva van Rooij and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer review reports are available.
Data availability
Processed snRNA-seq, snATAC-seq, and spatial transcriptomics data are available at cellxgene https://cellxgene.cziscience.com/collections/8191c283-0816-424b-9b61-c3e1d6258a77 and at the Zenodo data archive (https://zenodo.org/record/6578047). Raw data generated by CellRanger and SpaceRanger pipelines are available through the Human Cell Atlas Data Portal at https://data.humancellatlas.org/explore/projects/e9f36305-d857-44a3-93f0-df4e6007dc97 and at the Zenodo data archive (https://zenodo.org/record/6578553, https://zenodo.org/record/6578617 and https://zenodo.org/record/6580069). Source data are provided with this paper.
Code availability
All code used for analysis is available at https://github.com/saezlab/visium_heart and https://github.com/KramannLab/visium_heart.
Competing interests
The authors declare no competing interests directly related to this work. The authors however disclose unrelated funding, honorariums and ownership as follows: R.K. has grants from Travere Therapeutics, Galapagos, Chugai and Novo Nordisk and is a consultant for Bayer, Pfizer, Novo Nordisk and Gruenenthal. J.S.-R. reports funding from GSK and Sanofi and fees from Travere Therapeutics and Astex Therapeutics. K.L. has grants from Novartis and Amgen and receives consulting fees from Medtronic and Implicit Biosciences. I.G.C. has a grant from Illumina. L.S. has received grants from Bayer, Boehringer Ingelheim and Nattopharma, is consultant for ImmunoDiagnostics Systems (IDS) and is a shareholder in Coagulation Profile. L.W.V.L. reports consultancy fees to UMCU from Abbott, Medtronic, Vifor and Novartis.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Christoph Kuppe, Ricardo O. Ramirez Flores, Zhijian Li
These authors jointly supervised this work: Hendrik Milting, Ivan G. Costa, Julio Saez-Rodriguez, Rafael Kramann
Contributor Information
Julio Saez-Rodriguez, Email: pub.saez@uni-heidelberg.de.
Rafael Kramann, Email: rkramann@gmx.net.
Extended data
is available for this paper at 10.1038/s41586-022-05060-x.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-022-05060-x.
References
- 1.Wong ND. Epidemiological studies of CHD and the evolution of preventive cardiology. Nat. Rev. Cardiol. 2014;11:276–289. doi: 10.1038/nrcardio.2014.26. [DOI] [PubMed] [Google Scholar]
- 2.Niccoli G, et al. Optimized treatment of ST-elevation myocardial infarction. Circ. Res. 2019;125:245–258. doi: 10.1161/CIRCRESAHA.119.315344. [DOI] [PubMed] [Google Scholar]
- 3.Prabhu Sumanth D, Frangogiannis Nikolaos G. The biological basis for cardiac repair after myocardial infarction. Circ. Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Litviňuková M, et al. Cells of the adult human heart. Nature. 2020;588:466–472. doi: 10.1038/s41586-020-2797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang L, et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020;22:108–119. doi: 10.1038/s41556-019-0446-7. [DOI] [PubMed] [Google Scholar]
- 6.Tucker NR, et al. Transcriptional and cellular diversity of the human heart. Circulation. 2020;142:466–482. doi: 10.1161/CIRCULATIONAHA.119.045401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dodou E, Verzi MP, Anderson JP, Xu S-M, Black BL. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development. 2004;131:3931–3942. doi: 10.1242/dev.01256. [DOI] [PubMed] [Google Scholar]
- 8.Yan C, Zhu M, Staiger J, Johnson PF, Gao H. C5a-regulated CCAAT/enhancer-binding proteins β and δ are essential in Fcγ receptor-mediated inflammatory cytokine and chemokine production in macrophages. J. Biol. Chem. 2012;287:3217–3230. doi: 10.1074/jbc.M111.280834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kovary K, Bravo R. The jun and fos protein families are both required for cell cycle progression in fibroblasts. Mol. Cell. Biol. 1991;11:4466–4472. doi: 10.1128/mcb.11.9.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li S, Wang D-Z, Wang Z, Richardson JA, Olson EN. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc. Natl Acad. Sci. USA. 2003;100:9366–9370. doi: 10.1073/pnas.1233635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ge Y, et al. Switching macrophage gene expression from inflammation-resolution to hemorrhage-resolution by redirection of activating transcription factor 1 (ATF1) binding by SMARCA4, BACH1 and histone H3K9 acetylation. Atherosclerosis. 2020;315:e2. doi: 10.1016/j.atherosclerosis.2020.10.021. [DOI] [Google Scholar]
- 12.Pirruccello JP, et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat. Commun. 2020;11:2254. doi: 10.1038/s41467-020-15823-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruetten H, Dimmeler S, Gehring D, Ihling C, Zeiher AM. Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovasc. Res. 2005;66:444–453. doi: 10.1016/j.cardiores.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 14.Shook BA, et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science. 2018;362:eaar2971. doi: 10.1126/science.aar2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pakshir P, et al. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat. Commun. 2019;10:1850. doi: 10.1038/s41467-019-09709-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aoyagi T, Matsui T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 2011;17:1818–1824. doi: 10.2174/138161211796390976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mak TW, Hauck L, Grothe D, Billia F. p53 regulates the cardiac transcriptome. Proc. Natl Acad. Sci. USA. 2017;114:2331–2336. doi: 10.1073/pnas.1621436114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergmann MW. WNT signaling in adult cardiac hypertrophy and remodeling: lessons learned from cardiac development. Circ. Res. 2010;107:1198–1208. doi: 10.1161/CIRCRESAHA.110.223768. [DOI] [PubMed] [Google Scholar]
- 19.Ahern BM, et al. Myocardial-restricted ablation of the GTPase RAD results in a pro-adaptive heart response in mice. J. Biol. Chem. 2019;294:10913–10927. doi: 10.1074/jbc.RA119.008782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lotteau S, et al. Acute genetic ablation of cardiac sodium/calcium exchange in adult mice: implications for cardiomyocyte calcium regulation, cardioprotection, and arrhythmia. J. Am. Heart Assoc. 2021;10:e019273. doi: 10.1161/JAHA.120.019273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernandez-Caggiano M, et al. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat. Metab. 2020;2:1223–1231. doi: 10.1038/s42255-020-00276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hama N, et al. Rapid ventricular induction of brain natriuretic peptide gene expression in experimental acute myocardial infarction. Circulation. 1995;92:1558–1564. doi: 10.1161/01.CIR.92.6.1558. [DOI] [PubMed] [Google Scholar]
- 23.Waspe LE, Ordahl CP, Simpson PC. The cardiac β-myosin heavy chain isogene is induced selectively in α1-adrenergic receptor-stimulated hypertrophy of cultured rat heart myocytes. J. Clin. Invest. 1990;85:1206–1214. doi: 10.1172/JCI114554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beggah AT, et al. Reversible cardiac fibrosis and heart failure induced by conditional expression of an antisense mRNA of the mineralocorticoid receptor in cardiomyocytes. Proc. Natl Acad. Sci. USA. 2002;99:7160–7165. doi: 10.1073/pnas.102673599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bakker ML, et al. T-box transcription factor TBX3 reprogrammes mature cardiac myocytes into pacemaker-like cells. Cardiovasc. Res. 2012;94:439–449. doi: 10.1093/cvr/cvs120. [DOI] [PubMed] [Google Scholar]
- 26.Jiang J, et al. Cardiac myosin binding protein C regulates postnatal myocyte cytokinesis. Proc. Natl Acad. Sci. USA. 2015;112:9046–9051. doi: 10.1073/pnas.1511004112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piroddi N, et al. Myocardial overexpression of ANKRD1 causes sinus venosus defects and progressive diastolic dysfunction. Cardiovasc. Res. 2020;116:1458–1472. doi: 10.1093/cvr/cvz291. [DOI] [PubMed] [Google Scholar]
- 28.Hill C, et al. Inhibition of AP-1 signaling by JDP2 overexpression protects cardiomyocytes against hypertrophy and apoptosis induction. Cardiovasc. Res. 2013;99:121–128. doi: 10.1093/cvr/cvt094. [DOI] [PubMed] [Google Scholar]
- 29.Kramann R, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16:51–66. doi: 10.1016/j.stem.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hulsmans M, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169:510–522.e20. doi: 10.1016/j.cell.2017.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vieira JM, et al. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J. Clin. Invest. 2018;128:3402–3412. doi: 10.1172/JCI97192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ. Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 33.Deshpande SS, Angkeow P, Huang J, Ozaki M, Irani K. Rac1 inhibits TNF‐α‐induced endothelial cell apoptosis: dual regulation by reactive oxygen species. FASEB J. 2000;14:1705–1714. doi: 10.1096/fj.99-0910com. [DOI] [PubMed] [Google Scholar]
- 34.Kuppe C, et al. Decoding myofibroblast origins in human kidney fibrosis. Nature. 2021;589:281–286. doi: 10.1038/s41586-020-2941-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Z, et al. Chromatin-accessibility estimation from single-cell ATAC-seq data with scOpen. Nat. Commun. 2021;12:6386. doi: 10.1038/s41467-021-26530-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bugg D, et al. MBNL1 drives dynamic transitions between fibroblasts and myofibroblasts in cardiac wound healing. Cell Stem Cell. 2022;29:419–433.e10. doi: 10.1016/j.stem.2022.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis J, et al. MBNL1-mediated regulation of differentiation RNAs promotes myofibroblast transformation and the fibrotic response. Nat. Commun. 2015;6:10084. doi: 10.1038/ncomms10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kramann R, et al. Pharmacological GLI2 inhibition prevents myofibroblast cell-cycle progression and reduces kidney fibrosis. J. Clin. Invest. 2015;125:2935–2951. doi: 10.1172/JCI74929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J. Cell. Mol. Med. 2002;6:1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alexanian M, et al. A transcriptional switch governs fibroblast activation in heart disease. Nature. 2021;595:438–443. doi: 10.1038/s41586-021-03674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forte E, et al. Cross-priming dendritic cells exacerbate immunopathology after ischemic tissue damage in the heart. Circulation. 2021;143:821–836. doi: 10.1161/CIRCULATIONAHA.120.044581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dick SA, et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci. Immunol. 2022;7:eabf7777. doi: 10.1126/sciimmunol.abf7777. [DOI] [PubMed] [Google Scholar]
- 43.MacDonald L, et al. COVID-19 and RA share an SPP1 myeloid pathway that drives PD-L1+ neutrophils and CD14+ monocytes. JCI Insight. 2021;6:e147413. doi: 10.1172/jci.insight.147413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morse C, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019;54:1802441. doi: 10.1183/13993003.02441-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bevan L, et al. Specific macrophage populations promote both cardiac scar deposition and subsequent resolution in adult zebrafish. Cardiovasc. Res. 2020;116:1357–1371. doi: 10.1093/cvr/cvz221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeLeon-Pennell KY, et al. CD36 is a matrix metalloproteinase-9 substrate that stimulates neutrophil apoptosis and removal during cardiac remodeling. Circ. Cardiovasc. Genet. 2016;9:14–25. doi: 10.1161/CIRCGENETICS.115.001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ismahil MA, et al. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ. Res. 2014;114:266–282. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shiraishi M, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Invest. 2016;126:2151–2166. doi: 10.1172/JCI85782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bergmann O, et al. Dynamics of cell generation and turnover in the human heart. Cell. 2015;161:1566–1575. doi: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 50.Yekelchyk M, Guenther S, Preussner J, Braun T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res. Cardiol. 2019;114:36. doi: 10.1007/s00395-019-0744-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Habib N, et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods. 2017;14:955–958. doi: 10.1038/nmeth.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koenig, A. L. et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat. Cardiovasc. Res.1, 263–280 (2022). [DOI] [PMC free article] [PubMed]
- 53.Tippani, M. et al. VistoSeg: processing utilities for high-resolution Visium/Visium-IF images for spatial transcriptomics data. bioRxivhttps://www.biorxiv.org/content/10.1101/2021.08.04.452489v2 (2022). [DOI] [PMC free article] [PubMed]
- 54.Curaj A, Simsekyilmaz S, Staudt M, Liehn E, et al. Minimal invasive surgical procedure of inducing myocardial infarction in mice. J. Vis. Exp. 2015;99:e52197. doi: 10.3791/52197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Linkert M, et al. Metadata matters: access to image data in the real world. J. Cell Biol. 2010;189:777–782. doi: 10.1083/jcb.201004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bahry, E. et al. RS-FISH: precise, interactive, fast, and scalable FISH spot detection. Preprint at bioRxiv10.1101/2021.03.09.434205 (2021). [DOI] [PMC free article] [PubMed]
- 57.Greenwald NF, et al. Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. Nat. Biotechnol. 2021;40:555–565. doi: 10.1038/s41587-021-01094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hao Y, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Germain P-L, Lun A, Macnair W, Robinson MD. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 2021;10:979. doi: 10.12688/f1000research.73600.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Flanagan CH, et al. Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biol. 2019;20:210. doi: 10.1186/s13059-019-1830-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Korsunsky I, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Granja JM, et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021;53:403–411. doi: 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stuart T, et al. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Z, et al. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 2019;20:45. doi: 10.1186/s13059-019-1642-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fornes O, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Badia-i-Mompel P, et al. decoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinformatics Adv. 2022;2:vbac016. doi: 10.1093/bioadv/vbac016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ulirsch JC, et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet. 2019;51:683–693. doi: 10.1038/s41588-019-0362-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wolock SL, Lopez R, Klein AM. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hansen BB, Klopfer SO. Optimal full matching and related designs via network flows. J. Comput. Graph. Stat. 2006;15:609–627. doi: 10.1198/106186006X137047. [DOI] [Google Scholar]
- 75.Haghverdi L, Büttner M, Wolf FA, Buettner F, Theis FJ. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods. 2016;13:845–848. doi: 10.1038/nmeth.3971. [DOI] [PubMed] [Google Scholar]
- 76.Schep AN, Wu B, Buenrostro JD, Greenleaf W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017;14:975–978. doi: 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Freeman LC. Centrality in social networks conceptual clarification. Soc. Networks. 1978;1:215–239. doi: 10.1016/0378-8733(78)90021-7. [DOI] [Google Scholar]
- 78.Page L, et al. The PageRank Citation Ranking: Bringing Order to the Web. Stanford IfoLab; 1999. [Google Scholar]
- 79.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kleshchevnikov V, et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 2022;40:661–671. doi: 10.1038/s41587-021-01139-4. [DOI] [PubMed] [Google Scholar]
- 81.Schubert M, et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 2018;9:20. doi: 10.1038/s41467-017-02391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Holland CH, Szalai B, Saez-Rodriguez J. Transfer of regulatory knowledge from human to mouse for functional genomics analysis. Biochim. Biophys. Acta. 2020;1863:194431. doi: 10.1016/j.bbagrm.2019.194431. [DOI] [PubMed] [Google Scholar]
- 83.Zhu J, Sun S, Zhou X. SPARK-X: non-parametric modeling enables scalable and robust detection of spatial expression patterns for large spatial transcriptomic studies. Genome Biol. 2021;22:184. doi: 10.1186/s13059-021-02404-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liberzon A, et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gillespie M, et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2021;50:D687–D692. doi: 10.1093/nar/gkab1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grant AO. Cardiac ion channels. Circ. Arrhythm. Electrophysiol. 2009;2:185–194. doi: 10.1161/CIRCEP.108.789081. [DOI] [PubMed] [Google Scholar]
- 87.Tanevski, J., Flores, R. O. R., Gabor, A., Schapiro, D. & Saez-Rodriguez, J. Explainable multiview framework for dissecting spatial relationships from highly multiplexed data. Genome Biol.23, 97 (2022). [DOI] [PMC free article] [PubMed]
- 88.Pawlowsky-Glahn V, Buccianti A. Compositional Data Analysis: Theory and Applications. John Wiley & Sons; 2011. [Google Scholar]
- 89.Lun ATL, McCarthy DJ, Marioni JC. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res. 2016;5:2122. doi: 10.12688/f1000research.9501.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dimitrov, D. et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun.13, 3224 (2022). [DOI] [PMC free article] [PubMed]
- 91.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 92.Türei D, et al. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol. Syst. Biol. 2021;17:e9923. doi: 10.15252/msb.20209923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nagai JS, Leimkühler NB, Schaub MT, Schneider RK, Costa IG. CrossTalkeR: analysis and visualization of ligand–receptor networks. Bioinformatics. 2021;37:4263–4265. doi: 10.1093/bioinformatics/btab370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alquicira-Hernandez J, Powell JE. Nebulosa recovers single cell gene expression signals by kernel density estimation. Bioinformatics. 2021;37:2485–2487. doi: 10.1093/bioinformatics/btab003. [DOI] [PubMed] [Google Scholar]
- 95.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sampling location, macroscopic and microscopic images. a, Schematic of human cardiac tissue sampling locations. LV-free wall n= 20. Apex n=11. b, Macroscopic images of myocardial tissue specimens. Arrows point to sampled infarcted tissue areas. c, Histological images (trichrome staining) of cardiac tissues included in this study. Arrows highlight the fibrotic tissue areas identified by trichrome staining. Scale bar sizes as indicated.
Quality measurements of different data modalities and samples. a, snRNA-Seq QC. Included are the number of nucleus per sample, number of genes (per nucleus), and number of unique molecular identifiers (UMIs) (per nucleus). Samples from different tissue regions are represented in different colors. b, snATAC-Sq QC. Included are the number of nucleus per sample, number of fragments per sample (per nucleus) and transcription start site (TSS) enrichment scores (per nucleus). c, Spatial transcriptomics QC. Included are the number of spots per sample, the number of UMIs (per spot), the number of quantified nucleus (per spot) and the number of genes (per spot). d, Mean “pathway expression” of BioCarta’s “Death Pathway” and Reactome’s “Regulated Necrosis Pathway” per unfiltered slide, and number of recovered nucleus per sample in snRNA-Seq data separated by sample regions. Right panels show for an ischaemic sample the normalized mean gene expression of the previously mentioned pathways per spot and the number of UMIs. n = 4 for control, n = 5 for RZ, n = 3 for BZ, n = 6 for FZ, and n = 9 for IZ. e-g, Spatial transcriptomic histology (hematoxylin and eosin stainings, H&Es) and UMI distribution across the slide for controls (e), remote zones (f) and border zone (g). Box-Whisker plots showing median and inter-quartile range (IQR). Raw data for (a)-(c) are provide in the Supplementary Tables 2-4. Scale bars 8mm.
Spatial quality controls. a-b, Spatial transcriptomic histology (hematoxylin and eosin stainings, H&Es) and UMI distribution across the slide for ischaemic zones (a) and fibrotic zone (b) samples. Scale bars 8mm.
FACS-gating strategy. a, Gating strategy on DAPI-positive nuclei isolated from frozen human heart tissue.
Clinical data for all samples used in this study.
Meta information and quality check for snRNA-seq samples.
Meta information and quality check for snATAC-seq samples.
Meta information for visium samples
Meta information for imaging samples
Number of nuclei per major cell type for snRNA-seq and snATAC-seq.
Spatially variable genes
Cell-type-specific gene expression estimated from snRNA-seq
Cell-type-specific gene chromatin accessibility estimated from snATAC-seq
Marker genes for cardiomyocytes states
Gene regulatory network for cardiomyocytes
Marker genes for endothelial cell subtypes
Marker genes for fibroblasts states
Gene regulatory network for fibroblasts
Marker genes for myeloid cell subtypes
Statistical results of the sub-clusters for cardiomyocytes, endothelial cells, fibroblasts, and myeloid cells.
Ion-channel related gene sets
qPCR Primer
Data Availability Statement
Processed snRNA-seq, snATAC-seq, and spatial transcriptomics data are available at cellxgene https://cellxgene.cziscience.com/collections/8191c283-0816-424b-9b61-c3e1d6258a77 and at the Zenodo data archive (https://zenodo.org/record/6578047). Raw data generated by CellRanger and SpaceRanger pipelines are available through the Human Cell Atlas Data Portal at https://data.humancellatlas.org/explore/projects/e9f36305-d857-44a3-93f0-df4e6007dc97 and at the Zenodo data archive (https://zenodo.org/record/6578553, https://zenodo.org/record/6578617 and https://zenodo.org/record/6580069). Source data are provided with this paper.
All code used for analysis is available at https://github.com/saezlab/visium_heart and https://github.com/KramannLab/visium_heart.