

Abstract
Bone adapts to the mechanical forces that it experiences. Orthodontic tooth movement harnesses the cell- and tissue-level properties of mechanotransduction to achieve alignment and reorganization of the dentition. However, the mechanisms of action that permit bone resorption and formation in response to loads placed on the teeth are incompletely elucidated, though several mechanisms have been identified. Wnt/Lrp5 signaling in osteocytes is a key pathway that modulates bone tissue’s response to load. Numerous mouse models that harbor knock-in, knockout, and transgenic/overexpression alleles targeting genes related to Wnt signaling point to the necessity of Wnt/Lrp5, and its localization to osteocytes, for proper mechanotransduction in bone. Alveolar bone is rich in osteocytes and is a highly mechanoresponsive tissue in which components of the canonical Wnt signaling cascade have been identified. As Wnt-based agents become clinically available in the next several years, the a major challenge that lies ahead will be to gain a more complete understanding of Wnt biology in alveolar bone so that improved/expedited tooth movement becomes a possibility.
Keywords: Wnt, Lrp5, orthodontics, Sost, sclerostin, HBM
The emerging age of precision medicine is ushering in new opportunities to perform more refined and targeted procedures and therapies in a variety of health contexts. The practice of orthodontic tooth movement is among those fields that will benefit significantly from precision medicine approaches, but full realization will require a more detailed understanding of the signaling events in the different tissues involved in the process, e.g., alveolar bone-associated osteocytes and periodontal ligament-associated fibroblasts and neurovascular support cells. Alveolar bone undergoes a highly coordinated and rapid reorganization (i.e., remodeling) during tooth movement which is governed, at least in part, by the local osteocyte population. The stimulus driving alveolar bone remodeling during orthodontic tooth movement is, at the most basic level, the mechanical forces placed on the dentition by external hardware. The altered mechanical environment induces biochemical changes in the local cell populations that ultimately facilitate tooth movement through tissue space. These changes–collectively known as mechanotransduction—involve activation and inhibition of numerous pathways that coordinate to achieve an adapted structure. One of the key pathways that skeletal cells use to facilitate mechanotransduction is the Wnt signaling pathway, which is gaining recognition as a key player in orthodontic tooth movement process.
Wnt/Lrp5 signaling impacts bone mass and strength
Our understanding of osteocyte mechanotransduction took a significant leap forward when the importance of Wnt signaling in bone was identified. Prior to the turn of this century, research on the Wnt signaling cascade was confined largely to the cancer (e.g., tumor biology and proliferation) and developmental biology (e.g., axis patterning) fields. However, two key clinical discoveries changed our view on where and how Wnt signaling is important in the body, bringing the skeleton into direct focus as a major Wnt-dependent system. The first of these discoveries was the finding that loss-of-function mutations in the Wnt co-receptor Low density lipoprotein receptor-related protein 5 (LRP5) are a genetic cause of the rare, debilitating disease Osteoporosis Pseudoglioma (OPPG) (1). Patients with OPPG can present clinically with bone mineral density (BMD) values that are approximately 5 standard deviations below age-matched normal values, resulting in frequent fractures and impaired mobility. At around the same time, other groups reported that gain-of-function mutations in LRP5 are a genetic cause for endosteal hyperosteosis, a high bone mass (HBM) condition (2, 3). These patients have BMDs that are approximately 5 standard deviations above normal, but their skeletons are relatively normal in shape, and they present with none of the clinical manifestations associated with other, osteoclast-mediated sclerosing bone disorders (e.g., osteopetrosis). Thus, within a few short years, a single Wnt co-receptor was identified as causative for two very different rare skeletal diseases, one associated with very high bone mass and the other associated with very low bone mass. As LRP5 has no other well-characterized primary ligands beyond the Wnt family, these human genetic studies implicated Wnt signaling as a major regulator of bone metabolism.
Wnt/Lrp5 signaling participates in mechanotransduction
Shortly after the discoveries regarding LRP5’s role in regulating human skeletal properties, mouse models were developed to model the diseases in mice and open up experimental options not possible in human patients. Mice with loss-of-function mutations in Lrp5 (i.e., “knockout”) recapitulate the low bone mass phenotype observed in human OPPG patients (Fig. 1A). These mice have low BMD, compromised trabecular mass and architecture, reduced cortical bone size, and impaired biomechanical properties (4). We became interested in the role of Wnt signaling in mechanotransduction, and began experimenting with mechanoresponsiveness in Lrp5−/− mice. We performed in vivo mechanical loading experiments, using the rodent ulnar loading model, to assess the ability of Lrp5−/− mice to respond anabolically to a simulated vigorous exercise session. In both male and female Lrp5−/− mice, load-induced bone formation was significantly impaired compared to control (wild-type; WT) mice (4) (Fig. 1B). That conclusion was later confirmed by another group in an independently generated Lrp5 knockout model, using a different loading modality (tibia loading) (5). Thus the experimental data are consistent in implicating Wnt signaling through Lrp5 as a crucial process for skeletal mechanotransduction. i.e., without Lrp5 receptors, bone tissue cannot adapt properly to mechanical inputs. Those observations might explain a portion of the low bone mass phenotype among OPPG patients, i.e., that these individuals are unable to respond to otherwise stimulatory mechanical signals, and consequently, bone properties never reach their appropriate size and strength.
Figure 1:
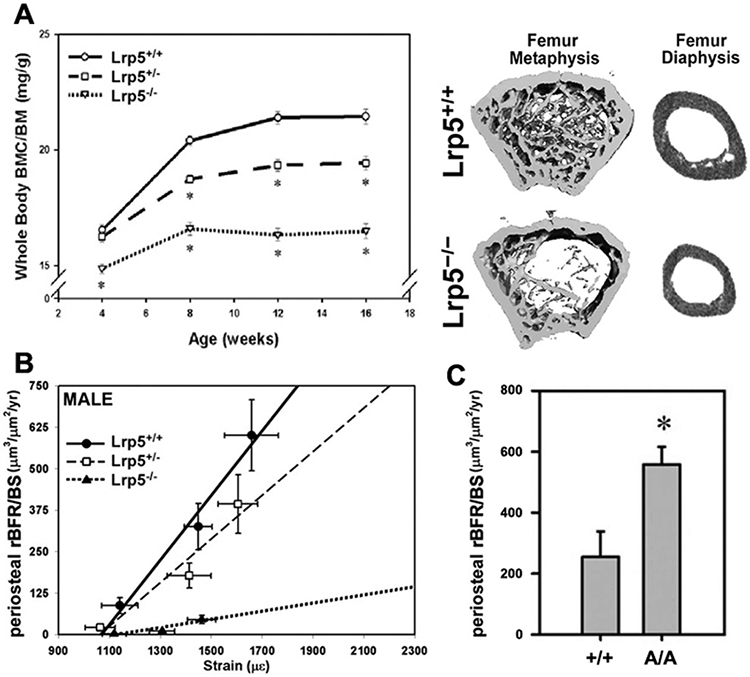
Mice with global homozygous loss-of-function mutations in the Lrp5 receptor have (A) reduced whole body bone mineral content per unit body mass (BMC/BM), and reduced trabecular and cortical bone mass in the femur diaphysis and metaphysis. (B) Mechanical stimulation of the ulna in Lrp5−/− mice results in significantly lower periosteal relative bone formation rates per unit bone surface (rBFR/BS) compared to wild type (Lrp5+/+) and heterozygous mice (Lrp5+/−), at strains greater than 1200 microstrain. (C) Mechanical stimulation of the tibia in mice with homozygous global high bone mass-causing missense mutations in Lrp5 results in significantly greater periosteal rBFR/BS compared to wild type (Lrp5+/+) mice.
While the Lrp5 knockout models are useful for exploring the role of the Wnt/Lrp5 axis in bone metabolism and mechanotransduction, we were also interested to learn whether the gain-of-function mutations in LRP5 found in several families with HBM might also confer improved mechanical signaling properties to the cell. To this end, we generated two different knock-in mouse lines, harboring one of two different HBM-causing missense mutations in Lrp5—G171V and A214V. Like the human patients that these mutations were modeled after, the HBM knock-in mice exhibited high BMD, dramatically increased trabecular and cortical bone mass, and improved biomechanical properties (6). More importantly, however, the gain-of-function mutant mice were more responsive to tibial loading than their WT littermates (Fig. 1C) (7). In summary, mice with either loss- or gain-of-function mutations in Lrp5 exhibit altered load-induced bone formation, suggesting that Lrp5 plays a major role in mechanotransduction signaling in bone.
Osteocytes are the cell type of action for Wnt/Lrp5-mediated modulation of bone mass.
While both the gain-of-function knock-ins, and the loss-of-function knockout, all model the phenotypes observed human patients with analogous LRP5 mutations (i.e., the mice and humans have altered Lrp5 expression globally), those mouse models do not reveal the specific cell type(s) of action that account for the Wnt-driven skeletal phenotypes. To address this issue, cell-selective deletion of Lrp5 or expression of Lrp5-HBM alleles in specific cell types can reveal the relative contributions of these cell types to the overall phenotype. Mice with floxed Lrp5 alleles (Lrp5f/f), where recombination of the loxP sites results in a null allele, have a normal bone mass in the absence of Cre recombinase. However, when crossed to mice that express Cre in osteocytes (Dmp1-Cre), the mice develop an early-onset osteopenic phenotype that mimics the global Lrp5−/− model (Fig 2A) (6). Likewise, mice engineered with a Cre-inducible heterozygous gain-of-function missense mutation in Lrp5, where recombination of a floxed Neo cassette in one of the introns releases the HBM allele from its dormant state, have normal bone mass in the absence of Cre recombinase. However, when crossed to mice that express Cre in osteocytes (Dmp1-Cre), the mice develop an early-onset hyperostosis phenotype that mimics the global Lrp5-HBM model (Fig 2B) (6). Thus, the osteocyte-selective deletion of Lrp5 and expression of Lrp5-HBM both recapitulate the global knockout and knock-in models, respectively, suggesting that mutation of Lrp5 in the osteocyte population is sufficient to recapitulate the bone phenotype of body-wide mutation. These data implicate the osteocyte as the cell type of action for Wnt/Lrp5-mediated signaling in the modulation of bone mass.
Figure 2:
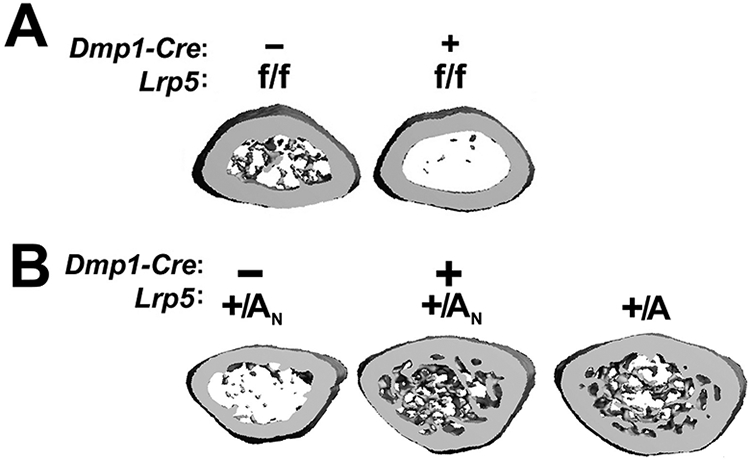
Distal femur metaphyseal μCT reconstructions reveal that (A) mice with Lrp5-HBM alleles activated only in Dmp1-expressing cells (Dmp1-Cre-mediated activation of dormant Lrp5-HBM alleles [+/AN]) recapitulate the HBM phenotype of mice with globally expressed HBM alleles (+/A). (B) Mice with floxed loss of function Lrp5 alleles recombined only in Dmp1-expressing cells exhibit a low bone mass phenotype, similar to that observed in mice carrying global loss-of-function alleles.
Osteocytes are the cell type of action for Wnt/Lrp5-mediated mechanotransduction
Given the discovery that Lrp5 signaling in the osteocyte population accounts for the skeletal effects of this receptor, we next evaluated whether Wnt/Lrp5-mediated mechanotransduction was localized to the osteocyte. As described above, crossing Dmp1-Cre mice to Lrp5f/f mice deletes Lrp5 selectively in the osteocyte population. When subjected to ulnar loading, these mice have a severe deficit in load-induced bone gain (8), much like the global Lrp5−/− mice (Fig. 3) (4). Because the osteocyte is a key cell type for Wnt/Lrp5-mediated mechanotransduction, we looked for cellular mechanisms that might control Lrp5 signaling specifically in osteocytes, potentially explaining the normal mode of action for the Lrp5 receptor in a typical mechanical signaling event. Here, we looked for Wnt Lrp5 modulators that were enriched in osteocytes, and the most obvious molecule is sclerostin. In the skeleton, sclerostin expression is localized largely to the osteocyte population, to the exclusion of other skeletal cells (9). Moreover, loss of sclerostin either by mutations in the SOST coding sequence (10), or by deletion of downstream SOST enhancer regions (11), results in very high bone mass. Those two attributes, in addition to the observation that sclerostin is a high-affinity Lrp5 antagonist (12), has fueled inquiries into whether sclerostin is a key mediator of Lrp5-mediated mechanotransduction. Sclerostin transcript and protein levels are dramatically reduced by mechanical loading, and the degree of down-regulation is closely associated with strain magnitude (13). Thus, a load-induced reduction in local sclerostin levels appears to be a reasonable explanation as to how Lrp5 achieves activation during loading. In that context, mechanical stimulation induces a down-regulation of sclerostin protein, which releases Lrp5 from inhibition, and promotes Wnt local Wnt signaling through Lrp5. If this model is true, it would require that sclerostin must undergo downregulation in order for mechanotransduction to occur. Testing this hypothesis directly with a functional study is challenging but not impossible, in that it would require applying mechanical loading to a mouse model in which sclerostin levels could be maintained at a high level, even in the presence of a mechanical stimulus (which normally causes Sost downregulation). Those experimental conditions were achieved by using the Dmp1-hSOST transgenic mouse model. This mouse harbors a transgene comprising a human SOST cDNA under the control of the mouse Dmp1 promoter (14). The Dmp1 promoter is load-responsive in the positive direction, i.e., Dmp1 expression is increased in response to a mechanical stimulus (15). In the Dmp1-hSOST transgenic mouse, mechanical loading induces the predicted downregulation of endogenous Sost expression (as would occur in a WT mouse), but at the same time induces an upregulation of transgenic hSOST expression, due to a load-induced increase in Dmp1 promoter activity. Therefore this mouse can maintain high levels of sclerostin protein even in the context of mechanical loading. When subjected to ulnar loading, the Dmp1-hSOST exhibits a nearly complete lack of mechanoresponsiveness (Fig. 4), which phenocopies the Lrp5-null mechanoresponsiveness (14). In summary, numerous functional studies suggest that osteocyte Wnt/Lrp5 signaling is crucial for skeletal mechanotransduction, and the process appears to be regulated by the secreted Lrp5 antagonist sclerostin.
Figure 3:
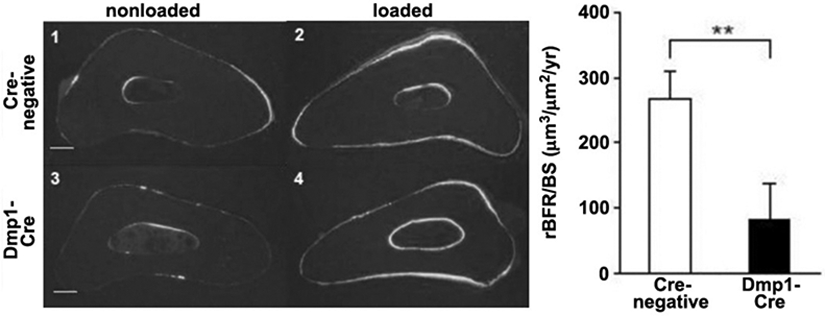
Mice with osteocyte-selective deletion of Lrp5 exhibit reduced responsiveness to ulnar loading than Cre-negative mice. (Left) Histological photomicrographs of the loaded and nonloaded ulnae in Cre-positive and Cre-negative mice, illustrating the paucity of labeling in Cre-positive mice. (Right) Quantification of periosteal relative bone formation rates per unit bone surface (rBFR/BS) in the ulnae was significantly reduced in Cre-positive mice.
Figure 4:
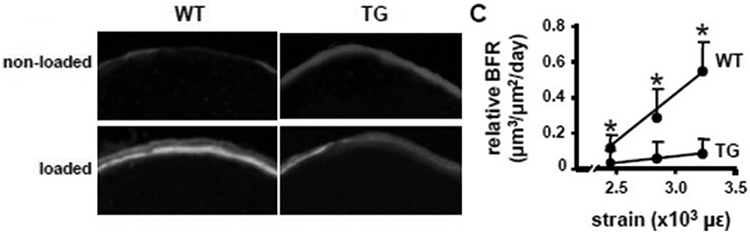
Mice carrying the Dmp1-hSOST transgene do not respond to mechanical stimulation of the ulna. (Left) Histological photomicrographs of the loaded and nonloaded ulnae in transgenic and nontransgenic mice, illustrating the paucity of labeling in transgenic mice. (Right) Quantification of periosteal relative bone formation rates per unit bone surface (rBFR/BS) in the ulnae was significantly reduced in transgenic mice.
More recently, sclerostin-mediated antagonism of Lrp5 appears to require a “facilitator” protein, without which the full inhibitory effects of sclerostin are not achieved. The facilitator protein has been identified as Lrp4 (16), another LDL-like family member that has important functions in the neuromuscular junction (17). Whether Lrp4 is involved in mechanotransduction, i.e., by altering the availability or localization of sclerostin during a mechanical loading event, is unknown. Lrp4 has a role in tooth development (18, 19), but its function in orthodontic tooth movement (or generalized skeletal mechanotransduction) are not known.
Wnt-mediated signaling in orthodontic tooth movement
It is clear that osteocytic Wnt signaling plays a major role in skeletal mechanotransduction, but most of those insights come from the study of cortical and trabecular bone in the limbs. Alveolar bone is rich with osteocytes, but less is known about the specific role of Wnt signaling in orthodontic tooth movement per se. However, accumulating data suggests that Wnt signaling is also important in alveolar bone for tooth movement. In mice subjected to tooth movement via and open-coil spring, Lrp5 and Fzd4 (a Wnt co-receptor) were upregulated in the periodontium near alveolar bone (20). In addition, the downstream canonical mediator β-catenin was also activated in the same tissues by tooth movement. Moreover, Lrp5-HBM mice exhibit an increase in alveolar bone formation, and narrowing of the PDL space (21). Interestingly, in the alveolar bone surrounding a tooth engaged in orthodontic tooth movement, Sost expression increases on the compression side and is briefly downregulated on the tension side (22). These dynamics are consistent with an increase in resorption in the area where the tooth is prompted to move (high sclerostin), and an increase in bone formation in the wake of the moving tooth (low sclerostin). It is likely that in the near future, Wnt-based therapies will be useful for facilitating more rapid orthodontic tooth movement, but more focused experiments are required to better understand the biology that is specific to alveolar bone. Currently, Wnt pathway activators (e.g., Sost antibody) and Wnt pathway inhibitors (e.g., small molecule Porcupine inhibitors) are currently under consideration by the FDA for clinical use. If these agents are successfully brought to market, an expanding toolbox of Wnt modulation will be available to the orthodontic community. Harnessing those tools to achieve a more favorable orthodontic outcome will remain a challenge until the basic science of Wnt specifically in alveolar bone and the PDL is more thoroughly elucidated.
References
- 1.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513–23. [DOI] [PubMed] [Google Scholar]
- 2.Van Wesenbeeck L, Cleiren E, Gram J, Beals RK, Benichou O, Scopelliti D, Key L, Renton T, Bartels C, Gong Y, et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet 2003;72(3):763–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet 2002;70(l):11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, Li J, Maye P, Rowe DW, Duncan RL, et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006;281(33):23698–711. [DOI] [PubMed] [Google Scholar]
- 5.Saxon LK, Jackson BF, Sugiyama T, Lanyon LE, and Price JS. Analysis of multiple bone responses to graded strains above functional levels, and to disuse, in mice in vivo show that the human Lrp5 G171V High Bone Mass mutation increases the osteogenic response to loading but that lack of Lrp5 activity reduces it. Bone. 2011;49(2):184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui Y, Niziolek PJ, MacDonald BT, Zylstra CR, Alenina N, Robinson DR, Zhong Z, Matthes S, Jacobsen CM, Conlon RA, et al. Lrp5 functions in bone to regulate bone mass. Nat Med. 2011;17(6):684–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niziolek PJ, Warman ML, and Robling AG. Mechanotransduction in bone tissue: The A214V and G171V mutations in Lrp5 enhance load-induced osteogenesis in a surface-selective manner. Bone. 2012;51(3):459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao L, Shim JW, Dodge TR, Robling AG, and Yokota H. Inactivation of Lrp5 in osteocytes reduces young's modulus and responsiveness to the mechanical loading. Bone. 2013;54(1):35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, and Reeve J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005; 19(13):1842–4. [DOI] [PubMed] [Google Scholar]
- 10.Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10(5):537–43. [DOI] [PubMed] [Google Scholar]
- 11.Loots GG, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, Ovcharenko D, Plajzer-Frick I, and Rubin EM. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15(7):928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semenov M, Tamai K, and He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280(29):26770–5. [DOI] [PubMed] [Google Scholar]
- 13.Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283(9):5866–75. [DOI] [PubMed] [Google Scholar]
- 14.Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, Stolina M, Turner CH, Robling AG, Plotkin LI, et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50(1):209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris SE, Gluhak-Heinrich J, Harris MA, Yang W, Bonewald LF, Riha D, Rowe PS, Robling AG, Turner CH, Feng JQ, et al. DMP1 and MEPE expression are elevated in osteocytes after mechanical loading in vivo: theoretical role in controlling mineral quality in the perilacunar matrix. J Musculoskelet Neuronal Interact. 2007;7(4):313–5. [PMC free article] [PubMed] [Google Scholar]
- 16.Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, Bouwmeester T, Schirle M, Bueno-Lozano M, Fuentes FJ, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286(22):19489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zong Y, and Jin R. Structural mechanisms of the agrin-LRP4-MuSK signaling pathway in neuromuscular junction differentiation. Cell Mol Life Sci. 2013;70(17):3077–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn Y, Sims C, Murray MJ, Kuhlmann PK, Fuentes-Antras J, Weatherbee SD, and Krumlauf R. Multiple modes of Lrp4 function in modulation of Wnt/beta-catenin signaling during tooth development. Development. 2017;144(15):2824–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohazama A, Porntaveetus T, Ota MS, Herz J, and Sharpe PT. Lrp4: A novel modulator of extracellular signaling in craniofacial organogenesis. Am J Med Genet A. 2010;152A(12):2974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu HD, Wang BK, Wan ZQ, Lin H, Chang ML, and Han GL. Wnt5a mediated canonical Wnt signaling pathway activation in orthodontic tooth movement: possible role in the tension force-induced bone formation. J Mol Histol. 2016;47(5):455–66. [DOI] [PubMed] [Google Scholar]
- 21.Lim WH, Liu B, Mah SJ, Yin X, and Helms JA. Alveolar bone turnover and periodontal ligament width are controlled by Wnt. J Periodontol. 2015;86(2):319–26. [DOI] [PubMed] [Google Scholar]
- 22.Odagaki N, Ishihara Y, Wang Z, Ei Hsu Hlaing E, Nakamura M, Hoshijima M, Hayano S, Kawanabe N, and Kamioka H. Role of Osteocyte-PDL Crosstalk in Tooth Movement via SOST/Sclerostin. J Dent Res. 2018:22034518771331. [DOI] [PubMed] [Google Scholar]