

Abstract
Experimental data accumulated during the last ten years strongly support the existence and functional significance of oligomerization of G protein-coupled receptors (GPCRs). In this essay, we discuss the unique biochemical properties of GPCR oligomers in the frame of ‘allosterism’ and how these properties can be used to identify GPCR oligomers in artificial systems and in native tissues. We also address how the significant advances in biophysical, computational and crystallization techniques have provided significant structural insights about the mechanisms behind allosterism in GPCRs, giving distinct clues about the mechanisms of allosteric interactions in GPCR homomers and heteromers. Finally, we elaborate on the emerging picture of the role of GPCR oligomers as components of pre-coupled macromolecular complexes that include different G proteins and effectors, such as adenylyl cyclase. Allosteric properties of GPCR oligomers can therefore extend to ligand interactions through the different components of the macromolecular complexes.
Keywords: G protein-coupled receptors, oligomerization, receptor heteromers, receptor homomers, allosterism, macromolecular complexes, artificial systems, native tissues
1. Introduction
Ten years have passed since a group of experts established by consensus a series of definitions and recommendation related to receptor oligomerization (Ferré et al., 2009). ‘Receptor heteromer’ was defined as “a macromolecular complex composed of at least two (functional) receptor units (protomers) with biochemical properties that are demonstrably different from those of its individual components”. This definition would be distinguished from a ‘heteromeric receptor’, defined as a dimeric or oligomeric receptor for which the minimal functional unit is composed of two or more different subunits that are not functional on their own. ‘Receptor homomer’ and ‘homomeric receptor’ would then be respectively defined the same as receptor heteromer and heteromeric receptor but composed of two or more identical protomers or identical non-functional subunits. Additional definitions were proposed, such as ‘allosteric interaction in the receptor heteromer’ and ‘biochemical fingerprint’ (see below), as part of recommendations for the identification of receptors oligomers in native tissues (Ferré et al., 2009).
These recommendations were meant to facilitate the research in the field, particularly, of G protein-coupled receptor (GPCR) oligomerization, homo and heteromerization, which was held with skepticism by several leaders in the GPCR research community. More precisely, although the evidence of GPCR oligomerization in artificial systems was generally accepted, their presence and functional significance in native tissues was clearly challenged (see open discussions by Bouvier and Hébert, 2014, and Lambert and Javitch, 2014). This would not be the case for family C GPCRs (which include glutamate and GABA metabotropic receptors), which are generally accepted to be obligatory homodimers, stabilized by disulfide bonds in their large extracellular domains or coiled-coil α helices in their C termini (Kniazeff et al., 2011). During the last ten years, with the development of new biophysical and computational methods, a series of studies led to the unequivocal demonstration of oligomerization in artificial systems of not only family C but also family A and family B GPCRs, as well as to the identification of their unique biochemical properties, which would allow the demonstration of GPCR oligomers in native tissues.
In this essay, we discuss the unique biochemical properties of GPCR oligomers in the frame of ‘allosterism’, which we believe it provides sufficient evidence for the demonstration of GPCR homo and heteromers in native tissues. We will also address how the significant advances in biophysical, computational and crystallization techniques, with the boost of crystal structures of GPCRs in different conformational states, including those bound to G proteins and to orthosteric and allosteric ligands, have provided significant structural insights about the mechanisms behind allosterism in GPCR. These insights provided distinct clues about the mechanisms of allosteric interactions in GPCR homomers and heteromers. Furthermore, we discuss how recent experiments challenge some classical pharmacological concepts of interactions between GPCR ligands believed to occur downstream in the signaling pathway, also known as interactions “via intracellular loops” or interactions at the “second-messenger level” (Zoli et al., 1993; Agnati et al., 1995; Ferré et al., 1997). These studies indicate that some of these interactions should be reevaluated as allosteric, mediated by macromolecular complexes of GPCR heteromers, G proteins and effectors.
2. Classical GPCR allosterism
Allosterism has been defined as the process by which the interaction of a chemical or protein at one location on a protein or macromolecular complex (the allosteric site) influences the binding or function of the same or another chemical or protein at a topographically distinct site (Smith and Milligan, 2010). The prefix ‘allo’ has a Greek origin meaning ‘other’ and the term allosterism was introduced by Jacques Monod and Francois Jacob (1961), to account for an ‘allosteric inhibition’. This implied a situation where an enzyme inhibitor (the allosteric ligand) was not a steric analog of the substrate. Their assumptions were largely based on results obtained by their graduate student Jean-Pierre Changeux with the enzyme L-threonine deaminase, its substrate L-threonine and the regulatory inhibitor L-isoleucine (Changeux, 1961). Allosterism would provide a new mechanism of regulatory proteins different to covalent modulation, such as phosphorylation. The introduction of these non-covalent modulations would initiate a revolution in Biochemistry, as Monod would anticipate when describing allosterism as “the second secret of life”, after the genetic code (Monod, 1971).
Interestingly enough, the concept of allosterism developed initially in the frame of what it seemed a common oligomeric structure of regulatory enzymes, with the Monod-Wyman-Changeux (MWC) model (Monod et al, 1965). Structurally, the MWC model implied that allosteric proteins are oligomers resulting from the assembly of ‘protomers’ associated in such a way that the molecule possesses at least one axis of symmetry. Functionally, it represented the first elaborated model of the two-state mechanism of pre-existing conformational states, a consequence of a selective, rather than an instructive, effect of ligands (Changeux, 2012). Oligomeric allosteric proteins would have access to at least two reversible states, which would be dependent on the conformational constraints imposed by the protomers. The affinity of both ligands, the substrate and the allosteric ligand, are altered with the transition of the protein from one to the other state and the molecular symmetry is always conserved. Therefore, the presence of the allosteric ligand is associated with a modification of the apparent affinity of the protein for the substrate, and conversely. As mentioned by Fenton (2008), allosterism can be more strictly defined as a comparison of how one ligand binds in the absence versus the presence of the second ligand.
A logical generalization would follow from allosterism of regulatory enzymes to allosterism of membrane receptors, which would be equivalent to regulatory proteins which activated state would not imply a modification of the substrate (enzymes) but a signal transduction (Changeux and Edelstein, 2005). An obvious parallelism could be established between substrate and orthosteric ligand (which binds to the same receptor site as the endogenous transmitter). The allosteric ligand, by binding to a non-orthosteric site, can then modify either of the two independent properties of the orthosteric ligand, affinity (the avidity to bind to the receptor) and intrinsic efficacy (the power of the bound ligand to induce a physiological response). As pointed out by Changeux (2012), “the notion that ligands selectively stabilize the state to which they preferentially bind and thereby mediate signal transduction via selection of conformational states arises directly from the formal description of the MWC model”.
3. Allosterism in GPCR monomers
The seminal studies by Nobel laureate Brian Kobilka and coworkers and other research groups (for recent review, see ref (Weis and Kobilka, 2018) provided, first, an example of a well-characterized allosteric mechanism within family A GPCRs, more specifically, of a process by which the interaction of a ligand at one location of the GPCR (the orthosteric site, but in this case, equivalent to the allosteric site in the definition of allosterism; see above) influences the binding of a protein at a topographically distinct site. With the more recent application of nuclear magnetic resonance (NMR) and double electron-electron resonance spectroscopy and single-molecule fluorescence resonance energy transfer (FRET) analysis, their studies also provided a strong support for the existence of pre-existing conformational states of GPCRs and the selection and stabilization of several different states (conformational selection), from more inactive to more active, from inverse agonists to neutral antagonists, partial agonists and full agonists, as well as their dependence on G protein and nucleotide binding (Liu et al., 2012; Ye et al., 2016; Manglik et al., 2015; Gregorio et al., 2017).
Thus, GPCRs are dynamic proteins that permit rapid small-scale structural fluctuations and pass through an energy landscape (Deupi and Kobilka, 2010) to adopt a number of conformations ranging from inactive to active (Figure 1A). The transition probability among conformations depends on their energy difference and the energy barrier between them. Ligand binding to the orthosteric site of a monomer (Figure 1A) changes the shape of the energy landscape relative to the unliganded landscape (black line), in such a manner that inverse agonists and antagonists (in red) stabilize inactive conformations (red line), agonists (in green) stabilize intermediate conformations, and the final formation of the agonist-receptor-active G protein complex stabilizes active conformations (green line) (adapted from Nygaard et al., 2013). The inactive and active conformations, at the molecular level, have been elucidated by X-ray crystallography. An agonist binding at the extracellular side, apparently, triggers small local structural changes near the binding site that are translated into larger-scale helix movements at the intracellular side (Rasmussen et al., 2011). We then have a significant knowledge about the basic molecular mechanisms of this GPCR allosterism, by which an agonist transfers a “signal” from the orthosteric ligand-binding pocket to a G protein binding to a specific portion of the GPCR intracellular domain, the Gα subunit-binding “intracellular cavity”.
Figure 1.
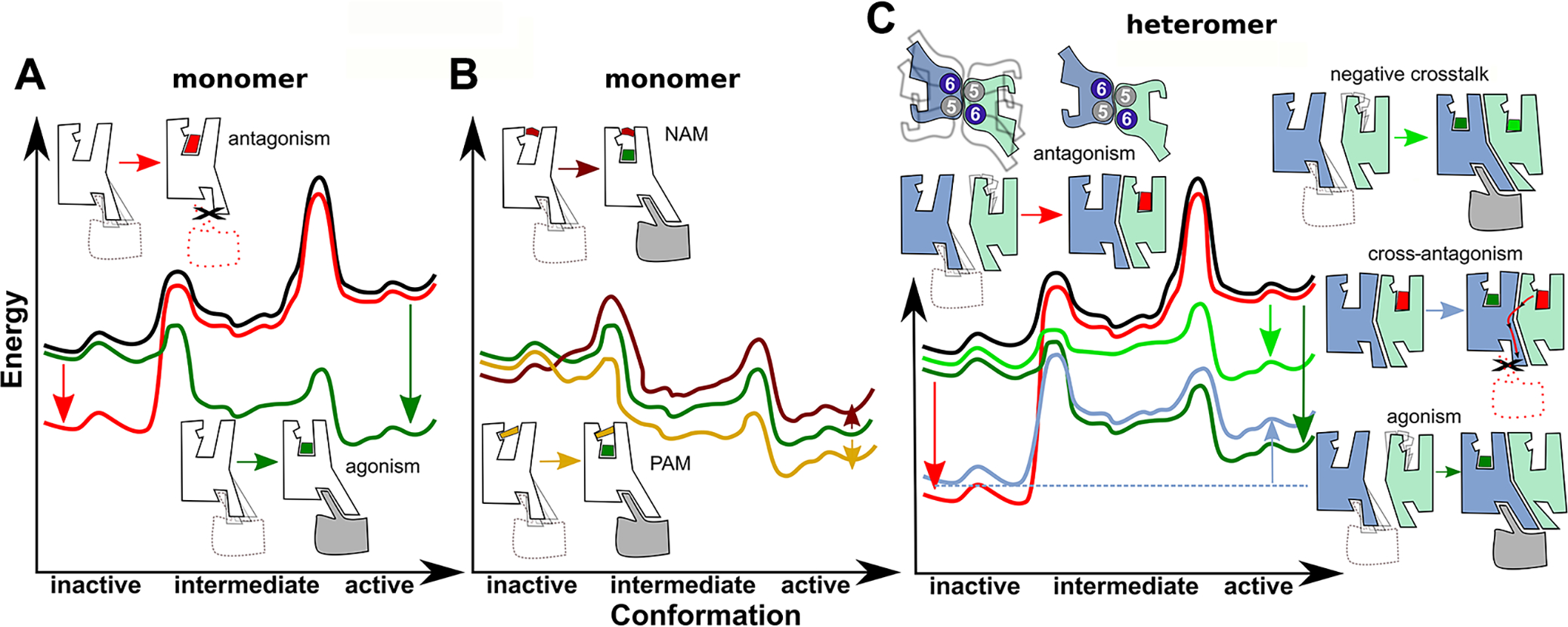
Dynamic properties of GPCR monomers and heteromers. (A) Energy landscape of a GPCR monomer in the unliganded form (black line), after the action of an inverse agonists/antagonists (in red) or an agonist (in green). (B) Energy landscape of a GPCR monomer in the presence of a negative allosteric modulator (NAM, in brown) and a positive allosteric modulator (PAM, in yellow). (C) Energy landscape of a GPCR heteromer in the unliganded form (black line), in the presence of an antagonist (in red), an agonist (in green), one antagonist and one agonist (in blue), and two agonists (in light green).
In a nutshell, the binding of the orthosteric agonist to the active conformation of the receptor is dependent on a very subtle rearrangement of some key residues of GPCRs (more specifically family A GPCRs), localized in three of the seven transmembrane domains (TMs) of the GPCR, TM 3, TM 5, and TM 6, in the orthosteric ligand-binding pocket, which leads to a mechanically amplified larger movement of particularly TM 6 in its intracellular site (Weis and Kobilka, 2018). This movement of TM 6 is simply mediated by a lever-arm effect, due to the presence of a kink (caused by an interruption in the intrahelical hydrogen bonds at conserved proline residues) close to its extracellular end. In this way, a small approximation of the extracellular side of TM 6 to TM 3 (which is a straight TM that forms the core of the GPCR) leads to a large separation of the intracellular side of TM 6 from TM 3 and also TM 5 and TM 7, creating an intracellular cavity for the helix 5 of the Ras-GTPase domain of the Gα subunit. Beautifully, with this TM 6 movement, some residues from the intracellular site of TM 3 and TM 5 that interact with TM 6 in the inactive state of the receptor are freed to form direct interactions with the C-terminal α5 helix of the G protein in the active state (Weis and Kobilka, 2018).
Considering GPCRs as monomeric entities, allosteric modulators are ligands that bind to sites that are topographically distinct from the orthosteric site and also alter the energy landscape of the receptor (Christopoulos, 2014; Figure 1B). Negative allosteric modulators (NAMs) (in brown) decrease the action (potency and/or efficacy) of the orthosteric agonist (in green) by destabilizing intermediate and active conformations. In contrast, positive allosteric modulators (PAMs) (in yellow) increase the action (potency and/or efficacy) of the orthosteric agonist by stabilizing intermediate and active conformations. Ago-PAMs are allosteric modulators than in addition to increase the potency and/or efficacy of orthosteric agonists, can induce receptor activation by themselves. There are numerous allosteric binding sites as elucidated by the increasing number of published crystal structures of classes A, B, C, and F of GPCRs in complex with small-molecule allosteric modulators (for a recent review, see Lu and Zhang, 2019). In addition to the prevailing site at the TMs near the extracellular side (classes A, C, and F), other binding sites are also observed at the TMs near the intracellular side (classes A and B), or at the extra-helical domains facing the membrane bilayer (classes A and B), among others.
We are also starting to understand the mechanistic aspects of these allosteric modulators. For instance, the crystal structure of the muscarinic M2 receptor (M2R) simultaneously bound to an orthosteric agonist and a PAM shows that the allosteric modulator binds at the extracellular vestibule inducing a slight contraction of the binding pocket, enhancing the potency without modifying the efficacy of the orthosteric agonist (Kruse et al., 2013). In contrast, a crystal structure of the β2-adrenergic receptor (β2R) also in complex with an orthosteric agonist and a PAM shows that the allosteric modulator binds on the receptor’s inner surface in a pocket created by intracellular loop 2 and TMs 3 and 4, stabilizing the loop in an α-helical conformation required to engage the G protein (Liu et al., 2019). Thus, the mechanisms by which PAMs elicit their function is, in contrast to orthosteric agonists, highly diverse. Going back to the definition of allosterism, these represent mechanistic explanations for a process by which the interaction of a ligand at one location of the GPCR (the allosteric site) influences the binding of another ligand at a topographically distinct site (the orthosteric site).
4. Allosterism in GPCR homomers
From radioligand binding experiments, it has been long recognized that agonists show ‘binding heterogeneity’, a dispersion of affinities for GPCRs. This phenomenon has been reproduced in experiments from membrane extracts from multiple native tissues and mammalian transfected cells as well as from artificial systems, including GPCR reconstituted in detergent micelles, liposomes (phospholipid vesicles with or without different proportions of cholesterol and integral membrane proteins) or nanodisks (phospholipid bilayer preparations held together by membrane scaffold proteins) (for review, see Ferré et al., 2014). The typical illustration is a competition experiment between a moderate concentration of a radio-labelled antagonist and increasing concentrations of the agonist, which results in a non-steep or even biphasic curve, apparently indicating the existence of two populations of GPCRs, with high and low affinities for the agonist (Figure 2A; solid curve). In the presence of a G protein, guanylyl nucleotides promote an ‘apparent’ inter-conversion from the high to the low affinity sites, with steepening and shifting to the right of the competition curve (Figure 2A; dotted curve), which led to the general assumption that the agonist binding heterogeneity was just a G protein-dependent phenomenon, counteracted by guanylyl nucleotides.
Figure 2.
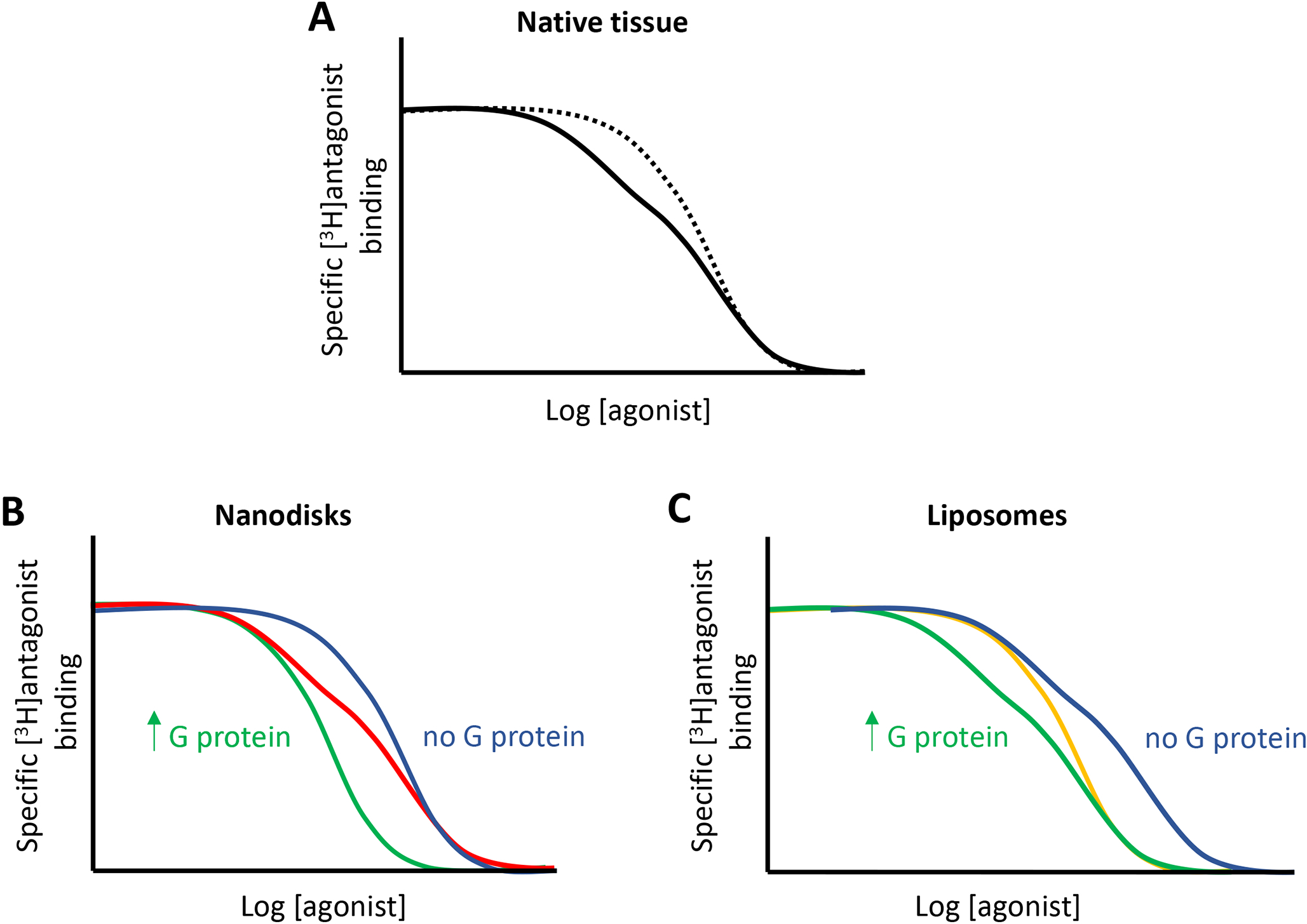
Theoretical [3H]antagonist/agonist competitive inhibition curves from radioligand binding experiments in membrane preparations from a native tissue (A), or from preparations of GPCRs reconstituted as monomers in nanodisks (B) or oligomers in liposomes (C), in excess (relative to the GPCR) or absence of G proteins. In A, the dotted curve represents the effect of guanylyl nucleotides. In B, the red curve represents the effect of an intermediate concentration of G proteins. In C, the orange curve represents the effect of guanylyl nucleotides in the presence of an excess of G proteins (see text and Redka et al., 2013, 2014)
In line with the classical view of GPCR signal transduction, a still common view of GPCR signaling holds that agonists promote the formation of a transient complex between the monomeric receptor and the G protein, with conformational changes in both molecules that lead to an increased affinity of the receptor for the agonist and for the heterotrimeric Gαβγ protein, which leads to G protein activation. The deep-rooted ‘ternary complex’ model of ligand-GPCR binding (De Lean et al., 1980) postulated that the receptor-G protein complex accounts for the high affinity site for the agonist, while the G protein-uncoupled receptor accounts for the low affinity site. Guanylyl nucleotides would promote uncoupling of the receptor-G protein complex, converting all receptors into low affinity sites. According to the model, a non-steep or biphasic antagonist-agonist competitive-inhibition curve implied the existence of two populations of receptors, coupled and non-coupled to the G protein. Implicitly, this interpretation assumed there is a limited pool of G proteins, an assumption difficult to reconcile with the fact that the expression of G proteins in native cell systems outnumbers that of GPCRs (Neubig, 1994).
However, a large amount of experimental data points to the existence of GPCR oligomerization and indicate that, in fact, oligomerization is a main property of GPCRs. This was initially suggested from a study by Limbird, Meyts and Robert Lefkowitz (who shared the Nobel prize with Brian Kobilka), that demonstrated negative cooperativity of the binding of a radio-labelled β2R agonist in membrane preparations from frog erythrocytes (Limbird et al., 1975). The existence of such a negative cooperativity was confirmed by kinetic experiments, which showed a significant increase in the rate of dissociation of the labelled β2R agonist by the non-labelled β2R, indicating the presence of a non-competitive negative interaction between labelled and non-labelled β2R agonist (Limbird et al., 1975). Thus, ligands that would compete for the same site on a monomeric GPCR should not influence one another’s dissociation kinetics. In contrast, allosteric modulations between two simultaneously bound and interacting sites, either within a receptor monomer or across a receptor homodimer or oligomer, should alter ligand dissociation (May et al., 2007). Many more experiments would follow which would be completely in line with the existence of allosteric modulations between different molecules of the same orthosteric agonist in oligomeric and most commonly homodimeric GPCRs, a ‘homodimer-cooperativity model’ (reviewed in Ferré et al. 2014).
According to the homodimer-cooperativity model, binding heterogeneity would be G protein-independent, at least under conditions of receptors in excess of G proteins. There are no high and low affinity states of a GPCR monomer for the agonist, but an affinity of the agonist for the binding to the first protomer in the homodimer, which allosterically modulates the affinity of the same ligand for the second protomer. In fact, the ‘monomer-ternary complex model’ could never explain some radioligand binding data, such as an apparent modulation of antagonist binding by guanine nucleotides (Burgisser et al., 1982; De Lean et al., 1982; Klotz et al., 1990), biphasic antagonist/antagonist competition curves (see below and Orrú et al., 2011), bell-shaped agonist/antagonist competitive curves (Casadó-Anguera et al., 2019) or a positive cooperativity (Albizu et al., 2006). Unfortunately, the initial data indicating cooperativity of the same orthosteric ligand and, therefore, indicating GPCR oligomerization, was overshadowed by the discovery of the powerful allosteric modulation by G proteins and guanylyl nucleotides and by the success of the pragmatic monomer-ternary complex model.
In apparent support for the monomer-ternary complex model, biphasic and guanylyl nucleotide-sensitive antagonist/agonist competitive inhibition curves were obtained with GPCR monomers (of β2R, μ opioid receptors [MOR], rhodopsin and M2R) either in solution or reconstituted in nanodisks, where they could also bind and activate G proteins (Bayburt et al. 2007; Ernst et al., 2007; Whorton et al., 2008; Kuszak et al., 2009; Redka et al., 2013, 2014). In these studies, the proportion of receptor with high affinity for agonists increased from zero to 100% by increasing the G protein pool, indicating that the low-affinity sites were receptors in excess of G proteins, that the agonist binding heterogeneity was totally G protein-dependent, implying a G protein-mediated allosteric modulation of an orthosteric ligand (Figure 2B). According to our definition of allosterism, this would be an example of the process by which the interaction of a protein (the α subunit of a G protein, which would act as the allosteric modulator), at one location of another protein (GPCR) influences the binding of a chemical (the orthosteric ligand) at a topographically distinct site. In these preparations the effect of G proteins on the binding of the orthosteric agonist was reversed by GTP and GTP analogues. Therefore, GPCR monomers in the presence of G proteins and guanylyl nucleotides are indistinguishable from GPCR monomers in the absence of G proteins (Figure 2B; blue curve). Guanylyl nucleotides, and GTP under physiological conditions, act as additional negative allosteric modulators, as potent inhibitors of the ability of helix 5 of the Ras-GTPase domain of the Gα subunit to interact with the intracellular cavity of the GPCR active conformation.
The results of isolated GPCR monomers were initially interpreted as a demonstration of their prevalence in native tissues, as main functional units, and as a demonstration of the main G protein dependence of agonist binding heterogeneity. But recent seminal studies by James Wells’s group demonstrated that, in the presence of enough G protein molecules, without receptors in excess of G proteins, GPCR oligomers show a significant G protein-independent binding heterogeneity, cooperativity (Redka et al., 2013, 2014) (Figure 2C; green curve). These studies compared the effect of G proteins and guanylyl nucleotides in radioligand binding experiments in purified M2R, M2R monomers reconstituted in nanodisks and M2R oligomers (mostly tetramers as the predominant species) reconstituted in liposomes (Figure 2C). Importantly, the experiments were also performed in cardiac muscle membrane preparations, which revealed that M2R in native tissues have the same biochemical characteristics than when reconstituted in liposomes as oligomers (Redka et al., 2013, 2014), demonstrating that a predominant proportion of M2R in cardiac muscle is forming oligomers. We are then closing the loop and going back to the initial evidence for negative cooperativity of β2R described by Limbird et al. (1975) to explain GPCR agonist binding heterogeneity.
The observation of GPCR agonist binding cooperativity therefore reveals that at least two GPCR protomers form part of a minimal GPCR functional unit. An important additional concept that then arises from the field of GPCR oligomerization is that the pentameric structure constituted by one GPCR homodimer and one heterotrimeric G protein provides a main functional unit and oligomeric entities can be viewed as multiples of dimers (Ferré et al., 2014). Thus, the relatively large size of the heterotrimeric G protein would make it unlikely to sustain a simultaneous binding of more than one G protein with a GPCR dimer. Additional evidence has been provided from a large number of studies by application of resonance energy transfer (RET), bimolecular complementation of fluorescent of bioluminescent molecules and other biophysical techniques in living cells, such as fluorescence correlation and cross-correlation spectroscopy (FCS and FCCS) and analysis of single fluorescence-labeled receptor molecules by total internal reflection fluorescence microscopy (TIRF) (Calebiro et al., 2013; Herrick-Davis et al., 2013; Teichmann et al., 2014). These methods differ in sensitivity and the population of receptors that they analyze. RET (which includes fluorescence and bioluminescence resonance energy transfer [FRET and BRET], fluorescence lifetime imaging [FLIM] and fluorescence recovery after photobleaching [FRAP]) and bimolecular complementation are proximity-based assays that monitor the distance between the fluorescent and bioluminescent probes, whereas TIRF, FCS and FCCS have near single molecule sensitivity.
Some of these studies have provided evidence for a receptor-dependent differential degree of oligomerization (oligomers versus monomers) and of the average stoichiometry of the oligomer species (number of monomers per oligomer), as recently supported by experiments in liposomes also using fluorescence-microscopy-based techniques (Walsh et al., 2018) and computational methods that characterize the energetics of oligomer interfaces, based on large-scale coarse-grained molecular dynamics simulations (MDS) (Periole et al., 2012; Meral et al., 2018). Those studies indicate a low degree of oligomerization for B1R and MOR, intermediate for β2R and predominant for cannabinoid CB1 receptor (CB1R) and rhodopsin. Nevertheless, apart from the fact that MDS only consider TM interfaces (see below for the role of intracellular domain interactions in GPCR heteromerization), the results of these studies performed in artificial systems most probably represent an underestimation of what occurs in native tissues. As also demonstrated in artificial systems, just the lipidic composition and membrane curvature can influence the degree of GPCR oligomerization (Botleho et al., 2006; Prasanna et al., 2014; Guixa-Gonzalez et al., 2016; Walsh et al., 2018). In addition, as discussed below, there is increasing evidence for an elaborated formation of protein complexes that include GPCR oligomers (homo and heteromers), signaling-related proteins and effectors. The emerging specific quaternary structure of these macromolecular complexes points to the existence of multiple simultaneous points of contact between their elements, calling for a significant stabilization of GPCR oligomers (Navarro et al., 2018).
5. Allosterism in GPCR heteromers
The first evidence for possible allosteric interactions in GPCR heteromers was labelled as “intramembrane receptor-receptor interactions” by Luigi Agnati and Kell Fuxe, based on observations from early radio-ligand binding experiments in the 1980’s, where a selective ligand for a GPCR changed the binding characteristics of another selective ligand for a different GPCR (reviewed in Agnati et al., 2003). Although, initially, allosteric mechanisms were implicitly assumed to be involved, it was not until recently than an operational definition of ‘allosteric interaction in the receptor heteromer’ was advanced: “an intermolecular interaction by which binding of a ligand to one of the receptor unit in the receptor heteromer changes the binding properties of another receptor unit” (Ferré et al., 2009). More recently, it has been explicitly formulated that GPCR heteromerization implies the possibility of multiple allosteric interactions between orthosteric ligands of different GPCRs (Ferré et al., 2014) (Figure 3A).
Figure 3.
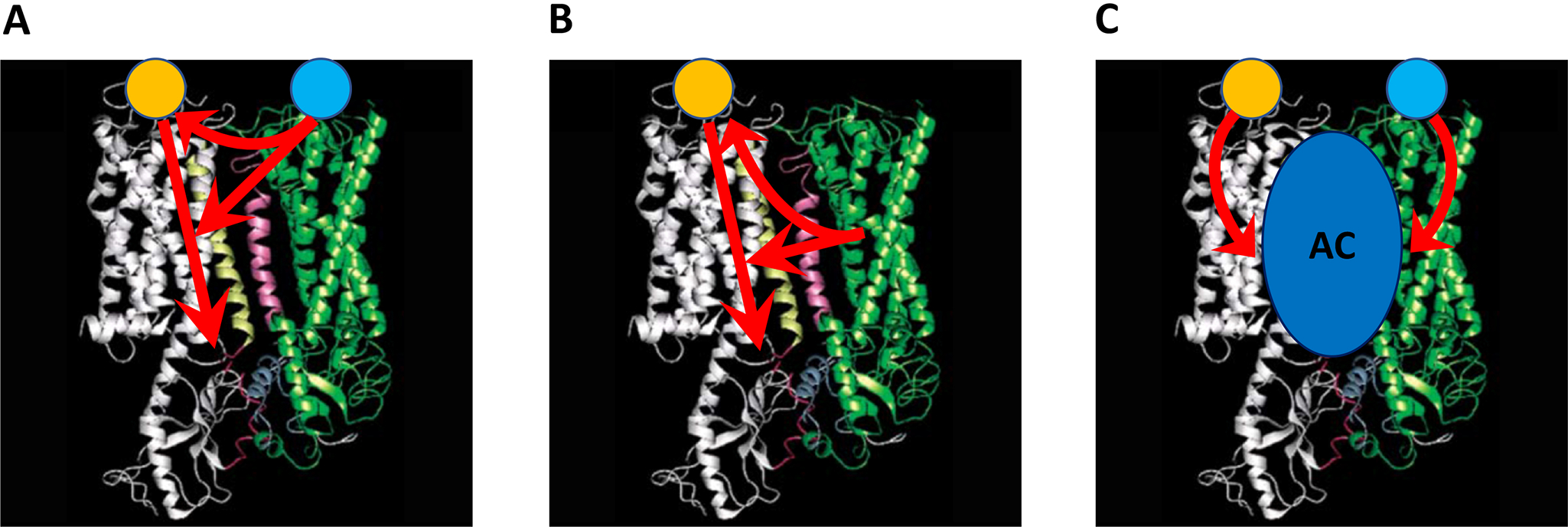
Allosteric interactions in GPCR heteromers: In A, allosteric interaction between ligands, where a ligand of one of the protomers changes the affinity or efficacy of a ligand for the other protomer. In B, ligand-independent allosteric interaction, where the binding of one protomer changes the properties of a ligand for the other protomer. In C, allosteric interaction through the effector. Homomeric interactions are not included and only one protomer of each different GPCR is shown. AC, adenylyl cyclase.
However, if there are still doubts about the existence of GPCR homomers, even more skepticism exists around the concept of GPCR heteromerization. Nevertheless, the number of putative functional and pharmacologically significant GPCR heteromers keeps increasing. It must however be acknowledged that many candidates do not follow the consensus criteria recently established for their identification in native tissues (Ferré et al., 2009, 2014; Gomes et al., 2016). The same as for GPCR homomers, a long list of biophysical techniques has been used to study GPCR heteromerization and, the same as for GPCR homomers, the most important challenge is their demonstration in native tissues (Ferré et al., 2009, 2014; Gomes et al., 2016). Although some attempts have been made, application of those or other techniques that imply a “direct” visualization of GPCR oligomers in native tissues remains problematic (reviewed in Ferré et al., 2014). An indirect but nevertheless straightforward demonstration depends on the identification of a ‘biochemical fingerprint of the GPCR heteromer’, which is currently defined as “a biochemical characteristic of a receptor heteromer, which can be used for its identification in a native tissue” (Ferré et al., 2009). To ascertain a biochemical fingerprint of the receptor heteromer, the putative biochemical property should be disrupted with molecular or chemical tools that significantly destabilize the quaternary structure of the heteromer (such as introducing mutations of key determinant residues at the GPCR heteromer interface or using competing peptides that harbor the same amino acid sequence as that of the heteromer interface).
Allosteric interactions in GPCR heteromers seem to constitute a universal property of GPCR heteromers which can then be commonly used as a biochemical fingerprint to identify the heteromer in native tissues. As an example, in this essay, we will address the allosteric interactions in the heteromer constituted by adenosine A2A receptors (A2AR) and dopamine D2 receptors (D2R), which is one of the few examples of a generally accepted GPCR heteromer (Gomes et al., 2016). Since the first report in 1991 (Ferré et al., 1991), A2AR ligands have been shown to modulate the binding properties of D2R ligands in membrane preparations from transfected cells and native tissues. Although initially thought to be restricted to a negative crosstalk between agonists, where an A2AR agonist reduced the affinity of dopamine and D2R agonists, more recent findings extended the interactions to any orthosteric ligand. This included the ability of selective A2AR antagonists or caffeine, a non-selective A1R-A2AR antagonist, to reduce the affinity of D2R ligands (Bonaventura et al., 2015; Ferré et al., 2016). That these interactions represent allosteric modulations in the A2AR-D2R heteromer was demonstrated by the ability of specific mutations or synthetic interfering peptides that selectively disrupt or destabilize the quaternary structure of the heteromer to selectively disrupt the interactions between A2AR and D2R ligands (for review, see Ferré et al., 2016). Importantly, the selective effect of interfering peptides could also be demonstrated in native tissue, in sheep brain (striatum) membrane preparations, demonstrating the existence of the A2AR-D2R heteromer in the brain and the dependence on the right quaternary structure of the heteromer to disclose allosteric interactions between orthosteric A2AR and D2R ligands (Bonaventura et al., 2015; Ferré et al., 2016).
The fact that both A2AR agonists and antagonists produced the same effect on D2R agonist binding and also signaling (Bonaventura et al., 2015) represented a challenge to our initial assumption of allosteric interactions in the A2AR-D2R heteromer being involved in the opposite and counteractive effects of A2AR agonists and antagonists observed in behavioral and functional studies. Those effects would include the psychomotor depression induced by A2AR agonists and the psychomotor activation induced by A2AR antagonists, including caffeine, as well as their competitive counteraction upon co-administration, which would be expected from their competition for the A2AR orthosteric site. The conundrum could be solved by the realization of the tetrameric structure of the A2AR-D2R heteromer, composed of A2AR and D2R homodimers, and by the existence of simultaneous heteromeric and homomeric allosteric interactions in the heterotetramer (Bonaventura et al., 2015). We could assume that, in the A2AR-D2R heterotetramer, occupancy of the A2AR homodimer with either an orthosteric agonist or an orthosteric antagonist is associated with a conformational state that determines the allosteric modulation of D2R ligands, while simultaneous occupancy of the A2AR homodimer by an orthosteric agonist and an orthosteric antagonist would not be possible with this conformational state. What could be demonstrated is that an A2AR agonist, but not an antagonist, allosterically modifies the dissociation kinetics of a radioactive A2AR antagonist ligand in striatal membrane preparations (Bonaventura et al., 2015). The functional significance of these simultaneous homomeric and heteromeric interactions in the brain was demonstrated with electrophysiological and locomotor activity experiments. We could show that, under specific experimental conditions, A2AR antagonists behaved as A2AR agonists, decreasing D2R function, and these effects were counteracted upon co-administration of both A2AR agonists and antagonists. For instance, at high doses, caffeine and A2AR antagonists produce locomotor depression, which can be counteracted by locomotor depressant doses of A2AR agonists (Bonaventura et al., 2015). Since under physiological conditions there is a tone of adenosine, this could in fact be the main mechanism by which caffeine and A2AR antagonists produce psychomotor stimulant activation, by counteracting the functional effects that depend on D2R signaling by the A2AR-D2R heterotetramer.
We are also beginning to explore the mechanisms behind the allosteric interactions through the heteromeric interface in GPCR heteromers. For instance, we have shown that in the heteromer of CB1R and serotonin 5-HT2A receptors (5HT2AR) a CB1R antagonist blocks the signalling triggered by a 5HT2AR agonist and that, conversely, a 5HT2AR antagonist blocks the signalling triggered by a CB1R agonist (Viñals et al., 2015). This cross-antagonism, the ability of an orthosteric antagonist of one of the protomers in a receptor heteromer to antagonize the signaling of an agonist of the other molecularly different protomer, has been described frequently, representing a common allosteric property of GPCR heteromers (Ferré et al., 2014). Thus, one of the protomers allosterically modulates the energy landscape of the interacting receptor (Figure 1C). We proposed, and demonstrated using interfering peptides, that this allosteric modulation is determined by the ability of CB1R and 5HT2AR to establish a symmetrical molecular interaction via TM 5 and TM 6 (Viñals et al., 2015). Relative to the unliganded landscape (black line), in which both TM 5 and TM 6 of both protomers are in a dynamic equilibrium of conformations, antagonist binding to either protomer stabilizes the closed, inactive, conformations of TM 5 and TM 6 (see above), promoting the interaction with TM 5 and TM 6 of the other protomer via the formation of a stable four-helix bundle, as described for the dimeric crystal of the MOR (Manglik et al., 2012) (red line). Importantly, this energy minimum of the antagonist-bound receptor is more stable in heteromers than in monomers, because of the additional formation of the four-helix bundle. These low free-energy states of the antagonist-bound heteromer (inactive states) impede the possibility of an agonist binding to the other protomer, which would require the energy minima of the active states (blue line), leading to cross antagonism (Figure 1C). The same as for the A2AR-D2R heteromer, a negative crosstalk could also be observed in the CB1R-5HT2AR heteromer, implying that an agonist binding to one of the protomers leads to a decrease in the signaling promoted by an agonist binding to the other molecularly different protomer. In this case, agonist binding to one protomer stabilizes the active conformations of TM 5 and TM 6 (open cavity) and the binding of the G-protein to the active protomer (dark green line). But simultaneous activation of both protomers by their corresponding agonists leads to energy minima less stable than activation of a single protomer (light green line), most probably because of a steric clash of the active conformations of TM 5 and TM 6 in both protomers, leading to negative crosstalk (Figure 1C). These putative mechanisms for cross-antagonism and negative crosstalk would apply irrespective of a dimeric or heterotetrameric structure of a GPCR heteromer, since these allosteric modulations would, after all, depend on the key role of the TM5 and TM6 on heteromerization and on receptor activation. Thus, restrictions of active conformations of the GPCR protomers involved in heteromerization would be directly imposed by molecular arrangements within the heteromeric interface.
In addition to providing the frame for allosteric interactions between ligands binding to their orthosteric sites, heteromerization can lead to changes in the properties of specific ligands for one of the protomers, independent of other ligands binding to the other protomer (Figure 3B). Indeed, the proof of concept came from experiments in transfected mammalian cells, where the potencies of different selective A2AR antagonists at binding to the A2AR alone or when co-expressed either with D2R or A1R were compared (Orrú et al., 2011). Interestingly, the most dramatic finding was that the A2AR antagonist SCH442416 showed a selective low affinity for the A2AR when co-expressed with D2R compared with its affinity for the A2AR alone or co-expressed with A1R. More specifically, SCH442416 showed a pronounced negative cooperativity of its binding to the A2AR homodimer within the A2AR-D2R heterotetramer, a biphasic antagonist/antagonist competition curve (Orrú et al., 2011). These results were in fact the first to reveal that the quaternary structure of the A2AR-D2R heteromer included A2AR homomers. The specific behavior of SCH442416 in the A2AR-D2R heteromer could then be used as a biochemical fingerprint of the heteromer in native tissues. Thus, SCH442416 negative cooperativity could be demonstrated in mouse striatal preparations, but not from mice with conditional striatal D2R-KO (Ferré et al., 2018).
The same type of ligand-independent allosteric interaction has recently been reported for the selective significant decrease in potency of the MOR agonist methadone, compared with morphine and fentanyl, within the MOR-galanin Gal1 receptor heteromer (Gal1R) (Cai et al., 2019). Since these heteromers are selectively localized in the ventral tegmental area (localization of cell bodies of dopaminergic cells involved in the processing of natural rewards), this could explain a selective dissociation of the clinical therapeutic versus euphoric/rewarding effects of methadone compared with the other opioids (Cai et al., 2019). We are obtaining data from computational modeling indicating that binding of the Gal1R to the MOR promotes a little displacement of TM6 which can selectively alter the stability of methadone in the MOR binding pocket (in preparation). Therefore, changes in the pharmacological properties of ligands induced by receptor heteromerization should constitute a very important strategy in the search for new therapeutic agents with further selectivity for their therapeutic versus side, unwanted effects.
6. Molecular architecture of GPCR heteromers
If the pentameric structure constituted by a GPCR homodimer and one heterotrimeric G protein provides a main functional unit, oligomeric entities should be expected to often represent multiples of dimers and GPCR heteromers would be often constituted by heteromers of homodimers (Ferré et al., 2014; Ferré, 2015). This seems to apply particularly to heteromers that include a GPCR homodimer preferentially coupling to Gs/olf (Gs for short) proteins and another GPCR homodimer preferentially coupled to Gi/o (Gi for short) proteins. It was postulated (Ferré, 2015) that such a ‘GPCR heterotetramer’ would sustain a functional pre-coupled macromolecular complex that includes two molecularly different GPCRs, their cognate G proteins and adenylyl cyclase (AC) and would provide the frame for a canonical interaction at the AC level. This canonical interaction implies the ability of an activated Gi-coupled receptor to inhibit a Gs-coupled receptor-mediated activation of certain subtypes of AC. More specifically, those AC isoforms than can be inhibited by the α subunits of Gi proteins, including AC1, AC5, and AC6 (Sadana et al., 2009a), and requires the simultaneous respective interaction of the Ras-GTPase domains of the α subunits of the Gs and Gi proteins with the C2 and C1 catalytic domains of AC (Dessauer et al., 1998).
The structural details of TM interfaces of GPCR heteromers, in the absence of crystal structures, cannot be predicted. Hence, in order to suggest the molecular interfaces of GPCR heteromers, we applied an indirect approach consisting on the application of synthetic peptides with the amino acid sequence of TMs of the corresponding GPCRs, to alter inter-protomer interactions in the heteromeric and homomeric interfaces in the A2AR-D2R heterotetramer. The disruptive peptides were fused to the HIV transactivator of transcription (TAT) peptide, which facilitates their insertion in the plasma membrane and determines its correct orientation (He et al., 2011). The TAT-TM peptides were then tested in bimolecular fluorescence complementation (BiFC) experiments, for their specific ability to disrupt BiFC of complementary halves of a fluorescent molecule separately fused to two different A2AR protomers, two different D2R protomers or to an A2AR and a D2R protomer. From the fourteen possible TAT-TM peptides corresponding to all TM domains of the A2AR and the D2R (abbreviated as TM1, TM2… of the respective GPCR), only TM6 of the A2AR and TM6 of the D2R disrupted BiFC of A2AR-A2AR and D2R-D2R homodimers, respectively (Navarro et al., 2018). These results therefore indicate a symmetrical TM 6/TM 6 interface for both homomers. The different apparent interface of D2R homomers reported in previous studies (which suggested the involvement of TM 4 and TM 5 (Guo et al., 2005) could be due to the different experimental approaches and, most likely, due to the presence of heteromeric partner receptors that influence the homomeric interfaces (Kofalvi et al., submitted). The involvement of TM 6 in D2R oligomerization was further substantiated by the selective ability of TM6 of D2R to completely counteract the decrease in binding of a bivalent ligand with selective picomolar affinity for the D2R homodimer (Pulido et al., 2018). Since the binding experiments were performed in membrane preparations of mammalian striatum, the bivalent ligand allowed to identify D2R homomers (homodimers or oligomers) in the native tissue. An important corollary of these findings is that they imply that D2R homomers are the predominant species in relation to D2R monomers in the brain, since a significant proportion of monomers would have masked the identification of the D2R homomers.
The fact that rearrangement of TM 6 constitutes a main ligand-induced conformational change that determines G protein activation and modulation of ligand affinity (see above), provides a frame for the understanding of allosteric communications, negative cooperativity, through the protomers in A2AR and D2R homomers. In fact, when constructing computational models of the structures for A2AR and D2R homodimers with a symmetrical TM 6/TM 6 interface, TM 6 in the inactive closed conformation of the unliganded protomer can only interact with TM 6 in the active open conformation of the G protein-bound protomer, but simultaneous outward movements of TM 6 in the homodimer is not feasible due to a steric clash between active open conformation of both TM 6 (Navarro et al., 2018). That is to say, an active conformation of one of the protomers makes very unlikely the active conformation of the other protomer. These interpretations are in agreement with those of a previous study by Jonathan Javitch’s group (Han et al., 2009), who used several constructs of wild-type and mutant D2R (binding- or G protein activation-deficient D2R) fused and non-fused to a reporter G protein. The potential ability of a non-fused protomer to activate the G protein fused to another protomer allowed evaluating the key role of each protomer in the D2R homodimer. The results of the study supported, first, that the minimal signaling D2R unit is composed of two protomers and a single G protein. Second, the study showed that the homodimer is more strongly activated by an agonist binding to a single protomer, whereas simultaneous binding to the second protomer blunts signaling (Han et al., 2009). But also, a constitutive activity of the second protomer decreases agonist-induced activation of the first protomer, since maximal activation of the homodimer is achieved upon agonist and inverse agonist binding to the first and second protomer, respectively (Han et al., 2009). In other words, the inactive conformation of one of the protomers makes very likely the active conformation of the other protomer. An additional, mathematical, interpretation was provided by Rovira et al. (2010), who explained the results by Han et al. (2009) by considering two active states of the D2R homodimer, which would be associated to different signaling pathways and whose corresponding protomers are singly or doubly activated.
From the fourteen possible TAT-TM peptides corresponding to all TM domains of the A2AR and the D2R, specifically, TM4 and TM5 of both the A2AR and the D2R disrupted BiFC of A2AR-D2R heteromers (Navarro et al., 2018). These results therefore indicated a symmetrical TM 4–5/TM 5–4 heteromeric interface. When taking also into account the results of TAT-TM peptides mentioned above on the A2AR and D2R homomeric interfaces, computational modeling resulted in one minimal solution that accommodates the TMs 4/5 interface for A2AR-D2R heterodimerization and the TM 6 interface for both A2AR-A2AR and D2R-D2R homodimerization (Navarro et al., 2018). The existence of these interfaces implies two internal interacting A2AR and D2R protomers and two external A2AR and D2R protomers in which the α-subunits of Gi and Gs bind to the corresponding external protomers of the D2R or A2AR homodimers, respectively (Navarro et al., 2018). This would be the only feasible configuration to avoid any steric clash between the two G proteins simultaneously bound to the complex. Finally, the model also predicts a large distance between both βγ subunits (Navarro et al., 2018).
Apart from the two large catalytic domains, C1 and C2, the topology of AC includes a long cytoplasmic N terminus and two membrane-spanning domains, M1 and M2, each with six TMs. Carmen Dessauer’s group has provided evidence for a pre-coupling of AC5 and G proteins involving the βγ subunits and the N terminus of AC (Sadana et al., 2009b). Using BRET and BiFC techniques with interfering synthetic peptides (with the amino acid sequence of the twelve domains of AC5) we were able to demonstrate that specific TMs of AC5 establish molecular interactions with TMs of A2AR and D2R in the A2AR-D2R heterotetramer (Navarro et al., 2018). We therefore found for the first time a functional role for the TMs of AC, their coupling to GPCRs. These interactions, which involve TMs of the receptors that do not form part of the homomeric and heteromeric interfaces, change upon the presence of A2AR or D2R ligands. Computational modeling supports that the ligands induce a rearrangement of the A2AR-D2R-AC5 complex which depends on the rearrangement of the βγ subunits. This movement of the βγ subunits uncovers the Ras-GTPase domain of the Gα subunit that interacts with the corresponding catalytic domain of AC (Cabrera-Vera et al., 2003) and direct the movement of AC to allowing this Gα-AC interaction (Navarro et al., 2018).
The utilization of TAT-TM peptides in striatal primary neuronal cultures allowed demonstrating that the canonical interaction at the AC level required the right quaternary structure of the A2AR-D2R heteromer in the A2AR-D2R-AC5 complex. Application of TM peptides that specifically interfere with the heteromeric interface or with the receptor-AC5 interface specifically counteracted the ability of a D2R agonist to inhibit AC5 activation by an A2AR agonist (Navarro et al., 2018). More generally, these results indicate that, although Gi-coupled receptors can artificially inhibit forskolin-induced AC activation, to inhibit a Gs-coupled receptor-mediated AC activation, they need to be part of a GPCR heterotetramer. This has been so far shown for the A2AR-D2R, A1R-D1R, and D1R-D3R heterotetramers, where destabilization of their heteromeric interface leads to the disruption of the canonical Gs-Gi antagonistic interaction at the AC level (Navarro et al., 2018, Rivera-Oliver et al., 2019, Guitart et al., 2019). When again considering the definition of allosterism in the frame of GPCRs, an important corollary to the concept of pre-coupled macromolecular complexes of heterotetramers, Gs and Gi proteins and the effector molecule AC is that the canonical Gs-Gi antagonistic interaction at the AC level also represents an allosteric interaction. In this case, the process by which the interaction of a chemical at one location (an orthosteric ligand of the Gi-coupled receptor) of a macromolecular complex (formed by heterotetramers, Gs and Gi proteins and the effector molecule AC) influences the function of another chemical at a topographically distinct site (an orthosteric ligand of the Gs-coupled receptor). It also becomes obvious that we should modify the definition of ‘allosteric interaction in the receptor heteromer’ [Ferré et al., 2009] (see above). Otherwise, it would probably be more appropriate to avoid a general definition and to describe the specific type of allosteric interaction in the GPCR heteromer: Heteromeric or homomeric allosteric interactions between ligands, ligand-independent allosteric interactions or allosteric interactions through the effector (Figure 3).
It is important to underline that the GPCR heterotetramer represents only a very plausible common architecture of GPCR heteromers, but growing experimental evidence suggests the existence of heterodimeric structures or of heteromeric complexes larger than GPCR heterotetramers. Those complexes should be also determined by their immediate intramembrane context, by their role as elements of specific GPCR signaling complexes.
7. Final remarks
There seems to be no doubt that GPCR oligomers are not only demonstrable in artificial systems, but also in native tissues, where their prevalence (versus monomeric entities) should also be out of question, at least for some GPCRs. Experimental tools are now available to determine the oligomeric interfaces and quaternary structure of the oligomers, as well as to dissect the specific biochemical properties of GPCR heteromers. In fact, the same techniques are allowing to disclose a much more elaborated picture, with GPCR oligomers just being the tip of the iceberg, constituting components of pre-coupled macromolecular complexes that include G proteins and effectors, such as AC. Allosteric properties can therefore extend to ligand interactions through the different components of the macromolecular complexes. Identification of specific heteromers in individual cells of native tissues, as well as their specific biochemical properties, should lead to new therapeutic approaches (as reviewed elsewhere; see for instance Ferré et al., 2014; Gomes et al., 2016).
8. Acknowledgements
Work supported by the intramural funds of the National Institute on Drug Abuse, FEDER/Ministerio de Ciencia, Innovación y Universidades-Agencia Estatal de Investigación (SAF2017-87349-R and SAF 2017-87629-R) and Generalitat de Catalunya (2017-SGR-1497).
9. References
- Agnati LF, Ferré S, Cortelli P, Fuxe K. A brief appraisal on some aspects of the receptor-receptor interaction. Neurochem Int. 1995;27:139–146. [DOI] [PubMed] [Google Scholar]
- Albizu L, Balestre MN, Breton C, Pin JP, Manning M, Mouillac B, Barberis C, Durroux T. Probing the existence of G protein-coupled receptor dimers by positive and negative ligand-dependent cooperative binding. Mol Pharmacol. 2006;70:1783–1791. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Leitz AJ, Xie G, Oprian DD, Sligar SG. Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J Biol Chem. 2007;282:14875–14881. [DOI] [PubMed] [Google Scholar]
- Bonaventura J, Navarro G, Casadó-Anguera V, Azdad K, Rea W, Moreno E, Brugarolas M, Mallol J, Canela EI, Lluís C, Cortés A, Volkow ND, Schiffmann SN, Ferré S, Casadó V. Allosteric interactions between agonists and antagonists within the adenosine A2A receptor-dopamine D2 receptor heterotetramer. Proc Natl Acad Sci USA. 2015;112:E3609–E3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho AV, Huber T, Sakmar TP, Brown MF. Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys J. 2006;91:4464–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier M, Hébert TE. CrossTalk proposal: Weighing the evidence for Class A GPCR dimers, the evidence favors dimers. J Physiol. 2014;592:2439–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgisser E, De Lean A, Lefkowitz RJ. Reciprocal modulation of agonist and antagonist binding to muscarinic cholinergic receptor by guanine nucleotide. Proc Natl Acad Sci USA. 1982;79:1732–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE. Insights into G protein structure, function, and regulation. Endocr Rev. 2003;24(6):765–81. [DOI] [PubMed] [Google Scholar]
- Cai NS, Quiroz C, Bonaventura J, Bonifazi A, Cole TO, Purks J, Billing AS, Massey E, Wagner M, Wish ED, Guitart X, Rea W, Lam S, Moreno E, Casadó-Anguera V, Greenblatt AD, Jacobson AE, Rice KC, Casadó V, Newman AH, Winkelman JW, Michaelides M, Weintraub E, Volkow ND, Belcher AM, Ferré S. Opioid-galanin receptor heteromers mediate the dopaminergic effects of opioids. J Clin Invest. 2019;129:2730–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Rieken F, Wagner J, Sungkaworn T, Zabel U, Borzi A, Cocucci E, Zürn A, Lohse MJ. Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc Natl Acad Sci USA. 2013;110:743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadó-Anguera V, Moreno E, Mallol J, Ferré S, Canela EI, Cortés A, Casadó V. Reinterpreting anomalous competitive binding experiments within G protein-coupled receptor homodimers using a dimer receptor model. Pharmacol Res. 2019;139:337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux JP. The feedback control mechanisms of biosynthetic L-threonine deaminase by L-isoleucine. Cold Spring Harb Symp Quant Biol. 1961;26:313–318. [DOI] [PubMed] [Google Scholar]
- Changeux JP. Allostery and the Monod-Wyman-Changeux model after 50 years. Annu Rev Biophys. 2012;41:103–133. [DOI] [PubMed] [Google Scholar]
- Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. [DOI] [PubMed] [Google Scholar]
- Christopoulos A Advances in G protein-coupled receptor allostery: from function to structure. Mol Pharmacol. 2014;86:463–478. [DOI] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- De Lean A, Kilpatrick BF, Caron MG. Dopamine receptor of the porcine anterior pituitary gland. Evidence for two affinity states discriminated by both agonists and antagonists. Mol Pharmacol. 1982;22:290–297. [PubMed] [Google Scholar]
- Dessauer CW, Tesmer JJ, Sprang SR, Gilman AG. Identification of a Gialpha binding site on type V adenylyl cyclase. J Biol Chem. 1998;273:25831–25839. [DOI] [PubMed] [Google Scholar]
- Deupi X, Kobilka BK. Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology. 2010;25:293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst OP, Gramse V, Kolbe M, Hofmann KP, Heck M. Monomeric G protein-coupled receptor rhodopsin in solution activates its G protein transducin at the diffusion limit. Proc Natl Acad Sci USA. 2007;104:10859–10864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW. Allostery: an illustrated definition for the ‘second secret of life’. Trends Biochem Sci. 2008;33:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, von Euler G, Johansson B, Fredholm BB, Fuxe K. Stimulation of high-affinity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proc Natl Acad Sci USA. 1991;88:7238–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Fredholm BB, Morelli M, Popoli P, Fuxe K. Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997;20:482–487. [DOI] [PubMed] [Google Scholar]
- Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, Mackie K, Milligan G, Pfleger KD, Pin JP, Volkow ND, Waldhoer M, Woods AS, Franco R. Building a new conceptual framework for receptor heteromers. Nat Chem Biol. 2009;5:131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ, Milligan G, Pin JP, Guitart X. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev. 2014;66:413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S The GPCR heterotetramer: challenging classical pharmacology. Trends Pharmacol Sci. 2015;36:145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Bonaventura J, Tomasi D, Navarro G, Moreno E, Cortés A, Lluís C, Casadó V, Volkow ND. Allosteric mechanisms within the adenosine A2A-dopamine D2receptor heterotetramer. Neuropharmacology. 2016;104:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Bonaventura J, Zhu W, Hatcher-Solis C, Taura J, Quiroz C, Cai NS, Moreno E, Casadó-Anguera V, Kravitz AV, Thompson KR, Tomasi DG, Navarro G, Cordomí A, Pardo L, Lluís C, Dessauer CW, Volkow ND, Casadó V, Ciruela F, Logothetis DE, Zwilling D. Essential Control of the Function of the Striatopallidal Neuron by Pre-coupled Complexes of Adenosine A(2A)-Dopamine D(2) Receptor Heterotetramers and Adenylyl Cyclase. Front Pharmacol. 2018;9:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Ayoub MA, Fujita W, Jaeger WC, Pfleger KD, Devi LA. G Protein-Coupled Receptor Heteromers. Annu Rev Pharmacol Toxicol. 2016;56:403–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, Zhou Z, Perez-Aguilar JM, Hauge M, Mathiasen S, Javitch JA, Weinstein H, Kobilka BK, Blanchard SC. Single-molecule analysis of ligand efficacy in β(2)AR-G-protein activation. Nature. 2017;547:68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitart X, Navarro G, Moreno E, Yano H, Cai NS, Sánchez-Soto M, Kumar-Barodia S, Naidu YT, Mallol J, Cortés A, Lluís C, Canela EI, Casadó V, McCormick PJ, Ferré S. Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: the dopamine D1-D3 receptor heterotetramer. Mol Pharmacol. 2014;86:417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitart X, Moreno E, Rea W, Sánchez-Soto M, Cai NS, Quiroz C, Kumar V, Bourque L, Cortés A, Canela EI, Bishop C, Newman AH, Casadó V, Ferré S. Biased G protein-independent signaling of dopamine D1-D3 receptor heteromers in the nucleus accumbens. Mol Neurobiol. 2019;56:6756–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guixà-González R, Javanainen M, Gómez-Soler M, Cordobilla B, Domingo JC, Sanz F, Pastor M, Ciruela F, Martinez-Seara H, Selent J. Membrane omega-3 fatty acids modulate the oligomerisation kinetics of adenosine A2A and dopamine D2 receptors. Sci Rep. 2016;6:19839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Shi L, Filizola M, Weinstein H, Javitch JA. Crosstalk in G protein-coupled receptors: changes at the transmembrane homodimer interface determine activation. Proc Natl Acad Sci USA. 2005;102:17495–17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol. 2009;5:688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He SQ, Zhang ZN, Guan JS, Liu HR, Zhao B, Wang HB, Li Q, Yang H, Luo J, Li ZY, Wang Q, Lu YJ, Bao L, Zhang X. Facilitation of μ-opioid receptor activity by preventing δ-opioid receptor-mediated codegradation. Neuron. 2011;69:120–131. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Cowan A, Mazurkiewicz JE. Fluorescence correlation spectroscopy analysis of serotonin, adrenergic, muscarinic, and dopamine receptor dimerization: the oligomer number puzzle. Mol Pharmacol. 2013;84:630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz KN, Keil R, Zimmer FJ, Schwabe U. Guanine nucleotide effects on 8-cyclopentyl-1,3-[3H]dipropylxanthine binding to membrane-bound and solubilized A1 adenosine receptors of rat brain. J Neurochem. 1990;54:1988–1994. [DOI] [PubMed] [Google Scholar]
- Kniazeff J, Prézeau L, Rondard P, Pin JP, Goudet C. Dimers and beyond: The functional puzzles of class C GPCRs. Pharmacol Ther. 2011;130:9–25. [DOI] [PubMed] [Google Scholar]
- Kuszak AJ, Pitchiaya S, Anand JP, Mosberg HI, Walter NG, Sunahara RK. Purification and functional reconstitution of monomeric mu-opioid receptors: allosteric modulation of agonist binding by Gi2. J Biol Chem. 2009;284:26732–26741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hübner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert NA, Javitch JA. CrossTalk opposing view: Weighing the evidence for class A GPCR dimers, the jury is still out. J Physiol. 2014;592:2443–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbird LE, Meyts PD, Lefkowitz RJ. Beta-adrenergic receptors: evidence for negative cooperativity. Biochem Biophys Res Commun. 1975;64:1160–1168. [DOI] [PubMed] [Google Scholar]
- Liu X, Ahn S, Kahsai AW, Meng KC, Latorraca NR, Pani B, Venkatakrishnan AJ, Masoudi A, Weis WI, Dror RO, Chen X, Lefkowitz RJ, Kobilka BK. Mechanism of intracellular allosteric β(2)AR antagonist revealed by X-ray crystal structure. Nature. 2017;548:480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Masoudi A, Kahsai AW, Huang LY, Pani B, Staus DP, Shim PJ, Hirata K, Simhal RK, Schwalb AM, Rambarat PK, Ahn S, Lefkowitz RJ, Kobilka B. Mechanism of β(2)AR regulation by an intracellular positive allosteric modulator. Science. 2019;364:1283–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Zhang J. Small Molecule Allosteric Modulators of G-Protein-Coupled Receptors: Drug-Target Interactions. J Med Chem. 2019;62:24–45. [DOI] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, Prosser RS, Kobilka BK. Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell. 2015;161:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. [DOI] [PubMed] [Google Scholar]
- Meral D, Provasi D, Prada-Gracia D, Möller J, Marino K, Lohse MJ, Filizola M. Molecular details of dimerization kinetics reveal negligible populations of transient μ-opioid receptor homodimers at physiological concentrations. Sci Rep. 2018;8:7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J. Chance and necessity: an essay on the natural philosophy of modern biology. New York: Alfred A Knopf; 1971. [Google Scholar]
- Monod J, Jacob F. Teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harb Symp Quant Biol. 1961;26:389–401. [DOI] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. [DOI] [PubMed] [Google Scholar]
- Navarro G, Cordomí A, Casadó-Anguera V, Moreno E, Cai NS, Cortés A, Canela EI, Dessauer CW, Casadó V, Pardo L, Lluís C, Ferré S. Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase. Nat Commun. 2018;9:1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig RR. Membrane organization in G-protein mechanisms. FASEB J. 1994;8:939–946. [DOI] [PubMed] [Google Scholar]
- Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, Thian FS, Kobilka TS, Shaw DE, Mueller L, Prosser RS, Kobilka BK. The dynamic process of β(2)-adrenergic receptor activation. Cell. 2013;152:532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrú M, Bakešová J, Brugarolas M, Quiroz C, Beaumont V, Goldberg SR, Lluís C, Cortés A, Franco R, Casadó V, Canela EI, Ferré S. Striatal pre- and postsynaptic profile of adenosine A(2A) receptor antagonists. PLoS One. 2011;6:e16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X, Knepp AM, Sakmar TP, Marrink SJ, Huber T. Structural determinants of the supramolecular organization of G protein-coupled receptors in bilayers. J Am Chem Soc. 2012;134:10959–10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanna X, Chattopadhyay A, Sengupta D. Cholesterol modulates the dimer interface of the β₂-adrenergic receptor via cholesterol occupancy sites. Biophys J. 2014;106:1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido D, Casadó-Anguera V, Pérez-Benito L, Moreno E, Cordomí A, López L, Cortés A, Ferré S, Pardo L, Casadó V, Royo M. Design of a True Bivalent Ligand with Picomolar Binding Affinity for a G Protein-Coupled Receptor Homodimer. J Med Chem. 2018;61:9335–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 2011;13;469:175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redka DS, Heerklotz H, Wells JW. Efficacy as an intrinsic property of the M(2) muscarinic receptor in its tetrameric state. Biochemistry. 2013;52:7405–7427. [DOI] [PubMed] [Google Scholar]
- Redka DS, Morizumi T, Elmslie G, Paranthaman P, Shivnaraine RV, Ellis J, Ernst OP, Wells JW. Coupling of g proteins to reconstituted monomers and tetramers of the M2 muscarinic receptor. J Biol Chem. 2014;289:24347–24365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Oliver M, Moreno E, Álvarez-Bagnarol Y, Ayala-Santiago C, Cruz-Reyes N, Molina-Castro GC, Clemens S, Canela EI, Ferré S, Casadó V, Díaz-Ríos M. Adenosine A(1)-Dopamine D(1) Receptor Heteromers Control the Excitability of the Spinal Motoneuron. Mol Neurobiol. 2019;56:797–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira X, Pin JP, Giraldo J. The asymmetric/symmetric activation of GPCR dimers as a possible mechanistic rationale for multiple signalling pathways. Trends Pharmacol Sci. 2010;31:15–21. [DOI] [PubMed] [Google Scholar]
- Sadana R, Dessauer CW. Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals. 2009a;17:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadana R, Dascal N, Dessauer CW. N terminus of type 5 adenylyl cyclase scaffolds Gs heterotrimer. Mol Pharmacol. 2009b;76:1256–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev. 2010;62:701–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann A, Gibert A, Lampe A, Grzesik P, Rutz C, Furkert J, Schmoranzer J, Krause G, Wiesner B, Schülein R. The specific monomer/dimer equilibrium of the corticotropin-releasing factor receptor type 1 is established in the endoplasmic reticulum. J Biol Chem. 2014;289:24250–24262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viñals X, Moreno E, Lanfumey L, Cordomí A, Pastor A, de La Torre R, Gasperini P, Navarro G, Howell LA, Pardo L, Lluís C, Canela EI, McCormick PJ, Maldonado R, Robledo P. Cognitive Impairment Induced by Delta9-tetrahydrocannabinol Occurs through Heteromers between Cannabinoid CB1 and Serotonin 5-HT2A Receptors. PLoS Biol. 2015;13:e1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh SM, Mathiasen S, Christensen SM, Fay JF, King C, Provasi D, Borrero E, Rasmussen SGF, Fung JJ, Filizola M, Hristova K, Kobilka B, Farrens DL, Stamou D. Single Proteoliposome High-Content Analysis Reveals Differences in the Homo-Oligomerization of GPCRs. Biophys J. 2018;115:300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis WI, Kobilka BK. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu Rev Biochem. 2018;87:897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Jastrzebska B, Park PS, Fotiadis D, Engel A, Palczewski K, Sunahara RK. Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J Biol Chem. 2008;283:4387–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Van Eps N, Zimmer M, Ernst OP, Prosser RS. Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature. 2016;533:265–268. [DOI] [PubMed] [Google Scholar]
- Zoli M, Agnati LF, Hedlund PB, Li XM, Ferré S, Fuxe K. Receptor-receptor interactions as an integrative mechanism in nerve cells. Mol Neurobiol. 1993;7:293–334. [DOI] [PubMed] [Google Scholar]