

Abstract
Cell-free expression systems provide a suite of tools that are used in applications from sensing to biomanufacturing. One of these applications is genetic circuit prototyping, where the lack of cloning is required and a high degree of control over reaction components and conditions enables rapid testing of design candidates. Many studies have shown utility in the approach for characterizing genetic regulation elements, simple genetic circuit motifs, protein variants or metabolic pathways. However, variability in cell-free expression systems is a known challenge, whether between individuals, laboratories, instruments, or batches of materials. While the issue of variability has begun to be quantified and explored, little effort has been put into understanding the implications of this variability. For genetic circuit prototyping, it is unclear when and how significantly variability in reaction activity will impact qualitative assessments of genetic components, e.g. relative activity between promoters. Here, we explore this question by assessing DNA titrations of seven genetic circuits of increasing complexity using reaction conditions that ostensibly follow the same protocol but vary by person, instrument and material batch. Although the raw activities vary widely between the conditions, by normalizing within each circuit across conditions, reasonably consistent qualitative performance emerges for the simpler circuits. For the most complex case involving expression of three proteins, we observe a departure from this qualitative consistency, offering a provisional cautionary line where normal variability may disrupt reliable reuse of prototyping results. Our results also suggest that a previously described closed loop controller circuit may help to mitigate such variability, encouraging further work to design systems that are robust to variability.
Graphical Abstract
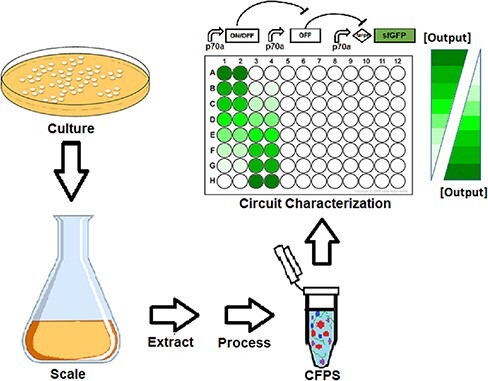
Keywords: reproducibility, cell-free expression, genetic circuit, control systems
1. Introduction
Cell-free expression (CFE) systems are a set of techniques that aim to reproduce complex cellular processes like transcription, translation and metabolism in a non-living format (1). Applications of CFE include sensing, biomanufacturing and education (2). In principle, CFE systems are simpler to engineer than cellular systems due to reduced complexity and facile adjustment of components normally protected by membranes, especially DNA. Nonetheless, the systems remain extremely complex. Many process variables exist for producing the cellular lysates frequently used to supply critical biological machinery, and the impacts of these variables remain only partially explored despite significant effort (3). Beyond the lysate, ∼30–40 additional components are added to fuel the reaction (4), and the impact of variations in reagent preparation is not well-understood.
As a result of this complexity, reproducibility remains a challenge, although only a small number of studies have attempted to quantify CFE variability outside of standard replicates using the same materials by the same scientist at the same time. For a single person in a single laboratory, reasonable variability has been demonstrated at 6–10%, including across days and batches of material (5, 6). Similarly, there is evidence that variability can be modest between individuals in the same laboratory (5, 7). However, others have reported high variability within different batches of materials in the same laboratory (8, 9) and with common materials shared between laboratories (5). Other works have developed methods with helpful details aimed at improving reproducibility, although they do not explicitly quantify the improvements compared to existing methods (10–12). These studies have only assessed variability for a small fraction of possible CFE recipes and process variables that may be important and thus only provide an early glimpse into the scope of the challenge.
Genetic prototyping is an important use case for CFE (9, 13, 14). Such efforts take advantage of the ease of varying the DNA constructs and their concentrations in CFE reactions to rapidly evaluate libraries of designs without cloning (15–17). This approach has been used to explore genetic parts for diverse organisms (18–26), probe promoter architecture (27), optimize metabolic pathways (28–30), predict cellular burden (31), screen sensor or CRISPR designs (32–36) and characterize genetic circuits like oscillators (37), feedforward loops (38), and logic gates (34, 39, 40). Some of these efforts have compared performance in CFE with performance in cells, with several showing correlation of relative performance (18, 19, 38); however, others have observed qualitative differences when varying the reaction composition (41). It remains an open question under what conditions CFE experiments can be used as a reliable indicator of behavior in cells. Moreover, it is unknown how much variability in activity of CFE reactions may impact the accuracy of prototyping for qualitative, let alone quantitative, performance.
Here, we explored the relationship between the variability of CFE and qualitative circuit performance. We performed experiments spanning three individual operators, three batches of CFE materials and three instruments, resulting in substantially different yields for ostensibly the same reaction. We used these conditions to test a set of increasingly complex circuits in a range of DNA concentrations for the underlying circuit components. The number of differences between conditions and limitations on quantity of materials within batches meant that direct comparisons provide little insight; however, by normalizing the data sets, we were able to compare qualitative circuit function between conditions. We found that for the simple circuits, qualitative performance was consistent across the experimental conditions, but for more complex circuits, performance varied considerably. Without normalization, variation is primarily explained by the condition rather than concentration of DNA encoding for circuit components. For the final circuit, which is a feedback controller designed to reject disturbances, we found evidence that indeed suggests robustness to variability. This work highlights the need to further improve reproducibility in CFE, better understand when and how circuit performance is impacted by changes in the CFE conditions and identify approaches to improve robustness.
2. Materials and methods
2.1. Preparation of lysates
Culturing of cells for production of lysates was performed in one of the following three conditions: In Condition 1, culturing was performed by the Noireaux Lab following a protocol that has been previously described in significant detail (42). Condition 2 followed a modified version of the protocol at Chemical Biological Center (CBC); the detailed protocol is described below. Conditions 3 and 4 followed the same protocol as Condition 1 as exactly as possible except that it was performed at CBC. A description of the method and modifications is as follows: in all cases, 2XYT (Yeast Extract Tryptone) media was supplemented to final concentrations of 40 mM dibasic phosphate, 22 mM monobasic phosphate and 34 µg/ml chloramphenicol; we refer to this as supplemented 2× YT media.
For lysates used in Conditions 1, 3 and 4, culturing began with inoculation of a fresh plate of supplemented 2× YT agar from a glycerol stock of Escherichia coli Rosetta2(DE3) harboring the DE3 phage lysogen, followed by incubation at 37°C for 17 h. The following day, starter cultures were made by selecting ∼5 colonies from the day 1 plate of E. coli and resuspending in 2.8 ml of supplemented 2× YT broth. The cultures were incubated with shaking at 225 rpm at 37°C for 8 h. Following this incubation, intermediate cultures were made by inoculating 56.5 ml of supplemented 2× YT broth with 3 ml of starter culture. The intermediate cultures were allowed to continue to grow at 37°C with shaking for 7 h. On the third day, final cultures were prepared by adding 60 ml of intermediate culture to 2.6 l of supplemented 2× YT across four 2.8-l culture flasks. The final cultures were incubated at 37°C with shaking until reaching an Optical Density (OD600) of ∼1.6 for both CBC Lysate Batches.
For lysates used in Condition 2, the inoculation steps were altered slightly. Culturing began identically with the inoculation of a fresh plate of supplemented 2× YT agar from a glycerol stock of E. coli Rosetta2 (DE3) followed by incubation at 37°C for 16 h. The following day, starter cultures were made by inoculating 60 ml of supplemented 2× YT broth with one colony from the day 1 plate of E. coli and growing overnight at 37°C with shaking at 225 rpm. On the third day, 3 l of supplemented 2× YT broth was inoculated with the overnight starter culture at a 1:100 ratio across four 2.8-l flasks. The final cultures were incubated at 37°C with shaking at 225 rpm until reaching an OD600 of ∼1.6.
Once the final culture reached the appropriate OD, all cultures were pelleted and washed in S30A buffer following the previously described protocol (42). For Condition 2 only, the cell pellet was flash-frozen in liquid nitrogen and stored at −80°C before thawing on ice and processing further. The final washed pellet for all conditions was resuspended in 1.025 ml of S30A buffer per gram of cell mass. For cell lysis, the protocol diverged from the previously described method by replacing bead beating with high-pressure homogenization. For Condition 1, cells were lysed using a French Press at 14 000 psi. For Conditions 2–4, the cells were lysed via high-pressure homogenization (Microfluidics 110-P) at 16 000 psi and the material was collected in 10 ml increments. The remainder of the lysate processing exactly followed the previously described method (42) for all conditions. Briefly, the extracted material was processed by centrifugation and the clarified lysate was collected. The lysates were then subjected to a run-off reaction at 37°C with shaking for 90 minutes followed by dialysis in S30B buffer for 1 hour. Post-dialysis materials were aliquoted and flash frozen in liquid nitrogen. A portion was retained and tested for protein content via Bradford assay.
2.2. Preparation of DNA
All plasmids, cell lines and plasmid preparation methods used in this study are listed in Table S1. The plasmids were maintained and routinely isolated from E. coli JM109 except for the p70a-degfp preparation used for Conditions 3 and 4 that used E. coli KL740 cl857+ (Daicel Arbor Biosciences) to increase plasmid yield for the p70a expression construct. The plasmids were isolated using either a Promega Plus Wizard Midi kit (Conditions 1 and 2) or a Qiagen Plasmid Midi kit (Conditions 3 and 4) following the manufacturer’s recommended protocol. The isolated plasmids were further processed with either a standard ethanol precipitation (Conditions 1 and 2) or a Zymo Clean and Concentrate kit (Conditions 3 and 4) again following the manufacturer’s recommended protocol. Purified, cleaned and concentrated plasmids were suspended in diH2O and stored at −20°C.
2.3. Cell-free reactions
The reagents used for CFE reactions under all four conditions were prepared as previously described (42) except for the amino acid mixture that was prepared following the protocol described by Caschera and Noireaux (43). The 14× energy mix consisted of 700 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 8, 21 mM Adenosine triphosphate (ATP), 21 mM Guanosine triphosphate (GTP), 12.6 mM Cytidine triphosphate, 12.6 mM Uridine triphosphate (UTP), 2.8 mg/ml Transfer ribonucleic acid (tRNA), 3.64 mM Coenzyme A (CoA), 4.62 mM Nicotinamide adenine dinucleotide (NAD), 10.5 mM Cyclic adenosine monophosphate (cAMP), 0.95 mM folinic acid, 14 mM Spermidine and 420 mM 3-phosphoglyceric acid (3-PGA). The final CFE reactions were composed of 9.5 mg/ml protein (from crude extract), 4.5–10.5 mM Mg-glutamate (optimized for each batch of extract), 40–160 mM K-glutamate (optimized for each batch of extract), 0.33–3.33 mM Dithiothreitol (DTT) (optimized for each batch of extract), 1.5 mM of each amino acid except leucine, which was 1.25 mM, 50 mM HEPES, 1.5 mM ATP and GTP, 0.9 mM CTP and UTP, 0.2 mg/ml tRNA, 0.26 mM CoA, 0.33 mM NAD, 0.75 mM cAMP, 0.068 mM folinic acid, 1 mM spermidine, 30 mM 3-PGA, 2% Polyethylene glycol (PEG)-8000 and 30 mM Maltodextrin and plasmid DNA at the desired concentration. For Conditions 1 and 2, DNA was dispensed using acoustic liquid handing (Labcyte Echo 525) followed by manual dispensing of the remaining components as a master mix into 96-well clear V-bottom plates. For Conditions 3 and 4, reactions were dispensed by hand in 96-well round bottom plates. Plates were immediately placed in a fluorescent plate reader at 29°C and read every 3 minutes for 16 hours; see Table S3 for additional details of plate reader settings for each experiment. Values were reported in µM of total apparent Green fluorescent protein (GFP) based on a standard curve using purified enhanced green fluorescent protein (eGFP) (Cell Biolabs, Inc STA-201).
2.4. Data analysis
Relative Fluorescence Unit (RFU) values were normalized to units that are comparable between experimental conditions in two different ways. To convert from RFUs to apparent GFP concentrations, standard curves were prepared in diH2O by two-fold dilutions from stock solutions of 1 mg/ml of eGFP for Conditions 3 and 4 or sfGFP as a proxy for Conditions 1 and 2. While the circuits all express deGFP, sfGFP (provided by Scott Walper, NRL, in a previous study) and eGFP (Cell Biolabs, Inc STA-201) were used due to availability in the laboratory. RFUs were measured using settings as described in Table S3. Resulting RFUs were fit via linear regression with the y-intercept fixed to the control with no GFP (Supplementary Figure S1). RFU values were converted to apparent GFP concentrations using the appropriate standard curve to enable comparison of data between conditions in statistical analyses and for presentation of Supplementary Figures S2–S8.
To compare qualitative performance across conditions, RFUs were normalized via Equation 1,
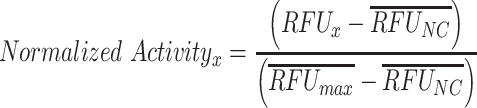 |
(1) |
where RFUNC is the appropriate negative control for that circuit and RFUmax is the condition with highest expected activity. In the case of the small RNA (sRNA) circuit where the no-DNA control should exhibit high activity, the no-DNA control for the small transcriptional activator (STAR) circuit was used for RFUNC since the STAR and sRNA circuits were performed on the same plate for each operator. For the integral controller circuits, the closed loop cases used the RFUmax values from the open loop case to facilitate comparison between the open and closed loop.
For Conditions 1 and 2, outliers were removed based on experience with the acoustic liquid handler. More specifically, we observe occasional wells where fluorescence of a replicate is similar to the background, while the other replicates are not; in these occasional cases, we assume that the DNA failed to dispense and exclude the well. These cases are indicated in the raw data available in the Supplementary Files. ANOVAs were performed in Matlab using the ‘anovan’ function. Raw data used and full statistical results for ANOVAs are provided in the Supplementary Files; in addition, all p-values for ANOVAs are presented in Table S4. Regression analyses were performed in GraphPad Prism, except for the RNA sequestration circuits, the fits for which were performed in Matlab. Sigmoid fits used Equation 2, while the RNA sequestration fits used Equation 3.
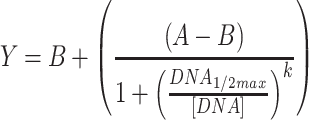 |
(2) |
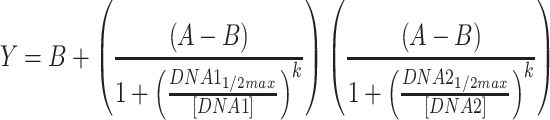 |
(3) |
Statistics for regressions are available in the Supplementary Files.
3. Results
The work presented here was part of an independent evaluation of designs coming from the Defense Advanced Research Projects Agency’s Biological Control program. One multi-institutional team led by the University of Minnesota (UMN) endeavored to develop PID controllers (proportional, integral and derivative controllers) in a CFE environment (44) by combining screening of components (36, 45), modeling (46, 47) and proteomic analysis (48), ultimately resulting in a functional integral controller that tracks an input signal and rejects external disturbances (49). We tested a subset of circuits developed under this program that range in complexity from constitutive expression to the full integral controller.
We evaluated the circuits using four different CFE conditions (Table 1). Reasons for the differences ranged from limited quantities of materials, personnel availability and pragmatism; we consider each condition to be a reasonable replication of the original work given practical limitations. Condition 1 used materials provided by UMN, except for circuit DNA that was prepared at CBC. Condition 2 used materials all prepared at CBC using published protocols provided by UMN with minor modifications to the starter culture preparation and lysis methods (Methods). Reactions using these two sets of materials were prepared by the same operator using acoustic liquid handling to dispense DNA. Condition 3 used materials prepared at CBC by a different operator following the same published protocol with modifications only to the lysis method (Methods). That same person then prepared all reactions by manual pipetting. In Condition 4, a third operator used the same materials in Condition 3 to also prepare reactions by hand. Standard curves of GFP were used to convert plate reader results to apparent molar concentrations (Methods, Supplementary Figure S1). While the circuits described here use deGFP as a reporter, due to available materials, other GFP variants were used to arrive at apparent GFP concentrations. Conditions 1 and 2 used sfGFP, while Conditions 3 and 4 followed existing protocols exactly by using eGFP. Known differences in fluorescence properties of these three variants limit confidence in the reliability of the standardized units. To initially characterize differences in these conditions, we used a p70a-degfp construct that constitutively expresses deGFP under the control of the housekeeping σ70 promoter at 0.5 nM; note that Condition 4 was left out as it only varied by operator (Figure 1a). We found that our yields varied up to 8-fold between these highly similar implementations of a single protocol (Figure 1b and Supplementary Figure S2). This large amount of variation is consistent with a previous report of high variability following a protocol based on the same method as here (9) and an interlaboratory study using an alternative CFE preparation protocol (5), but not with another report using the same alternative CFE preparation protocol by a single person in a single laboratory (6).
Table 1.
Differences in the preparation of each data set
| Condition 1 | Condition 2 | Condition 3 | Condition 4 | |
| Operator | Operator 1 | Operator 1 | Operator 2 | Operator 3 |
| Cell growth and preparation protocol | Sun et al. | Sun et al. with modified growth | Sun et al. | Sun et al. |
| Lysis method | French Press | Microfluidizer (Microfluidics M110-P) | Microfluidizer (Microfluidics M110-P) | Microfluidizer (Microfluidics M110-P) |
| Lysate prep location | UMN | CBC | CBC | CBC |
| Lysate batch | UMN | CBC-1 | CBC-2 | CBC-2 |
| Supplement mix prep location | UMN | CBC | CBC | CBC |
| Supplement mix prep batch | UMN | CBC-1 | CBC-2 | CBC-2 |
| DNA prep kit | Promega Plus Wizard Midi Kit | Promega Plus Wizard Midi Kit | Qiagen Plasmid Midi Kit | Qiagen Plasmid Midi Kit |
| DNA prep location | CBC | CBC | CBC | CBC |
| DNA prep batch | CBC-1 | CBC-1 | CBC-2 | CBC-2 |
| GFP standard prep batch | sfGFP-1 | sfGFP-1 | eGFP-1 | eGFP-1 |
| Dispensing method | Acoustic Liquid Handling (Labcyte Echo 525) | Acoustic Liquid Handling (Labcyte Echo 525) | Manual | Manual |
| Plate | 96-Well Clear V-bottom Polypropylene plates (Costar Cat# 3357) | 96-Well Clear V-bottom Polypropylene plates (Costar Cat# 3357) | 96-Well round bottom plates (Immulon 2HB U Bottom Thermo Fisher Cat# 3655) |
96-Well round bottom plates (Immulon 2HB U Bottom Thermo Fisher Cat# 3655) |
| Plate seal | Storage Mat for 96-well Polypropylene plates, Costar Cat# 3080 | Storage Mat for 96-well Polypropylene plates, Costar Cat# 3080 | 3 M Empore Sealing Tape MG Sci. Cat # E520-43 |
3 M Empore Sealing Tape MG Sci. Cat # E520-43 |
| Reaction prep location | CBC | CBC | CBC | CBC |
| Plate readera | BioTek Synergy H1A/BioTek Synergy HTb | BioTek Synergy H1A | BioTek Synergy H1B/BioTek NEO | BioTek Synergy H1B/BioTek NEO |
Two instruments of the same type (BioTek Synergy H1) were used and are indicated by A or B. See Table S2 for which circuit was tested on each plate reader.
No GFP standard curve at 5 µl is available for the data on the BioTek Synergy HT.
Figure 1.
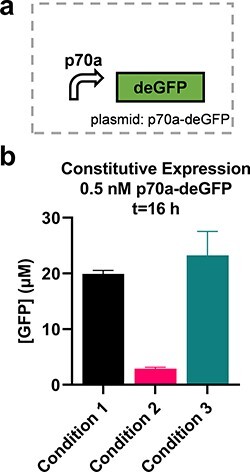
Different CFE reactions give different yields. (a) Yields from 0.5 nM p70a-degfp for the three material sets tested (i.e. Lysate Batch, Supplement Mix Batch and DNA Prep Batch as detailed in Table 1; Condition 4 used identical materials to Condition 3); n = 3. (b) Normalized yields for three conditions. Time series data are available in Supplementary Figure S2.
A long catalog of factors could potentially contribute to differences between laboratories in CFE, including different preparations of lysate, supplement mix or DNA; the individual operator preparing the reactions; method of converting RFUs to standardized units; materials like plates and plate seals; and different instruments, instrument settings or calibrations. Ensuring that these myriad factors are consistent poses a significant logistical challenge. We previously described an effort to control for as many of these factors as possible across three laboratories to isolate and quantify the contribution of specific factors to overall interlaboratory variability (5). Even in the most controlled case where prepared reactions were flash-frozen and shared between laboratories, variability was measurably higher between laboratories than within a laboratory (20% and 7.6% variability, respectively). Here, we take a different approach by normalizing RFU data to facilitate comparisons across data sets for each circuit despite variations in the exact protocols, batches of materials, operators and other details (Table 1). We perform this normalization within each circuit such that an appropriate no-DNA control corresponds to zero and the concentration predicted to have the highest activity corresponds to one (see Methods).
We proceeded to evaluate the performance of a set of circuits with increasing complexity. Data for Conditions 1 and 2 are presented only for some of the circuits due to a combination of limitations on materials in the same batches (i.e. extract or DNA) and instrument failures with the acoustic liquid handler. The first circuit is a transcriptional cascade where sigma factor 24 (σ24) is constitutively expressed, which in turn activates expression of deGFP from a σ24-responsive promoter (p24a) (Figure 2a) (44). A titration of each plasmid was performed (Figure 2b and c, Supplementary Figures S2–S3). Linear fits of plasmid concentration to normalized yield were reasonable for Conditions 2 and 3 (R2 = 0.8439 and 0.9033 for the p70a-σ24 titration and 0.9747 and 0.8072 for the p24a-degfp titration, respectively). For Condition 4, while the error bars were large and coefficient of determination is relatively weak (R2 = 0.6352 and 0.4900 for the p70a-σ24 and p24a-degfp titrations, respectively), means did closely track Condition 3, which uses the same materials, across plasmid concentrations. For the Condition 2 p70a-σ24 titration case, deGFP yields do not appear to increase linearly with the DNA concentration (Figure 2b), which could be indicative of saturation of the translational machinery, as has been described previously in CFE systems (50). We found that a 4-parameter sigmoid function improved the fit for that case but had little impact on the others despite the increased number of parameters (R2 = 0.9635, 0.9106, 0.6353 and 0.9779, 0.8076, and 0.4908 for sigmoid fits of Conditions 2–4 and p70a-σ24 and p24a-degfp, respectively). This result offers the first indication that circuit performance may vary with the CFE condition. Nonetheless, for the p70a-σ24 titration, an ANOVA analysis showed that 79% of the variation was explained by the plasmid concentration (p < 0.001), with just 2.7% explained by the condition p = 0.067. For the p24a-degfp titration, plasmid concentration showed 73% of variation (p < 0.001) compared to just 0.05% explained by condition p = 0.96. As expected, without normalization, the variability in activity between the conditions is such that 67% and 33% of the variation are explained by the condition for the p70a-σ24 and p24a-degfp titrations, respectively (Table S4). Overall, these results indicate that the normalized performance of a simple transcriptional cascade circuit can be consistent across different CFE conditions.
Figure 2.
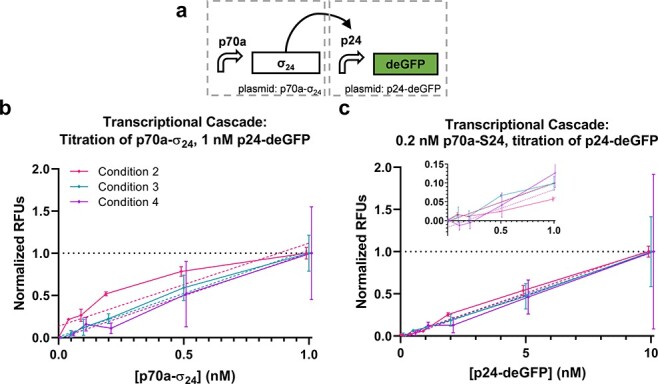
Transcriptional cascade. (a) Transcriptional cascade circuit and corresponding normalized performance data titrating (b) the S24 expression plasmid and (c) reporter plasmid. Data are normalized between the 0 and 1 nM points for p70-σ24 (b) and 0 and 10 nM points for p24a-degfp (c). Data points from each condition are presented slightly offset on the x-axis to improve plot visibility. Error bars indicate one standard deviation; n = 3 for Condition 2, n = 2 for Condition 3 and n = 2 for Condition 4. Solid lines are direct connections between data points. Dashed lines indicate linear fits of corresponding data as indicated by matching color. (c) includes an inset showing lower plasmid concentrations.
The second set of circuits utilize RNA regulation. The first regulator is a STAR that binds to a transcriptional termination hairpin in the 5ʹ UTR of a target gene to unfold the hairpin and allow transcription (Figure 3a and Supplementary Figure S4) (51). The second regulator uses a similar mechanism to introduce transcriptional termination in the 5ʹ UTR in the presence of a sRNA (Figure 3c and Supplementary Figure S) (52). Finally, an RNA that is reverse-complement to the sRNA, referred to here as sRNA’, is used to sequester the sRNA and alleviate the resulting repression (Figure 3e and Supplementary Figure S7). We subsequently refer to these three circuits as ‘STAR activation’, ‘sRNA repression’ and ‘RNA sequestration’. We found that sigmoid fits were superior to linear fits for the STAR activation circuit (R2 = 0.9274, 0.9053 and 0.9375 for the sigmoid fit vs. 0.8942, 0.8133 and 0.8166 for linear for Conditions 2–4, respectively) (Figure 3b). For the sRNA repression circuit, only Conditions 3 and 4 were evaluated due to lack of materials (specifically extract and template DNA) for Condition 1 and clear dispensing anomalies from the acoustic liquid handler for Condition 2. For Conditions 3 and 4, sigmoid fits again were superior (R2 = 0.8924 and 0.8355, respectively, for the sigmoid fit vs. 0.5297 and 0.4706 for linear) (Figure 3d). These results are consistent with a saturation effect of the STAR or sRNA effectors on a fixed concentration of reporter. As with the transcriptional cascade, an ANOVA found that most of the variation is explained by the DNA concentration for the STAR activation (90% for DNA concentration, p < 0.001; 0.9% condition, p = 0.045). As with the transcription (TX) cascade, when assessing the data in units of µM deGFP, most of the variance is instead explained by condition (37%; Table S4). The statistics for sRNA repression are less impactful because Conditions 3 and 4 vary only in operator; nonetheless, the statistics are available in Table S4.
Figure 3.
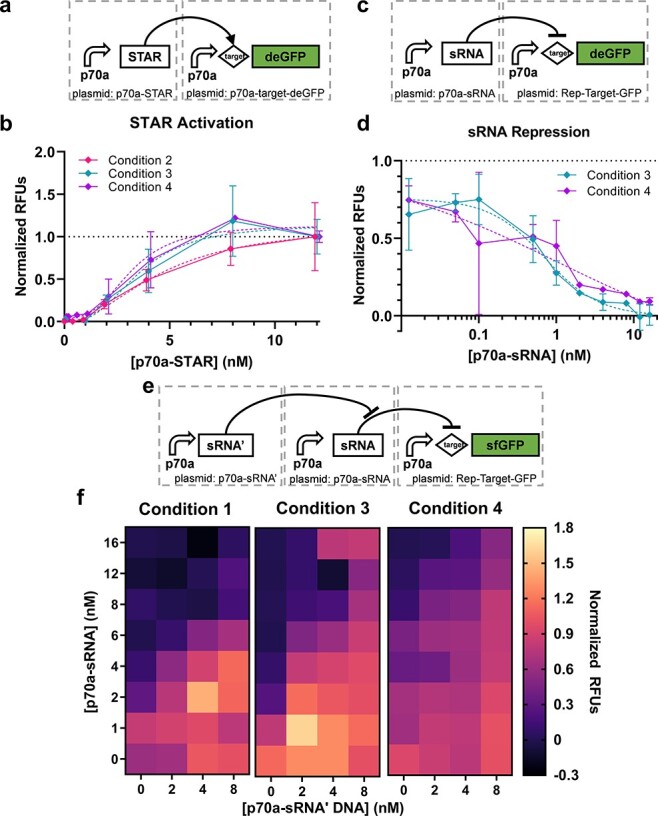
RNA regulation circuits. (a) The STAR activation circuit and (b) corresponding performance data (n = 2 or 3); data are normalized between 0 and 1 by the 0 and 16 nM cases, respectively. Error bars indicate one standard deviation. Dashed lines indicate sigmoid fits of corresponding data as indicated by matching color. (c) The sRNA repression circuit (n = 2) and (d) corresponding performance data; data are normalized between 0 and 1 by the 12 and 0 nM cases, respectively. Error bars indicate one standard deviation. Dashed lines indicate sigmoid fits of corresponding data as indicated by matching color. (e) The sRNA sequester circuit and (f) corresponding performance data (n = 3, 1 or 3 for Conditions 1, 3 and 4, respectively); data are normalized between 0 and 1 by the 16 nM p70a-sRNA + 0 nM p70a-sRNA’ and 0 nM p70a-sRNA + 8 nM p70a-sRNA’ cases, respectively.
For the RNA sequestration circuits, normalized performance appears qualitatively similar across three operator conditions (Figure 3f). Here, a 2-dimensional sigmoid function fits the Condition 1 data better than a plane (Methods; R2 = 0.9302 vs. 0.6361, respectively) but not for Condition 3 (R2 = 0.7895 vs. 0.7899) or Condition 4 (R2 = 0.7604 vs. 0.7620). This result again points to different saturation points for the materials batches, although it could also potentially be explained by undetected anomalies in the acoustic liquid dispensing method used for Condition 1. We performed an ANOVA to assess the relative contribution of concentration of the two circuits and the condition to overall variation. We found that the concentration of sRNA’ DNA was most important, explaining 54% of the variation (p < 0.001), followed by sRNA DNA (13%; p < 0.001) and operator (3%; p < 0.001). Once again, in the non-normalized case, the operator explains significantly more of the variation (72%; Table S4).
The final circuit tested is the complete integral controller (49). In this circuit, sigma factor 28 (σ28) controls expression of both deGFP and an anti-σ28 factor known as FlgM that in turn regulates the level of σ28 through negative feedback (Figure 4d). To assess the closed-loop vs. open-loop performance of the circuit, the p28a-flgM plasmid is replaced by a p70a-msa plasmid to break the negative feedback mediated via FlgM while expressing a similarly sized protein to limit changes in resource utilization (Figure 4a). Titrations of p70a-σ28 were performed for p28a-msa and p28a-flgM each at 1 and 2 nM (Figure 4b–c and e–f and Supplementary Figure S8). Here, a divergence of performance across different CFE conditions becomes more apparent, particularly for Condition 1 in the open-loop case. Since the circuits are more complex, we did not apply simple regression models here. An ANOVA shows that 42% of the variance is explained by condition, markedly higher than that for the previous circuits (Table S4). The closed-loop circuit presents an intriguing case as the purpose of the design is to reject variations in the system, such as DNA concentration and activity of the CFE reaction. Unlike the other circuits where the expectation is that most of the variance is explained by DNA concentration, a functional closed-loop controller should in theory exhibit variance due to primarily random noise. Indeed, we find that only 0.5% is explained by the condition and 76% of the variation is unexplained (Table S4), suggesting that the closed-loop circuit is likely performing as intended. However, more work would be required to verify this hypothesis, including multiple replicates with this circuit as well as replicates with a circuit of similar complexity.
Figure 4.
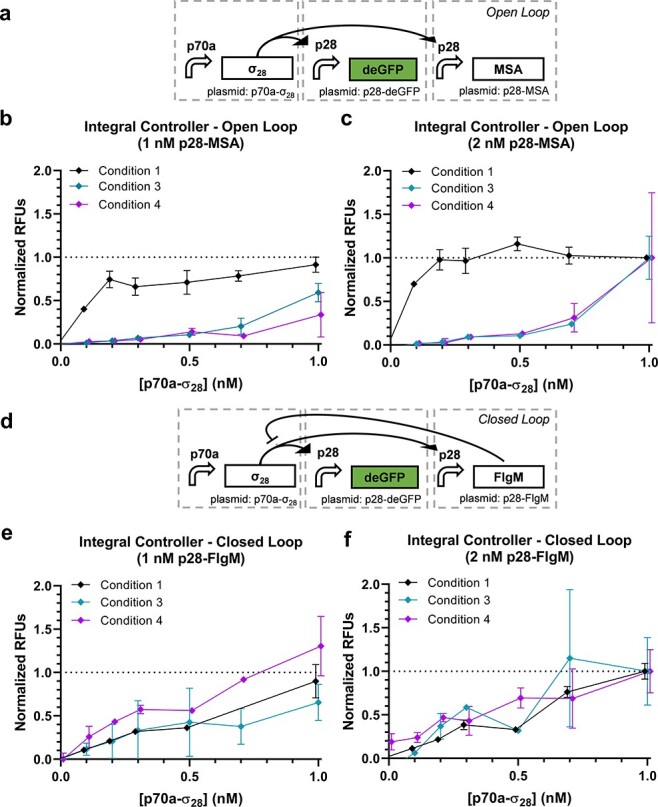
Integral controller. (a) The open-loop case of the integral controller and corresponding performance data with (b) 1 nM or (c) 2 nM p28a-msa (n = 3 for Condition 1 and n = 2 for Conditions 3 and 4). (d) The closed-loop case of the integral controller and corresponding performance data with (e) 1 nM or (f) 2 nM p28a-flgM (n = 3 for Condition 1 and n = 2 for Conditions 3 and 4). Open-loop data are normalized between 0 and 1 by the 0 nM p70a-σ28 + 1 nM p28a-msa case and 1 nM + p70a-σ28 + 2 nM p28a-msa case. Correspondingly, closed-loop data are normalized between 0 and 1 by the 0 nM p70a-σ28 + 1 nM p28a-flgM case and 1 nM + p70a-σ28 + 2 nM p28a-flgM case.
4. Discussion
As the field of CFE continues to grow, it is important to better understand issues of reproducibility. Other recent studies have made headway on quantifying variability; here, we explore the implications of variability in the context of genetic prototyping. We assessed seven genetic circuits in CFE reactions based ostensibly on the same protocol but varying material batches, personnel and measurement instruments. We find that while the activity levels of the different conditions varied widely, by normalizing RFU values within each circuit, qualitative performance was reasonably consistent for most circuits. For the simplest circuits, such as regulatory sequences controlling expression of a reporter, our results suggest that qualitative performance can be reliable even in the face of large quantitative variability. This conclusion is encouraging for the use case of sharing such results between laboratories where quantitative variability is known to be a challenge. However, when the complexity increased to expression of three separate proteins, we noted marked divergence in performance between conditions. It is unsurprising that variability in CFE activity becomes more problematic as complexity increases, but the tipping points had not previously been explored. This result offers a preliminary milepost for the field for genetic circuit complexity that can be considered reliable without careful calibration of raw CFE activity. Future work should enhance understanding of the trade-off between variability in CFE and the impacts for genetic prototyping and other CFE applications. Such understanding can help define what is acceptable quantitative variability for a particular use case, motivating and informing efforts to reduce variability. The use of automation is one commonly proposed approach to reduce variability and warrants mention here. While it is impossible to disentangle the effects of the use of acoustic liquid handling in Conditions 1 and 2 from other differences between conditions, we note that significant challenges with our instrument led to many failed experiments that consumed materials from single batches of limited quantities. Indeed, throughout the course of this work, and anecdotally in other experiments in our laboratory that used acoustic liquid handling to dispense CFE components, we have observed inconsistencies in accurate droplet dispensation by the liquid handler that are not always reported by the instrument. We recently reported an example of these issues and one approach to detect anomalies (16). It certainly may be that automation reduces variability, but it is not necessarily the most straightforward path to reproducibility and more work is needed to fully optimize and characterize these systems.
While it is important to understand and ultimately reduce variability in CFE and synthetic biology more broadly, in other disciplines, and indeed in natural genetic systems, the solution is often to utilize systems that operate reliably despite variability. The circuits tested here were part of an effort to develop exactly this type of functionality in CFE, resulting in the closed loop controller. While we did not thoroughly probe the ability of the circuit to reject disturbances, the low amount of variation explained by either DNA concentration or condition does suggest the intended functionality. As synthetic biology continues to progress toward application, methods to characterize and appropriately handle variability will be critical to success.
Supplementary Material
Acknowledgments
We would like to acknowledge Ryan Marshall and Dr Zachary Sun for technical advice with CFE material preparation, circuit design and construction, and CFE materials. We would also like to acknowledge Ms Caitlin Sharpes and Ms Vanessa Funk for assistance with plasmid preparations.
Contributor Information
Katherine A Rhea, US Army Combat Capabilities Development Command Chemical Biological Center, Aberdeen Proving Ground, MD, USA.
Nathan D McDonald, US Army Combat Capabilities Development Command Chemical Biological Center, Aberdeen Proving Ground, MD, USA.
Stephanie D Cole, US Army Combat Capabilities Development Command Chemical Biological Center, Aberdeen Proving Ground, MD, USA.
Vincent Noireaux, School of Physics and Astronomy, University of Minnesota, Minneapolis, MN, USA.
Matthew W Lux, US Army Combat Capabilities Development Command Chemical Biological Center, Aberdeen Proving Ground, MD, USA.
Patricia E Buckley, US Army Combat Capabilities Development Command Chemical Biological Center, Aberdeen Proving Ground, MD, USA.
Supplementary data
Supplementary data are available at SYNBIO Online.
Material availability
Materials used in this study can be made available upon request.
Funding
Funding was provided by the Defense Advanced Research Projects Agency’s Biological Technologies Office Biological Control program.
Conflict of interest statement.
None declared.
References
- 1. Garenne D., Haines M.C., Romantseva E.F., Freemont P., Strychalski E.A. and Noireaux V. (2021) Cell-free gene expression. Nat. Rev. Methods Prim., 1, 49.doi: 10.1038/s43586-021-00046-x. [DOI] [Google Scholar]
- 2. Silverman A.D., Karim A.S. and Jewett M.C. (2020) Cell-free gene expression: an expanded repertoire of applications. Nat. Rev. Genet., 21, 151–170.doi: 10.1038/s41576-019-0186-3. [DOI] [PubMed] [Google Scholar]
- 3. Cole S.D., Miklos A.E., Chiao A.C., Sun Z.Z. and Lux M.W. (2020) Methodologies for preparation of prokaryotic extracts for cell-free expression systems. Synth. Syst. Biotechnol., 5, 252–267.doi: 10.1016/j.synbio.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dopp J.L., Tamiev D.D. and Reuel N.F. (2019) Cell-free supplement mixtures: elucidating the history and biochemical utility of additives used to support in vitro protein synthesis in E. coli extract. Biotechnol. Adv., 37, 246–258.doi: 10.1016/j.biotechadv.2018.12.006. [DOI] [PubMed] [Google Scholar]
- 5. Cole S.D., Beabout K., Turner K.B., Smith Z.K., Funk V.L., Harbaugh S.V., Liem A.T., Roth P.A., Geier B.A., Emanuel P.A.. et al. (2019) Quantification of interlaboratory cell-free protein synthesis variability. ACS Synth. Biol., 8, 2080–2091.doi: 10.1021/acssynbio.9b00178. [DOI] [PubMed] [Google Scholar]
- 6. Silverman A.D., Kelley-Loughnane N., Lucks J.B. and Jewett M.C. (2019) Deconstructing cell-free extract preparation for in vitro activation of transcriptional genetic circuitry. ACS Synth. Biol., 8, 403–414.doi: 10.1021/acssynbio.8b00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dopp J.L., Jo Y.R. and Reuel N.F. (2019) Methods to reduce variability in E. coli-based cell-free protein expression experiments. Synth. Syst. Biotechnol., 4, 204–211.doi: 10.1016/j.synbio.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borkowski O., Koch M., Zettor A., Pandi A., Batista A.C., Soudier P. and Faulon J.L. (2020) Large scale active-learning-guided exploration for in vitro protein production optimization. Nat. Commun., 11, 1–8.doi: 10.1038/s41467-020-15798-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takahashi M.K., Hayes C.A., Chappell J., Sun Z.Z., Murray R.M., Noireaux V. and Lucks J.B. (2015) Characterizing and prototyping genetic networks with cell-free transcription-translation reactions. Methods, 86, 60–72.doi: 10.1016/j.ymeth.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 10. Levine M.Z., Gregorio N.E., Jewett M.C., Watts K.R. and Oza J.P. (2019) Escherichia coli-based cell-free protein synthesis: protocols for a robust, flexible, and accessible platform technology. J. Vis. Exp., 144, 1–11.doi: 10.3791/58882. [DOI] [PubMed] [Google Scholar]
- 11. Smith P.E.J., Slouka T. and Oza J.P. (2021) From cells to cell-free protein synthesis within 24 hours using cell-free autoinduction workflow. J. Vis. Exp., 11, 274–281.doi: 10.3791/62866. [DOI] [PubMed] [Google Scholar]
- 12. Romantseva E.F., Tack D.S., Alperovich N., Ross D. and Strychalski E.A. (2022) Best practices for DNA template preparation toward improved reproducibility in cell-free protein production. In: Karim, A.S., Jewett, M.C. (eds). Cell-Free Gene Expression. Methods in Molecular Biology. Vol. 2433, Humana, New York, NY, pp. 3–50. [DOI] [PubMed] [Google Scholar]
- 13. Moore S.J., MacDonald J.T. and Freemont P.S. (2017) Cell-free synthetic biology for in vitro prototype engineering. Biochem. Soc. Trans., 45, 785–791.doi: 10.1042/BST20170011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garenne D., Thompson S., Brisson A., Khakimzhan A. and Noireaux V. (2021) The all-E. coliTXTL toolbox 3.0: new capabilities of a cell-free synthetic biology platform. Synth. Biol., 5, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun Z.Z., Yeung E., Hayes C.A., Noireaux V. and Murray R.M. (2014) Linear DNA for rapid prototyping of synthetic biological circuits in an Escherichia coli based TX-TL cell-free system. ACS Synth. Biol., 3, 387–397.doi: 10.1021/sb400131a. [DOI] [PubMed] [Google Scholar]
- 16. McManus J.B., Bernhards C.B., Sharpes C.E., Garcia D.C., Cole S.D., Murray R.M., Emanuel P.A. and Lux M.W. (2021) Rapid characterization of genetic parts with cell-free systems. J. Vis. Exp., 2021, 1–18. [DOI] [PubMed] [Google Scholar]
- 17. Dopp J.L., Rothstein S.M., Mansell T.J. and Reuel N.F. (2019) Rapid prototyping of proteins: mail order gene fragments to assayable proteins within 24 hours. Biotechnol. Bioeng., 116, 667–676.doi: 10.1002/bit.26912. [DOI] [PubMed] [Google Scholar]
- 18. Moore S.J., MacDonald J.T., Wienecke S., Ishwarbhai A., Tsipa A., Aw R., Kylilis N., Bell D.J., McClymont D.W., Jensen K.. et al. (2018) Rapid acquisition and model-based analysis of cell-free transcription–translation reactions from nonmodel bacteria. Proc. Natl. Acad. Sci. U. S. A., 115, 4340–4349.doi: 10.1073/pnas.1715806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kelwick R., Webb A.J., MacDonald J.T. and Freemont P.S. (2016) Development of a Bacillus subtilis cell-free transcription-translation system for prototyping regulatory elements. Metab. Eng., 38, 370–381.doi: 10.1016/j.ymben.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 20. Kopniczky M.B., Canavan C., McClymont D.W., Crone M.A., Suckling L., Goetzmann B., Siciliano V., MacDonald J.T., Jensen K. and Freemont P.S. (2020) Cell-free protein synthesis as a prototyping platform for mammalian synthetic biology. ACS Synth. Biol., 9, 144–156.doi: 10.1021/acssynbio.9b00437. [DOI] [PubMed] [Google Scholar]
- 21. Krüger A., Mueller A.P., Rybnicky G.A., Engle N.L., Yang Z.K., Tschaplinski T.J., Simpson S.D., Köpke M. and Jewett M.C. (2020) Development of a Clostridia-based cell-free system for prototyping genetic parts and metabolic pathways. Metab. Eng., 62, 95–105.doi: 10.1016/j.ymben.2020.06.004. [DOI] [PubMed] [Google Scholar]
- 22. Li J., Wang H., Kwon Y.-C. and Jewett M.C. (2017) Establishing a high yielding streptomyces -based cell-free protein synthesis system. Biotechnol. Bioeng., 114, 1343–1353.doi: 10.1002/bit.26253. [DOI] [PubMed] [Google Scholar]
- 23. Moore S.J., Lai H.E., Needham H., Polizzi K.M. and Freemont P.S. (2017) Streptomyces venezuelae TX-TL – a next generation cell-free synthetic biology tool. Biotechnol. J., 12, 1–7.doi: 10.1002/biot.201600678. [DOI] [PubMed] [Google Scholar]
- 24. Wang H., Li J. and Jewett M.C. (2018) Development of a Pseudomonas putida cell-free protein synthesis platform for rapid screening of gene regulatory elements. Synth. Biol., 3, 1–7.doi: 10.1093/synbio/ysy003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yim S.S., Johns N.I., Park J., Gomes A.L., McBee R.M., Richardson M., Ronda C., Chen S.P., Garenne D., Noireaux V.. et al. (2019) Multiplex transcriptional characterizations across diverse bacterial species using cell‐free systems. Mol. Syst. Biol., 15, 1–15.doi: 10.15252/msb.20198875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McDonald N.D., Rhea K.A., Berk K.L., Zacharko J.L. and Miklos A.E. (2021) Cell-free protein systems from Yersinia pestis are functional and growth-temperature dependent. ACS Synth. Biol., 10, 3604–3607.doi: 10.1021/acssynbio.1c00505. [DOI] [PubMed] [Google Scholar]
- 27. McManus J.B., Emanuel P.A., Murray R.M. and Lux M.W. (2019) A method for cost-effective and rapid characterization of engineered T7-based transcription factors by cell-free protein synthesis reveals insights into the regulation of T7 RNA polymerase-driven expression. Arch. Biochem. Biophys., 674, 108045.doi: 10.1016/j.abb.2019.07.010. [DOI] [PubMed] [Google Scholar]
- 28. Dudley Q.M., Karim A.S., Nash C.J. and Jewett M.C. (2020) In vitro prototyping of limonene biosynthesis using cell-free protein synthesis. Metab. Eng., 61, 251–260.doi: 10.1016/j.ymben.2020.05.006. [DOI] [PubMed] [Google Scholar]
- 29. Karim A.S., Dudley Q.M., Juminaga A., Yuan Y., Crowe S.A., Heggestad J.T., Garg S., Abdalla T., Grubbe W.S., Rasor B.J.. et al. (2020) In vitro prototyping and rapid optimization of biosynthetic enzymes for cell design. Nat. Chem. Biol., 16, 912–919.doi: 10.1038/s41589-020-0559-0. [DOI] [PubMed] [Google Scholar]
- 30. Kelwick R., Ricci L., Chee S.M., Bell D., Webb A.J. and Freemont P.S. (2018) Cell-free prototyping strategies for enhancing the sustainable production of polyhydroxyalkanoates bioplastics. Synth. Biol., 3, 1–12.doi: 10.1093/synbio/ysy016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Borkowski O., Bricio C., Murgiano M., Rothschild-Mancinelli B., Stan G.B. and Ellis T. (2018) Cell-free prediction of protein expression costs for growing cells. Nat. Commun., 9, 1457.doi: 10.1038/s43586-021-00046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pandi A., Grigoras I., Borkowski O. and Faulon J.L. (2019) Optimizing cell-free biosensors to monitor enzymatic production. ACS Synth. Biol., 8, 1952–1957.doi: 10.1021/acssynbio.9b00160. [DOI] [PubMed] [Google Scholar]
- 33. de los Santos E.L.C., Meyerowitz J.T., Mayo S.L. and Murray R.M. (2016) Engineering transcriptional regulator effector specificity using computational design and in vitro rapid prototyping: developing a vanillin sensor. ACS Synth. Biol., 5, 287–295.doi: 10.1021/acssynbio.5b00090. [DOI] [PubMed] [Google Scholar]
- 34. Pardee K., Green A.A., Ferrante T., Cameron D.E., DaleyKeyser A., Yin P. and Collins J.J. (2014) Paper-based synthetic gene networks. Cell, 159, 940–954.doi: 10.1016/j.cell.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beabout K., Bernhards C.B., Thakur M., Turner K.B., Cole S.D., Walper S.A., Chávez J.L. and Lux M.W. (2021) Optimization of heavy metal sensors based on transcription factors and cell-free expression systems. ACS Synth. Biol., 10, 3040–3054.doi: 10.1021/acssynbio.1c00331. [DOI] [PubMed] [Google Scholar]
- 36. Marshall R., Maxwell C.S., Collins S.P., He Y., Beisel C.L., Noireaux V., Marshall R., Maxwell C.S., Collins S.P., Jacobsen T.. et al. (2018) Rapid and scalable characterization of CRISPR technologies using an E. coli cell-free transcription-translation system. Mol. Cell, 69, 146–157.e3.doi: 10.1016/j.molcel.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Niederholtmeyer H., Sun Z.Z., Hori Y., Yeung E., Verpoorte A., Murray R.M., Maerkl S.J. and Fe P. (2015) Rapid cell-free forward engineering of novel genetic ring oscillators. eLife., 4, 1–18.doi: 10.7554/eLife.09771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo S. and Murray R.M. (2019) Construction of incoherent feedforward loop circuits in a cell-free system and in cells. ACS Synth. Biol., 8, 606–610.doi: 10.1021/acssynbio.8b00493. [DOI] [PubMed] [Google Scholar]
- 39. Lehr F.X., Hanst M., Vogel M., Kremer J., Göringer H.U., Suess B. and Koeppl H. (2019) Cell-free prototyping of AND-logic gates based on heterogeneous RNA activators. ACS Synth. Biol., 8, 2163–2173.doi: 10.1021/acssynbio.9b00238. [DOI] [PubMed] [Google Scholar]
- 40. Takahashi M.K., Chappell J., Hayes C.A., Sun Z.Z., Kim J., Singhal V., Spring K.J., Al-Khabouri S., Fall C.P., Noireaux V.. et al. (2015) Rapidly characterizing the fast dynamics of RNA genetic circuitry with cell-free transcription-translation (TX-TL) systems. ACS Synth. Biol., 4, 503–515.doi: 10.1021/sb400206c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vezeau G.E. and Salis H.M. (2021) Tuning cell-free composition controls the time delay, dynamics, and productivity of TX-TL expression. ACS Synth. Biol., 10, 2508–2519.doi: 10.1021/acssynbio.1c00136. [DOI] [PubMed] [Google Scholar]
- 42. Sun Z.Z., Hayes C.A., Shin J., Caschera F., Murray R.M. and Noireaux V. (2013) Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. J. Vis. Exp., 79, 1–14.doi: 10.3791/50762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Caschera F. and Noireaux V. (2015) Preparation of amino acid mixtures for cell-free expression systems. Biotechniques, 58, 40–43.doi: 10.2144/000114249. [DOI] [PubMed] [Google Scholar]
- 44. Garamella J., Marshall R., Rustad M. and Noireaux V. (2016) The all E. coli TX-TL Toolbox 2.0: a platform for cell-free synthetic biology. ACS Synth. Biol., 5, 344–355.doi: 10.1021/acssynbio.5b00296. [DOI] [PubMed] [Google Scholar]
- 45. Maxwell C.S., Jacobsen T., Marshall R., Noireaux V. and Beisel C.L. (2018) A detailed cell-free transcription-translation-based assay to decipher CRISPR protospacer-adjacent motifs. Methods, 143, 48–57.doi: 10.1016/j.ymeth.2018.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Westbrook A., Tang X., Marshall R., Maxwell C.S., Chappell J., Agrawal D.K., Dunlop M.J., Noireaux V., Beisel C.L., Lucks J.. et al. (2019) Distinct timescales of RNA regulators enable the construction of a genetic pulse generator. Biotechnol. Bioeng., 116, 1139–1151.doi: 10.1002/bit.26918. [DOI] [PubMed] [Google Scholar]
- 47. Agrawal D.K., Tang X., Westbrook A., Marshall R., Maxwell C.S., Lucks J., Noireaux V., Beisel C.L., Dunlop M.J. and Franco E. (2018) Mathematical modeling of RNA-based architectures for closed loop control of gene expression. ACS Synth. Biol., 7, 1219–1228.doi: 10.1021/acssynbio.8b00040. [DOI] [PubMed] [Google Scholar]
- 48. Garenne D., Beisel C.L. and Noireaux V. (2019) Characterization of the all‐E. coli transcription‐translation system myTXTL by mass spectrometry. Rapid Commun. Mass Spectrom., 33, 1036–1048.doi: 10.1002/rcm.8438. [DOI] [PubMed] [Google Scholar]
- 49. Agrawal D.K., Marshall R., Noireaux V. and Sontag E.D. (2019) In vitro implementation of robust gene regulation in a synthetic biomolecular integral controller. Nat. Commun., 10, 5760.doi: 10.1038/s41467-019-13626-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Siegal-Gaskins D., Tuza Z.A., Kim J., Noireaux V. and Murray R.M. (2014) Resource usage and gene circuit performance characterization in a cell-free ‘breadboard’. ACS Synth. Biol., 3, 416–425.doi: 10.1021/sb400203p. [DOI] [PubMed] [Google Scholar]
- 51. Chappell J., Takahashi M.K. and Lucks J.B. (2015) Creating small transcription activating RNAs. Nat. Chem. Biol., 11, 214–220.doi: 10.1038/nchembio.1737. [DOI] [PubMed] [Google Scholar]
- 52. Takahashi M.K. and Lucks J.B. (2013) A modular strategy for engineering orthogonal chimeric RNA transcription regulators. Nucleic Acids Res., 41, 7577–7588.doi: 10.1093/nar/gkt452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.