

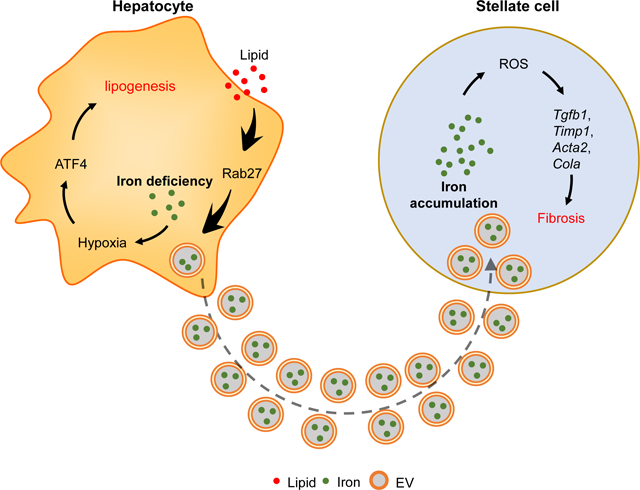
Summary
Hepatocytes have important roles in liver iron homeostasis, abnormalities in which are tightly associated with liver steatosis and fibrosis. Here, we show that non-alcoholic fatty liver disease (NAFLD) and steatohepatitis (NASH) are characterized by iron-deficient hepatocytes and iron overload in hepatic stellate cells (HSCs). Iron deficiency enhances hepatocyte lipogenesis and insulin resistance through HIF2α-ATF4 signaling. Elevated secretion of iron-containing hepatocyte extracellular vesicles (EVs), which are normally cleared by Kupffer cells, accounts for hepatocyte iron deficiency and HSC iron overload in NAFLD/NASH livers. Iron accumulation results in overproduction of reactive oxygen species that promote HSC fibrogenic activation. Conversely, blocking hepatocyte EV secretion or depleting EV iron cargo restore liver iron homeostasis, concomitant with mitigation of NAFLD/NASH-associated liver steatosis and fibrosis. Taken together, these studies show that iron distribution disorders contribute to the development of liver metabolic diseases.
Graphical Abstract
eTOC
Hepatocytes have important roles in liver iron homeostasis. Gao et al., report that hepatocyte-derived iron containing extracellular vesicles lead to hepatocyte iron deficiency and hepatic stellate cells iron overload, which contributes to the development of liver steatosis and fibrosis in Western diet fed mice.
Introduction
Iron is an essential trace element present at approximately 35 to 45 mg/kg of body weight in adult humans (Ganz, 2019). In a healthy individual, 20–30% of surplus iron is stored in hepatocytes, which serve as the most important reservoir and regulators of iron homeostasis (Meynard et al., 2014). Iron participates in many biological processes, such as oxygen transport, cellular energy metabolism, electron transport, and numerous enzymatic reactions (Muckenthaler et al., 2017). According to the World Health Organization, iron deficiency is the most common nutritional disorder (Brissot et al., 2018; McLean et al., 2009). Emerging evidence has uncovered unsuspected connections between iron homeostasis and metabolic disease (Britton et al., 2018; Britton et al., 2016; Hori et al., 2018; Ryan et al., 2018; Simcox and McClain, 2013). Clinical studies show that abnormal iron distribution in liver is tightly associated with the incidence of non-alcoholic fatty liver diseases (NAFLD) and non-alcoholic steatohepatitis (NASH) (Britton et al., 2018; Kim et al., 2012; Kowdley et al., 2012; Nelson et al., 2011), but the underlying mechanisms remain unknown. While hepatocytes play central roles in iron storage and nutrient metabolism, how hepatocyte iron homeostasis affects metabolic responses is unknown.
To maintain iron homeostasis, hepatocytes export excess iron via ferroportin (FPN) which is a well-established membrane-localized iron exporter (Abboud and Haile, 2000; Donovan et al., 2000; McKie et al., 2000). Hepcidin, a liver-secreted hormone, can block FPN function by inducing its internalization and degradation (Drakesmith et al., 2015; Nemeth et al., 2004; Nemeth et al., 2003). Liver inflammation enhances hepcidin production through activation of the interleukin 6 (IL-6)-STAT3 axis (Nemeth et al., 2003; Verga Falzacappa et al., 2007) that also plays a key role in obesity driven hepatocarcinogenesis (He et al., 2013; Park et al., 2010). In the context of obesity, one of the most common causes of chronic liver inflammation, circulating hepcidin levels are elevated, resulting in FPN degradation and systemic iron deficiency (Baumgartner et al., 2013; Stoffel et al., 2020; Tussing-Humphreys et al., 2010; Yanoff et al., 2007). These important findings led us to hypothesize that abnormalities in hepatocyte iron export could be one of the causal factors for iron distribution disorders and the pathogenesis of liver metabolic diseases.
Extracellular vesicles (EVs) and their cargos resemble an endocrine-like system transferring a variety of cellular materials and signals including RNA, DNA, proteins, and lipids between neighboring or distant cells, playing important roles in the development of metabolic diseases and cancer (Isaac et al., 2021; Kalluri and LeBleu, 2020; Luo et al., 2021; Mathieu et al., 2019). Previous studies, including ours, have shown that hepatocyte-derived EVs profoundly regulate insulin target cell functions during obesity-associated metabolic disorders (Ji et al., 2021; Zhao et al., 2020). Other recent studies suggest that EVs mediate iron expulsion in either cancer cells or macrophages (Brown et al., 2019; Truman-Rosentsvit et al., 2018). However, whether EVs provide a new mechanism whereby hepatocytes control the iron contents of other liver resident cells is unknown.
Here, we report that iron deficiency promotes lipogenesis in hepatocytes through activation of HIF2α-ATF4 signaling, contributing to the steatotic phenotype in NAFLD or NASH afflicted livers. Elevated secretion of iron containing EVs results in hepatocyte iron deficiency, whereas, blocking hepatocyte EV secretion preserves liver iron homeostasis in NAFLD/NASH mice. While Kupffer cells (KCs) clear hepatocyte EVs, the diminished KC population in NASH livers results in engulfment of excess hepatocyte EVs by hepatic stellate cells (HSCs) and subsequent iron overload. HSC iron overload enhances ROS accumulation and assumption of a pro-fibrotic phenotype. In contrast, depletion of iron cargos prevents the adverse effects of hepatocyte EVs.
Results
Hepatic iron homeostasis is impaired in human and mouse NAFLD/NASH.
Given the critical roles of hepatocytes in mediating iron storage and supply, we assessed hepatic iron balance in NAFLD and NASH. We found that total iron levels in human NAFLD/NASH hepatocytes were significantly lower than in healthy hepatocytes (Figure 1A). In addition, the abundance of both ferritin H (FTH) and L (FTL) subunits, which are essential iron storage proteins in mammals, was markedly reduced in human NAFLD/NASH hepatocytes (Figure 1B). In the Western diet (WD)-induced mouse NASH model, we observed that hepatocytes isolated from NASH (24 wks WD feeding) WT mice contained ~30% less total iron than lean WT mouse hepatocytes (Figures 1C and S1A). In addition, NASH mouse hepatocytes expressed less Fth and Ftl, compared to lean mouse hepatocytes (Figure S1B).
Figure 1. NAFLD/NASH is characterized by abnormal iron distribution in hepatic cells.
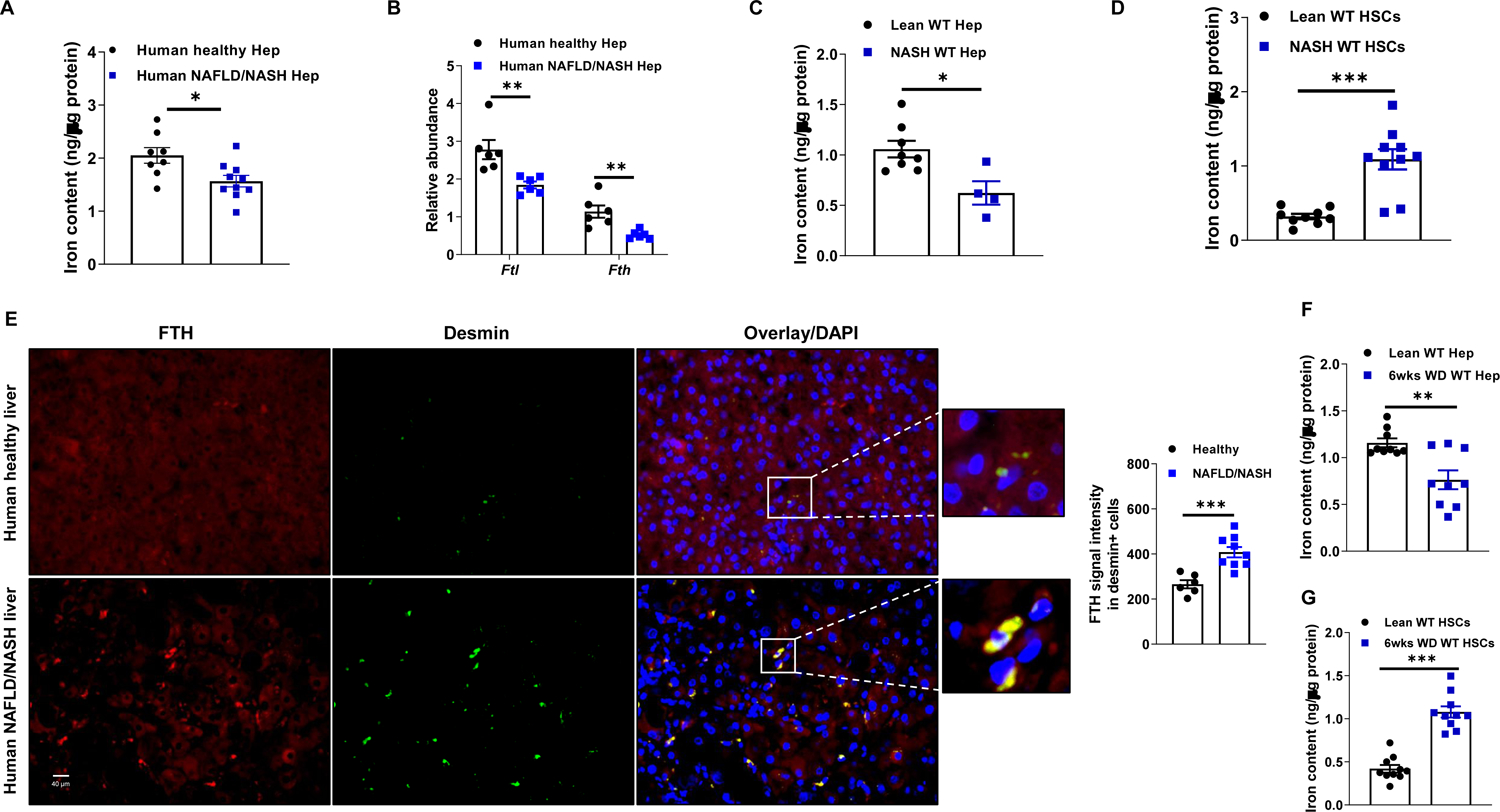
(A) Total iron content in human hepatocytes (Hep) of lean/healthy or NAFLD/NASH individuals.
(B) The abundance of ferritin in human healthy/lean vs. NAFLD/NASH hepatocytes.
Total iron levels in hepatocytes (C) and HSCs (D) of lean and 24 weeks Western diet-fed (24wks WD) NASH WT mice.
(E) Immunofluorescence staining of human liver sections of lean/healthy and NAFLD/NASH individuals to examine the abundance of FTH in Desmin+ HSCs. Total iron levels in hepatocytes (F) and HSCs (G) in WT mice after 6wks WD feeding.
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test. See also Figure S1.
Notably, the abundance of Fth and Ftl in NASH hepatic stellate cells (HSCs), which are the major collagen-producing cells that drive liver fibrosis, was greater than that in lean mouse HSCs (Figure S1C). Congruently, HSCs from NASH mouse liver contained ~2-fold more total iron than lean mouse HSCs (Figure 1D). Iron was also likely to be elevated in human NASH HSCs, as suggested by higher ferritin amounts in Desmin or α-SMA (smooth muscle actin) expressing cells in NASH livers than in healthy/lean human livers (Figures 1E and S1D).
We further examined whether abnormal iron distribution in hepatocytes and HSCs precedes the establishment of steatosis and fibrosis. After a short period (6 wks) of WD feeding, total iron and ferritin were significantly lower in hepatocytes, compared to hepatocytes from lean control mice (Figures 1F and S1E). Conversely, 6 wks WD-fed mouse HSC had higher iron and ferritin content than control mouse HSCs (Figures 1G and S1F). Expression of transferrin receptor (Tfrc1) and Fpn mRNAs was significantly repressed in HSCs after 6 wks of WD feeding (Figure S1G). Taken together, these results suggest that dysfunction of hepatic iron metabolism and homeostasis could be important for NAFLD and NASH pathogenesis.
Iron deficiency aggravates NAFLD-associated hepatic lipogenesis.
To investigate the effect of iron deficiency on hepatocyte functions, human healthy hepatocytes were treated with deferoxamine (DFO, 100 μM) to remove ~30% of their total iron content, thus mimicking iron levels in human NAFLD/NASH hepatocytes (Figure S2A). We assessed the impact of reduced iron content on the hepatocyte transcriptome. RNA sequencing (RNAseq) analysis showed that metabolic responses and fatty acid synthesis were among the top enriched pathways under iron deficiency (Figure 2A). Consistently, we found a significant increase in expression of key genes associated with lipogenesis in human healthy hepatocytes after DFO treatment (Figure S2B). In addition, in vitro lipogenesis assays using C14-labeled sodium acetate showed that iron deficiency resulted in more C14-acetate incorporation in DFO-treated human healthy hepatocytes, compared to control cells (Figure 2B). We also observed increased lipogenesis and lipogenic gene expression in DFO or Apo-transferrin-treated normal mouse hepatocytes (Figures S2C–S2E), suggesting that the lipogenic response to reduced hepatocyte iron is conserved and could have a causal role in hepatosteatosis. To assess the impact of iron deficiency on in vivo hepatic lipogenic activation, we used hepatocyte specific Tfrc1 KO (Tfrc1ΔHep) mice. After intravenous (i.v.) delivery of a lentivirus containing guide RNA (gRNA) for Tfrc1 (1×108 particles/mouse), hepatocyte specific Cas9 transgenic mice (AlbuminCre+Cas9+) had a lower Tfrc1 expression in hepatocytes than control AlbuminCre+Cas9+ mice injected with a lentivirus without gRNA (Figures 2C and S2F). Of note, Tfrc1 depletion caused decreased Ftl and Fth mRNA abundance, suggesting lower iron levels than lean WT mice (Figure 2C). More importantly, lean Tfrc1ΔHep mice showed higher expression of lipogenic genes than lean WT mice (Figure 2C).
Figure 2. Iron deficiency triggers lipogenesis and insulin resistance in hepatocytes.
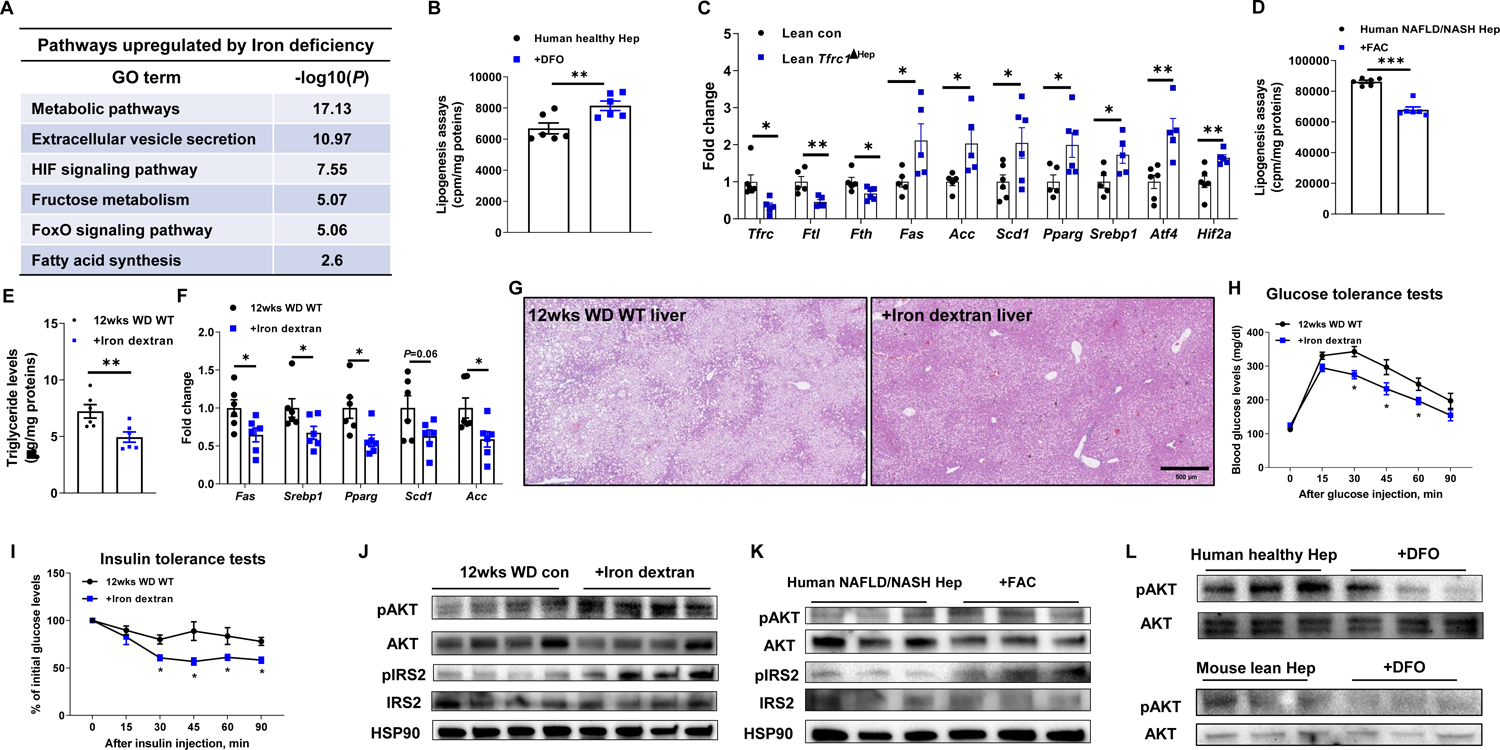
(A) Upregulated pathways in lean mouse hepatocytes after 24 hours deferoxamine (DFO; 100 μM)-induced iron deficiency.
(B) Effect of DFO treatment on lipogenesis in healthy human hepatocytes. Cells without DFO treatment were used as controls. cpm, count per minute.
(C) Expression of ferritin and lipogenic genes in the hepatocytes of lean AlbuminCre+Cas9+ mice after 2wks injection with lentivirus carrying gRNA-Tfrc1.
(D) Lipogenesis in human NAFLD/NASH hepatocytes after ferric ammonium citrate (FAC; 100 μM) treatment.
(E) Live triglyceride levels in 12wks WD WT mice after 6wks treatment with iron dextran (10 mg/Kg body weight). 12wks WD WT mice treated with saline were used as controls.
Effects of iron dextran treatment on the abundance of lipogenesis genes (F) and lipid accumulation (G) in the livers of 12wks WD WT mice. Representative images of n=6 per group.
(H and I) Glucose and insulin tolerance tests of 12wks WD WT mice after 6wks treatment with iron dextran.
(J) Effect of iron dextran treatment on AKT or IRS2 phosphorylation in the liver of 12wks WD mice.
(K) The levels of phosphorylated AKT or IRS2 in human NAFLD/NASH hepatocytes after in vitro treatment with FAC.
(L) Effect of DFO treatment on AKT phosphorylation in either human healthy hepatocytes or lean mouse hepatocytes.
Data are presented as mean ± SEM. n = 6 per group (G and H). *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test. See also Figure S2.
Activation of hepatic lipogenesis is essential for NAFLD development (Friedman et al., 2018). We, therefore, examined whether restoration of hepatocyte iron attenuates lipogenic activity in human NAFLD/NASH hepatocytes. After treatment with ferric ammonium citrate (FAC, 100 μM), iron levels in human NAFLD/NASH hepatocytes were comparable to those in human healthy hepatocytes, concomitant with downregulated expression of lipogenic genes and diminished C14-acetate incorporation (Figures 2D and S2G and S2H). We also confirmed that restoration of hepatocyte iron with FAC or holo-transferrin treatment reduced lipogenic gene expression in 12 wks WD WT mouse hepatocytes (Figures S2I and S2J). To validate the effect of hepatic iron restoration on hepatosteatosis, 6 wks WD fed WT mice were intraperitoneally injected with iron dextran (10 mg/Kg body weight, twice per week). Of note, iron dextran treatment restored hepatocyte iron content in WD-fed recipient mice to the same levels as in lean WT mice (Figures S2K and S2L). More importantly, after 6 wks of treatment, administration of iron dextran blunted hepatic lipogenic activity, as demonstrated by markedly lower levels of liver triglycerides and lipogenic mRNAs (Figures 2E and 2F). H&E staining of liver sections revealed that iron dextran-treated mouse livers contained fewer lipid droplets than untreated WD-fed livers (Figure 2G). These data suggest that restoration of hepatocyte iron can attenuate hepatosteatosis.
Hepatic insulin resistance accompanies NAFLD and NASH progression (Friedman et al., 2018; Koyama and Brenner, 2017). We, therefore, assessed the importance of iron homeostasis in modulation of hepatocyte insulin sensitivity. Compared to untreated 12 wks WD fed WT mice, iron dextran treated mice exhibited improved glucose tolerance and insulin sensitivity, as well as greater levels of insulin-stimulated phosphorylated AKT and IRS2 in the liver (Figures 2H–2J). While iron dextran treatment also led to increased ferritin abundance in KCs, the KC activation status was comparable between 12 wks WD fed WT control and iron dextran treated mice (Figures S2M and S2N). To further confirm the direct effect of iron content recovery on hepatocyte insulin sensitivity, human and mouse NAFLD/NASH hepatocytes were treated with FAC. This resulted in enhanced insulin induced AKT and IRS2 phosphorylation (Figures 2K and S2O). In contrast, DFO treatment of human or mouse healthy hepatocytes reduced insulin stimulated AKT phosphorylation (Figure 2L). Thus, these results show that iron homeostasis maintains hepatic insulin sensitivity.
HIF2α/ATF4 signaling mediates iron deficiency-induced lipogenesis and insulin resistance.
To further investigate how iron deficiency affects lipogenic gene expression in hepatocytes, we analyzed the transcriptomic profiles of lean mouse hepatocytes treated with DFO. Gene ontology analysis revealed that the differentially expressed gene sets induced by iron deficiency were tightly associated with cellular response to hypoxia and hypoxia-inducible factor (HIF) signaling (Figure 2A). Among genes overexpressed in DFO-treated hepatocytes was the gene coding for HIF2α (Figure S3A), which is the only Hif family member with iron-responsive elements in its untranslated region (Muckenthaler et al., 2017; Shah and Xie, 2014). Consistently, we observed that HIF2α protein abundance was markedly elevated in human and mouse NAFLD/NASH hepatocytes compared to lean hepatocytes (Figures 3A and S3B). To examine the importance of HIF2α in iron deficiency-induced hepatocyte dysfunction, we utilized HIF2a siRNA packaged into liposomes to silence HIF2α expression. RT-qPCR analysis indicated that reduced HIF2α amounts blunted iron deficiency-induced lipogenic gene expression in DFO-treated human or mouse hepatocytes (Figures 3B and S3C). Congruently, the lipogenesis assay showed that HIF2α siRNA transfection reduced 14C-acetate incorporation in human healthy hepatocytes (Figure 3C). In addition, iron deficient human NAFLD/NASH hepatocytes displayed reduced lipogenic activity after HIF2α silencing (Figures 3D and S3D).
Figure 3. Iron deficiency induces hepatocyte responses through the activation of HIF2α/ATF4 pathway.

(A) The abundance of HIF2α and ATF4 in hepatocytes isolated from lean and NAFLD/NASH humans or WT mice.
(B) Effect of HIF2a repression on the expression of genes associated with lipogenesis in DFO-treated lean mouse hepatocytes.
Lipogenesis in DFO-treated human healthy (C) or NAFLD/NASH (D) hepatocytes after transfection with siRNA-HIF2a.
(E) Effects of HIF2α knockdown on the abundance of phosphorylated AKT and ATF4 in DFO-treated healthy human hepatocytes.
Effect of DFO treatment on lipogenesis in lean ATF4KO mouse hepatocytes (F) and lipogenesis gene abundance in siRNA-Atf4 treated healthy human hepatocytes (G).
Levels of triglyceride (H) and H&E staining (I) in livers, glucose tolerance (J), and insulin sensitivity (K) of 16wks WD ATF4f/f and AlbuminCre+ATF4f/f (Atf4ΔHep) mice. Representative images of n=6 per group (I). n=7–10 per group (J&K). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test. See also Figure S3.
We also examined the role of HIF2α in regulation of hepatic insulin sensitivity in response to iron deficiency. After HIF2α knockdown in DFO-treated human healthy hepatocytes, the levels of insulin-stimulated AKT phosphorylation were significantly elevated (Figure 3E). In addition, NAFLD hepatocytes transfected with HIF2a siRNA contained more phosphorylated AKT, compared to control NAFLD cells (Figure S3E). These results suggest that HIF2α plays a critical role in modulating hepatic lipogenesis and insulin sensitivity in response to iron deficiency.
Previous studies have demonstrated that activating transcription factor 4 (ATF4) is a HIF target gene (Koditz et al., 2007; Lange et al., 2008). We found that DFO-treated lean/healthy hepatocytes or NAFLD/NASH hepatocytes expressed more ATF4 than control hepatocytes (Figures 3A and S3B). More importantly, concomitant with HIF2α repression, ATF4 expression was reduced in iron-deficient hepatocytes, demonstrating that HIF2α induces ATF4 expression (Figures 3B, 3E, and S3E). We next examined whether ATF4 ablation attenuates iron deficiency-induced hepatic dysfunctions. After treatment with DFO, the ATF4 target genes Chop and Pparg were upregulated in lean WT hepatocytes, whereas DFO treatment did not affect their expression in Atf4−/− mouse hepatocytes (Figure S3F). More importantly, Atf4−/− mouse hepatocytes had comparable lipogenic activity to untreated counterparts (Figure 3F). Atf4 knockdown in human healthy hepatocytes blunted effects of DFO on lipogenesis and insulin sensitivity (Figures 3G and S3G).
To further evaluate in vivo effects of ATF4 ablation on hepatocyte functions, hepatocyte specific Atf4ΔHep (AlbuminCre+Atf4f/f) mice were fed WD. While WT mice developed hepatosteatosis after 16 wks of WD feeding, Atf4ΔHep mice showed much fewer lipid droplets in their liver and less liver triglycerides (Figures 3H and 3I). In addition, in the WD-fed Atf4ΔHep liver lipogenic gene expression was lower than in WD-fed WT liver (Figure S3H). Also, after 16 wks of WD feeding, Atf4ΔHep mice showed improved glucose tolerance and insulin sensitivity, compared to WT mice (Figures 3J and 3K). The levels of phosphorylated AKT and IRS2 in the WD-fed Atf4ΔHep liver were greater than WD-fed WT livers (Figure S3I). Taken together, these data suggest that the HIF2α-ATF4 signaling axis mediates the effects of iron deficiency on hepatocyte lipid metabolism and insulin signaling.
Extracellular vesicle-dependent iron export underlies NAFLD/NASH-associated hepatocyte iron deficiency.
FPN is the main iron exporter in hepatocytes, and hepcidin is a small peptide hormone that directly binds FPN to block iron export (Drakesmith et al., 2015). Previous studies have shown that obesity is accompanied by elevated secretion of hepcidin (Stoffel et al., 2020). We also found that hepcidin levels in bloodstream and hepatocytes were upregulated after a short period (6 wks) of WD feeding and were higher in NASH mice (Figures 4A and S4A). Concomitant with high circulating hepcidin, WD-fed mouse hepatocytes had remarkably lower FPN abundance, compared to lean mouse hepatocytes (Figure 4B). We also observed a marked reduction in FPN expression in human NAFLD/NASH hepatocytes (Figure 4B). NASH hepatocytes expressed comparable amounts of mRNAs encoding iron importers (Tfrc1, Tfrc2, Slc39a14, and Dmt1) after 6 wks WD feeding (Figure S4B). However, the iron content of these hepatocytes was markedly reduced, suggesting FPN independent iron expulsion.
Figure 4. Extracellular vesicles play important roles in expulsing iron from hepatocytes in NAFLD/NASH livers.
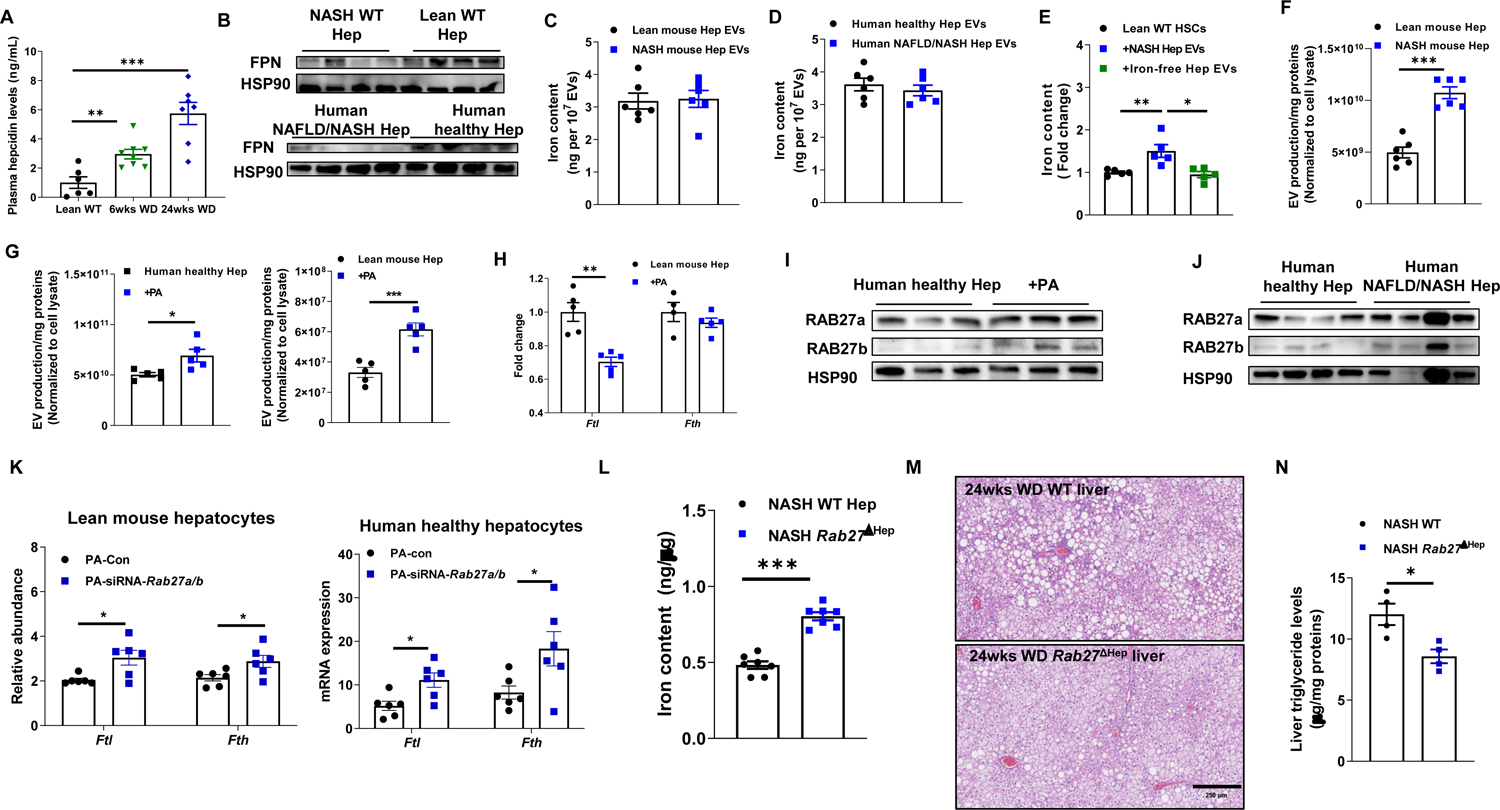
(A) Circulating hepcidin levels in WT mice after Western diet feeding.
(B) The abundance of ferroportin (FPN) in hepatocytes of lean and NAFLD/NASH livers.
Iron levels within extracellular vesicles (EVs) derived from mouse (C) and human (D) hepatocytes.
(E) Iron content in lean mouse HSCs after in vitro treatment with either NASH hepatocyte EVs (NASH Hep EVs) or iron-free NASH Hep EVs. Control cells were treated with empty liposomes.
(F) EV production of lean and NASH WT mouse hepatocytes.
(G) Effect of palmitic acid (PA; 500 μM) on EV production from either lean/healthy human or mouse hepatocytes.
(H) The abundance of ferritin in lean WT mouse hepatocytes after 24 hours PA treatment.
(I) The abundance of RAB27a and RAB27b in healthy human hepatocytes after 24 hours PA treatment.
(J) RAB27a and RAB27b expression in human healthy vs NAFLD/NASH hepatocytes.
(K) Effects of Rab27a/b depletion on ferritin expression in PA-treated lean mouse or healthy human hepatocytes.
(L) Hepatocyte iron levels in NASH WT vs. NASH hepatocyte-specific Rab27a/b KO mice (Rab27ΔHep).
Levels of liver steatosis (M) and triglyceride (N) of NASH WT vs. NASH Rab27ΔHep mice. Representative images of n=4 mice per group.
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test (C-L, N) or One-way ANOVA (A). See also Figure S4.
EVs transport various cargos between different cell types (Isaac et al., 2021; Kalluri and LeBleu, 2020). To examine whether iron is one of the components of hepatocyte-derived EVs, we purified EVs from hepatocyte culture media using the ultracentrifugation-based protocol described in our previous studies (Ji et al., 2021; Ying et al., 2021; Ying et al., 2017). Iron was detectable within human and mouse hepatocyte secreted EVs (Figures 4C and 4D). To further validate iron as hepatocyte EVs cargo, lean mouse hepatocyte derived EVs were co-cultured with mouse HSCs. After 24 hrs, EV addition resulted in elevated HSC total iron content (Figure 4E). In contrast, DFO-treated hepatocyte EVs did not increase HSC iron levels (Figures 4E and S4C). Although the iron content of EVs from lean and NASH mouse hepatocytes was nearly identical, NASH mouse hepatocytes produced more EVs than lean mouse hepatocytes (Figures 4C and 4F). Similarly, human NAFLD/NASH hepatocytes secreted more iron containing EVs, compared to human healthy hepatocytes (Figure S4D). Given that lipids are factors in NAFLD and NASH, we treated lean WT mouse and human healthy hepatocytes with palmitic acid (PA; 500 μM). Interestingly, PA treatment elevated the number of secreted EVs (Figure 4G), but the iron content was similar in EVs derived from control and PA-treated hepatocytes (Figure S4E). Concomitant with enhanced EV secretion, PA-treated hepatocytes had a significantly lower ferritin abundance, compared to control hepatocytes (Figures 4H and S4F).
To investigate the mechanisms underlying PA-induced EV secretion from hepatocytes, we examined a set of key regulators of EV export. Interestingly, expression of RAB27, a small GTPase that is critical for the secretion of small vesicles (Bobrie et al., 2012; Ji et al., 2021; Ostrowski et al., 2010; Song et al., 2019), was greater in PA or palmitic and oleic acid (PAOA)-treated lean mouse hepatocytes, compared to control cells (Figures 4I and S4G and S4H). PA or PAOA treatment, however, had a minimal effect on the abundance of other EV secretion regulators such as Prominin2 and Ncoa4 (Figure S4G and S4H). RAB27 expression was already elevated after a short period (6 wks) of WD feeding (Figure S4I). Consistently, human NAFLD/NASH hepatocytes expressed more RAB27 than healthy hepatocytes (Figure 4J). To test if PA-induced iron deficiency in hepatocytes is RAB27 dependent, either healthy/lean human or mouse hepatocytes were transfected with Rab27 siRNA prior to PA treatment. We found that, in the presence of PA, Rab27 silencing resulted in a great reduction in EV secretion, accompanied by greater ferritin abundance than the scramble RNA-treated cells (Figures 4K and S4J). Concomitantly, RAB27 downregulation repressed the expression of lipogenic genes in PA or PAOA-treated hepatocytes (Figures S4K and S4L).
To validate the role of RAB27-mediated EV secretion in regulation of hepatocyte iron homeostasis, we used a hepatocyte-specific Rab27 KO (Rab27ΔHep) mouse described in our previous studies (Ji et al., 2021). After intravenous (i.v.) delivery of a lentivirus containing guide RNAs (gRNAs) for Rab27a and Rab27b (1×108 particles/mouse), AlbuminCre+Cas9+ mice had lower RAB27a and RAB27b expression in hepatocytes than control mice injected with a lentivirus without gRNAs (Figure S4M). More importantly, Rab27ΔHep mice fed a NASH diet maintained higher iron levels than WT NASH mice (Figure 4L). In addition, hepatocyte specific Rab27 knockout mice exhibited less hepatosteatosis after 24wks WD feeding than WT mice, as evidenced by fewer lipid droplets, lower liver triglycerides and lower expression of lipogenic genes (Figures 4M, 4N and S4N). Hepatocyte Rab27 depletion also reduced glucose intolerance and insulin resistance after WD feeding (Figure S4O and S4P). DFO treatment reduced iron content of hepatocytes isolated from NASH Rab27ΔHep mice, concomitant with elevated expression of lipogenic genes (Figure S4Q). These results demonstrate that RAB27-mediated secretion of iron containing EVs accounts for hepatocyte iron deficiency thereby promoting the development of hepatosteatosis.
Kupffer cells are key players cleaning iron-containing hepatocyte EVs.
Kupffer cells (KCs) take up hepatocyte secreted iron to maintain microenvironmental iron homeostasis in the liver (Krenkel and Tacke, 2017). To assess whether KCs interact with hepatocyte EVs, we generated Cy3-labeled microRNA-223 (miR-223)-containing EVs as previously described (Figures S5A and S5B) (Ying et al., 2021). Flow cytometric analysis indicated enrichment of Cy3 signals in Clec4f+F4/80+ KCs after overnight i.v. injection of Cy3-miR-223 containing EVs (5×109 vesicles/mouse) into lean WT mice, compared to the controls without EV treatment (Figure 5A). These data demonstrate that KCs can efficiently engulf iron-containing hepatocyte EVs.
Figure 5. Kupffer cells engulf hepatocyte EVs.

(A) Intensity of Cy3 fluorescence in KCs (CD11blowF4/80high) of lean WT mice after 12 hours intravenous injection with Cy3-miR-223 containing NASH hepatocyte EVs. Control mice were treated with empty liposomes.
(B) The population of recruited hepatic macrophages (CD11bhighF4/80low; RHMs) and KCs (CD11blowF4/80high) after 24wks WD feeding or switching 24wks WD WT mice to normal chow diet (NCD) feeding for 9wks.
(C) The abundance of ferritin in HSCs of lean WT vs. KC-KO (diphtheria toxin-treated Clec4fCre+DTR+) mice.
(D) Iron content in the HSCs of lean KC-KO mice after 4wks treatment with either NASH Hep EVs or iron-free NASH Hep EVs. Control KC-KO mice were treated with empty liposomes.
(E) HSC iron levels after lean WT mice treated with NASH Hep EVs for 4 weeks. Levels of total iron (F) and ferritin abundance (G) in HSCs after switching 24wks WD WT mice to normal chow diet feeding for 9wks.
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test (A, C, E-G) or One-way ANOVA (B and D). See also Figure S5.
Previous studies demonstrated depletion of embryonic-derived KC population in human and mouse NAFLD/NASH (Seidman et al., 2020; Tran et al., 2020) and we made similar observations (Figure 5B). Thus, we assessed how KC loss affects liver iron homeostasis. We generated Clec4f+F4/80+ KC-depleted (KC-KO) mice by i.p. injecting diphtheria toxin (DT) into lean Clec4fCre+DTR+ mice (200 ng/mouse, one injection every two days) (Figure S5C). After 2 wks, we found that HSCs isolated from lean KC-KO mice contained significantly more ferritin than lean WT mouse HSCs (Figure 5C). To validate that the KC-mediated uptake of iron-containing hepatocyte EVs controls HSC iron accumulation, lean KC-KO mice were i.v. injected with either NASH mouse hepatocyte EVs or iron-free NASH hepatocyte EVs (5×109 vesicles/mouse, 2 injections per week). After 4 wks of treatment, we found that NASH hepatocyte EV treatment led to greater HSC iron than treatment with iron-free hepatocyte EVs (Figure 5D). In contrast, 4 wks of treatment with NASH hepatocyte EVs had little effect on HSC fibrosis and ferritin expression in lean WT mice (Figures 5E and S5D).
Previous studies showed that the KC population is recovered after switching WT mice from WD diet to a normal chow diet (diet-switch) (Tran et al., 2020). Accordingly, we observed elevated KC number 9 wks after diet-switch (Figure 5B). More importantly, diet-switch reduced HSC total iron and ferritin abundance (Figures 5F and 5G). Taken together, these data demonstrate that KCs play a critical role in preventing the transport of iron-containing hepatocyte EVs into HSCs.
Iron accumulation in HSCs enhances fibrogenic activation.
We observed that, concomitant with iron accumulation, fibrogenic gene expression was elevated in HSCs after 6 wks WD feeding (Figure S6A). To further assess HSC responses after iron overload, we analyzed the transcriptomic profiles in lean mouse HSCs after in vitro treatment with FAC (100 μM/1×106 cells). Among the genes upregulated by iron overload, many were associated with inflammatory responses, the cellular response to TGFβ stimulus, and collagen processing (Figure 6A), categories involved in NASH development. Since HSC iron overload is observed in both human and mouse NASH, we next evaluated whether iron enrichment exacerbates HSC fibrogenic activation and liver fibrosis. To obtain iron levels like those in NASH HSCs, FAC or holo-transferrin were added into the culture medium of healthy human and lean mouse HSCs (Figures S6B and S6C). We observed that iron accumulation resulted in greater levels of fibrogenic gene expression in human and mouse HSCs, compared to control cells (Figures 6B, S6D, and S6E). In contrast, treatment with DFO or Apo-transferrin attenuated fibrogenic activation in human and mouse NASH HSCs (Figures 6C, S6F, and S6G). To further test whether reduced HSC iron could blunt the development of liver fibrosis, 20 wks WD fed WT mice were treated with DFO (20 mg/Kg body weight, i.p. injection, twice per week). After 4 wks of treatment, we observed that DFO-treated mice had reduced liver fibrosis, including lower intrahepatic collagen in DFO-treated livers revealed by Sirius Red staining and lower expression of genes associated with fibrogenic activation (Figures 6D and S6H). We also observed that, after 2 wks injection of lentivirus carrying gRNA-Fpn, lean LratCre+Cas9+ mice expressed greater levels of ferritin and fibrogenic genes than lean WT control mice injected with the lentivirus without gRNA (Figures 6E and S6I). In addition, 12 wks WD fed FpnΔHSC mouse livers contained readily detectable intrahepatic collagen, whereas barely observed in 12 wks WD WT livers (Figure 6F). 12 wks WD fed FpnΔHSC mouse livers also expressed greater α-SMA levels than control mice (Figure S6J).
Figure 6. Iron overload triggers fibrogenic activation in HSCs.

(A) Pathways in lean mouse HSCs were significantly upregulated after treatment with FAC.
(B) Effect of FAC treatment (100 μM) on the expression of key genes associated with fibrogenic activation in healthy human HSCs.
(C) The abundance of fibrogenic genes in human NASH HSCs after 24 hours treatment with DFO (100 μM).
(D) Sirius Red staining of liver sections of 24wks WD WT and DFO-treated WT mice. Representative images of n=6 mice per group.
(E) Expression of Fth and fibrogenic genes in HSCs of lean LratCre+Cas9+ mice after 2wks injection with lentivirus carrying gRNA-Fpn.
(F) Sirius Red staining of liver sections of 12wks WD control vs. FpnΔHSC mice.
(G) Expression of fibrogenic genes in HSCs isolated from lean WT and KC-KO mice.
HSC total iron levels (H) and liver fibrosis (I) of 24wks WD WT and Rab27ΔHep mice. Representative images of n=5–6 mice per group.
(J) HSC fibrogenic activation in lean KC-KO mice after 4wks injection with either NASH Hep EVs or iron-free NASH Hep EVs.
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test. See also Figure S6.
In the KC-KO mouse model, concomitant with increased HSC iron, the levels of fibrogenic gene expression were higher than in lean WT liver (Figure 6G). To further demonstrate that hepatocyte EVs contribute to HSC iron and subsequent fibrogenic activation, Rab27ΔHep mice were fed WD. After 24 wks, loss of hepatocyte RAB27 prevented HSC iron accumulation, concomitant with lower levels of fibrogenic activation than WT mice (Figures 6H and S6K). Moreover, Sirius Red staining was significantly lower in 24 wks WD fed Rab27ΔHep mice (Figure 6I). Conversely, treatment with NASH hepatocyte EVs increased fibrogenic gene expression in HSCs of lean KC-KO mice compared to treatment with iron-free EVs (Figure 6J), suggesting that iron is the key EV cargo mediating the effect of NASH hepatocyte EVs on fibrogenic activation. In vitro experiments with lean WT mouse HSCs showed that the addition of NASH hepatocyte EVs enhanced fibrogenic activation to a greater extent than iron-free EVs (Figure S6L). Overall, these results demonstrate that iron accumulation triggers HSC fibrogenic activation.
Iron accumulation-induced HSC fibrogenic activation is ROS-dependent.
Iron is incorporated into ROS-producing enzymes, including subunits of the mitochondrial electron transport chain, which are tightly associated with oxidative stress (Dixon and Stockwell, 2014). Indeed, gene ontology analysis of our RNA-seq data revealed that top enriched pathways in significantly changed genes were related to oxidative stress (Figure 6A). As expected, ROS levels were elevated in healthy human and lean mouse HSCs after treatment with FAC (Figures 7A and S7A). Concomitant with iron overload, NASH mouse HSCs contained more ROS than lean WT HSCs (Figure S7B). Interestingly, Rab27ΔHep NASH mice had lower HSC ROS levels than HSC of WT NASH mice (Figure 7B), suggesting that hepatocyte EVs increase ROS accumulation in HSC. To further validate that iron is a key hepatocyte EV cargo needed for ROS production in HSCs, lean KC-KO mice were i.v. injected with either NASH hepatocyte EVs or iron-free EVs. After 4 wks of treatment, the HSCs of lean KC-KO mice treated with NASH hepatocyte EVs had higher ROS contents than HSC from iron-free EV-treated KC-KO mice (Figure 7C), thus demonstrating the importance of iron for the effect of hepatocyte EVs on HSC ROS production. To examine whether iron-induced fibrogenic activation is ROS-dependent, healthy human and lean mouse HSCs were treated with the combination of FAC and NAC (N-acetyl-cysteine; an anti-oxidant). We found that FAC failed to induce fibrogenic activation in the presence of NAC (Figures 7D and S7C), suggesting that ROS accumulation is critical for iron-induced fibrogenic activation.
Figure 7. Iron enrichment induces NASH-associated ROS accumulation in HSCs.

(A) Effect of FAC treatment on ROS content in healthy human HSCs.
(B) ROS abundance in HSCs isolated from 24wks WD fed WT and Rab27ΔHep mice.
(C) Effect of Hep EV treatment on HSC ROS levels in lean KC-KO mice.
(D) Effect of ROS chelator treatment on the expression of fibrogenic genes in FAC-treated healthy human HSCs.
HSC ROS content (E), fibrogenic gene abundance (F), and liver Sirius Red staining (G) after switching NASH WT mice to NCD diet for 9wks. Representative images of n=4–6 mice per group.
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t-test (A, B, D–G) or One-way ANOVA (C). See also Figure S7.
Weight loss is one of the current treatments for NAFLD/NASH patients (Promrat et al., 2010; Vilar-Gomez et al., 2015). In diet-switch WT mice, concomitant with reduced HSC iron levels, we observed a significant reduction in HSC ROS content and fibrogenic activation (Figures 5F and 5G, 7E and 7F). Consistently, these diet-switch WT mice exhibited lower levels of liver steatosis and fibrosis, compared to NASH WT controls (Figures 7G and S7D).
Discussion
Tight associations between impaired iron homeostasis and human and mouse liver metabolic disorders and fibrosis have been reported (Britton et al., 2016). The essential role of hepatocytes in maintaining liver iron balance underscores a critical need to investigate the impact of hepatocyte iron homeostasis on the pathogenic processes that underlie NAFLD and NASH progression. In this study, we find that human and mouse NAFLD/NASH hepatocytes are iron deficient, due to enhanced secretion of iron containing EVs. RAB27, which is a key regulator of EV secretion, is highly expressed in NAFLD/NASH hepatocytes and its ablation restores liver iron homeostasis and attenuates NASH progression. Transcriptomic analysis has shown that hepatocytes with iron deficiency have a greater lipid synthesizing ability and impaired insulin responsiveness linked to activation of HIF2α/ATF4 signaling. Restoration of iron content in NAFLD hepatocytes by iron dextran treatment or Rab27 ablation significantly reduced lipid accumulation and improved insulin sensitivity. In addition, taking up hepatocyte EVs leads to HSC iron overload, enhancing ROS accumulation and fibrogenic activation. Blocking hepatocyte EV secretion prevents HSC iron overload, concomitant with reduced liver fibrosis in WD-fed mice. KCs are also important players in hepatocyte to HSC iron transfer by clearing hepatocyte EVs. The marked reduction in the KC population during NAFLD/NASH results in elevated EV mediated iron transport into HSCs.
Hepatocytes are key regulators of iron metabolism, but the impact of iron deficiency on hepatocyte physiology is poorly understood. Our transcriptomic analysis revealed that iron-deficient hepatocytes exhibit marked alterations in pathways associated with lipogenesis and cellular insulin sensitivity. Hepatocyte iron deficiency resulted in activation of HIF2α signaling, which is already known to play critical roles in the development of NAFLD/NASH (Morello et al., 2018). We found that ATF4, a HIF target gene, was responsible for HIF2α-induced hepatocyte dysfunction. Consistent with previous studies (Li et al., 2016; Xiao et al., 2013), hepatocyte-specific ATF4 ablation mitigated hepatosteatosis in WD-fed mice. Moreover, restoration of hepatocyte iron blunted HIF2α/ATF4 axis activities in NAFLD hepatocytes. These findings support the hypothesis that hepatocyte iron deficiency is a key pathogenic factor contributing to NAFLD/NASH progression.
FPN-dependent iron export is the main pathway by which hepatocytes release iron (Drakesmith et al., 2015). However, in the inflamed liver, hepcidin can directly block FPN function (Drakesmith et al., 2015). In NAFLD patients circulating hepcidin levels are significantly greater than in healthy individuals (Auguet et al., 2017; Demircioglu et al., 2014) and we found that circulating hepcidin was elevated after WD feeding, concomitant with the reduction in hepatocyte FPN abundance. However, in our studies, FPN deficiency did not cause iron overload in human and mouse NAFLD hepatocytes. In addition, Zhang et al. reported that Fpn ablation in hepatocytes has minimal effects on hepatocyte iron content and systemic iron homeostasis (Zhang et al., 2012). We observed that NAFLD hepatocytes contained about ~30% less iron than normal healthy hepatocytes, suggesting that FPN-independent pathways mediate hepatocyte iron export during NAFLD/NASH development. Recent studies show that cancer cells can expulse iron through EV secretion (Brown et al., 2019). In addition, in previous studies, including ours (Ji et al., 2021; Zhao et al., 2020), obesity is accompanied by remarkably elevated hepatocyte EV secretion. To test whether EVs are important for hepatocytes iron export, we used mice with hepatocyte-specific ablation of RAB27, a key regulator of EV secretion, and found that hepatocyte iron content of WD-fed Rab27ΔHep mice was similar to that of lean mouse hepatocytes. Consistent with our findings, it was reported that macrophages can secrete iron-containing EVs and that RAB27 depletion reduces iron export (Truman-Rosentsvit et al., 2018). However, it is unclear whether RAB27 proteins may have effects on other secretory pathways besides extracellular vesicles, which could affect hepatocyte iron homeostasis.
We also found that healthy hepatocytes secreted a lower amount of iron-containing EVs compared to NAFLD/NASH hepatocytes. In addition, KCs remove hepatocyte iron-containing EVs in healthy liver, in line with their roles in maintaining liver microenvironmental iron homeostasis (Krenkel and Tacke, 2017). In contrast, the KC population is dramatically reduced in both human and mouse NAFLD/NASH liver, and transcriptomic analysis indicates that the NAFLD/NASH liver environment impairs KC functions such as the ability of iron recycling and phagocytosis (Seidman et al., 2020). In our studies, we used the KC-KO mouse model and found that iron overload occurred in hepatocyte neighboring cells such as HSCs after the loss of KCs. In addition, NASH hepatocyte EV treatment led to iron accumulation in HSCs in KC-KO mice, whereas administration of iron-free NASH hepatocyte EVs had minimal effects on HSC iron content in KC-KO mice. Thus, our findings support a critical role for KCs in preventing iron overload in hepatic cells.
Concomitant with elevated secretion of iron-containing hepatocyte EVs, HSCs in NAFLD afflicted liver exhibited elevated iron content. Previous studies observed iron accumulation in reticuloendothelial system cells in human NAFLD livers and correlated it with increased fibrosis (Nelson et al., 2011). Hereditary hemochromatosis patients who carry genetic mutations disrupting hepcidin production exhibit iron overload in different hepatic cells and a high risk for development of liver fibrosis (Britton et al., 2016; Falize et al., 2006; Valenti et al., 2010). Consistent with these observations, we found that iron overload resulted in fibrogenic activation in human and mouse HSCs. In addition, iron chelator treatment or depletion of RAB27-mediated hepatocyte EV secretion attenuated HSC fibrogenic activation and reduced liver fibrosis. It is well documented that iron accumulation leads to elevated ROS production, and excessive amounts of ROS in HSCs drive NASH fibrosis (Rolo et al., 2012). In summary, we find that maintenance of iron homeostasis is critical for hepatocyte metabolic responses and that hepatocytes affect neighboring cell functions through the secretion of iron-containing EVs. In contrast, impairment of hepatocyte iron homeostasis leads to iron distribution disorders, which contribute to NAFLD/NASH pathogenesis.
Limitations of study
These studies were performed in mice and human hepatocytes. It remains to be seen whether comparable results will be obtained in human studies. This study demonstrates the importance of iron homeostasis in hepatocytes and HSCs, but follow-up clinical studies are needed to determine how to adjust iron levels in these cells and whether that can be used to prevent and ameliorate NAFLD/NASH patients. In addition to HSCs, iron-containing hepatocyte EVs could spread into other cells where they can lead to iron overload and cellular abnormalities, which may also contribute to the development of liver diseases.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be direct to and will be fulfilled by the Lead Contact, Wei Ying (weying@health.ucsd.edu).
Material Availability
This study did not generate new unique reagents or materials. Most mouse lines in this study are available from the Jackson Laboratory. ATF4 flox/flox mice are maintained at Dr. Michael Karin’s laboratory and available for transfer request with a completed Material Transfer Agreement.
Data and Code Availability
RNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE185694.
This paper does not report original code.
Original data for creating all graphs in the paper are provided in Data S1. The RNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE185694.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice.
C57BL/6J (B6) mice were fed a AIN76A Western Diet (TestDiet® 5342; containing 50ppm iron) or a normal chow diet (TestDiet® 5TJS; containing 41ppm iron) ad libitum. After 12 wks WD feeding, mice developed hepatosteatosis phenotypes, and liver fibrosis was present after 24wks WD feeding. To generate hepatocyte-specific Cas9 transgenic mice, Rosa26-floxed STOP-Cas9 knockin mice were bred with Albumin-Cre mice. To generate hepatocyte-specific knockout of Rab27 or Tfrc1, hepatocyte-specific Cas9 transgenic mice were intravenously injected with lentivirus carrying gRNA-Rab27 or Tfrc1 (1×108 particles per mouse). Hepatocyte-specific Cas9 transgenic mice treated with lentivirus carrying empty vectors were used as controls. LratCre+Cas9+ mice were injected with lentivirus carrying gRNA-Fpn (1×108 particles per mouse) to generate hepatic stellate cell-specific Fpn knockout mice. To deplete Kupffer cells, diphtheria toxin (DT) was intraperitoneally (200 ng/mouse, i.p.) injected into Clec4fCre+DTR+ lean mice (KCKO) for three days, and these mice were treated with DT (200 ng/mouse) every two days to prevent KC recovery. All mice used in this study were male and maintained at 22°C in a 12/12-h light/dark cycle in a specific pathogen-free facility and given free access to food and water.
Study approval.
All animal procedures were done in accordance with University of California, San Diego Research Guidelines for the Care and Use of Laboratory Animals, and all animals were randomly assigned to cohorts when used.
Human specimens.
Deidentified livers declined for transplantation was used in the experiments of this paper, the patients’ consents were obtained by www.lifesharing.org. The studies in this paper have been reviewed by the Director of the UCSD HRPP, IRB Chair, or IRB Chair’s designee and is certified as not qualifying as human subjects research according to the Code of Federal Regulations, Title 45, part 46 and UCSD Standard Operating Policies and Procedures, and thus does not require IRB review. The levels of liver steatosis, inflammation, and fibrosis were evaluated by a pathologist using a double-blinded method and identified as NAFLD/NASH or normal.
METHOD DETAILS
Isolation of primary hepatocytes.
The method for primary hepatocyte isolation has been described previously (Ji et al., 2021). Mice were infused through vena cava with a buffer A (calcium and magnesium-free PBS containing 10 mM HEPES, 0.2 μM EGTA, 0.2% BSA, and 1 mM glucose, pH 7.4) for 3–5 min. After liver appeared beige or light brown color, the perfusion media was changed to collagenase-containing buffer B (PBS containing 1 mM calcium, 1 mM magnesium, 0.2% BSA, 30 mM HEPES, and 0.5 mg/mL collagenase D). After the liver surface showed signs of cracking, the liver was transferred into the ice-cold buffer A. Cells from the digested livers were then suspended in buffer A, passed through a 100-μm cell strainer, and spun at 50 × g for 6 min at 4 °C. The pellet was washed twice with buffer B (without collagenase) and then added with Percoll to a final concentration of 36% and centrifuged at 100 × g for 10 min, 4°C. The hepatocyte pellet was washed once with buffer B and finally resuspended in Williams Medium E containing 10% FBS on collagen-coated plates and antibiotics.
Isolation of hepatic stellate cell (HSC).
Mouse liver was sequentially perfused through vena cava first with 30~40 mL of calcium-free HEPES-phosphate buffer A, next with 30 mL of 0.5mg/mL pronase buffer, and finally with 30 mL of 0.5 mg/mL collagenase D containing buffer. After digestion of the clipped liver tissue in a buffer containing DNase I of collagenase D and pronase for 20 minutes, the cell suspension was centrifuged at 50 × g for 3 min to remove the residual liver parenchymal cells. The supernatant was collected and spun at 900 × g for 8 min (4°C) to pellet the non-parenchymal cells. The collected non-parenchymal cells were resuspended in 15% OptiPrep buffer and transferred to a 15 mL tube after sufficient mixing. Then, 5 mL of 11.5% OptiPrep and 2 mL of Gey’s Balanced Salt Solution (GBSS; Sigma-Aldrich, St. Louis, MO) were individually layered carefully onto the cell suspension. The tube was centrifuged at 2000 × g for 20 min (4°C, low acceleration), and the HSC layer between the 11.5% OptiPrep and GBSS was carefully collected. These cells were washed twice with centrifugation dilution solution (800 × g for 10 min, 4°C) and seeded with culture medium (1% Glutax, 10% FBS, and 2% penicillin/streptomycin) at 37°C.
Lipogenesis assay.
Incubation conditions: HEPES-Krebs-Ringer-bicarbonate buffer with 1 mM glucose, 0.2% BSA, 1 mM sodium acetate, and 1–14C-sodium acetate (10 uCi/mL). 1 mL of radioactive buffer per well in 6-well plates. After 2 hours incubation time in a cell incubator, the buffer was aspirated, and wells were washed with cold PBS. 0.5 mL of methanol was added to each well and kept swirling for 5 minutes. Methanol extracts were collected into microfuge tubes. 0.5 mL of methanol and chloroform (1:1) mixture was added to each well. After swirled for 5 minutes, solvents from these wells were transferred to the microfuge tubes containing methanol. All tubes were vortexed. Next, 0.5 mL of PBS was added to each tube, then vigorously vortexed. Tubes were centrifuged at 10,000 × g for 10 minutes. Two layers were separated. The lower chloroform layer from each tube was carefully transferred to scintillation vials. After solvents evaporated in a hood, a scintillation cocktail was added to each vial and counted for 14C-radioactivity. Plates were allowed to dry up inside the hood, and then 0.5 mL of 1 M NaOH was added to each well to dissolve cells. Plates were shaked vigorously for 20 mins. These NaOH extracts were used for protein assay.
Triglyceride detection.
Triglyceride levels were detected using the Triglyceride Colorimetric Assay kit according to the manufacturer’s instructions.
Glucose tolerance (GTT) and insulin tolerance tests (ITT).
Glucose tolerance tests were performed by intraperitoneally (i.p.) injecting dextrose (1 g/Kg body weight) into mice after 16 hr of fasting. After 6 hr fasting, insulin tolerance tests were performed by i.p. injecting insulin solution (0.35 or 0.175 units per kg of body weight for Western diet or NCD-fed mice, respectively).
Iron detection.
Iron in the cells was detected by using Iron Assay Kit (Sigma Aldrich) as protocol indicated. Then the iron contents were normalized to protein levels or tissue weight.
Extracellular vesicle (EV) purification and characterization.
As previously described (Ji et al., 2021), primary hepatocytes were isolated from either lean or NASH mice and cultured on 100mm collagen-coated plates (5 × 106 cells/plate) with 10–15 mL of William’s E medium containing 10% exosome-free FBS. After 24h of culture, medium was collected, and debris and dead cells were discarded by centrifugation at 1,000g for 10 min at 4 °C followed by filtration through a 0.2 μm filter. Then the medium was subjected to ultracentrifugation at 100,000g for 4h at 4 °C (Beckman Optima XPN with type 70 Ti fixed-angle rotor). After washing with PBS (100,000 × g for 20 min), the EV containing pellet was resuspended in 1 mL of PBS and filtered through a 0.2 μm filter to remove large particles. Vesicle particle size and concentration were measured using NanoSight analysis (Malvern Instruments). To deplete iron cargo (iron-free EVs), hepatocyte EVs (1~5×1010) were incubated with DFO (200 μM) for 4 hours on ice and then washed with PBS (100,000 × g for 20 min), and finally the EV concentration was measured using NanoSight analysis.
In vivo EV trafficking assays.
As previously described (Ying et al., 2021), lean mouse primary hepatocytes were transferred with Cy3 conjugated miR-223 mimic (100 pmol for 5 × 106 cells) with RNAiMAX transfection reagents. After 24 hours, culture medium was replaced with William’s E medium containing 10% exosome-free FBS. After another 24 hours culturing, Cy3-miR-223 containing EVs were prepared from the culture medium as described above and injected into the tail vein of lean WT recipient mice (5×109 EVs per mouse). After 12 hours, the abundance of Cy3 signal in Kupffer cells was measured by flow cytometry analysis.
In vivo and in vitro EV treatment.
For in vitro assays, 1×108 EVs as determined by NanoSight analysis were used to treat 0.5×106 cells for 36 h. For in vivo treatment, 5×109 EVs were injected into the tail vein of recipient mice (twice per week). The control groups were treated with empty liposomes or iron-free EVs in the in vitro and in vivo experiments.
In vivo and in vitro iron overload or deficiency experiments.
For in vitro experiments, cells were treated with ferric ammonium citrate (FAC) or deferoxamine (DFO) (100 μM/1×106 cells) in DMEM medium (containing 0.25 μM ferric nitrate) to mimic iron overload or deficiency, respectively. Alternatively, cells were treated with apo-transferrin or holo-transferrin (3 mg/mL for 1×106 cells) to induce iron deficiency or overload, respectively. To restore iron levels in vivo, iron dextran (10 mg/Kg body weight, twice per week) was injected intraperitoneally to 6wks Western diet-fed mice for 6 weeks. 20wks Western diet-fed mice were treated with DFO (20 mg/Kg body weight, i.p. injection, twice per week) for 4 weeks to reduce HSC iron levels.
siRNA transfection.
Recipient cells were transfected with siRNA (20 pmol siRNA/0.1×106 cells) using the RNAiMAX reagent. Control cells were treated with a non-targeting scramble siRNA. After mixing siRNAs with the RNAiMAX reagent and then incubating for 15 min at room temperature, the mixture was then added to the cell media.
Immunofluorescence staining assays.
Mouse livers were frozen in Optimum Cutting Temperature (Fisher Healthcare) with dry ice. Six-micrometer cryo-sections of liver slides were prepared and fixed with pre-cooled acetone for 20 mins. Immunostaining assays were performed as previously described (Luo et al., 2021). Slides were blocked with 5% normal donkey serum for 60 mins at room temperature. The slides were incubated with anti-ferritin, anti-desmin, or anti-α-SMA antibodies diluted in 1:100 in PBS at 4°C overnight. After washing, nuclei were stained for 10 mins with DAPI (4,6-diamidino-2-phenylindole) at room temperature. Finally, mounting media and coverslips were added to the slides. Images were acquired on a Keyence Fluorescent Microscope and were analyzed with the ImageJ (NIH).
ROS detection.
Reactive oxygen species (ROS) were measured using ROS-ID Total ROS/Superoxide Detection Kit (Enzo Life Sciences, ENZ-51010). Cells were seeded at 500,000 cells per well in 50 μL of resuspension medium into clear 96-well plates with black chimneys. The detection method was followed by the fluorescence microplate assay (adherent cells) protocol provided.
Gene expression analysis.
Total RNAs were extracted from tissues using an RNA extraction protocol according to the manufacturer’s instructions (Zymo Research). Complementary DNA was synthesized using SuperScript III and random hexamers. qPCR was performed in 10 μl of reaction using iTaq SYBR Green supermix on a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). The data are presented as the mean of 2-ΔΔCt from at least three independent experiments and normalized to β-actin or RPL13.
Western blot analysis.
Tissues or cells were homogenized in a RIPA buffer containing protease and phosphatase inhibitors. Equal amounts of cell lysate protein (30–40 μg of protein per lane) from each biological replicate were analyzed by western blotting assays. Using a ChemiDoc XRS imaging system (BioRad), protein bands on the blots were detected with the SuperSignal West Pico Chemiluminescent Substrate. The signal intensities of protein bands were analyzed using Image Lab software (BioRad) or ImageJ (NIH). The western blot data shown in the figures are all representative of more than three independent experiments.
RNA-seq library preparation and sequencing.
Total RNA was isolated from chow diet hepatocytes treated with DFO and chow diet HSCs treated with FAC. RNA purity was evaluated by an Agilent 2100 Bioanalyzer. Sequencing libraries were prepared using an mRNA Library Prep Kit (Illumina) according to the manufacturer’s protocol. Samples were ligated to unique adaptors and subjected to PCR amplification. Libraries were then validated using an Agilent 2100 Bioanalyzer, normalized, and pooled for sequencing processes. RNA-seq libraries prepared from biological replicates for each group as indicated were sequenced on an Illumina NovaSeq 6000 using barcoded multiplexing. The RNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession number: GSE185694.
Quantification and statistical analysis.
Samples were blinded whenever deemed appropriate and applicable. Sample description and identification were not available to the core laboratory personnel during data plotting and analyses. No samples or data were excluded from the study for statistical purposes. Each in vitro experiment was independently performed in duplicate or triplicate to ensure reproducibility. Group sizes of five mice or greater were sufficient to reach statistical power of at least 80%. Animals were assigned randomly to treatment groups for all studies. The tests used for statistical analyses are described in the figure legends. To determine whether the means of two groups are statistically different from each other, unpaired two-tailed Student’s t-tests were used for statistical analyses. All data passed the normality test using Prism8 software (GraphPad software v.8.0, Prism). P values of 0.05 or less were considered statistically significant. For the glucose and insulin tolerance tests, statistical comparisons between every two groups at each time point were performed with the unpaired two-tailed Student’s t-test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HIF2α | ABclonal | Cat# A7553; RRID: AB_2768078 |
| Anti-ATF4 | ABclonal | Cat# A18687; RRID: AB_2862422 |
| Anti-Rab27a | Cell signaling technology | Cat# 69295; RRID:AB_2799759 |
| Anti-Rab27b | ABclonal | Cat# A8897; RRID: AB_2771907 |
| Anti-HSP90 | Santa Cruz | Cat# sc-101494; RRID: AB_1124018 |
| Anti-Phospho-AKT Ser473 | Cell signaling technology | Cat# 9271S; RRID: AB_329825 |
| Anti-pan AKT | Cell signaling technology | Cat# 4691S; RRID: AB_915783 |
| Anti-FPN | ABclonal | Cat# A14884; RRID: AB_2761764 |
| Anti-FtH | ABclonal | Cat# A1144; RRID: AB_2758562 |
| Anti-Cd11b-FITC | BioLegend | Cat# 101205; RRID: AB_312788 |
| Anti-F4/80-PE Cy7 | BioLegend | Cat# 123113; RRID: AB_893490 |
| Anti-Cd45-APC Cy7 | BioLegend | Cat# 103115; RRID: AB_312980 |
| Anti-Desmin, eFluor 660 | eBioscience | Cat# 50-9747-80; RRID: AB_2574355 |
| Anti-phospho-eIF2a | Cell signaling technology | Cat# 3398; RRID: AB_2096481 |
| Anti-α-SMA (for immunofluorescent staining assays) | eBioscience | Cat# 14-9760-82; RRID: AB_2572996 |
| Anti-α-SMA (for Western blots) | Sigma Aldrich | Cat# A5228-25UL; RRID: AB_262054 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Live/Dead Fixable Aqua dead cell stain kit | ThermoFisher | Cat# L34966 |
| Novolin R regular human insulin used in ITTs | Novo-Nordisk | Cat# NDC 0169-1833-11 |
| Sodium acetate (1-14C) | APC0101A-50 μCi | American Radiolabeled Chemicals, Inc |
| Dextrose | Hospira, Inc | Cat# 0409-6648-02 |
| DT (Diphtheria Toxin) | Bio Academia | Cat# 01-517 |
| Percoll | GE Healthcare Life Sciences | Cat# 17-0891-01 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | ThermoFisher Scientific | Cat# 34095 |
| Halt Protease and Phosphatase Inhibitor Cocktail | ThermoFisher Scientific | Cat# 78440 |
| RIPA buffer (10x) | Cell Signaling Technology | Cat# 9806 |
| AIN76A Western Diet | TestDiet | Cat# 5342 |
| Collagenase D | Roche | Cat# 11088882001 |
| Pronase | Roche | Cat# 11459643001 |
| DNAnase I | Roche | Cat# 10104159001 |
| RBC lysis buffer | eBioscience | Cat# 00-4333-57 |
| High-capacity cDNA reverse transcription kit | ThermoFisher Scientific | Cat# 4368813 |
| iTaq SYBR Green supermix | Bio-Rad | Cat# 172-5125 |
| Exosome-depleted FBS | SBI | Cat# EXO-FBSHI-50A-1 |
| Lipofectamine RNAiMAX reagent | ThermoFisher Scientific | Cat# 13778-075 |
| Empty liposome | FormuMax | Cat# F70101-NH |
| Iron-Dextran | Sigma-Aldrich | Cat# D8517 |
| FAC (Ammonium iron citrate) | Sigma-Aldrich | Cat# F5879 |
| DFO (Deferoxamine mesylate salt) | Sigma-Aldrich | Cat# D9533 |
| Holo-transferrin | Sigma-Aldrich | Cat# T0665 |
| Apo-transferrin | Sigma-Aldrich | Cat# T5391 |
| Palmitic acid | Sigma-Aldrich | Cat# P5585 |
| Critical Commercial Assays | ||
| Quick-RNA microprep kit | Zymo research | Cat#R1051 |
| Triglyceride colorimetric assay kit | Cayman | Cat# 10010303 |
| Iron Assay Kit | Sigma Aldrich | MAK025 |
| Mouse Hepcidin ELISA Kit | BioVision | E4693-100 |
| ROS-ID Total ROS/Superoxide Detection Kit | Enzo Life Sciences | ENZ-51010 |
| Deposited Data | ||
| RNA seq data | GEO database | GSE185694 |
| Experimental Models: Organisms/Strains | ||
| Mouse: WT C57BL6/J | Jackson Laboratories | JAX: 000664 |
| Mouse: ATF4 flox/flox | Michael Karin lab | JAX: 006366 |
| Mouse: Rosa26-floxed STOP-Cas9 | Jackson Laboratories | JAX: 026175 |
| Mouse: Albumin cre | Jackson Laboratories | JAX: 003574 |
| Mouse: B6-iDTR, ROSA26iDTR | Jackson Laboratories | JAX: 007900 |
| Mouse: C57BL/6-Lratem1(2A-Cre)Smoc | Shanghai Model Organisms | NM-KI-190097 |
| Mouse: Clec4f-Cre-tdTomato | Jackson Laboratories | JAX: 033296 |
| Oligonucleotides | ||
| siRNA control | Horizon Discovery | Cat# D-001810-01-05 |
| siRNA-human Hif2a | Horizon Discovery | Cat# J-004814-06-0002 |
| siRNA-human ATF4 | Horizon Discovery | Cat# J-005125-10-0002 |
| siRNA-mouse Hif2a | Horizon Discovery | Cat# J-040635-09-0002 |
| siRNA-mouse Rab27a | Horizon Discovery | Cat# J-060970-09-0002 |
| siRNA-mouse Rab27b | Horizon Discovery | Cat# J-050808-09-0002 |
| siRNA-human Rab27a | Horizon Discovery | Cat# J-004667-05-0002 |
| siRNA-human Rab27b | Horizon Discovery | Cat# J-004228-05-0002 |
| Cy3-labeled miR-223 mimic | GE Dharmacon | Cat# SO-2465930G |
| gRNA-Rab27a plasmid | Sigma Aldrich | Sanger Clone ID: MM5000028879 |
| gRNA-Rab27b plasmid | Sigma Aldrich | Sanger Clone ID: MM5000025441 |
| gRNA-Tfrc1 plasmid | Sigma Aldrich | Sanger Clone ID: MMPD0000041027 |
| gRNA-Fpn plasmid | Sigma Aldrich | Sanger Clone ID: MMPD0000046431 |
| Software and Algorithms | ||
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | FlowJo | https://www.flowjo.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Image Lab | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
Highlights.
NAFLD/NASH livers present iron-deficient hepatocytes and iron overloaded HSCs
Hepatocyte iron deficiency enhances lipogenesis and insulin resistance via HIF2α-ATF4
Hepatocyte EVs shuttle iron into HSCs in NAFLD/NASH
Iron overload stimulates HSC ROS production and fibrogenic activation
Acknowledgments
We thank the UCSD IGM Genomics Center for RNA sequencing, the UCSD microscopy core for microscopic analysis, the UCSD Moore Cancer Center histology core for H&E and Sirius Red staining, and the Vector Development Core Laboratory for lentivirus preparation. This study was funded by National Institutes of Health grants (S10 OD026929 to UCSD IGM core), (NINDS P30 NS047101 to UCSD microscopy core), (R01DK101737, U01AA022614, R01DK099205, R01DK11866, R01DK099205 to T.K.), (P50AA011999 to T.K. and D.A.B.), (R01DK120714, R01CA234128, and P42ES010337 to M.K.), and (P30 DK063491, R00DK115998, and R01DK125560 to W.Y.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Data S1. Unprocessed source data underlying all blots and graphs. Related to Figures 1–7 and Supplemental Figures 1–7
References
- Abboud S, and Haile DJ (2000). A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 275, 19906–19912. [DOI] [PubMed] [Google Scholar]
- Auguet T, Aragones G, Berlanga A, Martinez S, Sabench F, Binetti J, Aguilar C, Porras JA, Molina A, Del Castillo D, et al. (2017). Hepcidin in morbidly obese women with non-alcoholic fatty liver disease. PLoS One 12, e0187065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner J, Smuts CM, Aeberli I, Malan L, Tjalsma H, and Zimmermann MB (2013). Overweight impairs efficacy of iron supplementation in iron-deficient South African children: a randomized controlled intervention. Int J Obes (Lond) 37, 24–30. [DOI] [PubMed] [Google Scholar]
- Bobrie A, Krumeich S, Reyal F, Recchi C, Moita LF, Seabra MC, Ostrowski M, and Thery C (2012). Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res 72, 4920–4930. [DOI] [PubMed] [Google Scholar]
- Brissot P, Pietrangelo A, Adams PC, de Graaff B, McLaren CE, and Loreal O (2018). Haemochromatosis. Nat Rev Dis Primers 4, 18016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton L, Bridle K, Reiling J, Santrampurwala N, Wockner L, Ching H, Stuart K, Subramaniam VN, Jeffrey G, St Pierre T, et al. (2018). Hepatic iron concentration correlates with insulin sensitivity in nonalcoholic fatty liver disease. Hepatol Commun 2, 644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton LJ, Subramaniam VN, and Crawford DH (2016). Iron and non-alcoholic fatty liver disease. World J Gastroenterol 22, 8112–8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, Baer CE, Dixon SJ, and Mercurio AM (2019). Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev Cell 51, 575–586 e574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demircioglu F, Gorunmez G, Dagistan E, Goksugur SB, Bekdas M, Tosun M, Kizildag B, and Kismet E (2014). Serum hepcidin levels and iron metabolism in obese children with and without fatty liver: case-control study. Eur J Pediatr 173, 947–951. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, and Stockwell BR (2014). The role of iron and reactive oxygen species in cell death. Nat Chem Biol 10, 9–17. [DOI] [PubMed] [Google Scholar]
- Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, et al. (2000). Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403, 776–781. [DOI] [PubMed] [Google Scholar]
- Drakesmith H, Nemeth E, and Ganz T (2015). Ironing out Ferroportin. Cell Metab 22, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falize L, Guillygomarc’h A, Perrin M, Laine F, Guyader D, Brissot P, Turlin B, and Deugnier Y (2006). Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology 44, 472–477. [DOI] [PubMed] [Google Scholar]
- Friedman SL, Neuschwander-Tetri BA, Rinella M, and Sanyal AJ (2018). Mechanisms of NAFLD development and therapeutic strategies. Nat Med 24, 908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T (2019). Anemia of Inflammation. N Engl J Med 381, 1148–1157. [DOI] [PubMed] [Google Scholar]
- He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, Shalapour S, Seki E, Yost SE, Jepsen K, et al. (2013). Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 155, 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S, Hara H, and Ishizuka S (2018). Marginal iron deficiency enhances liver triglyceride accumulation in rats fed a high-sucrose diet. Biosci Biotechnol Biochem 82, 2140–2148. [DOI] [PubMed] [Google Scholar]
- Isaac R, Reis FCG, Ying W, and Olefsky JM (2021). Exosomes as mediators of intercellular crosstalk in metabolism. Cell Metabolism 33, 1744–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Luo Z, Gao H, Dos Reis FCG, Bandyopadhyay G, Jin Z, Manda KA, Isaac R, Yang M, Fu W, et al. (2021). Hepatocyte-derived exosomes from early onset obese mice promote insulin sensitivity through miR-3075. Nat Metab 3 1163–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, and LeBleu VS (2020). The biology, function, and biomedical applications of exosomes. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CW, Chang Y, Sung E, Shin H, and Ryu S (2012). Serum ferritin levels predict incident non-alcoholic fatty liver disease in healthy Korean men. Metabolism 61, 1182–1188. [DOI] [PubMed] [Google Scholar]
- Koditz J, Nesper J, Wottawa M, Stiehl DP, Camenisch G, Franke C, Myllyharju J, Wenger RH, and Katschinski DM (2007). Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood 110, 3610–3617. [DOI] [PubMed] [Google Scholar]
- Kowdley KV, Belt P, Wilson LA, Yeh MM, Neuschwander-Tetri BA, Chalasani N, Sanyal AJ, Nelson JE, and Network N.C.R. (2012). Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 55, 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama Y, and Brenner DA (2017). Liver inflammation and fibrosis. J Clin Invest 127, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenkel O, and Tacke F (2017). Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 17, 306–321. [DOI] [PubMed] [Google Scholar]
- Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, and Ratan RR (2008). ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med 205, 1227–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Xiao Y, Yu J, Xia T, Liu B, Guo Y, Deng J, Chen S, Wang C, and Guo F (2016). Liver-specific Gene Inactivation of the Transcription Factor ATF4 Alleviates Alcoholic Liver Steatosis in Mice. J Biol Chem 291, 18536–18546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Ji Y, Gao H, Gomes Dos Reis FC, Bandyopadhyay G, Jin Z, Ly C, Chang YJ, Zhang D, Kumar D, et al. (2021). CRIg(+) Macrophages Prevent Gut Microbial DNA-Containing Extracellular Vesicle-Induced Tissue Inflammation and Insulin Resistance. Gastroenterology 160, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu M, Martin-Jaular L, Lavieu G, and Thery C (2019). Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol 21, 9–17. [DOI] [PubMed] [Google Scholar]
- McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, et al. (2000). A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 5, 299–309. [DOI] [PubMed] [Google Scholar]
- McLean E, Cogswell M, Egli I, Wojdyla D, and de Benoist B (2009). Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public Health Nutr 12, 444–454. [DOI] [PubMed] [Google Scholar]
- Meynard D, Babitt JL, and Lin HY (2014). The liver: conductor of systemic iron balance. Blood 123, 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello E, Sutti S, Foglia B, Novo E, Cannito S, Bocca C, Rajsky M, Bruzzi S, Abate ML, Rosso C, et al. (2018). Hypoxia-inducible factor 2alpha drives nonalcoholic fatty liver progression by triggering hepatocyte release of histidine-rich glycoprotein. Hepatology 67, 2196–2214. [DOI] [PubMed] [Google Scholar]
- Muckenthaler MU, Rivella S, Hentze MW, and Galy B (2017). A Red Carpet for Iron Metabolism. Cell 168, 344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp-Arida A, Kowdley KV, and Nonalcoholic Steatohepatitis Clinical Research, N. (2011). Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 53, 448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, and Kaplan J (2004). Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093. [DOI] [PubMed] [Google Scholar]
- Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, and Ganz T (2003). Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 101, 24612463. [DOI] [PubMed] [Google Scholar]
- Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, et al. (2010). Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 12, 19–30; sup pp 11–13. [DOI] [PubMed] [Google Scholar]
- Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, and Karin M (2010). Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, Fava JL, and Wing RR (2010). Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 51, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolo AP, Teodoro JS, and Palmeira CM (2012). Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med 52, 59–69. [DOI] [PubMed] [Google Scholar]
- Ryan JD, Armitage AE, Cobbold JF, Banerjee R, Borsani O, Dongiovanni P, Neubauer S, Morovat R, Wang LM, Pasricha SR, et al. (2018). Hepatic iron is the major determinant of serum ferritin in NAFLD patients. Liver Int 38, 164–173. [DOI] [PubMed] [Google Scholar]
- Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, Bruni CM, Ouyang Z, Li RZ, Sun X, et al. (2020). Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 52, 1057–1074 e1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah YM, and Xie L (2014). Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology 146, 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcox JA, and McClain DA (2013). Iron and diabetes risk. Cell Metab 17, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Tang S, Han X, Jiang Z, Dong L, Liu C, Liang X, Dong J, Qiu C, Wang Y, et al. 2019. KIBRA controls exosome secretion via inhibiting the proteasomal degradation of Rab27a. Nat Commun 10, 1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel NU, El-Mallah C, Herter-Aeberli I, Bissani N, Wehbe N, Obeid O, and Zimmermann MB (2020). The effect of central obesity on inflammation, hepcidin, and iron metabolism in young women. Int J Obes (Lond) 44, 1291–1300. [DOI] [PubMed] [Google Scholar]
- Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gelineau A, Marcelin G, Magreau-Davy E, Ouhachi M, Lesnik P, et al. (2020). Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity 53, 627–640 e625. [DOI] [PubMed] [Google Scholar]
- Truman-Rosentsvit M, Berenbaum D, Spektor L, Cohen LA, Belizowsky-Moshe S, Lifshitz L, Ma J, Li W, Kesselman E, Abutbul-Ionita I, et al. (2018). Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 131, 342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tussing-Humphreys LM, Nemeth E, Fantuzzi G, Freels S, Guzman G, Holterman AX, and Braunschweig C (2010). Elevated systemic hepcidin and iron depletion in obese premenopausal females. Obesity (Silver Spring) 18, 1449–1456. [DOI] [PubMed] [Google Scholar]
- Valenti L, Fracanzani AL, Bugianesi E, Dongiovanni P, Galmozzi E, Vanni E, Canavesi E, Lattuada E, Roviaro G, Marchesini G, et al. (2010). HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 138, 905–912. [DOI] [PubMed] [Google Scholar]
- Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, and Muckenthaler MU (2007). STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 109, 353–358. [DOI] [PubMed] [Google Scholar]
- Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, Friedman SL, Diago M, and Romero-Gomez M (2015). Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 149, 367–+. [DOI] [PubMed] [Google Scholar]
- Xiao G, Zhang T, Yu S, Lee S, Calabuig-Navarro V, Yamauchi J, Ringquist S, and Dong HH (2013). ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J Biol Chem 288, 25350–25361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanoff LB, Menzie CM, Denkinger B, Sebring NG, McHugh T, Remaley AT, and Yanovski JA (2007). Inflammation and iron deficiency in the hypoferremia of obesity. Int J Obes (Lond) 31, 1412–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Gao H, Dos Reis FCG, Bandyopadhyay G, Ofrecio JM, Luo Z, Ji Y, Jin Z, Ly C, and Olefsky JM (2021). MiR-690, an exosomal-derived miRNA from M2-polarized macrophages, improves insulin sensitivity in obese mice. Cell Metab 33, 781–790 e785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB, Ofrecio JM, Wollam J, Hernandez-Carretero A, Fu WX, et al. (2017). Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 171, 372–+. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhang F, Guo X, An P, Tao Y, and Wang F (2012). Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology 56, 961–971. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Zhao MF, Jiang S, Wu J, Liu J, Yuan XW, Shen D, Zhang JZ, Zhou N, He J, et al. (2020). Liver governs adipose remodelling via extracellular vesicles in response to lipid overload. Nat Commun 11, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE185694.
This paper does not report original code.
Original data for creating all graphs in the paper are provided in Data S1. The RNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE185694.