

Abstract
Background
Ischemic coronary heart disease (IHD) is the leading cause of death worldwide. Genetic variation is presumed to be a major factor underlying sex differences for IHD events, including mortality. The purpose of this study was to identify sex-specific candidate genes associated with all-cause mortality among people diagnosed with coronary artery disease (CAD).
Methods
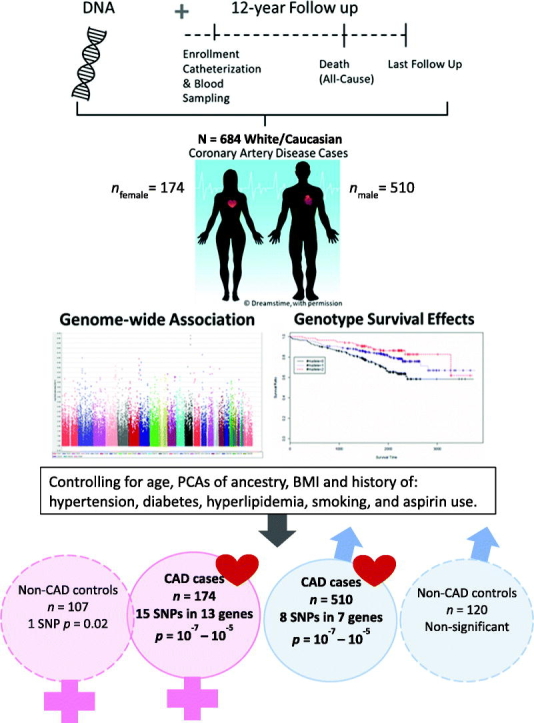
We performed a sex-stratified, exploratory genome-wide association (GWAS) screen using existing data from CAD-diagnosed males (n = 510) and females (n = 174) who reported European ancestry from the Duke Catheterization Genetics biorepository. Extant genotype data for 785,945 autosomal SNPs generated with the Human Omni1-Quad BeadChip (Illumina, CA, USA) were analyzed using an additive inheritance model. We estimated instantaneous risk of all-cause mortality by genotype groups across the 11-year follow-up using Cox multivariate regression, covarying for age and genomic ancestry.
Results
The top GWAS hits associated with all-cause mortality among people with CAD included 8 SNPs among males and 15 among females (p = 1 × 10−6 or 10−7), adjusted for covariates. Cross-sex comparisons revealed distinct candidate genes. Biologically relevant candidates included rs9932462 (EMP2/TEKT5) and rs2835913 (KCNJ6) among males and rs7217169 (RAP1GAP2), rs8021816 (PRKD1), rs8133010 (PDE9A), and rs12145981 (LPGAT1) among females.
Conclusions
We report 20 sex-specific candidate genes having suggestive association with all-cause mortality among CAD-diagnosed subjects. Findings demonstrate proof of principle for identifying sex-associated genetic factors that may help explain differential mortality risk in people with CAD. Replication and meta-analyses in larger studies with more diverse samples will strengthen future work in this area.
Keywords: Sex differences, Sex dimorphism, Coronary artery disease, Survival analysis, Genome-wide association study
Graphical abstract
Highlights
-
•
Genetic variation is a presumed factor underlying sex differences in MACE, yet the field lacks sex-stratified GWAS of longitudinal events.
-
•
We identified unique sex-associated candidate genes, adjusted for cardiovascular risk factors, for all-cause mortality among people with CAD.
-
•
Candidate genes among males (EMP2, KCNJ6) and females (LPGAT1, PDE9A, PRKD1, RAP1GAP2) have biologic relevance to CAD and/or mortality.
-
•
Future genetic research using sex-as-a-biological variable may help to explain how sex differences contribute to disparate CAD outcomes.
1. Introduction
Coronary artery disease (CAD) and the related conditions of acute coronary syndrome (ACS) and myocardial infarction (MI)—collectively known as ischemic coronary heart disease (IHD)—kill more people worldwide than any other disease [1]. Sex differences in IHD are well established in the literature with respect to biology (physiology, pathophysiology, biomarkers), epidemiology (age of onset, risk factors, symptomatology), and clinical phenotypes (angina etiology, ischemic events, and mortality) [2], [3]. For example, males tend to experience IHD events and death at earlier ages than women; however, after age 65, the female mortality rate from IHD rises steeply in comparison to that of males [4]. Women in the U.S. are at 50 % greater risk of a mis- or undiagnosed heart attack and 2–3 times greater risk for IHD-related mortality than men despite shared risk factors and similar access to care [2], [5], [6]. After MI, 35 % of women have another heart attack within 6 years, nearly double the incidence in males [7]. And one in five women assessed as being at moderate or intermediate risk of MI at initial evaluation will die within 30 days of seeking emergency care [7].
The known sex differences in biologic and epidemiologic risk factors, however, fail to fully explain the disparities in cardiac outcomes and mortality between men and women [2], [3], [8]. Genetic variation has been implicated as a major risk factor for IHD, as it precedes all other known factors. Yet sex differences in gene variation and the related mechanisms that contribute to IHD remain poorly understood. Observations of genetic effects in risk for IHD that are shared between the sexes or unique to them have been limited to candidate gene explorations using a case-control gene association design [9], [10], [11], [12], [13] or focused on genomic risk score prediction of CAD risk among the sexes [60]. While such studies have provided proof of principle for sex-dimorphic gene effects in IHD, the search for genetic associations for sex differences in IHD event outcomes has lagged.
We previously identified associations between single nucleotide polymorphisms (SNPs) within CAD candidate genes and longitudinal survivorship among people with prevalent CAD. These findings, however, replicated only among males [14]. In our recent genome-wide association study (GWAS) to identify novel candidate genes for survivorship outcomes among people with CAD (sex-combined analyses) [15], we noted evidence in the literature of dimorphic sex effects on lethal cardiovascular phenotypes for two of our top candidates. Our prior work revealed improved survival time among rs587936 C allele carriers with CAD [15]. This SNP annotated to DAB2IP (disabled homolog 2 interacting protein), a ras/GAP tumor suppressor gene which is highly expressed in vascular tissue and has multiple lines of evidence in the literature for relation to atherosclerosis, as previously reviewed [15]. In the sex-associated literature, allelic variation in DAB2IP (rs7025486, A allele) was associated with faster time to rupture of abdominal aortic aneurysm in women compared to men [16]. Our second top hit was rs13007553, with T allele carriers conferring higher risk for all-cause mortality [15]. This SNPresides between MYT1L (Myelin Transcription Factor 1 Like) and EIPR1 (EARP Complex And GARP Complex Interacting Protein 1; alias, TSSC1) on Chromosome 2; it is part of a female-specific linkage peak (2p25.3) associated with higher mitochondrial DNA levels among families with idiopathic thrombophilia cases (logarithm of the odds [LOD] score = 3.09) [17]. Of note, mitochondrial variation (inherited through maternal lineage) has been implicated in sexual dimorphism of cardiovascular diseases [18], and thrombophilia of any type is associated with increased risk of CAD and major adverse cardiac events (MACE). In our GWAS models that identified DAB2IP and MYT1L/EIPR1 as candidate genes for CAD survivorship, we controlled for sex as a covariate. In the present work, we employ a sex-stratified GWAS to screen for sex-specific candidate genes associated with all-cause mortality among people with CAD.
2. Methods
2.1. Design
To identify sex-specific gene associations with survivorship in people with CAD, we performed a sex-stratified, exploratory GWAS screen using existing GWAS data from the Duke Catheterization Genetics (CATHGEN) biorepository.
2.2. Participants/study population
The Institutional Review Board for an academic medical center in the southeastern U.S. approved the primary CATHGEN cohort biorepository (N = 9334) [19], the GWAS substudy of the first 2203 participants >18 years of age enrolled in CATHGEN [20], [21], and the present sex-stratified GWAS screen (N = 684). Briefly, the CATHGEN biorepository recruited participants who were undergoing evaluation for ischemic heart disease via cardiac catheterization at the academic medical center. Patients were ineligible for CATHGEN participation if valvular heart disease was either a primary or secondary indication for their coronary heart catheterization or if they had a pre-enrollment history of pulmonary hypertension, transplant, right heart catheterization, congenital heart disease, severe congestive heart failure (New York Heart Association [NYHA] class IV at baseline), or peripheral arterial disease intervention. Data available in the biorepository include genome-wide genotype data, demographic and clinical data abstracted from medical records, and follow-up data of mortality events at 6 months and then annually. A de-identified, anonymous data set containing the variables for our secondary analysis was curated for this project.
2.2.1. Inclusion criteria
We compiled the sample for the present analysis by selecting CAD cases from the CATHGEN GWAS substudy, resulting in 684 participants of self-reported White/European ancestry. To define positive CAD status, we followed the primary CATHGEN study's criterion of a Duke CAD index ≥32 (at least one vessel having at least 75 % stenosis), as determined by clinical coronary heart catheterization [19]. Of note, the Duke CAD index reflects both the extent and location of stenosis. It is used as an indicator of disease severity and includes a score for the presence of left main coronary artery disease.
2.2.2. Exclusion criteria
Our primary analysis excluded the non-CAD control participants due to our focus on all-cause mortality events among people with CAD (survivorship with CAD phenotype). We excluded participants with CAD from the survival analyses if they died within 14 days of their initial catheterization in order to mitigate any undue influence on the time-to-event results involving mortality due to procedural intervention. The limited number of Black/African American, Hispanic, Asian and Pacific Islanders present in the GWAS substudy sample provided inadequate power to include in the analyses, particularly as each group would need to be further stratified by sex for the present exploratory GWAS screen.
2.3. Data sources and variables
2.3.1. Sample collection and genotyping
All sample collection, processing, genotyping and quality control (QC) were performed for the primary CATHGEN study in same lab at the Molecular Genomic Core at the Duke Molecular Physiology Institute using the same protocols [20], [21]. Following informed consent, blood was obtained from the femoral artery, immediately processed to separate plasma, and frozen at −80 °C. Genomic DNA was extracted from blood using the Puregene system (Gentra Systems, Minneapolis, MN, USA). Genotyping was performed using 200 ng of DNA with the Illumina Human Omni1-Quad BeadChip (Illumina, San Diego, CA, USA) following the manufacturer's protocol. This BeadChip is designed to capture 95 % of genomic variation among people of European ancestry. After genotyping, BeadChips were imaged using the Illumina iScan system. Genotypes were called using Illumina's GenomeStudio V2010.2 software (version 1.7.4, Genotyping module). SNPs with <98 % call frequency or minor allele frequency (MAF) < 0.01 in all races or that were out of Hardy-Weinberg equilibrium (p < 5 × 10−6) were excluded, resulting in 785,945 autosomal SNPs for analysis. Samples with <98 % call frequency for all SNPs, mismatch between subject gender self-identification and sex chromosome makeup, or cryptic relatedness were excluded (172 samples) [20], [21]. Post-QC CATHGEN genotypes are stored in the Duke PEDIGENE® biorepository database.
2.3.2. Variables and outcomes
We defined the time-to-event outcome variable as number of days from study enrollment (baseline: time at coronary catheterization and blood collection) to death from any cause (event). Time-to-event for surviving individuals was censored on the date of last follow-up (censor), consistent with our previously defined “survivorship in CAD” phenotype [14], [22]. Clinical and medical history data came from the Duke Databank for Cardiovascular Disease, the data repository for the CATHGEN study. All patients in CATHGEN had one 6-month follow-up, then annual follow-up for all-cause mortality for a maximum of nearly 12 years. CATHGEN study staff adjudicated death events via vital-records searches (National Death Index and Social Security Death Index) [21].
Biological sex was determined using a standard genomic approach for gender-mismatch analysis via X-Chromosome zygosity status, as previously reported for the GWAS substudy [20]. Only data from gender matches with at least 98 % concordance were included. Age at time of event or censor was calculated based on date of birth.
2.4. Statistical analysis
Statistical analyses were performed using the R survival package [23]. We calculated means and frequencies for baseline demographic variables, diagnoses, and events. Individuals with CAD were first stratified by biological sex. Each SNP was analyzed individually, using an additive genetic inheritance model as informed by our prior work with survivorship with CAD [14]. The additive genetic model applies a value of zero to wild-type genotype carriers, a value of one to heterozygous genotype carriers, and a value of two to homozygous minor allele genotype carriers [24]. We employed Cox multivariate regression models to estimate instantaneous risk (hazard) of all-cause mortality by genotype groups. We fit a minimally adjusted model covarying age and four principal components of global genomic ancestry to account for this sample's European population admixture (PLINK program, V1.9) [25], [26]. Our base model included age and genomic ancestry principle components as covariates. We also tested the base model with the following additional covariates (adjusted model): body mass index (BMI) and history at baseline enrollment of the following: smoking, hypertension, diabetes mellitus, hyperlipidemia, and aspirin use. We constructed Kaplan-Meier curves to show survival probabilities by genotype. Post-hoc, we explored the sex-stratified base model in gene-centric analyses (all genotyped SNPs in the top candidate genes) and also tested the base model in non-CAD controls.
2.4.1. Statistical screening thresholds
Our target association level was the standard GWAS threshold, p = 1 × 10−8. Where this stringent threshold was not met, we accepted variants meeting the threshold of p = 1 × 10−5, indicating suggestive associations for candidate gene identification.
3. Results
3.1. Demographic characteristics
We present demographic and clinical characteristics in Table 1. Compared to males, on average, females tended to be older yet have slightly lower CAD severity (CAD index) with better ejection fraction and were more likely to have type 2 diabetes but less likely to be smokers. Comparing females younger than age 65 with those aged 65 and older, we observed very similar CAD severity and prevalence of risk factors, with the exception of lower prevalence of diabetes, hyperlipidemia, and smoking history among the older female group.
Table 1.
Demographic and clinical characteristics (N = 684).
| Characteristic | Male (n = 510) |
Female (n = 174) |
p | Female Age < 65 (n = 105) |
Female Age ≥ 65 (n = 69) |
|---|---|---|---|---|---|
| Age (years), mean ± SD | 63.9 ± 10.99 | 67.3 ± 10.78 | 0.0006 | 56.17 ± 5.50 | 74.5 ± 6.22 |
| CAD index, mean ± SD | 54.1 ± 18.8 | 49.5 ± 17.4 | 0.003 | 48.48 ± 17.2 | 50.10 ± 17.4 |
| BMI (kg/m2), mean ± SD | 29.1 ± 5.8 | 30.8 ± 7.9 | 0.11 | 32.26 ± 9.69 | 28.85 ± 7.18 |
| History of hypertension, % | 66.7 % | 64.9 % | 0.06 | 71.0 % | 77.1 % |
| History of type 2 diabetes mellitus, % | 30.2 % | 32.2 % | 0.69 | 44.9 % | 23.8 % |
| History of dyslipidemia, % | 68.8 % | 64.9 % | 0.39 | 75.4 % | 58.1 % |
| History of smoking, % | 55.3 % | 43.1 % | 0.007 | 52.2 % | 37.1 % |
| Ejection fraction (%), mean ± SD | 54.0 ± 13.3 | 58.4 ± 12.8 | 0.0001 | 58.46 ± 12.48 | 58.36 ± 13.07 |
| Creatinine (mg/dL), mean ± SD | 1.3 ± 0.87 | 1.1 ± 0.73 | 0.002 | 0.98 ± 0.89 | 1.09 ± 0.60 |
| History of myocardial infarct (MI), % | 39.0 % | 30.5 % | 0.05 | 34.8 % | 27.6 % |
| History of stroke, % | 8.4 % | 13.8 % | 0.06 | 15.9 % | 12.3 % |
| Aspirin use, % | 84.7 % | 86.2 % | 0.72 | 87.0 % | 85.7 % |
BMI = body mass index; CAD = coronary artery disease; SD = standard deviation. Histories of hypertension, type 2 diabetes mellitus, dyslipidemia, smoking, myocardial infarct (MI), stroke, and aspirin use defined elsewhere [21].
3.2. Events
We present the follow-up times and events in Table 2. The median and maximum follow-up times were 5.5 years and 10.8 years, respectively. A total of 159 all-cause mortality events were observed in the sample, representing a 23.3 % mortality rate. Males and females had similar median and maximum follow-up days; males had a slightly higher mortality rate (23.9 %) compared to females (21.3 %).
Table 2.
Follow-up and events.
| Follow-up and events | Total (N = 684) |
Male (n = 510) |
Female (n = 174) |
|---|---|---|---|
| Median follow-up, days (years) | 2004 (5.5) | 2002 (5.5) | 2028 (5.6) |
| Max. follow-up, days (years) | 3953 (10.8) | 3953 (10.8) | 3875 (10.6) |
| Death events, n (%) | 159 (23.3) | 122 (23.9) | 37 (21.3) |
3.3. Genome-wide screen of sex-associated survivorship with CAD
Quantile-quantile (Q-Q) plots by sex strata indicate that our observed genomic signals were consistent with the expected distribution under the null hypotheses (Fig. S1). The Manhattan plots (Fig. S2) show the negative log10(p-value) for each SNP by chromosome. Results of the sex-stratified models are presented in Tables 3 (male) and 4 (female).
Table 3.
Single nucleotide polymorphisms (SNPs) significantly associated with survival in males (n = 510) with coronary artery disease and European ancestry.
| SNP | Type | Gene | Chr | Minor Allele | MAF | p | p-adj | HR | 95 % CI | MAF | Female |
|---|---|---|---|---|---|---|---|---|---|---|---|
| p | |||||||||||
| rs2076780 | Intergenic | GREM2/RGS7 | 1q43 | A | 0.03 | 5.19e−06 | 1.61e−05 | 3.34 | [1.99, 5.62] | 0.03 | 0.49 |
| rs11252040 | ncRNA intronic | LOC105376360 | 10p15.2 | G | 0.07 | 7.82e−06 | 1.22e−05 | 2.35 | [1.61, 3.41] | 0.09 | 0.79 |
| rs17103766 | Intergenic | BRMS1L/LINC00609 | 14q13.2 | G | 0.04 | 8.33e−06 | 2.83e−05 | 2.89 | [1.81, 4.62] | 0.02 | 0.04 |
| rs2062640 | Intergenic | UNC13C/LOC105370829 | 15q21.3 | G | 0.11 | 1.79e−06 | 4.49e−07 | 2.35 | [1.66, 3.34] | 0.11 | 0.62 |
| rs4776247 | Intergenic | UNC13C/LOC105370829 | 15q21.3 | A | 0.10 | 3.44e−06 | 2.38e−06 | 2.40 | [1.66, 3.47] | 0.10 | 0.48 |
| rs9932462 | Intergenic | EMP2/TEKT5 | 16p13.13 | G | 0.01 | 1.41e−06 | 1.55e−06 | 4.92 | [2.57, 9.40] | 0.01 | 1.00 |
| rs12150051 | ncRNA intronic | LINC00670 | 17p12 | C | 0.41 | 3.12e−06 | 1.38e−06 | 0.51 | [0.39, 0.68] | 0.40 | 0.49 |
| rs2835913 | Intronic | KCNJ6 | 21q22.13 | G | 0.03 | 4.81e−06 | 1.66e−06 | 3.46 | [2.03, 5.90] | 0.03 | 0.30 |
Bold p-value indicates p < .05 in females. Chr = chromosome; CI = confidence interval; HR = hazard ratio; MAF = minor allele frequency; ncRNA = noncoding RNA. Full gene names provided in Table S5 of supplemental materials.
Table 4.
Single nucleotide polymorphisms (SNPs) significantly associated with survival in females (n = 174) with coronary artery disease and European ancestry.
| SNP | Type | Gene | Chr | Minor Allele | MAF | p | p-adj | HR | 95 % CI | Male |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
| MAF | p | ||||||||||
| rs10923243 | Intergenic | VTCN1/LINC01525 | 1p13.1 | G | 0.01 | 6.91e−06 | 2.49e−06 | 19.61 | [5.36, 71.74] | 0.02 | 0.53 |
| rs10494195 | ncRNA intronic | LOC101929099 | 1p13.1 | A | 0.06 | 9.55e−06 | 8.83e−06 | 5.23 | [2.51, 10.87] | 0.08 | 0.75 |
| rs12145981 | Intergenic | LOC91548/LPGAT1 | 1q32.3 | G | 0.17 | 3.18e−06 | 2.41e−06 | 3.40 | [2.03, 5.68] | 0.18 | 0.91 |
| rs17591646 | Intronic | SLC9A9 | 3q24 | G | 0.05 | 2.88e−06 | 1.28e−04 | 6.23 | [2.89, 13.40] | 0.08 | 0.80 |
| rs26445 | Intergenic | LOC102546299/LINC01947 | 5q34 | A | 0.07 | 7.75e−06 | 6.20e−05 | 4.05 | [2.19, 7.46] | 0.08 | 0.09 |
| rs9388813 | Intergenic | TMEM200A/SMLR1 | 6q23.1 | A | 0.04 | 5.85e−06 | 3.70e−06 | 5.71 | [2.69, 12.13] | 0.05 | 0.09 |
| rs1751291 | Intergenic | LINC00703/MANCR | 10p15.1 | G | 0.18 | 9.70e−06 | 1.05e−05 | 3.56 | [2.03, 6.25] | 0.15 | 0.51 |
| rs10768256 | Intergenic | C11orf74/LINC02760 | 11p12 | A | 0.09 | 9.28e−06 | 2.95e−05 | 3.80 | [2.11, 6.85] | 0.09 | 0.24 |
| rs7320901 | Intergenic | LINC00457/NBEA | 13q13.3 | A | 0.15 | 7.88e−07 | 3.10e−06 | 3.67 | [2.19, 6.15] | 0.13 | 0.24 |
| rs17051660 | Intergenic | LINC00457/NBEA | 13q13.3 | A | 0.05 | 1.02e−06 | 2.37e−07 | 6.23 | [2.99, 12.96] | 0.06 | 0.67 |
| rs9599764 | Intergenic | LINC00457/NBEA | 13q13.3 | G | 0.14 | 1.72e−06 | 5.45e−06 | 4.38 | [2.39, 8.03] | 0.14 | 0.44 |
| rs8021816 | Intronic | PRKD1 | 14q12 | C | 0.05 | 6.76e−06 | 1.27e−06 | 5.86 | [2.71, 12.65] | 0.07 | 0.60 |
| rs7217169 | Intronic | RAP1GAP2 | 17p13.3 | G | 0.08 | 4.98e−06 | 9.51e−06 | 4.06 | [2.22, 7.41] | 0.08 | 0.47 |
| rs8133010 | Intronic | PDE9A | 21q22.3 | G | 0.25 | 5.57e−06 | 2.02e−06 | 3.22 | [1.94, 5.33] | 0.27 | 0.64 |
| rs1771144 | Intronic | KLHL22 | 22q11.21 | A | 0.15 | 2.26e−06 | 4.15e−06 | 4.07 | [2.27, 7.27] | 0.19 | 0.72 |
Note. Chr = chromosome; CI = confidence interval; HR = hazard ratio; MAF = minor allele frequency; ncRNA = noncoding RNA. Full gene names provided in Table S5 of supplemental materials.
3.3.1. Males
We identified seven candidate loci (among eight SNPs) for all-cause mortality in our sample of males with European ancestry and a diagnosis of CAD. All SNPs identified in males had p ≤ 1 × 10−6, but none met the p = 1 × 10−8 GWAS threshold. All SNP effects remained significant after controlling for additional covariates (BMI and history of the following: smoking, hypertension, diabetes mellitus, hyperlipidemia, and aspirin use), as shown in Table 3 (p-adj). We observed a negative hazard ratio (HR = 0.51) in one SNP (rs12150051), indicating a potential protective effect. The average per-allele risk effect (HR) among the SNPs with HR > 1.00 was 3.10 (range 2.35–4.92). Kaplan-Meier survival curves for male-associated exemplar SNPs are presented in Fig. 1 (EMP2/TEKT5 and KCNJ6). These exemplars were selected because of their biological relevance identified via literature review and bioinformatic investigation of NCBI Entrez Gene [27], Weizmann Institute's GeneCards [28], and the UniProtKB/Swiss-Prot [29] databases during the gene annotation phase.
Fig. 1.

Exemplar Kaplan-Meier curves of survival time in days (x-axis) and all-cause survival probability (y-axis) by genotype category among males with CAD. The solid black line represents wild-type homozygous (AA) genotype carriers, the blue dashed line represents heterozygous (GA) genotype carriers, and the red dotted line represents carriers with two copies of the minor (“risk”) allele (GG; risk homozygous genotype). A) The frequency of rs9932462 risk homozygous genotype was extremely low in this group. Males with CAD having the heterozygous genotype had a 4.92-fold increased risk of all-cause mortality compared to males having the wild-type homozygous genotype (95 % CI [2.57, 9.40], p = 1.41e−06). B) The frequency of rs2835913 risk homozygous carriers was also very low; males with CAD having the heterozygous genotype had a 3.5-fold increase in risk of all-cause mortality compared to wild-type homozygous genotype carriers (HR = 3.46, 95 % CI [2.03, 5.90], p = 4.81e−06). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3.2. Females
In the analysis of our sample of females with European ancestry who had been diagnosed with CAD, we identified 14 candidate loci (among 15 SNPs) meeting p ≤ 1 × 10−6, but none met the p = 1 × 10−8 GWAS threshold. All female SNP effects remained significant after controlling for additional covariates (BMI and history of the following: smoking, hypertension, diabetes mellitus, hyperlipidemia, and aspirin use), as shown in Table 4 (p-adj). All SNPs identified in females were associated with increased risk of all-cause mortality, with per-allele risk effect ranging from 3.22 to 19.61. Kaplan-Meier survival curves for female-associated exemplar SNPs are presented in Fig. 2 (RAP1GAP2, PRKD1, PDE9A, and LPGAT1). These exemplars were selected because of their biological relevance identified during the gene annotation phase.
Fig. 2.

Exemplar Kaplan-Meier curves of survival time in days (x-axis) and all-cause survival probability (y-axis) by genotype category among females with CAD. A) rs7217169 (RAP1GAP2); B) rs8021816 (PRKD1); C) rs8133010 (PDE9A); and D) rs12145981 (LPGAT1). The black solid line represents wild-type homozygous genotype carriers, the blue dashed line represents heterozygous genotype carriers, and the red dotted line represents carriers with two copies of the minor (“risk”) allele (risk homozygous genotype). The frequency of rs7217169 and rs8021816 risk homozygous genotype was low. Compared to wild-type homozygous genotype carriers, A) each copy of the RAP1GAP2 rs7217169 G (risk) allele was associated with a 4.06-fold increased risk of all-cause mortality among females with CAD (95 % CI [2.22, 7.41], p = 4.98e−06); B) each copy of the PRKD1 rs8021816 C (risk) allele was associated with a 5.86-fold increased risk of all-cause mortality (95 % CI [2.71, 12.65], p = 6.76e−06); C) each copy of the PDE9A rs8133010 G (risk) allele was associated with a 3.22-fold increased event risk (95 % CI [1.94, 5.33], p = 5.57e−06); and D) each copy of the LPGAT1 rs12145981 G (risk) allele was associated with a 3.40-fold increased event risk (95 % CI [2.03, 5.68], p = 3.18e−06). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3.3. Cross-sex SNP comparisons
To explore potential for shared gene effects between sexes, we cross-checked all sex-associated SNPs in the alternate sex category (i.e., checked top male GWA hits in females and vice-versa). We summarize these comparisons in the last two columns of Table 3, Table 4 and in Fig. 3. One SNP identified in males (rs17103766; BRMS1L/ LINC00609) was detected at p < .05 among females. Conversely, none of the top SNPs identified in females were significant in males (p-value range .09–.91).
Fig. 3.

Venn diagram of top candidate genes and shared genetic variation, by sex. Genes in bold have biological relevance to cardiovascular disease or survival. †Top male SNP that also shows p < .05 among females (does not appear in list of top candidates for females).
3.3.4. Exploration of gene-centric SNPs
After identifying the top sex-specific candidate genes for all-cause mortality in males and females with CAD, we evaluated the total number of SNPs in each candidate gene that met p < .05 in Cox multivariate association with all-cause mortality, controlling for age and four principle components of global genomic ancestry. In Supplemental Tables S1 and S2, we note the additional SNPs for each candidate gene that met QC metrics and had p < .05. This analysis revealed additional signals among all candidate genes in both males and females.
3.3.5. Exploration of non-CAD controls
In a post-hoc analysis, we explored the top sex-associated SNPs among non-CAD control groups (stratified by sex, base model) to affirm lack of association with all-cause mortality in those without CAD. Non-CAD controls were defined as having no clinically appreciable CAD (Duke CAD index <23 and number of significantly obstructed vessels = 0), corresponding to no major epicardial vessel with >74 % occlusion as demonstrated by coronary angiography at enrollment, and no documented history of cerebrovascular or peripheral vascular disease, myocardial infarction, organ transplant, or interventional or surgical coronary revascularization (coronary artery bypass graft, stent, or intracoronary procedures) at enrollment [19]. Results are presented in Tables S3 and S4. None of the male-associated SNPs were associated with survival among male non-CAD controls; only one female SNP was marginally associated (p = .02) with all-cause mortality among female non-CAD controls (intergenic rs9599764 annotated to LINC00457/NBEA).
4. Discussion
Our sex-stratified, exploratory GWAS screen identified suggestive associations with 20 potential sex-specific candidate genes for SNP-wise sex effects on all-cause mortality with clinically defined CAD (Fig. 4). Prior researchers have reported sex-specific candidate genes for CAD-associated outcomes using sex-stratified analyses with a priori candidate genes. SCARB1, a known quantitative trait locus for HDL-cholesterol level (HDL-QTL6) [30], [31], was associated with increased risk for premature CAD among females (n = 574) but not males (n = 477) [11]. CPS1 (2q34) is a GWAS candidate validated in the CARDIoGRAM study as strongly associated with protection from CAD among females but not males [12]. The well-established 9p21 CAD risk locus demonstrated stronger effects in males than females in a GWAS re-analysis [13]. A genomic risk score approach also demonstrated sexual dimorphic effects on prediction of incident and prevalent CAD, and identified a novel gene-sex interaction at locus 21q22.11 [32]. The present GWAS scan of sex-associated gene effects on the longitudinal endpoint of all-cause mortality in CAD cases is a novel addition to the literature. That our candidate genes were different from those reported as sex-dimorphic in the literature reflects the distinct differences in candidate-gene versus GWAS approaches and dichotomous versus longitudinal event outcomes.
Fig. 4.

Forest plot of top SNP effect sizes. Circles indicate hazard ratio (HR), by male (blue) and female (pink). Horizontal lines indicate the 95 % confidence interval (CI), also provided in brackets, far right. Vertical dotted line indicates HR threshold value of 1. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Our only evidence of shared genetic association between men and women involved a single SNP (rs1703766) identified in males that also showed moderate significance among females. This variant is located on Chromosome 14q13.2 between BRMS1L (BRMS1 like transcriptional repressor) and LINC00609 [33]. BRMS1L is part of p53 cell-cycle arrest, apoptosis, and senescence functions. Variations in BRMS1L mRNA gene expression have been associated with invasion, migration, and poor patient outcomes in breast cancer [34]. In addition, we found eight SNPs across seven candidate genes or loci for all-cause mortality among males with CAD and 15 SNPs across 13 candidate genes or loci among females with CAD.
4.1. Candidates in males
All eight of the SNPs we identified in males were associated with increased risk of all-cause mortality (HR > 1; Table 3) and remained significant in models adjusted for multiple cardiovascular risk covariates. SNP-wise effects for two of these markers mapped to the intergenic region UNC13C/LOC105370829 (15q21.3). In a recent study, the nearby 15q21.1 region was identified as one of five major susceptibility loci for spontaneous coronary artery dissection (SCAD; OR = 1.75, 95 % CI [1.40–2.18], p = 7.23e−7); however, the investigators conducted their two-phase GWAS analysis in women only (n = 667), while we identified this association exclusively in males [35].
Among the SNPs we identified in males, the most biologically plausible candidates include those annotated to the genes EMP2/TEKT5 and KCNJ6. Presence of the heterozygous genotype of EMP2/TEKT5 SNP rs9932462 was associated with a 4.42-fold increased risk of all-cause mortality (95 % CI [2.57, 9.40], p = 1.4 × 10−6). As shown in Fig. 1a, the lack of GG homozygous risk carriers for rs9932462 (EMP2/TEKT5) indicates that the hazard estimate is primarily informing on the presence of a single risk allele among heterozygous genotype carriers. Notably, EMP2 is implicated in a wide array of atherosclerosis endophenotypes, as it has been shown to regulate migration of blood vessel endothelial cells [36], cell contraction [37], [38], focal adhesion density, F-actin conformation and cell adhesion capacity [39], and cellular proliferation [40]. EMP2 also promotes angiogenesis and vasculogenesis [41] and is involved in cell death and cell blebbing [42]. Interestingly, rs9932462, an intergenic SNP, is located adjacent to TEKT5, which encodes a protein suspected to be a structural component of the sperm flagellum [29]. EMP2 rs9932462 variation was not associated with survival among women with CAD (p = 1.00) [29].
Meanwhile, for each copy of the rs2835913 G risk allele within KCNJ6, we observed an approximately 3.5-fold increased risk of all-cause mortality among males (HR = 3.45, 95 % CI [2.03, 5.90], p = 4.8 × 10−6) compared to non-G carriers. The very limited representation of the risk homozygous genotype for rs2835913 (KCNJ6) again indicates that the hazard estimate is primarily informing on the presence of a single risk allele among heterozygous genotype carriers. Variation in rs2835913 was not associated with the outcome event among women (p = .30). KCNJ6 encodes a member of the G protein-coupled inwardly-rectifying potassium channel (GIRK) family [33]. The gene is expressed in cardiac and neuronal cells where it modulates heart rate and neuronal circuit activity, respectively [33]. An increase in intracellular potassium has homeostatic and physiologic effects that signal catecholamine release, which stimulates alpha 1 adrenergic receptor to cause potassium to shift out of the cells and into the blood. An increase in extracellular potassium induces arterial vasodilation in normal blood vessels, thereby increasing skeletal blood flow. Within endothelial cells, neurohumoral mediators and physical forces (such as vascular sheer stress) can cause potassium ions to be released. It may be that SNP variation in KCNJ6 leads to alterations of intra- and extracellular potassium balance and results in hypertension, a major risk factor for CAD and mortality. KCNJ6 polymorphisms have been positively associated with blood pressure response to variations in dietary sodium intake among 1906 participants of the GenSalt study [43]. Given that potassium channel activity is physiologically correlated with cardiac rhythm, KCNJ6 polymorphism may influence mortality risk via arrhythmias. In the literature, KCNJ6 variants were associated with long QT syndrome among a large Australian family [44]. KCNJ6 variation has also been explored for relation to various pain phenotypes, including pain tolerance and pain outcomes, with promising but largely inconclusive findings (as reviewed by Matic et al.) [45] Replication and further research are needed to better understand the influence of EMP2 and KCNJ6 SNP variation on survival outcomes in males with CAD.
4.2. Candidates in females
None of the 15 candidate SNPs identified in females with CAD met p < .05 among the male sample (Table 4). All of these SNPs conferred increased risk of death (HR range 3.2–19.6), even after controlling for multiple cardiac risk covariates. Of the top female candidate SNPs, four annotate to biologically relevant genes: rs7217169 (RAP1GAP2, Chr 17p13.3), rs8021816 (PRKD1, Chr 14q12), rs8133010 (PDE9A, Chr 21q22.3) and rs12145981 (LPGAT1, Chr 1q13.3). Among women with CAD, each copy of the G risk allele was associated with a 4.06-fold (rs7217169; RAP1GAP2), 3.22-fold (rs813133010; PDE9A), and 3.4-fold (rs12145981; LPGAT1) increased risk of all-cause mortality compared to non-carriers. Similarly, each copy of the C risk allele for rs8021816 (PRKD1) was associated with a 5.86-fold increased risk of all-cause mortality compared to non-C carriers. Fig. 2 shows a limited frequency of risk homozygous genotype for RAP1GAP2 (frequency of 0.005) and PRDK1 SNPs (frequency of 0.002).
RAP1GAP2 (RAP1 GTPase activating protein 2) encodes a GTPase-activating protein that activates the small guanine-nucleotide-binding protein rap1 in platelets [46]. The protein complex RAP1GAP2 is expressed in platelets and activates both rap1 protein and glycoprotein receptors GPIIb/IIIa to elicit maximum platelet aggregation responses [46]. Endothelial damage due to IHD thus activates RAP1GAP2 and causes the release of dense granules from platelets, leading to thrombosis, inflammation, and aggregation [47].
PDE9A (phosphodiesterase 9A) catalyzes the hydrolysis of cAMP and cGMP to their corresponding monophosphates [33]. PDE9A is linked to pathways related to platelet homeostasis and response to elevated platelet cytosolic calcium [48]. Variations in the expression of this gene have been reported in mice with diastolic dysfunction and in humans with heart failure with preserved ejection fraction (HFpEF) [49]. More recently, PDE9A inhibition was shown to improve diastolic dysfunction in murine models [50].
Located on Chromosome 14q12, rs8021816 maps to protein kinase D1 (PRDK1) and was present in 5 % of our sample of females of European ancestry with CAD. PRDK1 has many roles in various cellular processes, including cell migration, differentiation, and survival as well as regulation of cell shape and adhesion [51]. It has been associated with congenital heart defects and is part of the beta-adrenergic signaling pathway [52].
The candidate SNP rs12145981 annotates to LPGAT1, or lysophosphatidylglycerol acyltransferase 1, whose protein product is an important precursor for the synthesis of cardiolipin. Cardiolipin is a phospholipid exclusive to the inner membrane of the mitochondria and constitutes 20 % of the mitochondria's total lipids [53]. This phospholipid is essential to proper enzymatic function during mitochondrial energy metabolism [53]. Extracellular cardiolipin transfer has been implicated in apoptosis and cardiolipin may also serve as a proton trap for oxidative phosphorylation [54]. In a unique prothrombotic condition, anti-cardiolipin antibodies are associated with risk for recurrent thrombotic events that can occur as early as the teen years [55]. These autoantibodies have been detected among young women experiencing repeated spontaneous abortions [55]. Cardiolipin has also been associated with dilated cardiomyopathy and progressive familial heart block [56]. LPGAT1 itself has been implicated in cholesterol secretion and atherosclerosis [57].
4.3. Robustness and effect sizes of associations
Because authors have asserted that female-associated findings tend to be less robust in the literature of sex-dimorphic gene associations, we evaluated the robustness of statistical findings and effect sizes of the top GWAS hits among the male and female groups [13]. Comparing effect sizes of genetic associations in the male and female groups, the mean HR for males was 3.10 with an average 95 % CI width of 3.20, whereas among females the mean HR was 5.54 and average 95 % CI width was 10.38. The p-values were comparable between the sexes. The 95 % CI range for the tested SNPs was wider for females than males despite the fact that minor allele frequencies were similar between sexes (Fig. 4). These results are most likely due to the smaller sample size for females compared to males.
We report multiple additional SNPs meeting nominal significance (p < .05) for each sex-associated candidate gene in Tables S1–S2. These results strengthen the lines of genomic evidence for our candidates and support future meta-analyses and study comparisons.
4.4. Strengths and limitations
Our results are strengthened by additional lines of evidence from significant covariate-adjusted models and from demonstrated lack of significance among non-CAD control groups (with the exception of a single female SNP, rs9599764 having a marginal p-value). However, we caution inferences about the lack of association in controls, as it may be an artefact of limited mortality endpoints among the non-CAD control sample. The CATHGEN clinical cardiovascular biorepository provided exquisite phenotyping of clinically defined CAD along with 11-year longitudinal data on annual follow-up and mortality events to support the testing of our hypothesis of the existence of male and female sex-associated genetic effects in people with CAD. The repository's inclusion of death events adjudicated via national vital records is a strength. However, because the primary study's data on cause of death were too limited for use as an endpoint and causes of death are often inaccurate on death certificates [58], we were confined to the outcome of all-cause mortality within a subset of CAD cases as a proxy for CAD-related death. This study only examined sex as a biological variable of self-reported males and females. We were unable to evaluate other gender designations. Both the male and female groups had insufficient power, thus our GWAS screen should be considered exploratory and our results interpreted carefully. SNP-wise association with survival endpoints requires a minimum of 299 events to achieve 80 % power (α = 0.05), assuming a 20 % event rate [59]. In the present sample, we observed 159 total events. The small sizes of the sex-stratified group increases the likelihood of Type I error. For some top SNPs, the frequency of risk homozygous genotype was low (<1 %), therefore the additive genetic models were informing largely on the presence of the risk allele among heterozygous genotype carriers. Generalizability of our results is also limited because our sample of CAD-diagnosed individuals was confined to self-identified White individuals with European ancestry in the southeastern U.S. Despite retaining significance in models adjusted for multiple cardiovascular risk factors including aspirin use, the effect of medication use on survival presents a particular concern for confounding. Namely, statins, beta-blockers, and antiplatelet agents are well-established, independent predictors of survival and MACE among people with diagnosed CAD [60], [61], [62], and thus, are considered first-line therapy for prevention of MACE among people with CAD (including during the CATHGEN recruitment phase). However, detailed medication phenotyping and medical record adjudication for these additional drugs were not part of the initial study design and data collection, therefore, the observed SNP effects on survival may be influenced by the use of MACE prevention medications. Relatedly, sex differences in treatment, adherence, and response for MACE prevention medications represent a potential confounding concern; however, meta-analyses examining sex differences in efficacy of antiplatelet [63], antihypertensive [64], and statin therapies [65] revealed no major differences in outcomes between men and women. Regarding aspirin use (which we were able to control for in the fully adjusted model), a meta-analysis of aspirin efficacy by sex revealed reductions in composite MACE among both sexes; however, the effect in females was attributed to reductions in stroke, while the effect in males was attributed to reductions in MI [ [66]]. Addressing the influence of cardiac medications and their sex differences are future priorities in order to refine the sex-associated genetic contribution to CAD-related mortality. The present study does not include replication analyses, and all results should be interpreted with caution.
5. Conclusion
In the present study, we have identified numerous sex-specific GWA candidates having suggestive association with all-cause mortality risk among people with clinically diagnosed CAD. Our hypothesis that there are candidate genes for CAD unique to each biological sex was further supported by our demonstration of: minimal overlap in candidate genes between males and females, retained significance in models adjusted for cardiovascular risk factors, and lack of SNP associations among non-CAD control groups (save for one female SNP). This study demonstrates proof of principle for identifying sex-associated genetic factors that may help to explain differential mortality risk in people with CAD. Replication and meta-analyses will strengthen future work in this area.
Together with evidence from the literature, our sex-dimorphic candidate gene findings support the need for the expanded use of sex as a biological variable in research examining cardiovascular health, as supported by the NIH [67]. The present exploratory results require replication in larger studies with more diverse samples. The burden of CAD-related mortality falls heavily on Black and Brown people, particularly women [6], [8], [68]. Yet their limited representation in genomics research continues to deny them potential benefits of such research [69], [70]. Furthermore, it diminishes our ability to study whether and how population genomic effects interact with social determinants of health and discrimination as contributors to disparities in mortality [71], [72]. While the present study has limitations related to inclusion and representation, we are investigating approaches to adequately power research among trans-ethnic cohorts as next steps. The present and future investigations of the effects of sex-associated genomic variants in CAD will improve our understanding of sex-based disparities in CAD symptoms and outcomes and may lead to the development of personalized CAD therapeutics in males and females.
Funding sources
-
•
NINR (K99 NR011054/R00NR011054; JR Dungan, PI).
-
•
Duke University John A. Hartford Center for Excellence Jr. Faculty Fellowship (#2006-0109; JR Dungan, PI).
-
•
NHLBI (5RC2HL101621-02; WE Kraus, PI).
-
•
Small research grant, Duke University School of Nursing (No number; JR Dungan, PI).
CRediT authorship contribution statement
Jennifer R. Dungan: Conceptualization, Methodology, Writing-Original Draft, Review & Editing, Visualization, Funding acquisition, Supervision, Project Administration, Xue Qin: Methodology, Software, Data Curation, Validation, Formal analysis, Writing-Review & Editing, Visualization, Simon G. Gregory: Resources, Writing - Review & Editing, Rhonda Cooper-Dehoff: Writing - Review & Editing, Julio D. Duarte: Writing - Review & Editing, Huaizhen Qin: Writing - Review & Editing, Martha Gulati: Writing - Review & Editing, Jacquelyn Y. Taylor: Writing - Review & Editing, Carl J. Pepine: Writing - Review & Editing, *Elizabeth R. Hauser: Conceptualization, Methodology, Resources, Writing - Review & Editing & *William E. Kraus: Conceptualization, Methodology, Resources, Writing - Review & Editing, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The investigators would like to thank the participants of the Duke Catheterization Genetics (CATHGEN) study for their data and contribution to the research. We also wish to gratefully acknowledge Marnie Wiss for her editorial contributions and Myles Gibbs for his support as a research assistant.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ahjo.2022.100152.
Appendix A. Supplementary data
Supplementary figures S1, S2
Supplementary tables S1 - S5.
References
- 1.Virani S.S., et al. Heart disease and stroke statistics—2020 update: a report from the American Heart Association. Circulation. 2020:E139–E596. doi: 10.1161/CIR.0000000000000757. [DOI] [PubMed] [Google Scholar]
- 2.Izadnegahdar M., et al. Sex and ethnic differences in outcomes of acute coronary syndrome and stable angina patients with obstructive coronary artery disease. Circ. Cardiovasc. Qual. Outcomes. 2016;9(2 Suppl 1):S26–S35. doi: 10.1161/CIRCOUTCOMES.115.002483. [DOI] [PubMed] [Google Scholar]
- 3.Chandrasekhar J., Mehran R. Sex-based differences in acute coronary syndromes: insights from invasive and noninvasive coronary technologies. JACC Cardiovasc. Imaging. 2016;9(4):451–464. doi: 10.1016/j.jcmg.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Mikkola T.S., et al. Sex differences in age-related cardiovascular mortality. PLoS One. 2013;8(5) doi: 10.1371/journal.pone.0063347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham G. Acute coronary syndromes in women: recent treatment trends and outcomes. Clin. Med. Insights Cardiol. 2016;10:1–10. doi: 10.4137/CMC.S37145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graham G. Racial and ethnic differences in acute coronary syndrome and myocardial infarction within the United States: from demographics to outcomes. Clin. Cardiol. 2016;39(5):299–306. doi: 10.1002/clc.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benjamin E.J., et al. Heart disease and stroke Statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56–e528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 8.Graham G. Population-based approaches to understanding disparities in cardiovascular disease risk in the United States. Int. J. Gen.Med. 2014;7:393–400. doi: 10.2147/IJGM.S65528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamada Y., et al. Prediction of the risk of myocardial infarction from polymorphisms in candidate genes. N. Engl. J. Med. 2002;347(24):1916–1923. doi: 10.1056/NEJMoa021445. [DOI] [PubMed] [Google Scholar]
- 10.Silander K., et al. Gender differences in genetic risk profiles for cardiovascular disease. PloS one. 2008;3(10) doi: 10.1371/journal.pone.0003615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodarzynejad H., et al. The rs5888 single nucleotide polymorphism in scavenger receptor class B type 1 (SCARB1) gene and the risk of premature coronary artery disease: a case-control study. Lipids Health Dis. 2016;15(1):1–9. doi: 10.1186/s12944-016-0176-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartiala J.A., et al. Genome-wide association study and targeted metabolomics identifies sex-specific association of CPS1 with coronary artery disease. Nat. Commun. 2016;7(1):1–10. doi: 10.1038/ncomms10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L.Y., et al. Sex differences in disease risk from reported genome-wide association study findings. Hum. Genet. 2012;131(3):353–364. doi: 10.1007/s00439-011-1081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dungan J.R., et al. Case-only survival analysis reveals unique effects of genotype, sex, and coronary disease severity on survivorship. PloS one. 2016;11(5) doi: 10.1371/journal.pone.0154856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dungan J.R., et al. Genome-wide variants associated with longitudinal survival outcomes among individuals with coronary artery disease. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.661497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye Z., et al. A DAB2IP genotype: sex interaction is associated with abdominal aortic aneurysm expansion. J. Investig. Med. 2017;65(7):1077–1082. doi: 10.1136/jim-2016-000404. [DOI] [PubMed] [Google Scholar]
- 17.López S. 2012. Sex-specific Regulation of Mitochondrial DNA Levels: Genome-wide Linkage Analysis to Identify Quantitative Trait Loci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ventura-Clapier R., et al. Mitochondria: a central target for sex differences in pathologies. Clin. Sci. (Lond.) 2017;131(9):803–822. doi: 10.1042/CS20160485. [DOI] [PubMed] [Google Scholar]
- 19.Sutton B.S., et al. Comprehensive genetic analysis of the platelet activating factor acetylhydrolase (PLA2G7) gene and cardiovascular disease in case–control and family datasets. Hum. Mol. Genet. 2008;17(9):1318–1328. doi: 10.1093/hmg/ddn020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kraus W.E., et al. Metabolomic quantitative trait loci (mQTL) mapping implicates the ubiquitin proteasome system in cardiovascular disease pathogenesis. PLoS Genet. 2015;11(11) doi: 10.1371/journal.pgen.1005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kraus W.E., et al. A guide for a cardiovascular genomics biorepository: the CATHGEN experience. J. Cardiovasc. Transl. Res. 2015;8(8):449–457. doi: 10.1007/s12265-015-9648-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dungan J.R., et al. The genetic basis for survivorship in coronary artery disease. Front. Genet. 2013;4:191. doi: 10.3389/fgene.2013.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.R.C.D. Team . 2013. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 24.Balding D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006;7(10):781–791. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- 25.Purcell S.M., Chang C. 2021. PLINK 1.9. [Google Scholar]
- 26.Chang C.C., et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown G.R. Gene: a gene-centered information resource at NCBI. Nucleic Acids Res. 2015 Jan;43(Database issue) doi: 10.1093/nar/gku1055. D36-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weizmann Institute of Science GeneCards – the human gene database. 2022. www.genecards.org
- 29.The UniProt Consortium UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49 doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zanoni P., et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science. 2016;351(6278):1166–1171. doi: 10.1126/science.aad3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brunham L.R., et al. Novel mutations in scavenger receptor BI associated with high HDL cholesterol in humans. Clin. Genet. 2011;79(6):575–581. doi: 10.1111/j.1399-0004.2011.01682.x. [DOI] [PubMed] [Google Scholar]
- 32.Huang Y., Hui Q., Gwinn M., Hu Y.J., Quyyumi A.A., Vaccarino V., Sun Y.V. Sexual differences in genetic predisposition of coronary artery disease. Circ. Genom Precis. Med. 2021;14 doi: 10.1161/CIRCGEN.120.003147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Leary N.A., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koyama R., et al. Identification and characterization of a metastatic suppressor BRMS 1L as a target gene of p53. Cancer Sci. 2017;108(12):2413–2421. doi: 10.1111/cas.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turley T.N., et al. Identification of susceptibility loci for spontaneous coronary artery dissection. JAMA Cardiol. 2020;5(8):929–938. doi: 10.1001/jamacardio.2020.0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morales S.A., et al. Epithelial membrane protein 2 controls VEGF expression in ARPE-19 cells. Invest. Ophthalmol. Vis. Sci. 2013;54(3):2367–2372. doi: 10.1167/iovs.12-11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu M., et al. Epithelial membrane protein-2 promotes endometrial tumor formation through activation of FAK and src. PLoS One. 2011;6(5) doi: 10.1371/journal.pone.0019945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Morales S.A., et al. Anti-EMP2 diabody blocks epithelial membrane protein 2 (EMP2) and FAK mediated collagen gel contraction in ARPE-19 cells. Exp. Eye Res. 2012;102:10–16. doi: 10.1016/j.exer.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morales S.A., et al. Functional consequences of interactions between FAK and epithelial membrane protein 2 (EMP2) Invest. Ophthalmol. Vis. Sci. 2009;50(10):4949–4956. doi: 10.1167/iovs.08-3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gee H.Y., et al. Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am. J. Hum. Genet. 2014;94(6):884–890. doi: 10.1016/j.ajhg.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gordon L.K., et al. EMP2 regulates angiogenesis in endometrial cancer cells through induction of VEGF. Oncogene. 2013;32(46):5369–5376. doi: 10.1038/onc.2012.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson H.L., et al. Epithelial membrane proteins induce membrane blebbing and interact with the P2X7 receptor C terminus. J. Biol. Chem. 2002;277(37):34017–34023. doi: 10.1074/jbc.M205120200. [DOI] [PubMed] [Google Scholar]
- 43.Gong X., et al. Association of kir genes with blood pressure responses to dietary sodium intervention: the GenSalt study. Hypertens. Res. 2018;4:1045–1053. doi: 10.1038/s41440-018-0113-6. [DOI] [PubMed] [Google Scholar]
- 44.Summers K.M., et al. Mutations at KCNQ1 and an unknown locus cause long QT syndrome in a large australian family: implications for genetic testing. Am. J. Med. Genet. A. 2010;152A(3):613–621. doi: 10.1002/ajmg.a.33274. [DOI] [PubMed] [Google Scholar]
- 45.Matic M., et al. Analgesia and opioids: a pharmacogenetics shortlist for implementation in clinical practice. Clin. Chem. 2017;63(7):1204–1213. doi: 10.1373/clinchem.2016.264986. [DOI] [PubMed] [Google Scholar]
- 46.Schultess J., Danielewski O., Smolenski A.P. Rap1GAP2 is a new GTPase-activating protein of Rap1 expressed in human platelets. Blood. 2005;105(8):3185–3192. doi: 10.1182/blood-2004-09-3605. [DOI] [PubMed] [Google Scholar]
- 47.Schubert P., et al. A signaling pathway contributing to platelet storage lesion development: targeting PI3-kinase–dependent Rap1 activation slows storage-induced platelet deterioration. Transfusion. 2009;49(9):1944–1955. doi: 10.1111/j.1537-2995.2009.02224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Francis S.H., et al. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010;62(3):525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee D.I., et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. 2015;519(7544):472–476. doi: 10.1038/nature14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Methawasin M., et al. Phosphodiesterase 9a inhibition in mouse models of diastolic dysfunction. Circulation. Heart Failure. 2020;13(5) doi: 10.1161/CIRCHEARTFAILURE.119.006609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Durand N., Borges S., Storz P. Protein kinase D enzymes as regulators of EMT and cancer cell invasion. J. Clin. Med. 2016;5(2):20. doi: 10.3390/jcm5020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wood B.M., Bossuyt J. Emergency spatiotemporal shift: the response of protein kinase D to stress signals in the cardiovascular system. Front. Pharmacol. 2017;8:9. doi: 10.3389/fphar.2017.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paradies G. Role of cardiolipin in mitochondrial function and dynamics in health and disease: molecular and pharmacological aspects. Cells. 2019;8(7) doi: 10.3390/cells8070728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X.X., et al. Cardiolipin and its different properties in mitophagy and apoptosis. J. Histochem. Cytochem. 2015;63(5):301–311. doi: 10.1369/0022155415574818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wincup C., Ioannou Y. The differences between childhood and adult onset antiphospholipid syndrome. Front. Pediatr. 2018;6:362. doi: 10.3389/fped.2018.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dudek J., Hartmann M., Rehling P. The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim. Biophys. Acta Mol. basis Dis. 2019;1865(4):810–821. doi: 10.1016/j.bbadis.2018.08.025. [DOI] [PubMed] [Google Scholar]
- 57.Vickers K.C., Moore K.J. Small RNA overcomes the challenges of therapeutic targeting of microsomal triglyceride transfer protein. Circ. Res. 2013;113(11):1189–1191. doi: 10.1161/CIRCRESAHA.113.302732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lauer M.S., et al. Cause of death in clinical research: time for a reassessment? J. Am. Coll. Cardiol. 1999;34(3):618–620. doi: 10.1016/s0735-1097(99)00250-8. [DOI] [PubMed] [Google Scholar]
- 59.Schoenfeld D.A. Sample-size formula for the proportional-hazards regression model. Biometrics. 1983;39(2):499–503. [PubMed] [Google Scholar]
- 60.Antithrombotic Trialists' Collaboration Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. Br. Med. J. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dézsi C.A., Szentes V. The real role of β-blockers in daily cardiovascular therapy. Am. J. Cardiovasc. Drugs. 2017;17:361–373. doi: 10.1007/s40256-017-0221-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodriguez F., et al. Association of Statin Adherence with Mortality in patients with atherosclerotic cardiovascular disease. JAMA Cardiol. 2019;4:206–213. doi: 10.1001/jamacardio.2018.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lau E.S. Potent P2Y12 inhibitors in men versus women: a collaborative meta-analysis of randomized trials. J. Am. Coll. Cardiol. 2017;69(12):1549–1559. doi: 10.1016/j.jacc.2017.01.028. 28. [DOI] [PubMed] [Google Scholar]
- 64.Turnbull F., et al. And the blood pressure lowering treatment Trialists' collaboration. Do men and women respond differently to blood pressure-lowering treatment? Results of prospectively designed overviews of randomized trials. Eur. Heart J. 2008;29:2669–2680. doi: 10.1093/eurheartj/ehn427. [DOI] [PubMed] [Google Scholar]
- 65.Fulcher J., the Cholesterol Treatment Trialists' (CTT) Collaboration Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385(9976):1397–1405. doi: 10.1016/S0140-6736(14)61368-4. 11. [DOI] [PubMed] [Google Scholar]
- 66.Temizhan A. Kardiyovasküler korumada aspirin: cinsiyete göre farkli bir yaklaşim var mi? [Aspirin in cardiovascular prevention: does the approach differ by gender?] Anadolu Kardiyol. Derg. 2007;7(Suppl. 2):2–4. Turkish. [PubMed] [Google Scholar]
- 67.Clayton J.A., Collins F.S. Policy: NIH to balance sex in cell and animal studies. Nat. News. 2014;509(7500):282. doi: 10.1038/509282a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williams R.A. Cardiovascular disease in african american women: a health care disparities issue. J. Natl. Med. Assoc. 2009;101(6):536–540. doi: 10.1016/s0027-9684(15)30938-x. [DOI] [PubMed] [Google Scholar]
- 69.Landry L.G., et al. Lack of diversity in genomic databases is a barrier to translating precision medicine research into practice. Health Aff (Millwood) 2018;37(5):780–785. doi: 10.1377/hlthaff.2017.1595. [DOI] [PubMed] [Google Scholar]
- 70.Mudd-Martin G. American Heart Association Council on Genomic and Precision Medicine; Council on Cardiovascular and Stroke Nursing; and Council on Clinical Cardiology. Considerations for cardiovascular genetic and genomic research with marginalized racial and ethnic groups and indigenous peoples: a scientific statement from the American Heart Association. Circulation Genomic Precis. Med. 2021;14(4) doi: 10.1161/HCG.0000000000000084. [DOI] [PubMed] [Google Scholar]
- 71.Ibrahim B.B., et al. The association between neighborhood vulnerability and cardiovascular health risk among Black/African american women of childbearing age in the InterGEN study. Nurs. Res. 2021;70(5S Suppl 1):S3–S12. doi: 10.1097/NNR.0000000000000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Condon E.M., et al. Racial discrimination, mental health, and parenting among african american mothers of preschool-aged children. J. Am. Acad. Child Adolesc. Psychiatry. 2021;61(3):402–412. doi: 10.1016/j.jaac.2021.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures S1, S2
Supplementary tables S1 - S5.