

Abstract
Background
Smooth muscle cells in atherosclerotic plaque take on multiple non-classical phenotypes that may affect plaque stability and therefore the likelihood of myocardial infarction or stroke. However, the mechanisms by which these cells affect stability are only beginning to be explored.
Methods
In this study, we investigated the contribution of inflammatory MCP1 produced by both classical Myh11+ smooth muscle cells and smooth muscle cells that have transitioned through an Lgals3+ state in atherosclerosis using smooth muscle lineage-tracing mice that label all Myh11+ cells and a dual lineage tracing system that targets Lgals3-transitioned SMC only.
Results
We show that loss of MCP1 in all Myh11+ smooth muscle results in a paradoxical increase in plaque size and macrophage content, driven by a baseline systemic monocytosis early in atherosclerosis pathogenesis. In contrast, knockout of MCP1 in Lgals3-transitioned smooth muscle cells using a complex dual lineage-tracing system resulted in lesions with an increased Acta2+ fibrous cap and decreased investment of Lgals3-transitioned smooth muscle cells, consistent with increased plaque stability. Finally, using flow cytometry and single-cell RNAseq, we show that MCP1 produced by Lgals3-transitioned smooth muscle cells influences multiple populations of inflammatory cells in late stage plaques.
Conclusions
MCP1 produced by classical smooth muscle cells influences monocyte levels beginning early in disease and was atheroprotective, while MCP1 produced by the Lgals3-transitioned subset of smooth muscle cells exacerbated plaque pathogenesis in late stage disease. Results are the first to determine the function of Lgals3-transitioned inflammatory smooth muscle cells in atherosclerosis and highlight the need for caution when considering therapeutic interventions involving MCP1.
Keywords: Smooth muscle cell, inflammation, atherosclerosis, MCP1, CCL2
Keywords: Smooth Muscle Proliferation and Differentiation, Growth Factors/Cytokines, Inflammation, Atherosclerosis
Graphical Abstract
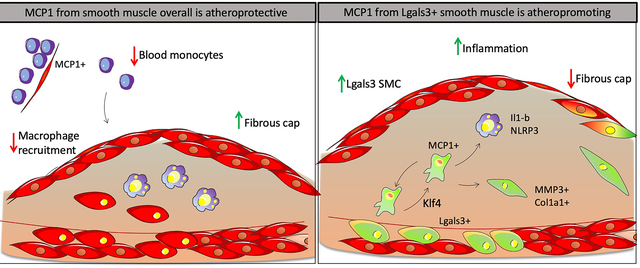
Introduction
Different smooth muscle cell (SMC) phenotypes have been hypothesized to have distinct roles in atherosclerotic lesion pathogenesis, some beneficial and some detrimental. The classical notion that SMC promote plaque stability by investment in the fibrous cap and production of matrix is supported by studies showing that SMC loss of Oct4 or Tcf21 result in decreased investment of SMC in the fibrous cap, increases in lesion size, and evidence of decreased stability, including decreased collagen production, increased lipid levels, and intraplaque hemorrhage.[1,2] In contrast, SMC loss of Klf4, a gene linked to increased risk for coronary disease (CAD) in human GWAS studies, resulted in lesions that were smaller, had increased fibrous cap thickness and decreased transition of SMC to an Lgals3+ state, implying that Lgals3+ phenotypically modulated SMC can have detrimental effects on lesion pathogenesis.[3] Extensive involvement of phenotypically modulated SMC in atherosclerosis has been documented by many groups[2,3] including subsets that take on osteogenic, macrophage-like, fibrocyte/ECM producing, and stem-like states, with each state potentially having different effects. In particular, the transition of SMC to a de-differentiated state in vitro and in vivo has been associated with expression of inflammatory genes including Vcam1, IL-6, and Mcp1/Ccl2, the protein products of which could influence both SMC and non-SMC in the lesion[4–6].
The cytokine MCP1, also known as CCL2, is one of several chemokines that play a key role in monocyte recruitment to atherosclerotic plaques.[7] There is evidence that MCP1 plays a critical role in the pathogenesis of atherosclerosis in humans, in that several polymorphisms in the MCP1 promoter thought to increase MCP1 expression have been associated with CAD, chronic stable angina, and myocardial infarction[7]. In addition, plasma levels of MCP1 are significantly associated with coronary artery atherosclerosis severity measured by angiography.[8] In mice, global MCP1 knockout on an LDLR−/− background results in reduced atherosclerotic lesion formation and recruitment of monocytes in models of acute injury.[9,10] Additionally, inhibition of MCP1 in ApoE−/− mice resulted in late stage plaque changes consistent with increased lesion stability such as increased collagen and a thicker fibrous cap.[11]
MCP1 expression is induced vigorously by monocytes stimulated in culture to differentiate into macrophages, leading to the belief that MCP1 is produced mostly by monocytes.[12] However, myeloid specific knockout of MCP1 fails to inhibit recruitment of monocytes to sites of acute inflammation in vivo, implying that non-myeloid cells play an important role.[13] Moreover, MCP1 produced by non-myeloid cells in the bone marrow vascular niche is crucial for recruitment of Ly6Chi monocytes into the blood in response to acute infection.[14] MCP1 transcript was first observed in aortic medial cells of primates more than a quarter century ago, and a recent study showed that MCP1 knockout in Nestin+ perivascular cells reduced fatty streak formation after 8 weeks of Western diet feeding.[6,15] However, the hypothesis that smooth muscle cells impact late stage plaque by producing MCP1 has never been tested.
Studies herein tested the hypothesis that SMC are a major source of MCP1 which exacerbates lesion pathogenesis by prolonging chronic inflammatory responses in late-stage atherosclerosis. However, completely contrary to expectations, SMC-specific conditional knockout of MCP1 resulted in larger atherosclerotic lesions with decreased indices of stability likely due to monocytosis resulting from loss of MCP1 from SMC and pericytes within hematopoietic cell niches. In contrast, selective knockout of MCP1 in a subset of SMC that transition through an Lgals3+ activation state within lesions induced the opposite phenotype, displaying smaller lesions that exhibited a thicker protective ACTA2+ fibrous cap and reduced Ly6Chi macrophages. Taken together, these results show that the same gene in different subsets of SMC exert different actions on lesion pathogenesis. In addition, results highlight the need for caution when considering systemically acting therapies in humans, as the effects will vary based on what cell types are dominant during the period of treatment.
Methods
All single-cell RNAseq data has been made publicly available at GEO [GSE204994] at [https://ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE204994]. All other data or materials in the study not available in the article and supplementary files is available on request from the authors.
Mice
All experiments followed guidelines of the University of Virginia Animal Care and Use Committee (Protocol 2400). Myh11-DreERT2 Lgals3-Cre Rosa-tdTomato-eGFP ApoE−/− mice and Myh11-CreERT2 eYFP ApoE−/− Klf4wt/wt and Klf4Δ/Δ mice have been previously described.[3,16,17] MCP1-mCherryflox/flox and MCP1flox/flox mice were purchased from The Jackson Laboratory (stock# 016849 and 023347, respectively). To generate true littermate controls avoiding the effect of genetic drift, we bred Myh11-CreERT2 eYFP ApoE−/− mice with MCP1-mCherryflox/flox mice to produce heterozygous breeder mice with genotypes Myh11-CreERT2 eYFP ApoE−/− MCP1wt/flox, used to generate Myh11-CreERT2 eYFP ApoE−/− MCP1wt/wt, MCP1wt/flox or MCP1flox/flox experimental mice. Similarly, Myh11-DreERT2 Lgals3-Cre Rosa-tdTomato-eGFP ApoE−/− mice and MCP1flox/flox were bred to produce Myh11-DreERT2 Rosa-tdTomato-eGFP ApoE−/− MCP1wt/flox and Lgals3-Cre Rosa-tdTomato-eGFP ApoE−/− MCP1wt/flox mice, used to generate Myh11-DreERT2 Lgals3-Cre Rosa-tdTomato-eGFP ApoE−/− MCP1wt/wt, MCP1wt/flox, or MCP1flox/flox experimental mice. All experimental mice were injected with 0.1mL of 1mg/mL tamoxifen dissolved in peanut oil (Sigma #T-5648) for 10 injections from weeks 6–8 of age. Mice were fed a Western diet containing 21% milk fat and 0.15% cholesterol (Tekland, TD.88137) or standard laboratory diet (Tekland, Irradiated LM-485) for 18 weeks. Since the Myh11-CreERT2 is located on the Y chromosome, only male mice were analyzed in these studies. However, because Myh11-DreERT2 does not have this limitation, both male and female mice were analyzed.
Immunohistochemistry and plaque morphometry
After 18 weeks of Western diet, brachiocephalic arteries were harvested, paraformaldehyde fixed, embedded in paraffin, and serially cut in 10-μm thick sections from the aortic arch to the bifurcation of the right subclavian artery. For morphometric analysis, we performed modified Russell–Movat staining on three locations along the BCA at 150 μm, 450 μm, and 750 μm from the aortic arch, as previously described[18].
Immunofluorescent staining and quantification
Paraffin BCA sections were deparaffinized, rehydrated, antigen retrieved (Vector Laboratories Antigen Unmasking Solution, Citrate-Based H-3300–250 ), and blocked with fish skin gelatin PBS (6 g/L) containing 10% horse serum for 1 hour at room temperature. Frozen sections were permeabilized with acetone at −20 degrees for 10 minutes. Slides were incubated with the antibodies shown in the Major Resources Table. Slides were imaged using a Zeiss LSM700 confocal microscope at 20x magnification and 0.5x zoom to acquire a series of z-stack images at 1-μm intervals. Quantification was done with manual DAPI+ single-cell counting in 1-μm z-stacks. Fibrous cap area was quantified as contiguous layers of ACTA2+ cells in contact with the lumen. Fibrous cap cell type was also quantified as DAPI+ cells in a uniform area 30 μm from the lumen along the length of the plaque. Maximal intensity projection was used to generate the representative images included in the figures, and Adobe Photoshop was used to process and format those images.
Monocyte bead tracing
Injection of fluorescent beads for monocyte tracing was done as previously described[18]. In brief, Myh11-CreERT2 eYFP ApoE−/− MCP1 mice fed Western diet for 16 weeks received an injection of Fluoresbrite Polychromic Red 1.0 μm beads (2.5% in solids-latex, Polysciences Inc., #18660). Beads were diluted 1:4 in sterile PBS and 250μl of the mixture was injected via tail vein. At 18 weeks of Western diet, animals were harvested, and 10-μm frozen sections were taken of the BCA. Lesional macrophage content was determined by single-cell counting of fluorescent beads as described above.
Flow cytometry
After 18 weeks of Western diet, experimental mice were euthanized with CO2 and peripheral blood was collected via cardiac puncture. Mice were then perfused with 20mL PBS, and aortas cleaned of peripheral adipose tissue were collected. Aortic samples were minced in an enzyme cocktail containing 4 U/ml Liberase TM (Roche 05401119001) and placed in a 37 °C incubator for 1.5 h. Cells were run through a 70-μm strainer and spun down at 500g for 5 min. Peripheral blood samples and aortic samples were resuspended in red blood cell lysis buffer (BD PharmLyse 555899) for two minutes and then inactivated using serum containing media, spun down again, and resuspended in 200 μl of PBS. Cells were stained with Live/Dead dye and a panel of fluorescent antibodies in the Major Resources Table, with aliquots of cells stained as fluorescent-minus-one controls. Samples were compensated and run on an LSR Fortessa flow cytometer and live, single cells were gated sequentially by CD45, CD11b, and Ly6C vs Ly6G and quantified using FlowJo software (https://www.flowjo.com).
Atherosclerotic plaque scRNAseq
Individual atherosclerotic plaques from the brachiocephalic artery were removed from underlying media with forceps and deposited into FACS buffer (1% BSA in PBS) plus 1 ug/mL Actinomycin D (Gibco, #11805017). Samples were then minced and digested with 1mL Liberase (Roche, #355374) plus 1ug/mL Actinomycin D for 1 hour at 37 degrees, then resuspended in PBS plus 0.04% UltraPure non-Acetylated BSA (ThermoFisher #AM2616) and filtered through 30μm filters on ice. Samples were then either counted and submitted directly for scRNAseq or sorted by flow cytometry. Samples for flow were stained with Sytox Blue viability dye and sorted with a BD FACSAria Fusion cell sorter with gating on live, single cells that were tdTomato+ or GFP+. Cells were collected into LoBind tubes containing 100ul 0.04% non-acetylated BSA. 2,000 cells in each group were targeted in Chromium 10X genomics libraries, which after barcoding, were pooled and sequenced on the Illumina NextSeq™, 400M paired-end reads. Reads were aligned against mouse genome mm10 using the CellRanger software (10x Genomics). Gene-barcode matrices were analyzed in R using Seurat v3. Cells were filtered for minimum (200) and maximum (5000) reads per UMI as well as for 10% or less mitochondrial and less than 5% hemoglobin gene content. Significant principal components of variation (PCs) were calculated using JackStraw test with 10000 repetitions, and clusters were calculated with 19 PCs. Raw data is available at GEO (submission pending). Differential expression analysis was done using MAST. Code is available upon request.
Statistical analysis
Statistics were performed using GraphPad Prism 7. For comparison of two groups of continuous variables with normal distribution and equal variances, two-tailed unpaired Student’s t-tests (with additional Welch’s correction for unequal variances, or non-parametric testing for non-normal distribution) were performed with a significance threshold of p ≤0.05. For multiple group comparison, we performed one-way or two-way analysis of variance (ANOVA) followed by the Sidak method of multiple pairwise comparisons. For data that was not normally distributed, we performed Kruskal Wallis test followed by Dunn’s test for multiple comparisons. For categorical variables, we used a X2 analysis. The number of mice used for each analysis is indicated in the figure legends.
Results
Vascular smooth muscle cells in Western diet-fed ApoE−/− mice express MCP1 in a Klf4-dependent manner
To test if smooth muscle cells express MCP1 in the context of atherosclerosis, we performed RNAscope fluorescent in situ hybridization analysis on brachiocephalic artery (BCA) sections from wild-type and ApoE−/− mice. While wild-type mice had no detectable hybridization signal with our MCP1 probe, medial cells from mice on an ApoE−/− background had noticeable staining at baseline, which was increased markedly in the medial and endothelial layer upon Western diet feeding (Supplemental Figure 1). Further, cultured mouse aortic SMC upregulated MCP1 transcription after 3 days of cholesterol loading (Figure 1A). Of major interest, SMC-specific Klf4 knockout completely abolished MCP1 production in response to cholesterol loading (Figure 1A). Previous work from our lab has shown that SMC-specific knockout of Klf4 resulted in smaller lesions with significant increases in Acta2+ fibrous cap thickness. As such, loss of MCP1 expression by SMC may have contributed to these beneficial changes. Immunostaining and high-resolution confocal microscopy of advanced BCA lesions sections from our Myh11-CreERT2 eYFP ApoE−/− Klf4wt/wt and Klf4Δ/Δ mice (Klf4SMC wt/wt, Klf4SMC Δ/Δ) showed a decrease in MCP1 protein expression in Klf4SMC Δ/Δ lesions (Figure 1 B–C), although this cannot be directly attributable to SMC production because MCP1 is a soluble protein. However, protein expression allows us to examine specifically the plaque area and account for turnover dynamics known to affect MCP1 regulation in other cell types. Taken together, these results suggest that SMC are a source of Klf4-dependent MCP1 production during progression of atherosclerosis.
Figure 1. Smooth muscle cells express MCP1 in an Klf4-dependent manner.
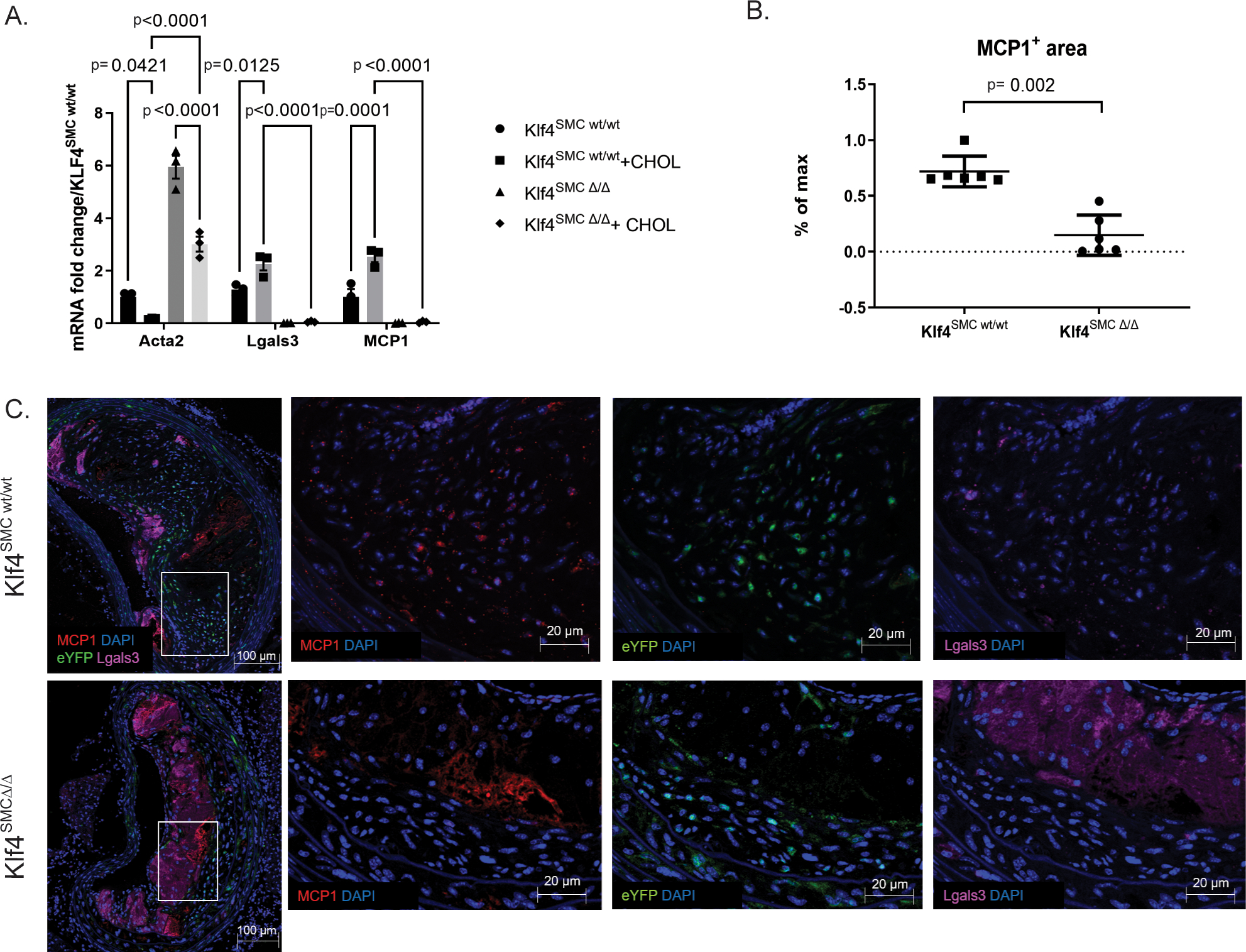
A) Cultured aortic SMC from SMC lineage-tracing mice on an ApoE background with or without SMC specific Klf4 knockout (Klf4SMC) were were treated with 0.2% FBS +/− 80 ng/μL cholesterol media for three days, and qRT-PCR was performed for SMC and macrophage marker genes. n=3, comparisons by two-way ANOVA and Tukey’s multiple comparisons test. B) After 18 weeks of Western diet feeding, brachiocephalic artery (BCA) lesions collected from Klf4SMC wt/wt and Klf4SMC Δ/Δ mice were stained for eYFP(marking lineage-traced SMC), LGALS3, MCP1, and DAPI. MCP1+ area per lesion quantified by pixilation. n=6 mean +/− SEM. p<0.001 by Mann Whitney test C) Representative images are shown from the experiment in B.
Heterozygous loss of MCP1 in SMC exacerbates atherosclerosis
To address the hypothesis that SMC exacerbate atherosclerotic lesion pathogenesis at least in part through secretion of MCP1, we crossed mice from our previously published smooth muscle cell specific lineage tracing system[3], Myh11-CreERT2 Rosa-eYFP ApoE−/−, with mice carrying MCP1-mCherry flanked by loxP sites (broadly referred to as MCP1SMC mice). We then induced MCP1 knockout in smooth muscle cells by tamoxifen injection at 6 weeks of age, and fed the mice a Western diet for 18 weeks to induce advanced atherosclerotic lesion formation. Myh11-CreERT2 Rosa-eYFP ApoE−/− MCP1fl/fl (MCP1SMCΔ/Δ) aortas showed tamoxifen-dependent recombination at the MCP1 locus (Supplemental Figure 2). Notably, MCP1 serum levels were not affected by SMC-specific MCP1 knockout (Supplemental Figure 3). We had hypothesized that loss of the inflammatory cytokine MCP1 in smooth muscle cells would result in smaller lesions with increased indices of stability. Surprisingly, we found the opposite result- an increase in BCA lesion size in the heterozygote Myh11-CreERT2 Rosa-eYFP ApoE−/− MCP1wt/Δ mice (MCP1SMC wt/Δ) (Figure 2A–B). MCP1SMC wt/Δ mice also had an increased number of LGALS3+ macrophages in lesions compared to MCP1SMCΔ/Δ (Figure 2C, F), while the total number of SMC-derived cells remained the same (Figure 2E). Finally, MCP1SMC wt/Δ mice had a significantly decreased fibrous cap thickness compared to wild type animals (Figure 2D). Total lesion collagen content was unchanged between the groups (Supplemental Figure 4). Interestingly, all of these changes were completely absent in the MCP1SMCΔ/Δ group, which was indistinguishable from wild type littermate control mice for all of the above measures. Given that SMC-specific MCP1 genetic knockout was highly efficient (Supplemental Figure 2), we reasoned that the MCP1SMCΔ/Δ group may have increased production of MCP1 from non-SMC sources and/or increased expression of other CC-family cytokines that have considerable sequence homology with MCP1, several of which can activate the MCP1 receptor.[7] However, our observations in the MCP1SMC wt/Δ mice may still be informative, because mutations in the MCP1 promoter have been associated with modest changes in expression that are better modeled in our heterozygous MCP1SMC wt/Δ knockout mice.[19]
Figure 2. MCP1SMC wt/Δ mice have significantly larger BCA lesions following 18 weeks Western diet.
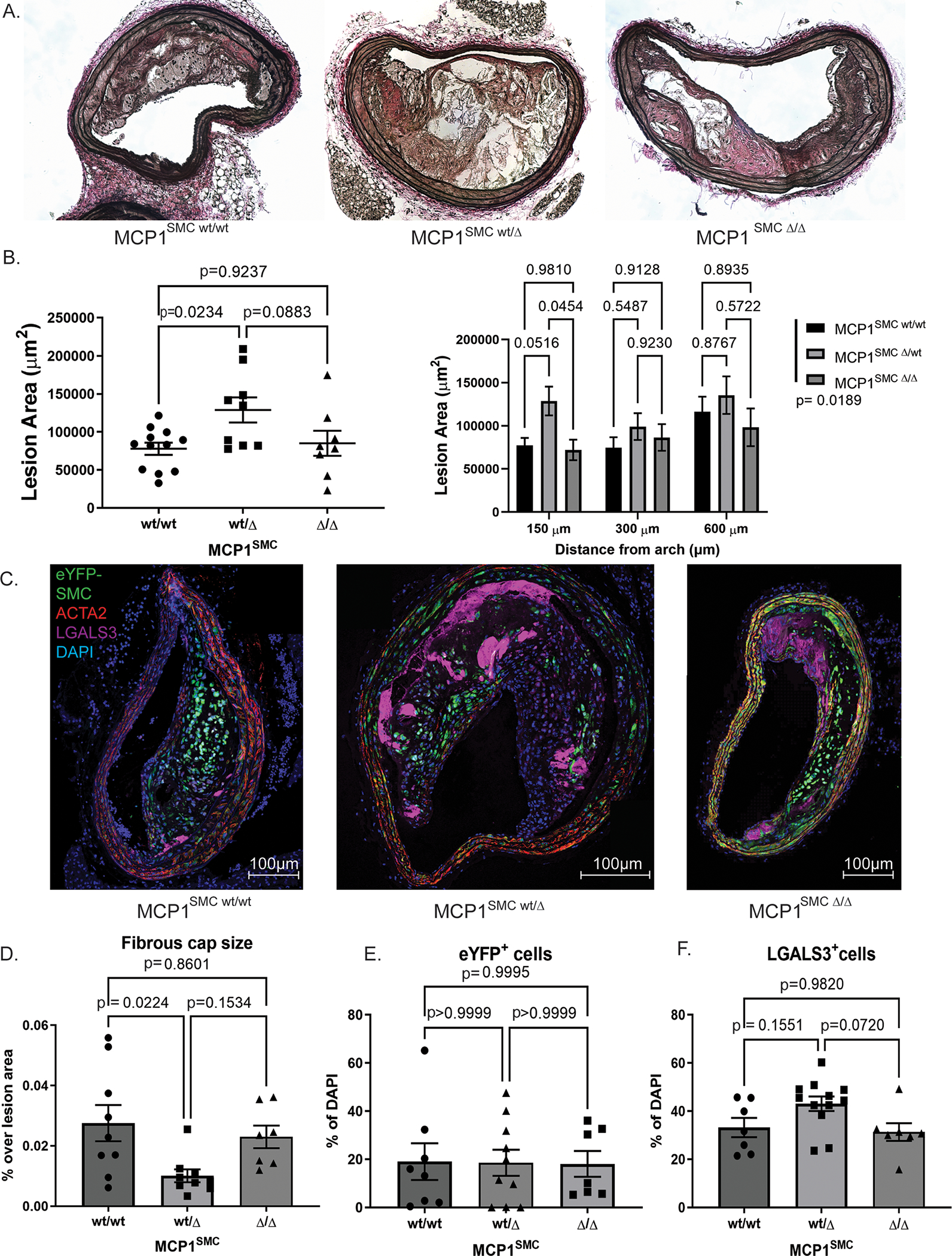
A) Representative images from serial sections of the BCA from MCP1SMC wt/wt, MCP1SMC wt/Δ and MCP1SMC Δ/Δ mice. B) Quantification of BCA area from mice in A. The left panel shows data at 150 μm from the arch analyzed by pairwise Sidak’s multiple comparison test after one way ANOVA. The right panel shows data from three locations along the BCA with pairwise comparisons analyzed by Sidak’s multiple comparison test after two way ANOVA. The legend p value represent total variation due to genotype analyzed by two way ANOVA. C) BCA sections from mice in A were immunostained for eYFP (marking lineage-traced SMC), ACTA2, LGALS3 and DAPI. C shows maximum intensity projections of representative images from each genotype. D) Fibrous cap size was measured as area of continuous ACTA2+ layers directly below the lumen (and endothelial layer) and normalized to total lesion area. E-F) Cells were quantified by single and double marker staining by single cell counting and normalized to total lesion DAPI stained cells. E shows that eYFP+ cells (SMC derived cells) were unchanged when normalized to total cells. F) MCP1SMC wt/Δ animals have greater numbers of LGALS3+ cells (eYFP+ or eYFP−) compared to MCP1SMC Δ /Δ animals. D-F) n=7–9, mean +/− SEM. P values represent pairwise Sidak’s multiple comparison test after one way ANOVA.
SMC-specific knockout of MCP1 results in sustained elevation of circulating Ly6Chi monocytes
Contrary to our expectations, the differences in MCP1SMC wt/Δ mouse lesions were not consistent with a detrimental inflammatory role for MCP1 in SMC but rather showed evidence of increased inflammation, with larger lesions and increase in non-SMC derived macrophage accumulation. Because MCP1 is known to influence migration of Ly6Chi inflammatory monocytes[14], peripheral blood samples from MCP1SMC mice were also taken at 18 weeks and examined by flow cytometry for leukocyte content. Of major interest, loss of smooth muscle cell MCP1 resulted in a dose dependent increase in Ly6Chi monocytes in peripheral blood, not only after 18 weeks of Western diet but also when blood was taken only one week after knockout induction (Figure 3A–C). Given that several recent studies of atherosclerosis have shown that systemic increases in circulating monocytes drives lesion expansion and macrophage content by increased recruitment[20–22], we reasoned that this was also a likely explanation for our findings that MCP1SMC wt/Δ mice had larger and less stable lesions. To study the dynamics of monocyte recruitment and efflux in MCP1SMC mice, we administered fluorescent beads by tail vein injection to MCP1SMC mice after 16 weeks of Western diet and measured bead content in lesions after 18 weeks of Western diet. MCP1SMC wt/Δ mice showed a significant increase in bead content of lesions, indicating increased recruitment of monocytes and suggesting that the increased number of monocytes in blood was not due to a generalized decrease in recruitment into tissues (Figure 3D–G). Interestingly, MCP1SMCΔ/Δ lesions did not show increased bead-traced monocytes in lesions (Figure 3E), although they exhibited the greatest increase in serum Ly6Chi monocytes (Figure 3A). This may indicate that some residual MCP1 production by SMC, as well as blood monocyte level, is important for Ly6Chi monocyte accumulation in the vessel wall.
Figure 3. MCP1SMC wt/Δ and MCP1SMC Δ/Δ mice have significantly more Ly6Chi monocytes in peripheral blood.
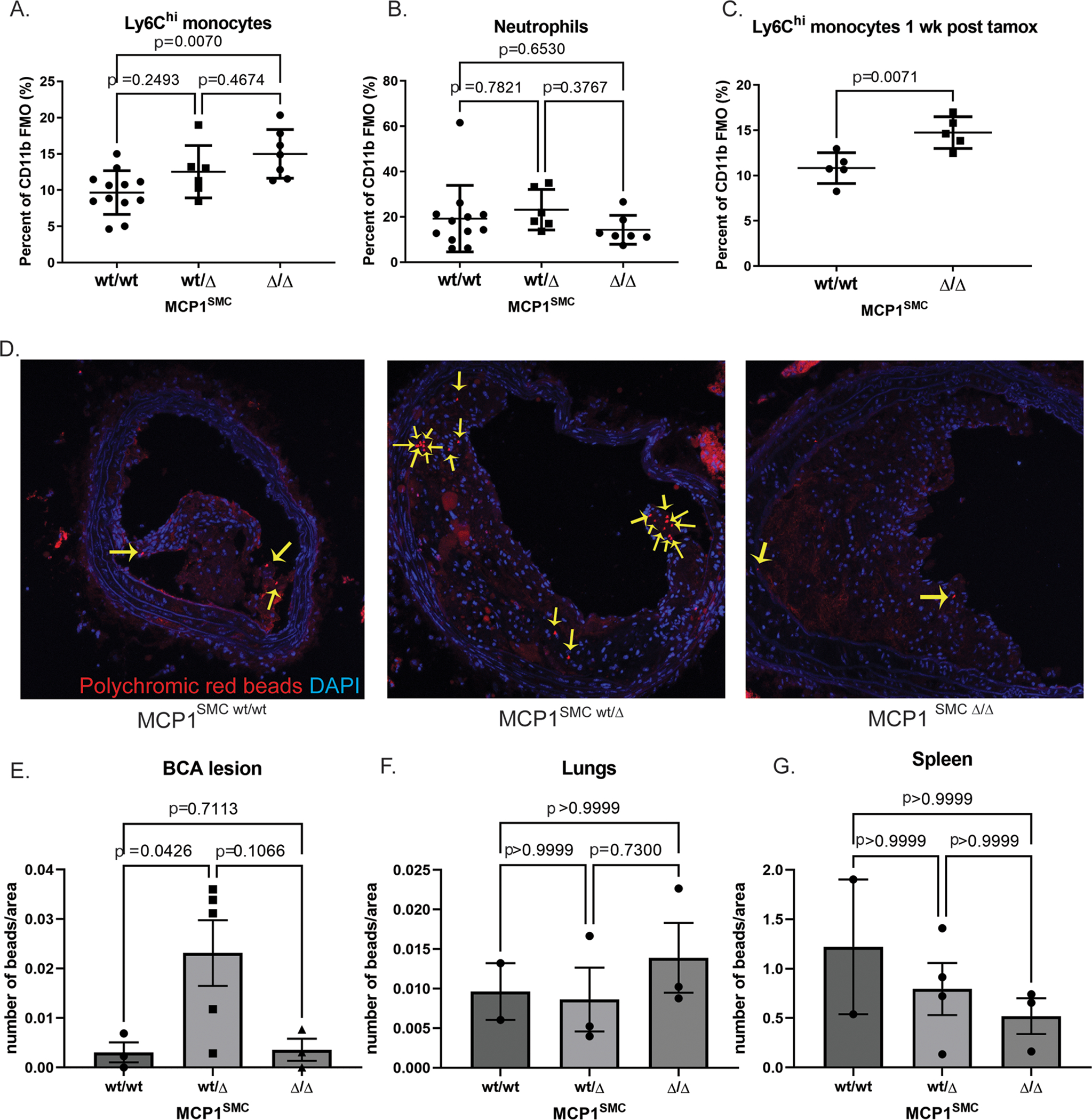
A-C Peripheral blood from MCP1SMC wt/wt, MCP1SMC wt/Δ and MCP1SMC Δ/Δ mice was collected after 18 weeks of Western diet (A-B) or by retro-orbital bleed 1 week after tamoxifen injection (C), stained for leukocyte markers, and analyzed by flow cytometry. Cells were gated on live, single CD45+CD11b+ Ly6G- (A,C) or Ly6G+ cells (B) with fluorescence-minus-one controls. n=5–9, mean +/− SEM. P values represent pairwise Sidak’s multiple comparison test after one way ANOVA for A and B, and C is a Mann-Whitney test. D) MCP1SMC mice fed 15 wks Western diet were tail-vein injected with fluorescent beads to label circulating monocytes. BCA plaques were collected 3 weeks later after 18 weeks of Western diet and frozen sections were imaged for beads (yellow arrows) within lesion cells. Representative images from this experiment are shown in D. E) Beads from the experiment in D were quantified as a percent of lesion area and analyzed by Kruskal-Wallace test with multiple comparisons. F-G) Bead content per area was also measured in non-atherosclerotic lung tissue to assess possible changes in overall tissue monocyte flux and in spleens to measure monocyte efflux. Three locations 100 μm apart were sampled and averaged for each animal.
Taken together, these experiments show that SMC-specific MCP1 knockout is associated with increased Ly6Chi monocyte levels in peripheral blood and that this likely contributes to exacerbation of atherosclerosis and increased macrophages in lesions. While the mechanism of the increase in Ly6Chi monocytes is not a peripheral recruitment defect (Figure 3F–G), and serum MCP1 levels were also unchanged (Supplemental Figure 3), it is possible that MCP1SMC knockout is acting at the level of monocyte entry into the vasculature from hematopoietic niches, a hypothesis supported by previous studies showing that Nestin+ perivascular cells regulate monocyte entry into the vasculature through MCP1.[14,15] It was previously thought that SMC did not participate in permeability or cell entry into sinusoidal vessels.[23] However, careful sectioning and immunofluorescence microscopy of bones from Myh11-CreERT2 SMC lineage tracing mice revealed SMC-derived cells separated from arterioles and visible blood vessels and in close contact with hematopoietic cells (Supplemental Figure 5), indicating that SMC-derived cells do exist in the hematopoietic niche and may overlap with the previously identified Nestin+ perivascular population. Regardless of the mechanism by which SMC-specific MCP1 expression may control monocyte trafficking, the Myh11-CreERT2 model of MCP1 knockout clearly exerted effects far before the appearance of phenotypically switched pro-inflammatory Lgals3+ SMC in late-stage atherosclerotic lesions which is the intended target of our studies.
MCP1 knockout in a subset of lesion SMC that transition through an Lgals3 activation state resulted in decreased lesion size and a thicker fibrous cap
We recently generated a dual recombinase lineage tracing mouse (Myh11-DreERT2 Lgals3-Cre Rosa-tdTomato-eGFP ApoE−/−, full validation in Alencar, Owsiany et al 2020)[16] that allows for lineage tracing and functional characterization of SMC that had lost expression of typical SMC markers including Acta2 but activated Lgals3. In brief, the Myh11 promoter drives a tamoxifen-inducible Dre recombinase, which removes a roxed stop cassette in the Rosa locus in front of a tdTomato-STOP-eGFP fluorescent reporter and a roxed stop cassette on the Lgals3 locus in front of an IRES-Cre. If this tdTomato+ SMC goes on to express Lgals3, it will express Cre and remove the tdTomato flanked by loxP sites, allowing expression of eGFP (a brief animation explaining this mouse model can be found at https://www.cvrc.virginia.edu/Owens/educationaltopics.html). We[3] and others[24,25] originally interpreted activation of Lgals3 as evidence that SMC had transitioned to a macrophage-like state. However, results of recent lineage tracing and scRNAseq analyses showed that Lgals3 expression in these cells does not signify their transition to a macrophage-like state. These Lgals3+ cells give rise to 3–4 distinct transcriptomic clusters including SMC-derived myofibroblast phenotype, osteochondrogenic cells, and of great interest to our studies, a pro-inflammatory phenotype[2,16,26,27].
To determine the role of MCP1 production by SMC-derived cells that go through an Lgals3 activation state (i.e., transition from tdTomato+ to GFP+) in late stage lesions, we crossed our Myh11 Lgals3 dual recombinase mouse with ApoE−/− mice containing floxed alleles of MCP1. In using the dual recombinase system, exons 1 and 2 of Mcp1 will be excised only in eGFP+, Lgals3-transitioned SMC. To avoid the effects of genetic drift and homozygous random insertion of transgenes, we performed extensive back-crosses of mice to generate breeder pairs containing all necessary Myh11 Lgals3 dual recombinase transgenes and having heterozygous floxed MCP1 alleles. As such, when Myh11-DreERT2+/− Rosa-tdTomato-eGFP+/+ ApoE−/− MCP1wt/fl mice were crossed with Lgals3-Cre+/− Rosa-Tomato-GFP+/+ ApoE−/− MCP1wt/fl mice, we generated experimental Myh11-DreERT2+/− Lgals3-Cre+/− Rosa-tdTomato-eGFP+/+ ApoE−/− mice only differing by having MCP1wt/wt, MCP1wt/fl, or MCP1fl/fl alleles (broadly refered to as MCP1SMC-Lgals3 mice). To test the specificity and efficiency of this knockout, eGFP+ cells were sorted from atherosclerotic aortas of MCP1SMC-Lgals3 wt/wt or MCP1SMC-Lgals3 fl/fl mice fed a Western diet for 18 weeks to induce advanced lesions. Results in Supplemental Figure 6 show knockout of MCP1 predominantly in eGFP+ cells from the knockout animals. Importantly, no GFP+ cells were observed in bone marrow sections from these mice (Supplemental Figure 5C) and no difference in circulating Ly6Chi monocytes were detected between MCP1 genotypes (Supplemental Figure 7). This mouse could thus test the hypothesis that MCP1 produced by SMCs phenotypically modified through a Lgals3-transitioned state in late stage lesions is detrimental for lesion pathogenesis.
After 18 weeks of Western diet, lesions were analyzed for size and surrogate indices of plaque stability, including fibrous cap area. Fibrous cap area, measured as continuous ACTA2+ layers beneath the endothelium, was significantly increased in MCP1SMC-Lgals3 Δ/Δ animals (Figure 4A–B). However, lesion size overall was unchanged between MCP1SMC-Lgals3 wt/wt, MCP1SMC-Lgals3 wt/Δ and MCP1SMC-Lgals3 Δ/Δ animals (Figure 4C–D). Similar to our findings in MCP1SMCΔ/Δ mice, the total number of lineage-traced SMC in lesions was unchanged with MCP1 knockout in the Lgals3 transitioned subset of SMC (Figure 4E). However, the frequency of Lgals3+ cells, both SMC and non-SMC derived, were not increased by MCP1SMC-Lgals3 knockout (Figure 4F–G). These findings contrast with MCP1 knockout in all Myh11+ SMC, which resulted in larger less stable lesions, and suggest that MCP1 produced by Lgals3+ SMC negatively affects atherosclerotic lesion pathogenesis.
Figure 4. Selective knockout of MCP1 in SMC that have gone through an Lgals3+ state resulted in lesions with a thicker fibrous cap.
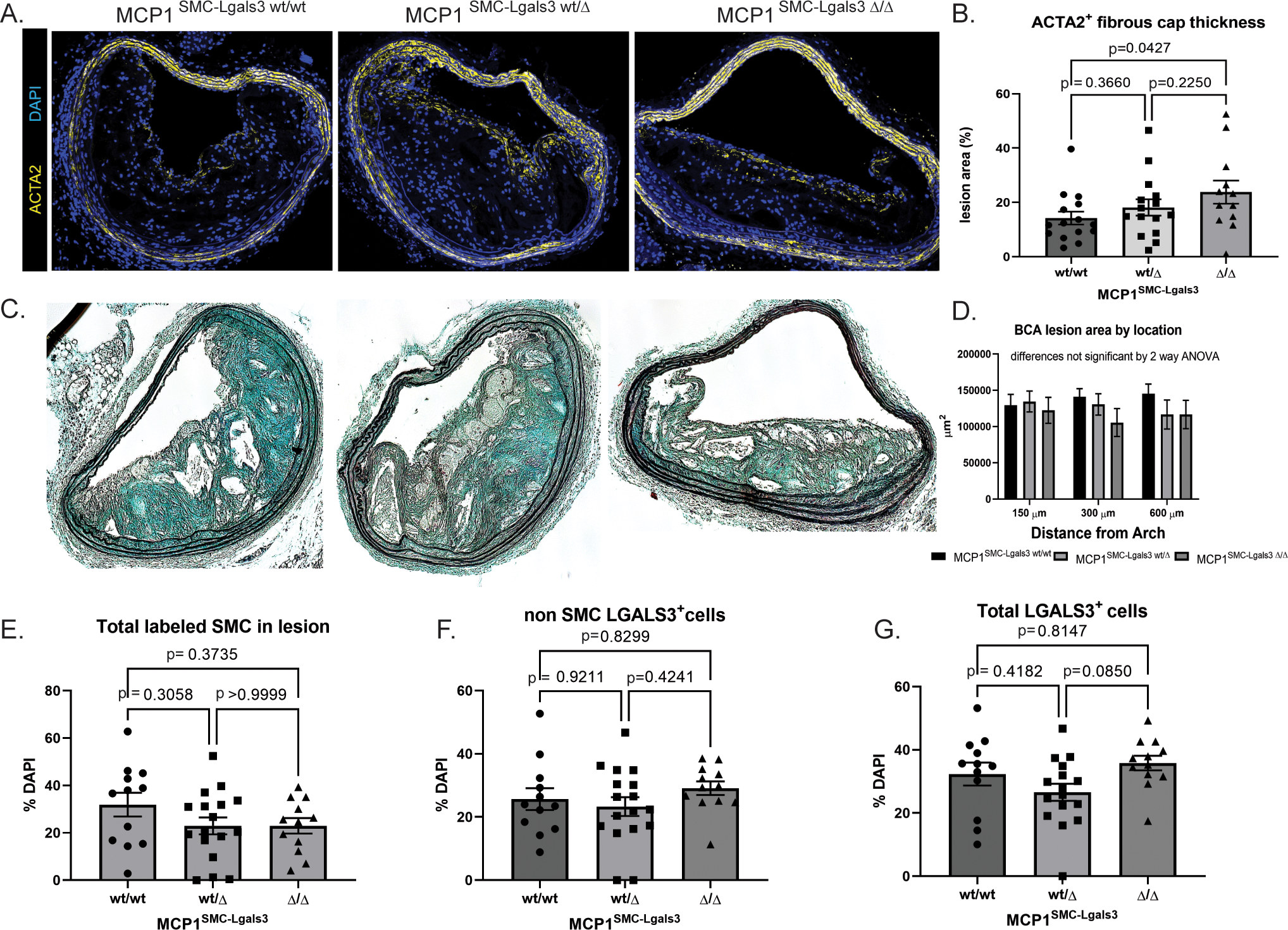
A) MCP1SMC-Lgals3 wt/wt, MCP1SMC-Lgals3 wt/Δ, and MCP1SMC-Lgals3 Δ/Δ mice were fed a Western diet for 18 weeks and 10μm sections from the BCA were immunostained with ACTA2 and DAPI. Panel A shows representative images from each genotype. B) Quantification of ACTA2+ cap thickness mice from A. Fibrous cap size was measured as area of continuous ACTA2+ layers directly below the lumen (and endothelial layer) and normalized over total lesion area. The p value represents comparison between MCP1SMC-Lgals3 wt/wt and MCP1SMC-Lgals3 Δ/Δ using Sidak’s multiple comparison test following one way ANOVA. The p value for linear trend is 0.0437. C) Representative images from Movat stained BCA sections from MCP1SMC-Lgals3 mice. D) BCA lesion area was quantified at three locations in mice from C, n=12–15, mean +/− SEM. E) BCA sections from animals in C were immunostained with GFP+, tdTomato+ (SMC), LGALS3, and DAPI. Total GFP+ and tdTomato+ SMC in lesions were quantified by single cell counting. F) GFP− tdTomato− LGALS3+ cells and G) total LGALS3+ cells were unchanged across the three MCP1SMC-Lgals3 genotypes. N=12–15 mice, error bars show SEM. No significant differences were observed in E-G by one way ANOVA.
Because MCP1SMC-Lgals3 mice are not restricted by Myh11-recombinase transgene placement on the Y chromosome that has limited sex comparison in previous SMC-specific mouse models[1,3,17], we included mice of both sexes in our experiments (Supplemental Figure 9). We did not observe any statistically significant differences in plaque size or inflammatory cell content based on sex. However, these studies did not intend to test sex-specific effects and were not powered to do so due to the extreme difficulty of generating these highly complex mice. Future studies will be needed to determine if there are significant sex differences.
MCP1 knockout in a subset of lesion SMC that transition through an Lgals3 activation state resulted in decreased SMC phenotypic transition and aortic inflammation
Dual lineage tracing allows for the observation of Myh11+ lineage traced SMC that undergo subsequent activation of Lgals3 by a color switch from tdTomato to GFP. In a previous study, our group showed that in wild-type animals >60% of SMC in lesions were GFP+, indicating they had activated Lgals3.[16] When we compared GFP+ SMC as a fraction of the SMC lineage traced (tdTomato or eGFP) total, we observed that although most lesions that contain lineage traced SMC are GFP-dominant in the MCP1SMC-Lgals3 wt/\wt group (Supplemental Figure 8), MCP1SMC-Lgals3 Δ/Δ lesions had fewer total GFP+ cells (Figure 5A–C). This decrease was significant for MCP1SMC-Lgals3 wt/wt vs MCP1SMC-Lgals3 Δ/Δ lesions when analyzing GFP+ cells in the lesion by simple percentage of all lesion cells (p=0.0169). As a categorical variable counting the number of lesions that contained or did not contain GFP+ cells, both MCP1SMC-Lgals3 wt/Δ and MCP1SMC-Lgals3 Δ/Δ lesions had fewer total GFP+ cells compared to lesions of MCP1 SMC-Lgals3 wt/wt mice (Figure 5C, chi-squared p-values <0.01). These results may indicate that MCP1 produced by Lgals3-transitioned, GFP+ SMC plays some part in driving their accumulation in the lesion, consistent with reports that MCP1 promotes the proliferation and migration of SMC into the neointima in a wire injury model.[28]
Figure 5.
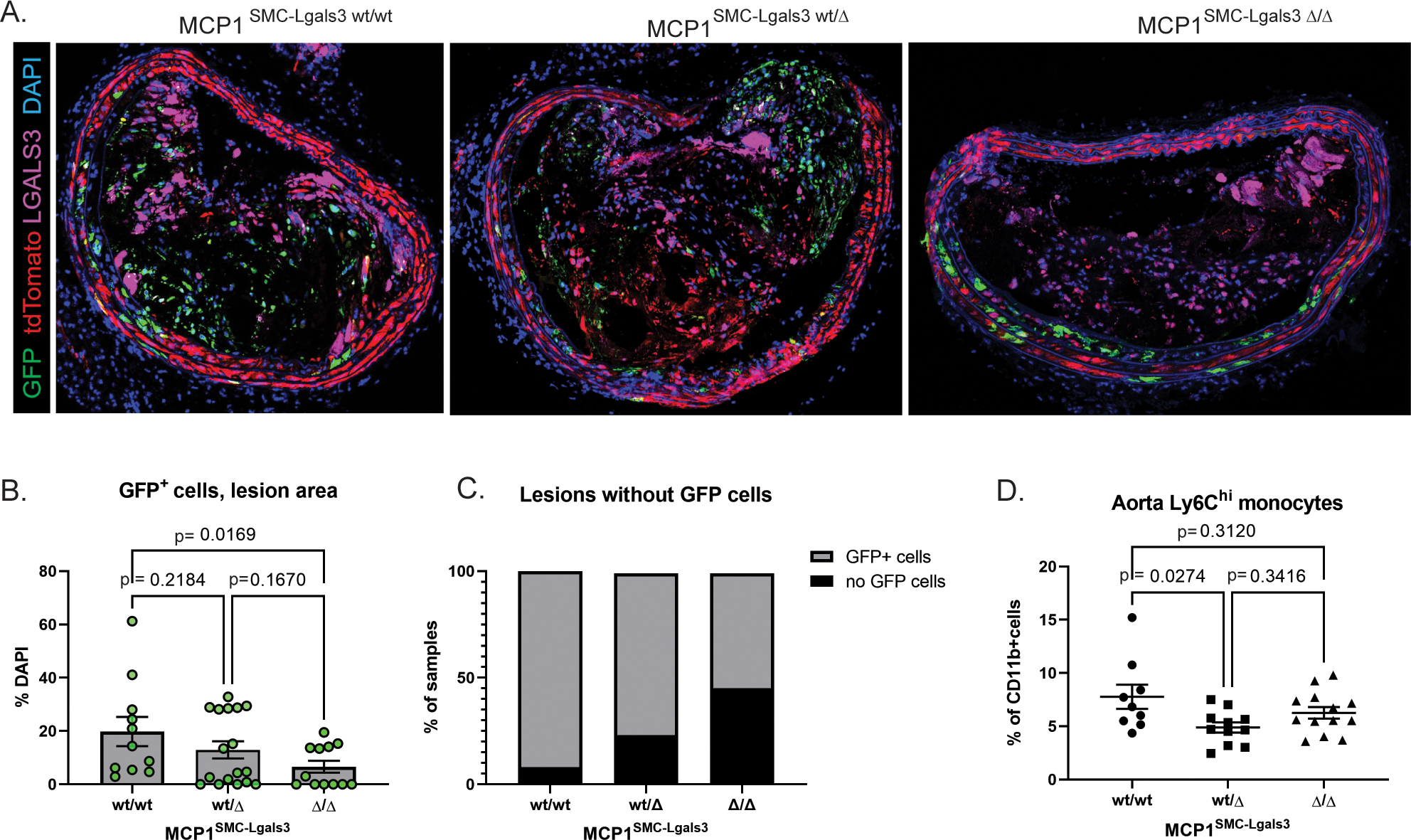
MCP1 knockout in a subset of lesion SMC that transition through an Lgals3+ activation state results in decreased SMC phenotypic transition and aortic inflammation. A) BCA lesions from MCP1SMC-Lgals3 wt/wt, MCP1SMC-Lgals3 wt/Δ, and MCP1SMC-Lgals3 Δ/Δ mice fed a Western diet for 18 weeks were immunostained for GFP, tdTomato, LGALS3, and DAPI. B)Total GFP+ cells per DAPI were counted manually in 1μm sections from confocal images in Panel A. n=12–15, p-values refer to pairwise comparisons with Kruskal-Wallace test. C) BCA sections from Panels A-B were classified as a categorical variable based on entry or non-entry of GFP+ cells into lesions. Chi-squared test p=0.0001. D) Aortas from MCP1SMC-Lgals3 wt/wt, MCP1SMC-Lgals3 wt/Δ, and MCP1SMC-Lgals3 Δ/Δ mice fed a Western diet for 18 weeks were digested, stained for lymphocyte markers, and analyzed by flow cytometry. Ly6Chi monocytes were gated out of live, single CD45+CD11b+LY6G− leukocytes using fluorescence-minus-one controls. Despite being unchanged in peripheral blood at 18 weeks of Western diet, Ly6Chi monocytes were decreased in aortas of MCP1SMC-Lgals3 wt/Δ mice. n=10–12, p-values refer to Sidak multiple comparison test after one way ANOVA.
Because of the known role for MCP1 in recruiting immune cells to sites of inflammation, we also performed flow cytometry on atherosclerotic aortas from MCP1SMC-Lgals3 mice to measure leukocyte recruitment to the vessel wall. Results showed a significant decrease in Ly6Chi monocytes in the aortas of MCP1SMC-Lgals3 wt/Δ mice compared to wild type (Figure 5D). Taken together, these results indicate that MCP1 produced by SMCs in late stage lesions acts to recruit inflammatory cells and may act as a feedback mechanism to favor accumulation of phenotypically switched SMC in lesions that perpetuate a cycle of inflammation.
MCP1 knockout in Lgals3 transition state SMC resulted in a decreased frequency of stem-like SMC and multiple immune cell clusters
To rigorously assess the influence of MCP1SMC-Lgals3 knockout on multiple phenotypes of SMC and non-SMC in late stage atherosclerosis, we performed single-cell RNAseq (scRNAseq) on advanced BCA lesion plaques and tdTomato+ and GFP+ cells sorted from plaques from MCP1SMC-Lgals3 wt/wt, MCP1SMC-Lgals3 wt/Δ, and MCP1SMC-Lgals3 Δ/Δ mice after 18 weeks of Western diet. The UMAP in Supplemental Figure 10 shows representation of multiple plaque cell types in our dataset, including those expressing canonical markers of SMC, endothelial cells, macrophages, and even B and T cells, as well as several previously-described groups of SMC-derived cells including Acta2+ cells, Sca1+/stem-like, ECM-producing, and osteochrondrogenic cells. Differences between MCP1SMC-Lgals3 wt/wt and MCP1SMC-Lgals3 Δ/Δ plaques are shown in the UMAP in Figure 6A (Data from MCP1dual SMC wt/Δ animals have changes similar to MCP1dual SMC Δ/Δ mice). MCP1SMC-Lgals3 Δ/Δ plaques showed a 30% decrease in Clusters 4–7 (range 27–35%), defined by expression of stem-like genes including Sca1/Ly6a, Cd34, and Vcam1, as well as matrix remodeling enzymes including Mmp2 and Mmp3(Figure 6 A–C). Clusters 5 and 6 express a number of inflammatory genes that may affect traditional immune cells, including Vcam1, Cxcl1 and IL-6. Cluster 5 is also defined by Col15a, a gene necessary for SMC investment into lesions.[29] Remarkably, although there were fewer stem-like and inflammatory SMC detected in MCP1SMC-Lgals3 Δ/Δ plaques, there was no significant increase of SMC in Myh11+ Acta2+ Clusters 0 and 1. However, there was a 60% increase in Clusters 9 and 10 (range 54–69%), which are Myh11 negative but which express multiple collagen and ECM genes (including Fn1, Col1a1, Col10a1, Col6a1, and Col1a2). While consistent with the idea that loss of MCP1 in late stage plaque results in greater lesion stability, this also suggests that transition through a Sca1+ state is either not necessary to take on those phenotypes or that loss of MCP1 simply changes the flux through this state.
Figure 6.
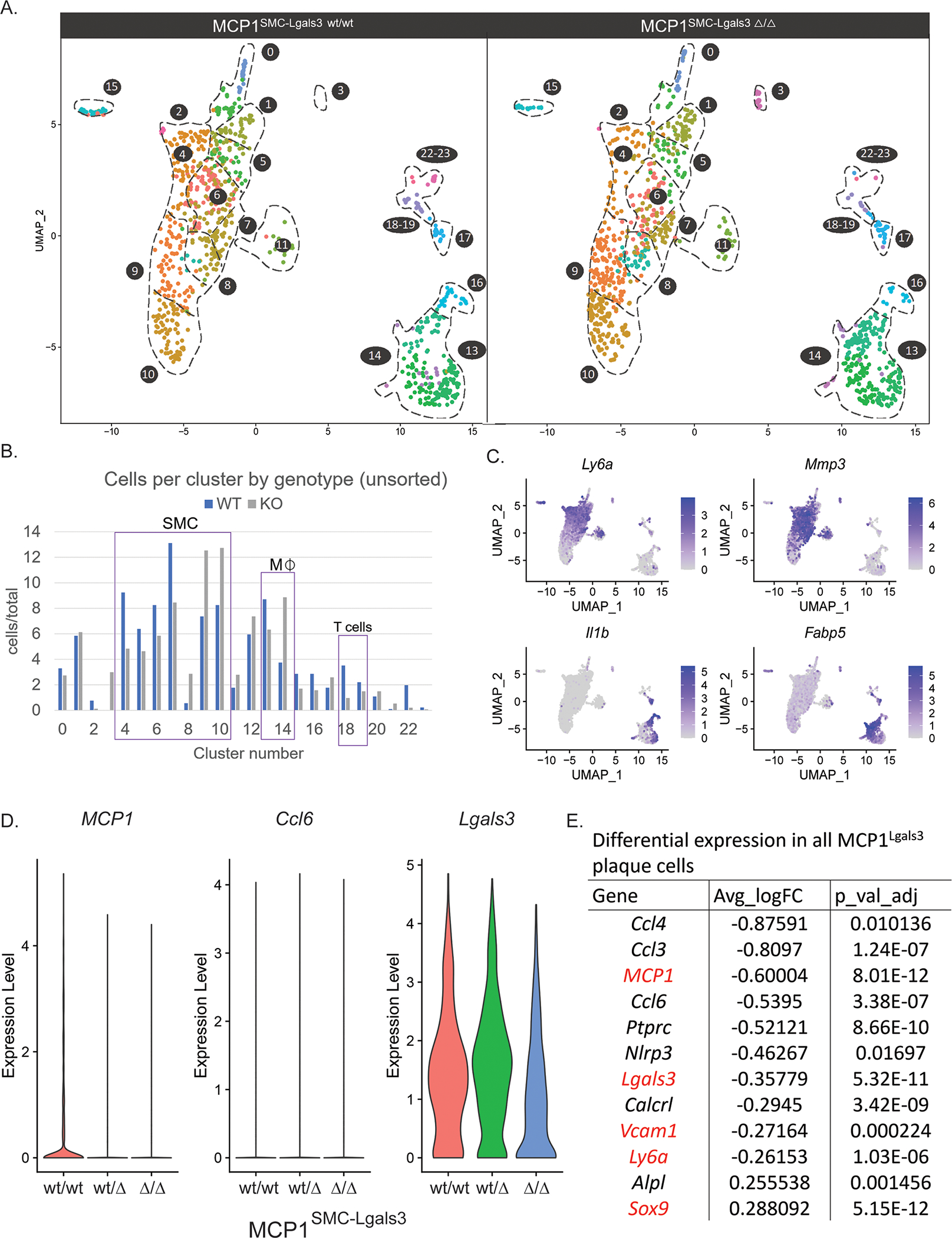
MCP1 knockout in Lgals3-transitioned SMC resulted in decreased frequency of stem-like SMC and changes to multiple immune cell clusters A) Atherosclerotic lesions from MCP1SMC-Lgals3 wt/wt, MCP1SMC-Lgals3 wt/Δ, and MCP1SMC-Lgals3 Δ/Δ mice fed a Western diet for 18 weeks were microdissected from the BCA and single-cell RNAseq was performed on unsorted lesion samples, as well as GFP+ and tdTomato+ cells sorted from those lesions. 4 animals per genotype were pooled to make 9 total libraries representing 12 animals. 5,111 total cells passed quality control for this experiment and were clustered together, with cells from MCP1SMC-Lgals3 wt/wt and MCP1SMC-Lgals3 Δ/Δ mice shown in UMAP graphs in A. B) Cluster frequency between MCP1SMC-Lgals3 wt/wt and MCP1SMC-Lgals3 Δ/Δ mice was compared by measuring total cells in each cluster normalized for total cells in each genotype, using unsorted lesion samples to avoid enrichment bias. C) Feature plot diagrams showing clusters with expression of stem-like (Ly6a, clusters 4–5), ECM-remodeling (Mmp3, cluster 6–7), and macrophage (Il1b, clusters 13 and 16, Fabp5, cluster 14) genes. D-E) Single cell data from from experiments in A was normalized and differential expression analysis was done using MAST. D shows violin plots of normalized expression levels of three inflammatory genes in plaque cells from MCP1SMC-Lgals3 wt/wt, MCP1SMC-Lgals3 wt/Δ, and MCP1SMC-Lgals3 Δ/Δ mice. E shows an abbreviated chart of significantly differentially expressed genes between MCP1SMC-Lgals3 wt/wt and MCP1SMC-Lgals3 Δ/Δ plaque cells.
In addition, MCP1SMC-Lgals3 knockout resulted in changes to non-SMC inflammatory populations. First, differential expression analysis between scRNAseq data from BCA plaques of MCP1SMC-Lgals3 wt/wt and MCP1SMC-Lgals3 Δ/Δ groups showed significant global decreases in Ccl2, Lgals3, Vcam1, and Ly6a (Figure 6D–E). Surprisingly, the total number of macrophages defined by expression of Ptprc and Adgre1 (commonly known as CD45 and F4/80, Clusters 12–16) were not universally decreased. However, MCP1SMC-Lgals3 Δ/Δ knockout resulted in decreases in Clusters 13 and 16 (Figure 6B), which includes macrophages expressing high levels of Il1β and Nlrp3, genes involved in inflammasome activation and response[30], as well as Cd36, a scavenger receptor involved in foam cell formation.[31,32] There was also a 46% increase of Cluster 14, which contains macrophages expressing Fabp5 and Gpnmb, markers associated with attenuation of the inflammatory response.[33,34] In addition, there was a marked four-fold decrease in Cluster 18–19 with MCP1SMC-Lgals3 knockout (Figure 6B), clusters that express markers of T cells, such as CD3e, IL7r, and CD4 (Supplemental Figure 10B), consistent with previous evidence that MCP1 promotes T cell chemotaxis in vitro.[35] Together, these observations suggest that MCP1 produced by Lgals3-transitioned SMC affects the late stage atherosclerotic plaque through regulation of SMC phenotype but also through regulation of multiple macrophage subsets and T cells.
Our findings provide evidence that SMC transitioning through an Lgals3+ state in late stage atherosclerotic lesions exacerbate lesion pathogenesis at least in part by production of MCP1, which drives ongoing aortic inflammation and promotes SMC phenotypic switching to an inflammatory stem-like state, leading to lesions with a reduced protective fibrous cap. Surprisingly, late stage immune changes appear to be manifested not only by monocyte recruitment, but by recruitment of T cells and expansion of inflammasome-activated macrophages.
Discussion
As one of the first genes studied in animal models of atherosclerosis[9,36], MCP1 has been extensively linked with atherosclerosis in humans and mice[7], but efforts to translate these findings into treatment have stumbled. Of critical interest, a Phase II clinical trial of MLN1202, a monoclonal antibody that blocks MCP1 binding to its receptor, CCR2,[37] showed lowered hsCRP in patients with previous cardiovascular risk carrying MCP1 promoter variants. These results take on even greater significance in the light of findings that lowered hsCRP in patients from the CANTOS trial was predictive of reduced risk of secondary cardiovascular events including MI and stroke.[38] However, MLN1202 failed to improve outcomes in large-scale, Phase III clinical trials of rheumatoid arthritis and multiple sclerosis, discouraging further development.[39] At the time, the well-characterized role of MCP1 in recruiting monocytes to sites of inflammation was thought to be important in the pathogenesis of both these diseases.[40,41] Originally, atherosclerosis studies followed the same logic, with several global knockout studies and local MCP1 inhibitors in animal models attributing decreased atherosclerotic lesion formation to early reductions in monocyte recruitment.[9,36,42,43] However, recent studies provide evidence that monocyte recruitment is less important in late stage lesions and that blocking monocyte responses to MCP1 has no effect on ongoing lesion pathogenesis.[44,45] In fact, Ly6Chi monocyte recruitment was recently shown to be necessary for atherosclerosis regression.[46] Results of studies herein show that knockout of MCP1 in a subset of Lgals3-transitioned SMC in lesions had multiple beneficial effects including increasing fibrous cap thickness and decreasing immune cell activation. However, we also show that knockout of just one allele of MCP1 in all SMC had opposite effects including mice having larger, less stable lesions likely due to prolonged monocytosis. In contrast, MCP1 inhibitor therapy in humans caused a transient decrease in circulating monocytes, which lasted about a week, whereas the decrease in hsCRP from the treatment persisted for months. Taken together, our results provide some explanation for the beneficial effects of MCP1 inhibition in late stage lesions, while highlighting the need for caution when applying findings based on monocytosis in mice to interventional human trials.
One of our most surprising findings was that Myh11-SMC knockout of MCP1 raises monocyte levels in blood. Although we did not attempt to definitively determine the mechanism for this effect, our results are consistent with several studies in the field that reported elevated monocyte levels with knockout or pharmacological blockade of MCP1 in atherosclerosis.[15,21,47] Originally, global knockout studies of MCP1 and CCR2 showed decreased mobilization of monocytes into the blood from the bone marrow in acute infection.[14,48] However, later studies observed a paradoxical increase in circulating monocytes in the absence of this cytokine in models of atherosclerosis, with authors reasoning that the chronic timescale of disease was responsible for this observation.[15] In contrast, endothelial knockout of MCP1 causes serum levels of MCP1 to drop and decreases blood monocyte content as expected.[15] Our study and others support the idea that MCP1 drives recruitment of monocytes into lesions. However, we did not find differences in recruitment of monocytes into other organs including the spleen and lung, making it unlikely that reduced uptake of monocytes by tissues is contributing to this systemic monocytosis. Rather, we postulate that production of MCP1 by perivascular cells represses release of monocytes from hematopoietic stem cell niches such that MCP1 knockout in Myh11-SMC results in increased release of monocytes into the bloodstream and monocytosis, although this would be an extremely difficult proposition to test since it would require selective loss of MCP1 exclusively in perivascular cells within HSC niches.
Our studies are the first to demonstrate a function of a specific phenotypically switched SMC subset in late-stage atherosclerosis through a rigorous loss of function (i.e., knockout) approach. Several SMC specific conditional knockout studies of transcription factors have provided important evidence that SMC-derived cells in advanced lesions can have detrimental or beneficial effects.[1–3] However, these studies relied on knockout of transcription factors in all SMC, which led to differences in prevalence of SMC subsets that were inferred to contribute to overall changes in lesion pathogenesis. Results of recent studies from our lab[16] and others[26,49] employing scRNAseq analysis of advanced lesions from SMC lineage tracing mice have provided compelling evidence that lesion SMC exhibit a wide range of complex transcriptomic phenotypes from which we can attempt to ascertain possible functions of these cells important in lesion pathogenesis. Moreover, based on the proximity of individual UMAP transcriptomic clusters one can draw inferences regarding how closely related cells are. However, such approaches provide no direct evidence that a cell within one transcriptomic cluster can transition to another and if so by what pathway. In contrast, our studies with dual lineage tracing mice directly show that Lgals3-transitioned SMC affect thickness of the fibrous cap and aortic inflammation through production of MCP1. Lgals3 is also known to activate the NLRP3 inflammasome in macrophages, consistent with our observation that loss of MCP1 in Lgals3-transitioned SMC was associated with decreases in not only Lgals3 GFP+ SMC but also a macrophage subset defined by Il1β and Nlrp3. The idea is also supported by findings that microvascular NG2+ pericytes promote chemotaxis of immune cells via MCP1 and NLRP3 inflammasome activation.[50] MCP1 itself may also play a feedback role in control of SMC phenotypic switching to an Lgals3+ stem-like and inflammatory state.
Finally, our studies provide a glimpse of SMC phenotypic switching in vivo in male and female animals. Our preliminary studies showed no change in BCA lesion size between male and female Myh11-DreERT2 Lgals3-Cre Rosa-tdTomato-eGFP ApoE−/− animals. We also did not observe a difference in or frequency of SMC-derived Lgals3-transitioned cells due to sex, confirming that the phenomenon of SMC phenotypic switching is present in males and females. However, due to the extreme difficulty in generating littermate knockout controls using the MCP1SMC-Lgals3 mouse model (with 8 different alleles, the dataset already consists of 54 animals harvested for tissue sections plus 12 animals for single cell RNAseq), we are underpowered for detecting small but perhaps important sex differences.[51] In wild-type animals, more rigorous analyses by sensitive techniques like scRNAseq in powered cohorts using dual lineage and new SMC lineage tracing models may be necessary to completely address the question of sex-specific differences in SMC phenotypic switching. Nevertheless, studies by Hartman et al[52] have provided evidence suggesting that sex differences in atherosclerosis involve phenotypic switching of plaque SMCs.
In conclusion, we show through SMC-specific MCP1 knockout that SMC are a major source of MCP1 in vivo and that loss of MCP1 in all Myh11+ SMC results in an increase in circulating Ly6Chi monocytes that favor plaque formation. However, using our novel dual lineage tracing mice, we show that MCP1 production by a subset of Myh11+ SMC that go on to express Lgals3 is detrimental for lesion pathogenesis by prolonging chronic inflammation and promoting SMC phenotypic switching and divestment from the fibrous cap. Our results provide insight into the unique nature of phenotypically switched SMC and their production of this highly drug-able target, a framework for the development of novel therapies, and justification for further clinical trials in this area that take into consideration the dichotomous detrimental and beneficial actions of MCP1 produced not only by SMC but also other lesion cell types.
Supplementary Material
Highlights.
Smooth muscle cells produce MCP1 in a Klf4-dependent manner
Heterozygous loss of MCP1 in all smooth muscle cells results in detrimental changes in plaque pathogenesis, including larger lesions with a decreased fibrous cap.
Loss of MCP1 in all smooth muscle cells also results in an early and sustained monocytosis, likely driving the observed changes in late stage plaque
Smooth muscle cells that have transitioned through an Lgals3-expressing state in late stage lesions can take on an inflammatory phenotype
Loss of MCP1 from Lgals3-transitioned inflammatory smooth muscle cells results in improved plaque parameters, including a thicker fibrous cap and changes in other inflammatory cell populations
Acknowledgements
We would like to thank the UVA Flow Cytometry Core and the UVA Genome Sequencing Core facilities for their role in generating data in this manuscript.
Sources of Funding
This work was funded by NIH R01 grants HL136314, HL 155165, and HL141425 to GKO, a Leducq Fondation Grant to GKO, and an AHA Clinical Health Profession Student Training Program Grant # 17CPRE33660350 to KO.
Nonstandard Abbreviations and Acronyms
- ACTA2
Actin alpha 2, smooth muscle
- ApoE
Apolipoprotein E
- BCA
Brachiocephalic artery
- CAD
Coronary artery disease
- ECM
Extracellular matrix
- eYFP
Enhanced yellow fluorescent protein
- eGFP
Enhanced green fluorescent protein
- IL1-b
Interleukin 1 beta
- Klf4
Kruppel-like factor 4
- Lgals3
Galectin 3
- MCP1
Monocyte chemoattractant protein 1
- Myh11
Myosin heavy chain 11
- scRNAseq
Single cell RNAseq
- SMC
Smooth muscle cell
- UMAP
Uniform manifold approximation and projection
Footnotes
Disclosures
None.
References
- 1.Cherepanova OA, Gomez D, Shankman LS, et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med 2016, 22:657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wirka RC, Wagh D, Paik DT, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med 2019. 25(8):1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shankman LS, Gomez D, Cherepanova O a, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 2015;21:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denger S, Jahn L, Wende P, et al. Expression of monocyte chemoattractant protein-1 cDNA in vascular smooth muscle cells: induction of the synthetic phenotype: a possible clue to SMC differentiation in the process of atherogenesis. Atherosclerosis 1999;144:15–23. [DOI] [PubMed] [Google Scholar]
- 5.Orr AW, Hastings NE, Blackman BR, et al. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res 2010;47:168–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu X, Dluz S, Graves DT, et al. Elevated expression of monocyte chemoattractant protein 1 by vascular smooth muscle cells in hypercholesterolemic primates. Proc Natl Acad Sci U S A 1992;89:6953–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deshmane SL, Kremlev S, Amini S, et al. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J Interf Cytokine Res 2009;29:313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahler SA, Register TC, Riley RF, et al. Monocyte Chemoattractant Protein-1 as a Predictor of Coronary Atherosclerosis in Patients Receiving Coronary Angiography. Crit Pathw Cardiol 2018;17:105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu L, Okada Y, Clinton SK, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell 1998;2:275–81. [DOI] [PubMed] [Google Scholar]
- 10.Lu B, Rutledge BJ, Gu L, et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med 1998;187:601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Waard Bot, Jager Egashira, Vries Quax, Blessen van B. Systemic MCP1/CCR2 blockade and leukocyte specific MCP1/CCR2 inhibition affect aortic aneurysm formation differently. Atherosclerosis 2010;211:84–9. [DOI] [PubMed] [Google Scholar]
- 12.Fantuzzi L, Borghi P, Ciolli V, et al. Loss of CCR2 expression and functional response to monocyte chemotactic protein (MCP-1) during the differentiation of human monocytes: role of secreted MCP-1 in the regulation of the chemotactic response. Blood 1999;94:875–83. [PubMed] [Google Scholar]
- 13.Yoshimura T, Galligan C, Takahashi M, et al. Non-Myeloid Cells are Major Contributors to Innate Immune Responses via Production of Monocyte Chemoattractant Protein-1/CCL2. Front Immunol 2014;4:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi C, Jia T, Mendez-Ferrer S, et al. Bone Marrow Mesenchymal Stem and Progenitor Cells Induce Monocyte Emigration in Response to Circulating Toll-like Receptor Ligands. Immunity 2011;34:590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Del Toro R, Chèvre R, Rodríguez C, et al. Nestin(+) cells direct inflammatory cell migration in atherosclerosis. Nat Commun 2016;7:12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Owsiany KM, Alencar GF, Karnewar S, et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020;142:2045–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomez D, Shankman LS, Nguyen AT, et al. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods 2013;10:171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez D, Baylis RA, Durgin BG, et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat Med 2018;24:1418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Zhang W, Li S, et al. Genetic variants of the monocyte chemoattractant protein-1 gene and its receptor CCR2 and risk of coronary artery disease: a meta-analysis. Atherosclerosis 2011;219:224–30. [DOI] [PubMed] [Google Scholar]
- 20.McAlpine CS, Kiss MG, Rattik S, et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 2019;566:383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winter C, Silvestre-Roig C, Ortega-Gomez A, et al. Chrono-pharmacological Targeting of the CCL2-CCR2 Axis Ameliorates Atherosclerosis. Cell Metab 2018;28:175–182.e5. [DOI] [PubMed] [Google Scholar]
- 22.Wall VZ, Barnhart S, Kanter JE, et al. Smooth muscle glucose metabolism promotes monocyte recruitment and atherosclerosis in a mouse model of metabolic syndrome. JCI insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramasamy SK. Structure and Functions of Blood Vessels and Vascular Niches in Bone. Stem Cells Int 2017;2017:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feil S, Fehrenbacher B, Lukowski R, et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res 2014;115. [DOI] [PubMed] [Google Scholar]
- 25.Allahverdian S, Chehroudi AC, McManus BM, et al. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014;129:1551–9. [DOI] [PubMed] [Google Scholar]
- 26.Pan H, Xue C, Auerbach BJ, et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020;:2060–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miano JM, Fisher EA, Majesky MW. Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation 2021;143:2110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu Wong, Potter Simpson, Karamariti Zhang, Zeng Warren, Hu Wang, Xu B. Vascular Stem / Progenitor Cell Migration Induced by Smooth Muscle Cell-Derived Chemokine Ligand 1 Contributes to Neointima Formation. 2016;2:2368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durgin BG, Cherepanova OA, Gomez D, et al. Smooth muscle cell-specific deletion of col15α1 unexpectedly leads to impaired development of advanced atherosclerotic lesions. Am J Physiol - Hear Circ Physiol 2017;312:H943–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflamasomes are required for atherogenesis and activated by cholesterol crystals that form early in disease. Nature 2010;464:1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med 2014;46:e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chistiakov DA, Melnichenko AA, Myasoedova VA, et al. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017;95:1153–65. [DOI] [PubMed] [Google Scholar]
- 33.Moore SM, Holt VV., Malpass LR, et al. Fatty acid-binding protein 5 limits the anti-inflammatory response in murine macrophages. Mol Immunol 2015;67:265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neal ML, Boyle AM, Budge KM, et al. The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. J Neuroinflammation 2018;15:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taub DD, Proost P, Murphy WJ, et al. Monocyte chemotactic protein-1 (MCP-1), −2, and −3 are chemotactic for human T lymphocytes. J Clin Invest 1995;95:1370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aiello RJ, Bourassa PA, Lindsey S, et al. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 1999;19:1518–25. [DOI] [PubMed] [Google Scholar]
- 37.Gilbert J, Lekstrom-Himes J, Donaldson D, et al. Effect of CC Chemokine Receptor 2 CCR2 Blockade on Serum C-Reactive Protein in Individuals at Atherosclerotic Risk and With a Single Nucleotide Polymorphism of the Monocyte Chemoattractant Protein-1 Promoter Region. Am J Cardiol 2011;107:906–11. [DOI] [PubMed] [Google Scholar]
- 38.Ridker PM, MacFadyen JG, Thuren T, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet (London, England) 2017;390:1833–42. [DOI] [PubMed] [Google Scholar]
- 39.Kalinowska A, Losy J. Investigational C-C chemokine receptor 2 antagonists for the treatment of autoimmune diseases. Expert Opin Investig Drugs 2008;17:1267–79. [DOI] [PubMed] [Google Scholar]
- 40.Szekanecz Z, Koch AE. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat Rev Rheumatol 2016;12:5–13. [DOI] [PubMed] [Google Scholar]
- 41.Miyabe Y, Lian J, Miyabe C, et al. Chemokines in rheumatic diseases: pathogenic role and therapeutic implications. Nat Rev Rheumatol 2019;15:731–46. [DOI] [PubMed] [Google Scholar]
- 42.Boring L, Gosling J, Cleary M, et al. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998;394:894–7. [DOI] [PubMed] [Google Scholar]
- 43.Combadière C, Potteaux S, Rodero M, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 2008;117:1649–57. [DOI] [PubMed] [Google Scholar]
- 44.Guo J, de Waard V, Van Eck M, et al. Repopulation of apolipoprotein E knockout mice with CCR2-deficient bone marrow progenitor cells does not inhibit ongoing atherosclerotic lesion development. Arterioscler Thromb Vasc Biol 2005;25:1014–9. [DOI] [PubMed] [Google Scholar]
- 45.Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med 2013;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rahman K, Vengrenyuk Y, Ramsey SA, et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest 2017;127:2904–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bot I, Ortiz Zacarías N V., de Witte WEA, et al. A novel CCR2 antagonist inhibits atherogenesis in apoE deficient mice by achieving high receptor occupancy. Sci Rep 2017;7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 2006;7:311–7. [DOI] [PubMed] [Google Scholar]
- 49.Wirka RC, Wagh D, Paik DT, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med 2019;25:1280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stark K, Eckart A, Haidari S, et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat Immunol 2013;14:41–51. [DOI] [PubMed] [Google Scholar]
- 51.Robinet P, Milewicz DM, Cassis LA, et al. Consideration of Sex Differences in Design and Reporting of Experimental Arterial Pathology Studies-Statement From ATVB Council. Arterioscler Thromb Vasc Biol 2018;38:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hartman RJG, Owsiany K, Ma L, et al. Sex-Stratified Gene Regulatory Networks Reveal Female Key Driver Genes of Atherosclerosis Involved in Smooth Muscle Cell Phenotype Switching. Circulation 2021;143:713–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.