

Study Design.
The effect of amlodipine (AM) on spinal cord injury (SCI) and autophagy was researched by establishing ventral spinal cord cells (VSC4.1) oxygen and glucose deprivation model and SCI mice model.
Objective.
To determine the neuroprotective effects of AM by upregulating autophagy during SCI repair.
Summary of Background Data.
AM, an antihypertensive medication, has been shown in several studies to inhibit neuronal apoptosis and exert neuroprotective effects in various central nervous system diseases. However, its effects on SCI are unexplored. Autophagy could inhibit cell apoptosis, which has been shown to promote SCI repair. However, the role of AM in autophagy remains unclear.
Methods.
We examined the relationship between AM, apoptosis, and autophagy in ventral spinal cord cells and the injured spinal cords of C57BL/6 female mice respectively, following histological, behavioral, microscopic, immunofluorescence, and western blotting analyses.
Results.
We found that AM could inhibit motor neuronal apoptosis in vitro. Furthermore, AM promoted locomotor recovery by upregulating autophagy and alleviating apoptosis, neuronal loss, and spinal cord damage after SCI.
Conclusion.
AM inhibited motoneuronal apoptosis by upregulating autophagy to improve SCI recovery.
Key words: amlodipine, apoptosis, autophagy, LC3-II, P62, spinal cord injury.
Level of Evidence: N/A
Traumatic spinal cord injury (SCI) is concomitant with severe neurological disability. 1 The pathological processes of SCI include primary and secondary injuries. Primary injury refers to the damage that is directly caused by trauma or other conditions, such as tumors or infection. Secondary injury includes a complex molecular cascade of events that result in more serious damage to the spinal cord and its functions. 2 Apoptosis is a crucial factor in the extended deterioration of the spinal cord following injury. 3 The inhibition of neuronal apoptosis caused by secondary injury has been found to be paramount to neural functional recovery. 4
Autophagy is an intracellular catabolic mechanism that causes the degradation of organelles by the autophagy-lysosomal pathway. 5 The autophagosome is a double-membrane structure that is formed during the initial stage of autophagy and encloses a portion of the cytoplasm. The autophagosome then fuses with a lysosome to become an autolysosome, leading to the degradation of the enclosed materials. 6 Autophagy takes place early after SCI, with significant augmentation of Beclin-1 starting at 4 hours, peaking at 3days, and lasting for 21 days. 7 The length of Beclin-1 expression is similar to the occurrence of apoptosis after SCI, indicating that autophagy is associated with apoptosis. 7 Autophagy can safeguard against apoptosis, playing a vital role in intracellular physiological homeostasis. 8
In the central nervous system, autophagy eliminates damaged mitochondria and reduces the accumulation of harmful substances. 9 Autophagy plays a key role in promoting neural function recovery in Alzheimer disease. 10 Ischemia is alleviated through the promotion of autophagy by suppressing p- Mammalian Target of Rapamycin expression, while autophagy indicators, such as Beclin-1 and Microtubule-associated protein 1 light chain 3 (LC3)-II, are upregulated. 11 Induced autophagy has also been reported to have neuroprotective effects against hypoxiaischemia neonatal brain injury in mice through decreased phosphorylation of Extracellular Signal-Regulated Kinase and Mammalian Target of Rapamycin. 12 Furthermore, autophagy participates in the pathological process of traumatic SCI. Studies have shown that autophagy is activated to protect the spinal cord. 13 Stimulating autophagy has neuroprotective effects, as it decreases neuronal apoptosis and accelerates functional recovery after SCI in rats. 14
Recent studies have shown the potential of long-acting dihydropyridine calcium channel blocker, amlodipine (AM), in neuronal apoptosis inhibition and nerve tissue repair. AM is currently a first-line drug for the treatment of hypertension. In addition to reducing blood pressure, the neuroprotective effects of AM in Alzheimer disease and acute ischemic neural damage have been recently discovered. 15,16 AM promoted the recovery of neural stem cells damaged by hydrogen peroxide. 17 It also inhibited apoptosis by upregulating antiapoptotic protein (Bcl-2) and downregulating cleaved caspase-9 and cleaved caspase-3 (herein Cle-Cas3). 18 However, the regulation of autophagy by AM in SCI has yet to be comprehensively elucidated. Thus, we investigated the therapeutic effects of AM following SCI and examined whether AM inhibits apoptosis by regulating autophagy.
Materials And Methods
Establishment of a Cell OGD Model
We followed the procedures of a previous study to establish the oxygen and glucose deprivation (OGD) model. 19 After AM treatment, ventral spinal cord (VSC4.1) cells were incubated in D-Hanks’ balanced salt solution without glucose and placed in an anoxic cell incubator for further culture. The conditions of the anoxic cell incubator were as follows: temperature: 37°C, oxygen content: 0%, and CO2 content: 5%.
Animals
Female C57BL/6 mice (8–12 wks, 20–25 g) were used for this study. The use of animals and the experimental protocols were approved by the Laboratory Animal Ethics Committee of Taizhou Enze Medical Center (group). Forty-five mice were randomly divided into three groups: sham, SCI, and AM. Mice in the AM group were intraperitoneally administered AM (3 mg/kg/d) dissolved in a 5% Dimethyl Sulfoxide normal saline solution for 7 days immediately after surgery. 20, 21
Surgical Procedures
The SCI model was induced by spinal cord hemisection as previously described. 22 Mice were anesthetized with inhaled isoflurane (1.5%), followed by laminectomy to expose the spinal cord at the segment of the 10th thoracic vertebra. We performed cross-cutting of the spinal cord right hemisphere at the corresponding segment of T10 with a microknife. The sham group underwent laminectomy without spinal cord hemisection. The incisions were closed by suturing the muscle and skin. Cefazolin sodium (50 mg/kg, i.p.) was administered to prevent infection.
The Basso Mouse Scale (BMS) Test
The Basso Mouse Scale (BMS) test was adopted to evaluate lower limb function recovery as previously documented, 23 on days 1, 3, 7, 14, 21, 28, and 35 postoperation, which was executed by two colleagues blinded to the design of the experiment. 24
Statistical Analysis
SPSS 22.0 software was used for statistical analysis. Data are described as the mean ± standard deviation. Statistical significance was determined by using one-way analysis of variance and Dunnett post hoc test. P < 0.05 was defined as statistically significant.
More detailed Materials and methods please find the document of Supplemental Data, http://links.lww.com/BRS/B847.
Results
AM Alleviates Motoneuronal Death
The injurious effects of different OGD durations (0, 2, 4, and 8 h) on VSC4.1 cells were evaluated by Cell Counting Kit-8 (CCK-8) assay and phalloidin-Fluorescein Isothiocyanate (FITC) staining. The viability of VSC4.1 cells after OGD treatment for 2, 4, and 8 hours decreased significantly compared with VSC4.1 cells treated with OGD for 0 hours (P < 0.001; Figure 1A). In addition, cell viability was reduced by approximately 40% after VSC4.1 cell treatment with OGD for 2 hours. The phalloidin-FITC staining results were consistent with those of the CCK-8 assay, showing that the viability of VSC4.1 cells dwindled after OGD for 2, 4, and 8 hours. Similarly, the number of living cells reduced significantly after 2 hours of OGD (Figure 1D). Therefore, we chose 2 hours of OGD for subsequent experiments. As such, we exposed cells to different AM concentrations (0, 1, 5, 10, 20, and 40 μM) for 24 hours and analyzed cell viability by CCK-8 assay. AM did not poison VSC4.1 cells when the concentration was less than 20 μM (P < 0.001; Figure 1B). To further explore the potential effects of AM on VSC4.1 cells, we pretreated VSC4.1 cells with AM for 2 hours before subjection to OGD (for 2h). AM (5 μM and 10 μM) showed significant protective effects against OGD-induced cell death (Figure 1C, P < 0.01).
Figure 1.
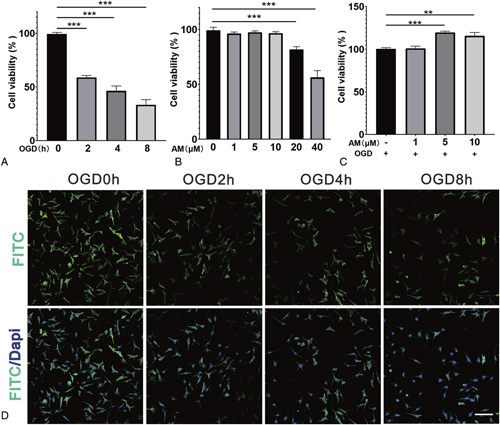
AM alleviates motoneuronal death. A, CCK-8 results of VSC4.1 cells treated for different OGD durations (0, 2, 4, and 8h). B, CCK-8 results of VSC4.1 cells treated with different concentrations of AM for 24 hours. C, CCK-8 results of VSC4.1 cells pretreated with AM and induced by OGD for 2 hours. D, Confocal laser scanning microscopy results of VSC4.1 cells treated for different OGD durations and stained with FITC-phalloidin and PI. Significant differences between groups are indicated as** P < 0.01,*** P < 0.001; n = 3. AM indicates amlodipine; FITC, fluorescein isothiocyanate; OGD, oxygen and glucose deprivation; VSC4.1, ventral spinal cord cells.
AM Promotes Locomotor Recovery and Alleviates Tissue Damage and Neuronal Loss After SCI
The BMS test was adopted during the 5 weeks after surgery to observe motor function recovery. Both the SCI and AM groups showed complete hindlimb paralysis on days 1 and 3 after SCI surgery. AM-treated mice displayed significantly better BMS performance than the SCI group starting on day 7 (Figure 2C, P < 0.05).
Figure 2.
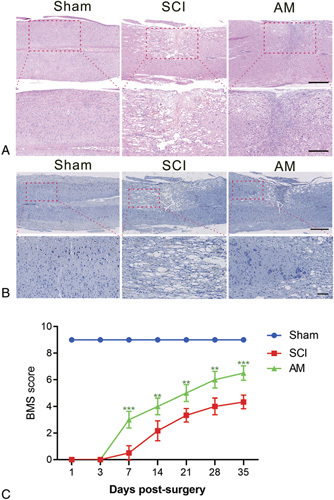
AM promotes locomotor recovery and alleviates tissue damage and neuronal loss after SCI. A, HE staining on day 14. Scale bars are 500 and 250 μm. B, Nissl staining of spinal cord tissue to evaluate neuronal loss on day 14. Scale bars are 500 and 100 μm. C, BMS scores.** P < 0.01 versus the SCI group,*** P < 0.001; n = 6. AM indicates amlodipine; BMS, Basso Mouse Scale; SCI, spinal cord injury.
Hematoxylin–eosin staining results showed that hemisection impaired the spinal cord structure, which was improved by AM treatment (Figure 2A), including a reduced cavity at the injured site. Nissl staining results showed that, right hemisphere cross-cutting of the spinal cord resulted in a marked decrease in neuron survival. However, the AM-treated group demonstrated a pronounced increase neuron survival (Figure 2B).
AM Inhibits Motoneuronal Apoptosis
aOGD generated a large number of Terminal deoxynucleotidyl transferase (TdT) dUTP nick end labeling (TUNEL)-positive cells (Figure 3A). Both the 5 μMAM- and10 μM AM-pretreated groups showed fewer TUNEL-positive cells than the OGD group. Furthermore, we evaluated apoptosis by Annexin V-FITC/PI double staining and flow cytometry. AM pretreatment (5 μM and 10 μM) significantly augmented the cell survival rate and decreased the cell apoptosis rate compared with untreated controls (Figure 3B, C). In addition, the therapeutic effects in the 5 μM group were better than that in the 10 μM group. We examined the expression levels of proapoptotic protein (Bax) and Bcl-2 and Cle-Cas3. The results showed that Bax/Bcl-2 and Cle-Cas3 expression decreased significantly in the AM-pre-treated group compared with the OGD group (Figure 3D–Figure 3F).
Figure 3.
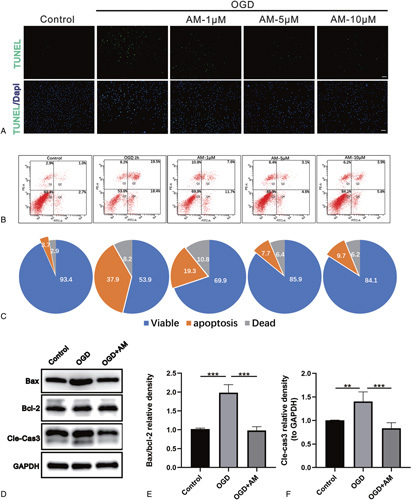
AM inhibits motoneuronal apoptosis. A, TUNEL assay was performed in motoneuron of control, OGD, AM-1 μM, AM-5 μM, AM-10 μM groups (scale bar: 100 μm). B, Flow cytometry analysis of the apoptosis rates of VSC4.1 cells induced by OGD for 2 hours after pretreatment with different concentrations of AM. C, Pie chart of the flow cytometry analysis exhibiting the proportion of viable, apoptotic, and dead cells. D–F, Protein expression of Cle-Cas3, Bax, and Bcl-2 in VSC4.1 cells. Significant differences between groups are indicated as** P< 0.01,*** P< 0.001; n = 4. AM indicates amlodipine; Bax, proapoptotic protein; Bcl-2, antiapoptotic protein; Cle-Cas3, cleaved caspase-3; OGD, oxygen and glucose deprivation; VSC4.1, ventral spinal cord cells.
AM Attenuates Apoptosis After SCI
Immunofluorescence staining of Cle-Cas3 was performed to detect Cle-Cas3-positive cells on day 7 after injury. An increased number of Cle-Cas3-positive cells were stained in the SCI group. Moreover, AM treatment resulted in a reduced number of Cle-Cas3-positive cells compared with the SCI group (Figure 4A). However, fewer Bcl-2-positive cells were found in the SCI group than in the sham group. Moreover, AM treatment resulted in a greater number of Bcl-2-positive cells (Figure 4B). Western blot analysis confirmed the above results, as AM significantly reversed the increased expression of Cle-Cas3 caused by SCI (Figure 4C, E). Similarly, the expression levels of the Bax and the Bcl-2 were correspondingly decreased and increased, respectively, and the ratio of Bax/Bcl-2 decreased after AM treatment (Figure 4C, D).
Figure 4.
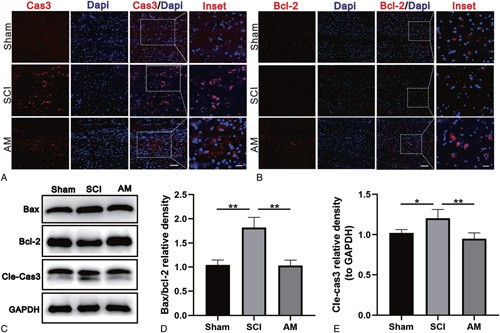
AM attenuates apoptosis caused by SCI. A and B, Immunofluorescence staining of Cle-Cas3 and Bcl-2 in spinal cord tissue sections from each group. Scale bars for caspase-3 are 50 and 20 μm; scale bars for Bcl-2 are 100 and 20 μm. C–E, Representative western blots and quantification data for Bax, Bcl-2, Cle-Cas3, and GAPDH in each group. Significant differences between groups are indicated as * P < 0.05,** P < 0.01; n = 4. AM indicates amlodipine; Bax, proapoptotic protein; Bcl-2, antiapoptotic protein; Cle-Cas3, cleaved caspase-3; SCI, spinal cord injury.
AM Upregulates Autophagy of Motoneuron
LC3, an indicator of autophagy formation, was upregulated after AM pretreatment detected by immunofluorescence. This effect was most obvious in the 5 μM AM group (Figure 5A). The western blot analysis results indicated that LC3-II expression increased significantly after cells were pretreated with 5 μM AM compared with the OGD group (P < 0.05, Figure 5B, E). Additionally, the expression of the autophagy-related protein Beclin-1 was higher in the group pretreated with 5 μM AM than in the OGD group (P < 0.01, Figure 5B, C). p62 accumulation is frequently used as a sign of autophagy impairment. Western blot analysis showed OGD-induced p62 accumulation; however, AM pretreatment decreased p62 protein expression (P < 0.001, Figure 5B, D).
Figure 5.
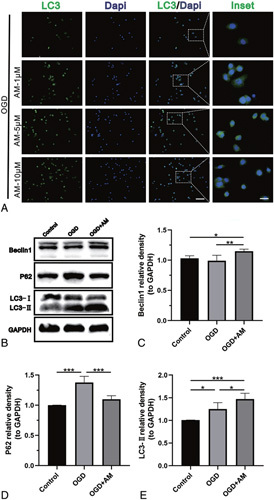
AM upregulates autophagy in motoneurons. A, Immunofluorescence staining of LC3 in VSC4.1 cells from each experimental group. The scale bars are 100 and 25 μm. B–E, Representative western blots and quantification data of Beclin-1, p62, LC3, and GAPDH in each group. Significant differences between groups are indicated as* P < 0.05,** P < 0.01,*** P < 0.001; n = 4. AM indicates amlodipine; LC3, Microtubule-associated protein 1 light chain 3; SCI, spinal cord injury; VSC4.1, ventral spinal cord cells.
AM Upregulates Autophagy After SCI
To confirm the induction of autophagy at the molecular level, the level of the autophagy protein LC3 was assessed by double staining for Chat (green)/LC3 (red) and western blot. After SCI, LC3 assembles in motor neurons, and this assembly further increases after AM treatment (Figure 6A). LC3-II is an indicator of autophagosome formation. Compared with the SCI group, LC3-II expression was weaker in the sham group but significantly stronger in AM-treated mice (Figure 6B, E, P < 0.01), suggesting that AM promoted autophagy. The autophagy protein Beclin-1 was also detected by western blot. After SCI, Beclin-1 expression was stronger, and after AM treatment, Beclin-1 expression was significantly higher than that in the SCI group (Figure 6B, C; P < 0.01). Additionally, p62 protein expression was upregulated after SCI but downregulated by AM treatment (Figure 6B, D; P < 0.01, n = 4).
Figure 6.
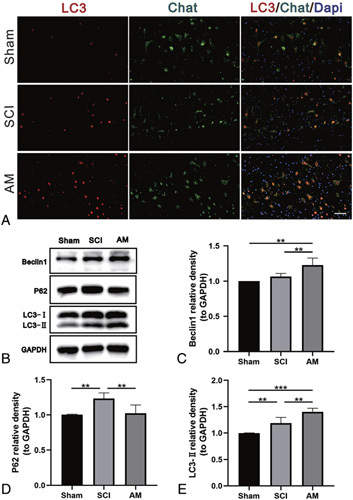
AM upregulates autophagy after SCI. A, Double staining for Chat (green)/LC3 (red) in spinal cord tissue sections from each group. Scale bars: 50 μm. B–E, Representative western blots and quantification data of Beclin-1, p62, LC3, and GAPDH in each group. Significant differences between groups are indicated as ** P < 0.01,*** P < 0.001; n = 4. AM indicates amlodipine; LC3, Microtubule-associated protein 1 light chain 3; SCI, spinal cord injury.
AM Inhibits Motoneuronal Apoptosis Through Autophagy Upregulation
To investigate whether autophagy was involved in the AM-induced protective effects against apoptosis, VSC4.1 cells were pretreated with the autophagy inhibitor 3-MA. AM significantly activated autophagic flux in VSC4.11 cells treated with OGD by downregulating the expression of p62. Pretreatment with 3-MA (10 mM) resulted in the marked accumulation of p62 compared with that in the AM group (Figure 7C, D; P < 0.001). Moreover, OGD significantly upregulated the expression ofthe protein Cle-Cas3. Pretreatment with AM markedly attenuated this increase in Cle-Cas3 compared with the untreated group. However, 3-MA reversed the effects of AM (Figure 7A, B; P < 0.05). The TUNEL assay results showed that the incidence of apoptosis was markedly decreased in cells treated with AM compared with the untreated group. Nonetheless, 3-MA abolished the antiapoptotic effects of AM. (Figure 7E).
Figure 7.
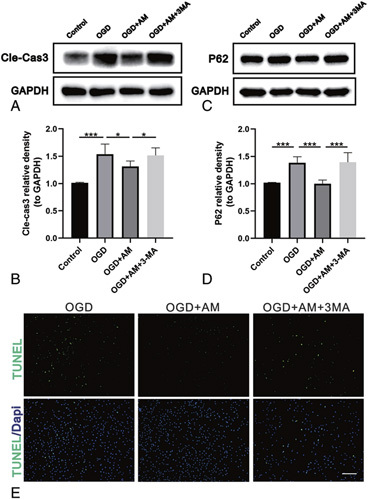
Autophagy inhibition counteracts the antiapoptotic effects of AM on motoneurons. A and B, Representative western blots and quantification data of Cle-Cas3 and GAPDH in each group. Significant differences between groups are indicated as* P < 0.05,*** P < 0.001; n = 4. C and D, Representative western blots and quantification data of p62 and GAPDH in each group. Significant differences between groups are indicated as *** P < 0.001; n = 4. E, TUNEL staining was performed to assess VSC4.1 cell apoptosis in each group (scale bar: 100 μm). AM indicates amlodipine; Cle-Cas3, cleaved caspase-3; TUNEL, Terminal deoxynucleotidyl transferase (TdT) dUTP nick end labeling; VSC4.1, ventral spinal cord cells.
Discussion
In the current study, we demonstrated that 1) AM promotes structural and functional recovery after nerve trauma, 2) AM inhibits apoptosis by upregulating autophagic flux, 3) a calcium channel blocker upregulates autophagy during SCI repair, and 4) AM reduces apoptosis and neuronal loss after nerve trauma.
Calcium is associated with cell fate, such as proliferation, differentiation, migration, and death. 25 Voltage-gated Ca2+ channel blockers have demonstrated protective effects on the central nervous system. Notably, voltage-gated Ca2+ channel blockers are beneficial for the treatment of Parkinson disease and chronic pain. 26,27 Similarly, diltiazem, an L-type calcium channel blocker, promoted axonal outgrowth of adult primary mouse dorsal root ganglia, and induced human sensory neurons. 28 In this study, we found that AM promoted structural and functional recovery following nerve trauma. Significant improvements in lower limb motor functions of mice treated with AM were observed starting at week 1 after SCI (Figure 2C). Similarly, HE staining showed fewer scars and vacuoles in the injured spinal cords of AM-treated mice than SCI mice (Figure 2A), indicating the beneficial effects of AM during nerve function recovery. A previous study reported the neuroprotective effects of AM besylate on neural stem cells injured by oxidative stress. 17 However, this is the first study to investigate the structural and functional recovery following nerve trauma.
The regenerative process after SCI is complex and involves a variety of cellular and molecular components. Recently, the study of autophagy, which was first discovered under conditions of starvation, has received increasing attention. 29 Autophagy is a tightly regulated process and a central component of the cellular stress response. 30 It plays a conserved role in maintaining cellular homeostasis by degrading dysfunctional proteins, lipids, and organelles through the autophagosome-lysosome pathway. 31 Several studies have reported the protective effects of autophagy on the nervous system. For instance, the activation of autophagic flux promoted axonal regeneration and nerve recovery in a C elegans model. 32 Some studies have demonstrated that dihydropyridine calcium channel blockers exert their neuroprotective effects by regulating autophagy, oxidative stress, and the nuclear factor-k-gene binding pathway. 33–35 In a recent study, injury caused by OGD to the mitochondria of neural stem cells was found to be reversed by AM. 36 In the study, we found that pretreatment with AM significantly downregulated the protein expression of Bax and Cle-Cas3 while reducing the apoptotic rate of cells under OGD (Figure 3). These data suggested the antiapoptotic effects of AM to protect nerve cells. Most importantly, we found that upregulation of autophagic flux might be involved in the antiapoptotic effects of AM. Our results showed that the autophagy indicators LC3 and Beclin-1 were upregulated, and this accumulation of LC3 and Beclin-1 could be due to either the formation of autophagosomes or autophagic flux blockade. The protein p62 (SQSTM1) is an indicator of autophagic flux and acts as a substrate during autophagy.
Accumulation of p62 implies autophagic flux inhibition. Our data showed that OGD blocked autophagic flux. However, autophagic flux was activated after pretreatment with AM through the downregulation of p62 (Figure 5). To demonstrate the relationship between autophagy and apoptosis, we used the classical autophagy inhibitor 3-MA. We found that the beneficial antiapoptotic effects were abolished when autophagy was inhibited by 3-MA (Figure 7), which further confirms that the neuroprotective effects of AM are mediated by autophagic flux stimulation.
Recent studies have revealed that the calcium channel blocker efonidipine can alleviate iron overload-induced brain neurotoxicity caused by apoptosis in both wild-type and thalassemic mice. 33 Similarly, verapamil can mitigate motor neuronal degeneration in a mouse model of amyotrophic lateral sclerosis. 35 However, only a handful of studies have reported the ability of calcium channel blockers to reduce apoptosis and neuronal loss after SCI. In our study, we showed that AM reduces apoptosis and neuronal loss after SCI. Nissl staining demonstrated that AM treatment triggered the survival of more neurons near the injured area after SCI (Figure 2B).
The precise role of autophagy in SCI is not yet clear. 37 Although controversial, the upregulation of autophagy after SCI is considered to be a neuroprotective out-come. 4,38 The expression of LC3, which is a marker of autophagy that is critical for autophagosome formation, is upregulated in the dorsal root ganglia after acute SCI. 39 In this study, we conducted immunofluorescence double staining of Chat/LC3 in spinal cord tissue sections. Chat was used to label motor neurons in the spinal cord. The results of our study demonstrated that LC3 expression was significantly increased in spinal cord motor neurons after treatment with the calcium channel blocker AM (Figure 6A). The expression of Beclin-1 and LC3-II increased after SCI and were further upregulated after AM treatment (Figure 6B, C, E), which were consistent with the immunofluorescence double staining. This result indicated that calcium channel blocker therapy promoted the formation of autophagosomes after SCI. The expression of the autophagy substrate p62 was augmented after SCI, but after treatment with the calcium channel blocker AM, the increased expression of p62 was reversed. All of the above results suggest that autophagic flux was blocked in spinal cord motoneurons after SCI but reactivated after treatment with the calcium channel blocker AM. For the first time, we found that calcium channel blockers could promote autophagic flux after SCI.
However, it remains unclear how AM activates autophagy. Moreover, the molecular mechanism of apoptosis downregulation due to autophagy induced by AM is still unknown. Therefore, we will continue to resolve these difficult problems in future studies.
In summary, we showed that AM administration following SCI can improve motor function and alleviate tissue damage and neuron loss via the upregulation of autophagy after SCI, suggesting that AM has therapeutic potential for SCI. This study facilitates a mechanistic clarification and clinical applications of AM for SCI.
Key Points.
AM promotes structural and functional recovery after nerve trauma.
AM inhibits apoptosis by upregulating autophagic flux.
A calcium channel blocker upregulates autophagy during SCI repair.
AM reduces apoptosis and neuronal loss after nerve trauma.
Supplementary Material
Footnotes
Y.H., H.R., and X.G. are co-first authors.
The manuscript submitted does not contain information about medical device(s)/drug(s).
The Science and Technology Program of Zhejiang Province (2018C37107); Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (2019KY782); National Natural Science Foundation of China (82172425, 81873995, 32000975); The Preponderant Discipline Supporting Project of the Second Affiliated Hospital of Soochow University (XKTJ-XK202003); The Suzhou Special Foundation for the Key Diseases Diagnosis and Treatment (LCZX201904, LCZX201708); The Social Development Key Programs of Jiangsu Province-Advanced Clinical Technology (BE2019662, BE2018656); The Key Laboratory for Spinal Cord Injury Regeneration of Suzhou (SZS201807); Guangdong Basic and Applied Basic Research Foundation (2019A1515010629) funds were received in support of this work.
No relevant financial activities outside the submitted work.
This is an open access article distributed under the terms of the Creative Commons Attribution-Non Commercial-No Derivatives License 4.0 (CCBY-NC-ND), where it is permissible to download and share the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal.
Supplemental digital content is available for this article. Direct URL citations appearing in the printed text are provided in the HTML and PDF version of this article on the journal’s Web site (www.spinejournal.com).
References
- 1.Ahuja CS, Wilson JR, Nori S, et al. Traumatic spinal cord injury. Nat Rev Dis Primers 2017;3:17018. [DOI] [PubMed] [Google Scholar]
- 2.Zhu S, Wang Z, Zhao Y, et al. Gelatin nanostructured lipid carriers incorporating nerve growth factor inhibit endoplasmic reticulum stress-induced apoptosis and improve recovery in spinal cord injury. Mol Neurobiol 2016;53:4375–4386. [DOI] [PubMed] [Google Scholar]
- 3.Choi D, Lee J, Moon Y, et al. Acupuncture-mediated inhibition of inflammation facilitates significant functional recovery after spinal cord injury. Neurobiol Dis 2010;39:272–282. [DOI] [PubMed] [Google Scholar]
- 4.Li H, Zhao X, Zhang X, et al. Exendin-4 enhances motor function recovery via promotion of autophagy and inhibition of neuronal apoptosis after spinal cord injury in rats. Mol Neurobiol 2016;53: 4073–4082. [DOI] [PubMed] [Google Scholar]
- 5.Yang Z, Klionsky D. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010;12:814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010;140:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanno H, Ozawa H, Sekiguchi A, et al. The role of autophagy in spinal cord injury. Autophagy 2009;5:390–392. [DOI] [PubMed] [Google Scholar]
- 8.Kliosnky D. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) (vol 12, pg 1, 2015). Autophagy 2016;12:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 2011;12: 437–452. [DOI] [PubMed] [Google Scholar]
- 10.Franco R, Martinez-Pinilla E, Navarro G, et al. Potential of GPCRs to modulate MAPK and mTOR pathways in Alzheimer’s disease. Prog Neurobiol 2017;149–150:21–38. [DOI] [PubMed] [Google Scholar]
- 11.Yang Y, Gao K, Hu Z, et al. Autophagy upregulation and apoptosis downregulation in DAHP and triptolide treated cerebral ischemia. Mediat Inflamm 2015;2015:120198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai X, Liu S, Yuan L, et al. Hydrogen-rich saline mediates neuroprotection through the regulation ofendoplasmic reticulum stress and autophagy under hypoxia-ischemia neonatal brain injury in mice. Brain Res 2016;1646:410–417. [DOI] [PubMed] [Google Scholar]
- 13.Tang P, Hou H, Zhang L, et al. Autophagy reduces neuronal damage and promotes locomotor recovery via inhibition of apoptosis after spinal cord injury in rats. Mol Neurobiol 2014;49:276–287. [DOI] [PubMed] [Google Scholar]
- 14.Zhang D, Xuan J, Zheng B, et al. Metformin improves functional recovery after spinal cord injury via autophagyflux stimulation. Mol Neurobiol 2017;54:3327–3341. [DOI] [PubMed] [Google Scholar]
- 15.Feldman L, Vinker S, Efrati S, et al. Amlodipine treatment of hypertension associates with a decreased dementia risk. Clin Exp Hypertens 2016;38:545–549. [DOI] [PubMed] [Google Scholar]
- 16.Kawai H, Deguchi S, Deguchi K, et al. Synergistic benefit of combined amlodipine plus atorvastatin on neuronal damage after stroke in Zucker metabolic rat. Brain Res 2011;1368:317–323. [DOI] [PubMed] [Google Scholar]
- 17.Choi N, Choi H, Park H, et al. Neuroprotective effects of amlodipine besylate and benidipine hydrochloride on oxidative stress-injured neural stem cells. Brain Res 2014;1551:1–12. [DOI] [PubMed] [Google Scholar]
- 18.Lee YJ, Park H, Koh S, et al. Amlodipine besylate and amlodipine camsylate prevent cortical neuronal cell death induced by oxidative stress. J Neurochem 2011;119:1262–1270. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Wang C, He T, et al. Mitochondrial transfer from bone marrow mesenchymal stem cells to motor neurons in spinal cord injury rats via gap junction. Theranostics 2019;9:2017–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egashira N, Okuno R, Abe M, et al. Calcium-channel antagonists inhibit marble-burying behavior in mice. J Pharmacol Sci 2008;108:140–143. [DOI] [PubMed] [Google Scholar]
- 21.Biala G, Kruk-Slomka M, Jozwiak K. Influence of acute or chronic calcium channel antagonists on the acquisition and consolidation of memory and nicotine-induced cognitive effects in mice. Naunyn Schmiedebergs Arch Pharmacol 2013;386:651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Wang X, Li W, et al. A Sensitized IGF1 treatment restores corticospinal axon-dependent functions. Neuron 2017;95:817–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y, Gu C, Wang Q, et al. The protective effort of GPCR kinase 2-interacting protein-1 in neurons via promoting Beclin1-Parkin induced mitophagy at the early stage of spinal cord ische-mia-reperfusion injury. FASEB J 2020;34:2055–2074. [DOI] [PubMed] [Google Scholar]
- 24.Allahdadi KJ, de Santana TA, Santos GC, et al. IGF-1 overexpression improves mesenchymal stem cell survival and promotes neurological recovery after spinal cord injury. Stem Cell Res Ther 2019;10:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanova H, Kerkhofs M, La Rovere RM, et al. Endoplasmic reticulum-mitochondrial Ca(2R) fluxes underlying cancer cell survival. Front Oncol 2017;7:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liss B, Striessnig J. The potential of L-type calcium channels as a drug target for neuroprotective therapy in parkinson’s disease. Annu Rev Pharmacol 2019;59:263–289. [DOI] [PubMed] [Google Scholar]
- 27.Snutch TP, Zamponi GW. Recent advances in the development of T-type calcium channel blockers for pain intervention. Brit J Pharmacol 2018;175:2375–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huebner EA, Budel S, Jiang Z, et al. Diltiazem promotes regenerative axon growth. Mol Neurobiol 2019;56:3948–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ravanan P, Srikumar IF, Talwar P. Autophagy: the spotlight for cellular stress responses. Life Sci 2017;188:53–67. [DOI] [PubMed] [Google Scholar]
- 30.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010;40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004;6:463–477. [DOI] [PubMed] [Google Scholar]
- 32.Ko S, Apple EC, Liu Z, et al. Age-dependent autophagy induction after injury promotes axon regeneration by limiting NOTCH. Autophagy 2020;16:2052–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sripetchwandee J, Khamseekaew J, Svasti S, et al. Deferiprone and efonidipine mitigated iron-overload induced neurotoxicity in wildtype and thalassemic mice. Life Sci 2019;239:116878. [DOI] [PubMed] [Google Scholar]
- 34.Teng T, Ridgley DM, Tsoy A, et al. Azelnidipine attenuates the oxidative and NFκB pathways in amyloid-β-stimulated cerebral endothelial cells. Acs Chem Neurosci 2019;10:209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Chen S, Lu K, et al. Verapamil ameliorates motor neuron degeneration and improves lifespan in the SOD1(G93A) mouse model of ALS by enhancing autophagic flux. Aging Dis 2019;10: 1159–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park H, Han M, Choi H, et al. Mitochondria damaged by oxygen glucose deprivation can be restored through activation of the PI3K/Akt pathway and inhibition of calcium influx by amlodipine camsylate. Sci Rep-Uk 2019;9:15717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Ren L, Cui X, et al. Muscular proteomic profiling of deep pressure ulcers reveals myoprotective role of JAK2 in ischemia and reperfusion injury. Am J Transl Res 2018;10:3413–3429. [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Y, Zhang H, Zheng B, et al. Retinoic acid induced-autophagic flux inhibits er-stress dependent apoptosis and prevents disruption of blood-spinal cord barrier after spinal cord injury. Int J Biol Sci 2016;12:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hou H, Zhang L, Zhang L, et al. Acute spinal cord injury could cause activation of autophagy in dorsal root ganglia. Spinal Cord 2013;51:679–682. [DOI] [PubMed] [Google Scholar]