

Abstract
We introduce Giraffe, a pangenome short read mapper, which can efficiently map to a collection of haplotypes threaded through a sequence graph. Giraffe maps sequencing reads to thousands of human genomes at a comparable speed to standard methods mapping to a single reference genome. The increased mapping accuracy enables downstream improvements in genome-wide genotyping pipelines for both small variants and larger structural variants. We use Giraffe to genotype 167 thousand structural variants, discovered in long-read studies, in 5,202 diverse human genomes sequenced using short reads. We conclude that pangenomics facilitates a more comprehensive characterization of variation and, as a result, has the potential to improve many genomic analyses.
Introduction
The field of genomics almost exclusively uses a single reference genome assembly as an archetype of a human genome. Reliance on comparing to the sequences within the reference assembly has created a pervasive bias toward the alleles it contains. This reference allele bias occurs because non-reference alleles are naturally harder to identify when mapping DNA sequencing data to the reference sequences. Reference allele bias is particularly acute for structural variations (SVs), which are complex alleles involving 50 or more nucleotides of divergent sequence. SVs affect millions of bases within each human genome. Due to reference allele bias, SVs are much more poorly characterized than single-nucleotide variants (SNVs) and short insertions and deletions (collectively termed indels) (1,2). Similarly, characterizing genetic variation in highly polymorphic and repetitive sequence has proven challenging (3).
Recent releases of the reference human genome assembly attempted to address these issues by adding additional sequences. These alternate sequences represent diversity in localized regions of the genome (4). However, to date these limited additions have not found widespread use. In contrast, pangenomes encode information about many complete genome assemblies and their homologies (the sequences that are shared between genomes by virtue of descending from a common ancestral sequence). Pangenomes are emerging as a replacement for linear reference assemblies to help mitigate these problems (5–7). They can particularly improve genotyping of structural variants (8).
Pangenomes are frequently formulated as sequence graphs (9): mathematical graphs that represent the homology relationships between multiple sequences. Several algorithms have been developed for mapping sequences to sequence graphs. None has yet made mapping the short sequencing reads from widely used DNA sequencers, such as those made by Illumina, to a structurally complex pangenome a practical option for large-scale applications. The original VG-MAP algorithm (10) maps to complex sequence graphs containing cycles produced by duplications and complex genomic rearrangements (10). However, VG-MAP is at least an order of magnitude slower than popular linear genome mappers that have comparable accuracy. Given that mapping is frequently a bottleneck in genome analysis, the cost of VG-MAP has proven prohibitive. Other pangenome mappers have different capabilities and limitations. Some are faster but limited to acyclic graphs containing variation at relatively low density (11), or can map to arbitrary sequence graphs but are designed for long reads (12). Other tools are not open source and thus unavailable for general testing and customization (13,14), and some additionally cannot run on commodity computing environments (14).
Results
Giraffe: Fast, Haplotype Aware Pangenome Mapping
When a sequence graph reference (5) (Fig. S1) is substituted for the traditional linear reference (Fig. 1A) it can reduce reference allele bias by including more alleles (10). However, it also expands the size of the alignment search space from a few linear chromosome strings to a combinatorially large number of paths in the graph. This has made our previous graph mappers slower than linear mappers (10). Giraffe solves this problem by considering the paths that are observed in individuals’ genomes: the reference haplotypes. We use the two haplotypes (one from each parent) that each individual has in their genome, and trace them as paths through the sequence graph. The graph describes which positions in the haplotypes are equivalent, while the haplotypes describe the subset of the possible paths in the graph to consider. Giraffe uses a Graph Burrows Wheeler Transform (GBWT) index (15) to store and query a graph’s haplotypes efficiently.
Figure 1.
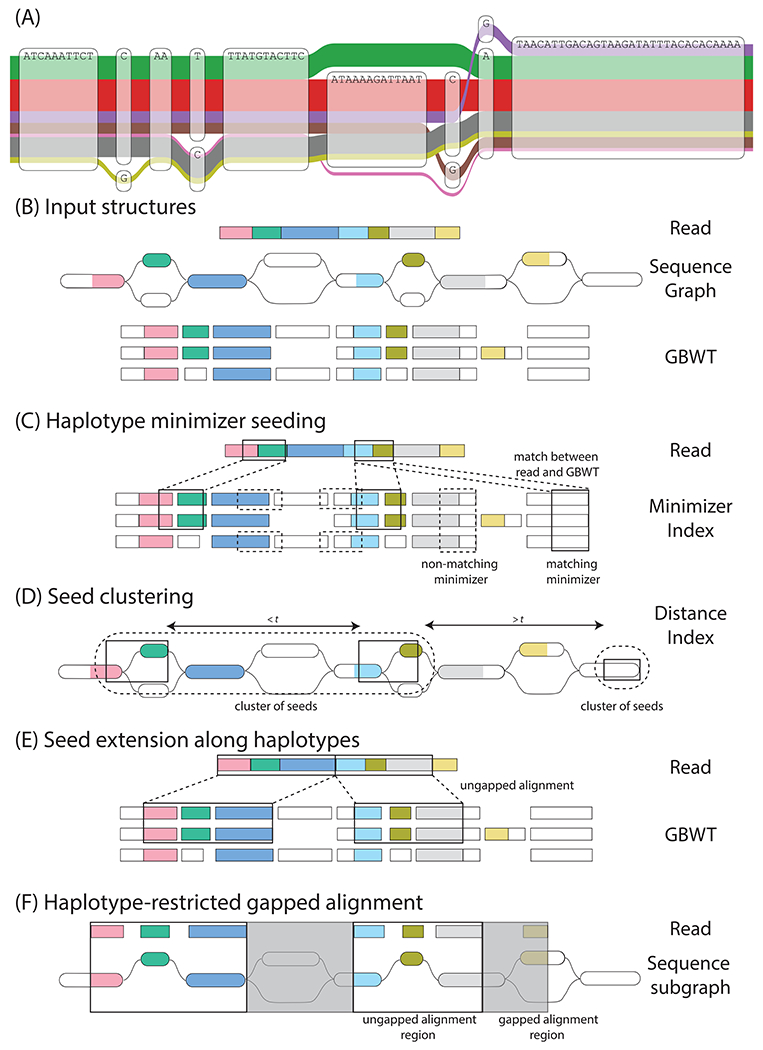
Haplotype mapping. (A) A region of the CASP12 gene in the 1000GP graph (17), illustrating complex local variation. The observed haplotypes (the colored ribbons of width log-proportional to population frequency) represent only a subset of the possible paths through the graph. (B-F) An overview of Giraffe. (B) Input structures: Giraffe takes as input each read to map, the sequence graph reference to map against, and the GBWT of known haplotypes to restrict to. The input read is represented as a series of colored rectangles. The haplotype sequences in the GBWT are similarly represented as series of rectangles, split according to the nodes they correspond to in the sequence graph. Nodes in the sequence graph and haplotypes in the GBWT are colored according to homology with the read. (C) Haplotype minimizer seeding: Seeds are identified using an index of minimizers (subsets of sequences of specified length k) (53) over the sequences of all the GBWT haplotypes. A matching minimizer between the read and the GBWT haplotypes constitutes a seed. The minimizers (black boxes) in the read are enumerated and the matching minimizers in the haplotypes are identified using the minimizer index. (D) Seed clustering: Minimizer instances in the graph are clustered by the minimum graph distance (t, measured in nucleotides) between them (54). (E) Seed extension along haplotypes: Minimizers in high scoring clusters are extended linearly to form maximal gapless local alignments. (F) Haplotype-restricted gapped alignment: Giraffe is designed on the assumption that, for most reads, it will be possible to gaplessly extend seed alignments all the way to the ends of the read, allowing the algorithm to stop at the previous step. However, any remaining gaps in the alignment between read and graph are resolved by gapped alignment in this final step.
Giraffe’s strategy of aligning to haplotype paths has two key benefits. First, it prioritizes alignments that are consistent with known sequences, avoiding combinations of alleles that are biologically unlikely. Second, it reduces the size of the problem by limiting the sequence space that the reads could be aligned to. This deals effectively with complex graph regions where most paths represent rare or nonexistent sequences.
We designed Giraffe to minimize the amount of gapped alignment that is performed. Computing gapped alignments, in which sequences are allowed to gain or lose bases relative to each other, is much more expensive than gapless alignment as it requires pairwise dynamic programming algorithms. Most Illumina sequencing errors are substitutions (16), and common true indels relative to the traditional linear reference should already be present in the haplotypes, therefore almost all reads will have a gapless alignment to some stored haplotype. Hence, we try to align each read without gaps before resorting to dynamic programming.
Giraffe follows the common seed-and-extend approach used by most existing mappers (see algorithm in (17)). In this framework, short seed matches between a sequencing read and a genomic reference are found with minimal work, and then only good seeds are extended into mappings of the entire read (18–20). A visual overview of Giraffe’s operation is given in (Fig. 1B–F). The Giraffe algorithm uses several heuristics for prioritizing alignments. These heuristics are configurable, and we present two presets: default Giraffe (written as just “Giraffe”) balances speed and accuracy, and fast Giraffe optimizes for speed at the expense of some accuracy.
Pangenome references for evaluation
To evaluate Giraffe we built two human genome reference graphs based on the GRCh38 reference assembly. One (the 1000GP graph) contained mostly small (<50 base pairs (bp)) variants from the 1000 Genomes Project (21). The other (the HGSVC graph) contained entirely SVs (≥50 bp) from the Human Genome Structural Variant Consortium (22,17). The 1000GP graph contained data from 2,503 individuals, with one (NA19239) held out for benchmarking. It was built from 76,749,431 SNVs, 3,177,111 small indels (<50 bp), and 181 larger SVs (≥50 bp). The HGSVC graph contained data from three individuals sequenced with long reads: HG00514, HG00733, and NA19240. The HGSVC graph contained 78,106 larger SVs (≥50 bp). Both graphs are available for re-use (see Data and materials availability).
Giraffe and VG-MAP map accurately to human pangenomes
We evaluated Giraffe for mapping human data by simulating paired-end reads for two individuals (17): NA19240, who has available genotypes for HGSVC variants (22), and NA19239, who has available genotypes for the 1000GP variants (21). Simulated read sets were mapped using Giraffe and competing tools (17). We examined the accuracy of single and paired-end mapping (Fig. 2). We looked at a variety of input read sets and evaluated the calibration of reported mapping quality, a standard measure of mapping uncertainty (Figs. S2, S3, S4, S5, S6, and S7 and Tables S1, S2, S3, S4, S5, and S6). Relative to other tools, at the highest reported mapping quality, VG-MAP and default Giraffe consistently have either higher precision or recall across all simulated read technologies and graphs. Their performance is generally similar. Relative to the linear mappers, the Giraffe and VG-MAP lead is larger for the HGSVC graph (Fig. 2C–D) than the 1000GP graph (Fig. 2A–B). This suggests that the gains from using a genome graph are higher when the graph facilitates alignment of genomic sequences from the sample that differ greatly from the linear reference.
Figure 2.
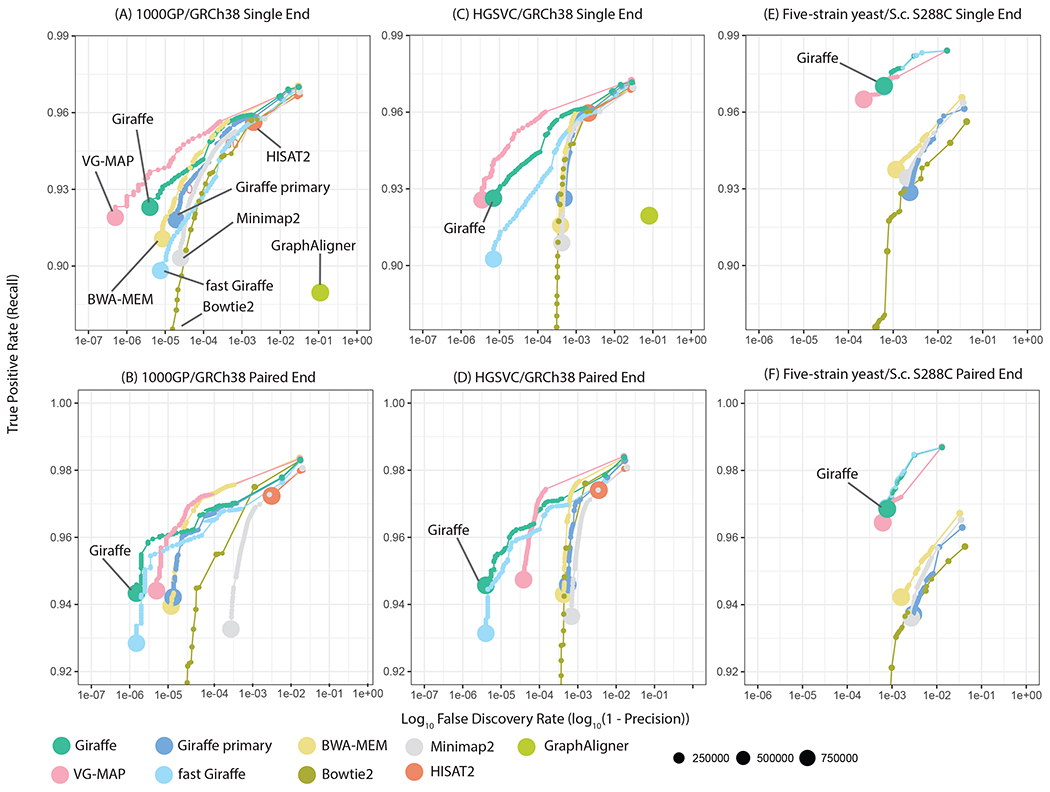
Simulated read mapping. Each panel shows recall vs. FDR (false discovery rate, or 1 minus precision) for a simulated read mapping experiment, comparing Giraffe to linear genome mappers (BWA-MEM, Bowtie2, Minimap2) and other genome graph mappers (VG-MAP, GraphAligner, HISAT2). Reads were simulated to match ~ 150 bp Illumina NovaSeq (for human) or HiSeq 2500 (for yeast) reads, either as single-ended reads (A-C) or as paired-end reads (D-F) (17). Results for each mapper are shown stratified by reported read mapping quality; the size of each point represents the log-scaled number of reads with the corresponding mapping quality. Three different mapping scenarios are assessed: (A,D) Comparing mapping to a graph derived from the 1000GP data to mapping to the linear reference genome assembly upon which it is based (GRCh38). (B,E) Comparing mapping to a graph containing larger structural variants from the HGSVC project to mapping to the GRCh38 assembly upon which it is based. (C,F) Comparing mapping to a multiple sequence alignment based yeast graph to mapping to the single S.c. S288C linear reference, for reads from the DBVPG6044 strain. For mapping with Giraffe, we used the full GBWT containing 6 haplotypes to map to the HGSVC graph and the 64-haplotype sampled GBWT to map to the 1000GP graph. “Giraffe primary” represents mapping with Giraffe to the linear reference.
Haplotype sampling improves read mapping
Having rare variants or errors in the graph and haplotypes may reduce mapping accuracy by creating opportunities for false positive mappings (23). Mapping reads to regions with many distinct local haplotypes can also be slow. Additionally, Giraffe needs a mechanism to synthesize haplotypes for graph components where no haplotype variation is known. To overcome these issues, Giraffe includes mechanisms for creating synthetic haplotype paths. When real haplotypes are available, these synthetic haplotype paths represent local haplotype variation sampled according to haplotype frequency, and we call the result a sampled GBWT (17). When no haplotypes are available, we call the result a path cover GBWT. In this case, the synthetic haplotypes represent random walks through the graph. We evaluated the effects of running our mapping evaluations with sampled and path cover GBWTs (Fig. S8, Tables S7 and S8, (17)). The mapping benefit of sampling more haplotypes plateaued at 64 haplotypes for the 1000GP graph (which contains around five thousand haplotypes), with higher accuracy than is achieved by mapping to the full haplotype set. We used the HGSVC graph (which contains just six haplotypes) for an experiment on generating path covers without known haplotypes. Path covers alone did not outperform the full underlying haplotype set for the HGSVC graph, but came close to matching its performance. We selected the 64-haplotype sampled GBWT for the 1000GP graph and the full GBWT for the HGSVC graph as the best-performing GBWTs, which we use in the rest of the analysis.
Giraffe improves pangenome mapping speed
We measured the runtime (Fig. 3A–B) and memory usage (Fig. 3C–D) of Giraffe and competing tools when mapping real reads (17). Giraffe was more than an order of magnitude faster than VG-MAP in all conditions. It was also faster at aligning to human graphs than Bowtie2 or BWA-MEM were at aligning to the corresponding linear reference. For the 1000GP graph using the 64-haplotype sampled GBWT for mapping instead of the full ~ 5,000-haplotype GBWT was much faster in every case. HISAT2 and fast Giraffe were both about equally fast and both faster than all other mappers.
Figure 3.
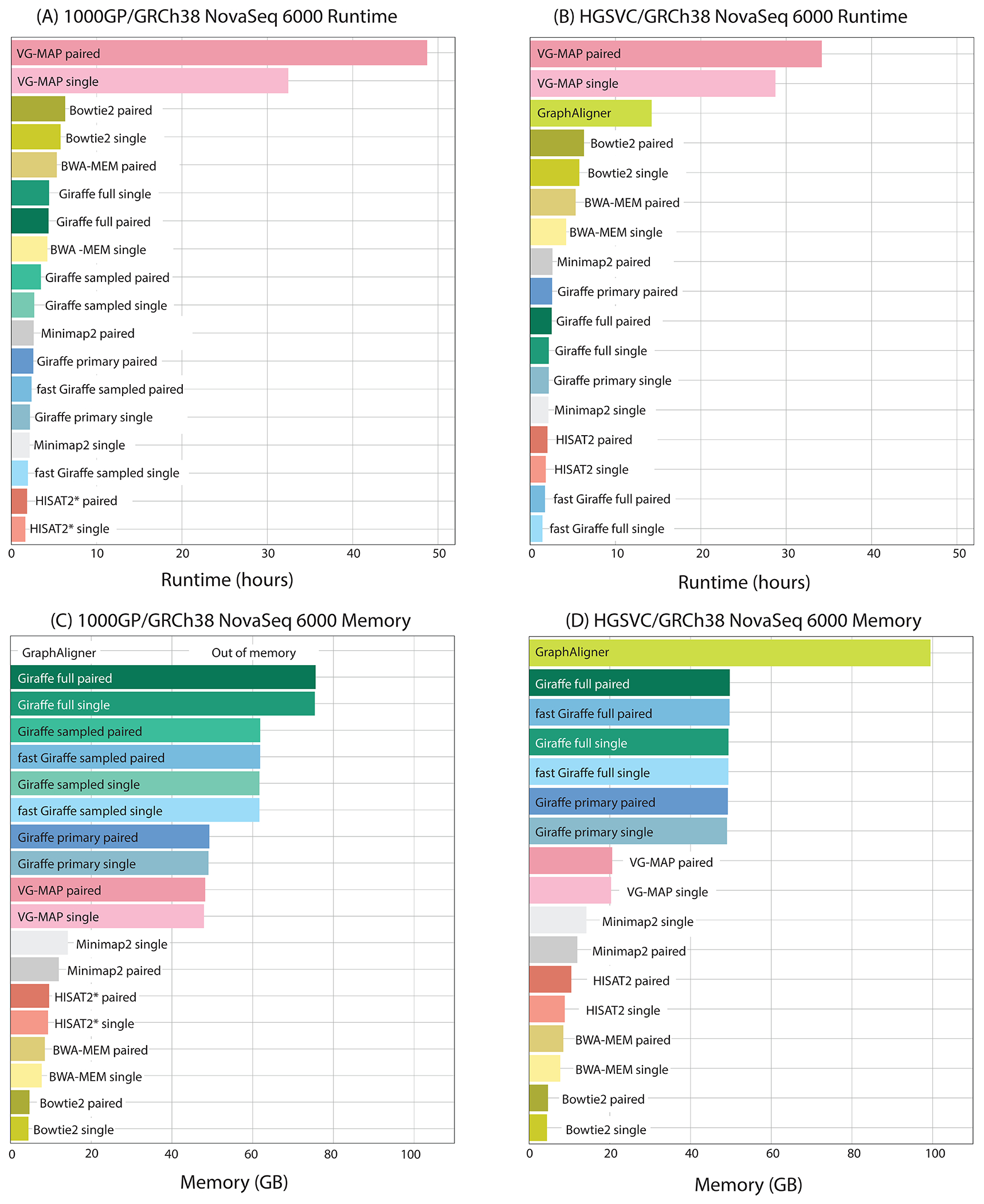
Runtime and memory usage. Total runtime (A,B) and peak memory use (C,D) for mapping ~600 million NovaSeq 6000 reads using 16 threads. Reads were mapped (A,C) to the 1000GP derived graph or (for linear mappers) the GRCH38 assembly, and (B, D) to the HGSVC graph or GRCh38 reference, respectively. HISAT2*: results are shown for the subset 1000GP graph (22). Giraffe full refers to mapping using the full GBWT of all haplotypes. Giraffe sampled refers to mapping using the 64-haplotype sampled GBWT.
Due to the in-memory indexes it uses, Giraffe’s memory consumption is higher than the other mappers, except for GraphAligner. However, it can map to the 1000GP graph with the full GBWT in ~ 80 gigabytes of memory—an amount readily available on compute cluster nodes (Fig. 3C–D).
Giraffe reduces allele mapping bias
We assessed Giraffe’s reference bias(17). We expected Giraffe to be able to use the extra variation information contained in the graph reference to achieve a lower level of bias than a linear mapper. For variants that were heterozygous in NA19239, we found the fraction of reads supporting alternate vs. reference alleles at each indel length (Fig. 4A). Giraffe and VG-MAP both show less bias towards the reference allele than a linear mapper and this difference becomes more pronounced as indel length increases, particularly for larger insertions.
Figure 4.
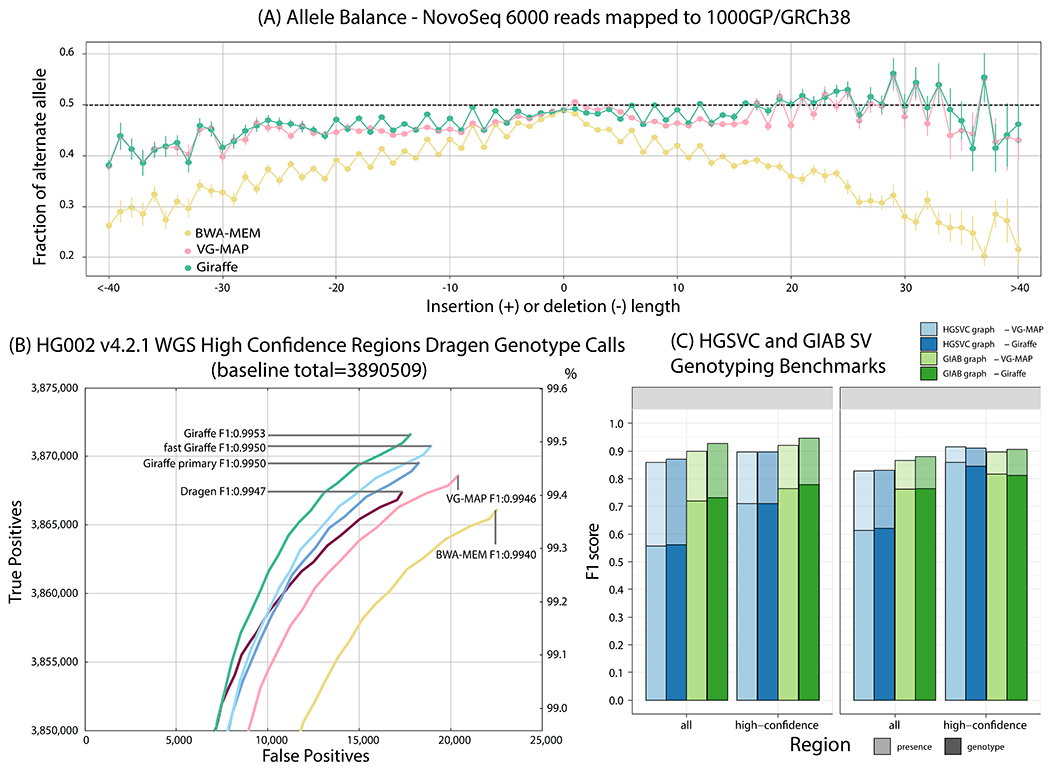
Evaluating Giraffe for genotyping. (A) The fraction of alternate alleles in reads detected for heterozygous variants in NA19239. Reads were mapped to the 1000GP graph with Giraffe and VG-MAP and to GRCh38 with BWA-MEM, and the fraction of reads supporting reference or alternate alleles was found for each indel length. (B) Assessing true positive and false positive genotypes made using the Dragen genotyper with mappings from Giraffe and other mappers. The line labeled Dragen represents the mapper included with the Dragen system itself. (C) Comparing Giraffe to VG-MAP for typing large insertions and deletions. “Presence” (lighter bars) evaluates the detection of SVs without regard to genotype; “genotype” (darker bars) requires the SV to be detected and its genotype to agree with the truth genotype. The y-axis shows the F1 score. For the HGSVC benchmark, we define high-confidence regions as regions not overlapping simple-repeats and segmental duplications. For the Genome in a Bottle consortium (GIAB) benchmark, we use the set high-confidence regions provided by GIAB.
Giraffe genotyping outperforms best practices
We used Illumina’s Dragen platform (14) to genotype SNV and short indels using Giraffe mappings to the 1000GP graph, projected onto the linear reference assembly. We compared these results to results using competing graph and linear reference mappers (17). No training or optimization was performed for any of the mappings other than those performed by default by Dragen itself. We evaluated the calls using the Genome In a Bottle (GIAB) v4.2.1 HG002 high confidence variant calling benchmark (24).
Out of the examined pipelines, Giraffe mappings to the 1000GP graph produce the highest overall F1 score (harmonic mean of precision and recall) at 0.9953 (Figure 4B and Tables S9 and S10). Similar but uniformly higher results were found with higher coverage, 250bp reads (Tables S11, S12 and Figure S9). Although one would expect longer reads and higher coverage to produce better variant calls, all else being equal, Giraffe has a slightly higher F1 score with the 150bp read set (0.9953) than BWA-MEM with the higher coverage 250bp read set (0.9952). Restricting comparison only to confident regions that overlap variant calls from the 1000GP variants used in graph construction, Giraffe has the highest F1 score at 0.9995 relative to the other methods (Table S13 and Figure S10). Perhaps surprisingly, Giraffe maintains the highest F1 score (0.9528) when performing the converse analysis, restricting the comparison to confident regions that do not overlap 1000GP variant calls (Table S14 and Figure S11).
DeepVariant is a highly accurate genotyping tool that requires training (25). We trained DeepVariant to use Giraffe mappings and evaluated it on the held-out sample HG003 (17). We compared it to the Dragen pipelines tested and DeepVariant using BWA-MEM with the BWA-MEM trained model they provide. The Giraffe/DeepVariant pipeline (F1: 0.9965) outperforms all other tested pipelines (Tables S15, S16 and Figure S12).
Previously, when we used VG-MAP to map reads to SV pangenomes, we found it to perform better than other methods for SV genotyping (8). We replicated that evaluation on the HGSVC and GIAB datasets (1,22) to confirm that the quality of the SV genotypes from Giraffe was competitive (17). We observe similar SV genotyping accuracy across SV types, genomic regions, and datasets (Fig. 4C). Of note, GraphTyper (26), which was published after our earlier benchmarking analysis (8), was also compared with vg as a variant caller, but showed lower genotyping performance across SV types, genomic regions, and datasets (Fig. S13).
Giraffe generalizes beyond human
We assessed Giraffe’s performance mapping to a yeast pangenome for five strains of the S. cerevisiae and S. paradoxus yeasts (17). This graph was substantially different from the human graphs. It proved challenging because it contains the cycles and duplications typical of graphs generated from genome-wide alignments of more divergent sequences. Using a graph decomposition technique (27), we find it contains 1,459,769 variant sites, 4 times the density of variation in the 1000GP graph. 90 of these sites are complex, meaning they are not directed, acyclic, and free of internal source and sink nodes.
Mapping accuracy results for reads from the held-out DBVPG6044 strain are displayed in Fig. 2 panels E (single end) and F (paired end). Speed results for mapping real reads are presented in Fig. S14. Neither HISAT2 nor GraphAligner could be map reads to the yeast graph; in the case of HISAT2, this was because it cannot map to graphs containing cycles. Giraffe is 28 times faster for paired end mapping than the only other tool that could map to this graph, VG-MAP, while achieving similar accuracy. Both graph mappers are much more accurate than the linear reference methods. Moreover, the gap between the graph methods and the linear reference is even larger on this graph than in the HGSVC graph (Fig. 2C–D). This lends further support to the hypothesis that graph mapping methods have the most benefit when facilitating alignment of genomic sequences that differ greatly from the reference, such as the sibling-subspecies-scale differences represented in the yeast graph.
Genotyping 5,202 samples’ structural variants
Building on our previous work to genotype SVs (8), we demonstrate the value of Giraffe by performing population-scale genotyping of an expanded compendium of SVs in large cohorts of samples sequenced with short reads. We built a comprehensive pangenome containing SVs combining variants from three catalogs of SVs discovered using long read sequencing (1, 22, 28). The combined catalog represents 16 samples from diverse human populations and is estimated to cover the majority of common insertions and deletions in the human population (28). Near-duplicate versions of variants (i.e. SVs with slightly different breakpoints) are often present within and across SV catalogs. A naïve integration of all these variants can lead to redundancy in the graph that can impact read mapping and variant genotyping. We remapped sequencing data and integrated variants iteratively into the graph to progressively build a non-redundant, compact SV graph (17). The final SV graph was constructed from 123,785 SVs from the original catalogs: 53,663 deletions and 70,122 insertions. Overall, the graph contained 26.2 Mbp of non-reference sequences in the form of insertions. Using a graph decomposition (27), we identified 228,405 subgraphs representing variant sites. Some of these correspond to smaller variants nested inside larger ones. Combining these cases, there were 96,644 non-nested, non-overlapping SV subgraphs.
Compared to Hickey et al. (8), we used a graph containing more SVs and a more recent version of the VG toolkit (see Data and materials availability), including the Giraffe mapper presented here. Our SV genotypes were as accurate as in (8) if not more so (Fig. S15A–C). Thanks to Giraffe and improvements in the variant calling approach, the genotyping workflow used about 12 times less compute on a sample sequenced at about 20x coverage.
SV genotyping was run using the NHLBI BioData Catalyst ecosystem (29) (Fig. S15D). We genotyped samples from the Multi-Ethnic Study of Atherosclerosis (MESA) cohort within the Trans-Omics for Precision Medicine (TOPMed) program. The MESA cohort is a longitudinal cohort study consisting of 6,814 participants at baseline (between the years 2000 and 2002). Participants were ascertained from 6 sites in the USA, and identified themselves as “Spanish/Hispanic/Latino” (22% at baseline), and/or “African American or Black” (28%), “Chinese” (12%), and “Caucasian or White” (39%) (30). Two thousand samples from the MESA cohort (30) were selected, using a criterion to maximize the sample diversity (17). Using the graph described above and fast Giraffe, it took around 4 days to genotype 2,000 samples from the MESA cohort. We used the same workflow to genotype the 3,202 diverse samples from the high-coverage 1000GP dataset (31) in around 6 days. On average, genotyping a sample took 194.4 CPU-hours of compute and cost between $1.11 and $1.56 (Fig. S15D, Table S17 and Table S18). The sequencing data were down-sampled in advance to ~20x coverage to reduce compute costs. Benchmarking indicates that this down sampling has a minimal impact on the genotyping accuracy (Fig. S16).
Diverse, clustered structural variants
Our SV graph construction approach preserves multiple alleles cataloged at a given SV site, and decomposes them into a parsimonious joint representation. In general, these SV representations require more alleles per site than is common for small variants. For example, there may be SNVs and indels within the sequence of an insertion or around a deletion’s breakpoints, or copy-number changes in Variable Number Tandem Repeat (VNTR) regions. The existence of these potentially recombining sub-variants implies the possibility of novel alleles.
We genotyped a total of about 1.7 million alleles clustered in 167,858 SV sites across the 2,000 MESA samples. In most SV sites, we only observed one or a few alleles (~90% of SV sites with 5 or fewer alleles, Fig. 5A). Additionally, most of the SV sites (~151 thousand, 90%) contained SV alleles that differed by only small variants (17), while the rest of the sites showed size variation from polymorphic VNTR regions (Fig. S17A). Examples of SV sites that illustrate these different profiles are given in Fig. 5B–C and Fig. S18. Fig.5D–E shows the size and frequency distributions of the most common allele at each site. SVs spanned the size spectrum (50 bp up to 125 kbp), with 89.8% shorter than 500 bp. For 84% of the SVs, simple repeats or low-complexity regions overlapped at least 50% of the SV region. Hence, the SVs genotyped in this study match the original SVs discovered in long-read sequencing studies (1,22,28) in terms of number, size distribution, and sequence context.
Figure 5.
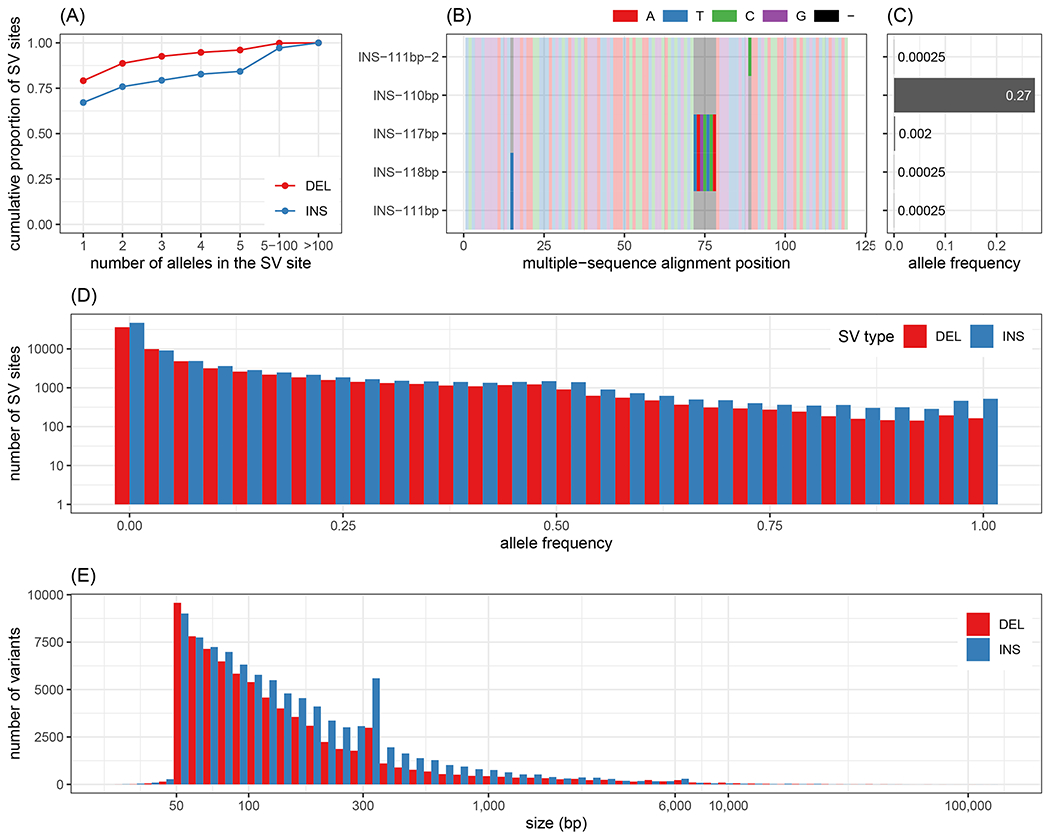
Structural variants in the Multi-Ethnic Study of Atherosclerosis (MESA) cohort. (A) Cumulative proportion of SV sites depending on the maximum number of alleles (x-axis) in the site. (B-C) illustrates an insertion site with 5 alleles. The alleles differ by 3 nested indels as shown by the multiple sequence alignment of the inserted sequences represented in (B). Only one allele is frequent in the population (AF=0.27) as highlighted by (C). (D) Allele frequency distribution of the major allele for each SV site. The y-axis, showing the number of SVs, is log-scaled. (E) Size distribution of the major allele for each SV site.
We observed similar patterns in the 1000 Genomes Project dataset. We identified 1.9 million alleles clustered in 167,188 SV sites, with a size and frequency distribution similar to that of the MESA cohort (Fig. S19). The 1000 Genomes Project dataset also provided 602 trios that we used to estimate the quality of our genotypes. First, we computed the rate of Mendelian error which was 5.2% for deletions and 4.7% for insertions when considering all variants. This error decreased as the confidence in the genotype increased. For example, the Mendelian error dropped to 2.1% and 2.5% for the ~70% of deletions and insertions, respectively, with the highest genotype qualities (Fig. S20A). The most common error by far occurred when a heterozygous variant was predicted in the offspring but both parents were predicted homozygous for the reference allele (Table S19). The transmission rate of heterozygous alleles was close to the expected 50%: 40-47% for deletions and 43-49% for insertions (Fig. S20B).
Comprehensive structural variant frequency estimates
The genotyped SVs were originally discovered with long read sequencing technology, and many are absent from the population scale SV catalogs that could provide frequency information. 93% of the SV sites genotyped using our pangenome approach are missing from the 1000 Genomes Project SV catalog (32), and 67% were missing from the gnomAD-SV catalog (33) (Table S20). This is consistent with the amount of novel structural variation described in the three studies from which our SV graph is derived (1,22,28). Our results provide frequency estimates across a large and diverse cohort for these SVs.
The frequency distribution resembled the allele frequency distributions in the 1000 Genomes Project SV catalog and gnomAD-SV (Fig. S21A). The frequencies of the subset of variants present in both our catalog and the mentioned public catalogs were largely concordant (Fig. S21B–C). Our frequency distribution looks rather different than that of SVPOP (28). However, we note that SVPOP’s frequency distribution is markedly different than the 1000 Genomes Project and gnomAD-SV (Fig. S21A), and has very different frequency estimates on matched variants (Fig. S21D–F).
Fine-tuning structural variants with frequencies
SVs in the input catalogs may contain errors. When multiple alleles co-occur at an SV site, we often observed that one allele was frequently present in the cohort while other similar alleles were not (Fig. 5B–C). The other alleles at these sites are either rare or erroneous. In either case, it is useful to identify the major alleles. In 7,520 SV sites, only one allele was called in more than 1% of the population while other alleles from the original catalogs were not. Further, the major allele was at least three times more frequent than the second most frequent allele in 6,175 of these sites (Fig. S17B). As a quality control, we verified that these alleles were more likely to match exactly with the alleles in the GIAB truth set (1), which is the SV catalog with the highest base-level confidence (permutation p-value < 0.0001, (17)). Our results thus help fine-tune the sequence resolution of these SVs. More generally, our results identify one major allele for 39,699 multi-allelic SV sites.
Structural variant frequency population signatures
Principal component analysis (PCA) of the allele counts at the 166,959 SV sites in the MESA cohort produces a low-dimensional embedding of the samples. This embedding appears similar to the TOPMed consortium’s PCA of SNV genotype data from all samples (Pearson correlation: 0.96-0.99 for the top three components, Fig. S22). This result is expected and provides confirmatory support for the accuracy of our SV genotypes.
We clustered samples with PCA, taking each cluster to be a population (17). Allele frequencies vary across these populations for thousands of SV sites (Fig. S23A–C). For example, we found 21,069 SV sites with strong inter-cluster frequency patterns, defined by a frequency in any population differing by more than 10% from the median frequency across all populations (Fig. S23D). The existence of structural variants with different frequencies across populations supports the need to develop and test genomic tools and references across multiple populations.
Since there is a risk of circularity when using the same genotype data to define populations and look for patterns across them, we replicated these observations in the high-coverage 1000 Genomes Project dataset (31). Here again, the PCA of the allele counts organized the samples in a way consistent with the known history of the 1000 Genomes Project “super population” groups (Fig. S24). In this analysis, we found 25,960 SV sites with strong inter-super-population frequency patterns, defined as for the MESA analysis, but with the 1000 Genomes super populations as the sample categories (Fig. S25). As a comparison, when the samples were randomly grouped into super populations, we observed only 14 SV sites with strong inter-group frequency patterns (17). More than 17 thousand SV sites with strong inter-superpopulation frequency patterns were enriched or depleted in the AFR super population, followed by about 10 thousand sites enriched or depleted in the EAS super population.
As an example of a newly annotated variant, a deletion of the RAMACL gene was genotyped with frequency 46.6% in the AFR super population, 4% in AMR, and less than 1% in other super populations. This deletion is not present in the 1000 Genomes Project SV catalog and was unresolved in version 2 of the gnomAD-SV catalog. It has been curated in gnomAD-SV v2.1 and shows similar population patterns there to what we found in our reanalysis of the 1000 Genomes Project dataset. Such variants could be falsely identified as putatively pathogenic if analyzed only in European-ancestry population where the frequency is low.
In addition, our approach is often capable of genotyping repeat-rich variants, such as short tandem repeats (STRs) that vary in length. For example, a 1 kbp expansion of an exonic variable-number tandem repeat (VNTR) in MUC6 with a frequency of 14% in the AFR super population was observed only rarely outside of it: 2.3% in AMR and <1% in other super populations (Fig. 6A). This repeat expansion is absent from gnomAD-SV and the SV catalog from the 1000 Genomes Project, despite its observed frequency.
Figure 6.
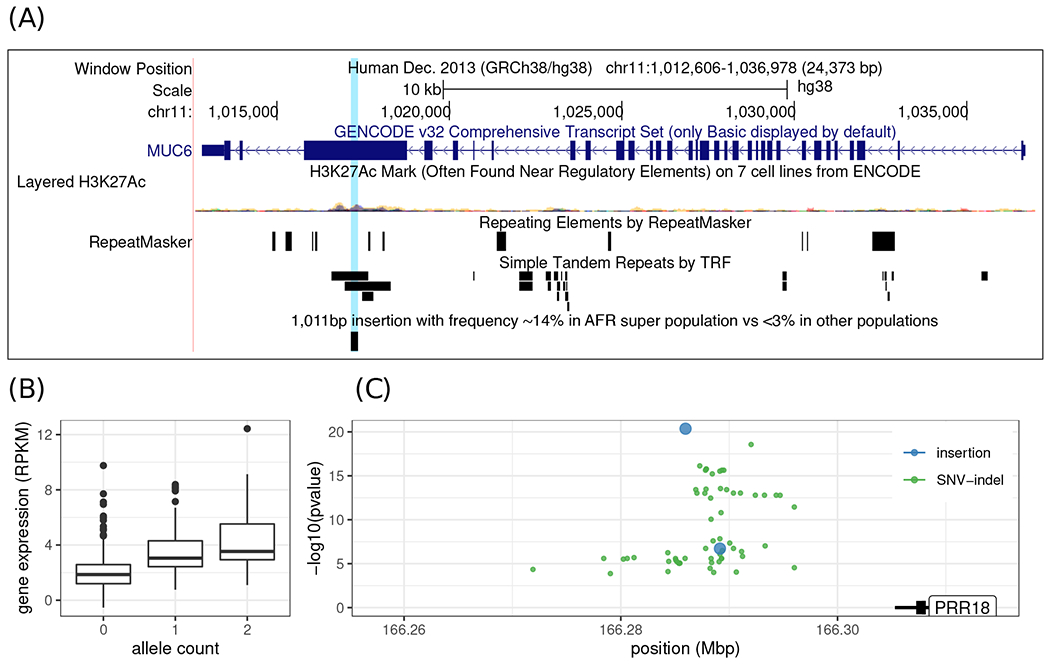
Population-specific structural variant and SV-eQTL in the 1000 Genomes Project dataset. (A) Example of an insertion at appreciable frequency (~ 14%) in the AFR super population while rare (< 3%) in the other super populations. The variant is a 1,011 bp expansion of a VNTR in the coding sequence of the MUC6 gene. (B-C) Association between a 10,083 bp insertion overlapping a predicted enhancer and the gene expression of the PRR18 gene. (B) Each allele is associated with an increase in gene expression. (C) shows the position of significant eQTLs (SNV/indels in green, insertions in blue). All the eQTLs are in the intergenic region downstream of the PRR18 gene. The y-axis represents the significance of the association, with the top eQTL being the highest point. Of note, the lead eQTL (the 10,083 bp insertion) overlaps a region predicted to be an enhancer by ENCODE.
Structural variants, genes, and expression
1,563 and 1,603 SVs overlapped coding regions of 408 and 380 protein-coding genes in the MESA and 1000 Genomes Project datasets respectively. When including promoters, introns and untranslated regions each dataset had overlaps between at least 78,290 SVs and 7,641 protein-coding genes. 10,640 of these SVs show strong inter-super-population frequency patterns in the 1000 Genomes Project dataset (see Fig. 6A).
We searched for associations between SVs and gene expression across 445 samples from the 1000 Genomes Project that have been RNA sequenced by the GEUVADIS consortium (34). These samples span four European-ancestry populations (CEU, FIN, GBR, and TSI), and the Yoruba in Ibadan, Nigeria (YRI) population (34). A pooled analysis identified 2,761 expression Quantitative Trait Loci (eQTLs) across 1,270 genes (false discovery rate of 1%, (17)). 878 of those genes are protein-coding genes. We note that 58% of the SV-eQTLs are located within simple repeats or low-complexity regions. The distribution of the p-values across all tests showed the expected patterns for genome-wide association studies (Fig. S26).
Genes with eQTLs, or eGenes, were enriched in gene families involved in immunity, as previously observed (35), but we also found significant enrichments in other families (Table S21). For example, 3 of the 10 genes in the anoctamins family have SV-eQTLs (adjusted p-value: 0.0006). This gene family is involved in the regulation of multiple processes including neuronal cell excitability, and mutations in some of its members have been linked to neurologic disorders (36). Other families enriched included the SMN complex family (3 out of 10 genes with SV-eQTLs, adjusted p-value: 0.0012) and aldehyde dehydrogenases genes (3 out of 19 genes with an SV-eQTLs, adjusted p-value: 0.008). As expected, SV-eQTLs were strongly enriched in coding, intronic, promoter, untranslated and regulatory regions (Fig. S27). Interestingly, SVs associated with decreased gene expression drove most of the enrichment in coding regions. Separate analysis of the four European-ancestry populations together, and the YRI population alone, identified, respectively, 44 and 139 SVs where an association with the expression of protein-coding genes was detected only in the smaller analysis (17). As expected, a number of these population-specific SV-eQTLs had shown strong inter-super-population frequency patterns (see above).
Finally, we performed a joint analysis with available SNVs and indels calls. Like previous studies (37,38), we found that the lead eQTLs (the strongest association for a gene) are enriched in SVs (permutation p-value: 0.022). For example, only 0.5% of the variants tested were SVs but SVs were the lead eQTL in 5.9% of the genes which had both SV and SNV/indel eQTLs. We didn’t observe a difference in relative effect size of SV-eQTLs compared to SNV/indel eQTLs, but we noticed that SV-eQTLs were 4-fold enriched in the genes with the highest expression (permutation p-value: 0.004; Fig. S28). Figs. 6B,C and Fig. S29 show two examples where the SV-eQTL is the strongest association: a 10,083 bp insertion associated with an increased expression of the PRR18 gene, and a 5,405 bp deletion associated with a reduced expression of the SLC44A5 gene. In addition, 39 genes had SV-eQTLs but no SNV/indel eQTLs (Table S21). These results show that the SV genotypes produced here can be used to test for phenotypic association.
Discussion
Pangenome references hold great potential as a replacement for standard linear reference genomes. They can represent diverse collections of human genomes, and they have been shown to reduce the bias that arises from using a linear reference (10). However, due to the significant complexity of the task, previous methods for mapping to pangenomes have been slow or not clearly better than comparable methods for linear genomes. In contrast, Giraffe can map to pangenome graphs consisting of thousands of aligned haplotypes, potentially with complex topologies, with accuracy comparable to the best previously published tools and speed surpassing linear reference mappers. Further, we have demonstrated that its mappings can improve genotyping.
Pangenome exchange formats have been co-evolving alongside pangenome methods. Giraffe is designed to meet and solidify these emerging standards while also interfacing with the broader genomics ecosystem. The GFA format for representing pangenome graphs has increasing tool support, including by vg (12,39–43). In addition, Giraffe can output the GAF read-to-pangenome-graph alignment format proposed by Li, et al. (39) and supported by other pangenome mappers (12). Giraffe also supports backwards compatibility to linear references by allowing mappings to be projected onto an embedded linear reference genome and output in standard formats. The state-of-the-art SNP and short indel genotyping results described in this study demonstrate the value of this support. These necessary technical advances are starting to nucleate an interoperable tool ecosystem for pangenomics.
For SVs, and particularly large insertions, we and others have shown that the benefits of pangenomes for genotyping are not merely incremental but transformative (8,39,44). Our approach allowed us to identify duplicate SVs, to refine the canonical definitions of SVs, and to establish the frequencies of these SVs in diverse human populations. Complementing previous surveys of SVs in diverse human populations (38, 45, 46), we demonstrate that many of the novel SVs studied here are also differentially distributed across human populations. This frequency information could be used, among other applications, for prioritizing variants to investigate for genomic medicine: variants common anywhere are unlikely to be pathogenic.
We expect accurate and unbiased SV genotyping to be one of pangenomics’ most impactful contributions. Among other applications, it will enable identifying more links from SVs to disease traits and other phenotypes. For example, we were able to detect thousands of associations between SVs and gene expression. Ebert et al. (38) recently performed a similar analysis using the same RNA-seq dataset from Geuvadis (complemented with 34 new deep RNA-sequencing experiments) and genotypes for SVs discovered in 32 haplotype-resolved genomes. Although we did not use this new sequencing data and SV catalog, we found a similar number of SV-eQTLs with our pangenomic approach (2,761 SV-eQTLs and 1,270 eGenes in this study; 2,109 SV-eQTLs and 1,526 eGenes in Ebert et al. (38)).
Soon, pangenomes will be built from larger collections of high-quality de novo assembled genomes using accurate long reads. We hope such human pangenomes will enable more comprehensive genotyping of common complex variants (including SVs) from existing catalogs of short-read sequencing data, allowing for the typing of such variants at the scale of existing catalogs of point variation. We expect that unlocking this latent information will ultimately aid with disease association studies and help us further understand how the architecture of the genome contributes to an individual’s phenotype.
Summary Methods
Evaluation
Read simulation:
To evaluate Giraffe for mapping human data, we obtained paired-end sequencing reads from a parent-child pedigree. Reads were obtained from an Illumina NovaSeq 6000 machine for parent NA19239 (accession ERR3239454) and from Illumina HiSeq 2500 and HiSeq X Ten machines for child NA19240 (accessions ERR309934 and SRR6691663, respectively). These samples were selected because NA19240 has genotypes for HGSVC variants (22), while NA19239 has genotypes for the 1000GP variants (21). NA19239 was excluded from the 1000GP graph (17). We simulated 1 million read pairs (2 million reads) from each individual’s haplotypes (17).
Read mapping accuracy:
Simulated read sets were mapped to the graphs using Giraffe, VG-MAP (10), HISAT2 (11), and GraphAligner (12). We were unable to build a HISAT2 index for the full 1000GP graph, and so instead we mapped it to a subset of the 1000GP data using a graph provided by the tool’s authors that contains approximately a third of the variants. In addition, we mapped the read sets to the primary graphs using Giraffe, and to the linear reference assemblies using the linear sequence mappers BWAMEM (19), Bowtie2 (18), and Minimap2 (20). Mapping accuracy was evaluated by comparing the positions along embedded, shared linear paths at which reads fell after mapping to similarly determined positions for their original simulated alignments.
Read mapping speed:
We compared mapping runtime, speed, and memory usage on an AWS EC2 i3.8xlarge node with 32 vCPUs and 244 GB of memory. To estimate real-world runtime and memory usage, we aligned a shuffled read set of 600 million NovaSeq 6000 reads from NA19239. We mapped reads to the 1000GP graph, the HGSVC graph, and the GRCh38 linear reference for comparison, and measured runtime and memory usage. For each tool we also separately measured reads mapped per thread per second, ignoring the start-up time of the mapper (Fig. S30). This measure gives an estimate of speed that is invariant to read set size or core/thread count, except for the effects of long-running work batches and thread synchronization overhead (17).
Read mapping bias:
To assess reference allele mapping bias, we mapped 600 million real paired-end NovaSeq 6000 reads for NA19239 to the 1000GP graph using default Giraffe and VG-MAP. For comparison, we mapped the same reads to GRCh38 with BWA-MEM.
Genotyping accuracy:
We compared the performance of Giraffe, VG-MAP, Illumina’s Dragen platform, and BWA-MEM for genotyping SNVs and short indels. The design of each calling pipeline is described in S4 in the supplement (17) and the parameters and indexes for each experiment is described in Table S22. The variants produced by each pipeline were compared against the Genome In a Bottle (GIAB) v4.2.1 HG002 high confidence variant calling benchmark (24) using the RealTimeGenomics vcfeval tool (47) and Illumina’s hap.py tool (48). This benchmark set covers 92.2% of the GRCh38 sequence.
We also evaluated a DeepVariant (25) pipeline that uses Giraffe mappings (17). Using the default DeepVariant 1.1.0 trained model, we tested genotyping the HG003 sample across the entire genome. This sample was not used in training the model.
Generalization to yeast:
To evaluate Giraffe’s performance on more diverged, non-human data, we used a yeast graph built from a Cactus multiple sequence alignment for five strains of the S. cerevisiae and S. paradoxus yeasts (8). For the corresponding negative control primary graph, we used the S.c. S288C assembly. We collected basic statistics about the yeast graph, and decomposed the graph for analysis using the method of (27). We simulated 500,000 read pairs from a held-out yeast strain, DBVPG6044, not included in the yeast graph, using an error and length model for Illumina HiSeq 2500 reads (17).
Structural variant genotyping
We built an SV pangenome from the HGSVC (22), GIAB (1), and SVPOP (28) sequence-resolved catalogs. After filtering erroneous duplicates using a re-mapping approach, the SVs were iteratively inserted in the genome graph to minimize the effect of errors and redundancy in the catalog. The SVs were then genotyped across 5,202 genomes by aligning short-read sequencing data using Giraffe with a Workflow Description Language (WDL) workflow that we deposited in Dockstore (49). 2,000 samples were selected from the Multi-Ethnic Study of Atherosclerosis (MESA) cohort to maximize sample diversity. The remaining 3,202 samples are from the 1000 Genomes Project and include 2,504 unrelated individuals. The trios available in this latter dataset were used to compute the rate of Mendelian concordance in the genotypes.
The different SV alleles observed in the population were clustered into SV sites based on their reciprocal overlap (for deletions) and sequence similarity (for insertions). We used the frequency profile across alleles within an SV site to identify the major allele and to fine-tune variants with near-duplicates in the combined catalog that may have been due to errors. Each variant was then annotated with its presence in existing SV databases (28,32,33), its repeat content, and its location relative to gene annotations. We also compared the frequency distributions across the SV databases and how well the frequency estimates matched for variants shared across databases.
Principal component analysis was performed on the SV genotypes and principal components were compared with those produced from SNV/indel genotypes. We defined strong inter-cluster or inter-super-population frequency patterns by a frequency in any cluster or super population differing by more than 10% from the median frequency across all of them. For the 2,000 MESA samples, the clusters were defined using hierarchical clustering on the first three principal components. For the 1000 Genomes Project, we used their “super population” assignments. Permutations were used to contrast the number of SVs with such patterns with an expected baseline.
Finally, we examined the SV genotypes in a subset of the samples that had gene expression data available from the Geuvadis consortium (34). MatrixEQTL (50) identified SV-eQTLs while controlling for sex and population structures, as summarized by the first four principal components. Separate analyses of the four European-ancestry populations together, and the YRI population alone were performed similarly. In addition, we performed a joint eQTL analysis with publicly available SNVs and indels (31). We used permutation to compute enrichment of SV-eQTLs in gene regions, gene families, or among lead-eQTLs (those with the strongest association for a gene).
Supplementary Material
Funding
Research reported in this publication was supported by the National Institutes of Health under Award Numbers U41HG010972, R01HG010485, U01HG010961, OT3HL142481, OT2OD026682, U01HL137183, and 2U41HG007234. Research reported in this publication was supported by the NHLBI BioData Catalyst Fellows Program of the National Institutes of Health through the University of North Carolina at Chapel Hill, under Award Number OT3HL147154. JAS was supported by the Carlsberg Foundation. Computational resources for the project were made available by the NIH and by Amazon Web Services, without full compensation at market value. The high coverage sequencing data for the 1000 Genomes Project were generated at the New York Genome Center with funds provided by NHGRI Grant 3UM1HG008901-03S1, and can be found on Terra. MESA and the MESA SHARe projects are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, UL1-TR-001420. Funding for SHARe genotyping was provided by NHLBI Contract N02-HL-64278. Genotyping was performed at Affymetrix (Santa Clara, California, USA) and the Broad Institute of Harvard and MIT (Boston, Massachusetts, USA) using the Affymetrix Genome-Wide Human SNP Array 6.0. Also supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. Whole genome sequencing (WGS) for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). WGS for “NHLBI TOPMed: Multi-Ethnic Study of Atherosclerosis (MESA)” (phs001416) was performed at the Broad Institute of MIT and Harvard (3U54HG003067-13S1 and HHSN268201500014C). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I).
We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute, the National Institutes of Health or the U.S. Department of Health and Human Services.
Footnotes
Competing interests
P.C. and A.C. are employees of Google and own Alphabet stock as part of the standard compensation package. The remaining authors declare no competing interests.
Data and materials availability
An overview of the data generated for this paper, and key input data to reproduce the analyses, is available at https://cglgenomics.ucsc.edu/giraffe-data/. The dataset is available via IPFS at https://ipfs.io/ipfs/QmVo4Q5hCKqUGJJZyYLGJTaiHZdK9JWhJtGJbKa9ojrSjh.
Archived copies of the code and final re-usable work products have been deposited in Zenodo as DOI 10.5281/zenodo.4721495, referenced here as (51). This archive also includes vg, toil-vg, and toil source code and Docker containers used in this work, as well as the giraffe-sv-paper orchestration scripts. “Final” versions of vg and toil-vg, including all features needed to reproduce this work, are 9907ab2 for vg and 99101f2 for toil-vg.
The latest version of the vg toolkit, including the Giraffe mapper, is customarily distributed at https://github.com/vgteam/vg. The scripts used for the analysis presented in this study were developed at https://github.com/vgteam/giraffe-sv-paper, a git bundle of which is archived in Zenodo (51).
Data used in the Giraffe read mapping experiments, including the 1000GP, HGSVC, and yeast target graphs, the linear control graphs, the graphs used to simulate reads, and the simulated reads themselves, can be found at https://cgl.gi.ucsc.edu/data/giraffe/mapping/.
The SV pangenomes and SV catalogs annotated with allele frequencies are hosted at https://cgl.gi.ucsc.edu/data/giraffe/calling/ and archived in (51). This repository also includes SVs with strong inter-super-population frequency patterns, SV-eQTLs, and SVs overlapping protein-coding genes.
To build the 1000GP and HGSVC graphs, we used the GRCh38 no-alt analysis set (accession GCA_000001405.15), and the hs38d1 decoy sequences (accession GCA_000786075.2), both available from NCBI, in addition to the variant call files distributed by the respective projects.
To train read simulation and evaluate speed, we used human read sets ERR3239454, ERR309934 and SRR6691663, and yeast read sets SRR4074256, SRR4074257, SRR4074394, SRR4074384, SRR4074413, SRR4074358, and SRR4074383, all available from SRA.
The public high-coverage sequencing dataset from the 1000 Genomes Project (31) is available at https://www.internationalgenome.org/data-portal/data-collection/30x-grch38, including ENA projects PRJEB31736 and PRJEB36890. The gene expression data was download from ArrayExpress E-GEUV-1
(GD462.GeneQuantRPKM.50FN.samplename.resk10.txt.gz). We downloaded the call sets from the ENCODE portal (52) (https://www.encodeproject.org/) with the following identifiers: ENCFF590IMH.
Individual whole-genome sequence data for TOPMed whole genomes are available through dbGaP. The dbGaP accession numbers for the Multi-Ethnic Study of Atherosclerosis (MESA) is phs001416. Data in dbGaP can be downloaded by controlled access with an approved application submitted through their website: https://www.ncbi.nlm.nih.gov/gap.
References
- (1).Zook JM, et al. , Nature Biotechnology 38, 1347 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mahmoud M, et al. , Genome Biology 20, 1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ebler J, Schönhuth A, Marschall T, Bioinformatics 33, 4015 (2017). [DOI] [PubMed] [Google Scholar]
- (4).Church DM, et al. , PLOS Biology 9, e1001091 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).The Computational Pan-Genomics Consortium, Briefings in Bioinformatics 19, 118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sherman RM, Salzberg SL, Nature Reviews Genetics 21, 243 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ballouz S, Dobin A, Gillis JA, Genome Biology 20, 159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hickey G, et al. , Genome Biology 21, 35 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Eizenga JM, et al. , Annual Review of Genomics and Human Genetics 21, 139 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Garrison E, et al. , Nature Biotechnology 36, 875 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kim D, Paggi JM, Park C, Bennett C, Salzberg SL, Nature Biotechnology 37, 907 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rautiainen M, Marschall T, Genome Biology 21, 253 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rakocevic G, et al. , Nature Genetics 51, 354 (2019). [DOI] [PubMed] [Google Scholar]
- (14).Illumina, Accuracy Improvements in Germline Small Variant Calling with the DRAGEN Platform. https://science-docs.illumina.com/documents/Informatics/dragen-v3-accuracy-appnote-html-970-2019-006/Content/Source/Informatics/Dragen/dragen-v3-accuracy-appnote-970-2019-006/dragen-v3-accuracy-appnote-970-2019-006.html.
- (15).Sirén J, Garrison E, Novak AM, Paten B, Durbin R, Bioinformatics 36, 400 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Schirmer M, D’Amore R, Ijaz UZ, Hall N, Quince C, BMC bioinformatics 17, 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Materials and methods are available as supplementary materials.
- (18).Langmead B, Salzberg SL, Nature methods 9, 357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li H, arXiv (2013). [Google Scholar]
- (20).Li H, Bioinformatics 34, 3094 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).1000 Genomes Project Consortium, et al. , Nature 526, 68 (2015).26432245 [Google Scholar]
- (22).Chaisson MJP, et al. , Nature Communications 10 (2019). [Google Scholar]
- (23).Pritt J, Chen N-C, Langmead B, Genome Biology 19, 220 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wagner J, et al. , bioRxiv (2020). [Google Scholar]
- (25).Poplin R, et al. , Nature Biotechnology 36, 983 (2018). [DOI] [PubMed] [Google Scholar]
- (26).Eggertsson HP, et al. , Nature Communications 10, 5402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Paten B, et al. , Journal of Computational Biology 25, 649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Audano PA, et al. , Cell 176, 663 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services, The NHLBI BioData Catalyst (2020). 10.5281/zenodo.3822858. [DOI]
- (30).Bild DE, et al. , American journal of epidemiology 156, 871 (2002). [DOI] [PubMed] [Google Scholar]
- (31).Byrska-Bishop M, et al. , bioRxiv (2021). [Google Scholar]
- (32).Sudmant PH, et al. , Nature 526, 75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Genome Aggregation Database Production Team, et al. , Nature 581, 444 (2020).32461652 [Google Scholar]
- (34).Lappalainen T, et al. , Nature 501, 506 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fagny M, et al. , Proceedings of the National Academy of Sciences 114, E7841 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Benarroch EE, Neurology 89, 722 (2017). [DOI] [PubMed] [Google Scholar]
- (37).Chiang C, et al. , Nature Genetics 49, 692 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ebert P, et al. , Science (2021). [Google Scholar]
- (39).Li H, Feng X, Chu C, Genome Biology 21, 265 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Koren S, et al. , Genome Research 27, 722 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Li H, Bioinformatics 32, 2103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wick RR, Schultz MB, Zobel J, Holt KE, Bioinformatics 31, 3350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A, Current Protocols in Bioinformatics 70 (2020). [DOI] [PubMed] [Google Scholar]
- (44).Chen S, et al. , Genome Biology 20, 291 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sudmant PH, et al. , Science pp. 1–16 (2015). [Google Scholar]
- (46).Sudmant PH, et al. , Nature 526, 75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cleary JG, et al. , bioRxiv (2015). [Google Scholar]
- (48).Illumina/hap.py (2020). https://github.com/Illumina/hap.py.
- (49).Monlong J, github.com/vgteam/vg_wdl/vg_mapgaffe_call_sv_cram (2020). 10.5281/zenodo.4290651. [DOI]
- (50).Shabalin AA, Bioinformatics 28, 1353 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Sirén J, et al. , Software and products for “Pangenomics enables genotyping known structural variants in 5,202 diverse genomes” (2021). 10.5281/zenodo.4774364. [DOI] [PMC free article] [PubMed]
- (52).Sloan CA, et al. , Nucleic Acids Research 44(D1), D726–D732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Roberts M, Hayes W, Hunt BR, Mount SM, Yorke JA, Bioinformatics 20, 3363 (2004). [DOI] [PubMed] [Google Scholar]
- (54).Chang X, Eizenga J, Novak AM, Sirén J, Paten B, Bioinformatics 36, i146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ferragina P, Manzini G, Journal of the ACM 52, 552 (2005). [Google Scholar]
- (56).Eizenga JM, et al. , Bioinformatics (2020). [Google Scholar]
- (57).Ghaffaari A, Marschall T, Bioinformatics 35, i81–i89 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zhao M, Lee W-P, Garrison EP, Marth GT, PLOS ONE 8, e82138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ewing B, Green P, Genome research 8, 186 (1998). [PubMed] [Google Scholar]
- (60).Durbin R, Eddy SR, Krogh A, Mitchison G, Biological sequence analysis: probabilistic models of proteins and nucleic acids (Cambridge university press, 1998; ). [Google Scholar]
- (61).Karlin S, Altschul SF, Proceedings of the National Academy of Sciences 87, 2264 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Beyer W, et al. , Bioinformatics 35, 5318 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Li H, Bioinformatics 27, 2987 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Krusche P, et al. , Nature biotechnology 37, 555 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).International HapMap Consortium, et al. , Nature 437, 1299 (2005).16255080 [Google Scholar]
- (66).Abyzov A, Urban AE, Snyder M, Gerstein M, Genome research 21, 974 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).International HapMap Consortium, et al. , Nature reviews. Genetics 5, 467 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Crysnanto D, Pausch H, Genome Biology 21, 184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Tange O, ;login: The USENIX Magazine 36, 42 (2011). [Google Scholar]
- (70).Treasures Hidden – Warm Up – precisionFDA. https://precision.fda.gov/challenges/1/view/results, retrieved on 2020-11-11, currently unavailable.
- (71).Truth Challenge V2: Calling Variants from Short and Long Reads in Difficult-to-Map Regions – precisionFDA. https://precision.fda.gov/challenges/10/view/results, retrieved on 2020-11-11, currently unavailable. [DOI] [PMC free article] [PubMed]
- (72).Chin C-S, et al. , Nature Communications 11, 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Mose LE, Perou CM, Parker JS, Bioinformatics 35, 2966 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Picard toolkit (2019). http://broadinstitute.github.io/picard/.
- (75).Sibbesen JA, et al. , bioRxiv (2021). [Google Scholar]
- (76).Monlong J, github.com/jmonlong/wdl-workflows/bcftools_merge (2020). 10.5281/zenodo.4290655 [DOI]
- (77).The ENCODE Project Consortium, Nature 489, 57 (2012).22955616 [Google Scholar]
- (78).Davis CA, et al. , Nucleic Acids Research 46, D794 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
An overview of the data generated for this paper, and key input data to reproduce the analyses, is available at https://cglgenomics.ucsc.edu/giraffe-data/. The dataset is available via IPFS at https://ipfs.io/ipfs/QmVo4Q5hCKqUGJJZyYLGJTaiHZdK9JWhJtGJbKa9ojrSjh.
Archived copies of the code and final re-usable work products have been deposited in Zenodo as DOI 10.5281/zenodo.4721495, referenced here as (51). This archive also includes vg, toil-vg, and toil source code and Docker containers used in this work, as well as the giraffe-sv-paper orchestration scripts. “Final” versions of vg and toil-vg, including all features needed to reproduce this work, are 9907ab2 for vg and 99101f2 for toil-vg.
The latest version of the vg toolkit, including the Giraffe mapper, is customarily distributed at https://github.com/vgteam/vg. The scripts used for the analysis presented in this study were developed at https://github.com/vgteam/giraffe-sv-paper, a git bundle of which is archived in Zenodo (51).
Data used in the Giraffe read mapping experiments, including the 1000GP, HGSVC, and yeast target graphs, the linear control graphs, the graphs used to simulate reads, and the simulated reads themselves, can be found at https://cgl.gi.ucsc.edu/data/giraffe/mapping/.
The SV pangenomes and SV catalogs annotated with allele frequencies are hosted at https://cgl.gi.ucsc.edu/data/giraffe/calling/ and archived in (51). This repository also includes SVs with strong inter-super-population frequency patterns, SV-eQTLs, and SVs overlapping protein-coding genes.
To build the 1000GP and HGSVC graphs, we used the GRCh38 no-alt analysis set (accession GCA_000001405.15), and the hs38d1 decoy sequences (accession GCA_000786075.2), both available from NCBI, in addition to the variant call files distributed by the respective projects.
To train read simulation and evaluate speed, we used human read sets ERR3239454, ERR309934 and SRR6691663, and yeast read sets SRR4074256, SRR4074257, SRR4074394, SRR4074384, SRR4074413, SRR4074358, and SRR4074383, all available from SRA.
The public high-coverage sequencing dataset from the 1000 Genomes Project (31) is available at https://www.internationalgenome.org/data-portal/data-collection/30x-grch38, including ENA projects PRJEB31736 and PRJEB36890. The gene expression data was download from ArrayExpress E-GEUV-1
(GD462.GeneQuantRPKM.50FN.samplename.resk10.txt.gz). We downloaded the call sets from the ENCODE portal (52) (https://www.encodeproject.org/) with the following identifiers: ENCFF590IMH.
Individual whole-genome sequence data for TOPMed whole genomes are available through dbGaP. The dbGaP accession numbers for the Multi-Ethnic Study of Atherosclerosis (MESA) is phs001416. Data in dbGaP can be downloaded by controlled access with an approved application submitted through their website: https://www.ncbi.nlm.nih.gov/gap.