

Abstract
Purpose:
Small cell lung cancer (SCLC) is an aggressive disease with an overall 5-year survival rate of less than 10%. Treatment for SCLC with cisplatin/etoposide chemotherapy (C/E) ± radiotherapy has changed modestly over several decades. The ubiquitin-proteasome system is an underexplored therapeutic target for SCLC. We preclinically evaluated TAK-243, a first-in-class small molecule E1 inhibitor against UBA1.
Experimental Design:
We assessed TAK-243 in 26 SCLC cell-lines as monotherapy and combined with C/E, the PARP-inhibitor, olaparib, and with radiation using cell viability assays. We interrogated TAK-243 response with gene expression to identify candidate biomarkers. We evaluated TAK-243 alone and in combination with olaparib or radiotherapy with SCLC patient-derived xenografts (PDX).
Results:
Most SCLC cell lines were sensitive to TAK-243 monotherapy (EC50 median 15.8 nmol/L; range 10.2 nmol/L–367.3 nmol/L). TAK-243 sensitivity was associated with gene-sets involving the cell cycle, DNA and chromatin organization, and DNA damage repair, while resistance associated with cellular respiration, translation, and neurodevelopment. These associations were also observed in SCLC PDXs. TAK-243 synergized with C/E and olaparib in vitro across sensitive and resistant SCLC cell lines. Considerable TAK-243–olaparib synergy was observed in an SCLC PDX resistant to both drugs individually. TAK-243 radiosensitization was also observed in an SCLC PDX.
Conclusions:
TAK-243 displays efficacy in SCLC preclinical models. Enrichment of gene sets is associated with TAK-243 sensitivity and resistance. TAK-243 exhibits synergy when combined with genotoxic therapies in cell lines and PDXs. TAK-243 is a potential therapeutic strategy to improve SCLC patient outcomes, both as a single agent and in combination with existing therapies.
Translational Relevance
Small cell lung cancer (SCLC) carries a poor prognosis and novel effective treatments are needed. The Ubiquitin-protease system (UPS) has been implicated and targeted effectively in other cancers such as multiple myeloma and leukemia. The Ubiquitin activating enzyme (UBA1) encoded by UBA1 is a critical E1 enzyme at the apex of the UPS. We demonstrate that UBA1 is a highly expressed and essential gene in SCLC and other cancers with experimental assays and public datasets. By inhibition of UBA1 with a first-in-class small molecule, TAK-243, we observed marked sensitivity of SCLC cell lines and patient-derived xenografts with identification of potential biomarkers. Furthermore, we demonstrated synergy of TAK-243 with standard-of-care chemotherapy, the PARP inhibitor olaparib, and with radiotherapy. These preclinical data provide a basis for the development of future clinical trials with TAK-243 along with biomarker evaluation or as a novel combination with currently utilized therapies for patients with SCLC.
Introduction
Lung cancer is the leading cause of cancer-related deaths world-wide with small cell lung cancer (SCLC), the most aggressive subset, accounting for an estimated 15% to 17% of all diagnosed lung cancer cases (1). SCLC is especially threatening due to its short doubling time, high growth fraction, and its association with early metastases (1). In 2019, a consensus review paper synthesized and defined molecular subtypes of SCLC based on the gene expression of four transcription regulators: achaete-scute homologue 1 (ASCL1), neuronal differentiation factor 1 (NEUROD1), POU class 2 homeobox 3 (POU2F3), and the yes-associated protein 1 (YAP1; ref. 2). Subsequently, the existence of a distinct YAP1 subtype is controversial (3). Thus, a separate fourth SCLC subtype has also been referred to as triple-negative (TN) SCLC and associated with an inflamed gene signature (4). A fifth subtype was also proposed by expression of atonal bHLH transcription factor 1 (ATOH1; ref. 5). Nevertheless, the standard-of-care treatment remains the same across all subtypes and consists of genotoxic cisplatin and etoposide chemotherapy (C/E) with the frequent addition of immunotherapy or radiotherapy (RT). Despite initial disease response to these first-line treatments, recurrence is frequent, resulting in an overall 5-year survival rate for patients with SCLC of less than 10% (1). There have been modest advancements in SCLC first-line therapy over the last 30 years (1), and novel treatments are needed to improve patient outcomes.
Targeting the ubiquitin-proteasome system (UPS) for cancer therapy is a novel treatment strategy that has improved patient outcomes in other cancer histologies (6). Various UPS members spanning the ubiquitin-conjugation pathway (UCP) and downstream proteasome system are implicated in the pathogenesis and progression of many cancers (7–9), including lung cancer (10). Together, these structures regulate many physiologic pathways including protein degradation, signal transduction, cell-cycle progression, apoptosis, immune response, and DNA damage repair (8, 11).
At the apex of the UCP are two E1 ubiquitin-activating enzymes, UBA1 and UBA6. UBA1 [a.k.a. ubiquitin-activating enzyme (UAE); encoded by UBA1], the most dominant human E1 enzyme, is responsible for initiating many of the downstream effects dysregulated in malignancies and is thereby an attractive anti-cancer strategy. However, effective targeting of UBA1 has been elusive until the recent development of TAK-243, a first-in-class small molecular inhibitor against UBA1/UAE (12–17).
TAK-243 irreversibly inhibits UBA1 by forming a covalent adduct to the E1-ubiquitin complex, which in turn prevents protein mono- and poly-ubiquitination (14, 18). TAK-243 is most effective against UBA1 as compared with other E1 enzymes and homologues (14). Previously, TAK-243 demonstrated antiproliferative activity across various models of human cancers including both hematologic and solid tumors (14). Of the 31 total cell lines tested, the most drug-sensitive cell lines were two SCLC cell lines (mean SCLC EC50: 0.0085 μmol/L vs. mean EC50 of cell lines from other cancers: 0.338 μmol/L) with a 40-fold drug dose concentration difference compared to the other 29 cell lines in vitro (14). Moreover, TAK-243 has an antineoplastic effect in in vivo xenograft models of multiple myeloma, diffuse large B-cell lymphoma, colon cancer, and non-SCLC (NSCLC), leading to tumor growth inhibition (14). However, SCLC in vivo models were not evaluated.
TAK-243 may elicit its antineoplastic effect through induction of DNA double-strand breaks (DSB) and impairment of nonhomologous end joining (NHEJ; ref. 13), an important DNA repair pathway. Inhibitors of PARP (PARPi) which target the DNA damage repair response, are among the few novel therapies with proven preclinical and clinical efficacy that are currently undergoing clinical investigation for the treatment of SCLC. Taken together, TAK-243 may be an effective monotherapy for SCLC or in combination with genotoxic therapy including platinum-based chemotherapy and PARPi.
To date, published data for SCLC comprises of only two cell lines and there has been no published in vivo assessment of TAK-243 in SCLC models, thus the translational potential of targeting UBA1 for SCLC is unknown. The limited cell line analyses also preclude biomarker interrogation. Herein, we leveraged a large panel of SCLC cell lines and in vivo patient-derived xenograft (PDX) models to interrogate TAK-243 efficacy as monotherapy or in combination with genotoxic therapies and potential biomarkers.
Materials and Methods
Cell lines and reagents
The cell lines used were generously provided by collaborators or purchased from the American Type Culture Collection. Cell line catalog information with short tandem repeat (STR) and Mycoplasma testing status can be found in Supplementary Table S1 and Supplementary Methods. TAK-243 (MLN-7243) from Active Biochem (#A-1384), C/E chemotherapy from Sigma-Aldrich (#232120 and #341205, respectively), olaparib from Selleck Chemicals (#S1060), were dissolved in either DMSO (in vitro) or 10% HP-β-CD[2-hydroxypropyl-β-cyclodextrin] (HPBCD; in vivo).
Immunoblotting
Immunoblotting was conducted as previously described (19) and methodologies are in Supplementary Methods. Antibodies utilized in this study include: anti-PARP1 (Santa Cruz, #sc-8007, 1:500), anti-cleaved PARP (Cell Signaling Technology, #: 9541S, 1:1000), anti-β actin (Cell Signaling Technology, #: 3700S, 1:15000), IRDye donkey-anti-rabbit 800CW (LI-COR Biosciences, P/N: 926–32213), and IRDye donkey-anti-mouse 680RD (LI-COR Biosciences, P/N: 926–68072) secondary antibodies.
Cell viability and drug response assays
SCLC cell line drug response to TAK-243 (0–1 μmol/L) was assessed using the CellTiter Glo 2.0 assay (Promega, #G9243). Cell-lines were plated using the Thermo Scientific Multidrop Combi Reagent Dispenser (#5840300). Drug was added using the Tecan D300e Digital Dispenser. After 3 days, cell viability was assessed by measuring luminescence using a microplate reader (CLARIOstar, BMG LABTECH).
Drug synergy was assessed using cell-line–specific concentration combinations of TAK-243 (0–2 μmol/L) and C/E (1:1 ratio, range = 0–15 μmol/L) or olaparib (range = 0–500 μmol/L). Cell viability was assessed using the alamarBlue resazurin conversion assay (Thermo Fisher Scientific, #DAL1025) by measuring fluorescence after 6 days using a microplate reader (CLARIOstar, BMG LABTECH) as described in the Supplementary Methods.
In vivo studies
Animal studies were conducted after protocol approval by the Animal Care Committee at the Princess Margaret Cancer Centre (Toronto, ON, Canada) and by the institutional Animal Resource Centre guidelines. Three SCLC PDX models (JHU-LX33, SCRX-Lu149 CN, SCRX-Lu149 CR; ref. 19), generously provided by Dr. Charles M. Rudin at Memorial Sloan Kettering Cancer Center (MSKCC; New York, NY), were used for this study. Chemoresistance (CR) models were previously established in the Rudin laboratory, from chemo-naive (CN) SCRX-Lu149 PDX model as described (19), whereby SCRX-Lu149 CN PDXs were exposed to repeated cycles of C/E to select PDX tumor cells that could survive chemotherapy.
For in vivo drug efficacy experiments, PDX cells were implanted subcutaneously into the right flank of 6- to 8-week-old NOD/SCID gamma (NSG; NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; Cancer Stemcell Colony) mice (male or female). For TAK-243 monotherapy: mice were treated with either the vehicle (10% HPBCD diluted in sterile water) or TAK-243 (20 mg/kg) biweekly, intravenously for 3 weeks (days 0, 3, 7, 10, 14). For TAK-243–olaparib combination: mice were treated with the vehicle control (as described, biweekly, intravenously, 5 weeks), TAK-243 (20 mg/kg, biweekly, intravenously, 5 weeks), olaparib (50 mg/kg, orally, until endpoint; 5× per week), or both TAK-243 and olaparib. For TAK-243–radiotherapy combination: mice were treated with vehicle (as described, biweekly, 3 weeks), TAK-243 (as described, biweekly, 3 weeks), radiotherapy (2 Gy × 4; days 0–3) or TAK + radiotherapy. Radiation was administered once daily over the first 4 days following treatment initiation. Treatment schedules remained the same for TAK-243–radiotherapy combination therapy; where, TAK-243 was administered 3 hours before irradiation. Prior to irradiation, animals were sedated using 2% to 5% isoflurane and restrained in a custom-built lead shielding device, purchased from the Princess Margaret Cancer Centre Machine Shop. Animals were treated in 2Gy fractions using the XRAD 320 X-Ray irradiator (Precision X-Ray).
Statistical analysis
Dose response analysis
Drug dose response was modelled by the LL.4 four-parameter log-logistic function. The half-maximal effective dose (EC50) and area under the dose response curve (AUC; range = 0%–100%; linear trapezoidal method) were measured to quantify the drug effect. ΔAUC was calculated using the following equation:
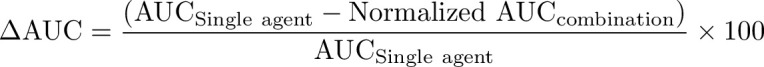 |
Synergy analysis
To evaluate synergy between drug combinations we assessed the cytotoxic effect of TAK-243 combined with C/E or olaparib using the Bliss Independence (20) synergy scoring model (carried out by the synergyfinder 2.0 web application with default parameters; ref. 21). Bliss synergy scores were defined as the extent to which the observed combination response differed from the expected response. The most synergistic area score (MSAS) defined as the mean Bliss synergy score across the most synergistic 3-by-3 dose-window in a dose response matrix (C/E n = 144; olaparib n = 56), was calculated for each cell line. MSAS > 10 was classified as synergistic, whereas antagonism was MSAS < −10, as recommended by synergyfinder (21, 22).
Gene essentiality interrogation
RStudio version 1.1.456 was used for data processing and statistical analysis. To evaluate UBA1 gene essentiality, Computational correction of copy-number Effect in CRISPR-Cas9 Essentiality Screens (CERES) gene dependency scores were retrieved from the Broad Institute (Cambridge, MA) Achilles database for 18,333 genes in 739 Cancer Cell Line Encyclopedia (CCLE) cell lines (23). UBA1 CERES scores were established using the previously described methods (24). Disease sites with data available for less than 10 cell lines were excluded from further analysis. A nonparametric Wilcoxon signed-rank test was used to determine whether the median UBA1 CERES score across all cancer cell lines evaluated was significantly different from the median CERES score for nonessential cancer genes (CERES score of 0).
Cell line biomarker analysis
Gene expression data for cancer cell lines was procured from the CCLE (DepMap release 21Q2; ref. 25) in transcripts per million RNA molecules [log2(1+TPM)] for protein-coding genes. Genes with expression correlating to the EC50 drug sensitivity scores for TAK-243 were determined using the Limma Bioconductor package (v. 3.46.0) to fit a linear model to each gene. Moderated t-statistics were generated, and P values were adjusted for multiple testing using Benjamini–Hochberg correction. This was performed for all SCLC cell lines as well as cell lines subdivided into the TN/YAP1-high, ASCL1-high, and NEUROD1-high subtypes. One TN/YAP1-high cell line, NCI-H196, was identified as a potential outlier based on its gene expression and TAK-243 response. Therefore, all subsequent analyses were performed with and without NCI-H196. Genes positively correlating with TAK-243 EC50 are defined as resistors and those negatively correlating with TAK-243 EC50 are defined as sensitizers. Resistor and sensitizer genes from each regression analysis were compared to identify genes unique to each group of cell-lines (i.e., TN/YAP1-high, ASCL1-high, and NEUROD1-high subgroups).
Genes were ranked according to significance and their resistor/sensitizer classification for a preranked gene-set enrichment analysis (GSEA) using the fgsea and gage Bioconductor packages (v. 1.16.0 and v. 2.40.2, respectively) and manually curated pathway gene-sets obtained from http://baderlab.org/GeneSets (September 2021). Pairwise comparisons of the normalized enrichment scores (NES) of significant pathways (FDR < 0.05) for each set of cell lines was performed to identify shared and unique pathways important for TAK-243 resistance/sensitivity. Potential TAK-243 biomarkers were identified from the leading-edge genes of enriched pathways that were identified both with and without outlier NCI-H196. Enrichment of potential biomarker gene-sets identified from each analysis of three subsets of SCLC cell lines representing transcription factor subtypes with ≥3 cell lines available [n = 3 (NEUROD1), n = 6 (TN/YAP1), n = 13 (ASCL1); Supplementary Fig. S1) was then calculated for all SCLC cell lines (n = 24) using a single sample GSEA (ssGSEA) as a form of validation.
PDX biomarker analysis
An ssGSEA was performed on PDX expression (Reads Per Kilobase Million) using the GSVA Bioconductor package (v. 1.38.2) to independently validate the candidate biomarker gene sets identified from the SCLC cell-line whole transcriptome regression analyses. Row Z-score ssGSEA enrichment score was plotted in a heatmap to visualize relative gene-set enrichment and identify the response of PDX models to TAK-243. Gene-sets were considered validated when the absolute mean scaled enrichment score was greater than 0.5 in one of the models.
Kaplan–Meier survival analysis
Time to reach a prespecified volumetric endpoint of 1,000 mm3 was defined as the event, while animals that were euthanized before the volumetric endpoint were censored as no event. Secondary endpoint analyses of additional volumetric endpoints are as labeled. Log-rank tests were used to evaluate differences between groups.
Tumor growth inhibition analysis
Tumor growth inhibition (TGI) was calculated by comparing the average normalized tumor volume (TV) of the treatment group with that of the normalized control group on the timepoint at which the first control animal reached the endpoint (day 14/15).
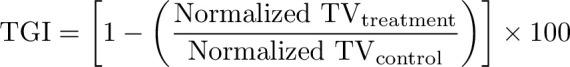 |
Data availability statement
The data generated in this study are available within the article and supplementary data files. Computational code for biomarker analysis is available on Code Ocean (https://codeocean.com/capsule/6678351/tree/v1). CERES gene dependency scores and gene expression data for cancer cell lines was procured from the CCLE at https://depmap.org/portal/download/ (DepMap release 21Q2; Achilles Avana Public 17Q4 v2).
Results
UBA1 is a pan-cancer essential, highly expressed gene, and a pharmacologic target for SCLC
We evaluated the essentiality of UBA1 for cancer cell survival to characterize UBA1 as an anticancer target. We queried UBA1 CERES gene dependency scores across 20 different cancer subtypes and 11 disease sites. Based on previously reported analysis methodologies, CERES scores of 0 and −1 are the median scores of pan-cancer nonessential and essential genes, respectively (24). UBA1 CERES scores across all cancer cell lines (median = −2.07, range: −3.90 to −1.13) were significantly lower than 0, indicating that UBA1 is essential across all 11 disease sites evaluated [Fig. 1A; nonparametric Wilcoxon signed-rank test, P < 0.001 for all cancer cell lines (n = 423) when compared with the nonessential value of 0].
Figure 1.

UBA1 is an essential, highly expressed gene target in SCLC. A, Box and whisker plot of UBA1 (top) and UBA6 (bottom, representative control gene) dependency (CERES) scores by cancer subtype (n = 20). Colored boxes indicate disease site (n = 11). CERES scores of 0 and -1 represent the median pan-cancer nonessential and essential gene scores, respectively. A nonparametric Wilcoxon signed-rank test revealed that the median UBA1 CERES score across all cancer cell lines evaluated (n = 423) was significantly different from the median CERES score for nonessential cancer genes of 0. B, Box and whisker plot displaying relative UBA1 mRNA expression across cancer cell lines (SCLC n = 50; colorectal n = 56; neuroblastoma n = 16; AML n = 36; NSCLC n = 128) and immortalized normal cell lines (n = 6) using a log-arithmic (log10) scale. A Student t test revealed UBA1 expression was high in SCLC cell lines compared with other cancer subtypes. Normal cell lines originated from ovarian, breast, fibroblast, kidney, lung, and prostate tissues. Blue asterisks represent the immortalized normal lung cell lines evaluated. C, Dose response curves of SCLC cell lines after treatment with TAK-243 for 3 days, with individual EC50 indicated in the legend. Individual points on the plots indicate the mean of three technical replicates with SD represented by error bars. Outliers were excluded from analysis. Colors represent sensitive (red-yellow) and resistant (green) SCLC cell lines. GI, gastrointestinal; AML, acute myeloid leukemia; SCLC, small cell lung cancer; NSCLC, non small cell lung cancer.
Next, we evaluated UBA1 mRNA expression in cancer cell lines with available data from the CCLE (Fig. 1B). Relative UBA1 mRNA expression ranged from 35.28 to 422.64 TPM across cell-lines belonging to five cancer subtypes (n = 286). In lung cancer, SCLC cell-lines had a significantly higher mean expression (mean = 206.09, range: 112.62–368.04 TPM, n = 50, t test P < 0.005) compared with NSCLC cell lines (mean = 124.68, range: 35.28–422.64, n = 128), suggesting relative upregulation of UBA1 in SCLC. Moreover, the mean expression level of UBA1 was significantly higher (approximately 34.1% greater) in SCLC compared with immortalized normal cell lines across various tissues (mean = 153.06, range: 94.36–199.89 TPM, n = 6, t test P = 0.02), suggesting this upregulation may be cancer site-specific in SCLC.
Subsequently, we sought to determine the feasibility of pharmacologically targeting UBA1 in SCLC with TAK-243. We assessed the anticancer activity of TAK-243 across 26 SCLC cell lines with a treatment duration of 3 days (Fig. 1C). EC50 and AUC values were compared. Cell lines demonstrated variable response to TAK-243 whereby NCI-H1184 was most sensitive (EC50 = 0.010 μmol/L, AUC = 39%) and NCI-H196 was most resistant to TAK-243 (EC50 = 0.367 μmol/L, AUC = 76%). Moreover, treatment with TAK-243 (0.5 μmol/L and 1 μmol/L) demonstrated a dose-dependent increase in cellular apoptosis (Supplementary Fig. S2), as indicated by cleaved-PARP1 after 24 hours of treatment across three SCLC cell lines (NCI-H146, SBC-5, and SHP77).
Candidate gene-sets are putative biomarkers of TAK-243 response in SCLC cell lines
We sought to identify genes that can determine SCLC sensitivity or resistance to TAK-243. CCLE gene expression data were available for 24 of 26 SCLC cell lines treated with TAK-243 for analysis to identify candidate genes and biological processes. Potential biomarker genes were identified as significantly negatively or positively correlated with TAK-243 drug response and were classified as sensitizer or resistor genes, respectively (Fig. 2A). NCI-H196 demonstrated extreme resistance to TAK-243 and showed a different pattern in gene expression from other TAK-243 resistant cell-lines. Thus, eight sets of potential biomarkers were identified by performing a linear regression analysis on all 24 SCLC cell lines, and then also by subsetting the cell lines based on SCLC subtype: ASCL1-high (13 cell lines), TN/YAP1-high (6 cell lines), and NEUROD1 (3 cell lines; Fig. 2B; Supplementary Fig. S1; Supplementary Tables S2–S7). Across all SCLC cell lines, significantly enriched sensitizer biological processes identified through gene ontology analysis of the regression analysis (without NCI-H196) are related to cell cycle, DNA and chromatin organization, and DNA damage repair. Contrastingly, no biological processes were found to be enriched across resistor genes.
Figure 2.

Candidate gene-sets are putative biomarkers of TAK-243 response in SCLC cell lines. A, Volcano plot showing results of TAK-243 EC50 regression analysis where the x-axis represents the regression coefficient assigned to each gene and the y-axis represents significance. Genes highlighted in blue represent sensitizer genes (expression is significantly negatively correlated with EC50; FDR < 0.05) and red genes represent resistor genes (expression is significantly positively correlated with EC50; FDR < 0.05). The dotted black horizontal line represents the significance threshold (Padj < 0.05). B, Venn diagram showing overlaps of sensitizer (left) and resistor (right) genes identified from each of the four regression analyses: All SCLC cell lines, ASCL1-high cell lines only, TN/YAP1-high cell lines only, and NEUROD1-high cell lines only. C, Representative example of how leading-edge genes were selected through GSEA to identify sensitizer (left) and resistor (right) pathways from across SCLC cell lines and subtypes. Leading-edge genes (highlighted orange bar) were used as potential biomarkers for TAK-243 response. MYC target (sensitizer) and aerobic respiration (cellular respiration; resistor) are depicted. D, Heatmap of scaled ssGSEA enrichment score for each set of biomarker genes identified from the regression or pathway analyses used to identify TAK-243 response in subsets of SCLC cell lines and subsequently applied to each SCLC cell line.
In parallel, candidate biological processes were also determined using a preranked GSEA and leading-edge genes of negatively and positively enriched processes were classified as sensitizer and resistor genes, respectively (Fig. 2C; Supplementary Fig. S3; Supplementary Table S8). Sensitizer genes were associated with negative enrichment in ribonucleoprotein complex assembly and MYC targets, while candidate resistor genes were associated with positive enrichment in cellular respiration, protein translation, and neurodevelopment pathways after accounting for extreme resistance demonstrated by NCI-H196. Candidate biomarker gene-sets from both regression analyses and GSEA and their respective Z-scores determined by an ssGSEA across all 24 SCLC cell lines are shown in Fig. 2D. Validated gene-sets derived from regression analysis were associated with both sensitivity and resistance across 24 SCLC cell lines, while positive enrichment of the gene-sets corresponding to cellular respiration pathways appeared to be most associated with TAK-243 resistance (Fig. 2D).
TAK-243 sensitivity and resistance in SCLC PDX models are associated with candidate gene-sets identified in silico
ssGSEA was performed using PDX gene expression data from MSKCC (19) to independently validate the candidate biomarker gene-sets identified from the SCLC cell-line regression analyses (Fig. 3A). These results indicated that cell line–derived gene-sets when applied to the JHU-LX33 and SCRX-LU149 PDX models associated with TAK-243 resistance and sensitivity, respectively. Only gene-sets identified by the regression analysis were confirmed to be associated with TAK-243 sensitivity while GSEA gene-sets primarily involved in cellular respiration, translation, and neurodevelopment were found to be associated with TAK-243 resistance.
Figure 3.

TAK-243 sensitivity and resistance across two SCLC PDX models can be identified by candidate gene-sets derived in vitro.A, Heatmap of mean scaled single-sample GSEA enrichment score for each set of validated biomarker genes identified from in vitro cell line analyses and their independent associations with TAK-243 response in SCLC PDX models. ‘S’ denoting that SCRX-Lu149 CN and CR PDX models were TAK-243 sensitive and ‘R’ denoting that JHU-LX33 PDX model was TAK-243 resistant. Individual PDX tumor growth curves (left graphs) and Kaplan–Meier survival plots (right graphs) of SCRX-LU149 CN and CR (B and C); JHU-LX33 CN (D); PDX models were treated with either vehicle control or 20 mg/kg of TAK-243 (biweekly X 3 weeks, intravenously) alone. Freedom from volumetric endpoint (1,000 mm3, indicated by the dotted line in each growth curve) was determined by Kaplan–Meier survival analysis. For the Kaplan–Meier curves, shaded areas represent the 95% confidence intervals (CI) around each curve, and the dotted line, the median freedom from volumetric endpoint. The log-rank test was used to evaluate statistical significance, with adjusted P values to account for multiple tests.
To further investigate TAK-243 as a novel treatment strategy for SCLC, we evaluated its anticancer activity in vivo with SCRX-Lu149 and JHU-LX33 PDX models. In concordance with our in vitro biomarker findings, the SCRX-Lu149 CN and CR PDX models were sensitive to TAK-243. Significant tumor growth inhibition was observed in both the SCRX-Lu149 CN– and CR TAK-243–treated PDXs compared with the vehicle control group (Fig. 3B and C). Significantly increased freedom from the volumetric endpoint (SCXR-Lu149 CN, P = 0.03; SCXR-Lu149 CR, P = 0.0003) was observed for TAK-243 treated groups in both models, when compared with control groups, suggesting TAK-243 sensitivity is independent of chemoresistance in these paired PDX models. The mean maximal response to TAK-243 in each model was −32% (SCRX-Lu149 CN) and −51% (SCRX-Lu149 CR), compared with tumor volume immediately prior to initiating treatment (Supplementary Fig. S4A). The median time to the prespecified volumetric endpoint of 1,000 mm3 for the TAK-243–treated SCXR-Lu149 CN PDXs could not be estimated. However, using a secondary volumetric endpoint (900 mm3, the median time to endpoint was 38 days for the TAK-243–treated group compared with 21 days for the controls (Supplementary Fig. S4B). Similarly, for the SCXR-Lu149 CR PDX, the median time to a 1,000-mm3 volumetric endpoint of 42 days and 21 days was calculated for the TAK-243–treated and control-treated groups, respectively.
In contrast, the JHU-LX33 CN PDX model was resistant to TAK-243 (Fig. 3D). No significant difference in tumor growth or freedom from volumetric endpoint was observed [P = 0.81; median time to volumetric endpoint (1,000 mm3: control = 24 days and TAK-243 = 27.5 days]. Animal body weights were maintained or recovered in the TAK-243–treated and control-treated groups compared with baseline across all SCLC PDX models (Supplementary Fig. S5).
TAK-243 synergizes with standard-of-care C/E chemotherapy and the PARPi olaparib
Among its many cellular functions, the UPS regulates DNA damage response (DDR) mediators (26). Previously, impaired DNA repair was observed after treatment with TAK-243 in cancer (13, 14). We sought to evaluate the potential of TAK-243 to synergize with DNA damage inducing chemotherapy and PARPi as an inhibitor of DNA repair. For this study, synergy was defined as a combination cytotoxic effect of TAK-243 with C/E or PARPi greater than the additive cytotoxic effect expected from each of TAK-243, C/E alone, or PARPi.
Drug synergy matrix analyses
We administered SCLC cell lines (C/E n = 7; PARPi n = 5) with combination TAK-243 and C/E chemotherapy or TAK-243 and PARPi treatment (olaparib) and determined synergy by the Bliss Independence model (20). High positive synergy scores represent drug combinations that produce a greater response than expected (i.e., synergy) while large negative scores suggest the opposite (antagonism). Synergy scores near 0, where the observed response is similar to the expected response, indicate an additive effect. We calculated the MSAS to enable comparisons among cell lines.
Mean MSASs greater than 10 (synergy) were found for all SCLC cell lines evaluated in both combinations of TAK-243 with C/E and with olaparib, with the exception of NCI-H146 (TAK-243-C/E). NCI-H69 (MSAS = 41), NCI-H209 (MSAS = 29), and NCI-H82 (MSAS = 16) cell lines demonstrated the largest maximum chemosynergistic combination effects, while SBC-5 (MSAS = 38), NCI-H1092 (MSAS = 31), and NCI-H446 (MSAS = 21) showed the most TAK-243–olaparib synergy. Synergy matrices for TAK-243-C/E and TAK-243–olaparib are in Fig. 4A and Supplementary Figs. S6 and S7. Importantly, MSAS analysis showed most cell lines exhibited significant synergy between both TAK-243 and C/E or TAK-243 and olaparib at several drug combinations.
Figure 4.

TAK-243 synergizes with standard of care C/E chemotherapy and olaparib. A, The NCI-H69 and SBC-5 SCLC cell lines serve as a representative example of the dose response matrices utilized to evaluate TAK-243 synergy with C/E chemotherapy and olaparib, respectively. Relative synergy scores are color-coded where red indicates synergy, white indicates lack of synergy, and green indicates antagonism based on respective synergy scoring. NCI-H69 and SBC-5 demonstrated the most synergy with TAK-243 in combination with chemotherapy and olaparib, respectively, as determined by the Bliss MSAS synergy metric. MSAS was calculated using the values outlined in gray boxes. B, Schematic of experimental design for TAK-243–C/E and TAK-olaparib dose response and analysis. SCLC cell-lines were treated with either various or fixed doses of TAK-243, C/E, or olaparib over 6 days. C, Dose response curves of representative NCI-H889 and SBC-5 cell lines after combination therapy consisting of either a fixed dose of TAK-243 with various doses of C/E (top left), a fixed dose of C/E with various doses of TAK-243 (bottom left), a fixed dose of TAK-243 with various doses of olaparib (top right), or a fixed dose of olaparib with various doses of TAK-243 (bottom right). Gray shaded region indicates the change in AUC measured. Individual points on the plots indicate the mean of three technical replicates with SD represented by error bars. Outliers were excluded from analysis. D, Observed TAK-243–C/E and TAK-243–olaparib synergy in SCLC was independent of specific cell line sensitivity or resistance to single-agent TAK-243. Top, Heatmap depicting SCLC cell-lines and their respective EC50 (μmol/L) and AUC value (%) in order of sensitivity (left to right). Middle, Heatmap illustrating the difference in AUC between single agent (TAK-243, C/E, or olaparib) and combination therapies (TAK-243 + C/E or TAK-243 + olaparib) across labeled SCLC cell lines. Bottom, Heatmap depicting Bliss MSAS as determined by TAK-243–C/E and TAK-243–olaparib synergy analysis for labeled SCLC cell lines. Gray nonnumbered boxes indicate cell lines for which combination experiments or analyses were not conducted. MSAS, most synergistic area score.
Synergy analyses with a fixed TAK-243 dose combined with a range of C/E chemotherapy or olaparib doses
To further evaluate TAK-243 in combination with chemotherapy and with olaparib, we assessed the relative contributions of chemotherapy or olaparib and TAK-243 to the overall synergistic effect. We first applied a fixed dose of TAK-243 in combination with varying dose range of either C/E 1:1 chemotherapy or olaparib and evaluated cell viability after 6 days (Fig. 4B and C). The difference in AUC was evaluated to determine whether synergy was observed. NCI-H889 demonstrated the most chemosynergy (ΔAUC = 13%), while NCI-H1092 demonstrated the most synergy with olaparib (ΔAUC = 26%). Mean ΔAUCs across the cell lines evaluated was 3.71% (range: −3%–13% units, n = 8) for the TAK-243(fixed)–C/E condition and 15% for the TAK-243(fixed)–olaparib group (range: 7%–26% units, n = 5; Fig. 4D). Dose response curves can be found in Supplementary Figs. S8 and S9.
Synergy analyses with a fixed C/E chemotherapy or olaparib dose combined with a range of TAK-243 doses
We subsequently conducted the inverse experiment where we applied a fixed dose of chemotherapy or olaparib to varying doses of TAK-243. NCI-H889 consistently revealed increased sensitivity to TAK-243 by C/E, while SBC-5 demonstrated the most synergy with olaparib and TAK-243 (Fig. 4C). The ΔAUC after C/E combination treatment ranged from −4% to 13%, with a mean ΔAUC of 3.38% (Fig. 4D; n = 7). For olaparib in combination with TAK-243 the ΔAUC ranged from 2% to 19% with a mean ΔAUC of 9% (Fig. 4D; n = 5). Dose response curves are in Supplementary Figs. S10 and S11.
Taken together, synergistic effects of TAK-243 with genotoxic therapy (C/E or olaparib), as assessed by MSAS or ΔAUC, were observed across multiple cell lines. In addition, synergy was independent of whether cell lines were sensitive or resistant to single-agent TAK-243 (Fig. 4D). Specifically, most SCLC cell lines that demonstrated the greatest resistance to single-agent TAK-243 (SHP77, SBC-5, NCI-H526, and NCI-H446) synergized with either C/E chemotherapy, olaparib (PARPi), or both, while a few other TAK-243 resistant cell lines, NCI-H196, and NCI-H146, demonstrated additive drug combination effects (Fig. 4D). Overall, these data provide a basis for potential novel therapeutic combinations.
TAK-243 synergizes most with olaparib in the TAK-243, olaparib, monotherapy resistant JHU-LX33 CN SCLC PDX model
We further investigated the TAK-243 and PARPi combination in vivo with PDX models. While the TAK-243 sensitive SCRX-Lu149 CN PDX model demonstrated significant tumor growth inhibition to olaparib (adjusted P = 0.0069) and TAK-243 (adjusted P = 0.0053) individually as single agents compared with controls, only modest tumor growth inhibition after combination treatment [TAK-243 (20 mg/kg), olaparib (50 mg/kg)] was observed (Fig. 5A). Median time to volumetric endpoint for the combination group was 31.5 days compared with 26 and 28 days for the animals that received either olaparib (50 mg/kg) or TAK-243 alone, respectively (adjusted P = 0.024–0.031). Treatment-related (TAK-243 and/or olaparib) body weight loss was observed that recovered starting 10 days after treatment initiation for most animals (Supplementary Fig. S12).
Figure 5.

TAK-243 synergizes with genotoxic therapies in TAK-243 monotherapy sensitive and resistant SCLC PDX models. Individual PDX tumor growth curves (left) and Kaplan–Meier survival plots (middle) of SCRX-LU149 CN (A and C) and JHU-LX33 CN (B). PDX models after treatment with either vehicle control, olaparib (50 mg/kg, 5 times/week until termination, oral), TAK-243 (20 mg/kg, biweekly × 5 weeks, intravenous), and TAK-243–olaparib (TAK-243 20 mg/kg, olaparib 50 mg/kg; A and B) or vehicle control, TAK-243 (20 mg/kg, biweekly × 3 weeks, intravenous), radiation (2G x 4), and TAK-243 + radiotherapy combination (TAK-243 20 mg/kg, radiotherapy 2G x 4, C). Dosing schedule is indicated below x-axis of growth-response curves: the horizontal green and purple lines represent period of TAK-243 and olaparib or radiation dosing, respectively. Freedom from volumetric endpoint (1,000 mm3, indicated by the dotted line in each growth curve) was determined by Kaplan–Meier survival analysis. For the Kaplan–Meier curves shaded areas represent the 95% CIs around each curve, and the dotted line, the median freedom from volumetric endpoint. The log-rank test was used to evaluate statistical significance, with adjusted P values to account for multiple tests. B, TAK-243 synergizes most with olaparib in the TAK-243, olaparib monotherapy resistant JHU-LX33 CN SCLC PDX model. Middle, Kaplan–Meier curve analysis for the control, TAK-243, and olaparib single agent groups was calculated by creating variance between groups artificially (+ or −1 day per each event) to enable visualization of each curve. Right, Average tumor volume of olaparib, TAK-243-, and combination-treated mice relative to control on day 15 of treatment. TAK-243 + olaparib–treated mice had significantly smaller tumors 15 days following treatment compared with all other groups (unpaired t test, P < 0.0001). C (right, top, TAK-243 + radiotherapy–treated mice had significantly smaller tumors 14 days following treatment compared with all other groups (unpaired t test, P < 0.0005). Waterfall plot (right, bottom) depicting the best response of individual mice treated with control, TAK-243, radiotherapy, or TAK-243 + radiotherapy. Best response was considered as the smallest tumor volume (compared with baseline) over the course of the study. The gray shaded area and the purple horizontal line of radiation indicates duration of time in which combination therapy overlapped. RT, radiotherapy; PDX, patient derived xenograft.
In contrast, significant TAK-243–olaparib synergy was observed after combination treatment in the JHU-LX33 SCLC PDX model which demonstrated resistance to TAK-243 and olaparib when administered individually (Fig. 5B). Average tumor growth of the combination group was inhibited by 66% compared with control mice, 15 days following treatment (unpaired t test, P < 0.0001). Median time to the volumetric endpoint for the combination group could not be calculated, compared with 18 days across all other groups (TAK-243–olaparib vs. olaparib P = 0.049; TAK-243-olaparib vs. TAK-243 P = 0.065; TAK-243-olaparib vs. control P = 0.065). However, as various mice across all treatment groups reached humane endpoint prior to the prespecified 1,000 mm3 volumetric endpoint due to the aggressive invasive behavior of the JHU-LX33 tumors in this experiment, we also performed a secondary analysis to a 400-mm3 volumetric endpoint. This demonstrated a median time to 400-mm3 endpoint for the combination group of 29 days compared with 11 days for the animals that received either olaparib (50 mg/kg; adjusted P = 0.0035) or TAK-243 alone (adjusted P = 0.0035), respectively (Supplementary Fig. S13). Treatment-related (TAK-243 and/or olaparib) body weight loss recovered shortly after treatment initiation for most animals (Supplementary Fig. S14).
TAK-S243 administered with radiotherapy demonstrates synergy in the SCRX-Lu149 CN SCLC PDX model
Finally, we interrogated TAK-243 and its effects on DNA repair through combination experiments with radiotherapy as a genotoxic therapeutic in SCRX-Lu149 CN PDX models. We treated SCRX-Lu149 CN tumor-bearing mice with either the vehicle control, TAK-243 (20 mg/kg), radiotherapy (2 Gy × 4), or combination TAK-243 + radiotherapy (TAK-243 20 mg/kg, 2 Gy × 4). SCRX-Lu149 demonstrated sensitivity to all three treatments compared with control (Fig. 5C). However, average tumor growth was inhibited most in the TAK-243 + radiotherapy combination group by 91% compared with control mice, 14 days following treatment (unpaired t test, P = 0.0002). Median time to volumetric endpoint for the combination group was 52 days, compared with 28 days and 35 days for the animals that received radiation or TAK-243 alone (adjusted P = 0.01), respectively. Treatment-related (TAK-243 and/or radiotherapy) body weight loss was observed that recovered starting 10 days after treatment initiation for most animals (Supplementary Fig. S15).
Discussion
Herein, our results demonstrate that inhibiting the UPS using a selective first-in-class UBA1 inhibitor, TAK-243, has the potential to be an effective anticancer strategy for SCLC. Our work determines that UBA1, the most abundant E1 enzyme, is essential for SCLC viability and extends previous findings that identified other E1 enzymes and homologues as key players in the pathogenesis of NSCLC (27–30). We support and methodically expand the initial preliminary findings by Hyer and colleagues which identified SCLC as a candidate cancer-type sensitive to TAK-243 monotherapy in two SCLC cell lines in vitro, without in vivo evaluation in SCLC. The present study broadens these findings to fully characterize drug response of more than 25 SCLC cell lines in vitro and three SCLC PDX models in vivo. Our in vitro and in vivo SCLC TAK-243 drug efficacy results were in a similar range as the other histologies (i.e., colon, breast, NSCLC) assessed and reported by Hyer and colleagues (14). While Hyer and colleagues found relative in vivo resistance in the Calu-6 NSCLC xenograft model, we identified greater in vivo resistance in the JHU-LX33 SCLC PDX model. This highlights the importance of our exploratory biomarker analysis that our much larger SCLC sample size allowed and may improve the translational success of TAK-243 monotherapy for SCLC. Strikingly, TAK-243 monotherapy resistance in the JHU-LX33 SCLC PDX model was overcome with combination treatment with PARPi. Accordingly, we identified synergy of TAK-243 with genotoxic therapies (i.e., chemotherapy and PARPi).
Putative biomarkers for TAK-243 response in SCLC
Identifying biomarkers of TAK-243 response in SCLC can highlight underlying biological mechanisms that cause or prevent anticancer cytotoxicity. In our study, candidate gene-sets identified through regression analysis were associated with TAK-243 response in SCLC cell lines and PDX models. Sensitizer gene-sets related to processes involving the cell cycle, DNA and chromatin organization, and DNA damage repair, all of which are known to be interrupted by TAK-243. TAK-243 promotes G1 and G2–M cell cycle arrest, disrupting cell-cycle progression and yielding part of its anticancer effect (14). Thus, subsets of cancer cells which upregulate and depend on the cell cycle are more vulnerable to the disruptive effects of TAK-243 and may in turn be more sensitive to its downstream cytotoxicity.
Another TAK-243 sensitizer gene-set we identified is associated with DNA and chromatin organization. Ubiquitination is involved in chromatin functions as a form of posttranslational modification that can result in downstream cellular signaling (31). Inhibition of UBA1 by TAK-243 can disrupt these cellular processes, altering downstream signaling of many chromatin-associated processes. Interestingly, BEN domain-containing protein 3 (BEND3), a regulator of chromatin organization and transcriptional repressor, has been implicated in TAK-243 resistance across acute myeloid leukemia (AML) cancer cells (32). Although our study did not directly characterize the effect of TAK-243 on regulating of chromatin and DNA organization, we speculate that the sensitive SCLC cells may have higher expression of other regulation pathways that can promote or enhance the cellular effects of TAK-243, making them more vulnerable to UBA1 inhibition.
As previously reported, TAK-243 diminishes the DNA damage repair response (13, 14, 33). Although the downstream mechanistic effects of TAK-243 in SCLC and other cancer types have yet to be fully ascertained, there is a strong rationale to suggest that alterations in DNA damage repair machinery may contribute significantly to its anticancer effect in SCLC. SCLC is a disease characterized by the deleterious loss-of-function mutations of TP53 and RB1, DNA repair and cell-cycle regulators, respectively. Loss-of-function molecular alterations both enable rapid cancer proliferation and produce a genomic environment encumbered by replicative stress (34) whereby the expression of other functional DDR mediators is elevated to manage this stress (35, 36). This is further corroborated by the cellular enrichment in DNA repair identified in our study, specifically associated with the TAK-243 sensitive preclinical models (cell lines and PDX). Like other cancers, SCLC relies extensively on such DNA repair mediators for genomic stability and may therefore be more vulnerable to pharmacologic inhibition of such pathways by TAK-243 and other genotoxic therapies. Moreover, the inhibitory effects of TAK-243 on DNA damage repair enables us to leverage the anticancer effects of this vulnerability in SCLC as a single agent and importantly, paired with genotoxic therapies like chemotherapy, PARPi, or radiation.
Resistor gene-sets were primarily nominated by ssGSEA, whereby the positive enrichment of cellular respiration genes was most strongly associated with TAK-243 resistance across cell line and PDX SCLC models. These genes were primarily associated with the electron transport chain (i.e., cytochromes, NADH ubiquinone oxidoreductase, etc.). Interestingly, increased mitochondrial respiration has been reported to promote cellular adaptation and survival in response to endoplasmic reticulum (ER) stress, particularly by increasing electron transport and respiration (37). As a result, cytotoxic reactive oxygen species (ROS) accumulation from ER stress may be circumvented by increased efficiency of electron transport, promoting cell survival and resistance to ER stress (38). As activation of ER stress and unfolded protein response is one of the mechanisms by which TAK-243 elicits its cytotoxic effect (13, 14, 33), enrichment of genes related to cellular respiration may be a survival mechanism underlying the TAK-243 resistant SCLC population we observed.
The gene-sets identified in our study, nominated by regression analysis and GSEA, are consistent across our preclinical models of SCLC. Based on existing literature, the mechanisms underlying their action fit within the current paradigm of TAK-243 activity. As such, there may be clinical-translational potential to stratify patients for TAK-243 drug sensitivity based on molecular characteristics. However, our findings are limited to in silico correlative analysis and future functional studies are needed to mechanistically interrogate these potential gene-set response biomarkers.
TAK-243 synergy with genotoxic therapies, effects on DNA repair, and other novel combinations
The ability of TAK-243 to target and confer cytotoxic effects on SCLC, while also synergizing with the current SCLC standard-of-care treatments offers a translational path for this novel therapeutic to clinical trial development. We demonstrate that TAK-243 enhances the effect of genotoxic C/E chemotherapy, radiotherapy, and inhibition of PARP, the former virtually all patients with SCLC receive in the first-line setting. An important consideration for the management of SCLC is that while first-line therapy is effective, relapse and acquired treatment resistance are frequent, and most patients eventually die from their recurrent disease.
Platinum chemoresistance can occur through the upregulation of DDRs to shield against the DNA damaging treatment (36, 39, 40), particularly in SCLC (36). This dependence on DNA repair pathways is likely exacerbated in SCLC and may make these tumors particularly vulnerable to TAK-243. Inducing ER stress has also been reported to reverse chemoresistance in SCLC while promoting autophagy and apoptosis (41). As TAK-243 has been shown to activate ER stress (13, 14), this approach may have benefit in for patients with chemoresistant SCLC and should be further explored.
PARPi in combination with other agents, including radiation are currently being evaluated clinically as a therapeutic option in first-line and relapsed settings for patients with SCLC (42). Interestingly, we observed striking synergy when pairing TAK-243 with olaparib in some SCLC preclinical models. However, the mechanism by which TAK-243 potentiates the genotoxic effects of olaparib, may be different from that observed with C/E chemotherapy. Olaparib inhibits DNA repair by arresting base excision repair, leading to destabilization of the replication fork and subsequent DNA DSBs (43). Olaparib and other PARPi also have variable potency to trap PARP enzymes at damaged DNA with associated downstream cytotoxic effects (44). Thus, olaparib is particularly effective in schlafen11 (SLFN11)-high cancers, whereby SLFN11 inhibits homologous recombination and blocks replication fork progression (45–47). In 2021, Murai and colleagues reported that TAK-243 inhibits DNA replication by activating CHK1 via Claspin independently of ataxia telangiectasia and RAD3-related (ATR) and, further, that deficiency of SLFN11 sensitizes cancer cells to TAK-243 (33). Together, these findings provide a rationale for TAK-243 and olaparib synergy. Perhaps, these drugs in combination can target multiple forms of DNA replication and repair leading to a synergistic, complementary, and lethal reduction in DDR. Future studies should more fully interrogate the interaction between PARPi and TAK-243 along with mechanistic studies to characterize how these two novel drugs synergize.
Although we observed modest synergy with TAK-243 and olaparib in the SCRX-Lu149 model, this model responded dramatically to TAK-243-RT combination therapy. We hypothesize that this observed effect may be occurring similarly to that with chemotherapy whereby cell death is enhanced when DNA damage inducing agents (i.e., C/E or RT) are combined with a therapeutic agent that can dysregulate the DDR (i.e., TAK-243).
Of further interest, YAP1-deficient cancers, including the majority of SCLC, are selectively sensitive to XPO1, NAMPT, BCL2, HDAC, EIF4, AURK, and TERT inhibitors, and forced YAP1 expression induces drug resistance (48). Whether these inhibitors synergize with TAK-243 or enhance the synergy we observed with PARPi and C/E chemotherapy may be promising therapeutic combinations to investigate.
Conclusion
Here, we determined that TAK-243 has potential anticancer efficacy against SCLC. The evaluation of TAK-243 in the setting of SCLC provides insight into the role of UPS in SCLC oncogenesis and progression, and as a novel target for SCLC therapy. Future studies should further: (i) validate the effect of TAK-243 in a larger set of SCLC PDX models, (ii) elucidate downstream pathways altered by TAK-243, (iii) further characterize the putative gene-set response biomarkers and their molecular mechanisms, and (iv) extend combination treatment findings by optimizing the TAK-243-C/E and TAK-243-PARPi drug–drug dosing relationships and evaluating the systemic and/or radiotherapy treatment regimens for combination therapy (i.e., concurrent, adjuvant, or neoadjuvant TAK-243 delivery and treatment duration). The findings from our study provide a starting foundation to inform future preclinical research and clinical trial development with TAK-243 to improve outcomes for our patients with SCLC.
Authors' Disclosures
A.D. Schimmer reports grants from Takeda during the conduct of the study; grants and personal fees from Takeda; grants from Medivir AB; and personal fees from Bristol-Myers Squibb, Novartis, Jazz, and Otsuka outside the submitted work; in addition, A.D. Schimmer has a patent for Use of DNT cells for the treatment of AM: pending. B.H. Lok reports grants from Pfizer and grants, personal fees, and nonfinancial support from AstraZeneca outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
Research in the B.H. Lok laboratory is supported by funding from the Conquer Cancer Foundation of American Society of Clinical Oncology (ASCO), International Association for the Study of Lung Cancer (IASLC), Ontario Molecular Pathology Research Network of the Ontario Institute for Cancer Research (Toronto, ON, Canada), Lung Cancer Research Foundation, Canada Foundation for Innovation, Cancer Research Society, Canadian Institutes of Health Research, NIH/NCI (U01CA253383), Clinical and Translational Science Center at Weill Cornell Medical Center (New York, NY) and MSKCC (UL1TR00457). S. Majeed is funded in part by the Ontario Graduate Scholarship, the Department of Medical Biophysics fellowship, and the Graduate & Life Sciences Education Awards from the Temerty Faculty of Medicine, University of Toronto (Toronto, ON, Canada). G. Liu acknowledges the Princess Margaret Foundation (Lusi Wong Family Fund, Alan B. Brown Chair in Molecular Genomics). A.D. Schimmer holds the Ronald N. Buick Chair in Oncology Research.
The authors thank Dr. Rima Al-Awar, Dr. Richard Marcellus, and Ratheesh Subramaniam from the Ontario Institute of Cancer Research for their generous support and technical assistance; members of the Tsao, Liu, and Lok labs for their guidance and critical discussion.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
S. Majeed: Conceptualization, data curation, software, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. M.K. Aparnathi: Data curation, formal analysis, validation, investigation, visualization, methodology, writing–review and editing. K.C.J. Nixon: Data curation, software, formal analysis, investigation, visualization, methodology, writing–review and editing. V. Venkatasubramanian: Validation, investigation, methodology, writing–review and editing. F. Rahman: Formal analysis, validation, methodology, writing–review and editing. L. Song: Formal analysis, validation, investigation, methodology, writing–review and editing. J. Weiss: Software, formal analysis, visualization, methodology, writing–review and editing. R. Barayan: Formal analysis, visualization, methodology, writing–review and editing. V. Sugumar: Conceptualization, investigation, writing–original draft, writing–review and editing. S.H. Barghout: Methodology, writing–review and editing. J.D. Pearson: Methodology, writing–review and editing. R. Bremner: Resources, methodology, writing–review and editing. A.D. Schimmer: Conceptualization, resources, supervision, writing–review and editing. M.S. Tsao: Conceptualization, resources, supervision, methodology, writing–review and editing. G. Liu: Conceptualization, resources, supervision, funding acquisition, visualization, methodology, writing–original draft, project administration, writing–review and editing. B.H. Lok: Conceptualization, resources, data curation, supervision, funding acquisition, validation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Prim 2021;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019;19:289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baine MK, Hsieh MS, Lai WV, Egger JV, Jungbluth AA, Daneshbod Y, et al. SCLC subtypes defined by ASCL1, NEUROD1, POU2F3, and YAP1: a comprehensive immunohistochemical and histopathologic characterization. J Thorac Oncol 2020;15:1823–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021;39:346–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Simpson KL, Stoney R, Frese KK, Simms N, Rowe W, Pearce SP, et al. A biobank of small cell lung cancer CDX models elucidates inter- and intratumoral phenotypic heterogeneity. Nat Cancer 2020;1:437–51. [DOI] [PubMed] [Google Scholar]
- 6. Mcbride A, Ryan PY. Proteasome inhibitors in the treatment of multiple myeloma. Expert Rev Anticancer Ther 2013;13:339–58. [DOI] [PubMed] [Google Scholar]
- 7. Gelmann E, Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. Expert Rev Anticancer Ther 2013;13:339–58.23477520 [Google Scholar]
- 8. Tu Y, Chen C, Pan J, Xu J, Zhou ZG, Wang CY. The ubiquitin proteasome pathway (UPP) in the regulation of cell cycle control and DNA damage repair and its implication in tumorigenesis. Int J Clin Exp Pathol 2012;5:726–38. [PMC free article] [PubMed] [Google Scholar]
- 9. Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer 2018;18:69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fan Q, Wang Q, Cai R, Yuan H, Xu M. The ubiquitin system: orchestrating cellular signals in non-small-cell lung cancer. Cell Mol Biol Lett 2020;25:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin–proteasome system as drug targets. Nat Rev Drug Discov 2011;10:29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Best S, Liu T, Bruss N, Kittai A, Berger A, Danilov AV. Pharmacologic inhibition of the ubiquitin-activating enzyme induces ER stress and apoptosis in chronic lymphocytic leukemia and ibrutinib-resistant mantle cell lymphoma cells. Leuk Lymphoma 2019;60:2946–50. [DOI] [PubMed] [Google Scholar]
- 13. Barghout SH, Patel PS, Wang X, Xu GW, Kavanagh S, Halgas O, et al. Preclinical evaluation of the selective small-molecule UBA1 inhibitor, TAK-243, in acute myeloid leukemia. Leukemia 2019;33:37–51. [DOI] [PubMed] [Google Scholar]
- 14. Hyer ML, Milhollen MA, Ciavarri J, Fleming P, Traore T, Sappal D, et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med 2018;24:186–93. [DOI] [PubMed] [Google Scholar]
- 15. Aggarwal S. Targeted cancer therapies. Nat Rev Drug Discov 2010;96:427–8. [DOI] [PubMed] [Google Scholar]
- 16. Liu G, Yu J, Wu R, Shi L, Zhang X, Zhang W, et al. GRP78 determines glioblastoma sensitivity to UBA1 inhibition-induced UPR signaling and cell death. Cell Death Dis 2021;12:733. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17. Liu Y, Awadia S, Delaney A, Sitto M, Engelke CG, Patel H, et al. UAE1 inhibition mediates the unfolded protein response, DNA damage and caspase-dependent cell death in pancreatic cancer. Transl Oncol 2020;13:100834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barghout SH, Schimmer AD. E1 enzymes as therapeutic targets in cancers. Pharmacol Rev 2021;73:1–56. [DOI] [PubMed] [Google Scholar]
- 19. Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, et al. Chemosensitive relapse in small cell lung cancer proceeds through an EZH2- SLFN11 axis. Cancer Cell 2017;31:286–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bliss CI. The toxicity of poisons applied jointly. Ann Appl Biol 1939;26:585–615. [Google Scholar]
- 21. Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res 2020;48:488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Synergyfinder.fimm.fi [homepage on the Internet]. SynergyFinder - User documentation. SynergyFinder, 2020. [cited 2020 May 24]. Available from: https://synergyfinder.fimm.fi/synergy/synfin_docs/.
- 23. DepmapBroad. DepMap Achilles 19Q3 Public. Version 3. figshare. Fileset,2019.
- 24. Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR- Cas9 essentiality screens in cancer cells. Nat Genet 2017;49:1779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ulrich HD, Walden H. Ubiquitin signalling in DNA replication and repair. Nat Rev Mol Cell Biol 2010;11:479–89. [DOI] [PubMed] [Google Scholar]
- 27. McLaughlin PMJ, Helfrich W, Kok K, Mulder M, Hu SW, Brinker MGL, et al. The ubiquitin-activating enzyme E1-like protein in lung cancer cell lines. Int J Cancer 2000;85:871–6. [DOI] [PubMed] [Google Scholar]
- 28. Li L, Wang M, Yu G, Chen P, Li H, Wei D, et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst 2014;106:dju083. [DOI] [PubMed] [Google Scholar]
- 29. Jiang B, Fan X, Zhang D, Liu H, Fan C. Identifying UBA2 as a proliferation and cell cycle regulator in lung cancer A549 cells. J Cell Biochem 2019;120:12752–61. [DOI] [PubMed] [Google Scholar]
- 30. He X, Riceberg J, Soucy T, Koenig E, Minissale J, Gallery M, et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat Chem Biol 2017;13:1164–71. [DOI] [PubMed] [Google Scholar]
- 31. Vaughan RM, Kupai A, Rothbart SB. Chromatin regulation through ubiquitin and ubiquitin-like histone modifications. Trends Biochem Sci 2021;46:258–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barghout SH, Aman A, Nouri K, Blatman Z, Arevalo K, Thomas GE, et al. A genome-wide CRISPR/Cas9 screen in acute myeloid leukemia cells identifies regulators of TAK-243 sensitivity. JCI Insight 2021;6:e141518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murai Y, Jo U, Murai J, Jenkins LM, Huang SN, Chakka S, et al. SLFN11 inactivation induces proteotoxic stress and sensitizes cancer cells to ubiquitin activating enzyme inhibitor TAK-243. Cancer Res 2021;81:3067–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. George J, Lim JS, Jang SJ, Cun Y, Ozretia L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Foy V, Schenk MW, Baker K, Gomes F, Lallo A, Frese KK, et al. Targeting DNA damage in SCLC. Lung Cancer 2017;114:12–22. [DOI] [PubMed] [Google Scholar]
- 36. Sen T, Gay CM, Byers LA. Targeting DNA damage repair in small cell lung cancer and the biomarker landscape. Transl Lung Cancer Res 2018;7:50–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Knupp J, Arvan P, Chang A. Increased mitochondrial respiration promotes survival from endoplasmic reticulum stress. Cell Death Differ 2018;26:487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hijazi I, Knupp J, Chang A. Retrograde signaling mediates an adaptive survival response to endoplasmic reticulum stress in Saccharomyces cerevisiae. J Cell Sci 2020;133:jcs241539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: The role of DNA repair pathways. Clin Cancer Res 2008;14:1291–5. [DOI] [PubMed] [Google Scholar]
- 40. Postel-Vinay S, Vanhecke E, Olaussen KA, Lord CJ, Ashworth A, Soria JC. The potential of exploiting DNA-repair defects for optimizing lung cancer treatment. Nat Rev Clin Oncol 2012;9:144–55. [DOI] [PubMed] [Google Scholar]
- 41. Yu XS, Du J, Fan YJ, Liu FJ, Cao LL, Liang N, et al. Activation of endoplasmic reticulum stress promotes autophagy and apoptosis and reverses chemoresistance of human small cell lung cancer cells by inhibiting the PI3K/AKT/mTOR signaling pathway. Oncotarget 2016;7:76827–39. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42. Barayan R, Ran X, Lok BH. PARP inhibitors for small cell lung cancer and their potential for integration into current treatment approaches. J Thorac Dis 2020;12:6240–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992;356:356–8. [DOI] [PubMed] [Google Scholar]
- 44. Murai J, Shar-yin NH, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 2012;72:5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, et al. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin Cancer Res 2017;23:523–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mu Y, Lou J, Srivastava M, Zhao B, Feng XH, Liu T, et al. SLFN11 inhibits checkpoint maintenance and homologous recombination repair. EMBO Rep 2016;17:94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Murai J, Tang SW, Leo E, Baechler SA, Redon CE, Zhang H, et al. SLFN11 blocks stressed replication forks independently of ATR. Mol Cell 2018;69:371–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pearson JD, Huang K, Pacal M, McCurdy SR, Lu S, Aubry A, et al. Binary pan-cancer classes with distinct vulnerabilities defined by pro- or anti-cancer YAP/TEAD activity. Cancer Cell 2021;39:1115–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and supplementary data files. Computational code for biomarker analysis is available on Code Ocean (https://codeocean.com/capsule/6678351/tree/v1). CERES gene dependency scores and gene expression data for cancer cell lines was procured from the CCLE at https://depmap.org/portal/download/ (DepMap release 21Q2; Achilles Avana Public 17Q4 v2).