

Abstract
Purpose:
Tislelizumab is an anti–programmed cell death protein 1 (anti–PD-1) monoclonal antibody specifically designed to minimize binding to Fcγ receptors (FcγR).
Patients and Methods:
Here, we present the extended 3-year follow-up of a phase II study of tislelizumab in 70 patients with relapsed/refractory classical Hodgkin lymphoma (cHL) who failed or were ineligible for autologous stem cell transplantation.
Results:
With a median follow-up of 33.8 months, the overall response rate by the independent review committee was 87.1%, and the complete response (CR) rate was 67.1%. Responses were durable as shown by a median duration of response of 31.3 months, and median progression-free survival (PFS) of 31.5 months. The 3-year PFS and overall survival rates were 40.8% and 84.8%, respectively. Treatment-related adverse events (TRAEs) of any grade occurred in 97.1% of patients; the grade ≥3 TRAE rate was low (31.4%), and only 8.6% of patients experienced adverse events leading to treatment discontinuation. Correlative biomarker analysis showed that FcγRΙ-expressing macrophages had no observed impact on either the CR rate or PFS achieved with tislelizumab, which may be potentially related to its engineered Fc region.
Conclusions:
With extended follow-up, tislelizumab yielded long-term benefits and demonstrated a favorable safety profile for patients with relapsed/refractory cHL. This trial was registered at clinicaltrials.gov as NCT03209973.
Translational Relevance.
Tislelizumab, a humanized immunoglobulin G4 anti–programmed cell death protein 1 antibody, has an engineered Fc region that minimizes binding to Fcγ receptor (FcγR) on macrophages, thereby abrogating antibody-dependent phagocytosis (ADCP)–induced T-cell clearance. Tislelizumab demonstrated an overall response rate of 87.1% with a high complete response (CR) rate (62.9%) and with a median progression-free survival (PFS) of 31.5 months and 3-year PFS rate of 40.8% in Chinese patients with relapsed/refractory (R/R) classical Hodgkin lymphoma (cHL). Correlative biomarker analysis showed that FcγRΙ-expressing macrophages had no observed impact on either the CR rate or PFS achieved with tislelizumab, which may be potentially related to its engineered Fc region.
Introduction
Classical Hodgkin lymphoma (cHL) is characterized by rare malignant Hodgkin Reed—Sternberg (HRS) cells surrounded by extensive inflammatory and immune cell infiltrate, such as T cells, macrophages, neutrophils, and B cells (1). HRS cells evade antitumor immunity via several mechanisms, including genetic amplification at 9p24.1, leading to programmed death ligand 1 (PD-L1) and programmed death ligand 2 (PD-L2) overexpression on the tumor cell surface (2–4). Therefore, cHL is a unique disease entity in its thriving lymphocyte-rich microenvironment.
Although most newly diagnosed patients with cHL are likely to be cured, approximately 5% to 10% of patients will have primary refractory disease, and an additional 10% to 30% will relapse after having achieved a complete response (CR; ref. 5). Despite salvage therapies, including high-dose chemotherapy (HDT)/autologous hematopoietic stem cell transplant (ASCT) or brentuximab vedotin (BV), clinical outcome for these relapsed/refractory (R/R) cHL patients remained poor (6–8). Recent clinical evidence has revealed promising sensitivity of cHL to programmed cell death protein 1 (PD-1) blockade. Nivolumab and pembrolizumab demonstrated clinically meaningful activity in patients with cHL following failure of HDT/ASCT, BV, or both (9–12). Both drugs received approval from the U.S. Food and Drug Administration (FDA) for treatment of R/R cHL after ≥3 lines of therapy. In China, sintilimab and camrelizumab were approved in patients with R/R cHL based on single-arm, phase II studies (13, 14). However, in these clinical studies, only a minority of patients achieved CR, and most patients developed disease progression within 18 months. Therefore, there remained huge unmet needs for patients with cHL.
Predictive biomarkers that are potentially associated with response to anti–PD-1 treatment in patients with R/R cHL have been explored in several studies. In CheckMate-205, patients with higher levels of PD-L1 and major histocompatibility complex class II expression on HRS cells had superior PFS with nivolumab treatment (15). In Keynote-013, biomarker analyses demonstrated a high prevalence of PD-L1 and PD-L2 expression, treatment-induced expansion of T cells and natural killer cells, and activation of interferon gamma (IFNγ), T-cell receptor, and expanded immune-related signaling pathways, but none of these exploratory biomarkers seems significantly associated with response to pembrolizumab (11). Other than tumor tissue biomarkers, Shi and colleagues reported that CHD8 mutation frequency was significantly higher in patients with longer PFS by circulating tumor DNA sequencing in blood (16). Notably, these analyses were exploratory and in small cohorts. Larger cohort studies are needed to further confirm the findings.
Tislelizumab is a humanized immunoglobulin (Ig) G4 monoclonal antibody that binds to the extracellular domain of human PD-1 with high specificity and affinity to block binding to both PD-L1 and PD-L2. Unlike other anti–PD-1 antibodies, tislelizumab has a mutated Fc region and is specifically engineered to minimize FcγR binding on macrophages, thereby abrogating antibody-dependent phagocytosis, which is a potential mechanism of T-cell clearance and resistance to anti–PD-1 therapy (17). Preclinical models demonstrated that this Fc engineering led to better antitumor activity in vivo than anti–PD-1 antibodies that lacked the mutated Fc region. It was hence hypothesized that tislelizumab may induce deeper responses and longer duration of response (DOR), especially in tumor types with high macrophage infiltration, such as cHL (18, 19).
Efficacy and safety data of tislelizumab in patients with R/R cHL from this phase II study have been reported previously. Tislelizumab demonstrated high antitumor activity, with an overall response rate (ORR) of 87.1% and CR rate of 62.9% at 9.8-month median follow-up (20). Here, we presented the results from the extended 3-year follow-up to evaluate the durability of response and long-term safety of tislelizumab in patients with R/R cHL. In addition, we conducted exploratory biomarker analysis [including CD8 T cells, FcγRΙ-expressing macrophages by multiplexed immunohistochemistry (mIHC) and gene expression profiles of 1,392 immuno-oncology–related genes] and their association with clinical outcomes of tislelizumab.
Patients and Methods
Study design and patients
This was a multicenter, single-arm, open-label, phase II study. Patients were enrolled from 11 sites in China. Eligible patients had cHL with measurable disease that was histologically confirmed by central pathologic review. Patients must have had R/R disease and must have met one of the following criteria: (i) failed to achieve a response or progressed after ASCT; (ii) received ≥2 prior systemic chemotherapy regimens for cHL and were considered ineligible for ASCT. Other inclusion and exclusion criteria have been reported previously (20).
This study was designed and monitored in accordance with sponsor procedures and in compliance with the ethical principles of the International Conference on Harmonization Good Clinical Practice guidelines, the Declaration of Helsinki, and applicable local regulatory requirements. All patients provided written informed consent. The protocol, any amendments, and informed consent forms were approved by the local institutional review boards or independent ethics committees as appropriate.
Procedures and assessments
All enrolled patients received a fixed dose of tislelizumab 200 mg intravenously every 3 weeks until disease progression, unacceptable toxicity, or study termination. Dose reductions or escalations were not permitted during the study.
Contrast-enhanced computed tomography (CT) or magnetic resonance imaging scans were performed at weeks 12, 18, 30, and 42 in the first year of study, and every 15 weeks thereafter. Positron emission tomography (PET) scans were performed at weeks 12, 24, 42, and 57, and every 30 weeks thereafter. Disease assessment was as per an independent review committee (IRC; Bioclinica), which assessed disease response for each patient according to the Lugano classification (21).
Patients were allowed to continue study treatment if pseudo-progression was suspected based on a CT/PET scan, provided there was no concurrent clinical evidence of disease progression; if a subsequent CT/PET scan demonstrated disease progression, however, the study treatment was permanently discontinued (21).
Patients were monitored for the occurrence of adverse events (AEs), serious AEs, and immune-mediated AEs. The AEs were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0. Immune-mediated AEs were based on a list of terms specified by the sponsor and included by the investigator regardless of attribution to study treatment or of immune relatedness.
Outcomes
The primary endpoint was ORR, defined as either a partial response (PR) or complete response (CR) as assessed by the IRC and according to the Lugano classification (22). Secondary endpoints included CR rate, PFS, DOR, and time to response, as assessed by the IRC and per the Lugano classification, as well as safety and tolerability. Overall survival (OS) and biomarker analysis were exploratory endpoints (21).
Tissue samples
For the exploratory biomarker study, biomarker tests were performed retrospectively on formalin-fixed, paraffin-embedded (FFPE) tumor tissue collected prior to study treatment.
Multiplex immunohistochemistry
mIHC was performed using an Opal automation Multiplex IHC kit (PerkinElmer NEL801001KT or NEL821001KT, or equivalent) on Leica BOND Rx platform followed by IF 6-colorWJJ-CD30 protocol in CAP-controlled area within the Oncology and Immunology Unit of WuXi AppTec. Human FFPE specimens were labeled with different primary antibodies (CD30 Ber-H2, Dako M0751; FcγRΙ+ OTI3D3, Abcam ab140779; CD68 KP-1, Ventana 790–2931; PD-L1 SP263, Ventana 790–4905; CD8 SP57, Ventana 790–4460), followed by appropriate secondary antibodies (Polymer HRP from Opal automation Multiplex IHC Kit) and different Opal dyes, and finally counterstained with DAPI (spectral 4′,6-diamidino-2-phenylindole), rabbit immunoglobulin G (IgG; Abcam ab172730, EPR25A), and mouse IgG1 (Abcam ab18443, kappa monoclonal MOPC-21) were used as isotype control. Whole slide images were acquired for each case using a Leica Aperio VERSA 8 automated microscope. Image analysis was performed using the HALO software package (Indica Labs), and segmentation and mark-up of individual cells were performed, reviewed, and scored blinded by two pathologists using the IndicaLabs-HighPlex FL module.
Gene expression profiling
For gene expression profiling analyses, the Precision Immuno-Oncology Panel assay consisting of 1,392 genes was used (HTG Molecular Diagnostics, Inc.), according to the manufacturer's specifications. The library was sequenced on the Illumina NextSeq 500 platform (Illumina, Inc.), and data were processed by HTG EdgeSeq parser software. Log2CPM (counts per million) was used as the normalized gene expression value in the analysis.
Statistical analysis
A binomial exact test indicated that a sample of 68 patients would provide 91% power to detect a difference in the ORR of 35% (H0: ORR, 0.35; the minimal threshold for clinically meaningful benefit) versus 55% (HA: ORR, 0.55) at a one-sided alpha level of 0.025 and a 95% confidence interval (CI) of 0.425–0.671, when the observed ORR was 55%.
Efficacy and safety analyses included all patients who received at least one dose of tislelizumab. ORR and CR rate were assessed and the 95% CIs were conducted using two-sided Clopper–Pearson estimations. Forest plot for the subgroup analyses of patients achieving an ORR or CR were provided with 95% CI. Time-to-event, including DOR, PFS, and OS, was estimated using the Kaplan–Meier method, with 95% CIs calculated by the Brookmeyer and Crowley method (23). Event-free rates at landmark time points were calculated by the Kaplan–Meier method, and Greenwood's formula (24) was used for the 95% CIs. Patient follow-up was censored at the last adequate disease assessment before the initiation of subsequent anticancer therapy for PFS and DOR estimates. For patients who did not have either a baseline or at least one postbaseline response, assessments were censored on the first day of study treatment.
Expression levels of biomarkers were dichotomized according to the median value, where levels below median were assigned to the “low group” and those above or equal to median to the “high group.” For the mIHC analysis, the distribution of patients with CR and non-CR was compared across different biomarker groups using Fisher exact test. For the gene expression profiling analysis, the univariate Cox regression model was conducted to analyze the association between single genes and PFS. The genes with a P value <0.05 by the Wald test were selected as candidates for the biomarker analysis.
Results
Patient characteristics and disposition
A total of 70 patients were enrolled and treated. All patients were evaluable for the efficacy and safety analyses. The median age was 32.5 years (range, 18–69), and 57% were male. Patients had received a median of three prior lines of therapy (range, 2–11), with 60% receiving ≥3 prior lines. Twenty-one (30.0%) patients received previous radiotherapy. Thirteen (18.6%) patients had undergone prior ASCT, while the remaining patients (81.4%) were ineligible for prior ASCT and had received at least two prior systemic regimens for cHL. Four (5.7%) patients were exposed to BV before study enrollment. Detailed baseline characteristics have been reported previously (20) and can be found in Supplementary Table S1. At the final data cut-off date (November 2, 2020), the 33 (47.1%) patients who were still on treatment were transferred to the long-term extension study (NCT04164199). Of the 37 (52.9%) patients who discontinued treatment, the most common causes for discontinuation were disease progression (n = 24, 34.3%) and AEs (n = 6, 8.6%). The median study follow-up was 33.8 months (range, 3.4–38.6), and the median duration of exposure was 119.93 weeks (range, 6.0–167.9).
Efficacy
The ORR was 87.1% (95% CI, 77.0–93.9), with 67.1% of patients achieving CR and 20.0% achieving PR. Responses were observed across all subgroups analyzed (Fig. 1). Of the 13 patients with prior ASCT, 11 (84.6%) achieved a CR and one achieved a PR. CR occurred in all four patients with previous BV treatment. All patients had reductions in target lesion burden (Supplementary Fig. S1).
Figure 1.

Forest plots of subgroup analyses of tislelizumab. A, Overall response rate. B, Complete response rate according to demographic and baseline disease characteristics. ASCT, autologous hematopoietic stem cell transplant; cHL, classical Hodgkin lymphoma; CI, confidence interval; CR, complete response; ECOG PS, Eastern Cooperative Oncology Group performance status; ORR, overall response rate.
Median PFS according to the Lugano classification was 31.5 months [95% CI, 16.53–not evaluable (NE); Fig. 2A]. The estimated 2-year and 3-year PFS rates were 55.4% and 40.8%, respectively. A clear separation of PFS curves was observed between patients with CR and patients achieving PR or stable disease (Fig. 2B). The median PFS was not reached in patients with CR. The estimated 2-year and 3-year PFS rates in patients with a CR were 70.7% and 52.1%, respectively. No difference in PFS was observed between the groups with and without prior ASCT (Fig. 2C). PFS was similar regardless of the number of prior lines of therapy (Fig. 2D).
Figure 2.
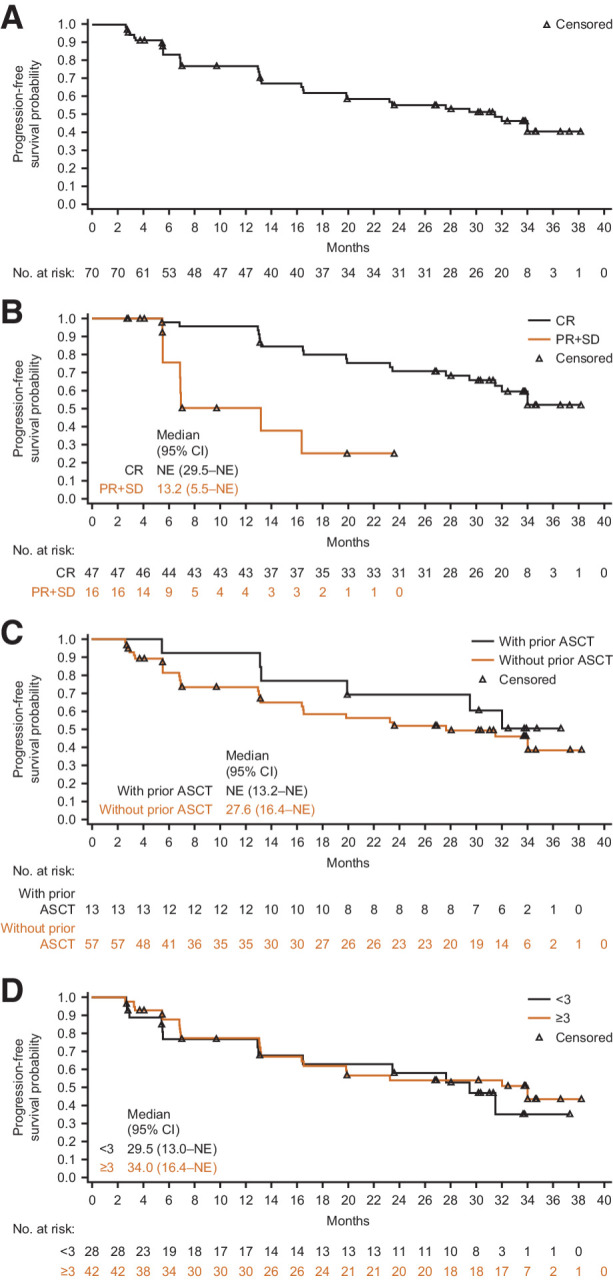
Progression-free survival by independent review committee per the Lugano classification. A, All treated patients. B, Patients achieving CR or PR + SD. C, Patients with/without prior ASCT. D, Patients with different number of lines of prior therapies (<3 vs. ≥3). ASCT, autologous hematopoietic stem cell transplant; CI, confidence interval; CR, complete response; NE, not estimable; PR, partial response; SD, stable disease.
Median time to response was 12 weeks (range, 8.9–57.0). Median DOR was 31.3 months (range, 0.0+–35.4+) in all responders (Fig. 3A). The estimated DOR rates at 2-year and 30-month time points were 63.9% and 52.5%, respectively. The majority of patients in this study had very good OS, as the median OS was not reached (Fig. 3B). The estimated 2-year and 3-year OS rates were 93.9% and 84.8%, respectively.
Figure 3.

Duration of response and overall survival. A, Duration of response in patients with an objective response by independent review committee per the Lugano classification. B, Overall survival for all treated patients. CI, confidence interval.
Safety
Almost all (97.1%) patients experienced ≥1 treatment-emergent AEs (TEAE), with grade ≥3 TEAEs reported in 41.4% of patients (Table 1). The most frequently reported TEAEs (occurring in ≥20% patients) were pyrexia (57.1%), upper respiratory tract infection (38.6%), hypothyroidism (37.1%), weight increased (34.3%), cough (21.4%), white blood cell count decreased (21.4%), and alanine aminotransferase increased (20.0%). Treatment-related AEs (TRAEs) occurred in 68 (97.1%) patients; 22 (31.4%) patients experienced grade ≥3 TRAEs. The most common TRAEs were pyrexia (54.3%), hypothyroidism (37.1%), weight increased (27.1%), upper respiratory tract infection (24.3%), and white blood cell count decreased (20.0%). The most common grade ≥3 TRAEs (occurring in ≥2% patients) were weight increased, blood creatine phosphokinase increased, lipase increased, pneumonitis, neutropenia, and hypertension (Table 1).
Table 1.
Adverse events.
| Tislelizumab (N = 70) | ||||
|---|---|---|---|---|
| TEAE | TRAE | |||
| N (%) | All grades | Grade ≥3 | All grades | Grade ≥3 |
| Patients with ≥1 AE | 68 (97.1) | 29 (41.4) | 68 (97.1) | 22 (31.4) |
| Pyrexia | 40 (57.1) | 0 | 38 (54.3) | 0 |
| Upper respiratory tract infection | 27 (38.6) | 2 (2.9) | 17 (24.3) | 0 |
| Hypothyroidism | 26 (37.1) | 0 | 26 (37.1) | 0 |
| Weight increased | 24 (34.3) | 2 (2.9) | 19 (27.1) | 2 (2.9) |
| White blood cell count decreased | 15 (21.4) | 1 (1.4) | 14 (20.0) | 0 |
| Cough | 15 (21.4) | 0 | 9 (12.9) | 0 |
| Alanine aminotransferase increased | 14 (20.0) | 0 | 14 (20.0) | 0 |
| Pruritus | 13 (18.6) | 0 | 12 (17.1) | 0 |
| Weight decreased | 12 (17.1) | 0 | 8 (11.4) | 0 |
| Aspartate aminotransferase increased | 11 (15.7) | 0 | 8 (11.4) | 0 |
| Rash | 11 (15.7) | 1 (1.4) | 11 (15.7) | 1 (1.4) |
| Neutrophil count decreased | 10 (14.3) | 2 (2.9) | 9 (12.9) | 1 (1.4) |
| Hyperuricemia | 10 (14.3) | 0 | 7 (10.0) | 0 |
| Diarrhea | 10 (14.3) | 0 | 6 (8.6) | 0 |
| Anemia | 9 (12.9) | 1 (1.4) | 4 (5.7) | 0 |
| Blood creatine phosphokinase increased | 8 (11.4) | 2 (2.9) | 8 (11.4) | 2 (2.9) |
| Blood thyroid stimulating hormone increased | 8 (11.4) | 0 | 8 (11.4) | 0 |
| Blood bilirubin increased | 7 (10.0) | 0 | 7 (10.0) | 0 |
| Platelet count decreased | 7 (10.0) | 0 | 6 (8.6) | 0 |
| Pneumonia | 7 (10.0) | 2 (2.9) | 6 (8.6) | 1 (1.4) |
| Vomiting | 7 (10.0) | 0 | 5 (7.1) | 0 |
| Headache | 7 (10.0) | 1 (1.4) | 5 (7.1) | 1 (1.4) |
| Neutropenia | 4 (5.7) | 2 (2.9) | 3 (4.3) | 2 (2.9) |
| Hypertension | 3 (4.3) | 2 (2.9) | 3 (4.3) | 2 (2.9) |
| Lipase increased | 2 (2.9) | 2 (2.9) | 2 (2.9) | 2 (2.9) |
| Pneumonitis | 2 (2.9) | 2 (2.9) | 2 (2.9) | 2 (2.9) |
Note: All grades include AEs reported in ≥10% of patients; grade ≥3 events include those reported in ≥2% of patients. AE, adverse event; TEAE, treatment-emergent adverse event; TRAE, treatment-related adverse event.
Immune-mediated AEs were reported in 45.7% of patients, including grade ≥3 immune-mediated AEs in 11.4%. The most commonly reported (occurring in ≥5% of patients) immune-mediated AEs were hypothyroidism (28.6%), skin adverse reaction (8.6%), and pneumonitis (7.1%). A total of 16 patients (22.9%) received corticosteroid therapy for immune-mediated AE management, and none received immunosuppressive drug. Infusion-related reactions were reported in 38.6% of patients.
Serious AEs were observed in 18 (25.7%) patients during the conduct of this study. TEAEs leading to treatment discontinuation occurred in six (8.6%) patients, including pneumonitis in two patients, and focal segmental glomerulosclerosis, organizing pneumonia, psychomotor skills impaired, and seizure in one patient, respectively. There was one death due to TEAE. The patient with impaired psychomotor skills who discontinued from treatment died after cycle 49, 26 days after receiving the last dose of tislelizumab.
Exploratory biomarkers
We conducted exploratory biomarker analysis, including CD8 T cells, macrophage markers by mIHC and gene expression profiles of 1,392 immuno-oncology related genes and their association with clinical outcomes. Baseline demographic characteristics and clinical outcomes in biomarker subgroups were generally consistent with the overall population (Supplementary Table S1).
Tislelizumab, with its specifically engineered Fc region, did not crosslink with Fcγ receptor I (FcγRI) on macrophages and did not cause macrophage-induced antibody-dependent cellular phagocytosis (ADCP) and T-cell elimination; thus, hypothetically its clinical outcome may not be compromised by FcγRI+ macrophages in the tumor microenvironment. To test this hypothesis, an mIHC panel (including CD8, CD68, FcγRI, PD-L1, and CD30 markers) was applied to the pretreated cHL tissue samples of 41 patients. A representative case of mIHC is shown in Fig. 4A. Total numbers of CD8 T cells (CD8+ cells), total FcγRI levels (FcγRI+ cells), macrophages (CD68+ cells), FcγRI+ macrophages (FcγRI; CD68 double-positive cells) by mIHC-based image analysis were quantified by Halo software. FcγRI+ macrophages are macrophages that can interact with the Fc region of the antibody and thus have ADCP potency. Using median value of cell percentage of CD8 T and FcγRI+ macrophages as the cutoff, four subgroups were identified by the relative abundance of CD8 T cells and FcγRI+ macrophages (Fig. 4B). Each subgroup was then associated with the clinical outcomes of tislelizumab treatment. Under the CD8 T-cell-high condition, the CR rate was 76.9% in FcγRI+ macrophage-high compared with 87.5% in FcγRI+ macrophage-low population (not significant by Fisher test). While under the CD8 T-cell-low condition, the CR rate was 75.0% in FcγR1+ macrophage-high population compared with 50.0% in FcγRI+ macrophage-low population (not significant by Fisher test), suggesting that tislelizumab showed high CR rate regardless of FcγRI+ macrophage abundance. In addition, similar PFSs were also observed in the four subgroups (Fig. 4C). These data show that FcγRI+ macrophages in the cHL microenvironment exerted little or no effect on the clinical activity of tislelizumab in this R/R cHL cohort we analyzed, which may be related to its Fc-null molecular characteristic.
Figure 4.

Associations of FcγRI+ macrophages in cHL tumor microenvironment with clinical outcomes of tislelizumab. A, Representative figure showing multiplex immunofluorescence staining (20× resolution) for CD8 T cells (CD8+ cells), total Fcγ receptor I (FcγRI+ cells), macrophages (CD68+ cells), FcγRI+ macrophages (FcγRI; CD68 double-positive cells), PDL1+, and CD30+ cells. B, CD8+ T cells, FcγRΙ+ macrophage infiltration, and their associations with complete response rates to tislelizumab. C, PFS by CD8+ T cells and FcγRΙ+ macrophages, divided by median cell percentage value of CD8 T cells and FcγRΙ+ macrophages separately. All mIHC images were acquired using Leica Aperio VERSA 8 auto-scanner. Note: (i) Make and model of microscope: LEICA DM6 B; (ii) Type, magnification, and numerical aperture of the objective lenses: ∞/0.17/OFM25/C, HC PL APO, 20×/0.80; (iii) Temperature: 20°C–30°C; (iv) Imaging medium: sCMOS: 23CAM014; (v) Fluorochromes: PerkinElmer Opal automation Multiplex IHC kit; (vi) Camera make and model: Andor Zyla 5.5; (vii) Acquisition software: Aperio VERSA. CR, complete response.
Figure 5.

Heatmap of genes significantly associated with PFS by univariate Cox proportional hazards regression model. BOR, best overall response; CR, complete response; PFS, progression-free survival.
In addition, it was found that patients who achieved CR tended to have a higher percentage of CD8 T-cell infiltration (Supplementary Fig. S2A). No significant differences in CD68+ macrophages were observed between CR patients and non-CR patients (Supplementary Fig. S2B). Notably, patients with higher FcγRI expression tended to achieve CRs (Supplementary Figs. S2C and S2D), indicating that the effect of tislelizumab may not be compromised by either total FcγRI expression or FcγRI expressed on macrophages, consistent with its Fc-null molecular characteristics.
Gene expression profiles of baseline tumor tissues from 36 patients were analyzed. Univariate Cox models were applied to assess the relationship between PFS and gene expression levels. Among the 150 genes identified that significantly correlated with PFS (P < 0.05, Supplementary Table S2), several functional groups emerged by unsupervised hierarchical clustering. Overall, the “B-cell markers” cluster is associated with shorter PFS. This cluster included CD19, CD22, CD72, and CD79B genes. Among them, CD19 and CD79B were pan B-cell markers expressed in all stages of B cells and these markers were previously reported as prognostic markers for B-cell neoplasms (25, 26). All three genes (i.e., S100A12, S100A9, and S100A8), which belong to the “proinflammatory” cluster, were associated with prolonged PFS. The “immune modulators” and “cytokine and receptors” clusters consisted of both immune activator and inhibitor genes, and thus, some of them are associated with longer PFS and others with shorter PFS. Immune genes positively associated with PFS included IL1A, IL1R1, IL4, IL9, CCR10, CCL11, CXCL8, and IFNL3; while genes negatively associated with PFS included IL6ST, IL6R, IL32, IRF1, IRF2, IRF3, IRF8, IRF9, CSF2RB, and IFNAR2. Interferon regulatory factors IRF1, IRF2, IRF3, IRF8, and IRF9 were all significantly associated with shorter PFS, indicating a role in upstream regulation of IFNγ secretion and their contribution to anti–PD-1 response.
Discussion
Here, we reported the high and deep response of tislelizumab in patients with R/R cHL in this study. With extended follow-up, the majority of responses observed were CR. In previous literature for nivolumab and pembrolizumab, it was noted that the CR rate appeared to correlate with better long-term efficacy parameters (e.g., DOR, PFS, and OS), suggesting that increasing the CR rate had potential to lead to better long-term outcomes (10, 12). However, one big limitation of anti–PD-1 antibodies was that the CR rate remained low in R/R cHL, ranging from 12% to 34% across study designs and different cohorts (10, 11, 13, 14). In our study, the CR rate was numerically higher (67.1%), and the median PFS reached 31.5 months. The prolonged PFS was largely driven by patients achieving CR (as shown in Fig. 2B). Acknowledging differences in study design and patient population, the results reported herein compared positively with other PD-1 studies published to date, particularly in the depth and duration of response.
Response rates were generally consistent across subgroups, and the trend to achieve high response rates was observed even in those subgroups that have traditionally responded poorly to therapy, including heavily pretreated patients (≥3 prior lines of therapy) and those who had prior ASCT. In this study, the proportion of patients with prior ASCT and/or BV was lower, which will limit the comparison of its data with that of nivolumab and pembrolizumab. However, the enrolled population in our study was representative of the characteristics and treatment patterns in China (13, 14). Furthermore, studies of other PD-1 inhibitors have not shown a significant difference in response based on prior BV treatment, supporting the theory that prior therapy is not a major determinant of response to immune checkpoint inhibition. In our study, the ORR and CR rates were numerically higher in patients with prior ASCT and/or BV, as was demonstrated by forest plot analysis in Fig. 1. In addition, patients receiving prior ASCT had a trend of longer PFS (median: not reached vs. 27.6 months, Fig. 2C).
Most of the anti–PD-1 antibodies investigated in clinical studies are of the IgG4 isotype, which can bind to FcγR1 with high affinity and mediate cross-linking between PD-1 and FcγRI. A previous in vivo study revealed a PD-1 antibody can be transferred from CD8+ T cells to macrophages by Fc–FcγR interaction and then degraded by macrophage activity, leading to compromised aPD-1 monoclonal antibody activity in mice (19). Zhang and colleagues compared BGB-A317 with engineered Fc region (e.g., tislelizumab), with BGB-A317/IgG4 s228p [with wild-type (WT) Fc region] directly in the preclinical setting. Both in vitro and in vivo results demonstrated that A317/IgG4S228 induced more ADCP and CD8 T-cell clearance than the A317 isoform and resulted in a compromised antitumor activity (18). In this study, we investigated Fc–FcγRI interaction and the potential outcomes in the R/R cHL clinical setting for the first time. cHL was distinct from other tumor types in that its microenvironment was highly inflamed and composed of different types of noncancerous normal immune cells (27). Among them, a significant proportion of macrophages was found to be in close contact with T cells in cHL, which increased the potential for PD-1 T-effector and macrophage cross-linking by anti–PD-1 antibodies with WT Fc region (28). FcγRΙ was reported to be highly expressed in macrophages under inflammatory conditions (29). Our in vitro results also showed that FcγRΙ expression was induced by IFNγ and interleukin 10 (IL10; Supplementary Fig. S3A). Consistently, in cHL clinical samples, we also found that Fc fragment of IgG receptor Ia (FcGR1A) expression was highly correlated with IFNγ and IL10 expression (Supplementary Fig. S3B and S3C). This high level of FcγRI expression in cHL may cross-link PD-1+ T cells and FcγRI+ macrophages, resulting in macrophage-mediated phagocytosis of PD-1+ T cells and thereby dampening T-cell–mediated immune responses of anti–PD-1 antibody was in WT IgG4 (S228P) form (18, 19). This may help explain why tislelizumab demonstrated a high CR rate and relatively long PFS, especially in cHL.
In terms of safety, tislelizumab was generally well tolerated. The spectrum of tislelizumab-associated toxicities was similar to those reported among patients with cHL treated with anti–PD-1 antibodies. The majority of events were grade 1 or 2, and no new safety concern was identified. Most TEAEs were either not treatment-limiting or resulted in transient treatment delays. Only six patients required discontinuation of tislelizumab.
There were certain limitations to our study. From a clinical perspective, this study was designed as a single-arm study, which limited direct comparison with controlled interventions. An ongoing, randomized, controlled, phase III confirmatory study (NCT04486391) is being conducted to compare tislelizumab to physician's choice of salvage chemotherapy in R/R cHL patients with prior ASCT or who were ineligible for transplantation. Also, interpretation of this long-term survival follow-up result was limited due to decreasing patient numbers at risk around 30 to 34 months. Although the “survival plateau” was a known trait for immunotherapy (30) and this similar trend was observed in the Kaplan–Meier curve shown in PFS and OS, the 3-year survival data should be interpreted with caution due to the smaller sample size.
In addition, the association between high CR rate/long PFS and the engineered Fc region of tislelizumab should be interpreted cautiously due to several limitations of this biomarker study. Firstly, we did not show whether clinical activity of other anti–PD-1 agents with WT IgG4 was influenced by FcrRI+ macrophages in the tumor microenvironment. Secondly, as Carey and colleagues reported, some PD-1+ CD4 T cells, together with CD8 T cells, are enriched in the vicinity of, and in contact with, PD-L1+ macrophages and HRS cells and may play important roles in cHL immunosuppression (15). Because CD4 was not included in our mIHC panel, we are unable to evaluate the potential role of CD4 T cells in the activity of tislelizumab. Finally, the sample size of this study was rather small, which precludes a firm conclusion that the high and deep response in cHL of tislelizumab is the consequence of its engineered Fc structure. Thus, the exploratory biomarker results of this study need to be further validated in larger cohorts in the future.
In summary, treatment with tislelizumab led to high ORR and CR rates in R/R cHL patients in this phase II study. Tislelizumab demonstrated a similar toxicity profile to other anti–PD-1 therapies, with no new safety signals. The majority of AEs were mild or moderate, manageable, and generally did not limit treatment duration. In summary, tislelizumab showed a favorable benefit–risk profile, and has potential as a new treatment option for patients with R/R cHL. Correlative biomarker analysis showed that FcγR+ macrophages in the tumor microenvironment had no observed impact on clinical outcomes of tislelizumab, which may be potentially related to its engineered Fc region.
Data sharing statement
On request, and subject to certain criteria, conditions, and exceptions, BeiGene will provide access to individual deidentified participant data from BeiGene-sponsored global interventional clinical studies conducted for medicines (1) for indications that have been approved or (2) in programs that have been terminated. Data requests may be submitted to DataDisclosure@beigene.com.
Supplementary Material
Acknowledgments
The authors thank the patients who participated in the study, their supporters, and the investigators and clinical research staff from the study centers. Funding for this research was provided by BeiGene (Beijing) Co., Ltd., Beijing, China, and BeiGene USA, Inc., San Mateo, CA. Medical writing and editorial assistance were funded by BeiGene and provided by Ify Sargeant, Kathryn Quinn, and Maxine Skipp on the behalf of Twist Medical.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article is featured in Highlights of This Issue, p. 1053
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
Y. Song reports other support from BeiGene Company during the conduct of the study. H. Guo reports other support from BeiGene outside the submitted work. X. Zhao reports nonfinancial support from Twist Medical during the conduct of the study and other support from BeiGene outside the submitted work. J. Huang reports other support from BeiGene during the conduct of the study and other support from BeiGene outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
Y. Song: Conceptualization, data curation, validation, investigation, methodology, writing–review and editing. Q. Gao: Data curation, writing–review and editing. H. Zhang: Data curation, writing–review and editing. L. Fan: Data curation, writing–review and editing. J. Zhou: Data curation, writing–review and editing. D. Zou: Data curation, writing–review and editing. W. Li: Data curation, writing–review and editing. H. Yang: Data curation, writing–review and editing. T. Liu: Data curation, formal analysis, writing–review and editing. Q. Wang: Data curation, writing–review and editing. F. Lv: Data curation, writing–review and editing. H. Guo: Conceptualization, formal analysis, validation, investigation, methodology, writing–original draft, writing–review and editing. X. Zhao: Formal analysis, writing–original draft. D. Wang: Formal analysis, writing–original draft, writing–review and editing. P. Zhang: Formal analysis, writing–review and editing. Y. Wang: Formal analysis. L. Wang: Formal analysis. T. Liu: Formal analysis, writing–original draft, writing–review and editing. Y. Zhang: Conceptualization, formal analysis, validation, investigation, methodology, writing–original draft, writing–review and editing. Z. Shen: Formal analysis, writing–original draft, writing–review and editing. J. Huang: Conceptualization, data curation, validation, investigation, methodology, writing–review and editing. J. Zhu: Conceptualization, data curation, formal analysis, validation, investigation, methodology, writing–review and editing.
References
- 1. Marafioti T, Hummel M, Foss HD, Laumen H, Korbjuhn P, Anagnostopoulos, et al. Hodgkin and Reed-Sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood 2000;95:1443–50. [PubMed] [Google Scholar]
- 2. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010;116:3268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MGM, Xu ML, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res 2013;19:3462–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ansell SM. Hodgkin lymphoma: 2012 update on diagnosis, risk-stratification, and management. Am J Hematol 2012;87:1096–103. [DOI] [PubMed] [Google Scholar]
- 6. Stathis A, Younes A. The new therapeutical scenario of Hodgkin lymphoma. Ann Oncol 2015;26:2026–33. [DOI] [PubMed] [Google Scholar]
- 7. Younes A, Connors JM, Park SI, Fanale M, O'Meara MM, Hunder NN, et al. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin's lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncol 2013;14:1348–56. [DOI] [PubMed] [Google Scholar]
- 8. Chen R, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. Five-year survival and durability results of brentuximab vedotin in patients with relapsed or refractory Hodgkin lymphoma. Blood 2016;128:1562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Younes A, Santoro A, Shipp M, Zinzani PL, Timmerman JM, Ansell S, et al. Nivolumab for classical Hodgkin's lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: a multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol 2016;17:1283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Armand P, Engert A, Younes A, Fanale M, Santoro A, Zinzani PL, et al. Nivolumab for relapsed/refractory classic Hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: extended follow-up of the multicohort single-arm phase II CheckMate 205 trial. J Clin Oncol 2018;36:1428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Radford J, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol 2017;35:2125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen R, Zinzani PL, Lee HJ, Armand P, Johnson NA, Brice P, et al. Pembrolizumab in relapsed or refractory Hodgkin lymphoma: 2-year follow-up of KEYNOTE-087. Blood 2019;134:1144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song Y, Wu J, Chen X, Lin T, Cao J, Liu Y, et al. A single-arm, multicenter, phase II study of camrelizumab in relapsed or refractory classical Hodgkin lymphoma. Clin Cancer Res 2019;25:7363–9. [DOI] [PubMed] [Google Scholar]
- 14. Shi Y, Su H, Song Y, Jiang W, Sun X, Qiaan W, et al. Safety and activity of sintilimab in patients with relapsed or refractory classical Hodgkin lymphoma (ORIENT-1): a multicentre, single-arm, phase 2 trial. Lancet Haematol 2019;6:e12–9. [DOI] [PubMed] [Google Scholar]
- 15. Roemer MGM, Redd RA, Cader FZ, Pak CJ, Abdelrahman S, Ouyang J, et al. Major histocompatibility complex class II and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic Hodgkin lymphoma. J Clin Oncol 2018;36:942–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi Y, Su H, Song Y, Jiang W, Sun X, Qian W, et al. Circulating tumor DNA predicts response in Chinese patients with relapsed or refractory classical hodgkin lymphoma treated with sintilimab. EBioMedicine 2020;54:102731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dahan R, Sega E, Engelhardt J, et al. FcgammaRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell 2015;28:285–95. [DOI] [PubMed] [Google Scholar]
- 18. Zhang T, Song X, Xu L, Ma J, Zhang Y, Gong W, et al. The binding of an anti-PD-1 antibody to FcγRΙ has a profound impact on its biological functions. Cancer Immunol Immunother 2018;67:1079–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med 2017;10:eaal3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song Y, Gao Q, Zhang H, Fan L, Zhou J, Zou D, et al. Treatment of relapsed or refractory classical Hodgkin lymphoma with the anti-PD-1, tislelizumab: results of a phase 2, single-arm, multicenter study. Leukemia 2020;34:533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheson BD, Ansell S, Schwartz L, Gordon LI, Advani R, Jacene HA, et al. Refinement of the Lugano classification lymphoma response criteria in the era of immunomodulatory therapy. Blood 2016;128:2489–96. [DOI] [PubMed] [Google Scholar]
- 22. Cheson BD, Fisher RI, Barrington SF, Cavalli F, Schwartz LH, Zucca E, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brookmeyer R, Crowley J. A confidence interval for the median survival time. Biometrics 1982;38:29–41. [Google Scholar]
- 24. Kalbfleisch JD, Prentice RL. The statistical analysis of failure time data. 1st ed.New York, NY: John Wiley & Sons Inc; 1980. [Google Scholar]
- 25. Korkolopoulou P, Cordell J, Jones M, Kaklamanis L, Tsenga A, Gatter KC, et al. The expression of the B-cell marker mb-1 (CD79a) in Hodgkin's disease. Histopathology 1994;24:511–5. [DOI] [PubMed] [Google Scholar]
- 26. Chu PG, Arber DA. CD79: a review. Appl Immunohistochem Mol Morphol 2001;9:97–106. [DOI] [PubMed] [Google Scholar]
- 27. Aldinucci D, Borghese C, Casagrande N. Formation of the immunosuppressive microenvironment of classic Hodgkin lymphoma and therapeutic approaches to counter it. Int J Mol Sci 2019;20:2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carey CD, Gusenleitner D, Lipschitz M, Roemer MGM, Stack EC, Gjini E, et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood 2017;130:2420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Swisher JFA, Feldman GM. The many faces of FcγRI: implications for therapeutic antibody function. Immun Rev 2015;268:160–74. [DOI] [PubMed] [Google Scholar]
- 30. Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res 2013;19:5300–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.