

Abstract
Opioids are potent analgesics, but their pain-relieving effects diminish with repeated use. The reduction in analgesic potency is a hallmark of opioid analgesic tolerance, which hampers opioid pain therapy. In the central nervous system, opioid analgesia is critically modulated by adenosine, a purine nucleoside implicated in the beneficial and detrimental actions of opioid medications. Here, we focus on the A3 adenosine receptor (A3AR) in opioid analgesic tolerance. Intrathecal administration of the A3AR agonist MRS5698 with daily systemic morphine in male rats attenuated the reduction in morphine antinociception over 7 days. In rats with established morphine tolerance, intrathecal MRS5698 partially restored the antinociceptive effects of morphine. However, when MRS5698 was discontinued, these animals displayed a reduced antinociceptive response to morphine. Our results suggest that MRS5698 acutely and transiently potentiates morphine antinociception in tolerant rats. By contrast, in morphine-naïve rats MRS5698 treatment did not impact thermal nociceptive threshold or affect antinociceptive response to a single injection of morphine. Furthermore, we found that morphine-induced adenosine release in cerebrospinal fluid was blunted in tolerant animals, but total spinal A3AR expression was not affected. Collectively, our findings indicate that spinal A3AR activation acutely potentiates morphine antinociception in the opioid tolerant state.
Keywords: adenosine, analgesia, opioids, opioid tolerance, RRID:AB_10711040, RRID:AB_10751536, RRID:AB_141844, RRID:AB_2039711, RRID:AB_2535792, RRID:AB_2636996, RRID:AB_449329, RRID:AB_476743, RRID:AB_521594, spinal cord
Graphical Abstract
1 |. INTRODUCTION
Opioid medications are used to manage a variety of pain conditions, but their pain-relieving effects diminish with repeated use. The reduction in analgesic potency is a feature of opioid analgesic tolerance and is a major barrier to effective pain management (Mercadante et al., 2019). Few strategies exist to address the problem of opioid tolerance, and as patients struggle to manage pain, they are often prescribed higher doses or switched to more potent opioid medications. Dose escalation increases opioid use liability and may contribute to opioid use disorder (Babu et al., 2019; Hayes et al., 2020). Novel strategies for overcoming opioid tolerance are urgently needed to improve the clinical utility and safety of opioid medications for treating pain.
Interactions between the opioid and adenosine systems critically modulate opioid analgesia within the central nervous system. Adenosine is a purine nucleoside that exerts its effects via four receptor subtypes: Gi-protein coupled A1AR and A3AR, and Gs-protein coupled A2AAR and A2BAR (Boison et al., 2010; Fredholm et al., 2005, 2011). Early evidence indicated that acute morphine administration in rats drives adenosine release in the spinal cord (Cahill et al., 1993; M. Sweeney et al., 1987) and adenosine signaling through A1AR is critical for morphine antinociception (Suh et al., 1997; M. Sweeney et al., 1987; M. I. Sweeney et al., 1987; Wu et al., 2005). By contrast, prolonged morphine treatment decreases extracellular adenosine and impairs adenosine A1AR signaling (Nelson et al., 2009; Zarrindast et al., 1999). Reduced signaling at A1AR and A2AAR contributes to adverse opioid effects, including opioid tolerance, dependence, and withdrawal (Kaplan & Sears, 1996; Wu et al., 2013; Zarrindast et al., 1999). However, targeting A1AR and A2AAR as opioid adjuncts or standalone analgesics is not viable because of negative side effects, most notable of which are adverse cardiovascular events (Boison, 2013; Kiesman et al., 2009; Zylka, 2011).
In search for adenosine-targeted therapies, attention is shifting to A3AR because of its low expression in cardiac tissue and its cardioprotective and cerebroprotective effects (Abbracchio et al., 1997; Fishman et al., 2012; Lopes et al., 2003; Ru et al., 2011; Von Lubitz et al., 1999). Phase II/III clinical trials for cancer and several autoimmune conditions have reported good safety outcomes with small molecule A3AR selective agonists, such as IB-MECA and MRS5698 (Fishman et al., 2002, 2012; Silverman et al., 2008; Stemmer et al., 2013; Tosh et al., 2012). Preclinical studies reveal that A3AR is expressed along key pain signaling pathways in the spinal cord and brainstem, and its activity has anti-inflammatory effects within the nervous system (Chen et al., 2012; Little et al., 2015; Salvemini & Jacobson, 2017; Trang et al., 2015). In nerve-injured rats, selective A3AR agonists alleviate neuropathic pain without disrupting normal nociceptive processing (Chen et al., 2012; Janes et al., 2014, 2015; Little et al., 2015). Recent evidence also indicates that chronic morphine disrupts A3AR-dependent signaling in the spinal cord (Doyle et al., 2020). Intervention with A3AR selective agonists to normalize adenosine signaling effectively attenuates morphine-induced hyperalgesia, tolerance, and withdrawal (Doyle et al., 2020). In the present study, we further examine the interaction between chronic morphine treatment and A3AR, uncovering that spinal A3AR activation transiently restores morphine antinociception in opioid tolerant animals.
2 |. METHODS
2.1 |. Animals
Adult male Sprague Dawley rats (aged 6–8 weeks, Charles River Laboratories, Sherbrooke, QC, Canada) were housed in standard cages under a 12:12 light/dark cycle (lights on at 08:00 hr) with ad libitum access to standard rodent chow pellets and water. A total of 321 rats were used in experiments contributing to this paper. Sample size calculation using G*Power revealed target group size of 5 (effect size 0.5, α = 0.05). An average sample size of six per group was used dependent on animal cohort sizes and variability in results. Rats were lightly anesthetized with a 1.5% isoflurane O2 mixture during intrathecal injections. Rats were handled and exposed to equipment prior to behavioral experiments for acclimatization and were randomly allocated to different test groups. All experiments were approved by the University of Calgary Animal Care Committee and were in accordance with the guidelines of the Canadian Council on Animal Care and the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
2.2 |. Drugs
Morphine sulfate (Professional Compounding Centers of America, London, ON, Canada) was prepared in saline (Sodium Chloride solution 0.9% w/v, Sigma-Aldrich, St. Louis, MO, USA). Drugs were commercially purchased: IB-MECA (1-deoxy-1-[6-[((3-iodophenyl)methyl) amino]-9H-purin-9-yl]-N-methyl-β-D-ribofuranuronamide, N6-(3-Iodobenzyl)adenosine-5’-N-methyluronamide; Sigma-Aldrich, St. Louis, MO, USA), MRS5698 ((1S,2R,3S,4R,5S)-4-(6-((3-chlorobenzyl) amino)-2-((3,4-difluorophenyl)ethynyl)-9H-purin-9-yl)-2, 3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide; R&D Systems, Minneapolis, MN, USA), and MRS1523 (3-propyl-6-ethyl-5[(ethylthio)carbonyl]-2-phenyl-4-propyl-3-pyridinecarboxylate; Sigma-Aldrich, St. Louis, MO, USA). Drugs were dissolved in DMSO with saline.
2.3 |. Intrathecal drug administration
Intrathecal injections of IB-MECA, MRS5698, or vehicle control were performed as described by De la Calle and Paino (De la Calle & Paíno, 2002). Briefly, animals were placed on a cylindrical Falcon tube such that the spinal column was curved at the level of the L3-L5 vertebrae. Drugs or vehicle control were delivered to the intrathecal space between L3-L4 and L4-L5 vertebrae via lumbar puncture with a 30-gauge needle connected to a Hamilton syringe. Intrathecal delivery of drugs or vehicle (3% DMSO) was administered in a 10 μl volume. To control for intrathecal delivery, morphine-treated rats were intrathecally injected with vehicle, while saline-treated (morphine-naïve) rats were intrathecally injected with IB-MECA or MRS5698 as drug controls.
2.4 |. Morphine treatment and nociceptive testing
Morphine sulfate was administered intraperitoneally (i.p.) once daily for 7 consecutive days at 15 mg/kg body weight to establish systemic antinociceptive tolerance (Burma et al., 2017; Leduc-Pessah et al., 2017). Control animals were given an equivalent volume of saline daily for 7 consecutive days. On each day, baseline nociceptive threshold was measured 15 min before, and then assessed 30 min after, intraperitoneal injection of morphine or saline using the thermal tail-flick test (Ugo Basile, Varese, Italy). In this test, an infrared thermal stimulus was applied to the ventral surface of the tail. The latency, in seconds (s), for removal of the tail from the stimulus was measured and expressed as tail-flick latency. A maximum cut-off time of 10 s was used to prevent tissue damage. The intensity of the heat stimulus was set to 80 infrared intensity. Parameters for assessment remained consistent for each experimental measurement. The tail-flick latency for each animal was calculated as the average of three consecutive measurements.
2.5 |. Intrathecal IB-MECA or MRS5698 injection and acute morphine antinociception
Acute response to a single injection of morphine was performed on day 1 to characterize the timecourse of morphine-induced antinociception at 30, 60, 90, and 120 min after morphine injection. Intrathecal IB-MECA (3 or 50 nmol, i.t.) or MRS5698 (3 or 50 nmol, i.t.) was administered at the time of systemic morphine injection. A submaximal dose of morphine (7.5 mg/kg; i.p.) was given to discern whether IB-MECA or MRS5698 potentiates acute morphine antinociception. As control, rats received intrathecal vehicle injection with a submaximal dose of systemic morphine.
2.6 |. Daily intrathecal IB-MECA or MRS5698 injections and chronic morphine antinociception
In another animal cohort, intrathecal IB-MECA (50 nmol, i.t.) or MRS5698 (3 or 50 nmol i.t.) was co-administered with daily systemic morphine (15 mg/kg, i.p.) over 7 days. Control animals received systemic saline (i.p.) and/or intrathecal vehicle injection. Thermal antinociception was assessed prior to (baseline) and at 30 min post-injections each day using the tail-flick test.
2.7 |. Intrathecal MRS5698 in animals with established morphine antinociceptive tolerance
To establish morphine tolerance, rats were injected daily with morphine (15 mg/kg, i.p.) for 7 consecutive days. On days 8–12, morphine-tolerant rats continued to receive systemic morphine (15 mg/kg, i.p.), but also daily intrathecal injection of either MRS5698 (3 or 50 nmol) or vehicle. Control animals received systemic saline (i.p.) and/or intrathecal vehicle injections.
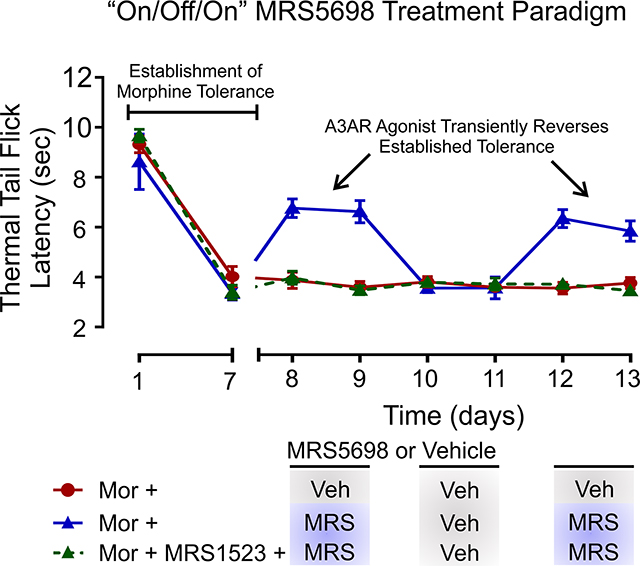
In separate experiments, we tested the transient effects of MRS5698 (50 nmol) using an “on/off/on” drug administration paradigm in morphine-tolerant rats. On days 8 and 9, rats received intrathecal MRS5698 (“on”) and systemic morphine; on days 10 and 11, these rats were injected with intrathecal vehicle (“off”) and systemic morphine; and on days 12 and 13, intrathecal MRS5698 (‘on”) was again administered with systemic morphine. A3AR selective antagonist MRS1523 (25 nmol) was intrathecally administered 15 min prior to MRS5698. As an injection control, intrathecal vehicle was administered with systemic morphine on days 8–13. In all experiments, thermal antinociception was assessed using the tail-flick test prior to (baseline) and at 30 min post-injections each day.
2.8 |. Dose-response experiments
To determine morphine potency, escalating doses of morphine (2.5, 5, 15, 30, 50, 75 mg/kg) were administered intraperitoneally every 30 min and the response to morphine was assessed by the thermal tail-flick test until a maximal level of antinociception (10 s, cut-off time) was achieved. The tail-flick latencies obtained at baseline and at each dose were used to calculate the percent of maximum possible effective dose (% MPE) in order to produce morphine dose-response curves and calculate the median effective dose (ED50) for each animal. Dose-response curves and ED50 values were compared between treatment groups. Morphine dose-response experiments were conducted on day 8 or day 13 of experiments. Rats were injected with either intrathecal vehicle or MRS5698 concomitantly with the first injection of systemic morphine; subsequent injections for the dose-response were performed only with escalating doses of systemic morphine.
2.9 |. Adenosine measurements in cerebral spinal fluid (CSF)
CSF was collected from adult male Sprague Dawley rats (175–225 g) treated with either a single subcutaneous (s.c.) injection of morphine (6 mg/kg) or after 7 days of daily morphine treatment (6mg/kg; s.c.). As a control, rats received either a single or 7 day s.c. injection of saline. CSF samples were collected from the cisterna magna 30 min after morphine or saline injection (Mousseau et al., 2018; Pegg et al., 2010). In brief, rats were anesthetized with 2% isoflurane and positioned in a stereotaxic frame. Ear bars were placed in the external auditory meatus, the scalp was shaved, and a midline scalp incision was performed to expose the cervico-spinal muscle and the atlanto-occipital membrane. Using a 1 ml syringe connected to a 27G needle, the atlanto-occipital membrane was punctured, and CSF was gently aspirated into an Eppendorf tube containing ABT-702 (50 nM, Tocris Biosciences), ARL-67156 (1 μM, Sigma), and protease inhibitors (Sigma) and immediately flash frozen on dry ice. Adenosine levels were analyzed using a fluorometric Adenosine Assay kit (Abcam) according to the manufacturer’s instructions and fluorescence levels were measured using a FilterMax F5 Multimode Microplate Reader (Molecular Devices). For calculations, CSF adenosine levels were corrected to the mean fluorescent value of a blank and the amount of adenosine/well (pmol) was derived from a trendline equation based on standard curve data. All samples were diluted to fit within standard value range and are presented as adenosine concentration (μM) in CSF.
2.10 |. Western blotting
Spinal cords were isolated from rats on the last day of treatment (D7 or D12) using hydraulic extrusion. The lumbar region (L3–L5) was isolated and hand homogenized with microfuge tube homogenization sticks in 250 μl of cold RIPA buffer containing 50 mM TrisHCl, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 1 mM Na3VO4, 1 U/ml aprotinin, 20 μg/ml leupetin, 20 μg/ml pepstatin A, protease inhibitors, and phosphatase inhibitors. Samples were incubated on ice for 30 min with periodic vortexing, and then centrifuged at 4°C for 30 min at 12,000 rpm. Supernatants were collected from samples and total protein was measured using a Pierce BCA Protein Assay Kit (Thermo Scientific). Samples were heated at 95°C for 5 min in loading buffer (350 mM Tris, 30% glycerol, 1.6% SDS, 1.2% bromophenol 25 blue, 6% β-mercaptoethanol) then electrophoresed on a 10% polyacrylamide gel and transferred onto nitrocellulose membrane. After blocking, membranes were probed with primary antibodies. Rabbit α-A3AR (1:1,000, RRID:AB_2039711), and mouse α-β-Actin (1:10,000, RRID:AB_476743).
Membranes were washed in TBST (20 mM Tris, 137 mM NaCl, 0.05% Tween 20) and incubated for 1 hr at room temperature in fluorophore-conjugated secondary antibodies (α-rabbit and α-mouse conjugated IR Dyes 1:5,000, Mandel Scientific, Guelph, ON). Membranes were imaged using the LICOR Odyssey Clx Infrared Imaging System (Mandel Scientific). Band intensities were quantified using Image J from original full-length images. Band intensities were first normalized to β-actin as a loading control and expressed relative to the average of the control samples (Sal or Mor + Veh).
2.11 |. Immunohistochemistry
Rats were anesthetized with pentobarbital (Bimeda-MTC Animal Health Inc.) and perfused transcardially with phosphate-buffered saline (PBS) followed by ice-cold 4% paraformaldehyde (PFA, Sigma, P6148). The lumbar spinal cord (L3–L5) was removed, postfixed overnight at 4°C, and placed in 30% sucrose solution at 4°C. Spinal cord was sectioned using a microtome (Leica, SM2010R) at a thickness of 40 μm. Free-floating spinal cord sections were incubated in blocking solution (3% normal donkey serum) for 2 hr at room temperature and then incubated 48 hr at 4°C with primary antibodies: rabbit anti-A3AR (1:100, RRID:AB_10751536) mouse anti-GFAP (1:200, RRID:AB_449329), mouse anti-NeuN (1:500, RRID:AB_10711040), and goat anti-Iba1 (1:500, RRID:AB_521594). Sections were then incubated with flurochrome-conjugated secondary antibodies: donkey anti-rabbit Alexa 488 (1:1,000, Invitrogen, A-21206, RRID:AB_2535792), donkey anti-mouse Alexa 568 (1:1,000, abcam, ab175472, RRID:AB_2636996), and donkey anti-goat Alexa 647 (1:1,000, Invitrogen, A-21447, RRID:AB_141844) for 3 hr at room temperature. Images were obtained using a Leica SP8 confocal microscope (Table 1).
TABLE 1.
Primary antibodies
| RRID | Company | Cat no. | Host | Reactivity | Target | |
|---|---|---|---|---|---|---|
| A3AR(IHC) | AB_10751536 | Biorbyt, St. Louis, MO, USA | orbl0053 | Rabbit | Human, Mouse, Rat | KLH conjugated synthetic peptide derived between 95 andl70 amino acids of human Adenosine Receptor A3 |
| a3ar (WB) | AB_2039711 | Alomone Labs Ltd, Jerusalem, Israel | AAR-004 | Rabbit | Rat, Human | Polyclonal antibody–Peptide (C) KETGAFYGREFKTAK, corresponding to amino acid residues 216–230 of human A3AR |
| β-Actin (WB) | AB_476743 | Sigma-Aldrich, St. Louis, MO, USA | A5316 | Mouse | Human, Mouse, Rat and others | Monoclonal antibody recognizes an epitope located on the N-terminal end of the β-isoform of actin, peptide Ac-DDDIAALVINGSGK |
| GFAP(IHC) | AB_449329 | Abeam, Cambridge, UK | ab4648 | Mouse | Human, Mouse, Rat, Pig | Monoclonal [2A5] to full-length native protein (purified) corresponding to Pig GFAP |
| Ibal(IHC) | AB_521594 | Novus Biological, Centennial, CO, USA | NB100–1028 | Goat | Human, Mouse, Rat and others | Polyclonal antibody recognizes peptide C-TGPPAKKAISELP, from the C Terminus of the protein sequence |
| NeuN (IHC) | AB_10711040 | Abeam, Cambridge, UK | ab!04224 | Mouse | Human, Mouse, Rat | Monoclonal [1B7] recombinant fragment corresponding to Human NeuN aa 1–100 (N terminal) |
2.12 |. Data and statistical analysis
All data are presented as the mean ± SEM. Statistical analyses of results were performed with GraphPad Prism 6 software using unpaired t test (two-sided), or ordinary one-way ANOVA with post hoc Sidak’s test. Timecourse and daily antinociception experiments were analyzed using a two-way repeated measure ANOVA with post hoc Dunnett, Tukey or Sidak’s test. p < 0.05 was deemed significant. p values were automatically adjusted for multiple comparisons in Graphpad. No outliers were identified and no data were excluded from analysis. Further statistical details are reported in figure legends.
3 |. RESULTS
3.1 |. Spinal A3AR activation does not alter acute morphine antinociception in naïve animals
To investigate the importance of spinal A3AR in morphine antinociception, rats were treated with an intrathecal injection of selective A3AR agonist, MRS5698 (Figure 1a). First, we examined whether intrathecal MRS5698 affects acute morphine antinociception. In these experiments, a submaximal dose of morphine (7.5 mg/kg, i.p.) produced an antinociceptive response in the thermal tail-flick test that peaked at 30 min post-injection and declined over the course of 120 min (Figure 1b). Neither the magnitude nor timecourse of acute morphine-induced antinociception was affected by an intrathecal injection of MRS5698 (3 nmol and 50 nmol) (Figure 1b). In morphine-naïve (saline-treated) rats, intrathecal MRS5698 had no impact on baseline thermal nociceptive responses, indicating that MRS5698 alone is not antinociceptive (Figure 1b).
FIGURE 1.
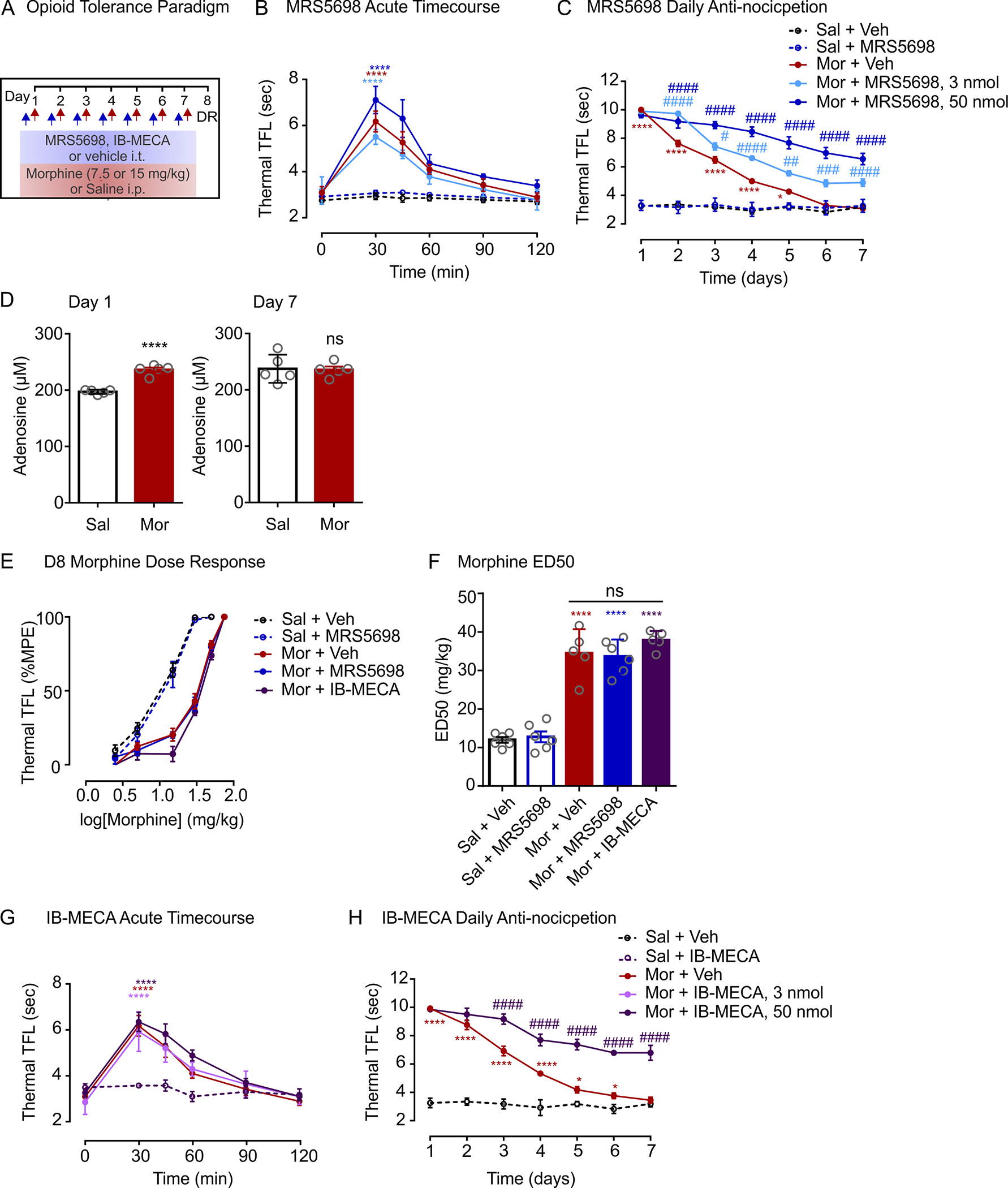
Spinal A3AR activation potentiates morphine antinociception. (a) Schematic of drug administration paradigm. (b) Timecourse of antinociceptive response to an acute injection of morphine (7.5 mg/kg) or saline. Sal + Veh n = 6, Sal + MRS5698 n = 6, Mor + Veh n = 6, Mor + MRS5698 3 nmol n = 6, Mor + MRS5698 n = 5. Two-way ANOVA, Tukey’s post hoc test (Interaction F(20,120) = 11.94, p < 0.0001; Time F(5,120) = 81.35, p < 0.0001; Treatment F(4,24) = 19.55, p < 0.0001). *Compared to Sal + MRS5698 at 30 min (Mor + MRS5698 50 nmol p < 0.0001, Mor + MRS5698 3 nmol p < 0.0001, Mor + Veh p < 0.0001). Compared to Mor + Sal at 30 min (Mor + MRS5698 3 nmol p = 0.345, Mor + MRS5698 50 nmol p = 0.1038). (c) Daily morphine (15 mg/kg, i.p.) antinociception. Sal + Veh n = 6, Sal + MRS5698 n = 6, Mor + Veh n = 10, Mor + MRS5698 3 nmol n = 6, Mor + MRS5698 n = 10. Two-way ANOVA, Dunnett’s post hoc test (Interaction F(24,198) = 22.87, p < 0.0001; Time F(6,198) = 113.5, p < 0.0001; Treatment F(4,33) = 278.1, p < 0.0001). *Represents Mor + Veh compared to Sal + Veh (D1–4 p < 0.0001, D5 p = 0.0127). #Represents Mor + MRS5698 50 nmol (D2–7 p < 0.0001) and Mor + MRS5698 3 nmol (D3 p = 0.0370, D4 p < 0.0001, D5 p = 0.0021, D6 p < 0.0001, D7 p < 0.0001) compared to Sal + MRS5698. (d) ATP (μM) levels in CSF 30 min after saline or morphine injection on day 1 (left panel; Unpaired two-tailed t test, n = 5 p < 0.0001, t = 3.937, df = 8) or day 7 (right panel; Unpaired two-tailed t test, n = 5 p = 0.8976, t = 0.1329, df = 8). NS, not significant. (e) Morphine dose-response curves and (f) median effective dose (ED50) calculated on day 8. No pre-treatment with MRS5698 or IB-MECA. Sal + Veh n = 6, Sal + MRS5698 n = 6, Mor + Veh n = 5, Mor + MRS5698 50 nmol n = 6, Mor + IB-MECA 50 nmol n = 5. One-way ANOVA, Sidak’s post hoc test F(4,23) = 60.69, p < 0.0001 *Comparison to saline controls (Mor + Veh p < 0.0001, Mor + MRS5698 p < 0.0001, Mor + IB-MECA p < 0.0001). (g) Timecourse of antinociceptive response to an acute injection of morphine or saline. For all groups n = 6. Two-way ANOVA, Dunnett’s post hoc test (Interaction F(15,100) = 7.595, p < 0.0001; Time F(5,020) = 80.37, p < 0.0001; Treatment F(3,20) = 7.421, p = 0.0016). *Compared to Sal + IB-MECA at 30 min (Mor + IB-MECA 50 nmol p < 0.0001, Mor + IB-MECA 3 nmol p < 0.0001, Mor + Veh p < 0.0001). (h) Daily morphine antinociception. Sal + IB-MECA n = 6, Mor + Veh n = 6, Mor + IB-MECA 50 nmol n = 5. Two-w ay ANOVA, Tukey’s post hoc test (Interaction F(12,84) = 29.78, p < 0.0001; Time F(6,84) = 96.08, p < 0.0001; Treatment F(2,14) = 392.3, p < 0.0001). *Represents Mor + Veh compared to Sal + IB-MECA (D1–4 p < 0.0001, D5 p = 0.0121, D6 p = 0.0208, D7 p = 0.7373). #Represents Mor + IB-MECA 50 nmol (D2–7 p < 0.0001) compared to Mor + Veh. All data are presented as mean ± SEM
3.2 |. Spinal A3AR activation potentiates morphine antinociception in tolerant animals without preventing the loss of morphine potency
To establish morphine antinociceptive tolerance, rats were treated with morphine (15 mg/kg, i.p.) once daily for 7 consecutive days (Figure 1a). Thermal tail-flick latency was assessed before and 30 min after morphine injection. On day 1, morphine treatment induced a significant increase in tail-flick latency (Figure 1c) and elevated cerebrospinal fluid (CSF) levels of adenosine as compared with saline-treated control rats (Figure 1d). The antinociceptive effect decreased with repeated morphine exposure, such that on days 6 and 7 an injection of morphine had no significant impact on thermal nociceptive threshold (Figure 1c) and CSF adenosine levels 30 min after morphine injection were no different from saline-treated rats (Figure 1d). Furthermore, thermal tail-flick latency following morphine injection in these tolerant rats was indistinguishable from saline-treated control rats (Figure 1c). By contrast, when intrathecal MRS5698 (3 or 50 nmol) was co-administered daily with morphine, antinociception persisted to day 7 (Figure 1c).
Repeated morphine exposure results in a significant reduction in antinociceptive potency, which is a hallmark of morphine tolerance. To assess morphine potency, we performed a cumulative dose-response in which escalating doses of systemic morphine were administered every 30 min until a maximal antinociceptive effect was reached in the thermal tail-flick test (Figure 1e,f). Determination of potency occurred on day 8, 24 hr after the last injection of morphine and intrathecal MRS5698 or vehicle. We found that rats treated with morphine for 7 days required higher doses of morphine to achieve a maximal antinociceptive response than morphine-naïve (saline-treated) rats. This reduction in antinociceptive potency was reflected by a rightward shift in the morphine dose-response curve (Figure 1e) and a significant increase in the morphine ED50 (Figure 1f). Likewise, rats treated with morphine and MRS5698 over 7 days required higher morphine doses, as demonstrated by a similar shift in morphine dose-response and increased ED50. We confirmed these results using another A3AR agonist IB-MECA (3 or 50 nmol), which attenuated the progressive decline in antinociception resulting from daily morphine treatment (Figure 1h), but also did not prevent the increase in morphine ED50 (Figure 1e,f) or affect acute morphine response (Figure 1g). Thus, daily co-administration of MRS5698 or IB-MECA with morphine did not alter the potency or efficacy of morphine in rats that were opioid tolerant.
In determining morphine potency, rats in the above experiments were not given an intrathecal injection of MRS5698 or IB-MECA immediately prior to escalating doses of morphine on day 8. It is possible that the actions of these A3AR selective agonists may be acute, and therefore, transiently augment morphine-induced antinociception in tolerant rats. We tested this possibility in dose-response experiments wherein MRS5698 (50 nmol) was administered with the first escalating dose of systemic morphine (Figure 2a). MRS5698 was used in subsequent experiments because of its greater A3AR selectivity: MRS5698 is >10,000-fold more selective for A3AR than A1AR or A2AAR subtypes (Tosh et al., 2012), while IB-MECA has 50-fold greater selectivity for A3AR compared to A1AR or A2AAR subtypes (Gallo-Rodriguez et al., 1994; Jacobson et al., 1993). In morphine-tolerant rats, we found that an intrathecal injection of MRS5698 on day 8 reduced the rightward shift in morphine dose-response (Figure 2b) and attenuated the increase in morphine ED50 (Figure 2c). By contrast, morphine-tolerant rats that received intrathecal vehicle injection prior to dose-response experiments displayed a blunted antinociceptive response to escalating doses of morphine. The response to morphine was independent of whether rats had received prior 7 days of MRS5698 or vehicle. In morphine-naïve (saline-treated) rats, intrathecal MRS5698 had no effect on morphine antinociceptive potency. Taken together, our findings indicate that activating spinal A3AR acutely potentiates morphine antinociception in tolerant animals.
FIGURE 2.
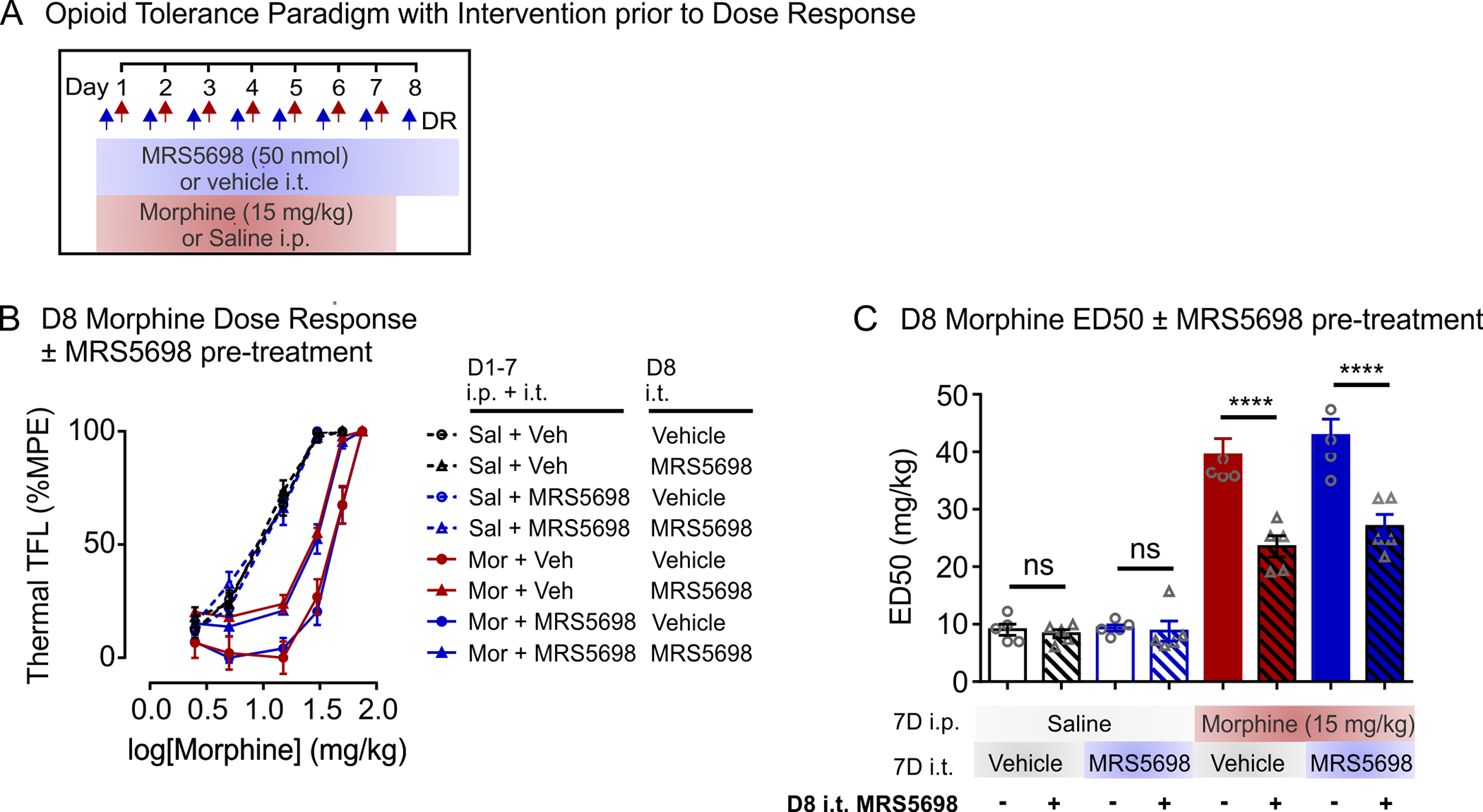
Spinal A3AR activation potentiates morphine antinociception in tolerant animals. (a) Schematic of drug administration paradigm. (b,c) Morphine dose-response was performed on day 8. MRS5698 (50 nmol; i.t) or vehicle (i.t) was administered with the first morphine injection. (b) Morphine dose-response curves and (c) median effective dose (ED50). Data are presented as mean ± SEM. N = 5 for all groups. One-way ANOVA, Sidak’s post hoc test F(7,32) = 58.11, p < 0.0001 *Comparisons as indicated ****p < 0.0001, NS, not significant
3.3 |. Spinal A3AR activation partially restores morphine antinociception in tolerant animals
We next examined whether spinal A3AR activation restores morphine antinociception in rats with established tolerance (Figure 3). Rats were rendered morphine tolerant with 7 days of morphine treatment. On days 8–12, rats received a systemic injection of morphine (15 mg/kg) with intrathecal MRS5698 (3 or 50 nmol) or vehicle (Figure 3a). In morphine tolerant rats, the combination of morphine and MRS5698 produced a dose-dependent increase in tail-flick latency; this antinociceptive effect was comparable across days 8–12 (Figure 3b). By contrast, rats treated with morphine and intrathecal vehicle continued to display a blunted morphine response (Figure 3b). Intrathecal MRS5698 or vehicle injection had no effect on nociceptive threshold in morphine-naïve (saline-treated) rats (Figure 3c).
FIGURE 3.
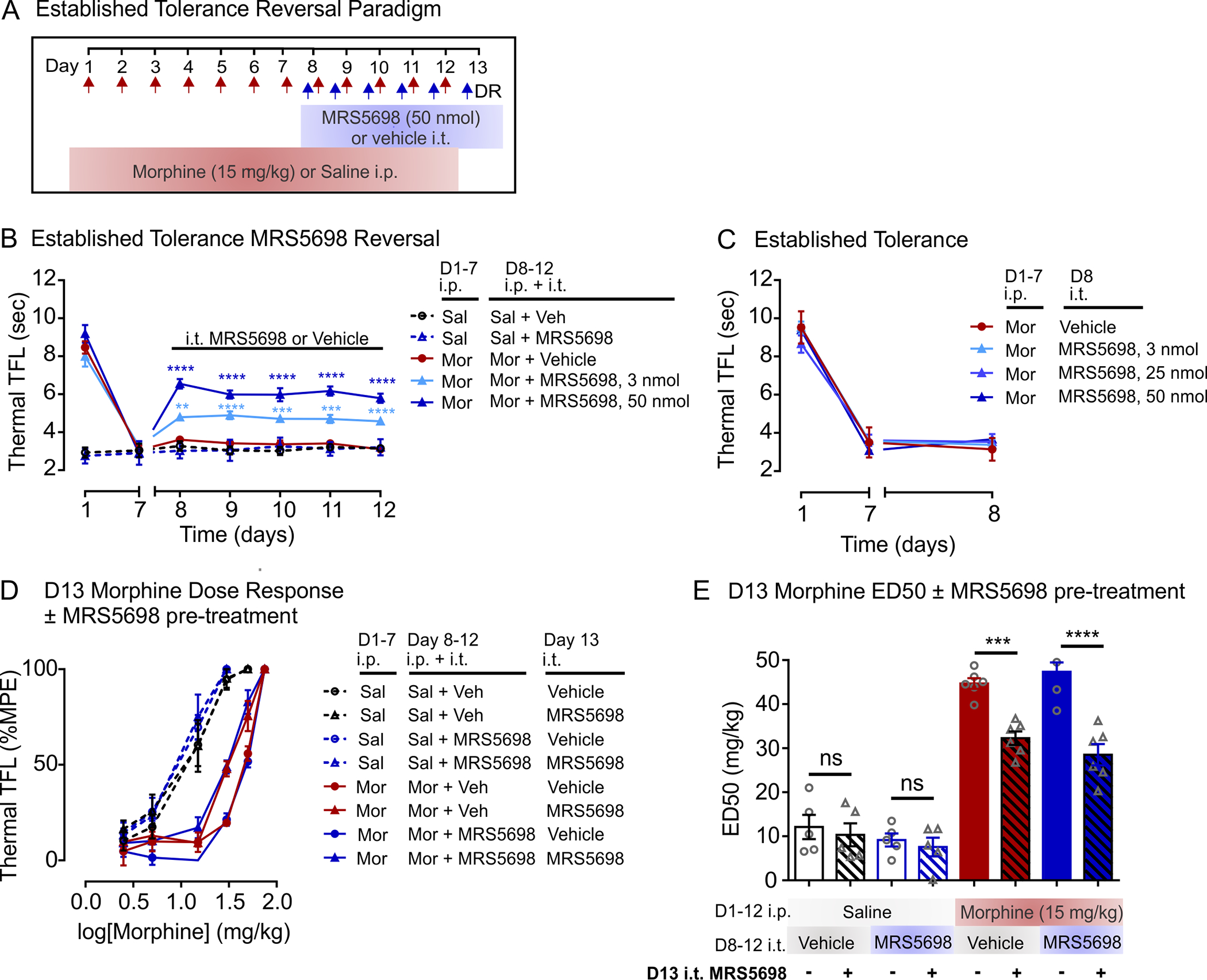
Spinal A3AR activation partially restores morphine antinociception in morphine-tolerant rats. (a) Schematic of drug administration paradigm. (b,c) Effect of intrathecal injection of MRS5698 in rats with established morphine tolerance. (b) Co-treatment of intrathecal MRS5698 or vehicle injection with morphine (15 mg/kg; i.p) or saline (i.p) on days 8–12. Sal + Veh n = 6, Sal + MRS5698 n = 6, Mor + Veh n = 8, Mor + MRS5698 3 nmol n = 7, Mor + MRS5698 50 nmol n = 7. Two-way ANOVA, Dunnett’s post hoc test (Interaction F(24,174) = 23.34, p < 0.0001; Time F(6,174) = 97.94, p < 0.0001; Treatment F(4,29) = 90.15, p < 0.0001). *Significance compared to Sal + MRS5698 (Mor + MRS5698 50 nmol (D8–12 p < 0.0001) and Mor + MRS5698 3 nmol (D8 p = 0.0014, D9 p < 0.0001, D10 p = 0.0002, D11 p = 0.0005, D12 p < 0.0001). (c) Day 8 intrathecal MRS5698 injection in the absence of morphine. Mor + Veh n = 5, Mor + MRS5698 3 nmol n = 6, Mor + MRS5698 25 nmol n = 5, Mor + MRS5698 50 nmol n = 6. Two-way ANOVA, Dunnett’s post hoc test (Interaction F(6,36) = 1.404, p = 0.2398; Time F(2,36) = 503.9, p < 0.0001; Treatment F(3,18) = 0.2733, p = 0.8438). No significance at each timepoint. (d) Morphine dose-response curves and (e) median effective dose (ED50) calculated on day 13. Intrathecal MRS5698 (50 nmol) or vehicle was administered with the first morphine injection. D1–12 ip Saline n = 5 for all groups, D1–12 ip Morphine n = 6 for all groups. One-way ANOVA, Sidak’s post hoc test F(7,36) = 63.13, p < 0.0001. *Comparisons as indicated (Mor + Vehicle + D13 MRS5698 i.t. vs. Mor + Vehicle + D13 Vehicle i.t. p = 0.0003, Mor + MRS5698 + D13 MRS5698 i.t. vs. Mor + MRS5698 + D13 Vehicle i.t. p < 0.0001). NS, not significant. All data are presented as mean ± SEM
Morphine dose-response experiments were performed on day 13 (Figure 3d,e). Consistent with the above results (Figure 2), morphine tolerant rats given intrathecal MRS5698 with the first escalating dose of morphine displayed a greater antinociceptive response than rats receiving intrathecal vehicle (Figure 3d,e). Prior MRS5698 treatment on days 8–12 had no impact on the morphine dose-response, and MRS5698 administered in the absence of morphine did not affect thermal nociceptive threshold (Figure 3b) or alter morphine dose-response (Figure 3d,e).
Next, we asked whether repeated morphine exposure or MRS5698 treatment affects spinal A3AR protein levels. Western blot analysis of L3-L5 spinal cord homogenates revealed that total A3AR expression is comparable in rats treated with saline or morphine for 7 days (Figures 4a and S1). Spinal A3AR expression was also not affected by co-administration of MRS5698 with morphine on days 1–7 or 8–12 (Figure 4b,c).
FIGURE 4.
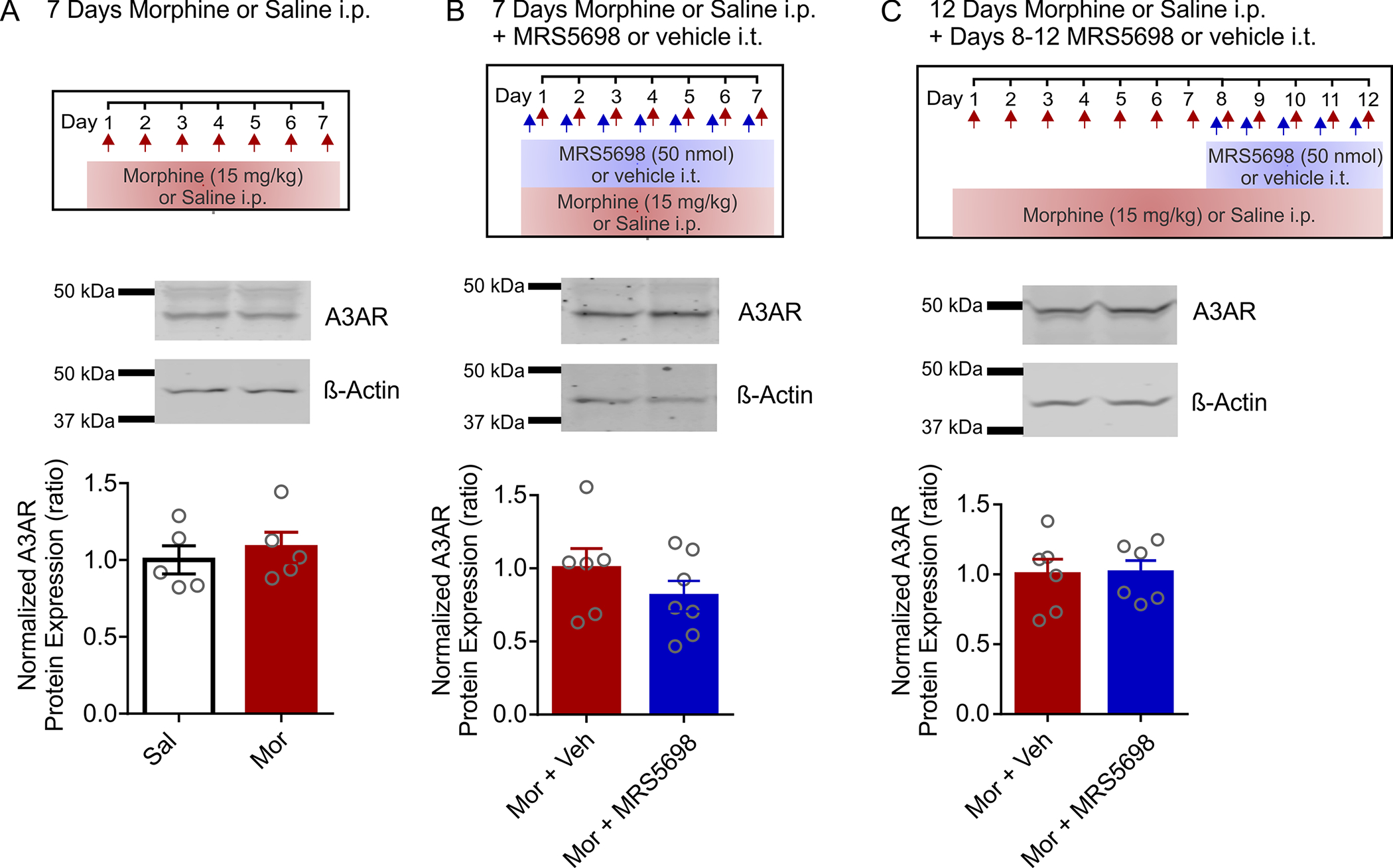
Spinal A3AR protein expression is not affected by morphine or MRS5698. Western blot analysis of A3AR protein expression in rat spinal cord homogenates. Rat spinal cord was collected and processed for western blotting on D7 or D12 of treatment. Band intensity has been normalized to β-Actin as a loading control and expressed relative to Sal (a) or Mor + Veh (b,c) controls. All quantification was done on full-length unmodified images (Figure S1). (a) Day 7 saline versus morphine treatment Unpaired two-tailed t test; n = 5, p = 0.5650, t = 0.6002, df = 8; (b) Day 7 morphine and intrathecal vehicle versus MRS5698 Unpaired two-tailed t test; Mor + Veh n = 6, Mor + MRS5698 n = 7, p = 0.2814, t = 1.133, df = 11; (c) Day 12 morphine and intervention on days 8–12 with intrathecal vehicle versus MRS5698 Unpaired two-tailed t test, n = 6, p = 0.9224, t = 0.09987, df = 10
3.4 |. MRS5698 transiently potentiates morphine antinociception in morphine-tolerant animals
Our results indicate that MRS5698 acutely restores morphine antinociceptive response in opioid tolerant rats. To further test this, we used an “on/off/on” MRS5698 treatment paradigm (Figure 5a). In these experiments, rats were exposed to systemic morphine over 7 days to induce antinociceptive tolerance. On days 8 and 9, morphine-tolerant rats receiving intrathecal MRS5698 (“on”) together with systemic morphine displayed a partial antinociceptive response (Figure 5b). This response to morphine was absent when intrathecal vehicle (“off”) was administered on days 10 and 11, whereas MRS5698 injection (“on”) on days 12 and 13 reinstated the morphine antinociceptive response. The actions of MRS5698 were blocked by intrathecal injection of A3AR antagonist MRS1523, indicating acute MRS5698 potentiation of morphine antinociception is A3AR dependent.
FIGURE 5.
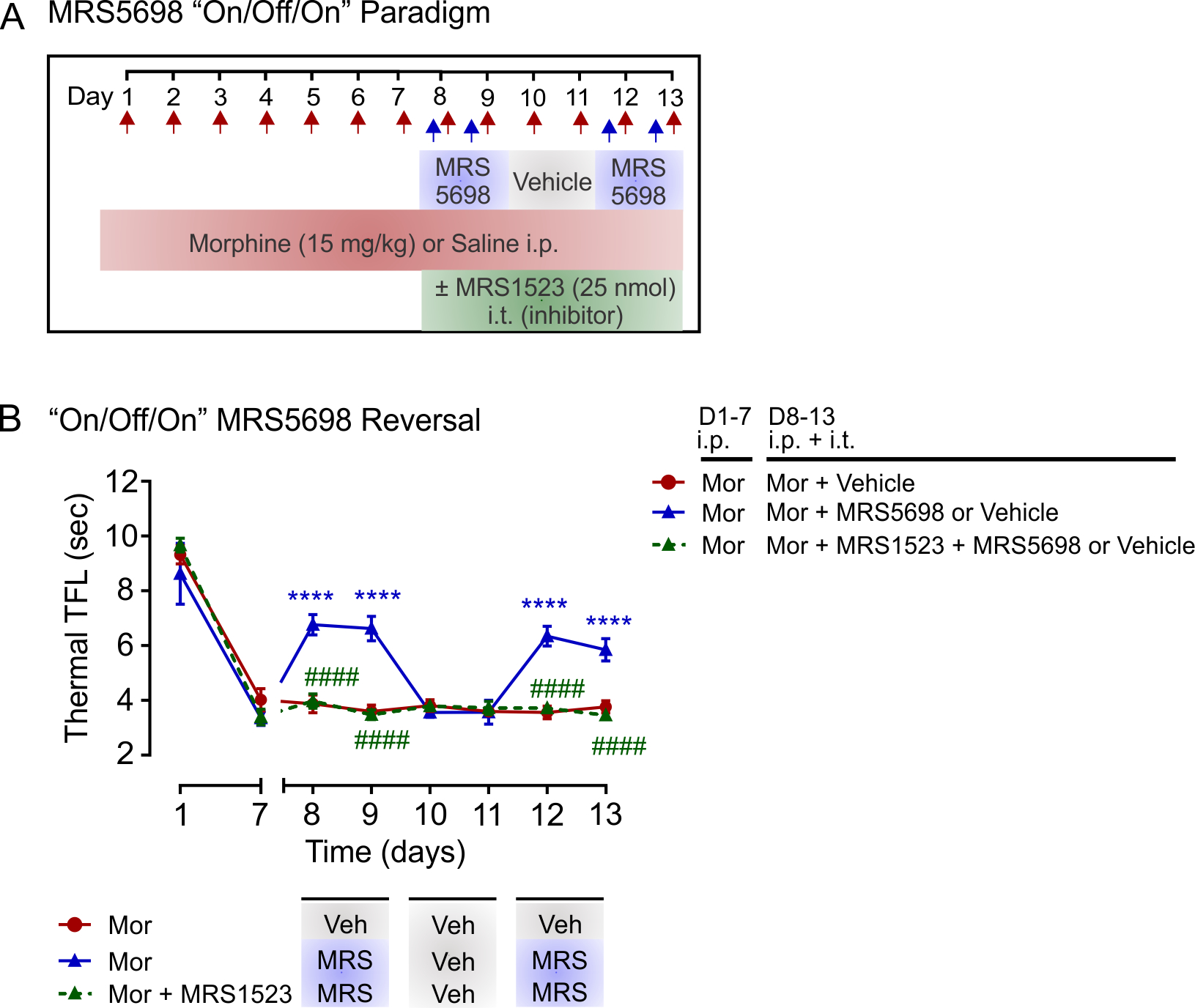
MRS5698 potentiates morphine antinociception in morphine-tolerant rats. (a) Schematic of drug administration paradigm. (b) Effect of intrathecal MRS5698 on morphine (15 mg/kg; i.p) antinociception in rats with established morphine tolerance. On days 8–9 and 12–13, intrathecal MRS5698 (50 nmol) was administered with morphine. On days 10–11, intrathecal vehicle was administered with morphine. Effects of MRS5698 were blocked by intrathecal treatment with the A3AR antagonist MRS1523 (25 nmol). N = 6 for all groups. Two-way ANOVA, Sidak’s post hoc test (Interaction F(14,105) = 19.60, p < 0.0001; Time F(7,105) = 203.9, p < 0.0001; Treatment F(2,15) = 36.29, p < 0.0001). *Significance compared to Mor + Vehicle control (D8,9,12,13 p < 0.0001). #Significance compared to Mor + MRS5698/vehicle (D8,9,12,13 p < 0.0001). All data are presented as mean ± SEM
4 |. DISCUSSION
Opioids produce potent analgesia, but their pain-relieving effects diminish with repeated use. Here, we show that treatment with A3AR agonists, IB-MECA, or MRS5698, transiently restores antinociception in morphine-tolerant rats. These compounds possess high A3AR selectivity (Jacobson et al., 1993; Tosh et al., 2012) and demonstrate good safety and efficacy profiles in phase II/III clinical trials for cancer and inflammatory conditions (Fishman et al., 2012; Silverman et al., 2008; Stemmer et al., 2013). Our findings suggest that A3AR-targeted approaches may circumvent the need for dose escalation in opioid tolerance, possibly decreasing the risk of adverse effects associated with higher opioid doses.
A key observation in this study is that intrathecal MRS5698 and IB-MECA treatment attenuated the progressive decline in antinociception when administered daily with morphine and transiently restored antinociceptive responses when given to rats with established morphine tolerance. Rats treated with morphine and an A3AR selective agonist for 7 days required greater amounts of morphine than naïve rats to achieve maximal antinociception. The requirement for higher doses was comparable to rats exposed to morphine only, indicating that neither daily MRS5698 nor IB-MECA treatment prevented the reduction in morphine potency. It is important to note that these dose-response experiments were performed on day 8 by exposing rats to progressively higher doses of morphine in the absence of MRS5698 and IB-MECA. We therefore suspected that selective A3AR agonist treatment may be exerting an acute effect on morphine antinociception in the tolerant state. Along these lines, when MRS5698 was administered with morphine in dose-response experiments, it attenuated the increase in ED50. By using an intrathecal MRS5698 “on/off/on” dosing paradigm, we confirmed in tolerant rats that morphine response was partially and transiently restored in the presence of MRS5698, an effect blocked by the A3AR antagonist MRS1523. We ruled out a direct A3AR-mediated antinociceptive effect because neither compound administered in the absence of morphine affected thermal nociceptive threshold, consistent with previous reports that MRS5698 and IB-MECA are not antinociceptive per se (Chen et al., 2012; Little et al., 2015).
Another important observation is that intrathecal MRS5698 and IB-MECA differentially potentiated morphine antinociception in tolerant rats: there was no impact on morphine response when A3AR agonists were administered to naïve rats. Similarly, spinal delivery of MRS5698 or IB-MECA suppresses opioid-induced hyperalgesia and alleviates withdrawal without altering acute morphine antinociception (Doyle et al., 2020). This state-dependent effect has also been reported in neuropathic rats wherein A3AR agonist treatment augmented morphine antinociceptive efficacy and potency (Doyle et al., 2020), as well as prevented or reversed mechanical allodynia in the absence of opioids (Chen et al., 2012; Little et al., 2015). These findings are consistent with the notion that opioid tolerance and neuropathic pain share a common adenosine-mediated mechanism (Joseph et al., 2010; Mayer et al., 1999). Indeed, Sandner-Kiesling et al. (2001) reported that impaired opioid-induced adenosine release in the spinal cord may account for diminished opioid efficacy and potency in the treatment of neuropathic pain (Sandner-Kiesling et al., 2001). By contrast, interventions that increase spinal adenosine levels by inhibiting adenosine kinase, or that supplement A3AR adenosine signaling, augment morphine antinociceptive responses in neuropathic animals (Doyle et al., 2020). These interventions also prevent or reverse neuropathic pain in the absence of opioid treatment (Chen et al., 2012; Little et al., 2015).
In rodents, μ-receptor activation critically contributes to morphine-induced spinal adenosine release and this release is modulated by interactions between μ-and δ-receptors (Cahill et al., 1995; Cahill et al., 1996; Sweeney et al., 1989). Neither δ- nor κ-receptor activation alone is sufficient to cause adenosine release from spinal cord synaptosomes (Cahill et al., 1995, 1996). In humans, adenosine release has also been reported following intrathecal administration of morphine and fentanyl, potent μ-receptor agonists (Eisenach et al., 2004). While acute morphine exposure induces adenosine release (Cahill et al., 1993), repeated exposure pathologically alters spinal pain processing and opioid signaling (Ferrini et al., 2013; Gintzler & Chakrabarti, 2006; Rivat & Ballantyne, 2016). In opioid tolerant rats, we found that morphine-induced CSF adenosine release and antinociceptive responses were blunted. However, total CSF adenosine levels were comparable with saline control rats, suggesting that it is morphine-induced adenosine release rather than a change in basal adenosine levels that is affected in morphine-tolerant rats. However, previous studies have reported that chronic opioid administration decreases extracellular levels of adenosine in the brain (Nelson et al., 2009; Wu et al., 2013).
Although we found that total spinal A3AR protein levels were not affected by repeated morphine or MRS5698 treatment, it is possible that cell-specific changes in A3AR expression, localization, and/or Gi-coupled signaling may contribute to the A3AR agonist response in morphine-tolerant rats. A3AR is expressed on endothelial cells, inflammatory cells, neurons, and glia (Abbracchio et al., 1997; Jacobson, 1998; Lopes et al., 2003). Within the rat spinal dorsal horn, A3AR may be found on neurons and astrocytes with lower levels detected in microglia (Figure S2), whereas higher A3AR expression has been reported in brain microglia (Butovsky et al., 2014). However, single-cell RNA sequencing failed to detect A3AR in spinal sensory neurons (Häring et al., 2018). A3AR agonists are known to suppress microglia reactivity (Hammarberg et al., 2003; Lee et al., 2006), reduce neuronal excitability and dampen astrocyte activity (Brand et al., 2001; Janes et al., 2015; Wahlman et al., 2018; Wittendorp et al., 2004). Interactions among these various cell types within the spinal circuitry underlies the development of opioid tolerance. Thus, future studies directed at teasing apart the cell-specific effects of A3AR will provide a more detail mechanistic understanding.
Another limitation of this study is that experiments were performed only in male rats. However, our group recently determined that chronic morphine impairs spinal A3AR signaling in male and female rats. In both sexes, inhibiting spinal adenosine kinase or activating A3AR preserved morphine antinociception, attenuated opioid-induced hyperalgesia, and reduced withdrawal (Doyle et al., 2020). Thus, A3AR activity critically modulates response to repeated opioid treatment and it is a convergent mechanism in both males and females.
In conclusion, our findings show that A3AR activation transiently restores morphine antinociception in opioid tolerant rats. These results provide a mechanistic twist—the presence of MRS5698 is required to preserve morphine antinociception. This information could optimize A3AR-targeted therapies and address the need for dose escalation in opioid tolerant individuals, decreasing the risk of adverse effects associated with higher opioid doses. Another notable advantage of targeting A3AR is that agonists of this receptor do not engage reward centers, which means their addiction liability may be low, and A3AR-mediated antinociceptive effects do not diminish over time (Little et al., 2015). These are important therapeutic features when considering A3AR agonists as adjuncts for restoring or enhancing opioid pain relief in opioid tolerant individuals.
Supplementary Material
Full-length Western blots featured in Figure 4
Immuno-labeling of A3AR in rat spinal cord. Naïve rat spinal cord sections immuno-labeled for A3AR and cell type markers. (a) A3AR expression in the spinal dorsal horn. Scale bar = 500 μm. (b) A3AR co-labeling with GFAP (astrocytes), NeuN (neurons), and Iba1 (microglia). Arrows indicate co-labeling. Scale bar = 50 μm
Significance.
Tolerance to the pain-relieving effects of opioids leaves patients struggling to manage their pain. This limits the effectiveness of opioid pain therapy and can lead to dose escalation, which increases the risk of adverse effects. Here, we show that activating the adenosine A3 receptor (A3AR) acutely restores morphine antinociception in opioid tolerant rats. The findings have implications for developing A3AR-targeted therapies as adjuvants to restore or augment opioid response in tolerant individuals.
ACKNOWLEDGMENTS
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN06289-2019), the Canadian Institutes of Health Research (CIHR PJ8-169697), NIDDK Intramural Research (ZIADK31117), and the Vi Riddell Program for Pediatric Pain. C.A. holds a Canada Research Chair Tier 2 in inflammatory pain. H.L.P. is supported by an Alberta Innovates (AI) Health Solutions Graduate Scholarship, C.X. is funded by an NSERC Canada Graduate Scholarship, R.D. by a CIHR Canada Graduate Scholarship, C.F. is funded by an Eyes High University of Calgary Scholarship, and N.N.B. by an AI Postgraduate Fellowship. We would like to thank Livui Luongo for advising on immunohistochemistry experiments.
Funding information
National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Number: ZIADK31117; Canada Research Chairs; Canadian Institutes for Health Research, Grant/Award Number: CIHR PJ8-169697; Natural Sciences and Engineering Research Council of Canada, Grant/Award Number: NSERC RGPIN06289-2019
Footnotes
COMPETING INTERESTS
Dr. Salvemini is founder of BioIntervene, Inc., a company developing A3AR agonists for clinical use. Dr. Trang is co-founder of AphioTx Inc., which is developing pannexin-1channel-targeted therapies. All other authors have declared that no conflict of interest exists.
DECLARATION OF TRANSPARENCY
The authors, reviewers and editors affirm that in accordance to the policies set by the Journal of Neuroscience Research, this manuscript presents an accurate and transparent account of the study being reported and that all critical details describing the methods and results are present.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
Transparent Peer Review Report
Transparent Science Questionnaire for Authors
DATA AVAILABILITY STATEMENT
Data sets and materials generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- Abbracchio MP, Rainaldi G, Giammarioli AM, Ceruti S, Brambilla R, Cattabeni F, Barbieri D, Franceschi C, Jacobson KA, & Malorni W (1997). The A3 adenosine receptor mediates cell spreading, reorganization of actin cytoskeleton, and distribution of Bcl-XL: Studies in human astroglioma cells. Biochemical and Biophysical Research Communications, 241(2), 297–304. 10.1006/bbrc.1997.7705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu KM, Brent J, & Juurlink DN (2019). Prevention of opioid overdose. New England Journal of Medicine, 380(23), 2246–2255. 10.1056/NEJMra1807054 [DOI] [PubMed] [Google Scholar]
- Boison D (2013). Role of adenosine in status epilepticus: A potential new target? Epilepsia, 54(s6), 20–22. 10.1111/epi.12268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Chen J-F, & Fredholm BB (2010). Adenosine signalling and function in glial cells. Cell Death and Differentiation, 17(7), 1071–1082. 10.1038/cdd.2009.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A, Vissiennon Z, Eschke D, & Nieber K (2001). Adenosine A(1) and A(3) receptors mediate inhibition of synaptic transmission in rat cortical neurons. Neuropharmacology, 40(1), 85–95. 10.1016/s0028-3908(00)00117-9 [DOI] [PubMed] [Google Scholar]
- Burma NE, Leduc-Pessah H, & Trang T (2017). Genetic deletion of microglial Panx1 attenuates morphine withdrawal, but not analgesic tolerance or hyperalgesia in mice. Channels, 11(5), 487–494. 10.1080/19336950.2017.1359361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, & Weiner HL (2014). Identification of a unique TGF-β dependent molecular and functional signature in microglia. Nature Neuroscience, 17(1), 131–143. 10.1038/nn.3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, White TD, & Sawynok J (1993). Morphine activates ω-conotoxin-sensitive Ca2+ channels to release adenosine from spinal cord synaptosomes. Journal of Neurochemistry, 60(3), 894–901. 10.1111/j.1471-4159.1993.tb03234.x [DOI] [PubMed] [Google Scholar]
- Cahill CM, White TD, & Sawynok J (1995). Spinal opioid receptors and adenosine release: Neurochemical and behavioral characterization of opioid subtypes. Journal of Pharmacology and Experimental Therapeutics, 275(1), 84–93. [PubMed] [Google Scholar]
- Cahill CM, White TD, & Sawynok J (1996). Synergy between mu/delta-opioid receptors mediates adenosine release from spinal cord synaptosomes. European Journal of Pharmacology, 298(1), 45–49. 10.1016/0014-2999(95)00775-x [DOI] [PubMed] [Google Scholar]
- Chen Z, Janes K, Chen C, Doyle T, Bryant L, Tosh DK, Jacobson KA, & Salvemini D (2012). Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB Journal, 26(5), 1855–1865. 10.1096/fj.11-201541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Calle JL, & Paíno CL (2002). A procedure for direct lumbar puncture in rats. Brain Research Bulletin, 59(3), 245–250. 10.1016/S0361-9230(02)00866-3 [DOI] [PubMed] [Google Scholar]
- Doyle TM, Largent-Milnes TM, Chen Z, Staikopoulos V, Esposito E, Dalgarno R, Fan C, Tosh DK, Cuzzocrea S, Jacobson KA, Trang T, Hutchinson MR, Bennett GJ, Vanderah TW, & Salvemini D (2020). Chronic morphine-induced changes in signaling at the A3 adenosine receptor contribute to morphine-induced hyperalgesia, tolerance, and withdrawal. Journal of Pharmacology and Experimental Therapeutics, 374(2), 331–341. 10.1124/jpet.120.000004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenach JC, Hood DD, Curry R, Sawynok J, Yaksh TL, & Li X (2004). Intrathecal but not intravenous opioids release adenosine from the spinal cord. Journal of Pain, 5(1), 64–68. 10.1016/j.jpain.2003.10.001 [DOI] [PubMed] [Google Scholar]
- Ferrini F, Trang T, Mattioli T-A, Laffray S, Del’Guidice T, Lorenzo L-E, Castonguay A, Doyon N, Zhang W, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu J-M, Cahill CM, Salter MW, & De Koninck Y (2013). Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl− homeostasis. Nature Neuroscience, 16(2), 183–192. 10.1038/nn.3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman P, Bar-Yehuda S, Liang BT, & Jacobson KA (2012). Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discovery Today, 17(7), 359–366. 10.1016/j.drudis.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman P, Bar-Yehuda S, Madi L, & Cohn I (2002). A3 adenosine receptor as a target for cancer therapy. Anti-Cancer Drugs, 13(5), 437–443. 10.1097/00001813-200206000-00001 [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Chen J-F, Cunha RA, Svenningsson P, & Vaugeois J-M (2005). Adenosine and brain function. International Review of Neurobiology, 63, 191–270. 10.1016/S0074-7742(05)63007-3 [DOI] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Linden J, & Müller CE (2011). International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacological Reviews, 63(1), 1–34. 10.1124/pr.110.003285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo-Rodriguez C, Ji X, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu Q, Olah ME, van Galen PJM, Stiles GL, & Jacobson KA (1994). Structure-activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. Journal of Medicinal Chemistry, 37(5), 636–646. 10.1021/jm00031a014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gintzler AR, & Chakrabarti S (2006). Post-opioid receptor adaptations to chronic morphine; Altered functionality and associations of signaling molecules. Life Sciences, 79(8), 717–722. 10.1016/j.lfs.2006.02.016 [DOI] [PubMed] [Google Scholar]
- Hammarberg C, Schulte G, & Fredholm BB (2003). Evidence for functional adenosine A3 receptors in microglia cells. Journal of Neurochemistry, 86(4), 1051–1054. 10.1046/j.1471-4159.2003.01919.x [DOI] [PubMed] [Google Scholar]
- Häring M, Zeisel A, Hochgerner H, Rinwa P, Jakobsson JET, Lönnerberg P, La Manno G, Sharma N, Borgius L, Kiehn O, Lagerström MC, Linnarsson S, & Ernfors P (2018). Neuronal atlas of the dorsal horn defines its architecture and links sensory input to transcriptional cell types. Nature Neuroscience, 21(6), 869–880. 10.1038/s41593-018-0141-1 [DOI] [PubMed] [Google Scholar]
- Hayes CJ, Krebs EE, Hudson T, Brown J, Li C, & Martin BC (2020). Impact of opioid dose escalation on the development of substance use disorders, accidents, self-inflicted injuries, opioid overdoses and alcohol and non-opioid drug-related overdoses: A retrospective cohort study. Addiction, 115(6), 1098–1112. 10.1111/add.14940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA (1998). Adenosine A3 receptors: Novel ligands and paradoxical effects. Trends in Pharmacological Sciences, 19(5), 184–191. 10.1016/s0165-6147(98)01203-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Nikodijević O, Shi D, Gallo-Rodriguez C, Olah ME, Stiles GL, & Daly JW (1993). A role for central A3-adenosine receptors. Mediation of behavioral depressant effects. FEBS Letters, 336(1), 57–60. 10.1016/0014-5793(93)81608-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K, Esposito E, Doyle T, Cuzzocrea S, Tosh DK, Jacobson KA, & Salvemini D (2014). A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain, 155(12), 2560–2567. 10.1016/j.pain.2014.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K, Wahlman C, Little JW, Doyle T, Tosh DK, Jacobson KA, & Salvemini D (2015). Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain, Behavior, and Immunity, 44, 91–99. 10.1016/j.bbi.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Reichling DB, & Levine JD (2010). Shared mechanisms for opioid tolerance and a transition to chronic pain. Journal of Neuroscience, 30(13), 4660–4666. 10.1523/JNEUROSCI.5530-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan GB, & Sears MT (1996). Adenosine receptor agonists attenuate and adenosine receptor antagonists exacerbate opiate withdrawal signs. Psychopharmacology, 123(1), 64–70. 10.1007/BF02246282 [DOI] [PubMed] [Google Scholar]
- Kiesman WF, Elzein E, & Zablocki J (2009). A1 adenosine receptor antagonists, agonists, and allosteric enhancers. Handbook of Experimental Pharmacology, 193, 25–58. 10.1007/978-3-540-89615-9_2 [DOI] [PubMed] [Google Scholar]
- Leduc-Pessah H, Weilinger NL, Fan CY, Burma NE, Thompson RJ, & Trang T (2017). Site-specific regulation of P2X7 receptor function in microglia gates morphine analgesic tolerance. Journal of Neuroscience, 37(42), 10154–10172. 10.1523/JNEUROSCI.0852-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Jhun BS, Oh YT, Lee JH, Choe W, Baik HH, Ha J, Yoon K-S, Kim SS, & Kang I (2006). Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-α production through inhibition of PI 3-kinase/Akt and NF-κB activation in murine BV2 microglial cells. Neuroscience Letters, 396(1), 1–6. 10.1016/j.neulet.2005.11.004 [DOI] [PubMed] [Google Scholar]
- Little JW, Ford A, Symons-Liguori AM, Chen Z, Janes K, Doyle T, Xie J, Luongo L, Tosh DK, Maione S, Bannister K, Dickenson AH, Vanderah TW, Porreca F, Jacobson KA, & Salvemini D (2015). Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain, 138(Pt 1), 28–35. 10.1093/brain/awu330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes LV, Rebola N, Pinheiro PC, Richardson PJ, Oliveira CR, & Cunha RA (2003). Adenosine A3 receptors are located in neurons of the rat hippocampus. NeuroReport, 14(12), 1645–1648. 10.1097/00001756-200308260-00021 [DOI] [PubMed] [Google Scholar]
- Mayer DJ, Mao J, Holt J, & Price DD (1999). Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proceedings of the National Academy of Sciences of the United States of America, 96(14), 7731–7736. 10.1073/pnas.96.14.7731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercadante S, Arcuri E, & Santoni A (2019). Opioid-induced tolerance and hyperalgesia. CNS Drugs, 33(10), 943–955. 10.1007/s40263-019-00660-0 [DOI] [PubMed] [Google Scholar]
- Mousseau M, Burma NE, Lee KY, Leduc-Pessah H, Kwok CHT, Reid AR, O’Brien M, Sagalajev B, Stratton JA, Patrick N, Stemkowski PL, Biernaskie J, Zamponi GW, Salo P, McDougall JJ, Prescott SA, Matyas JR, & Trang T (2018). Microglial pannexin-1 channel activation is a spinal determinant of joint pain. Science Advances, 4(8), eaas9846. 10.1126/sciadv.aas9846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AM, Battersby AS, Baghdoyan HA, & Lydic R (2009). Opioid-induced decreases in rat brain adenosine levels are reversed by inhibiting adenosine deaminase. Anesthesiology, 111(6), 1327–1333. 10.1097/ALN.0b013e3181bdf894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg CC, He C, Stroink AR, Kattner KA, & Wang CX (2010). Technique for collection of cerebrospinal fluid from the cisterna magna in rat. Journal of Neuroscience Methods, 187(1), 8–12. 10.1016/j.jneumeth.2009.12.002 [DOI] [PubMed] [Google Scholar]
- Rivat C, & Ballantyne J (2016). The dark side of opioids in pain management: Basic science explains clinical observation. Pain Reports, 1(2), e570. 10.1097/PR9.0000000000000570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru F, Surdenikova L, Brozmanova M, & Kollarik M (2011). Adenosine-induced activation of esophageal nociceptors. American Journal of Physiology-Gastrointestinal and Liver Physiology, 300(3), G485–G493. 10.1152/ajpgi.00361.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, & Jacobson KA (2017). Highly selective A3 adenosine receptor agonists relieve chronic neuropathic pain. Expert Opinion on Therapeutic Patents, 27(8), 967. 10.1080/13543776.2017.1341018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandner-Kiesling A, Li X, & Eisenach JC (2001). Morphine-induced spinal release of adenosine is reduced in neuropathic rats. Anesthesiology, 95(6), 1455–1459. 10.1097/00000542-200112000-00026 [DOI] [PubMed] [Google Scholar]
- Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, … Fishman P (2008). Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase II clinical trial. Journal of Rheumatology, 35(1), 41–48. [PubMed] [Google Scholar]
- Stemmer SM, Benjaminov O, Medalia G, Ciuraru NB, Silverman MH, Bar-Yehuda S, Fishman S, Harpaz Z, Farbstein M, Cohen S, Patoka R, Singer B, Kerns WD, & Fishman P (2013). CF102 for the treatment of hepatocellular carcinoma: A phase I/II, open-label, dose-escalation study. Oncologist, 18(1), 25–26. 10.1634/theoncologist.2012-0211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh HW, Song DK, & Kim YH (1997). Differential effects of adenosine receptor antagonists injected intrathecally on antinociception induced by morphine and β-endorphin administered intracerebroventricularly in the mouse. Neuropeptides, 31(4), 339–344. 10.1016/S0143-4179(97)90069-X [DOI] [PubMed] [Google Scholar]
- Sweeney MI, White TD, Jhamandas KH, & Sawynok J (1987). Morphine releases endogenous adenosine from the spinal cord in vivo. European Journal of Pharmacology, 141(1), 169–170. 10.1016/0014-2999(87)90428-6 [DOI] [PubMed] [Google Scholar]
- Sweeney M, White T, & Sawynok J (1987). Involvement of adenosine in the spinal antinociceptive effects of morphine and noradrenaline. Journal of Pharmacology and Experimental Therapeutics, 243, 657–665. [PubMed] [Google Scholar]
- Sweeney MI, White TD, & Sawynok J (1989). Morphine, capsaicin and K+ release purines from capsaicin-sensitive primary afferent nerve terminals in the spinal cord. The Journal of Pharmacology and Experimental Therapeutics, 248(1), 447–454. [PubMed] [Google Scholar]
- Tosh DK, Phan K, Gao Z-G, Gakh AA, Xu F, Deflorian F, Abagyan R, Stevens RC, Jacobson KA, & Katritch V (2012). Optimization of adenosine 5′-carboxamide derivatives as adenosine receptor agonists using structure-based ligand design and fragment screening. Journal of Medicinal Chemistry, 55(9), 4297–4308. 10.1021/jm300095s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang T, Al-Hasani R, Salvemini D, Salter MW, Gutstein H, & Cahill CM (2015). Pain and poppies: The good, the bad, and the ugly of opioid analgesics. Journal of Neuroscience, 35(41), 13879–13888. 10.1523/JNEUROSCI.2711-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Lubitz DKJE, Lin RC-S., Bischofberger N, Beenhakker M, Boyd M, Lipartowska R, & Jacobson KA (1999). Protection against ischemic damage by adenosine amine congener, a potent and selective adenosine A1 receptor agonist. European Journal of Pharmacology, 369(3), 313–317. 10.1016/S0014-2999(99)00073-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlman C, Doyle TM, Little JW, Luongo L, Janes K, Chen Z, Esposito E, Tosh DK, Cuzzocrea S, Jacobson KA, & Salvemini D (2018). Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. Pain, 159(6), 1025–1034. 10.1097/j.pain.0000000000001177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittendorp MC, Boddeke HWGM, & Biber K (2004). Adenosine A3 receptor-induced CCL2 synthesis in cultured mouse astrocytes. Glia, 46(4), 410–418. 10.1002/glia.20016 [DOI] [PubMed] [Google Scholar]
- Wu M, Sahbaie P, Zheng M, Lobato R, Boison D, Clark JD, & Peltz G (2013). Opiate-induced changes in brain adenosine levels and narcotic drug responses. Neuroscience, 228, 235–242. 10.1016/j.neuroscience.2012.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W-P, Hao J-X, Halldner L, Lövdahl C, DeLander GE, Wiesenfeld-Hallin Z, Fredholm BB, & Xu X-J (2005). Increased nociceptive response in mice lacking the adenosine A1 receptor. Pain, 113(3), 395–404. 10.1016/j.pain.2004.11.020 [DOI] [PubMed] [Google Scholar]
- Zarrindast M-R, Naghipour B, Roushan-zamir F, & Shafaghi B (1999). Effects of adenosine receptor agents on the expression of morphine withdrawal in mice. European Journal of Pharmacology, 369(1), 17–22. 10.1016/S0014-2999(99)00021-7 [DOI] [PubMed] [Google Scholar]
- Zylka MJ (2011). Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends in Molecular Medicine, 17(4), 188–196. 10.1016/j.molmed.2010.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full-length Western blots featured in Figure 4
Immuno-labeling of A3AR in rat spinal cord. Naïve rat spinal cord sections immuno-labeled for A3AR and cell type markers. (a) A3AR expression in the spinal dorsal horn. Scale bar = 500 μm. (b) A3AR co-labeling with GFAP (astrocytes), NeuN (neurons), and Iba1 (microglia). Arrows indicate co-labeling. Scale bar = 50 μm
Data Availability Statement
Data sets and materials generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.