

Summary
Eukaryotic genomes vary in terms of size, chromosome number, and genetic complexity. Their temporal organization is complex, reflecting coordination between DNA folding and function. Here, we used fused karyotypes of budding yeast to characterize the effects of chromosome length on nuclear architecture. We found that size-matched megachromosomes expand to occupy a larger fraction of the enlarged nucleus. Hi-C maps reveal changes in the three-dimensional structure corresponding to inactivated centromeres and telomeres. De-clustering of inactive centromeres results in their loss of early replication, highlighting a functional correlation between genome organization and replication timing. Repositioning of former telomere-proximal regions on chromosome arms exposed a subset of contacts between flocculin genes. Chromatin reorganization of megachromosomes during cell division remained unperturbed, and it revealed that centromere-rDNA contacts in anaphase, extending over 0.3 Mb on wild-type chromosome, cannot exceed ∼1.7 Mb. Our results highlight the relevance of engineered karyotypes to unveiling relationships between genome organization and function.
Keywords: karyotype engineering, nuclear and chromosome architecture, spatiotemporal DNA replication, Hi-C, replication origin, FLO genes
Graphical abstract
Highlights
-
•
Nuclear size is influenced by chromosome size and their relative length
-
•
Chromosome-core repositioning of subtelomeres unveils specific gene contacts in 3D
-
•
Centromere inactivation delays replication firing in their vicinity
-
•
The rDNA-centromere bridge of contacts expands over 1.5 Mb linear genomic distance
The work of Lazar-Stefanita et al. uses genetically engineered fused chromosomes to investigate chromosome size effect on nuclear architecture and function. The gigantic chromosomes were found to occupy more space within enlarged nuclei and maintain DNA reorganization during cell division. Site-specific, three-dimensional changes were functionally linked to temporal variations of the DNA replication program.
Introduction
Over billions of years of evolution, genomes have acquired distinct characteristics. In eukaryotes, chromosome size, number, and structural organization within the nuclear space are diverse, reflecting a history of dynamic evolutionary changes. The 12-Mb budding yeast genome, distributed among 16 chromosomes, encodes ∼6,000 genes.1 By contrast, flies, worms, and humans have much larger genomes (8–250 times as large as yeast, distributed across 4, 6, and 23 pairs of chromosomes, respectively), but only carry 2–3 times as many genes as yeast do. This indicates a lack of strong functional correspondence between genome size, chromosome number, and gene content.2 Nevertheless, DNA content has been linked to a number of phenotypic traits: its increase is concomitant with the cell size across a wide variety of taxa while it correlates inversely with the rate of cell division. Hence, a high DNA content is typically found in nuclei of large and slowly dividing cells.2,3
Imaging and chromosome conformation capture (3C; Hi-C) studies have revealed complex architectures. Distinct nuclear territories of various sizes occasionally interact with one another; yeast centromeres and telomeres cluster into discrete foci, whereas the nucleolus occupies a distinct territory opposite the spindle pole body (SPB).4 Ergo, the DNA molecule is not strictly informational, but can also structure the nucleus. Nuclear repositioning has been linked to local gene regulation in yeast,5,6 and highly transcribed genes result in boundaries in chromosome contact maps delimiting small microdomains along interphase chromosomes.7,8 As yeast cells enter mitosis, their chromosomes are reorganized into arrays of relatively small (∼10–40 kb) chromatin loops, mediated by the cohesin complex.9, 10, 11, 12, 13 The mechanism of cohesin-mediated loops is conserved in mammals, where they maintain large topologically associating domains (TADs) in interphase.14, 15, 16, 17
Here, we deploy a new strategy to investigate nuclear DNA architecture and function on karyotype-engineered yeasts that carry megachromosomes produced by sequential rounds of telomere to telomere fusion and simultaneous removal of one centromere.18,19 These genomes, which carry massive “designer rearrangements,” represent new resources for investigating functional consequences of genome three-dimensional (3D) restructuring. We show that inactivation of pericentromeric regions during megachromosome engineering modifies the higher order organization and abolishes their early replication firing, demonstrating a functional link between overall genome organization and regulation of replication. Likewise, reposition of formerly telomere-proximal regions along megachromosome arms reveals unexposed interactions between discrete chromosomal sites that evaded detection in the native yeast genome. Despite that the relative size of the fused megachromosomes was found to differentially affect nuclear size, initial studies revealed a surprising lack of major fitness defects, pointing to a resilient genome structure that strongly argues against the idea that intranuclear position is a major determinant of gene expression.18, 19, 20 In addition, the mitotic characteristics of native chromosomes are maintained in megachromosomes, showing that chromosome length regulates neither chromatin compaction nor segregation efficiency, underscoring the robustness of mitosis in yeast. These results reinforce the relevance of local 3D nuclear architecture on genome structure and function and highlights the importance of finding alternative ways to investigate it.
Results
Size-matched versus unmatched megachromosomes
Yeast strains carrying massively reorganized karyotypes have been engineered and thoroughly described.18,19 Briefly, these studies showed that chromosome size can be massively increased through multiple rounds of telomere-telomere fusion and simultaneous centromere deletion (to prevent the formation of dicentric chromosomes) without incurring major fitness defects.
The present work focuses on yeasts whose 16 native chromosomes have been sequentially merged into 2 final megachromosomes (Figures 1A and S1A), which, for the sake of clarity, we named alphabetically from the largest to the smallest (e.g., chrA and chrB) in each strain. In earlier work by Luo et al.,18 we described an n = 2 strain, JL402, with two approximately equal size chromosomes. Here, to further investigate the potential importance of the “size-matched” chromosomes, we designed and constructed an “unmatched” n = 2 karyotype, in which one chromosome is 3 times larger than the other (Figure S1A). Interestingly, the unmatched n = 2 strain (JL498) has a severe growth defect relative to the size-matched strain (Figure S1B). Additional studies were performed on two n = 3 strains (JL381 and JL410; growth rates reported by Luo et al.18) that served as progenitors of the two n = 2 strains (Key resources table; Figure S1A).
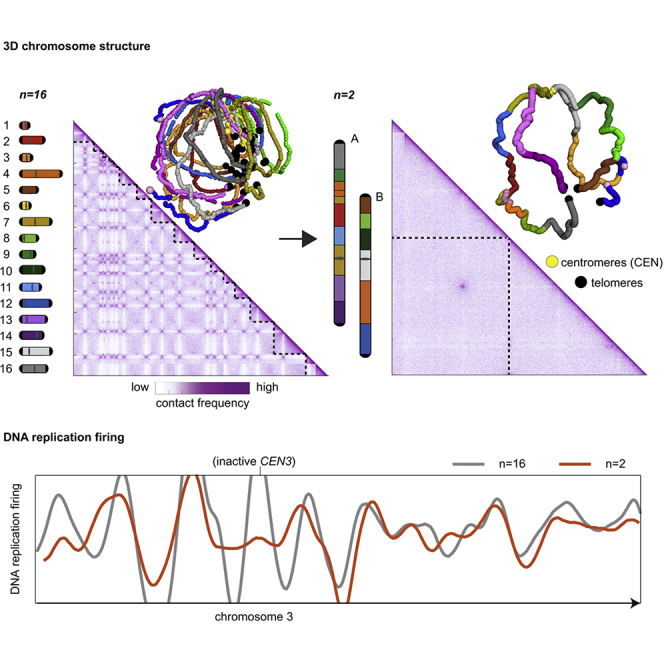
Figure 1.

Genome structure of 2 megachromosomes
(A) Overview of the chromosome fusion design from n = 16 to n = 2. The 16 native chromosomes are uniquely colored and ordered numerically, while the 2 fused megachromosomes are alphabetically ordered (A and B). Length of chromosome arms is indicated as a function of distance from the centromere position (Mb).
(B) Measurements of DNA surface (μm2) in cells stained with SYTOX Green. Violin plots display: Median values of DNA surface in haploid (n = 16 and n = 2) and diploid (n = 16, positive control for size increase) strains, and their relative (%) increments when compared to n = 16 haploid. p values obtained through Kolmogorov-Smirnov test.
(C) Contact map comparison of n = 16 and n = 2 in G1 phase. Left and right Hi-C maps (5 kb binned) were generated by aligning both n = 16 and n = 2 reads on a reference sequence containing only 2 chromosomes. Bottom left map shows all 16 native chromosomes underlined by dotted lines, while the top right map displays only 2 chromosomes (chrA and chrB, atop the map). Black arrowheads point to inter-pericentromeric contacts. Violet to white color scale reflects high to low contact frequencies (log10).
(D) 3D average representations of G1 contact maps of n = 16 and n = 2. Color code reflects the 16 native chromosomes, and centromeres, telomeres, and rDNA are highlighted.
(E) p(s) derivative represents the average decay of the intra-chromosomal contact frequency p between loci with respect to their genomic distance s. Two G1 replicates of both n = 16 from Lazar-Stefanita et al.21 and n = 2 were plotted together.
(F) Cumulative log ratios of active and inactivated pericentromeric regions. Blue to red color scale reflects contact enrichment in 200-kb windows in n = 2 versus n = 16 (log2).
Transcriptional regulation is affected in the size-unmatched n = 2
We previously showed that the transcriptome of the size-matched n = 2 strain is not severely changed compared to n = 16, and was mostly attributed to the relocation of previously telomeric genes to internal chromosomal arm locations.18 We analyzed RNA sequencing (RNA-seq) libraries of the n = 2 size-unmatched strain (Method details), which has a stronger growth defect compared to n = 2 size-matched (Figure S1B). Consistent with the slow growth phenotype, we detected hundreds of differentially expressed genes (DEGs; log2 fold change >2 or <−2 and p < 1E10−5); however, no coherent pattern of gene ontology (GO) term enrichment could be identified among those DEGs (Figure S2; Table S1. Gene ontology of biological process (log2 fold change > 2 or <−2 and p < 1E10−5), related to RNA-seq method and Figure S2, Table S2. Gene ontology of cellular component (log2 fold change > 2 or <−2 and p < 1E10−5), related to RNA-seq method and Figure S2, Table S3. Gene ontology of molecular function (log2 fold change > 2 or <−2 and p < 1E10−5), related to RNA-seq method and Figure S2). The transcription of the former pericentromeric genes, located ∼20 kb upstream and downstream of the inactivated centromeres, remained largely unaffected and independent of chromosome size, suggesting that transcription is not influenced by centromere positioning nor the structure of the pericentromeric chromatin. The disturbed transcriptome of the n = 2 unmatched strain could be directly related to its slow growth phenotype. Alternatively, it is possible that one or more novel mutations, which are absent from the size-matched strain, arose in the unmatched affecting its fitness.
3D nuclear organization of megachromosomes
We reasoned that a ∼5-fold increase in chromosome length, relative to the longest native chromosome, may alter nuclear occupancy of the megachromosomes. To investigate this hypothesis, we started by measuring the surface area occupied by the genomic DNA in cells labeled with SYTOX Green (Method details). Our results showed an increase of ∼26% in DNA occupancy in the n = 2 size-matched compared to n = 16 (Figure 1B), which was undetected in the n = 2 unmatched strain (Figure S3; Table S4). Finally, the increase in DNA occupancy in the n = 2 size-matched was directly correlated with a ∼25% enlargement of the nuclear surface itself (Figure S5; Table S5). These results suggest a link between the size of the nucleus and the size of chromosomes. However, it remains unknown how only the size-matched megachromosomes but not the unmatched ones have this effect on nuclear size.
To characterize the spatial organization of megachromosomes, Hi-C experiments were performed on G1 synchronized n = 16 and n = 2 cells (Method details). In contrast to the contact map of the reference strain with 16 chromosomes, which displayed a characteristic 15-spot off-diagonal pattern reflecting centromere clustering associated with contact enrichment between pericentromeric regions,22 the n = 2 map displays only 2 long chromosomes and a single off-diagonal spot of inter-pericentromeric contacts (Figure 1C). Although 2D maps and the corresponding 3D representations of n = 2 and n = 16 are highly distinct (Figures 1D and S6), the key known architectural elements of yeast nuclear organization remain conserved and independent of chromosome length. Most noticeable are the trans contacts between the remaining active centromeres (Figures 1C, black arrowheads, and S6), reflecting their clustering adjacent to the SPB (yeast microtubule organizing center), as well as telomere clustering.
Chromatin folding is independent of chromosome size
We expected that the process of chromosome fusion would alter the overall balance between intra- and inter-chromosomal contacts, that we evaluated by quantifying their relative contact percentages in the 2D maps of both n = 2, size-matched and unmatched strains, and compared to n = 16. A gain of up to ∼30% of intrachromosomal contacts was observed in n = 2 strains (Figure S7A). We next asked whether this increase could affect the internal structure of megachromosomes during G1 compared to their n = 16 counterparts. Modifications of chromatin folding can be assessed, to some extent, by computing the contact probability p as a function of the genomic distance (s) along chromosome arms.21,23 Because chromosome size is considerably enlarged in n = 2 compared to n = 16 strains, a direct comparison between their p(s) curves is made problematic by the high variation in the inter-/intra-contact ratio between the 2 genomes. To address this issue, we computed the p(s) in 300-kb windows over the chromosomes of both strains (Method details). No significant differences were detected along the megachromosomes compared to native ones in G1 in this local analysis (Figure S7B). We then calculated the local derivative of the full p(s) curve in log-log space for both n = 16 and n = 2 genomes to magnify their differences9,24 (Method details). For the most part, the derivatives displayed similar slopes, with the exception of long distances (>150 kb), as small chromosome arms introduce variability in the n = 16 plot (Figure 1E). This result indicates that within the nuclear space, the long chromosomal arms of fused strains display an average chromatin folding state similar to that of the 32 shorter arms in G1.
Centromere inactivation leads to the reorganization of the pericentromeric chromatin
Due to centromere clustering, chromosomal arms adopt a “polymer brush”-like conformation25 that leads to local contact variations around centromere positions in both cis and trans.26,27 Given that the number of active centromeres in n = 2 is reduced 8-fold compared to n = 16, we thought that this may affect the brush structure, which is known to depend on the grafting density (e.g. the number of polymer end per unit area [4 for n = 2 karyotype, compared to 32 for n = 16 karyotype]).28 A decreased polymer brush effect was observed in n = 2 contact maps when compared to n = 16, as highlighted in the log ratios maps between 200-kb regions from n = 2 and n = 16 strains centered on either the retained active centromeres CEN7 and CEN15 or the inactivated ones (Figure 1F). In the n = 2 strain, the four pericentromere regions around CEN7 and CEN15 engage in longer range contacts than in the n = 16 strain (red stripes), as they become unconstrained by the loss of the brush-like effect. The chromatin flanking the 14 inactivated centromeres completely lose the polymer brush conformation (Figure S7C, log2-ratio maps). In other words, the larger the collective bulk of centromeric sequences at the kinetochores, the higher their degree of insulation.
Telomere deletion reveals inter- and intra-chromosomal FLO gene contacts
In yeast, heterochromatin is observed at and near the 32 telomeres and at the 2 silent mating type loci, HML and HMR, located near the left and right telomere of chromosome 3, respectively.4 These heterochromatic loci are thought to co-localize, forming subnuclear compartments enriched in silencing proteins Sir2-3-4.29 As each chromosome fusion deletes two telomeres and relocates the subtelomeric regions internal to a chromosome arm, it was not surprising to observe that the 28 formerly subtelomeric regions in the n = 2 strain stopped being in contact. However, the 2D maps revealed that a subset of these regions continued to exhibit discrete contacts (Figure 2). Among these contacts were the mating type loci (HML and HMR) with the retained subtelomeres (Figure 2A, red arrowheads), probably a result of their Sir3-dependent anchoring to the nuclear envelope.30 In addition, a set of distinct contacts involving three loci, formerly positioned within subtelomeric regions, appeared on the fused chromosomal maps. These previously unexposed contacts correspond to members of the FLO gene family, a co-regulated set of genes that trigger yeast flocculation in response to adverse conditions.31 FLO1, 5, 9, 10, and 11 are telomere-adjacent (∼10–40 kb) genes encoding 5 cell-wall glycoproteins or flocculins.32 The expression of the flocculins is regulated both genetically and epigenetically, through recombination33,34 and histone deacetylase-mediated silencing, respectively.35 To eliminate the possibility that these contacts correspond to sequence misalignments due to the repetitive nature of the genes, we masked their repetitive components and investigate contacts only between the unique regions flanking these genes (Method details; Figure S8). The contact maps binned at 2-kb windows display interactions between all 10-kb regions adjacent to either the 5′ or 3′ UTR of each FLO gene (Figure 2B). Surprisingly, the 3′ UTR flanking region of FLO9 engages in strong cis contacts with both regions adjacent to FLO1 and in trans contacts with a third position located at the FLO5 5′ UTR (Figure 2B). The same pattern of contacts was also observed in other fusion strains and is further illustrated by 4C-like analysis (Figure S9). Finally, a closer inspection of the n = 16 contact map reveals their presence in the wild-type yeast chromosomes, where they are largely masked by the contact signal resulting from telomere clustering (Figure 2B).
Figure 2.

Repositioning of subtelomeric genes internally to chromosomal arms reveals their contact in 3D
(A) Contact map of chrA in n = 2 (5 kb-binned). Red arrowheads point at cis contact enrichment between HML, HMR, and TEL. Violet to white color scale reflects high to low contact frequencies as in Figure 1.
(B) Dotted lines indicate locations of the FLO genes both on n = 2 and n = 16 chromosomes. Contact maps of unique FLO-flanking regions in n = 2 and n = 16 (2 kb-binned). y and x axes indicate gene name and the orientation of the unique 10-kb sequences flanking the FLO5′ and-3′ UTRs) in the n = 2 genome. The resulting 20-kb regions adjacent to each gene are underlined by dotted lines.
Megachromosomes therefore unveil contacts between these functionally related genes, which were difficult to observe in wild-type n = 16 strains due to their original subtelomeric position. Whether the FLO genes are actively clustered (and why), or whether these contacts are an indirect consequence of nuclear envelope targeting of silenced chromatin remains unknown. No additional intra- and inter-arm off-diagonal contacts were detected in the n = 2 contact maps, suggesting that robust trans contacts in the yeast genome are rare.
Structural and functional outcomes of 3D megachromosome organization
Spatiotemporal alteration of the DNA replication program
Replication of eukaryotic chromosomes is regulated both spatially and temporally. In yeast, replication initiates at ∼260 discrete origins, which are classified according to their firing time (early or late).36 The resulting replication program is regulated by limiting amounts of pre-replication complex factors that are recruited preferentially to early origins during early S phase and recycled to late origins throughout S phase. Replication factors are preferentially recruited to pericentromeric origins by their interaction with kinetochore components and to non-pericentromeric origins by forkhead transcription factors.37,38 This observation was proposed to account for the early firing of origins located in the vicinity of centromeres (spanning as much as 100 kb).37,39
Given that the former pericentromeric regions of inactive centromeres lose their spatial proximity to the SPB in the megachromosome strains, this new configuration provided an opportunity to directly test the interplay between genome organization and replication timing. Flow cytometry of DNA contents of cells synchronized in G1 with α-factor and released into S phase showed a delay in n = 2 compared to n = 16 (Figure S10; Method details). A prolonged S phase suggested a potential lower number of early replicated regions in n = 2. To test this hypothesis, we computed the replication profile as well as origin mapping using marker frequency analysis (MFA) approaches.40 n = 2 and n = 16 cells were arrested in G1 using α-factor and released synchronously into S phase in the presence of 200 mM hydroxyurea (HU) (Figure S11; Method details). HU treatment induces deoxynucleoside triphosphate (dNTP) starvation that results in the early S phase arrest. The mapping of early firing origins was done by computing chromosome sequence coverages in S phase that were normalized to G1 (unreplicated) and plotted along the reference genome at 1-kb resolution (Figures 3A, 3B, S12, and S13). In parallel, the replication timing profiles were generated from asynchronous cultures normalized to G1 read coverage (Figure S14).40 Replication timing profiles and origin mapping of homologous regions in n = 2 and n = 16 remained highly conserved along chromosomal arms. However, both plots displayed differences near the former pericentromeric positions in n = 2 compared to n = 16, with a loss of early firing in regions flanking the inactive centromeres (Figure 3A, brackets), whereas the replication of active centromeres remained unaffected (Figure 3B). A quantification of this effect is illustrated in the firing ratio of the active pericentromeres (n = 16) versus the inactive (n = 2) within 100-kb centromere-flanking regions (Figures 3C and S15). These results reinforced the essential role of the ∼125-bp centromere sequence in defining early firing and provides direct evidence of the 4D regulation of the replication program in yeast, with the SPB-centromere-kinetochore complex titrating essential components for origin firing and acting as an “early replication factory.”11
Figure 3.

3D de-cluster of inactive centromeres delays their replication timing
Each replication timing plot41,42 in (A) and (B) is the average representation of 3 independent replicates and shows the sequencing coverage ratio of S phase (HU) synchronized cells normalized on the G1 (α-factor) non-replicating cells (1 kb-binned). Replication timing profiles of the n = 16 (BY4741) are shown in gray, while those of n = 2 size-matched (JL402) are in orange.
(A) Representative comparison profiles of chromosomes with inactive centromeres in n = 2, but active in n = 16 (brackets).
(B) Comparison profiles of chromosome 15 with active centromere in both n = 2 and n = 16.
(C) Pericentromeric firing in n = 16 versus n = 2. The ratio plot shows the early firing of pericentromeric regions (∼100 kb) in n = 16 in respect to n = 2, in which centromeres were inactivated. Centromere position appears in brackets and is indicated with a dotted line.
Mitotic folding and segregation
After replication, the two sister chromatids (SCs) must be equally distributed during mitosis in the two daughter cells. In metaphase, the structural maintenance of chromosomes (SMC) cohesin complex not only holds SCs together but also promotes compaction through the formation of chromatin loops along their lengths.10, 11, 12 Cohesins are subsequently released, while the SMC condensin complex promotes chromosome segregation during anaphase.11,43, 44, 45, 46
The n = 2 strain enables the investigation of chromosome size effect on metaphase and anaphase chromosome structures. Hi-C experiments of n = 2 size-matched were performed in metaphase-arrested cells using nocodazole, and late-anaphase arrest using a thermosensitive allele (cdc15-2 t45) (Method details). Comparisons of contact maps and frequency p(s) show that megachromosomes undergo a cell-cycle-dependent reorganization similar to n = 16 chromosomes (Figures 4A and S16–S18).
Figure 4.

Cell-cycle reorganization of the megachromosomes
(A) p(s) derivatives of G1, metaphase, and anaphase synchronized cells. Top and bottom panels display slope decays along n = 2 chromosomes and n = 16 chromosomes, respectively (Hi-C data relative to n = 16 from Dauban et al. and Lazar-Stefanita et al.9,11).
(B) Pile-up ratio plots of 80kb windows (2-kb bins) centered on pairs of Scc1-enriched or randomly chosen positions during G1, metaphase, and anaphase. Ratios are ordered according to the distance between Scc1 binding sites in n = 2 and n = 16 strains. Blue to red color scale indicates contact enrichment between Scc1 binding sites compared to random genomic positions (log2).
(C) Centromere-rDNA contacts extend over Mb distance in fused genomes. Comparison of log-ratio contact maps between G1 and anaphase in n = 16, n = 3, and n = 2 (50 kb-binned). Size of the chromosome and centromere-rDNA distance increase progressively from left to right contact maps. Centromere (yellow) and rDNA (pink) positions, as well as their relative distances (Mb), are indicated at the top of the maps. Blue to red color scale reflects contact enrichment in anaphase compared to G1 (log2). For clarity, n = 16. Data reproduced from Lazar-Stefanita et al.21
(D) 3D average representations of n = 2 G1 and anaphase contact maps. Color code reflects the 16 native chromosomes, and centromeres, telomeres, and rDNA are highlighted.
Given the high degree of sequence similarity between n = 2 and n = 16 strains (>99.9%, not counting deletions of subtelomeres), we speculated that the sites of SMC enrichment would be conserved. To verify this hypothesis, we computed log ratios of pile-up plots that display the Scc1 cohesin deposition pattern47 during metaphase compared to non-cohesin sites in 80-kb windows (Method details).24,48 The pile-up displayed conserved patterns of contact enrichment at cohesin sites in n = 2 and n = 16 from G1 to anaphase (Figures 4B and S16, top panel), suggesting that cohesin distribution on metaphase chromosomes was chromosome size independent. In addition, cohesion loop scores computed using Chromosight49 were similar in n = 16 and n = 2 strains (Figure S16, bottom panel).
In a previous work, we showed that during yeast anaphase, a specific type of SMC complex, the condensin, mediates contact enrichment between the rDNA and the pericentromeric regions of the 16 chromosomes of wild-type yeast.21 We hypothesized that condensin-mediated loop extension would promote, directly or indirectly, the formation of a large (∼300 kb) loop/domain on chr12 between the two putative “roadblocks,” the nucleolus and the centromere (CEN12). In this work, we took advantage of the increased distance between the rDNA and the centromere to measure the size of this potential loop/domain of contacts within megachromosomes. As a consequence of chromosome fusion, the rDNA-centromere distance was increased by 5- to 10-fold in n = 3 (LS381) and n = 2 (size-matched, strain: LS402) fused strains (Key resources table), respectively. The extension of the rDNA-CEN contact domain was measured by comparing 2D maps obtained from n = 16, n = 3, and n = 2 cells arrested in either G1 (when no contacts are expected) or in late anaphase using the cdc15-2 allele (rDNA-CEN contacts are expected). In all of the strains, the ratio maps showed an increase in short-/medium-range contacts that span from the left-flanking region adjacent to the rDNA locus toward the centromere, reaching up to ∼1.7 Mb in n = 2 (Figures 4C and S18). In n = 3, the rDNA-CEN contacts extend over the entire 1.5-Mb enclosed region, whereas in n = 2, where the rDNA-CEN distance is 2.8 Mb, the intensity of contact enrichment emerging from the rDNA gradually decreases and eventually vanishes at approximately 1 Mb from the centromere without forming a clear chromatin boundary. These results suggest that the condensin-mediated loop/domain is highly dependent on the rDNA locus, which appears to represent a fixed roadblock that could, for instance, provide a reservoir of condensins that may extrude chromatin loops while moving toward the centromere. The 3D representations of the contact maps show no loop formation between the centromere and the rDNA in n = 2 (Figure 4D). Therefore, we concluded that the structural transitions of chromatin known to occur during the cell cycle are conserved in the megachromosomes.
Discussion
In this work, we investigated the regulation of chromosome folding of yeast strains whose genomes carry massive chromosome fusions. The normal karyotype of yeast (containing 16 chromosomes, n = 16) was reduced up to 8 times (in n = 2) through serial telomere-telomere fusions and centromere deletion.18 During this process, the size of the relatively small yeast chromosomes was dramatically increased. Megachromosomes represent a unique resource to investigate aspects of nuclear architecture using Hi-C, independently of the reverse genetics approach. The Hi-C protocol, especially the crosslinking step, favors cis over trans contacts.50 As a result, the underrepresented trans contacts may be overlooked in the Hi-C analysis.51 Therefore, we reasoned that this technical limitation could at least partially be circumvented in the fused genomes where many trans contacts became cis. It is worth noticing that this simple working model does not account for contact variability that may result from differences in chromatin properties, when loci in trans are repositioned in cis, nor the increased chromosome length. These are potential factors that may play key roles in their contact detection. Chromosome organization of a distinct but related fusion configuration was recently studied using a similar approach.19 However, this study was performed in asynchronous cells and did not address some functional aspects of these novel 3D genome organizations. Here, we tackle this question and compare two n = 2 strains with size-matched versus unmatched versions of megachromosome and characterize their properties compared to the regular n = 16 parental genome.
Nuclear occupancy and genome organization of fused karyotypes during the cell cycle
We observed that size-matched megachromosomes occupy a larger fraction of the total nuclear space than do standard chromosomes. Why this trend was not seen in the unmatched megachromosome strain remains unknown. We can only speculate that cells are able to perceive total number and length of chromosomes and regulate their nuclear volume accordingly. One hypothesis is that the size discordance between the two unmatched megachromosomes (∼3 versus ∼9 Mb) is not efficient to induce a nuclear increase as in the size-matched cells. Further studies are required to test this hypothesis and the molecular mechanisms involved in this chromosome-nucleus communication. Furthermore, the n = 2 unmatched strain also contained a larger population of cells carrying excess DNA content compared with the size-matched n = 2 and n = 16 strains (Figure S4), consistent with a slight increase in autopolyploidization, which was mainly observed by sensitivity to canavanine (toxic analog of arginine). Given these observations we speculate that a size-matched karyotype in the context of a reduced chromosome number is required to maintain ploidy.
We have shown that the structure of chromatin and its dynamic reorganization during the cell cycle is maintained independently of fused chromosome length. In addition, key landmarks of yeast chromosome configuration, such as centromere and telomere clusters, were conserved throughout the entire cell cycle. These results agree with the retained contact enrichment among Scc1 binding sites genome-wide along the large chromosomes, suggesting that SMC deposition is independent of the length of the DNA molecule. This conservation in G2/M cohesin-dependent folding suggests that the length of the condensed yeast chromosomes does not affect their segregation. Instead, condensin-mediated pulling of chromatin is likely to be the main driving force promoting efficient segregation of the genetic material between daughter cells in M phase.52
Finally, the fused chromosomes allowed assessment of the potential expansion of the condensin-dependent CEN-rDNA anaphase loop, reported on chr12 of wild-type yeasts.11,53 We show that in mitosis, the centromere and rDNA contact each other over distances that do not exceed 1.7 Mb. Our hypothesis is that condensins may extrude DNA loops of various sizes that gradually merge but, eventually, unload from the chromosome before they reach the other major condensin loading center and/or roadblock, the centromere. A similar scenario was envisioned during dosage compensation of the X chromosome in Caenorhabditis elegans.54
Effects on centromeres and telomeres
We show that, despite the tremendous increase in the size of the chromosomes, the better known aspects of yeast chromosome folding are overall preserved in n = 2 genomes. However, local contact variations, especially surrounding the inactivated centromeres and deleted telomeres, were observed. Relocation of formerly pericentromeric and subtelomeric regions to internal chromosome arm positions revealed direct functional aspects associated with their 3D localization. Centromeres have long been known to be early-replicating regions.39,55,56 Their temporal regulation was shown to depend on preferential recruitment of a limiting pre-replication factor, Dbf4, at pericentromeres through its physical interaction with Ctf19, a kinetochore component.38,39 Consequently, it was proposed that origins located in the immediate vicinity of centromeres fire early because of their Dbf4 enrichment. Here, we showed that the structural inactivation of centromeres (some of which were limited to 3-bp deletions; see detailed design by Luo et al.18) leads to the loss of early firing in their environs. Therefore, our results are consistent with the suggestion that the centromeres cluster acts as a replication factory that titrate limiting factors to ensure synchronous firing of pan-chromosome replication.
We also unveiled interactions involving a subset of heterochromatin-prone loci and members of a functionally related gene family, the FLO genes. Contacts between the silent mating type loci HML and HMR and the distant remaining telomeres were also shown in our earlier work to depend on the silencing complex, SIR.30 Here, we describe additional contacts between HML and FLO10 (Figure S9), another Sir-dependent complex.35 FLO10 is a member of the flocculin gene family encoding cell-surface glycoproteins (FLO1, 5, 9, 10, and 11) that confer adherence to agar, solid surfaces, and other yeast cells. FLO genes are subtelomeric, a location suggested to play important roles in their evolution.33,57 We have shown that FLOs and their flanking sequences remain in contact with one another both in cis and in trans, revealing their intrinsic ability to co-localize, even when no longer subtelomeric. As these genes are not expressed in our strains,58 we speculate that their co-localization is independent of their functional activity and may be linked to their epigenetic silencing. It is noteworthy that FLO1 and FLO5, found to be in spatial proximity with FLO9, were both deleted in the design of the single chromosome karyotype n = 1, published by Shao et al.19 As a matter of fact, neither we nor the authors of this latter work were able to detect contacts between the flanking regions of these genes that were partially retained after their deletion.
In summary, studies of megachromosomes have revealed a wealth of new information about the plasticity of genome organization, control of nuclear volume and DNA replication, and previously undetected interchromosomal contacts.
STAR Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Certified Megabase Agarose | BioRad | Cat#161-3108 |
| SYTOX green | Thermo Fischer | Cat# S7020 |
| Alpha-Factor | Zymo Research | Cat#Y1001 |
| RNAse A, DNase and protease-free | Thermo Fischer | Cat#EN0531 |
| Concanavalin a from Canavalia ensiformis (Jack bean),Type VI, lyophilized powder | Sigma-Aldrich | Cat#L7647-250MG |
| Formaldehyde 37% | Sigma-Aldrich | Cat#F8775-4X25ML |
| Nocodazole | Sigma-Aldrich | Cat#M1404 |
| DpnII | NEB | Cat#R0543M |
| Biotin-14-dCTP | Invitrogen | Cat#19518018 |
| DNA Polymerase I Klenow Fragment | NEB | Cat#M0210L |
| T4 DNA ligase | Thermo Fischer | Cat#EL0014 |
| Proteinase K | Thermo Scientific | Cat#EO0492 |
| T4 DNA Polymerase | NEB | Cat#M0203L |
| Dynabeads MyOne Streptavidin C1 | Invitrogen | Cat#65001 |
| Hydroxyurea,98%, powder | Sigma-Aldrich | Cat# H8627-25G |
| Glass beads, acid-washed | Sigma-Aldrich | Cat#G8772-100G |
| Critical commercial assays | ||
| DNA size markers S. pombe chromosomal DNA | BioRad | Cat#170-3633 |
| VK05 Lysing KIT | Ozyme | Cat#0042 |
| NEBNext Ultra II FS kit | NEB | Cat#E7805L |
| Nextseq 500/550 High Output Kit v2.5 (150 Cycles) | Illumina | Cat# 20024907 |
| NextSeq High-output 75-cycle V2.5 Kit | Illumina | Cat#20024906 |
| Deposited data | ||
| Raw microscopy images | This study; Mendeley | https://data.mendeley.com/datasets/swdfkxzzk7/1 |
| Raw FASQ files | This study | BioProject: PRJNA757122 https://www.ncbi.nlm.nih.gov/bioproject/PRJNA757122 Submission ID: SUB10236799 |
| Experimental models: Organisms/strains | ||
| S. cerevisiae. Genotype: MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0; chromosome# n = 16 | Brachmann et al., 1998 | BY4741 |
| S. cerevisiae. Genotype: MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0; chromosome# n = 3 | Luo et al., 2018 | JL381 |
| S. cerevisiae. Genotype: MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0; chromosome# n = 3 | Luo et al., 2018 | JL410 |
| S. cerevisiae. Genotype: MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0; chromosome# n = 2 size-matched size | Luo et al., 2018 | JL402 |
| S. cerevisiae. Genotype: MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0; chromosome# n = 2 unmatched size | This study | JL498 |
| S. cerevisiae. Genotype: MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 cdc15-2(ts); chromosome# n = 3 | This study | LS381 |
| S. cerevisiae. Genotype: MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 cdc15-2(ts); chromosome# n = 2 size-matched size | This study | LS402 |
| S. cerevisiae. Genotype: MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 NUP49::mScarlet-S.p.HIS5; chromosome# n = 16 | This study | LS125 |
| S. cerevisiae. Genotype: MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 NUP49::mScarlet-S.p.HIS5; chromosome# n = 2 size-matched size | This study | LS126 |
| Oligonucleotides | ||
| chr11R-gRNA for CRISPR-Cas9 AAATGAAGAAGTGCCATGGG |
This study | n/a |
| chr5L-gRNA for CRISPR-Cas9 TTTCACATGCTCGACCGTTT |
This study | n/a |
| cen7-gRNA TATTTTATTGTCGGTGTTTG |
This study | n/a |
| Recombinant DNA | ||
| Plasmid: TEF1p-CRISPR-Cas9 | Luo et al., 2018 | |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Matlab 2018 | MATLAB 2018a, The MathWorks, Inc., Natick, Massachusetts, United States. | https://www.mathworks.com/products/matlab.html |
| mat2pdb: https://fr.mathworks.com/matlabcentral/fileexchange/42957-read-and-write-pdb-files-using-matlab | ||
| FlowJo v10.0.7 | Becton, Dickinson and Company; 2021 | https://www.flowjo.com/ |
| Bowtie 2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Clustal Omega | Sievers et al., 2011 | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| PyMOL | Molecular Graphics System, Version 2.0 Schrödinger, LLC | https://pymol.org/edu/ |
| DEseq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Other | ||
| Saccharomyces Genome Database | SGD for S288c reference genome files | https://www.yeastgenome.org/ | |
| Yeast genome annotation pipeline - YGAP | Proux-Wera et al., 2012 | http://wolfe.ucd.ie/annotation/index.html#questioncheck1 |
Resource availability
Lead contact
Further information and requests for resources should be directed to Jef D. Boeke (jef.boeke@nyulangone.org).
Materials availability
Yeast strains generated in this study can be requested directly by contacting the lead contact.
This study did not generate new unique reagents.
Experimental model and subject details
Yeast strains used in this work are listed in STAR Methods - Key resources table. JL498 was generated using a CRISPR-Cas9 method from a parent that was generated in a previous study JL410.18 This involved fusing the right arm of chromosome 14 to the left arm of chromosome 5 and deleting CEN7 using a combination of three gRNAs: chr14R-gRNA, chr5L-gRNA and cen7-gRNA (refer to the oligonucleotides section in the Key resources table).
Method details
Media and culture conditions
All strains were grown in rich medium (YPD: 1% bacto peptone (Difco), 1% bacto yeast extract (Difco) and 2% glucose). Cells were grown at either 30°C or 23–25°C (the latter temperature corresponding to the permissive temperature of cdc15-2; see below).
Spot assays
Spot assays were performed as previously described by Luo et al.18 except that the culture was initially diluted to A600 = 0.1.
Pulsed-field gel electrophoresis
Pulsed-field gel electrophoresis (PFGE) was carried out as we previously described,18 with running conditions recommended for S. pombe chromosomes (BioRad No.170-3633) to achieve separation of the large fused chromosomes.
DNA staining
Approximately 107 cells/sample were fixed in 70% ethanol and stored at 4°C overnight. Cell pellets were washed twice with RNAse solution (10 mM Tris pH 8.0, 15 mM NaCl) and treated with 0.1 mg/mL RNAse A for 3–4 h at 37°C. Cells were centrifuged, washed once with 50 mM Tris pH 7.5 and resuspended in labeling solution (1 μM SYTOX Green in 50 mM Tris pH 7.5; ThermoFisher) for 1 h at 4°C. Before imaging and flow cytometry data acquisition, cells were washed twice and resuspended in 50 mM Tris pH 7.5.
Imaging DNA occupancy and nuclear size
Estimating DNA occupancy. To minimize the variability introduced by experimental procedure, the surface area occupied by DNA was calculated in groups of samples manipulated simultaneously (on the same day). Exponentially-growing cultures were treated for DNA staining (see above). For Z-stack acquisition, concanavalin A (Sigma-Alderich) coated glass slides were used to immobilize cells. Imaging was performed using the total internal reflection fluorescence (TIRF) microscope (objective: Plan Apo VC 100× Oil DIC N2; camera: Andor Zyla VSC-03679). NIS element AR software was used for image acquisition: 470 nm excitation wavelength (3% power), 10 ms exposure time, z stack 1.8 μm range (relative positions in Z −0.9 to +0.9 μm, 7 steps of 0.3 μm). Images were imported and analyzed with ImageJ software.59 The surface area occupied by the DNA was estimated using the “3D object counter” option after image segmentation and thresholding set at 3500-pixel intensity. Approximately 800–1500 DNA surfaces/strain (Table S4) were plotted using the violin function and the p-values were calculated using the Kolmogorov–Smirnov test (K–S test) functions available in Matlab R2018.
The increase in DNA occupancy in the size-matched n = 2 strain was correlated with an enlargement of the nuclear size in this strain compared to the wild type, n = 16. The surface area of the nucleus was measured in n = 2 (LS126) and n = 16 (LS125) yeasts, in which the nuclear envelope protein, Nup49, was tagged with mScarlet. Live cells in exponential growing phase were imaged using the EVOS M7000 microscope (Olympus X-APO 100X Oil, 1.45NA/WD 0.13mm oil objective). Images were analyzed with ImageJ software.59 Spherical nuclei (∼150 for n = 16; ∼300 for n = 2) were manually selected and, their circumference and surface area were estimated Table S5. Experiments were performed on two and three independent clones of LS125 and LS126.
Cell cycle progression
G1 arrested cells were obtained by incubating exponentially-growing cultures (∼108 cells) with 37 μM α-factor (Zymo Research) for 150 min at 23°C. An aliquot of ∼1.5 × 107 cells G1 cells was fixed in 70% ethanol while the remaining culture was centrifuged and washed twice with fresh medium. Pelleted cells were resuspended in fresh medium and incubated at room temperature. G1-released cultures were sampled at multiple time points throughout the entire S-to-M cell cycle progression. The corresponding aliquots were treated for DNA staining (see above). Flow cytometry was performed on a BD Accuri C6 Flow Cytometer (BD CSampler Software) and data were analyzed using FlowJo v10.0.7 software.
Cell synchronization
G1 elutriation. G1 daughter cells were recovered from exponentially-growing cultures through elutriation - a physical method of cell cycle synchronization, used to separate cells according to their density and sedimentation velocity. For a detailed protocol see Lazar-Stefanita et al.21 After elutriation, ∼1.5 × 109 G1 cells were suspended in 150 mL fresh YPD at 30°C for 30 min and then crosslinked with 3% formaldehyde (Sigma-Aldrich) for Hi-C library preparation (see below).
Nocodazole. Metaphase cells were chemically synchronized using the microtubule disrupting drug, nocodazole (Sigma-Aldrich). Exponentially-growing cultures (7 × 106 cells/mL) were grown for 3 h at 30°C in YPD supplemented with 15 μg/mL nocodazole. Cells were then crosslinked with 3% formaldehyde (Sigma-Aldrich) for Hi-C library preparation.
M phase ts mutants. Anaphase-synchronized cells were obtained through the thermosensitive (ts) mutant, cdc15-2.60 Exponentially-growing cultures (∼7 × 106 cells/mL) of strains, carrying the mutated cdc15-2 allele, were transferred from the permissive temperature (25°C) to the non-permissive growing temperature (37°C). Cultures were incubated for 3 h at 37°C before being crosslinked with 3% formaldehyde (Sigma-Aldrich) for Hi-C library preparation (see below).
Hi-C: Library preparation
Hi-C experiments (Table S6) were performed following the protocol described by Belton et al.61 For each library preparation 1–3 × 109 cells in 150 mL culture medium were first fixed for 20 min in 3% formaldehyde (Sigma-Aldrich), then quenched with 0.4 M glycine for other 20 min at room temperature. Cells were harvested, washed with culture medium and resuspended in chilled TE buffer supplemented with protease inhibitors. The VK05 Lysing KIT (Ozyme) at 6700 rpm was adopted to break down yeast cell wall. Lysates containing 0.5% SDS were incubated for 20 min at 65°C then digested overnight at 37°C with DpnII (final concentration 500 U/pellet; NEB). Digestion pellets were resuspended in cold water and the 5′ DpnII overhangs were filled in using a biotin-labeled 30 μM dNTP mix (dATP, dGTP, dTTP and Biotin-14-dCTP Invitrogen) and Klenow enzyme (NEB). Biotinylated DpnII restriction fragments were ligated using T4 DNA ligase (final concentration of 250 Weiss U/pellet; Thermo Fischer) for 4 h at 16°C. Cross-linking was reversed through overnight treatment in the presence of 250 μg/mL Proteinase K (Thermo Scientific) at 50°C. DNA was extracted by phenol/chloroform, precipitated and treated with 500 μg/mL RNAse A (Thermo Fischer). The effective digestion and ligation reactions were verified on agarose gel 1% (controls: non-digested and non-religated). Biotin was removed from the non-ligated DNA fragments using the T4 DNA polymerase (final concentration of 5 U/pellet; NEB). Finally, ligated fragments were sheared to a size of 500 bp using Covaris S220 and pulled down using Dynabeads MyOne Streptavidin C1 (Invitrogen). Resulting libraries were amplified by PE-PCR primers for standard paired-end deep-sequencing on NextSeq500 Illumina platform (2 × 75 bp cycles).
Hi-C: Data processing
Generating contact maps. Raw paired-end reads were processed using the HICLib algorithm,62 adapted for the S. cerevisiae genome. PCR duplicates were removed before either read-pair was independently mapped using Bowtie 263 (mode: --very-sensitive --rdg 500,3 --rfg 500,3) on the corresponding reference sequence (S288C from SGD and fusion versions of it) indexed for DpnII restriction site. An iterative alignment with a 20 bp seed length was used to maximize the yield of uniquely mapped reads (mapping quality, MAPQ >30). The aligned reads were classified based on their assignment and orientation on the DpnII indexed reference genome. To generate contact frequency maps, the unwanted events were filtered out (e.g., loops, non-digested fragments, etc.; for details see Cournac et al.64) while the valid Hi-C reads were binned into units of single restriction fragment (RF). Then consecutive RFs were assigned to fixed size bins of either 5 kb or 50 kb. Bins with a high variance in contact frequency (<1.5 Standard Deviation or 1.5–2 S.D.) were discarded. To remove potential biases resulting from the uneven distribution of restriction sites and variation in GC content and mappability, filtered contact maps were normalized using the sequential component normalization procedure with L1 norm (SCN).64 Approximately 15 million valid contacts were used to generate a genomic contact map (∼400 contacts/RF; ∼6500 contacts/5kb bin).
FLO contact maps. Contact enrichment maps between FLO genes were detected by aligning Hi-C reads on unique sequences flanking both 5′ and-3′ ends of the FLO coding regions. Sequence identity was verified using the multiple alignment option available for DNA in Clustal Omega on the EMBL – EBI website. Default parameters were used to align a total number of 12 10-kb long sequences upstream and downstream all FLO coding sequences. The alignments were used to determine the degree of % identity between all sequences (gap-exclusion identity) and to compute the “distance” between pairs (tree). Using this approach, we chose 10-kb long FLO-flanking sequences that display ∼35% sequence identity to assemble a new reference sequence (100-kb long). Most of these DNA sequences were immediately adjacent to the coding sequence except for the 3′ UTRs of FLO1 and FLO5 that were placed 20 and 25 kb away, respectively. The new reference was used to generate 2D FLO contact maps (2kb-binned).
Contact probability on the genomic distance, p(s). Hi-C contact frequency (p) decreases with as genomic distance between restriction fragments increases. p(s) functions were computed on intrachromosomal read pairs from which self-circularizing and uncut events – identified as reads having different orientations and separated by less than 1.5 kb - were discarded. The retained reads were log-binned in function of their distance along chromosome arms, according to the following formula:
Therefore, p(s) function is a histogram representation of the sum of contacts weighted by both bin-size 1.1(1+bin) and chromosome length (s).
Comparison of the degree of p(s) decay is indicative of a change in polymer state. Derivatives of the p(s) were computed to compare chromatin states in native versus fused chromosomes.
Contact probability was computed by “chunk,” with each chunk corresponding to a 300 kb window measured as distance from the centromere (e.g., 0, 300, and 600 kb from the centromere). n = 16 and n = 2 chromosomes were divided into multiple chunks and then the intra-chunk p(s) decay was computed following the method described above. Finally, the average of chunk p(s) decay per genome was extrapolated.
Log-ratio comparison maps. To assess genome-wide contact map similarities and differences, data were first binned at 50 kb and normalized. Finally, they were compared by computing the log2 ratio between each pair.
Similarly, local contact differences at pericentromeres and SMC binding sites were computed based on 5 kb-binned contact maps.
Centromere agglomerated log ratios. Centromere positions were partitioned into centromeres retained in n = 2 (CEN7 and CEN15) and deleted centromeres. For each group, in 2 kb resolution contact maps, windows surrounding the centromeres were extracted and averaged. The resulting observed signal in the n = 2 strain was divided by the signal in the n = 16 stain. The log of these ratios was then computed.
Cohesin agglomerated plots. Data from Petela et al.47 were used to generate Scc1 ChIP-Seq profiles with a 2 kb resolution. Bins with a signal over 1.5 were labeled as cohesin binding sites (CBS). All possible pairs of CBS within chromosomal arms were determined and partitioned according to their genomic distance. In 2 kb resolution contact maps, windows surrounding these positions were extracted and averaged. The resulting observed signal was divided by the expected signal, generated by averaging windows around random positions maintaining the same genomic distance as the pairs of CBS. For each window, under-covered bins were defined as bins with a total number of reads under the median (number of reads/bin) –SD and excluded from the averaging operations to reduce noise. Eventually log2 ratio plots that display the average contact signal of all possible pairs of Scc1 enrichment sites over the average contacts of random sites in 80 kb windows were computed.
4C-like profiles. To obtain 4C-like contact profiles of a given locus, filtered-normalized contact maps at 5 kb resolutions were indexed and the selected position/s annotated. Profiles of the selected bins were extracted and plotted (log10) using Matlab 2018 (no smoothing was applied).
3D representations. For visualization purposes, Hi-C contact frequency maps can be represented as 3D structures generated using the “Shortest-path Reconstruction in 3D″ algorithm, ShRec3D.65,66 The algorithm computes a distance matrix by inverting element-wise the corresponding 5 kb-binned contact map that was previously filtered and normalized (see Hi-C: Data processing). The resulting distance matrix is supplemented by computing shortest path distances. This procedure removes infinite values and yields values that satisfy the triangular inequality. To obtain the coordinates in 3D (x, y, z) the Sammon mapping algorithm was applied on the distance matrix.67 Finally, the “mat2pdb” function in Matlab 2018 was used to generate pdb files, which were rendered using PyMOL (Molecular Graphics System, Version 2.0 Schrödinger, LLC). The resulting 3D structures are average representations of the contact maps and, therefore, do not represent exact structures found in individual cells. These maps must be interpreted in light of contact frequencies across large cell populations. In this work, 3D structures were not used to compare datasets; all computational analyses were performed on contact maps.
Replication timing
Each profile of replication timing was generated from three independent clones by sort-seq analysis as described previously.40 Briefly, fractions of replicating and non-replicating cells were obtained by arresting cells in α-factor (see cell cycle progression) and then releasing them in 200 mM hydroxyurea (HU; Sigma-Aldrich) for 90 min at 30°C. Synchronization efficiency was checked by flow cytometry (see DNA staining and cell cycle progression). Pellets of ∼6 × 108 cells were used to extract genomic DNA using acidic wash beads (Sigma-Aldrich) and phenol-chloroform. Library preparation was performed using the NEBNext Ultra II FS kit (NEB) according to Illumina protocol. Resulting libraries were paired-end deep-sequenced on NextSeq500 Illumina platform (2 × 36 bp cycles). Reads were mapped to the corresponding reference genome using Bowtie 263 in its --very-sensitive mode. Profiles of replication timing were generated by normalizing the replicating (S phase - HU) sample to the non-replicating (G1 - α-factor) sample in 1-kb bins. The resulting ratios were gaussian-smoothed and plotted by genomic coordinate. They measure relative DNA copy number as a proxy of replication time.
RNA-seq
Total RNA extraction, library preparation and sequencing followed a previously-described method17. Briefly, RNA-seq reads were aligned to S288C reference genome (sacCer2) using STAR with default parameters.68 Differential expression analysis was performed using DEseq2.69 We consider genes with log2 fold-change > 2 or < −2 and p-value < 1E10−5 to be differentially expressed (Table S1. Gene ontology of biological process (log2 fold change > 2 or <−2 and p < 1E10−5), related to RNA-seq method and Figure S2, Table S2. Gene ontology of cellular component (log2 fold change > 2 or <−2 and p < 1E10−5), related to RNA-seq method and Figure S2, Table S3. Gene ontology of molecular function (log2 fold change > 2 or <−2 and p < 1E10−5), related to RNA-seq method and Figure S2).
Genome annotation
Fused genomes were annotated using a combination of different software packages. The yeast genome annotation pipeline (YGAP)70 online tool was used to annotate coding regions and generate general feature format (GFF) files. Other genome features (e.g., centromeres, telomeres, ARS, transposons etc.) were first mapped to the fused reference genome, and the resulting alignment files used to generate GFF3 files.
Quantification and statistical analysis
Information on the number of biological replicates, statistical tests and p-values is provided in the Method details and Figure legends.
Acknowledgments
We thank Vittore Scolari for discussion and advice on the contact analysis of the flocculin genes. We thank Laura McCulloch and Vittore Scolari for their comments on the manuscript. This work was supported in part by National Science Foundation grants MCB-1616111 and MCB-1921641 to J.D.B. L.L.-S. was supported in part by a fellowship from the Fondation pour la Recherche Médicale. This research was supported by funding to R.K. from the European Research Council under the Horizon 2020 Program (ERC grant agreement 771813).
Author contributions
L.L.-S., R.K., and J.D.B. designed the research. L.L.-S., A.T., and J.L. performed the experiments. L.L.-S., R.M., and X.S. analyzed the data, with contributions from A.T. J.M. generated the 3D representations. L.L.-S., R.K., and J.D.B. wrote the manuscript.
Declaration of interests
J.D.B. is a founder and director of CDI Labs, a founder of Neochromosome, Inc, a founder of and consultant to Re-Open Diagnostics, and serves or served on the Scientific Advisory Boards of Modern Meadow, Rome Therapeutics, Sample6, Sangamo, Tessera Therapeutics, and the Wyss Institute. The remaining authors declare no competing interests.
Published: July 27, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100163.
Contributor Information
Romain Koszul, Email: romain.koszul@pasteur.fr.
Jef D. Boeke, Email: jef.boeke@nyulangone.org.
Supplemental information
(A) Summary table of average measurements calculated from single events in (B)–(K). (B and C) Single-cell measurements of independent isolates of the diploid, n = 16 (BY4743). (D and E) Single-cell measurements of independent isolates of the haploid, n = 16 (BY4741). (F–H) Single-cell measurements of independent isolates of the haploid, n = 2 size-matched (JL402). (I–K) Single-cell measurements of independent isolates of the haploid, n = 2 size unmatched (JL498)
(A) Summary table of nuclear and cell sizes measurements calculated from single events in (B) and (C). (B) Single-cell measurements of the nuclear size in independent haploid isolates of n = 16 (BY4741) and n = 2 size-matched (JL402). (C) Single-cell measurements of the cell size in independent isolates of n = 16 (BY4741) and n = 2 size-matched (JL402).
Data and code availability
Raw microscopy images were deposited on Mendeley at: https://data.mendeley.com/datasets/swdfkxzzk7/1.
FASTQ files of GWS (HiC datasets and RNA sequencing) in were deposited in the NCBI GEO database.
BioProject: PRJNA757122 https://www.ncbi.nlm.nih.gov/bioproject/PRJNA757122.
Submission ID: SUB10236799.
No new code was generated in this study.
References
- 1.Goffeau A., Barrell B.G., Bussey H., Davis R.W., Dujon B., Feldmann H., Galibert F., Hoheisel J.D., Jacq C., Johnston M., et al. Life with 6000 genes. Science. 1996;274:546–563-7. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 2.Gregory T.R., Nicol J.A., Tamm H., Kullman B., Kullman K., Leitch I.J., Murray B.G., Kapraun D.F., Greilhuber J., Bennett M.D. Eukaryotic genome size databases. Nucleic Acids Res. 2007;35:D332–D338. doi: 10.1093/nar/gkl828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gregory T.R. Coincidence, coevolution, or causation? DNA content, cellsize, and the C-value enigma. Biol. Rev. Camb. Philos. Soc. 2001;76:65–101. doi: 10.1017/s1464793100005595. [DOI] [PubMed] [Google Scholar]
- 4.Taddei A., Gasser S.M. Structure and function in the budding yeast nucleus. Genetics. 2012;192:107–129. doi: 10.1534/genetics.112.140608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brickner D.G., Sood V., Tutucci E., Coukos R., Viets K., Singer R.H., Brickner J.H. Subnuclear positioning and interchromosomal clustering of the GAL1-10 locus are controlled by separable, interdependent mechanisms. Mol. Biol. Cell. 2016;27:2980–2993. doi: 10.1091/mbc.E16-03-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taddei A., Van Houwe G., Nagai S., Erb I., van Nimwegen E., Gasser S.M. The functional importance of telomere clustering: global changes in gene expression result from SIR factor dispersion. Genome Res. 2009;19:611–625. doi: 10.1101/gr.083881.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh T.-H.S., Fudenberg G., Goloborodko A., Rando O.J. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat. Methods. 2016;13:1009–1011. doi: 10.1038/nmeth.4025. [DOI] [PubMed] [Google Scholar]
- 8.Ohno M., Ando T., Priest D.G., Kumar V., Yoshida Y., Taniguchi Y. Sub-nucleosomal genome structure reveals distinct nucleosome folding motifs. Cell. 2019;176:520–534.e25. doi: 10.1016/j.cell.2018.12.014. [DOI] [PubMed] [Google Scholar]
- 9.Dauban L., Montagne R., Thierry A., Lazar-Stefanita L., Bastié N., Gadal O., Cournac A., Koszul R., Beckouët F. Regulation of cohesin-mediated chromosome folding by Eco1 and other partners. Mol. Cell. 2020;77:1279–1293.e4. doi: 10.1016/j.molcel.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Luis J., Lazar-Stefanita L., Gutierrez-Escribano P., Thierry A., Cournac A., García A., González S., Sánchez M., Jarmuz A., Montoya A., et al. FACT mediates cohesin function on chromatin. Nat. Struct. Mol. Biol. 2019;26:970–979. doi: 10.1038/s41594-019-0307-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lazar-Stefanita L., Scolari V.F., Mercy G., Muller H., Guérin T.M., Thierry A., Mozziconacci J., Koszul R. Cohesins and condensins orchestrate the 4D dynamics of yeast chromosomes during the cell cycle. EMBO J. 2017;36:2684–2697. doi: 10.15252/embj.201797342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schalbetter S.A., Goloborodko A., Fudenberg G., Belton J.-M., Miles C., Yu M., Dekker J., Mirny L., Baxter J. SMC complexes differentially compact mitotic chromosomes according to genomic context. Nat. Cell Biol. 2017;19:1071–1080. doi: 10.1038/ncb3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naumova N., Imakaev M., Fudenberg G., Zhan Y., Lajoie B.R., Mirny L.A., Dekker J. Organization of the mitotic chromosome. Science. 2013;342:948–953. doi: 10.1126/science.1236083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haarhuis J.H.I., van der Weide R.H., Blomen V.A., Yáñez-Cuna J.O., Amendola M., van Ruiten M.S., Krijger P.H.L., Teunissen H., Medema R.H., van Steensel B., et al. The cohesin Release factor WAPL Restricts chromatin loop extension. Cell. 2017;169:693–707.e14. doi: 10.1016/j.cell.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J., et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wutz G., Várnai C., Nagasaka K., Cisneros D.A., Stocsits R.R., Tang W., Schoenfelder S., Jessberger G., Muhar M., Hossain M.J., et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017;36:3573–3599. doi: 10.15252/embj.201798004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo J., Sun X., Cormack B.P., Boeke J.D. Karyotype engineering by chromosome fusion leads to reproductive isolation in yeast. Nature. 2018;560:392–396. doi: 10.1038/s41586-018-0374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao Y., Lu N., Wu Z., Cai C., Wang S., Zhang L.-L., Zhou F., Xiao S., Liu L., Zeng X., et al. Creating a functional single-chromosome yeast. Nature. 2018;560:331–335. doi: 10.1038/s41586-018-0382-x. [DOI] [PubMed] [Google Scholar]
- 20.Di Stefano M., Di Giovanni F., Pozharskaia V., Gomar-Alba M., Baù D., Carey L.B., Marti-Renom M.A., Mendoza M. Impact of chromosome fusions on 3D genome organization and gene expression in budding yeast. Genetics. 2020;214:651–667. doi: 10.1534/genetics.119.302978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lazar-Stefanita L., Scolari V.F., Mercy G., Muller H., Guérin T.M., Thierry A., Mozziconacci J., Koszul R. Cohesins and condensins orchestrate the 4D dynamics of yeast chromosomes during the cell cycle. EMBO J. 2017;36:2684–2697. doi: 10.15252/embj.201797342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan Z., Andronescu M., Schutz K., McIlwain S., Kim Y.J., Lee C., Shendure J., Fields S., Blau C.A., Noble W.S. A three-dimensional model of the yeast genome. Nature. 2010;465:363–367. doi: 10.1038/nature08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gassler J., Brandão H.B., Imakaev M., Flyamer I.M., Ladstätter S., Bickmore W.A., Peters J.-M., Mirny L.A., Tachibana K. A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. EMBO J. 2017;36:3600–3618. doi: 10.15252/embj.201798083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Gennes P.G. Conformations of polymers attached to an interface. Macromolecules. 1980;13:1069–1075. [Google Scholar]
- 26.Tjong H., Gong K., Chen L., Alber F. Physical tethering and volume exclusion determine higher-order genome organization in budding yeast. Genome Res. 2012;22:1295–1305. doi: 10.1101/gr.129437.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong H., Marie-Nelly H., Herbert S., Carrivain P., Blanc H., Koszul R., Fabre E., Zimmer C. A predictive computational model of the dynamic 3D interphase yeast nucleus. Curr. Biol. 2012;22:1881–1890. doi: 10.1016/j.cub.2012.07.069. [DOI] [PubMed] [Google Scholar]
- 28.Zhao B., Brittain W.J. Polymer brushes: surface-immobilized macromolecules. Prog. Polym. Sci. 2000;25:677–710. [Google Scholar]
- 29.Miele A., Bystricky K., Dekker J. Yeast silent mating type loci form heterochromatic clusters through silencer protein-dependent long-range interactions. PLoS Genet. 2009;5:e1000478. doi: 10.1371/journal.pgen.1000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo J., Mercy G., Vale-Silva L.A., Sun X., Agmon N., Zhang W., Yang K., Stracquadanio G., Thierry A., Ahn J.Y., et al. Synthetic chromosome fusion: effects on genome structure and function. bioRxiv. 2018 doi: 10.1101/381137. Preprint at. [DOI] [Google Scholar]
- 31.Sampermans S., Mortier J., Soares E.V. Flocculation onset in Saccharomyces cerevisiae: the role of nutrients. J. Appl. Microbiol. 2005;98:525–531. doi: 10.1111/j.1365-2672.2004.02486.x. [DOI] [PubMed] [Google Scholar]
- 32.Teunissen A.W., Steensma H.Y. Review: the dominant flocculation genes of Saccharomyces cerevisiae constitute a new subtelomeric gene family. Yeast. 1995;11:1001–1013. doi: 10.1002/yea.320111102. [DOI] [PubMed] [Google Scholar]
- 33.Ogata T., Izumikawa M., Kohno K., Shibata K. Chromosomal location of Lg-FLO1 in bottom-fermenting yeast and the FLO5 locus of industrial yeast. J. Appl. Microbiol. 2008;105:1186–1198. doi: 10.1111/j.1365-2672.2008.03852.x. [DOI] [PubMed] [Google Scholar]
- 34.Verstrepen K.J., Jansen A., Lewitter F., Fink G.R. Intragenic tandem repeats generate functional variability. Nat. Genet. 2005;37:986–990. doi: 10.1038/ng1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halme A., Bumgarner S., Styles C., Fink G.R. Genetic and epigenetic regulation of the FLO gene family generates cell-surface variation in yeast. Cell. 2004;116:405–415. doi: 10.1016/s0092-8674(04)00118-7. [DOI] [PubMed] [Google Scholar]
- 36.Nieduszynski C.A., Hiraga S.i., Ak P., Benham C.J., Donaldson A.D. OriDB: a DNA replication origin database. Nucleic Acids Res. 2007;35:D40–D46. doi: 10.1093/nar/gkl758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang D., Lengronne A., Shi D., Forey R., Skrzypczak M., Ginalski K., Yan C., Wang X., Cao Q., Pasero P., Lou H. Dbf4 recruitment by forkhead transcription factors defines an upstream rate-limiting step in determining origin firing timing. Genes Dev. 2017;31:2405–2415. doi: 10.1101/gad.306571.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Natsume T., Müller C.A., Katou Y., Retkute R., Gierliński M., Araki H., Blow J.J., Shirahige K., Nieduszynski C.A., Tanaka T.U. Kinetochores coordinate pericentromeric cohesion and early DNA replication by Cdc7-Dbf4 kinase recruitment. Mol. Cell. 2013;50:661–674. doi: 10.1016/j.molcel.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pohl T.J., Brewer B.J., Raghuraman M.K. Functional centromeres determine the activation time of pericentric origins of DNA replication in Saccharomyces cerevisiae. PLoS Genet. 2012;8:e1002677. doi: 10.1371/journal.pgen.1002677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Müller C.A., Hawkins M., Retkute R., Malla S., Wilson R., Blythe M.J., Nakato R., Komata M., Shirahige K., de Moura A.P.S., Nieduszynski C.A. The dynamics of genome replication using deep sequencing. Nucleic Acids Res. 2014;42:e3. doi: 10.1093/nar/gkt878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvino G.M., Collingwood D., Murphy J.M., Delrow J., Brewer B.J., Raghuraman M.K. Replication in hydroxyurea: it’s a matter of time. Mol. Cell Biol. 2007;27:6396–6406. doi: 10.1128/MCB.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yabuki N., Terashima H., Kitada K. Mapping of early firing origins on a replication profile of budding yeast. Gene Cell. 2002;7:781–789. doi: 10.1046/j.1365-2443.2002.00559.x. [DOI] [PubMed] [Google Scholar]
- 43.D’Amours D., Stegmeier F., Amon A. Cdc14 and condensin control the dissolution of cohesin-independent chromosome linkages at Repeated DNA. Cell. 2004;117:455–469. doi: 10.1016/s0092-8674(04)00413-1. [DOI] [PubMed] [Google Scholar]
- 44.Guacci V., Hogan E., Koshland D. Chromosome condensation and sister chromatid pairing in budding yeast. J. Cell Biol. 1994;125:517–530. doi: 10.1083/jcb.125.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guérin T.M., Béneut C., Barinova N., López V., Lazar-Stefanita L., Deshayes A., Thierry A., Koszul R., Dubrana K., Marcand S. Condensin-mediated chromosome folding and internal telomeres drive dicentric severing by cytokinesis. Mol. Cell. 2019;75:131–144.e3. doi: 10.1016/j.molcel.2019.05.021. [DOI] [PubMed] [Google Scholar]
- 46.Hirano T. Capturing condensin in chromosomes. Nat. Genet. 2017;49:1419–1420. doi: 10.1038/ng.3962. [DOI] [PubMed] [Google Scholar]
- 47.Petela N.J., Gligoris T.G., Metson J., Lee B.-G., Voulgaris M., Hu B., Kikuchi S., Chapard C., Chen W., Rajendra E., et al. Scc2 is a potent activator of cohesin’s ATPase that promotes loading by binding Scc1 without Pds5. Mol. Cell. 2018;70:1134–1148.e7. doi: 10.1016/j.molcel.2018.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muller H., Scolari V.F., Agier N., Piazza A., Thierry A., Mercy G., Descorps-Declere S., Lazar-Stefanita L., Espeli O., Llorente B., et al. Characterizing meiotic chromosomes’ structure and pairing using a designer sequence optimized for Hi-C. Mol. Syst. Biol. 2018;14:e8293. doi: 10.15252/msb.20188293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matthey-Doret C., Baudry L., Breuer A., Montagne R., Guiglielmoni N., Scolari V., Jean E., Campeas A., Chanut P.H., Oriol E., et al. Computer vision for pattern detection in chromosome contact maps. Nat. Commun. 2020;11:5795. doi: 10.1038/s41467-020-19562-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scolari V.F., Mercy G., Koszul R., Lesne A., Mozziconacci J. Kinetic signature of cooperativity in the irreversible collapse of a polymer. Phys. Rev. Lett. 2018;121:057801. doi: 10.1103/PhysRevLett.121.057801. [DOI] [PubMed] [Google Scholar]
- 51.Maass P.G., Barutcu A.R., Weiner C.L., Rinn J.L. Inter-chromosomal contact properties in live-cell imaging and in Hi-C. Mol. Cell. 2018;70:188–189. doi: 10.1016/j.molcel.2018.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guérin T.M., Béneut C., Barinova N., López V., Lazar-Stefanita L., Deshayes A., Thierry A., Koszul R., Dubrana K., Marcand S. Condensin-mediated chromosome folding and internal telomeres drive dicentric severing by cytokinesis. Mol. Cell. 2019;75:131–144.e3. doi: 10.1016/j.molcel.2019.05.021. [DOI] [PubMed] [Google Scholar]
- 53.Paul M.R., Markowitz T.E., Hochwagen A., Ercan S. Condensin depletion causes genome decompaction without altering the level of global gene expression in Saccharomyces cerevisiae. Genetics. 2018;210:331–344. doi: 10.1534/genetics.118.301217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ercan S., Dick L.L., Lieb J.D. The C. elegans dosage compensation complex propagates dynamically and independently of X chromosome sequence. Curr. Biol. 2009;19:1777–1787. doi: 10.1016/j.cub.2009.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCarroll R.M., Fangman W.L. Time of replication of yeast centromeres and telomeres. Cell. 1988;54:505–513. doi: 10.1016/0092-8674(88)90072-4. [DOI] [PubMed] [Google Scholar]
- 56.Raghuraman M.K., Winzeler E.A., Collingwood D., Hunt S., Wodicka L., Conway A., Lockhart D.J., Davis R.W., Brewer B.J., Fangman W.L. Replication dynamics of the yeast genome. Science. 2001;294:115–121. doi: 10.1126/science.294.5540.115. [DOI] [PubMed] [Google Scholar]
- 57.Verstrepen K.J., Klis F.M. Flocculation, adhesion and biofilm formation in yeasts. Mol. Microbiol. 2006;60:5–15. doi: 10.1111/j.1365-2958.2006.05072.x. [DOI] [PubMed] [Google Scholar]
- 58.Liu H., Styles C.A., Fink G.R. Saccharomyces cerevisiae S288c has a mutation in Flo8, a gene required for filamentous growth. Genetics. 1996;144:967–978. doi: 10.1093/genetics/144.3.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hartwell L.H., Mortimer R.K., Culotti J., Culotti M. Genetic control of the cell division cycle in yeast: V. Genetic analysis of cdc mutants. Genetics. 1973;74:267–286. doi: 10.1093/genetics/74.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Belton J.-M., McCord R.P., Gibcus J.H., Naumova N., Zhan Y., Dekker J. Hi–C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods. 2012;9:999–1003. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cournac A., Marie-Nelly H., Marbouty M., Koszul R., Mozziconacci J. Normalization of a chromosomal contact map. BMC Genom. 2012;13:436. doi: 10.1186/1471-2164-13-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lesne A., Riposo J., Roger P., Cournac A., Mozziconacci J. 3D genome reconstruction from chromosomal contacts. Nat. Methods. 2014;11:1141–1143. doi: 10.1038/nmeth.3104. [DOI] [PubMed] [Google Scholar]
- 66.Morlot J.-B., Mozziconacci J., Lesne A. Network concepts for analyzing 3D genome structure from chromosomal contact maps. EPJ Nonlinear Biomed. Phys. 2016;4:2–15. [Google Scholar]
- 67.Sammon J.W. A nonlinear mapping for data structure analysis. IEEE Trans. Comput. 1969;C-18:401–409. [Google Scholar]
- 68.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Proux-Wéra E., Armisén D., Byrne K.P., Wolfe K.H. A pipeline for automated annotation of yeast genome sequences by a conserved-synteny approach. BMC Bioinformatics. 2012;13:237. doi: 10.1186/1471-2105-13-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Summary table of average measurements calculated from single events in (B)–(K). (B and C) Single-cell measurements of independent isolates of the diploid, n = 16 (BY4743). (D and E) Single-cell measurements of independent isolates of the haploid, n = 16 (BY4741). (F–H) Single-cell measurements of independent isolates of the haploid, n = 2 size-matched (JL402). (I–K) Single-cell measurements of independent isolates of the haploid, n = 2 size unmatched (JL498)
(A) Summary table of nuclear and cell sizes measurements calculated from single events in (B) and (C). (B) Single-cell measurements of the nuclear size in independent haploid isolates of n = 16 (BY4741) and n = 2 size-matched (JL402). (C) Single-cell measurements of the cell size in independent isolates of n = 16 (BY4741) and n = 2 size-matched (JL402).
Data Availability Statement
Raw microscopy images were deposited on Mendeley at: https://data.mendeley.com/datasets/swdfkxzzk7/1.
FASTQ files of GWS (HiC datasets and RNA sequencing) in were deposited in the NCBI GEO database.
BioProject: PRJNA757122 https://www.ncbi.nlm.nih.gov/bioproject/PRJNA757122.
Submission ID: SUB10236799.
No new code was generated in this study.