

Abstract
The SARS-CoV-2 3CL protease (3CLpro) is an attractive therapeutic target, as it is essential to the virus and highly conserved among coronaviruses. However, our current understanding of its tolerance to mutations is limited. Here, we develop a yeast-based deep mutational scanning approach to systematically profile the activity of all possible single mutants of the 3CLpro and validate a subset of our results within authentic viruses. We reveal that the 3CLpro is highly malleable and is capable of tolerating mutations throughout the protein. Yet, we also identify specific residues that appear immutable, suggesting that these may be targets for future 3CLpro inhibitors. Finally, we utilize our screening as a basis to identify E166V as a resistance-conferring mutation against the clinically used 3CLpro inhibitor, nirmatrelvir. Collectively, the functional map presented herein may serve as a guide to better understand the biological properties of the 3CLpro and for drug development against coronaviruses.
Keywords: COVID-19, SARS-CoV-2, 3CL protease, deep mutational scanning, protease inhibitors, nirmatrelvir, drug resistance
Graphical abstract
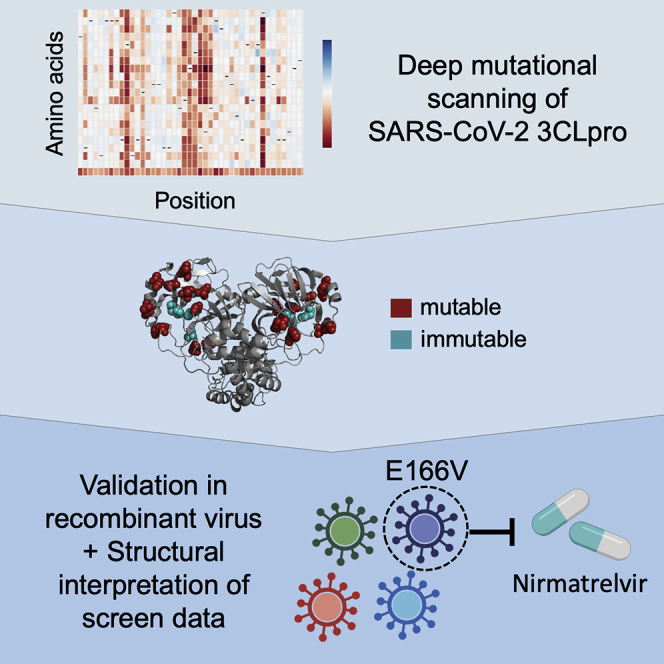
Iketani et al. conducted a screen to examine the malleability of the SARS-CoV-2 3CL protease, finding it is highly tolerant to mutations. Nevertheless, several conserved sites were identified, which may serve as drug targets. Combining their data with existing structural information, they find E166V to confer resistance to nirmatrelvir.
Main text
SARS-CoV-2 has transformed life around the globe and caused widespread socioeconomic damage (Wu et al., 2020; Zhou et al., 2020). To curtail the ongoing pandemic, researchers have intensively studied SARS-CoV-2 in hopes of identifying effective avenues for therapeutic intervention. To that end, several antibodies and antivirals have been identified and authorized for use in COVID-19 patients (Beigel et al., 2020; Chen et al., 2021b; Gupta et al., 2021; Hammond et al., 2022; Jayk Bernal et al., 2022; Weinreich et al., 2021). Current treatments are directed toward three viral targets: the spike glycoprotein (monoclonal antibodies), the RNA-dependent RNA polymerase (remdesivir and molnupiravir), and the 3-chymotrypsin-like protease (3CLpro) (nirmatrelvir). Despite the promise of these therapies, previous experiences with the rapid evolution of viruses suggest that resistance is likely to quickly arise (Strasfeld and Chou, 2010). Indeed, spike variants with enhanced resistance against antibody neutralization underscore this unfortunate possibility (Liu et al., 2022b; Wang et al., 2021; Wang et al., 2022).
Furthermore, studies have demonstrated that the adaptation of the virus to remdesivir-mediated polymerase inhibition can be readily achieved both in vitro and in vivo, suggesting that despite the conserved nature of a viral target, resistance can still arise (Gandhi et al., 2022; Stevens et al., 2022; Szemiel et al., 2021). Although no reports exist for SARS-CoV-2 3CLpro resistance within the clinic, examples of resistance to protease inhibitors in the case of HIV-1 and HCV foreshadow this likely event for the coronavirus (Clavel and Hance, 2004; Strasfeld and Chou, 2010).
The 3CLpro is an attractive therapeutic target as it is essential for viral replication, and there is a history of using protease inhibitors to successfully treat other viral illnesses (Chen and Njoroge, 2009; Ghosh et al., 2016; Jin et al., 2020a). To date, numerous SARS-CoV-2 3CLpro inhibitors have been reported, with a few advancing into clinical trials, and one (nirmatrelvir, utilized in combination with ritonavir and marketed as PAXLOVID™ by Pfizer) being authorized for clinical use to mitigate against severe disease and deaths (Boras et al., 2021; Dai et al., 2020; Hammond et al., 2022; Iketani et al., 2021; Jin et al., 2020a, 2020b; Ma et al., 2020; Owen et al., 2021; Zhang et al., 2020). Such drug discovery efforts are enticing, particularly as the 3CLpro exhibits significant conservation across coronaviruses, suggesting that broad-spectrum or ready-made “off-the-shelf” antivirals may be achievable for this and future epidemics. Indeed, several 3CLpro inhibitors developed for other coronaviruses have been found to also hold activity against SARS-CoV-2 3CLpro (Iketani et al., 2021; Resnick et al., 2021). However, the tolerance of the 3CLpro to mutations remains unknown. Such knowledge could allow for the rational design of inhibitors with improved potency and breadth, as well as provide insight into the biology of the enzyme.
Here, we sought to systematically evaluate the plasticity of SARS-CoV-2 3CLpro by interrogating the activity of all possible point mutants. We speculated that such a comprehensive analysis would reveal the malleability of the enzyme, as well as uncover conserved sites that could be used to rationally design inhibitors that are resistant to viral escape. Such extensive profiling of mutants of a gene, also known as deep mutational scanning (DMS), has been conducted successfully for the spike gene of SARS-CoV-2 by several groups, revealing the impact of mutations on expression, binding to its cognate receptor angiotensin converting enzyme 2 (ACE2), and binding by antibodies, underscoring the applicability of this approach (Garrett et al., 2021; Starr et al., 2020, 2021).
To conduct DMS, a scalable assay that can report on the properties under study must first be designed (Fowler and Fields, 2014). We chose to work with Saccharomyces cerevisiae (budding yeast) due to their ease of manipulation, successful use in previous SARS-CoV-2 DMS studies, and similarity to humans with regard to their folding, post-translational modification, and turnover of proteins (Botstein and Fink, 2011; Starr et al., 2020, 2021). Previous reports have identified that exogenous expression of viral proteases in cells can lead to cellular toxicity (Blanco et al., 2003; Resnick et al., 2021). We speculated that a similar approach could be used with SARS-CoV-2 3CLpro in yeast, and so, we cloned the SARS-CoV-2 3CLpro gene into an inducible yeast expression vector and generated a yeast strain containing this construct. As compared with cells containing a non-toxic control protein, enhanced yellow fluorescent protein (EYFP), cells expressing the SARS-CoV-2 3CLpro showed a significant growth deficiency (Figure S1A). To determine if the observed decrease in growth was due to the activity of the protease or a cryptic function performed by the exogenously expressed protease, we generated a catalytically inactive C145A mutant (Lee et al., 2020; Resnick et al., 2021). When expressed in yeast, the C145A mutant grew similarly to the non-toxic EYFP control, suggesting that the observed toxicity was a direct effect of 3CLpro activity (Figure S1A).
This growth defect upon expressing the 3CLpro in yeast formed the basis of our assay and suggested that we could profile libraries of 3CLpro mutants by growing them within a mixed pool and analyzing their relative abundances before and after inducing their expression. In this paradigm, we expected active variants to deplete and inactive variants to enrich within the pool upon induction. To generate our library of 3CLpro variants, we used degenerate oligonucleotides containing an NNK codon to generate all possible amino acids at each targeted residue (see STAR Methods for details). Each primer also contained a degenerate codon(s) upstream and/or downstream of the NNK stretch. This additional site(s) of degeneracy enabled the same amino acid mutation to be represented by a much larger number of DNA sequences than in conventional mutagenesis approaches, allowing us to average across the various DNA codings and by doing so better control for the noise inherent to assays performed at this scale (Schmierer et al., 2017; Zhu et al., 2019; Figure S1B). Once the mutant plasmids were generated, they were transformed into yeast, and cells were grown with or without induction of protease expression, followed by the analysis of the abundance of the various mutants under each condition.
We conducted the DMS in biological duplicate for each condition. Upon observing the robust correlation between the replicates, we proceeded to aggregate the effects between the two replicates, as done before (Figure S1C; Starr et al., 2020). The representation of mutations at each residue was as expected, with mostly non-synonymous mutations created by the site-directed mutagenesis method employed (Figure S1D). With regard to overall coverage, we observed most variants, with 94.3% (6,060 of 6,426 possibilities) of all possible variants assessed for activity.
To determine the activity of each 3CLpro variant, we compared its enrichment in the induced versus uninduced conditions and calculated an activity score (see STAR Methods for details). We note that these scores by definition incorporate not just the enzymatic activity of the protease but additional factors, such as their stability and expression. These activity scores were then normalized to the wild type and stop coding, set at 0 and −1, respectively, and visualized as a heatmap (Figure 1 A). Significant heterogeneity in tolerance to mutation across the protein was observed, with some specific residues and regions appearing highly constrained and intolerant to mutation (i.e., mutations resulted in a loss of activity).
Figure 1.

Activity of SARS-CoV-2 3CLpro mutants
(A) Heatmap delineating protease activity of all point mutants in SARS-CoV-2 3CLpro. Wild type at each residue is denoted with a black circle, and the asterisk designates the insertion of a stop codon at the residue. The activity score is bounded from −2 to 2. NF, N-finger; SBL, substrate-binding loop. Residues without a score are colored gray.
(B) Overlay of histograms of all activity scores in the screen (n = 6,060) and for those observed in clinical isolates (n = 862).
(C) Correlation between protease activity and evolutionary conservation of residues. Each residue was categorized as low (n = 95), medium (n = 73), or high (n = 138) conservation. Data are shown as a box and whiskers plot with whiskers denoting minimum to maximum. Dashed line is shown at 0. Statistical significance was determined by Kruskal-Wallis test followed by Dunn’s multiple comparisons test. ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
(D) Tolerance of residues within SARS-CoV-2 3CLpro for mutations overlaid onto the crystal structure.
See also Figure S1.
As an initial validation of our data, we first examined known characteristics of the 3CLpro, either in SARS-CoV-2 or in its close relative, SARS-CoV. First, mutations to the catalytic dyad, His41 and Cys145, resulted in the loss of activity, as would be expected given their essential role in protease activity (Lee et al., 2020). Additional critical residues within the active site were also generally conserved. These included Asp187, which stabilizes His41, and Arg40, which forms a salt bridge with Asp187 to stabilize its position (Kneller et al., 2020; Suárez and Díaz, 2020). Residues previously shown to directly bind one of the natural substrates of the 3CLpro, such as Phe140, Glu143, His163, His164, and Gln192, were mostly conserved and intolerant to mutations, although other substrate-binding residues such as Glu166 and Gln189 were not (Lee et al., 2020; Zhao et al., 2022). These latter nonconserved residues have previously been implicated in conferring substrate flexibility for SARS-CoV 3CLpro and therefore may tolerate mutation (Muramatsu et al., 2016; Zhao et al., 2022). These observations suggested that our data recapitulated known interactions and could also reveal complexities in the properties of the protease.
To further validate the data, we curated a list of mutations within the 3CLpro that have been identified within patient samples and mapped them onto our DMS data. We expected that clinical variants should have high activity scores within our dataset, as viruses with inactive proteases would not be expected to replicate sufficiently to be found in clinical isolates. We identified 932 single mutants from clinical samples (with ≥3 occurrences) and found that all of these mutants clustered around the wild-type score of 0, suggesting that our data were in line with what is observed in natural infections (Figure 1B).
Although the 3CLpro is largely conserved, there is still variability that can be observed throughout the coronavirus 3CL proteases. We speculated that residues that are less conserved across evolution should overlap with mutation-tolerant sites as denoted by our DMS data. To investigate this, we utilized the ConSurf server to determine the rate of evolution for each residue, with residues that were poorly conserved being scored as having a high rate of evolution (Glaser et al., 2003; Landau et al., 2005). We then grouped residues as having a high, medium, or low rate of evolution and plotted the average activity score for each of the residues in the various groups. As expected, residues with high or low rates of evolution were generally tolerant or intolerant to mutation, respectively (Figure 1C). Of note, although we do observe an association between these two metrics, there are additional factors that influence evolutionary conservation, such as compensatory or enabling mutations that were not considered in our screening and likely limit the strength of our association.
Finally, we considered whether our data aligned with the known structural features of SARS-CoV-2 3CLpro. When we mapped the average activity score at each position onto the crystal structure of the protein, we observed that surface-exposed residues were significantly more tolerant to mutations than buried residues (Figure 1D). Furthermore, regions of importance, such as those forming the catalytic pocket or the dimerization interface, strongly stood out as areas that were intolerant of mutations. Collectively, the above analyses suggest that our data are robust and reflect the true biological properties of the protease.
We next sought to further validate our data by investigating specific mutants indicated to be active or inactive by our screen. We chose 24 mutants throughout the gene with varying scores (14 predicted to be active and 10 predicted to be inactive) and produced isogenic yeast strains for each mutation. Each mutant was then individually tested for protease activity, and all were found to behave as predicted from our DMS (Figure 2 A). Unexpectedly, various C-terminal truncations, which lack residues that are believed to be critical for protein dimerization, exhibited activity (Hsu et al., 2005). To verify this observation, we also generated an independent strain with a truncated 3CLpro variant (deletion of residues 301–306). In agreement with our screening data, this truncated mutant showed protease activity (Figure S2A). We note that such a mutant is likely inviable in nature in which the C-terminal residues are critical for the release of the 3CLpro from the polyprotein, whereas the direct expression of the protease in our system circumvents the need for polyprotein processing.
Figure 2.

Validation of mutations and critical interactions identified in this study
(A) Validation in yeast of individual SARS-CoV-2 3CLpro mutants that scored as active or inactive in the screen. Strains were grown for 48 h, and then the growth was quantified by the measurement of OD at 595 nm. Data are shown as mean ± SEM of technical triplicates.
(B) Testing of the recombinant live infectious virus with selected individual 3CLpro mutations through a reverse genetics system. Generated mutants were used to infect Vero E6 cells for 72 h, and then the infection was quantified by the measurement of luciferase activity. Dashed line is shown at the mean value of mock. Data are shown as the mean ± SEM of biological triplicates.
(C–I) Conserved residues identified in the screen and their possible interactions and roles in 3CLpro activity. The following colors are used to denote specific regions: yellow (domain I), pink (domain II), green (domain III), gray (N-finger), and cyan (substrate-binding loop). The substrate is shown in dark blue. A previously solved structure of the 3CLpro with its native substrate is shown (PDB: 7KHP). Residues within the other protomer are denoted with a prime symbol (′).
(J–L) Testing of the inhibition of the wild type and E166V live infectious virus by nirmatrelvir and remdesivir. Viruses were used to infect Huh7-ACE2 cells for 24 h, and the infection was quantified by the measurement of luciferase activity. Data are shown as the mean ± SEM of technical triplicates.
See also Figure S2.
Although the correlation of our DMS data with clinical sequencing of SARS-CoV-2 isolates suggests that our high-throughput approach captures the true tolerance of the protease to mutation, as further validation, we tested several mutants, as well as the wild type and C145A as controls, in the context of authentic SARS-CoV-2 virus by reverse genetics. We found that the results recapitulated the screening data, with mutations predicted to retain an active protease resulting in a fully viable virus, whereas those predicted to inactivate the protease led to an inviable virus, with the exception of one mutant, E288F (Figure 2B). This mutant was scored to be inactive in the screen and validated as such within yeast yet exhibited activity within the recombinant virus. This difference in phenotype may be due to the slightly different environmental conditions between yeast and mammalian cells as well as the differences in substrates in the two contexts. Nevertheless, 14 of the 15 SARS-CoV-2 strains tested led to comparable results between the yeast and virus assays, suggesting that our screening largely recapitulates what is observed in a functional SARS-CoV-2 virus (Figures 2A and 2B).
For the 10 inactive protease variants that were independently validated, we wondered if a loss of 3CLpro dimerization, protein instability, or deficient catalytic activity might explain their lack of function. To address the question of dimerization, we designed a yeast two-hybrid assay to test whether the mutations were still competent for dimerization and found that seven of the 10 could still dimerize at varying levels, suggesting that a loss of enzymatic activity, reduction in dimerization efficiency, or a combination of both could explain the observed phenotypes (Figure S2B). For the three that no longer seemed to be able to dimerize, we tested protein expression levels by immunoblotting, observing that two of the three (L250G and L268N) were poorly detected, suggesting that they formed unstable proteins. The final variant, D295G, was well-expressed, and hence, its lack of signal in the yeast two-hybrid assay suggests it to be a dimerization-defective mutant, although additional experiments are required to further investigate this mutant’s dimerization properties (Figure S2C). These results suggest that despite the multiple mechanisms by which the protease can be inactivated, our DMS robustly reflects the protease activity, regardless of the nature of the mutation.
Altogether, we believe that the activity scores we derived for each mutant are reflective of their true properties given the correlations we observed with clinical data, evolutionary data, and structure, in combination with the validation of individual mutations, both in yeast and authentic viruses. Therefore, we speculated that by examining residues defined to be invariant in our screen, we could identify critical interactions within the protease that could serve as targets for the next generation of protease inhibitors.
As numerous sites appeared to have varying tolerance to mutation, we focused our subsequent structural analysis on the small subset that was highly intolerant to mutations. Upon analyzing these residues within a crystal structure of SARS-CoV-2 3CLpro complexed with its physiological substrate (the C-terminal autocleavage sequence, Ser301 to Gln306), we found that these sites formed critical interactions with their adjacent neighbors (Lee et al., 2020). Within domain I, several residues likely contribute to the stabilization of the protein structure. As shown in Figure 2C, the side chains of Asn95 and Cys16 form hydrogen bonds with the backbones of Trp31 and Lys12, respectively. Backbone hydrogen bonds are also identified between Val13 and Cys16, and Met17 and Trp31. In addition, the side chains of Asn95 and Trp31 interact favorably via the π electrons of their amide and aromatic moieties, respectively. As such, Lys12, Val13, Cys16, Met17, Trp31, and Asn95 contribute to the critical intra-domain interactions to stabilize the tertiary structure within domain I, which may explain the loss of activities when these residues are mutated. Examining the interface between domain I and domain II (Figure 2D), the side chains of Val13, Leu115, Phe150, Pro122, and Pro9′ of the opposing protomer form strong hydrophobic interactions. Mutating these residues to larger or more polar ones causes a reduction in protease activity, suggesting they are important inter-domain anchors. At the dimer interface between domains I and I′ (Figure 2E), an extensive hydrogen bond network is observed among Ser10, Gly11, and Glu14 and Ser10′, Gly11′, and Glu14′ from the second protomer of the homodimer. These same residues have been shown to be critical for dimerization in SARS-CoV 3CLpro (Chen et al., 2008a, 2008b). Looking at the catalytic domain II with a focus on the P1 pocket of the substrate-binding site (Figure 2F), besides the catalytic Cys145, residues including Ser144 and His163 form a multitude of hydrogen bond interactions with the substrate. The side chains of Phe140 and His163 also form a π-π stacking interaction. Moreover, Phe140 interacts with S1′ in the N-finger (NF) domain of the neighboring protomer via hydrogen bonds. This network of interactions affords structural stability near the substrate-binding site, which could explain the sensitivities of the enzymatic activities to the mutations in this region. Consistently, these residues have previously been shown to be important for SARS-CoV 3CLpro catalytic activity (Barrila et al., 2006; Cheng et al., 2010; Hu et al., 2009; Huang et al., 2004; Tan et al., 2005). During the close examination of our DMS data and the interface between domain II and domain III, we observed an interaction that our data suggest may help to stabilize this interface (Figure 2G). Specifically, residues Gly109 and Thr111 of domain II form hydrogen bonds with Asn203 and Asp295 of domain III, respectively. Previous studies have implicated the importance of the NF in dimerization, and we observed similar results (Chen et al., 2008b; Hu et al., 2009). The NF folds between domains II, III, and II′ of the abutting protomer (Figures 2H and 2I). The aromatic side chain of Phe3 is completely encapsulated in the hydrophobic pocket composed of side chains of Trp207, Ala210, Phe291, and Val296 (Figure 2H). As such, the biological activity of SARS-CoV-2 3CLpro is largely preserved when Phe3 is mutated to other hydrophobic residues including isoleucine, leucine, methionine, and tryptophan. However, mutations to residues of polar side chains completely abolish protein activity, corroborating the structural role of hydrophobic encapsulation. The residue Ala7 also engages interactions at the interface of NF, domain II and II′ (Figure 2I). Hydrogen bonds are observed between the backbones of Ala7 and Val125′. The enzymatic activities are similar to wild type when Ala7 is mutated to valine, cysteine, serine, or threonine but considerably lower for other mutants, suggesting the volume of the pocket is spatially constrained. In general, the structural insights support the results from our mutagenesis study and begin to explain the mechanism by which various residues are recalcitrant to mutation.
A recent study, which explored the binding interactions of all compounds co-crystallized with 3CLpro to date, identified that 185 from a total of 233 unique molecules bind within the active site, engaging the following 13 residues: Thr25, His41, Met49, Asn142, Ser144, Cys145, His163, His164, Glu166, Pro168, His172, Gln189, and Ala191 (Cho et al., 2021). Our analysis (Figure 1A) suggests that although some of these positions are unlikely to evolve (e.g., Ser144, His163, and His172), others are more flexible (e.g., Met49, Asn142, Glu166, Pro168, Gln189, and Ala191) and may therefore harbor the potential to confer resistance against many of the existing therapeutic candidates and lead compounds. This is of particular concern as the only authorized 3CLpro inhibitor, nirmatrelvir (PF-07321332), is such a therapeutic, targeting the catalytic Cys145 and nestling within the substrate-binding pocket (Owen et al., 2021). Although most of the contact sites of this compound are intolerant to mutation, several demonstrate significant plasticity in our data. These include Glu166 at the S1 subsite, which through its side chain forms a hydrogen bond with the lactam ring of nirmatrelvir at the P1 position. From our observations, Glu166 variants that convert the acidic residue into aliphatic, basic, sulfur-containing, and even aromatic amino acids retain wild-type-like activity. Residues Met49 and Gln189, which contribute to the formation of the lipophilic S2 subsite via their side chains and engage in hydrophobic interactions with the P2 dimethylcyclopropylproline group of nirmatrelvir, are also notably flexible, in accordance with other studies (Zhao et al., 2022). These analyses suggest that resistance to nirmatrelvir may readily arise.
To demonstrate this possibility, we generated a recombinant SARS-CoV-2 virus harboring the E166V mutation in 3CLpro and tested its inhibition by nirmatrelvir and remdesivir as a control. Relative to the wild-type SARS-CoV-2, the inhibition of the E166V mutant by nirmatrelvir was significantly hampered, with a 265-fold loss in EC50 (Figures 2J and 2L). By contrast, inhibition by remdesivir was unaffected, suggesting that this mutation specifically enables escape from nirmatrelvir (Figures 2K and 2L). Recent work by another group suggests that E166V results in a fitness loss for the virus, so this mutation may benefit from an additional compensatory mutation for full functionality in vivo (Zhou et al., 2022). These data underscore the risk of resistance developing against nirmatrelvir as it becomes widely used and further highlight the value of our screen as a resource for drug design.
In summation, we have mapped the functional landscape of the SARS-CoV-2 3CLpro using a DMS approach. Our work reveals that despite the strongly conserved nature of the gene across coronaviruses, the enzyme is highly malleable and can tolerate a plethora of mutations, even within the catalytic pocket, as also recently described by others during the preparation of this manuscript (Flynn et al., 2022). These empirical results differ from bioinformatic predictions based on available sequences, emphasizing the value of this approach (Krishnamoorthy and Fakhro, 2021). Consequently, these data suggest that resistance to 3CLpro inhibitors may readily arise and affirm the need for combinations of therapeutics to help reduce the rate of viral escape. Yet, despite this plasticity, we were able to identify highly conserved regions within the protein. Our results suggest that many of these sites are indispensable for catalytic activity or the overall structural integrity of the protein. Given their critical importance, it is natural to speculate that such residues may serve as ideal anchor points for the next generation of 3CLpro inhibitors developed to address the current and future coronavirus pandemics.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal GAL4 antibody (Clone RK5C1) | Santa Cruz Biotechnology | Catalog #sc-510, RRID: AB_627655 |
| Anti-mouse IgG Kappa light chain binding protein HRP-conjugated | Santa Cruz Biotechnology | Catalog #sc-516102, RRID: AB_2687626 |
| Bacterial and virus strains | ||
| pBeloBAC11 bacterial artificial chromosome containing the SARS-Cov-2 genome with the ORF7a replaced with NanoLuc luciferase reporter, with the following mutations as independent constructs: WT, C145A, T25C, Q69C, C160F, P168R, H172Y, N180G, G258Q, T292M, V114W, L250G, L268N, E288F, D295G, or E166V | Ye et al., 2020; this manuscript | N/A |
| Recombinant SARS-CoV-2 NanoLuc reporter viruses (with ORF7a deletion), with the following mutations as independent viruses: WT, C145A, T25C, Q69C, C160F, P168R, H172Y, N180G, G258Q, T292M, V114W, L250G, L268N, E288F, D295G, or E166V | Ye et al., 2020; this manuscript | N/A |
| 10-beta Electrocompetent E. coli | New England Biolabs | Catalog #C3020K |
| MDS42 E. coli | Umenhoffer et al., 2010 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| PF-07321332 (nirmatrelvir) | Aobius | Catalog #AOB14800 |
| Remdesivir | Selleckchem | Catalog #S8932 |
| Critical commercial assays | ||
| Nucleobond Xtra Midi kit | Macherey-Nagel | Catalog #740410.50 |
| Q5® High-Fidelity DNA Polymerase | New England Biolabs | Catalog #M0491L |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Catalog #34094 |
| Nucleobond BAC 100 kit | Takara Bio | Catalog #740579 |
| TRIzol™ Reagent | Thermo Fisher Scientific | Catalog #15596026 |
| TURBO™ DNase | Thermo Fisher Scientific | Catalog #AM2238 |
| TaqMan™ Fast Virus 1-Step Master Mix | Thermo Fisher Scientific | Catalog #4444432 |
| TaqMan™ 2019nCoV Assay Kit v1 | Thermo Fisher Scientific | Catalog #A47532 |
| Nano-Glo® Luciferase Assay System | Promega | Catalog #N1130 |
| Deposited data | ||
| Raw sequencing data | NCBI Short Read Archive | BioProject ID PRJNA813935 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Catalog #CRL-3216, RRID: CVCL_0063 |
| Vero E6 | ATCC | Catalog #CRL-1586, RRID: CVCL_0574 |
| Huh7-ACE2 | Liu et al., 2022a | N/A |
| Experimental models: Organisms/strains | ||
| Yeast, BY4741 strain (S288C background) | Brachmann et al., 1998 | N/A |
| Yeast, PJ69-4a/ɑ strain | James et al., 1996 | N/A |
| Oligonucleotides | ||
| Oligos for mutagenizing SARS-CoV-2-3CL | This manuscript | Table S1 |
| Oligos for amplifying SARS-CoV-2 3CL amplicons | This manuscript | Table S2 |
| Recombinant DNA | ||
| pAG416-GAL-SARS-CoV-2-3CL | This manuscript | N/A |
| Software and algorithms | ||
| Pipeline for calculation of activity scores of each 3CLpro variant | This manuscript | https://github.com/alejandrochavezlab/3CL_protease_DMS |
| BWA mem | Li and Durbin, 2009 | N/A |
| SAMtools | Li et al., 2009 | N/A |
| ConSurf Server | Glaser et al., 2003; Landau et al., 2005 | N/A |
| GraphPad Prism v9.2 | GraphPad Software | N/A |
Resource availability
Lead contact
Further information and requests for materials should be directed to and will be fulfilled by the lead contact, Alejandro Chavez (ac4304@cumc.columbia.edu).
Materials availability
Materials used in this study will be made available under an appropriate Materials Transfer Agreement.
Experimental model and subject details
Yeast
The yeast strains used for activity profiling were constructed in a BY4741 (S288C background) (Brachmann et al., 1998) and contained additional drug sensitizing mutations (Δpdr1 Δprd3 Δsnq2). For yeast two-hybrid studies, we used the PJ69-4a/ɑ background without the addition of the drug sensitizing mutations (James et al., 1996). All yeast were grown at 30 °C in a rotary shaker (for culture tubes) or in a plate shaker at 1000 RPM (for 96 deep well-plates) in growth media as appropriate.
The following yeast media were used in this study: yeast extract-peptone-dextrose medium (YPD), synthetic complete medium deficient in uracil with glucose (SC -ura GLU), synthetic complete medium deficient in uracil with galactose (SC -ura GAL), synthetic complete medium deficient in leucine and tryptophan with glucose (SC -leu -trp GLU), synthetic complete medium deficient in leucine, histidine, and tryptophan with glucose (SC -leu –his -trp GLU). YPD was formulated with 10 g/L yeast extract, 20 g/L peptone, and 20 g/L D-(+)-glucose. Synthetic complete media were formulated with 1.5 g/L yeast synthetic drop-out mix without yeast nitrogen base (US Biological), 1.7 g/L yeast nitrogen base without amino acids, carbohydrate, and without ammonium sulfate (US Biological), 5 g/L ammonium sulfate, 20 g/L D-(+)-glucose or galactose, and supplemented with the appropriate amino acids at the following concentrations: 90 mg/L histidine, 180 mg/L leucine, 90 mg/L lysine, 90 mg/L methionine, 90 mg/L adenine, and 18 mg/L uracil. For plates, the same recipe was used with the addition of 2% (w/v) agar and 600 μL of 5 M NaOH/L to help the plates solidify.
For yeast transformations, a modified lithium acetate-heat shock method was used (Gietz and Schiestl, 2007). Briefly, the appropriate parental strain was first grown to saturation in YPD, then diluted 1:100 in YPD (∼1 mL per reaction) and grown for 4 h at 30 °C to achieve log-phase growth. A plate mix was prepared by combining the following reagents per reaction: 71 μL 50% (w/v) PEG 3350, 8.8 μL 10X TE, 1.47 μL freshly boiled salmon sperm ssDNA (5 mg/mL), 8.8 μL 10X LiAc, 8.8 μL DMSO, and 1 μg of plasmid DNA to be transformed. The yeast were centrifuged at 4000 RPM for 5 min, the supernatant discarded, and then resuspended in 1X LiAc. The yeast were centrifuged again and then resuspended in 10 μL 1X LiAc per reaction. The plate mix and yeast were then combined into individual wells of a 96 well-plate. The plate was sealed and incubated for 20 min at 42 °C in a thermocycler. The plate was centrifuged at 4000 RPM for 5 min, the supernatant discarded, and then each pellet was resuspended in 20 μL sterile PBS. The yeast were then plated onto appropriate plates and grown for 48 h at 30 °C.
Mammalian cells
HEK293T cells and Vero E6 cells were obtained from ATCC (Catalog #CRL-3216 and #CRL-1586, respectively). Huh7-ACE2 cells (Huh7 cells overexpression human ACE2) were generated previously by transduction of Huh7 cells with lentivirus encoding human ACE2, packaged using pLEX307-ACE2-blast (Addgene plasmid #158449), pMD2.G (Addgene plasmid #12259, gift of Didier Trono), and psPAX2 (Addgene plasmid #12260, gift of Didier Trono), and selecting for stable expression with 5 μg/mL blasticidin (Liu et al., 2022a). Morphology was visually confirmed prior to use and all cell lines tested mycoplasma negative.
Method details
Measurement of SARS-CoV-2 3CL protease-induced toxicity
The full-length SARS-CoV-2 3CL protease gene (Genbank ID: MN908947) was synthesized (Twist Biosciences) and cloned into the yeast expression vector pAG416-GAL (Alberti et al., 2007) by Gateway cloning (Thermo Fisher Scientific). The N-terminus of the gene has the addition of a Met for translation initiation and the C-terminus is native.
Mutations were generated by site-directed mutagenesis (further detailed below). The plasmids were then transformed into the parental strain, and grown to saturation in SC -ura GLU. The yeast were then diluted 1:500 into 1 mL of SC -ura GLU or SC -ura GAL and grown at 30 °C with shaking. Growth was quantified by removing 100 μL from wells after 48 h and measuring OD at 595 nm with a spectrophotometer.
Deep mutational scanning (DMS) of SARS-CoV-2 3CL protease
For the deep mutational scanning, libraries were prepared as biological duplicates by conducting all steps with two independent replicates.
The yeast expression vector pAG416-GAL-SARS-CoV-2-3CL was freshly prepared by Midiprep (Macherey-Nagel), and variants were introduced by a modified single primer site-directed mutagenesis method (Shenoy and Visweswariah, 2003). Oligos were designed to introduce the degenerate codon NNK at the residue of interest to introduce all possible amino acids. In addition, each oligo was designed to introduce four or more synonymous mutations at the codon(s) immediately preceding or following the residue of interest to be used as genetic redundancies to average observed effects, analogous to screening approaches in which unique molecular identifiers (UMIs) have been used (Schmierer et al., 2017; Zhu et al., 2019) (see the following STAR Methods sections for further details and Table S1 for all oligos). Mutagenesis was conducted by mixing the following for each reaction: 150 ng template DNA, 1.25 μL 10 μM primer, 0.5 μL 10 mM dNTPs, 5 μL 5X Q5® Reaction Buffer, 0.25 μL Q5® polymerase, and H2O to 25 μL. All mutagenesis reactions were conducted in technical duplicate. The following cycling conditions were used:
1.98 °C, 45 s
2.98 °C, 15 s
3.57 °C, 15 s
4.72 °C, 4 min 30 s
5.Return to step #2 for 29 additional cycles
6.72 °C, 2 min
7.Hold at 4 °C
Each reaction was then digested by adding 1 μL DpnI and incubating at 37 °C for 1 h. Technical duplicates were then combined. Multiple reactions were then further combined into sets that could be sequenced together in one amplicon (see Table S2 for sets). The combined reactions were then cleaned and concentrated by a PCR purification column.
Each set of mutagenesis reactions was then transformed into 10-beta electrocompetent E. coli (New England Biolabs) according to the manufacturer’s instructions. Transformations were conducted in quadruplicate to ensure adequate coverage. Following recovery and outgrowth, the bacteria was directly inoculated into liquid culture, again in quadruplicate for each transformation to avoid bottlenecking (e.g. 16 culture tubes were inoculated per set). Simultaneously, some of the bacteria was serially plated on agar plates to check transformation efficiency and to send for Sanger sequencing to confirm that mutagenesis was successful. Typical transformations resulted in 3 to 4 million colonies and 10 to 40% mutation rate when 30 colonies were sequenced.
The bacterial cultures were combined for each set and the plasmid libraries purified from these cultures by Midiprep. The libraries were transformed into the parental yeast strain. To ensure appropriate coverage, each set was independently transformed 96 times. The transformed yeast were then combined, plated on SC -ura GLU agar plates, and grown at 30 °C for 48 h. Serial dilutions were performed to check transformation efficiency. Typical transformations resulted in approximately 10,000 colonies per transformation, producing on average a million colonies across all the transformations for a given set of mutants.
Yeast were then scraped off all plates into sterile PBS. The concentration of yeast was determined by measuring OD at 595 nm with a spectrophotometer and diluted to be equivalent to a saturated culture (∼40,000 cells/μL). The yeast were then diluted 1:1000 into 1 mL of SC -ura GLU or SC -ura GAL, and grown at 30 °C for 48 h. To control against “jackpotting” events, each outgrowth condition was repeated across 24 replicates.
Following growth, DNA was extracted from the yeast using a lithium-acetate (LiOAc)-SDS lysis method (Lõoke et al., 2011). Briefly, plates were centrifuged at 4000 RPM for 5 min and the supernatant discarded. Pellets were resuspended in 200 μL of 200 mM LiOAc + 1% SDS, then incubated at 70 °C for 20 min. Then, 600 μL of 100% ethanol was added, and mixed well. Plates were centrifuged at 4000 RPM for 10 min, the supernatant discarded, and then allowed to air-dry until the residual ethanol evaporated. Pellets were resuspended in 200 μL of 1X TE, incubated at 42 °C for 30 min, then centrifuged again at 4000 RPM for 10 min. The supernatant, containing the extracted DNA, was removed and transferred to a new plate. DNA was stored at -20 °C until further processing.
Each set was independently amplified and indexed (see Table S1 for primers used for amplification of each set) and then combined and sequenced. DNA from the 24 replicates of each condition were pooled together and used as the template for first-round PCR. The following mix was used for each reaction: 0.5 μL template DNA, 0.1 μL 100 μM forward primer, 0.1 μL 100 μM reverse primer, 0.4 μL 10 mM dNTPs, 4 μL 5X Q5® Reaction Buffer, 0.1 μL Q5® polymerase, 14.8 μL H2O. All PCRs were conducted with six technical replicates to reduce sampling bias. The following cycling conditions were used:
-
1.
98 °C, 45 s
-
2.
98 °C, 15 s
-
3.
57 °C, 20 s
-
4.
72 °C, 1 min
-
5.
Return to step #2 for 27 additional cycles
-
6.
72 °C, 3 min
-
7.
Hold at 4 °C
PCR products were run on a gel to confirm size, and then technical replicates were combined and purified with AMPure XP beads (Beckman Coulter) according to the manufacturer’s instructions. The purified products were used as template for second-round PCR with the following mix: 0.5 μL template DNA, 0.1 μL 100 μM forward primer, 0.1 μL 100 μM reverse primer, 0.4 μL 10 mM dNTPs, 2 μL 10X Taq Reaction Buffer, 0.1 μL Taq polymerase, 16.8 μL H2O. All second-round PCRs were conducted in technical duplicate. The following cycling conditions were used:
-
1.
94 °C, 3 min
-
2.
94 °C, 30 s
-
3.
57 °C, 20 s
-
4.
72 °C, 30 s
-
5.
Return to step #2 for 11 additional cycles
-
6.
72 °C, 3 min
-
7.
Hold at 4 °C
PCR products were gel purified and sequenced on an Illumina NextSeq system with 75 bp single-end reads, except for Sets #8 – 10 which were sequenced on an Illumina NextSeq system with 150 bp single-end reads along with residues 113, 114, 180, 187, 196, 198, 199, and 233.
Determination of catalytic activity of 3CLpro variants from DMS
The computational pipeline for the calculation of activity scores of each 3CLpro variant is available at https://github.com/alejandrochavezlab/3CL_protease_DMS. First, the raw FASTQ files were preprocessed to determine the abundances of mutations compatible with the designed oligos under each of the conditions. This was achieved by aligning the reads to the original SARS-CoV-2 3CLpro sequence using BWA mem (version 0.7.17-r1188) (Li and Durbin, 2009), converting the aligned reads to bam files with SAMtools view, sort, and index (version 1.7-2) (Li et al., 2009), and then determining the counts for sequences consistent with the oligos used for mutagenesis. The raw counts for both replicates in both conditions (induced and non-induced) are available at https://github.com/alejandrochavezlab/3CL_protease_DMS.
After the preprocessing, activity scores were calculated for each variant. We first filtered out counts corresponding to sequences that differed only by one nucleotide from the native wild-type SARS-CoV-2 3CLpro sequence as these counts have a possibility of being artificially inflated due to PCR or sequencing error. Additionally, we applied a count threshold of 10 for the glucose condition for each variant (no threshold for the galactose condition). Following the filtering, a pseudocount of 1 was added to all raw counts, and the resulting counts were normalized to the native wild-type read counts of each set to determine the relative abundance values of all mutant variants within the pool. Using these values, the log fold change (LFC) between the glucose and galactose conditions for each variant was calculated and an activity score determined by performing a one-sided t-test that compared the set of LFC values representing a single amino acid variant against the set of LFC values representing the recoded wild-type variant of the corresponding set. The t-statistic of the t-test was taken to be the activity score of each mutant. This t-statistic represents the difference between the LFC values of a variant and those of the wild-type variants divided by the estimated pooled standard deviation of two sets of LFC values. These obtained scores were then normalized set by set, such that wild-type = 0 and stop codon = -1. We note that the reported wild-type activity score at each residue (Table S3) is derived only from the synonymous codings of the wild-type protease, and does not use the native wild-type DNA sequence which was only used for raw count normalization. The scores are available at https://github.com/alejandrochavezlab/3CL_protease_DMS and also provided as Table S3.
Identification of clinical single nucleotide 3CLpro variants
To obtain a list of 3CLpro variants derived from clinical specimens, we made use of the COVID-CG database (Chen et al., 2021a; Shu and McCauley, 2017). The database was accessed on 3/25/2022 and used to download all amino acid variants from all geographic regions within the 3CLpro, corresponding to nucleotides 10,055 to 10,972. At the time the database was accessed, there were a total of 10,441,890 3CLpro sequences within the database. As it is possible for interactions between mutations to convert a variant that alone is detrimental into one that is well tolerated, we curated the data provided by COVID-CG to identify mutations that occur in isolation within the 3CLpro (≥ 3 occurrences in the database), as these mutants are most similar to the variants we tested within our high-throughput study. Insertions are not considered by COVID-CG so did not need to be removed from the processed list. Deletions were removed as there is no equivalent in our data.
Analysis of residue conservation within 3CLpro
Residue level evolutionary conservation was estimated by using the ConSurf server, run with the default settings on 3/25/2022 (Glaser et al., 2003; Landau et al., 2005). PDB 7JST was used as the input sequence. MAFFT was used as the MSA method. Homologs were collected from the UNIREF90 database using HMMER with settings of E-value 0.0001, 3 iterations, 95% maximal %ID between sequences, and 35% minimal %ID for homologs. 150 sequences closest to the query were chosen for sequence selection. Rate calculation was performed using the Bayesian method. Best fit was used as the model of substitution for proteins. The conservation score from 1 to 9 for each residue as calculated by ConSurf was used to group residues, with low conservation being scores 1 to 3, medium conservation being scores 4 to 6, and high conservation being scores 7 to 9.
Yeast two-hybrid to examine protease dimerization
Yeast two-hybrid experiments were conducted by generating haploid PJ69-4a and PJ69-4ɑ strains expressing SARS-CoV-2 3CLpro C145A with or without additional mutations and fused at the N-terminus with the Gal4 activation domain or DNA binding domain, respectively. The wild-type 3CLpro could not be used for these experiments as it is toxic to the yeast when expressed (Figure S1A). The MATa and MATɑ haploid strains were mated on a YPD agar plate overnight to generate MATa/MATɑ diploids, and then grown to saturation in liquid SC -leu -trp GLU media. Yeast were subsequently diluted 1:500 into 1 mL SC -leu -trp -his GLU (selection media), and grown at 30 °C. Growth was quantified by removing 100 μL from wells after 38 h and measuring OD at 595 nm to assess the dimerization capability of the mutants.
To confirm that these proteases were expressed and that the proteins were not unstable, each of the strains were also examined by immunoblotting. Strains were grown to saturation to equilibrate input cell number and then proteins were extracted from 1 mL of culture by a tricholoroacetic acid (TCA) method (Chun et al., 2019; McCullough et al., 2015). Briefly, cultures were spun down, washed once with 20% TCA, snap frozen and thawed, and then resuspended in 20% TCA with glass beads (acid-washed, 425-600 μm, Sigma). After incubation on ice for 5 min, the mixture was repeatedly vortexed and left on ice to disrupt cells, and then the pellet of extracted proteins were washed with 5% TCA. The pellet was then washed with ice-cold 100% ethanol, and then the pellet was resuspended in 1 M Tris-HCl pH 8.0. Samples were run on SDS-PAGE and transferred to PVDF membranes. Membranes were then washed, blocked with Blotting-Grade Blocker (Bio-Rad), washed, incubated overnight at 4 °C with mouse monoclonal GAL4 antibody (1:100 dilution, Clone #RK5C1, Santa Cruz Biotechnology), washed, incubated with anti-mouse IgG kappa light chain binding protein HRP-conjugated (1:1000 dilution, Santa Cruz Biotechnology) for 1 h at RT, washed, and then developed using SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
Validation of 3CLpro mutations in a SARS-CoV-2 reverse genetics system
Individual 3CLpro mutations were introduced into the pBeloBAC11 bacterial artificial chromosome containing the SARS-CoV-2 genome in which ORF7a was replaced with a NanoLuc luciferase reporter as previously described (Ye et al., 2020). The BAC containing the wild-type 3CLpro was kindly provided by Luis Martinez-Sobrido, and was propagated in the MDS42 E. coli strain to prevent insertion of transposable elements within the viral genome (Umenhoffer et al., 2010). To introduce 3CLpro mutants, the intact BAC was digested with PacI and MluI to remove the WT 3CLpro sequence, overlapping fragments were amplified using primers encoding the desired mutation (Table S1), and the mutant BAC was generated by Gibson assembly (New England Biolabs). The resulting mutant BAC was electroporated into NEB 10-beta cells and purified using the Nucleobond BAC 100 kit (Takara).
These BACs were transfected into HEK293T cells using Lipofectamine 3000® (Thermo Fisher Scientific) in 12 well-plates (2 μg DNA per well) and incubated at 37 °C under 5% CO2 for 6-8 h. A mock transfection in which DNA was not added was included with each set of transfections. The media was changed to fresh complete media (DMEM + 10% FCS + P/S), and further incubated for an additional 40-42 h. These cells were then co-cultivated with naïve Vero E6 cells in media with reduced serum (DMEM + 2% FCS + P/S) and incubated for 7 days. The supernatant was then collected, clarified by centrifugation, and aliquoted and stored at -80 °C until further use.
To normalize the amount of virus used during our subsequent assays quantifying the replication of each mutant, RNA was first extracted from an aliquot from each viral sample using TRIzol™ Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. The extracted RNA was treated with TURBO™ DNase (Thermo Fisher Scientific), and then quantified by qRT-PCR using the TaqMan™ Fast Virus 1-Step Master Mix (Thermo Fisher Scientific) with the TaqMan™ 2019nCoV Assay Kit v1 (Thermo Fisher Scientific). These Ct values were then utilized for normalization of all of the viruses to equilibrate the input volume by RNA, such that adequate signal could be observed by the wild-type SARS-CoV-2 infection. The determined normalized volumes were used to infect naïve Vero E6 cells in complete media in a 24 well-plate. After 72 h, cells were lysed and the luciferase activity was quantified using the Nano-Glo® Luciferase Assay System (Promega) according to the manufacturer’s instructions.
Inhibition assay with recombinant SARS-CoV-2
The E166V mutant was generated as described above. As Vero E6 cells express high levels of the efflux transporter P-glycoprotein, making them unsuitable for this assay, Huh7-ACE2 cells were utilized instead (Owen et al., 2021). Viruses were first titrated to equilibrate input by the Reed-Muench method by infecting Huh7-ACE2 cells at serial dilutions and scoring for positive wells (Reed and Muench, 1938). One day prior to infection, Huh7-ACE2 cells were seeded at a density of 20,000 cells per well in 96-well plates in complete media. The following day, serial five-fold dilutions of nirmatrelvir or remdesivir were prepared in complete media, and then added to cells in triplicate. Cells were then infected with 0.05 MOI of either wild-type or E166V. After 24 h, cells were lysed and the luciferase activity was quantified using the Nano-Glo® Luciferase Assay System according to the manufacturer’s instructions. Inhibition was calculated by comparison to uninfected and infected but untreated cells. EC50 values were determined by nonlinear regression using GraphPad Prism v9.2.
Quantification and statistical analysis
Statistical details of experiments are denoted in the figure legends, including the sample size (n) depicted in each figure. P values between groups in Figure 1C were determined by Kruskal-Wallis test followed by Dunn’s multiple comparisons test using GraphPad Prism v9.2.
Acknowledgments
This work was supported by funding from the Jack Ma Foundation to Y.S., S.P.G., D.D.H., and A.C. and by funding from the JPB Foundation, Andrew and Peggy Cherng, Samuel Yin, Carol Ludwig, and David and Roger Wu to D.D.H. A.C. is also supported by a Career Awards for Medical Scientists from the Burroughs Wellcome Fund and a pilot grant from the Columbia HICCC. S.I. was supported by NIH grant T32AI106711. We thank Luis Martinez-Sobrido and Chengjin Ye for the bacterial artificial chromosome system to generate recombinant SARS-CoV-2; Jesse D. Bloom, Li Xing, and Fang-Yu Lin for their help with the analysis of the data; Sagi Shapira and Barry Honig for their useful discussions; and Ayako Matsuda for assistance with preparation of Figure 2.
Author contributions
A.C. conceived this project. S.I. and S.J.H. conducted the deep mutational scan. J.S., R.T., and A.C. conducted initial studies to develop the screening method. F.F.A., F.B., and H.M. developed and conducted the computational analyses of the screening data. J.S. and A.K.A. provided support to improve the computational analyses. S.I., S.J.H., B.C., M.I.L., Y.S., and A.C. developed the method to generate recombinant SARS-CoV-2 variants and tested those viruses. S.I., S.J.H., J.S., B.C., A.F.K., and A.C. analyzed the results. S.P.G. and D.D.H. contributed to discussions of the data and analysis. H.M., Y.S., D.D.H., and A.C. directed and supervised the project. S.I., S.J.H., B.C., and A.C. wrote the manuscript with input from all authors.
Declaration of interests
S.I., D.D.H., and A.C. are inventors on patent applications related to the development of inhibitors against the SARS-CoV-2 3CL protease.
Published: August 11, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2022.08.003.
Supplemental information
Data and Code Availability
-
•
All raw data tables used for calculating activity scores are available at https://github.com/alejandrochavezlab/3CL_protease_DMS. The activity scores which are plotted in Figures 1A and S1C are also available as Table S3. The raw sequencing data is available for download from the NCBI Short Read Archive under BioProject ID PRJNA813935. The raw data for Figures 1B, 1C, 2A, 2B, 2J, 2K, S1A, S1B, S1D, S2A, and S2B, and the raw Western Blot in Figure S2C are available from Mendeley Data at https://doi.org/10.17632/ktgwchw4n4.1.
-
•
The code for analysis is available at https://github.com/alejandrochavezlab/3CL_protease_DMS.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- Alberti S., Gitler A.D., Lindquist S. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast. 2007;24:913–919. doi: 10.1002/yea.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrila J., Bacha U., Freire E. Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry. 2006;45:14908–14916. doi: 10.1021/bi0616302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., Hohmann E., Chu H.Y., Luetkemeyer A., Kline S., et al. Remdesivir for the treatment of Covid-19 – preliminary Report. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco R., Carrasco L., Ventoso I. Cell killing by HIV-1 protease. J. Biol. Chem. 2003;278:1086–1093. doi: 10.1074/jbc.M205636200. [DOI] [PubMed] [Google Scholar]
- Boras B., Jones R.M., Anson B.J., Arenson D., Aschenbrenner L., Bakowski M.A., Beutler N., Binder J., Chen E., Eng H., et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021;12:6055. doi: 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D., Fink G.R. Yeast: an experimental organism for 21st Century biology. Genetics. 2011;189:695–704. doi: 10.1534/genetics.111.130765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Chen A.T., Altschuler K., Zhan S.H., Chan Y.A., Deverman B.E. COVID-19 CG enables SARS-CoV-2 mutation and lineage tracking by locations and dates of interest. eLife. 2021;10:e63409. doi: 10.7554/eLife.63409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.X., Njoroge F.G. A review of HCV protease inhibitors. Curr. Opin. Investig. Drugs. 2009;10:821–837. [PubMed] [Google Scholar]
- Chen P., Nirula A., Heller B., Gottlieb R.L., Boscia J., Morris J., Huhn G., Cardona J., Mocherla B., Stosor V., et al. SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with Covid-19. N. Engl. J. Med. 2021;384:229–237. doi: 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Hu T., Zhang J., Chen J., Chen K., Ding J., Jiang H., Shen X. Mutation of Gly-11 on the dimer interface results in the complete crystallographic dimer dissociation of severe acute respiratory syndrome coronavirus 3C-like protease: crystal structure with molecular dynamics simulations. J. Biol. Chem. 2008;283:554–564. doi: 10.1074/jbc.M705240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Zhang J., Hu T., Chen K., Jiang H., Shen X. Residues on the dimer interface of SARS coronavirus 3C-like protease: dimer stability characterization and enzyme catalytic activity analysis. J. Biochem. 2008;143:525–536. doi: 10.1093/jb/mvm246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S.C., Chang G.G., Chou C.Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 2010;98:1327–1336. doi: 10.1016/j.bpj.2009.12.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E., Rosa M., Anjum R., Mehmood S., Soban M., Mujtaba M., Bux K., Moin S.T., Tanweer M., Dantu S., et al. Dynamic profiling of beta-coronavirus 3CL M(pro) protease ligand-binding sites. J. Chem. Inf. Model. 2021;61:3058–3073. doi: 10.1021/acs.jcim.1c00449. [DOI] [PubMed] [Google Scholar]
- Chun Y., Joo Y.J., Suh H., Batot G., Hill C.P., Formosa T., Buratowski S. Selective kinase inhibition shows that Bur1 (Cdk9) phosphorylates the Rpb1 linker in vivo. Mol. Cell. Biol. 2019;39 doi: 10.1128/MCB.00602-18. e00602–e00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel F., Hance A.J. HIV drug resistance. N. Engl. J. Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J.M., Samant N., Schneider-Nachum G., Barkan D.T., Yilmaz N.K., Schiffer C.A., Moquin S.A., Dovala D., Bolon D.N.A. Comprehensive fitness landscape of SARS-CoV-2 M(pro) reveals insights into viral resistance mechanisms. eLife. 2022;11:e77433. doi: 10.7554/eLife.77433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler D.M., Fields S. Deep mutational scanning: a new style of protein science. Nat. Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S., Klein J., Robertson A.J., Peña-Hernández M.A., Lin M.J., Roychoudhury P., Lu P., Fournier J., Ferguson D., Mohamed Bakhash S.A.K., et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: a case report. Nat. Commun. 2022;13:1547. doi: 10.1038/s41467-022-29104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett M.E., Galloway J., Chu H.Y., Itell H.L., Stoddard C.I., Wolf C.R., Logue J.K., McDonald D., Weight H., Matsen F.A.t., et al. High-resolution profiling of pathways of escape for SARS-CoV-2 spike-binding antibodies. Cell. 2021;184:2927–2938.e11. doi: 10.1016/j.cell.2021.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.K., Osswald H.L., Prato G. Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. J. Med. Chem. 2016;59:5172–5208. doi: 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R.D., Schiestl R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- Glaser F., Pupko T., Paz I., Bell R.E., Bechor-Shental D., Martz E., Ben-Tal N. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. doi: 10.1093/bioinformatics/19.1.163. [DOI] [PubMed] [Google Scholar]
- Gupta A., Gonzalez-Rojas Y., Juarez E., Crespo Casal M., Moya J., Falci D.R., Sarkis E., Solis J., Zheng H., Scott N., et al. Early treatment for Covid-19 with SARS-CoV-2 neutralizing antibody sotrovimab. N. Engl. J. Med. 2021;385:1941–1950. doi: 10.1056/NEJMoa2107934. [DOI] [PubMed] [Google Scholar]
- Hammond J., Leister-Tebbe H., Gardner A., Abreu P., Bao W., Wisemandle W., Baniecki M., Hendrick V.M., Damle B., Simón-Campos A., et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N. Engl. J. Med. 2022;386:1397–1408. doi: 10.1056/NEJMoa2118542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M.F., Kuo C.J., Chang K.T., Chang H.C., Chou C.C., Ko T.P., Shr H.L., Chang G.G., Wang A.H., Liang P.H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T., Zhang Y., Li L., Wang K., Chen S., Chen J., Ding J., Jiang H., Shen X. Two adjacent mutations on the dimer interface of SARS coronavirus 3C-like protease cause different conformational changes in crystal structure. Virology. 2009;388:324–334. doi: 10.1016/j.virol.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wei P., Fan K., Liu Y., Lai L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- Iketani S., Forouhar F., Liu H., Hong S.J., Lin F.Y., Nair M.S., Zask A., Huang Y., Xing L., Stockwell B.R., et al. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun. 2021;12:2016. doi: 10.1038/s41467-021-22362-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P., Halladay J., Craig E.A. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayk Bernal A., Gomes da Silva M.M., Musungaie D.B., Kovalchuk E., Gonzalez A., Delos Reyes V., Martín-Quirós A., Caraco Y., Williams-Diaz A., Brown M.L., et al. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N. Engl. J. Med. 2022;386:509–520. doi: 10.1056/NEJMoa2116044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Jin Z., Zhao Y., Sun Y., Zhang B., Wang H., Wu Y., Zhu Y., Zhu C., Hu T., Du X., et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020;27:529–532. doi: 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- Kneller D.W., Phillips G., O'Neill H.M., Jedrzejczak R., Stols L., Langan P., Joachimiak A., Coates L., Kovalevsky A. Structural plasticity of SARS-CoV-2 3CL M(pro) active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020;11:3202. doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy N., Fakhro K. Identification of mutation resistance coldspots for targeting the SARS-CoV2 main protease. IUBMB Life. 2021;73:670–675. doi: 10.1002/iub.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Worrall L.J., Vuckovic M., Rosell F.I., Gentile F., Ton A.T., Caveney N.A., Ban F., Cherkasov A., Paetzel M., et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 2020;11:5877. doi: 10.1038/s41467-020-19662-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Iketani S., Zask A., Khanizeman N., Bednarova E., Forouhar F., Fowler B., Hong S.J., Mohri H., Nair M.S., et al. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nat. Commun. 2022;13:1891. doi: 10.1038/s41467-022-29413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Iketani S., Guo Y., Chan J.F., Wang M., Liu L., Luo Y., Chu H., Huang Y., Nair M.S., et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature. 2022;602:676–681. doi: 10.1038/s41586-021-04388-0. [DOI] [PubMed] [Google Scholar]
- Lõoke M., Kristjuhan K., Kristjuhan A. Extraction of genomic DNA from yeasts for PCR-based applications. BioTechniques. 2011;50:325–328. doi: 10.2144/000113672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough L., Connell Z., Petersen C., Formosa T. The abundant histone chaperones Spt6 and FACT collaborate to assemble, inspect, and maintain chromatin structure in Saccharomyces cerevisiae. Genetics. 2015;201:1031–1045. doi: 10.1534/genetics.115.180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu T., Takemoto C., Kim Y.T., Wang H., Nishii W., Terada T., Shirouzu M., Yokoyama S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA. 2016;113:12997–13002. doi: 10.1073/pnas.1601327113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D.R., Allerton C.M.N., Anderson A.S., Aschenbrenner L., Avery M., Berritt S., Boras B., Cardin R.D., Carlo A., Coffman K.J., et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938;27:493–497. [Google Scholar]
- Resnick S.J., Iketani S., Hong S.J., Zask A., Liu H., Kim S., Melore S., Lin F.Y., Nair M.S., Huang Y., et al. Inhibitors of coronavirus 3CL proteases protect cells from protease-mediated cytotoxicity. J. Virol. 2021;95:e0237420. doi: 10.1128/JVI.02374-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmierer B., Botla S.K., Zhang J., Turunen M., Kivioja T., Taipale J. CRISPR/Cas9 screening using unique molecular identifiers. Mol. Syst. Biol. 2017;13:945. doi: 10.15252/msb.20177834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy A.R., Visweswariah S.S. Site-directed mutagenesis using a single mutagenic oligonucleotide and DpnI digestion of template DNA. Anal. Biochem. 2003;319:335–336. doi: 10.1016/s0003-2697(03)00286-0. [DOI] [PubMed] [Google Scholar]
- Shu Y., McCauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017;22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.N., Greaney A.J., Addetia A., Hannon W.W., Choudhary M.C., Dingens A.S., Li J.Z., Bloom J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science. 2021;371:850–854. doi: 10.1126/science.abf9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.N., Greaney A.J., Hilton S.K., Ellis D., Crawford K.H.D., Dingens A.S., Navarro M.J., Bowen J.E., Tortorici M.A., Walls A.C., et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295–1310.e20. doi: 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens L.J., Pruijssers A.J., Lee H.W., Gordon C.J., Tchesnokov E.P., Gribble J., George A.S., Hughes T.M., Lu X., Li J., et al. Mutations in the SARS-CoV-2 RNA dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci Transl. Med. 2022;14:eabo0718. doi: 10.1126/scitranslmed.abo0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasfeld L., Chou S. Antiviral drug resistance: mechanisms and clinical implications. Infect. Dis. Clin. North Am. 2010;24:809–833. doi: 10.1016/j.idc.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Suárez D., Díaz N. SARS-CoV-2 main protease: A Molecular Dynamics study. J. Chem. Inf. Model. 2020;60:5815–5831. doi: 10.1021/acs.jcim.0c00575. [DOI] [PubMed] [Google Scholar]
- Szemiel A.M., Merits A., Orton R.J., MacLean O.A., Pinto R.M., Wickenhagen A., Lieber G., Turnbull M.L., Wang S., Furnon W., et al. In vitro selection of Remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLoS Pathog. 2021;17:e1009929. doi: 10.1371/journal.ppat.1009929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J., Verschueren K.H., Anand K., Shen J., Yang M., Xu Y., Rao Z., Bigalke J., Heisen B., Mesters J.R., et al. PH-dependent conformational flexibility of the SARS-CoV main proteinase (M(pro)) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 2005;354:25–40. doi: 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umenhoffer K., Fehér T., Balikó G., Ayaydin F., Pósfai J., Blattner F.R., Pósfai G. Reduced evolvability of Escherichia coli MDS42, an IS-less cellular chassis for molecular and synthetic biology applications. Microb. Cell Fact. 2010;9:38. doi: 10.1186/1475-2859-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Nair M.S., Liu L., Iketani S., Luo Y., Guo Y., Wang M., Yu J., Zhang B., Kwong P.D., et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature. 2021;593:130–135. doi: 10.1038/s41586-021-03398-2. [DOI] [PubMed] [Google Scholar]
- Wang Q., Guo Y., Iketani S., Nair M.S., Li Z., Mohri H., Wang M., Yu J., Bowen A.D., Chang J.Y., et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4, and BA.5. Nature. 2022;608:603–608. doi: 10.1038/s41586-022-05053-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich D.M., Sivapalasingam S., Norton T., Ali S., Gao H., Bhore R., Musser B.J., Soo Y., Rofail D., Im J., et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19. N. Engl. J. Med. 2021;384:238–251. doi: 10.1056/NEJMoa2035002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C., Chiem K., Park J.G., Oladunni F., Platt R.N., 2nd, Anderson T., Almazan F., de la Torre J.C., Martinez-Sobrido L. Rescue of SARS-CoV-2 from a single bacterial artificial chromosome. mBio. 2020;11 doi: 10.1128/mBio.02168-20. e02168–e02120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Zhu Y., Liu X., Jin Z., Duan Y., Zhang Q., Wu C., Feng L., Du X., Zhao J., et al. Structural basis for replicase polyprotein cleavage and substrate specificity of main protease from SARS-CoV-2. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2117142119. e2117142119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Gammeltoft K.A., Ryberg L.A., Pham L.V., Fahnoe U., Binderup A., Hernandez C.R.D., Offersgaard A., Fernandez-Antunez C., Peters G.H.J., et al. Nirmatrelvir resistant SARS-CoV-2 variants with high fitness in vitro. bioRxiv. 2022 doi: 10.1101/2022.06.06.494921. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Cao Z., Liu Z., He Y., Wang Y., Yuan P., Li W., Tian F., Bao Y., Wei W. Guide RNAs with embedded barcodes boost CRISPR-pooled screens. Genome Biol. 2019;20:20. doi: 10.1186/s13059-019-1628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All raw data tables used for calculating activity scores are available at https://github.com/alejandrochavezlab/3CL_protease_DMS. The activity scores which are plotted in Figures 1A and S1C are also available as Table S3. The raw sequencing data is available for download from the NCBI Short Read Archive under BioProject ID PRJNA813935. The raw data for Figures 1B, 1C, 2A, 2B, 2J, 2K, S1A, S1B, S1D, S2A, and S2B, and the raw Western Blot in Figure S2C are available from Mendeley Data at https://doi.org/10.17632/ktgwchw4n4.1.
-
•
The code for analysis is available at https://github.com/alejandrochavezlab/3CL_protease_DMS.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.