

Abstract
Bivalent ligands have emerged as chemical tools to study G protein-coupled receptor dimers. Using a combination of computational, chemical, and biochemical tools, here we describe the design of bivalent ligand 13 with high affinity (KDB1=21 pM) for the dopamine D2 receptor (D2R) homodimer. Bivalent ligand 13 enhances the binding affinity relative to monovalent compound 15 by 37-fold, indicating simultaneous binding at both protomers. Using synthetic peptides with amino acid sequences of transmembrane (TM) domains of D2R, we provide evidence that TM6 forms the interface of the homodimer. Notably, the disturber peptide TAT-TM6 decreased the binding of bivalent ligand 13 by 52-fold and had no effect on monovalent compound 15, confirming the D2R homodimer through TM6 ex vivo. In conclusion, using a versatile multivalent chemical platform, we have developed a precise strategy to generate a true bivalent ligand that simultaneously targets both orthosteric sites of the D2R homodimer.
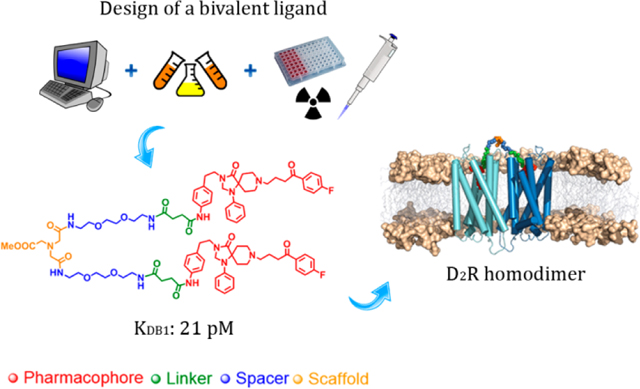
Graphical Abstract
INTRODUCTION
It is now well accepted that many G protein-coupled receptors (GPCRs) form, in addition to functional monomers,1 dimers and higher-order oligomeric complexes constituted by a number of equal (homo) or different (hetero) monomers.2 Oligomerization plays an important role in terms of receptor function and structure, introducing changes in signaling pathways which are due to the allosteric mechanisms of these complexes. Thus, these oligomers present functional properties different from those of the constituent monomers (protomers), making oligomerization a biological resource to generate pharmacological diversity.3 Considering the involvement of GPCRs in the regulation of many physiological processes, these novel functional units have recently received special attention as new targets for drug development.4 Besides the set of existing biochemical and biophysical tools,5 to gain insight into the mechanisms by which oligomers signal, specific chemical tools can also contribute to evaluate their pharmacology and to assess their potentiality as drug targets.
One of these tools are bivalent ligands, defined as single chemical entities composed of two pharmacophore units covalently linked by an appropriate spacer. These ligands are designed to interact simultaneously with a (homo/hetero) GPCR dimer to enhance affinity and subtype selectivity.6 Homobivalent ligands contain two copies of the same pharmacophore,7 whereas heterobivalent ligands link two different pharmacophores.8 A requirement for bivalent ligands is the simultaneous binding of the two pharmacophores at the orthosteric sites of the (homo/hetero) dimer. Thus, the spacer length is a key factor in these ligands and depends on the dimer interface, the structure of the pharmacophores, and the geometry of the attachment points.9 If the spacer length is not suitable to cover the distance between the orthosteric sites of both GPCR dimer protomers these ligands act in a non-simultaneous interaction mode, with a dual-acting profile.10 Other types of ligands composed by two pharmacophores connected by a spacer, but designed to interact simultaneously with orthosteric and allosteric sites, are referred as bitopic ligands.11
Considering the above-mentioned diversity of interaction modes of these types of compounds, the generation of a bivalent ligand requires not only a precise design, but also an accurate validation of its type of interaction. Using a combination of computational, chemical and biochemical tools, here we describe the design of a true bivalent ligand with high affinity for the dopamine D2 receptor (D2R) homodimer. We have selected the D2R as a test case for two reasons: a) because it forms homo/heterodimers12 and higher-order oligomers13 implicated in several neuropsychiatric disorders, such as Parkinson disease or schizophrenia;14 and b) due to the existing controversy regarding the interaction mode of some of the described D2R homodimer bivalent ligands.15 D2 receptors are present in several tissues and cell lines and, as other class A GPCRs, exist in a dynamic equilibrium between monomers, dimers and higher order oligomers. It has been suggested that bivalent ligands act stabilizing preexisting dimers;16 however, recent data shows that some of them can modulate the dynamics of oligomerization shifting the equilibrium towards the dimeric state.17
RESULTS AND DISCUSSION
Design.
The design of bivalent ligands requires the selection of: i. a scaffold that contains at least two chemical functionalities that can be properly derivatized; ii. a ligand that binds the orthosteric binding site with high affinity (pharmacophore unit); iii. an appropriate length spacer to cover the distance between both protomers; and, finally, iv. if necessary, a linker between this pharmacophore and the bivalent system, adequate in terms of both the topological position of the attachment point and the chemistry used for the conjugation (Figure 1).9
Figure 1.

A) Components for the design of bivalent ligands. B) Bivalent ligands (12–13) and their corresponding monovalent counterparts (14–15).
Herein, the selected scaffold (i) is the nitrilotriacetic acid (NTA), which contains three symmetric carboxylic acids and permits the controlled desymmetrization of each of these functional groups.18 This multivalent platform allows not only the attachment of two pharmacophore units, but also the introduction of a reporter molecule for imaging studies or another pharmacophore unit to study higher-order oligomers, such as trimers.19
As a proof of concept, a neutral antagonist has been selected as pharmacophore with the aim to design bivalent ligands whose potential simultaneous interaction with the D2R homodimer would result only in increased affinity values, avoiding cooperative mechanisms that could encumber the evaluation of the binding interaction. The selected pharmacophore unit (ii) is a derivative of the D2R antagonist spiperone, namely the N-(p-aminophenethyl)spiperone 6 (NAPS),20 which was functionalized with an extra succinic acid linker (iv) to facilitate its incorporation to the bivalent system (Figure 1). The resulting pharmacophore-linker derivative 7 was docked into a D3-based homology model of D2R, and its stability was assessed by 1μs of unbiased molecular dynamic (MD) simulations. (Figure S1). Results showed that the pharmacophore unit (red) remains highly stable at the binding site during the simulation, whereas the linker moiety (green) is very flexible and achieves diverse conformations between extracellular loops (ECL) 2 and 3, always at the extracellular aqueous environment, which makes the selected attachment point (purple) adequate to link the spacer moieties.
The selected spacers (iii) were different length oligoethylene glycol (OEG) moieties with the aim to increase water solubility of the final bivalent ligands. A key factor in the design of bivalent ligands is the spacer length, which depends on the dimer interface. Crystal structures of GPCRs display several dimerization interfaces21 that can be grouped into three clusters, depending on the transmembrane helices (TMs) involved: TMs 1 and 2 (TM1/2 interface), TMs 4 and 5 (TM4/5 interface), and TMs 5 and 6 (TM5/6 interface). Using a computational tool,9b developed in house for the Molecular Operating Environment (MOE) software (Chemical computing group Inc., Montreal QC, Canada), we calculated the preferred spacer length for the different interfaces via the shortest pathway along the D2R homodimer van der Waals surface (Table S1). This surface represents a favorable interaction between the dimer and the spacer/linker/pharmacophore moieties of the bivalent ligand. We constructed sets of molecules formed by the pharmacophore (starting at the N atom of the amide bond of the triazaspiro moiety) / linker / spacer (-OCH2CH2-)n=2–4 / scaffold / spacer (-OCH2CH2-)n=2–4 / linker / pharmacophore (ending at the N atom of the amide bond of the triazaspiro moiety) as inputs for our MOE-based tool. The tool predicts the favorable conformation of these input molecules, and the interaction energy between these atoms and the rest of the system, calculated for each of the input molecules after a stepped energy minimization protocol.9b The lengths of energetically favorable spacers were used as recommendations for synthesis.
The TM5/6 dimerization interface led to the ligand with the shortest spacer/scaffold/spacer (25-atoms, calculated between attachment atoms shown in purple in Figure 1), because this interface has the shortest distance between orthosteric sites (33 Å), and also the attachment point directs toward TMs 5 and 6 (Table S1). The TM4/5 interface gave the largest distance (43-atoms, 43 Å), whereas the TM1/2 interface is in between (31 atoms, 36 Å) (Table S1). Based on these data, we designed two bivalent ligands: 13 (25-atoms between both attachment atoms), representing the shortest possible bivalent interaction (via TM5/6), and a longer alternative, 12 (35-atoms), which could also interact at other dimer interaction interfaces, excluding TM4/5, which is on the opposite side to the direction of the linker elongation, and therefore implausible to reach it.
Chemical synthesis.
The NTA-based core 2 was prepared according to literature procedures22 starting from glycine methyl ester hydrochloride, which was dialkylated with benzyl bromoacetate, and then hydrogenated to remove the benzyl protecting groups, affording desired compound in 83% yield (Scheme S1).
The pharmacophore-linker derivative 7 was prepared following the described methodology with minor modifications.20a Briefly, the N-alkylation of spiperone with 4-(N-tert-butyloxycarbonyl)aminophenethyl bromide afforded 5 in 65% yield. Removal of Boc group using HCl (2 M in dioxane) provided NAPS (6, 66% yield) which was subsequently acylated with succinic anhydride to afford compound 7 (76% yield).
OEG-based precursors 8, 10 and 9, 11 were prepared from commercially available OEGs in good yields (88% for 8, 87% for 9, and 57% for 10, 56% for 11) (Scheme S3). The final bivalent ligands 12 and 13 were synthesized by acylation of two carboxylic acids of the NTA scaffold (2) with compounds 10 or 11, respectively (Scheme 1). Finally, the synthesis of monovalent ligands 14 and 15 required differentiation between the free carboxylic acids of 2. This desymmetrization was accomplished by means of the favored formation of a six-membered cyclic anhydride between the pair of carboxylic acids, which reacted selectively with only one equivalent of 8 or 9 to form the amide. Then, the resulting free carboxylic acid could be acylated in a further step using the corresponding compounds 10 or 11 respectively (Scheme 1).
Scheme 1.

a Synthesis of the bivalent ligands 12 and 13 and the monovalent ligands 14 and 15.
a Reagents and conditions: (a) 10, EDC·HCl, HOBt·H2O, DIEA, DMF, rt, 16 h (49%); (b) 11, EDC·HCl, HOBt·H2O, DIEA, DMF, rt, 16 h (64%); (c) EDC·HCl, dry DMF, rt, 2 h, then 8, DIEA, dry DMF, rt, 90 min (79%); (d) 10, EDC·HCl, HOBt·H2O, DIEA, DMF, rt, 16 h (25%); (e) EDC·HCl, dry DMF, rt, 2 h, then 9, DIEA, dry DMF, rt, 90 min (80%); (f) 11, EDC·HCl, HOBt·H2O, DIEA, DMF, rt, 16 h (24%).
Biological assays.
In vitro binding affinities of the bivalent ligands (12 and 13) and the corresponding monovalent counterparts (14 and 15) were obtained from [3H]YM-09151–2 radioligand competition-binding assays using membranes from sheep brain striatum that naturally express D2R. Data were analyzed according to a ‘two-state dimer model’ (Table 1).23 The model assumes GPCR dimers as a main functional unit and provides a more robust analysis of parameters obtained from saturation and competition experiments with orthosteric ligands, as compared with the commonly used ‘two-independent-site model’.23,24 In competition experiments the model analyzes the interactions of the radioligand with a competing ligand and it provides the affinity of the competing ligand for the first protomer in the unoccupied dimer (KDB1) and the affinity of the competing ligand for the second protomer when the first protomer is already occupied by the competing ligand (KDB2). All studied compounds show monophasic non-cooperative curves, as expected for an antagonist with a non-cooperative binding to D2R dimer. In these conditions, KDB1 is enough to characterize the binding of these compounds.
Table 1.
Affinity constants (KDB1) of the D2R ligands 7, 12–15 with or without TM6 peptides.
| Compound | KDB1 (nM) | + TM6 D2R | + TM6 A2AR |
|---|---|---|---|
| 7 | 0.70±0.06 | ||
| 12 | 0.07±0.03*### | ||
| 13 | 0.021±0.003**### | 1.1±0.3^^ | 0.05±0.01 |
| 14 | 1.5±0.6* | ||
| 15 | 0.77±0.04 | 0.8±0.2 | 0.8±0.2 |
Values are mean±SEM from 3–10 determinations. Statistical significance was calculated by one-way ANOVA followed by Bonferroni’s post hoc test.
p<0.05
p<0.01 compared with 7.
p<0.001 compared with the corresponding monovalent ligand.
p<0.01 compared with the respective control without TM peptides.
Compound 7 has high affinity for D2R (KDB1=0.70 nM). Monovalent compound 15 (25-atoms, KDB1=0.77 nM) has similar affinity for D2R than compound 7, whereas monovalent compound 14 (35-atoms, KDB1=1.5 nM) shows a slightly less favorable binding affinity. These results are remarkable since attachment of the spacer should decrease binding affinity. This suggests that the OEG spacer favorably interacts with residues at the groove connecting both protomers. Notably, bivalent ligands 12 (35-atoms, KDB1=0.07 nM) and 13 (25-atoms, KDB1=0.021 nM) significantly enhance the binding affinity relative to monovalent counterparts 14 and 15 (21-fold and 37-fold, respectively). Clearly, addition of the second pharmacophore unit increases binding affinity due to its higher local concentration in a close radius above the second protomer. Thus, compounds 12 and 13 seem to act as bivalent ligands, that is, both pharmacophores simultaneously target both orthosteric sites of the homodimer.
To further test that the antagonistic nature of these compounds on D2R signaling remains unaltered, we resolved the real-time signaling signature by using a label-free method (DMR)25 in CHO cells stably co-expressing A2AR and D2R to mimic the pattern receptor expression of brain striatum, where a high proportion of D2R form heteromers with A2AR.13 This approach detects changes in local optical density due to cellular mass movements induced upon receptor activation (see Experimental Section). The magnitude of the signaling by sumanirole, a highly selective D2R full agonist, significantly decreased in the presence of both bivalent ligands 12 and 13 as much as when adding spiperone (Figures 2A and 2B). Because the affinity of compound 13 is 3.5-fold higher than 12, additional biochemical experiments were carried out with 13 and its corresponding monovalent counterpart 15. Compound 13 had a better inhibitory potency antagonizing sumanirole signal (15±3 nM) than the corresponding monovalent ligand 15 (280±70 nM) due to its higher affinity (Figure 2C).
Figure 2.

Antagonistic effect of the studied compounds on global cellular response induced by sumanirole. Dynamic mass redistribution (DMR) assays were performed in CHO cells stably expressing D2R and A2AR. A) Quantification of the antagonist effect of all D2R ligands on DMR. Values are mean±SEM from 3 determinations carried out in triplicates. Statistical significance was calculated by one-way ANOVA followed by Dunnett’s post hoc test. **p<0.01 compared to sumanirole alone. B) Representative DMR curves from one of these experiments in which cells were treated with medium (control) (black), with 1μM of spiperone (grey), with 1μM of monovalent compounds 14 (light blue) or 15 (dark blue) or with 500 nM of bivalent compounds 12 (orange) or 13 (red) for 30 minutes. After that, cells were treated with 100 nM of sumanirole. Each curve is the mean of a representative optical trace experiment carried out in triplicates. The resulting shifts of reflected light wavelength (pm) were monitored over time. C) Dose-response of the antagonistic effect of bivalent compound 13 (black) (IC50=15±3 nM) and monovalent 15 (red) (IC50=280±70 nM) on the DMR induced by 100 nM sumanirole. Data are mean±SEM from 3–8 experiments and are presented as percentage of the maximal effect of sumanirole.
Because of the higher affinity of 13, we predicted the TM5/6 interface for homodimerization of D2R. To validate this hypothesis with a bimolecular fluorescence complementation (BiFC) assay in HEK-293T cells, we used synthetic peptides with the amino acid sequence of TMs 5 and 6 and TM7 (negative control) of D2R fused to the cell-penetrating HIV transactivator of transcription (TAT) peptide to alter inter-protomer interactions.13,26 In this assay, two complementary halves of YFP (Venus variant; cYFP and nYFP) are separately fused to the D2 receptor and the fluorescence is obtained after reconstitution of the functional YFP when the D2 receptors homodimerize. Only the transmembrane peptide TAT-TM6 bound to the receptor and disturbed the quaternary structure of the homodimer, causing a significant fluorescence decrease (Figure 3), indicating that only TM6 forms the interface of the D2R homodimer, according to the recently reported results.26 We also tested the ability of compounds 13 and 15 to modulate the dynamics of oligomerization, as it has been suggested for other bivalent compounds using TIRF microscopy.17 With this aim, we treated the cells transfected with the two complementary halves of YFP with 100 nM of compounds 13 or 15 for 10 minutes before reading the fluorescence. Under our conditions, neither bivalent compound 13 nor monovalent 15 significantly altered the dimerization state in fluorescence complementation assays (p > 0.793) (Figure S2).
Figure 3.

Effect of TAT-TM peptides on disturbance of the D2R homodimer, determined by BiFC experiments in HEK-293T cells transfected with D2R-nYFP and D2R-cYFP cDNA. Values are mean±SEM from 6–9 determinations. Statistical significance was calculated by one-way ANOVA followed by Bonferroni’s post hoc test. ***p<0.001 compared to non TAT-TM treated complementation.
Because we have identified the TAT-TM6 peptide as a disturber of the inter-protomer interaction, we tested the binding affinity of compounds 13 and 15 in the presence of TAT-TM6 peptides of D2R and adenosine A2R (negative control) in native tissue (Figure 4). Neither TAT-TM6 peptide of D2R nor A2R influenced the binding of monovalent compound 15. In contrast, TAT-TM6 peptide of D2R, but not TAT-TM6 peptide of A2R, decreased the binding of the bivalent ligand 13 (KDB1 (13)=0.021nM vs. KDB1 (13+TM6)=1.1nM). Remarkably, in the presence of the TAT-TM6 peptide of D2R, bivalent compound 13 performed as the monovalent compound 15 (KDB1 (13+TM6)=1.1nM vs. KDB1 (15)=0.77nM).
Figure 4.

Effect of TAT-TM6 peptides of D2R and A2AR on competition experiments of [3H]YM-09151–2 vs. D2R ligands. Competition curves with increasing concentrations of monovalent 15 (in A) or bivalent 13 (in B) D2R ligands in the absence (black) or in the presence of TAT-TM6 of A2AR (red) or TAT-TM6 of D2R (blue), using membranes from sheep brain striatum. Data are mean±SEM from 3 experiments performed in triplicate.
This suggests that the TAT-TM6 peptide alters the homodimer in such a way that compound 13 binds the orthosteric binding site of the first protomer without reaching the second one. These results show the importance of the simultaneous binding of the two pharmacophore units at both orthosteric sites of the homodimer for obtaining an improvement in affinity, and confirm the inter-protomer interaction of D2R homodimer through TM6. These results also ratify the bivalent interaction mode of compound 13, validating it as a true bivalent ligand. Interestingly, in brain striatal tissue, the effect caused by the TAT-TM6 peptide seems enough to avoid the simultaneous occupancy of both orthosteric binding sites of the homodimer by the bivalent ligand but, in the BiFC assay performed with HEK-293T cells, the homodimer is also bound by the complemented fluorescent protein and this may hamper the disruption caused by the transmembrane peptide. In fact, at the beginning of the usage of TAT-TM peptides to disturb oligomers, some authors hypothesized that the complementation was irreversible. However, more recently, different papers have reported that it is actually reversible.26–28
Molecular modelling of bivalent ligand 13 into D2R homodimer model.
Accordingly, we constructed a computational model of the D2R homodimer, using exclusively TM6 as the molecular interface (see Experimental Section), and performed 1μs of unbiased MD simulations to evaluate the stability of compound 13 in the model (Figures 5 and S3-S4). This TM6 interface predicts similar distances between orthosteric binding sites than the TM5/6 interface, thus, leading to the same number of atoms for the spacer. The MD simulations showed that compound 13 comfortably fulfills and maintains simultaneous binding of the two pharmacophoric units at both orthosteric sites throughout the simulation (Figure S3), thus, providing further confidence in the bivalent interaction and the picomolar binding affinity.
Figure 5.

Evolution of bivalent ligand 13 in the D2R homodimer (TM1 and TM4, ECL1 and part of ECL2 are omitted for clarity), constructed via the TM6 interface, as devised from MD simulations. The structures of 13 (the color code of the atoms is as in Figure 1) are extracted from the simulations (20 structures collected every 50 ns), whereas the structure of the D2R homodimer corresponds to the initial model. A detailed analysis of the simulation (Figure S3) confirms that the designed bivalent ligand 13 remains stable at the orthosteric binding cavities through the unbiased 1 μs MD simulation.
CONCLUSIONS
We have developed a precise strategy to create bivalent ligands of GPCR (homo/hetero) dimers based on a versatile multivalent chemical platform. The use of computational tools that consider the TM interfaces, distances between orthosteric binding sites and mode of interaction of the pharmacophore units, allows a reduction in the number of synthesized bivalent ligands, yet a high success in the affinity results. Bivalent ligand 13 showed picomolar binding affinity, and the use of different TAT-TM6 disturber peptides allowed the confirmation, in native tissue, of its simultaneous interaction with both orthosteric sites of the D2R homodimer, constituted through TM6. Furthermore, our results confirm the recently described interface interaction of D2R homodimer through TM6.
This strategy can be applied to other GPCR oligomers, thus allowing the generation and validation of novel ligands with a clear bivalent interaction mode. These ligands can be used as pharmacological tools in combination with disturber TAT-TM peptides to validate inter-protomer GPCR interactions, both in vitro and in native tissue, and this information could be potentially used for the design of new therapeutic compounds targeting GPCR oligomers.
EXPERIMENTAL SECTION
General Methods.
Reagents and solvents were purchased from commercial sources and were used without further purification. TLC was performed on Merck 60F254 silica plates were visualized by UV light (254 nm), or by potassium permanganate stains. Flash chromatography on silica was carried out on a Teledyne Isco Combiflash Rf instrument using Redisep Rf silica columns. 1H-NMR (400 MHz) and 13C-NMR (101 MHz) spectroscopy was performed on a Varian Mercury 400 MHz instrument at the NMR unit of the Scientific and Technological Centers of the University of Barcelona (CCiTUB). Chemical shifts (δ) are expressed in ppm relative to tetramethylsilane (TMS). Coupling constants (J) are expressed in Hertz (Hz). The following abbreviations are used to indicate multiplicity: s: singlet; d: doublet, t: triplet, m: multiplet, and br: broad signal. Analytical RP-HPLC and mass spectra were performed on a Waters Alliance 2795 with an automated injector and a photodiode array detector Waters 2996 coupled to an electrospray ion source (ESI-MS) Micromass ZQ mass detector, using a XSelect™ C18 reversed-phase analytical column (4.6 mm×50 mm, 3.5 μm), and the MassLynx 4.1 software. The instrument was operated in the positive ESI (+) ion mode. Analyses were carried out with several elution systems. System A: a linear gradient 5–100% CH3CN (0.07% HCOOH) in H2O (0.1% HCOOH) over 4.5 min at a flow rate of 2 mL/min; and System B: a linear gradient 5–100% CH3CN (0.07% HCOOH) in H2O (0.1% HCOOH) over 3.5 min at a flow rate of 1.6 mL/min. Purity of all test compounds was determined by HPLC analysis to be ≥95 %. High-Resolution Mass Spectroscopy (HRMS) was carried out using an LC/MSD-TOF spectrometer from Agilent Technologies, at the molecular characterization mass spectrometry unit of the Scientific and Technological Centers of the University of Barcelona (CCiTUB). Semi-preparative RP-HPLC purification was performed on a Waters system with a 2545 binary gradient module, a 2767 manager collector and a 2489 UV detector, coupled to an electrospray ion source (ESI-MS) Micromass ZQ mass detector, and the MassLynx 4.1 software. Gradients and columns used are detailed in each case.
Synthesis.
4-((4-(2-(8-(4-(4-fluorophenyl)-4-oxobutyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-3-yl)ethyl)phenyl)amino)-4-oxobutanoic acid (7).
To a solution of 6 (267 mg, 519 μmol, 1.0 eq) in CH3CN (15 mL) was added succinic anhydride (62.3 mg, 623 μmol, 1.2 eq) and the mixture was stirred at room temperature overnight (16 h). After completion of the reaction the mixture was evaporated to dryness. The resulting crude was dissolved in CH2Cl2 (20 mL) and immediately washed with brine (2×20 mL). The resulting organic phase was dried over MgSO4 and evaporated. The crude was purified by flash chromatography on silica using CH2Cl2 and MeOH as solvents (0 to 15% MeOH in CH2Cl2) to afford compound 7 as a white solid (244 mg, 397 μmol, 76%). 1H NMR (400 MHz, DMSO-d6, 298 K) δ 1H NMR (400 MHz, DMSO-d6) δ 9.89 (s, NH), 8.10 – 8.01 (m, 2H), 7.52 – 7.45 (m, 2H), 7.39 – 7.30 (m, 2H), 7.22 – 7.12 (m, 4H), 6.80 – 6.71 (m, 3H), 4.57 (s, 2H), 3.54 (t, J = 7.2 Hz, 2H), 3.02 (t, J = 6.9 Hz, 2H), 2.83 (t, J = 7.2 Hz, 2H), 2.78 – 2.60 (m, 4H), 2.56 – 2.45 (m, 4H), 2.45 – 2.30 (m, 4H), 1.87 – 1.76 (m, 2H), 1.45 – 1.36 (m, 2H); 13C NMR (101 MHz, DMSO-d6, 298 K) δ 198.5, 173.8, 173.0, 169.9, 164.8 (d, J = 251.1 Hz), 143.0, 137.6, 133.8 (d, J = 2.7 Hz), 132.9, 130.8 (d, J = 9.3 Hz), 129.0, 128.8, 118.9, 118.0, 115.6 (d, J = 21.9 Hz), 114.6, 62.8, 59.5, 57.0, 49.0, 41.2, 35.8, 32.0, 31.0, 28.9, 28.4, 21.3; HPLC: System A, tR: 2.02 min, 99% (214 nm), 99% (240 nm); LRMS: calculated mass for C35H40FN4O5: 615.3 [M+H]+, found by HPLC-MS (ESI): 615.2.
2-oxo-7,10,13-trioxa-3-azahexadecan-16-aminium chloride (8).
To a solution of 1-(tert-butoxycarbonyl-amino)-4,7,10-trioxa-13-tridecanamine (401 mg, 1.25 mmol, 1.0 eq) in CH2Cl2 (4 mL) was added acetic anhydride (130 μL, 1.38 mmol, 1.1 eq) and DIEA (470 μL, 2.76 mmol, 2.2 eq). The resulting mixture was stirred at room temperature for 3 h. After this time, the crude was washed with saturated NaHCO3 (2×5 mL) and brine (1×5 mL). The organic phase was dried over MgSO4 and evaporated. Subsequent treatment of the crude with a 2 M solution of HCl in dioxane (10 mL, 20 mmol, 16 eq) at room temperature for 1 h, followed by the evaporation of dioxane and HCl to dryness, afforded compound 8 as a pale yellow oil (352 mg, 1.10 mmol, 88%). 1H NMR (400 MHz, D2O, 298 K) δ 3.73 – 3.64 (m, 10H), 3.58 (t, J = 6.4 Hz, 2H), 3.25 (t, J = 6.8 Hz, 2H), 3.12 (t, J = 7.2 Hz, 2H), 2.01 – 1.92 (m, 5H), 1.83 – 1.75 (m, 2H); 13C NMR (101 MHz, D2O, 298 K) δ 173.9, 69.5, 69.4, 69.3, 69.2, 68.3, 68.2, 37.6, 36.4, 28.1, 26.4, 21.8; LRMS: calculated mass for C12H27ClN2O4 (hydrochloride): 298.2, calculated mass for C12H27N2O4 (amine): 263.2 [M+H]+, found by HPLC-MS (ESI): 263.0.
2-(2-(2-acetamidoethoxy)ethoxy)ethan-1-aminium chloride (9).
To a solution of 1-(tert-butoxycarbonyl-amino)-3,6-dioxa-8-octanamine (120 mg, 0.48 mmol, 1.0 eq) in CH2Cl2 (4 mL) was added acetic anhydride (50 μL, 0.53 mmol, 1.1 eq) and DIEA (181 μL, 1.06 mmol, 2.2 eq). The resulting mixture was stirred at room temperature for 3 h. After this time, the crude was washed with saturated NaHCO3 (2×5 mL) and brine (1×5 mL). The organic phase was dried over MgSO4 and evaporated. Subsequent treatment of the crude with a 2 M solution of HCl in dioxane (10 mL, 20 mmol, 16 eq) at room temperature for 1 h, followed by the evaporation of dioxane and HCl to dryness, afforded compound 9 as a pale yellow oil (95.3 mg, 0.42 mmol, 87%). 1H NMR (400 MHz, D2O, 298 K) δ 3.76 (t, J = 5.0 Hz, 2H), 3.70 (s, 4H), 3.63 (t, J = 5.4 Hz, 2H), 3.38 (t, J = 5.4 Hz, 2H), 3.21 (br t, J = 5.1 Hz, 2H), 1.99 (s, 3H); 13C NMR (101 MHz, D2O, 298 K) δ 174.3, 69.5, 69.4, 68.7, 66.3, 39.0, 38.9, 21.8; LRMS: calculated mass for C8H19ClN2O3: 226.1 (hydrochloride), calculated mass for C8H19N2O3 (amine): 191.1 [M+H]+, found by HPLC-MS (ESI): 190.9.
18-((4-(2-(8-(4-(4-fluorophenyl)-4-oxobutyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-3-yl)ethyl)phenyl)amino)-15,18-dioxo-4,7,10-trioxa-14-azaoctadecan-1-aminium chloride (10).
To a mixture of 7 (50.7 mg, 82.5 μmol, 1.0 eq), EDC·HCl (23.8 mg, 0.12 mmol, 1.5 eq) and HOBt·H2O (19.0 mg, 0.12 mmol, 1.5 eq) was added a solution of 1-(tert-butoxycarbonyl-amino)-4,7,10-trioxa-13-tridecanamine (39.7 mg, 0.12 mmol, 1.5 eq) in DMF (5 mL). The resulting mixture was stirred at room temperature overnight (18 h). After this time the solvent was evaporated to dryness. The crude was dissolved in AcOEt (15 mL) and washed with saturated NaHCO3 (3×15 mL), 0.5% w/v citric acid (3×15 mL) and brine (1×15 mL). The organic phase was dried over MgSO4 and evaporated to obtain the Boc-protected compound (44.3 mg, 48.3 μmol). This compound was dissolved in dioxane (1 mL) and a 4 M solution of HCl in dioxane (0.5 mL, 2.0 mmol, 41 eq) was added. The mixture was stirred at room temperature for 1 h. Then, the dioxane and HCl were evaporated to dryness. Finally, the crude was dissolved in H2O (1 mL) and lyophilized to afford compound 10 (40.1 mg, 47.0 μmol, 57%). 1H NMR (400 MHz, D2O, 298 K) δ 8.05 – 7.97 (m, 2H), 7.43 – 7.32 (m, 4H), 7.32 – 7.19 (m, 4H), 7.10 – 7.02 (m, 1H), 7.02 – 6.93 (m, 2H), 5.45 (s, NH), 4.64 (s, 2H), 3.76 – 3.67 (m, 2H), 3.68 – 3.57 (m, 8H), 3.57 – 3.50 (m, 2H), 3.52 – 3.34 (m, 6H), 3.28 – 3.02 (m, 8H), 2.93 (br t, J = 6.5 Hz, 2H), 2.70 – 2.55 (m, 2H), 2.57 – 2.38 (m, 4H), 2.13 – 1.96 (m, 2H), 1.96 – 1.89 (m, 2H), 1.83 – 1.60 (m, 4H); 13C NMR (101 MHz, D2O, 298 K) δ 200.7, 174.3, 173.2, 172.7, 165.9 (d, J = 253.6 Hz), 141.7, 135.8, 134.9, 132.4, 131.0 (d, J = 9.6 Hz), 129.6, 129.6, 121.9, 121.2, 118.4, 115.8 (d, J = 22.0 Hz), 69.4, 69.3, 69.2, 68.2, 63.5, 59.1, 56.0, 48.8, 41.5, 37.6, 36.2, 34.9, 32.0, 31.8, 31.0, 28.2, 27.0, 26.4, 18.0; HPLC: System B, tR: 1.67 min, 98% (214 nm), 97% (240 nm); LRMS: calculated mass for C45H62ClFN6O7: 852.4 (hydrochloride), calculated mass for C45H62FN6O7 (amine): 817.5 [M+H]+, found by HPLC-MS (ESI): 817.3.
2-(2-(2-(4-((4-(2-(8-(4-(4-fluorophenyl)-4-oxobutyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-3-yl)ethyl)phenyl)amino)-4-oxobutanamido)ethoxy)ethoxy)ethan-1-aminium chloride (11).
To a mixture of 7 (50.6 mg, 82.3 μmol, 1.0 eq), EDC·HCl (23.6 mg, 0.12 mmol, 1.5 eq) and HOBt·H2O (18.8 mg, 0.12 mmol, 1.5 eq) was added a solution of 1-(tert-butoxycarbonyl-amino)-3,6-dioxa-8-octanamine (30.5 mg, 0.12 mmol, 1.5 eq) in DMF (5 mL). The resulting mixture was stirred at room temperature overnight (18 h). After this time the solvent was evaporated to dryness. The crude was dissolved in AcOEt (15 mL) and washed with saturated NaHCO3 (3×15 mL), 0.5% w/v citric acid (3×15 mL) and brine (1×15 mL). The organic phase was dried over MgSO4 and evaporated to obtain the Boc-protected compound (50.9 mg, 60.2 μmol). This compound was dissolved in dioxane (1 mL) and a 4 M solution of HCl in dioxane (0.5 mL, 2.0 mmol, 39 eq) was added. The mixture was stirred at room temperature for 1 h. Then, the dioxane and HCl were evaporated to dryness. Finally, the crude was dissolved in H2O (1 mL) and lyophilized to afford compound 11 (36.0 mg, 46.1 μmol, 56%). 1H NMR (400 MHz, D2O, 298 K) δ 8.04 – 7.94 (m, 2H), 7.42 – 7.29 (m, 4H), 7.31 – 7.17 (m, 4H), 7.09 – 7.02 (m, 1H), 7.00 – 6.92 (m, 2H), 4.63 (s, 2H), 3.77 – 3.65 (m, 4H), 3.66 – 3.58 (m, 4H), 3.54 (t, J = 5.5 Hz, 2H), 3.47 – 3.33 (m, 4H), 3.32 (t, J = 5.5 Hz, 2H), 3.25 – 3.04 (m, 6H), 2.92 (br t, J = 6.6 Hz, 2H), 2.69 – 2.57 (m, 2H), 2.57 – 2.50 (m, 2H), 2.52 – 2.38 (m, 2H), 2.11 – 1.93 (m, 2H), 1.65 (d, J = 14.5 Hz, 2H); 13C NMR (101 MHz, D2O, 298 K) δ 200.7, 174.6, 173.2, 172.8, 165.9 (d, J = 253.8 Hz), 141.6, 135.7, 134.9, 132.4, 131.0 (d, J = 9.9 Hz), 129.6, 129.6, 121.8, 121.2, 118.4, 115.8 (d, J = 22.2 Hz), 69.5, 69.4, 68.8, 66.3, 63.4, 59.1, 56.0, 48.7, 41.5, 39.0, 38.8, 34.9, 32.0, 31.6, 30.7, 27.0, 18.0; HPLC: System B, tR: 1.60 min, 99% (214 nm), 97% (240 nm); LRMS: calculated mass for C41H54ClFN6O6: 780.4 (hydrochloride), calculated mass for C41H54FN6O6 (amine): 745.4 [M+H]+, found by HPLC-MS (ESI): 745.2.
Methyl 24-((4-(2-(8-(4-(4-fluorophenyl)-4-oxobutyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-3-yl)ethyl)phenyl)amino)-3-(21-((4-(2-(8-(4-(4-fluorophenyl)-4-oxobutyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-3-yl)ethyl)phenyl)amino)-2,18,21-trioxo-7,10,13-trioxa-3,17-diazahenicosyl)-5,21,24-trioxo-10,13,16-trioxa-3,6,20-triazatetracosanoate (12).
Compound 2 (2.6 mg, 12.7 μmol, 1.0 eq), compound 10 (21.6 mg, 25.4 μmol, 2.0 eq), EDC·HCl (7.3 mg, 38.0 μmol, 3.0 eq) and HOBt·H2O (5.8 mg, 38.0 μmol, 3.0 eq) were dissolved in DMF (2 mL) and DIEA (7.0 μL, 41.1 μmol, 3.2 eq) was added. The resulting mixture was stirred at room temperature overnight (16 h). After this time the solvent was evaporated to dryness, and the crude was purified by semi-preparative reversed-phase HPLC (45 to 72% acetonitrile in aqueous 10 mM NH4HCO3 in 8 min, XBridge C18 19×150 mm 5μm) affording compound 12 (11.3 mg, 6.27 μmol, 49%). 1H NMR (400 MHz, CDCl3, 298 K) δ 9.21 (s, 2 NH), 8.05 – 7.94 (m, 4H), 7.76 – 7.65 (m, 2 NH), 7.52 – 7.42 (m, 4H), 7.33 – 7.21 (m, 4H), 7.20 – 7.08 (m, 8H), 7.05 – 6.97 (m, 2 NH), 6.91 – 6.78 (m, 6H), 4.58 (s, 4H), 3.77 – 3.65 (m, 7H), 3.65 – 3.42 (m, 30H), 3.42 – 3.23 (m, 16H), 3.12 (t, J = 6.7 Hz, 4H), 3.09 – 2.95 (m, 8H), 2.91 (t, J = 7.1 Hz, 4H), 2.71 – 2.50 (m, 8H), 2.17 (br s, 4H), 1.82 – 1.66 (m, 8H), 1.54 (d, J = 14.4 Hz, 4H); 13C NMR (101 MHz, CDCl3, 298 K) δ 172.6, 172.2, 171.1, 170.8, 166.0 (d, J = 255.4 Hz), 142.2, 137.5, 133.0, 132.9, 130.8 (d, J = 9.1 Hz), 129.7, 129.3, 120.2, 119.7, 115.9 (d, J = 21.9 Hz), 114.9, 77.2, 70.6, 70.2, 70.1, 69.9, 69.8, 69.4, 63.6, 58.7, 56.6, 56.0, 52.0, 48.8, 41.8, 38.0, 37.2, 35.6, 33.1, 31.7, 29.4, 29.0, 27.4; HPLC: System A, tR: 2.18 min, >99% (214 nm), >99% (240 nm); LRMS: calculated mass for C97H130F2N13O18: 1803.0 [M+H]+, found by HPLC-MS (ESI): 1803.0, 902.3 [M+2H]2+, 601.9 [M+3H]3+. HRMS (ESI): calculated exact mass for C97H130F2N13O18 [M+H]+: 1802.9619, found 1802.9618.
Methyl 19-((4-(2-(8-(4-(4-fluorophenyl)-4-oxobutyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-3-yl)ethyl)phenyl)amino)-3-(16-((4-(2-(8-(4-(4-fluorophenyl)-4-oxobutyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-3-yl)ethyl)phenyl)amino)-2,13,16-trioxo-6,9-dioxa-3,12-diazahexadecyl)-5,16,19-trioxo-9,12-dioxa-3,6,15-triazanonadecanoate (13).
Compound 2 (2.7 mg, 13.1 μmol, 1.0 eq), compound 11 (20.5 mg, 26.2 μmol, 2.0 eq), EDC·HCl (7.5 mg, 39.3 μmol, 3.0 eq) and HOBt·H2O (6.0 mg, 39.3 μmol, 3.0 eq) were dissolved in DMF (2 mL) and DIEA (7.0 μL, 41.1 μmol, 3.1 eq) was added. The resulting mixture was stirred at room temperature overnight (16 h). After this time the solvent was evaporated to dryness, and the crude was purified by semi-preparative reversed-phase HPLC (45 to 67% acetonitrile in aqueous 10 mM NH4HCO3 in 8 min, XBridge C18 19×150 mm 5μm) affording compound 13 (14.0 mg, 8.44 μmol, 64 %).
1H NMR (400 MHz, CDCl3, 298 K) δ 9.11 (s, 2 NH), 8.04 – 7.93 (m, 4H), 7.81 (br t, J = 5.7 Hz, 2 NH), 7.51 – 7.41 (m, 4H), 7.30 – 7.23 (m, 4H), 7.22 – 7.16 (m, 2 NH), 7.16 – 7.09 (m, 8H), 6.93 – 6.87 (m, 4H), 6.87 – 6.81 (m, 2H), 4.57 (s, 4H), 3.74 – 3.63 (m, 7H), 3.58 – 3.31 (m, 38H), 3.12 (t, J = 6.7 Hz, 4H), 3.05 – 2.94 (m, 8H), 2.91 (t, J = 6.9 Hz, 4H), 2.70 – 2.54 (m, 8H), 2.24 – 2.11 (m, 4H), 1.50 (d, J = 14.2 Hz, 4H); 13C NMR (101 MHz, CDCl3, 298 K) δ 197.1, 173.2, 172.9, 172.1, 171.1, 171.0, 166.0 (d, J = 255.0 Hz), 142.3, 137.4, 133.1, 133.0, 130.8 (d, J = 9.3 Hz), 129.7, 129.3, 120.3, 119.6, 115.9 (d, J = 21.9 Hz), 115.1, 70.4, 69.8, 63.6, 59.0, 58.5, 56.3, 55.9, 52.0, 48.4, 41.7, 39.5, 39.2, 35.7, 33.0, 31.6, 27.2, 18.8; HPLC: System A, tR: 2.08 min, 98% (214 nm), >99% (240 nm); LRMS: calculated mass for C89H114F2N13O16: 1658.8 [M+H]+, found by HPLC-MS (ESI): 1658.9, 830.3 [M+2H]2+, 553.9 [M+3H]3+. HRMS (ESI): calculated exact mass for C89H114F2N13O16 [M+H]+: 1658.8469, found: 1658.8454.
Methyl 3-(2,18-dioxo-7,10,13-trioxa-3,17-diazanonadecyl)-24-((4-(2-(8-(4-(4-fluorophenyl)-4-oxobutyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-3-yl)ethyl)phenyl)amino)-5,21,24-trioxo-10,13,16-trioxa-3,6,20-triazatetracosanoate (14).
Compound 2 (45.0 mg, 219 μmol, 1.0 eq) and EDC·HCl (42.0 mg, 219 μmol, 1.0 eq) were dissolved in dry DMF (1 mL) and the mixture was stirred at room temperature for 2 h under Ar atmosphere. Then, a solution of compound 8 (65.5 mg, 219 μmol, 1.0 eq) and DIEA (75 μL, 441 μmol, 2.0 eq) in dry DMF (1 mL) was added and the resulting mixture was stirred at room temperature for 90 min. After this time the solvent was evaporated to dryness, and the crude was purified by Waters Porapak™ Rxn RP column (aqueous 10 mM NH4HCO3) to afford the intermediate 3-(2-methoxy-2-oxoethyl)-5,21-dioxo-10,13,16-trioxa-3,6,20-triazadocosanoic acid (78.4 mg, 174 μmol, 79%). 1H NMR (400 MHz, D2O, 298 K) δ 3.70 (s, 3H), 3.68 – 3.59 (m, 8H), 3.59 – 3.50 (m, 6H), 3.37 (s, 2H), 3.34 – 3.24 (m, 4H), 3.21 (t, J = 6.8 Hz, 2H), 1.95 (s, 3H), 1.85 – 1.70 (m, 4H); 13C NMR (101 MHz, D2O, 298 K) δ 178.7, 174.0, 173.9, 173.8, 69.5, 69.3, 69.2, 68.3, 68.2, 58.2, 58.2, 55.4, 52.0, 36.4, 35.9, 28.2, 28.1, 21.8; MS: calculated exact mass for C19H36N3O9: 450.2 [M+H]+, found by HPLC-MS (ESI): 450.2. This intermediate (8.0 mg, 17.8 μmol, 1.0 eq), compound 10 (16.7 mg, 19.6 μmol, 1.1 eq), EDC·HCl (5.1 mg, 26.7 μmol, 1.5 eq) and HOBt·H2O (4.1 mg, 26.7 μmol, 1.5 eq) were dissolved in DMF (1.5 mL) and DIEA (7.0 μL, 41.1 μmol, 2.2 eq) was added. The resulting mixture was stirred at room temperature overnight (15 h). After this time the solvent was evaporated to dryness, and the crude was purified by semi-preparative reversed-phase HPLC (37 to 45% acetonitrile in aqueous 10 mM NH4HCO3 in 8 min, XBridge C18 19×150 mm 5μm) affording compound 14 (5.5 mg, 4.40 μmol, 25%). 1H NMR (400 MHz, CDCl3, 298 K) δ 9.19 (br s, NH), 8.03 – 7.94 (m, 2H), 7.68 (br s, 2 NH), 7.51 – 7.43 (m, 2H), 7.31 – 7.22 (m, 2H), 7.20 – 7.09 (m, 4H), 7.00 (br s, NH), 6.90 – 6.78 (m, 3H), 6.56 (br s, NH), 4.58 (s, 2H), 3.76 – 3.66 (m, 5H), 3.66 – 3.42 (m, 28H), 3.42 – 3.26 (m, 14H), 3.22 – 2.97 (m, 6H), 2.93 (t, J = 7.0 Hz, 2H), 2.70 – 2.54 (m, 4H), 2.27 – 2.11 (m, 2H), 1.95 (s, 3H), 1.82 – 1.67 (m, 8H), 1.52 (d, J = 14.8 Hz, 2H); 13C NMR (101 MHz, CDCl3, 298 K) δ 172.7, 172.1, 171.1, 170.8, 170.5, 137.5, 132.8, 130.8 (d, J = 9.4 Hz), 129.7, 129.3, 120.2, 119.7, 116.0 (d, J = 21.8 Hz), 114.8, 70.6, 70.2, 70.1, 70.0, 69.8, 69.6, 69.5, 63.6, 58.8, 58.7, 56.0, 52.0, 48.8, 41.7, 38.1, 38.0, 37.3, 37.3, 35.5, 33.2, 33.1, 32.1, 31.8, 29.9, 29.8, 29.4, 29.1, 29.0, 27.1, 23.4, 22.8; HPLC: System A, tR: 1.98 min, 99% (214 nm), 98% (240 nm); LRMS: calculated mass for C64H95FN9O15: 1248.7 [M+H]+, found by HPLC-MS (ESI): 1248.5, 624.9 [M+2H]2+. HRMS (ESI): calculated exact mass for C64H95FN9O15: 1248.6926 [M+H]+, found: 1248.6936.
Methyl 3-(2,13-dioxo-6,9-dioxa-3,12-diazatetradecyl)-19-((4-(2-(8-(4-(4-fluorophenyl)-4-oxobutyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-3-yl)ethyl)phenyl)amino)-5,16,19-trioxo-9,12-dioxa-3,6,15-triazanonadecanoate (15).
Compound 2 (42.1 mg, 205 μmol, 1.0 eq) and EDC·HCl (39.3 mg, 205 μmol, 1.0 eq) were dissolved in dry DMF (1 mL) and the mixture was stirred at room temperature for 2 h under Ar atmosphere. Then, a solution of compound 9 (46.5 mg, 205 μmol, 1.0 eq) and DIEA (70 μL, 412 μmol, 2.0 eq) in dry DMF (1 mL) was added and the resulting mixture was stirred at room temperature for 90 min. After this time the solvent was evaporated to dryness, and the crude was purified by Waters Porapak™ Rxn RP column (aqueous 10 mM NH4HCO3) to afford the intermediate 3-(2-methoxy-2-oxoethyl)-5,16-dioxo-9,12-dioxa-3,6,15-triazaheptadecanoic acid (62.1 mg, 164 μmol, 80%). 1H NMR (400 MHz, D2O, 298 K) δ 3.71 (s, 3H), 3.68 – 3.56 (m, 10H), 3.44 (t, J = 5.5 Hz, 2H), 3.41 (s, 2H), 3.36 (t, J = 5.3 Hz, 2H), 3.30 (s, 2H), 1.98 (s, 3H); 13C NMR (101 MHz, D2O, 298 K) δ 178.6, 174.3, 174.1, 174.0, 69.4, 68.7, 68.7, 58.1, 58.1, 55.2, 52.0, 38.9, 38.5, 21.7; LRMS: calculated mass for C15H28N3O8: 378.2 [M+H]+, found by HPLC-MS (ESI): 378.1. This intermediate (7.25 mg, 19.2 μmol, 1.0 eq), compound 11 (16.5 mg, 21.1 μmol, 1.1 eq), EDC·HCl (5.5 mg, 28.8 μmol, 1.5 eq) and HOBt·H2O (4.4 mg, 28.8 μmol, 1.5 eq) were dissolved in DMF (1.5 mL) and DIEA (7.5 μL, 44.1 μmol, 2.3 eq) was added. The resulting mixture was stirred at room temperature overnight (15 h). After this time the solvent was evaporated to dryness, and the crude was purified by semi-preparative reversed-phase HPLC (35 to 43% acetonitrile in aqueous 10 mM NH4HCO3 in 8 min, XBridge C18 19×150 mm 5μm) affording compound 15 (5.0 mg, 4.53 μmol, 24%). 1H NMR (400 MHz, CDCl3, 298 K) δ 8.87 (br s, NH), 8.04 – 7.95 (m, 2H), 7.69 – 7.57 (m, 2 NH), 7.50 – 7.41 (m, 2H), 7.31 – 7.20 (m, 2H), 7.19 – 7.10 (m, 4H), 6.97 (br s, NH), 6.90 – 6.79 (m, 3H), 6.55 (br s, NH), 4.57 (s, 2H), 3.75 – 3.65 (m, 5H), 3.66 – 3.27 (m, 34H), 3.10 (br s, 6H), 2.92 (t, J = 6.8 Hz, 2H), 2.70 – 2.57 (m, 4H), 2.14 (br s, 2H), 1.98 (s, 3H), 1.51 (d, J = 14.3 Hz, 2H); 13C NMR (101 MHz, CDCl3, 298 K) δ 172.8, 172.2, 170.9, 170.9, 170.8, 170.7, 166.0 (d, J= 254.9 Hz), 137.3, 130.8 (d, J = 9.4 Hz), 129.6, 129.3, 120.2, 119.6, 115.9 (d, J = 21.9 Hz), 115.1, 70.4, 70.3, 70.1, 69.9, 69.8, 63.7, 58.7, 56.0, 52.1, 49.0, 39.6, 39.5, 39.2, 39.2, 33.1, 32.1, 31.7, 29.8, 23.3, 22.8; HPLC: System A, tR: 1.90 min, 96% (214 nm), 96% (240 nm); LRMS: calculated mass for C56H79FN9O13: 1104.6 [M+H]+, found by HPLC-MS (ESI): 1104.8, 552.9 [M+2H]2+. HRMS (ESI): calculated exact mass for C56H79FN9O13 [M+H]+: 1104.5776, found: 1104.5799.
TM with TAT peptides.
A peptide derived from the HIV transactivator of transcription, HIV TAT (YGRKKRRQRRR), was fused to peptides with the amino acid sequences of human A2AR or D2R TM domain 6, human D2R TM domain 5 and human D2R TM domain 7 (Genemed Synthesis), to promote integration of the TM domains in the plasma membrane. Because HIV TAT binds to the phosphatidylinositol-(4, 5)-bisphosphate found on the inner surface of the membrane, HIV TAT peptide was fused to the N-terminus of TM6 and to the C-terminus of TM5 and TM7 to obtain the right orientation of the inserted peptide. The amino acid sequences were:
TAT-TM6 of D2R: YGRKKRRQRRR M374LAIVLGVFIICWLPFFITHIL395;
TAT-TM6 of A2AR: YGRKKRRQRRRL235AIIVGLFALCWLPLHIINCFTFF258;
TM5-TAT of D2R: F189VVYSSIVSFYVPFIVTLLVYIKIY213YGRKKRRQRRR;
TM7-TAT of D2R: A410FTWLGYVNSAVNPIIYTTFNI431YGRKKRRQRRR.
Radioligand binding experiments.
Brains of male and female sheep of 4–6 months old were freshly obtained from the local slaughterhouse. Striatal brain tissues were disrupted with a Polytron homogenizer (PTA 20 TS rotor, setting 3; Kinematica, Basel, Switzerland) for two 5 s-periods in 10 volumes of 50 mM Tris-HCl buffer at pH 7.4, containing a proteinase inhibitor cocktail (Sigma, St. Louis, MO, USA). Membranes were obtained by centrifugation, twice at 105000 g for 45 min at 4°C. The pellet was stored at −80°C, washed once more as described above and resuspended in 50 mM Tris-HCl buffer for immediate use. Membrane protein was quantified by the bicinchoninic acid method (Pierce Chemical Co., Rockford, IL, USA) using bovine serum albumin dilutions as standard. Binding experiments were performed with membrane suspensions at room temperature in 50 mM Tris-HCl buffer at pH 7.4, containing 10 mM MgCl2. For D2R competition-binding assays, membrane suspensions (0.2 mg of protein/mL) were incubated for 2 h with a constant free concentration of 0.8 nM of the D2R antagonist [3H]YM-09151–2 (KDA1 = 0.15 nM) and increasing concentrations of each tested ligand. Non-specific binding was determined in the presence of 30 μM of dopamine, because at this concentration dopamine does not displace the radioligand from sigma receptors. Competition-binding assays using TAT-TM peptides were performed as described previously, but preincubating peptides and membranes for 1 h before the addition of the ligands and the radioligand. In all cases, free and membrane-bound ligands were separated by rapid filtration of 500 μL aliquots in a cell harvester (Brandel, Gaithersburg, MD, USA) through Whatman GF/C filters embedded in 0.3% polyethylenimine, that were subsequently washed for 5 s with 5 mL of ice-cold 50 mM Tris-HCl buffer. The filters were incubated with 10 mL of Ecoscint H scintillation cocktail (National Diagnostics, Atlanta, GA, USA) overnight at room temperature and radioactivity counts were determined using a Tri-Carb 2800 TR scintillation counter (PerkinElmer) with an efficiency of 62%. Radioligand competition curves were analyzed by nonlinear regression using the commercial Grafit curve-fitting software (Erithacus Software, Surrey, UK) by fitting the binding data to the mechanistic two-state dimer receptor model.23 The macroscopic equilibrium dissociation constants from competition experiments were determined applying the following general equation:
. where A represents the radioligand concentration, B the assayed competing compound concentration and KDAB the hybrid allosteric modulation between A and B. For A and B non-cooperative and non-allosteric modulation between A and B, the equation can be simplified due to the fact that ;
Cell culture.
CHO cells stably co-expressing the human cDNAs of A2AR and D2R were obtained and tested as described in Orru et al. (2011).29 This clone was grown in Minimum Essential Medium (MEMα; Gibco) supplemented with 2 mM L-glutamine, 100 μg/mL sodium pyruvate, MEM nonessential amino acid solution (1/100), 100U/mL penicillin/streptomycin, 5% (vol/vol) of heat-inactivated FBS (all supplements from Invitrogen) and with 600 mg/mL Geneticin (G 418 Sulfate, Calbiochem) and 300 mg/mL Hygromycin B (Invitrogen). HEK-293T cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 2 mM L-glutamine, 100 μg/mL sodium pyruvate, MEM nonessential amino acid solution (1/100), 100U/mL penicillin/streptomycin, 5% (vol/vol) of heat-inactivated FBS. All cells were cultured at 37°C and 5% CO2.
Dynamic Mass Redistribution (DMR) Assay.
The global cell signaling profile was measured using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, Massachusetts, US). This label-free approach uses refractive waveguide grating optical biosensors, integrated into 384-well microplates. Changes in local optical density are measured in a detection zone up to 150 nm above the surface of the sensor. Cellular mass movements induced upon receptor activation are detected by illuminating the underside of the biosensor with polychromatic light and measured as changes in the wavelength of the reflected monochromatic light. These changes are a function of the refraction index. The magnitude of this wavelength shift (in picometers) is directly proportional to the amount of DMR. CHO cells stably co-expressing A2AR and D2R were used to perform the DMR. Briefly, 24 h before the assay, cells were seeded at a density of 7,000 cells per well in 384-well sensor microplates with 30 μL growth medium and cultured for 24 h (37°C, 5% CO2) to obtain 70%–80% confluent monolayers. Previous to the assay, cells were washed twice with assay buffer (media with 20 mM HEPES, pH 7.15, 0.1% DMSO and 0.1% BSA) and incubated 2 h in 30 μL per well of non assay buffer in the reader at 24°C. Hereafter, the sensor plate was scanned, and a baseline optical signature was recorded for 10 min before adding 10 μL of the antagonist compound dissolved in the assay buffer at different concentrations. The DMR response was recorded for 30 min. Finally, 10 μL of a 100 nM solution of the agonist (sumanirole) dissolved in the assay buffer was added and recorded for at least 90 min. The resulting shifts of reflected light wavelength (pm) were monitored over time. Kinetic results were analyzed using EnSpire Workstation Software v 4.10.
Expression vectors and fusion proteins.
For bimolecular fluorescence complementation experiments, in order to obtain receptors fused to the hemitruncated Venus variant of the YFP, sequences encoding the amino acid residues 1–155 (nYFP) and 156–238 (cYFP) of the YFP Venus, were subcloned in pcDNA3.1 vector. Moreover, the human cDNA for D2R cloned into pcDNA3.1 was subcloned to be in-frame with restriction sites EcoRI and BamHI of the pcDNA3.1-nYFP and the pcDNA3.1-cYFP. Between the receptor and the hemitruncated fluorescence protein there is a linker of 36 nucleotides: TTCTGCAGATATCCAGCACAGTGGCGGCCGCTCGAG.
Bimolecular fluorescence complementation (BiFC).
HEK-293T cells were transiently co-transfected with lipofectamine with the cDNA encoding D2R fused to nYFP and/or with the same amount of the receptor fused to cYFP. After 48 h, cells were treated or not with the indicated TAT-TM peptides (4 μM) for 4 h at 37°C. To quantify protein reconstructed YFP Venus expression, cells resuspended in HBSS (Hank’s Balanced Salt Solution) supplemented with glucose 0.1 % were distributed (20 μg protein; 50,000 cells/well) in 96-well microplates (black plates with a transparent bottom, Porvair, King’s Lynn, UK), and emission fluorescence at 530 nm was determined in a FLUOstar Optima Fluorimeter (BMG Labtechnologies, Offenburg, Germany) equipped with a high-energy xenon flash lamp, using a 10-nm bandwidth excitation filter at 400 nm reading. In order to study the effect of our compounds in the dynamics of oligomerization, we treated the cells with 5 μL of HBSS or with 100 nM of ligands 13 or 15 for 10 minutes before reading the fluorescence. Protein fluorescence expression was determined as the fluorescence of the sample minus the fluorescence of cells not expressing the fusion proteins.
Computational models of the D2R monomer and homodimer.
A homology model of D2R (Uniprot code P14416) was constructed from the crystal structure of D3R (PDB id 3PBL)30 using Modeller 9.12.31 The structure of D2R has recently been revealed32 and showed a remarkably similar structure with the homology model (root mean-square deviation between Cα atoms of 0.9Å). As noted by the authors the major differences relative to D3R are in ECL1, the much longer ECL2, and the extracellular end of TM 6 that shows an outward movement.32 Three computational models of the D2R homodimer were built using alternative transmembrane (TM) helix interfaces: the TM1/2 (involving TMs1 and 2 and helix 8) and TM5/6 (involving TMs 5 and 6) interfaces using the crystal of the μ-opioid receptor (4DKL)33 as a template, and the TM4/5 (involving TMs 4 and 5) interface using the crystal structure of the β1-adrenergic receptor (4GPO).34 Nevertheless, the results with disturber peptides indicate a direct interaction exclusively between TM6 of D2R in the homodimer (Figure 3). Due to the absence of crystal structures of oligomers using exclusively the TM6 interface, the D2R homodimer was additionally modelled with HADDOCK2.235 using residues K3676.29 – I3846.46 as directly involved in the interaction. The stability of this TM6 interface homodimer was evaluated by molecular dynamic (MD) simulations.
Docking of ligands.
The pharmacophore-linker derivative 7 was docked into D2R using MOE (Chemical computing group Inc., Montreal, QC, Canada). Inspired by computational scripts that link fragments in a binding site for fragment-based drug discovery, we developed a MOE-based computational tool to design the optimal spacer size connecting the attachment points of the pharmacophore-linker derivative 7 (Table S1). This tool was used to model bivalent ligands 12 and 13 into the D2R homodimer. The selection of the preferred spacer was based upon the interaction energy between ligand and protein, internal energy of the ligand, and visual inspection.
Molecular dynamic simulations.
The pharmacophore-linker derivative 7, in complex with the D2R monomer, and bivalent ligand 13, in complex with the D2R homodimer constructed via the TM6 interface, were embedded in a pre-equilibrated box (9×9×9 nm3 for monomers and 12×12×10 nm3 for homodimers) containing a lipid bilayer (∼205 or ∼300 molecules of POPC) with explicit solvent (∼14000 or ∼30.000 water molecules) and 0.15 M concentration of Na+ and Cl- ions (∼140 or ∼330 ions). Model systems were energy minimized and subjected to a 6 step MD equilibration (10+5+2+2+2+2 ns) in which constraints on hydrogen atoms, protein loops, and protein and ligand atoms were subsequently relaxed. Next, these restraints were released, and unrestrained MD trajectories were produced for 0.5 μs for compound 7 in complex with the D2R monomer and for 1 μs for compound 13 in complex with the D2R homodimer. A 2 fs time step and constant temperature of 300K was used. All bonds and angles were kept frozen using the LINCS algorithm. Lennard-Jones interactions were computed using a cutoff of 10 Å, and electrostatic interactions were treated using PME with the same real-space cutoff. The AMBER99SD-ILDN force field was used for the protein, the parameters described by Berger and co-workers for lipids, the general Amber force field (GAFF) and HF/6–31G*-derived RESP atomic charges for ligands. This combination of protein and lipid parameters has previously been validated.36 All simulations were performed using GROMACS software v5.1.4.37
Supplementary Material
ACKNOWLEDGMENT
This study was funded by grants from the Spanish Ministerio de Economía, Industria y Competitividad (SAF2014-60138-R to MR, SAF2014-54840-R to VC, SAF2016-77830-R to LP, and SAF2015-74627-JIN to AC; those grants may include FEDER funds), CIBER-BBN (CB06-01-0074), CIBERNED (CB06/05/0064), Generalitat de Catalunya (2014SGR137, 2014SGR1236, 2017SGR1439 and 2017SGR1497), “Fundació La Marató de TV3” Grant 20140610, and the intramural funds of the National Institute on Drug Abuse (SF).
ABBREVIATIONS
- A2AR
adenosine A2A receptor
- BiFC
bimolecular fluorescence complementation
- CHO
Chinese hamster ovary
- D2R
dopamine D2 receptor
- DIEA
diisopropyl ethylamine
- DMR
dynamic mass redistribution
- ECL
extracellular loop
- EDC·HCl
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- HOBt·H2O
1-Hydroxybenzotriazole hydrate
- MOE
molecular operating environment
- NAPS
N-(p-aminophenethyl)spiperone
- NTA
nitrilotriacetic acid
- OEG
oligoethylene glycol
- TAT
transactivator of transcription
- TM
transmembrane
- YFP
yellow fluorescent protein
Footnotes
ASSOCIATED CONTENT
The following files are available free of charge. Synthetic schemes for compounds 1–11, experimental procedures for compounds 1–6, supporting figures S1-S4, table S1, 1H- and 13C-NMR spectra of compounds 1–15 and HPLC traces of compounds 7 and 12–15 (PDF). Molecular formula strings for all synthesized compounds (CSV).
The authors declare no competing financial interest.
REFERENCES
- (1).Whorton MR; Bokoch MP; Rasmussen SGF; Huang B; Zare RN; Kobilka B; Sunahara RK A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. USA 2007, 104, 7682–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ferré S; Casadó V; Devi LA; Filizola M; Jockers R; Lohse MJ; Milligan G; Pin JP; Guitart X. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol. Rev 2014, 66, 413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Farran B. An update on the physiological and therapeutic relevance of GPCR oligomers. Pharmacol. Res 2017, 117, 303–327. [DOI] [PubMed] [Google Scholar]
- (4).(a) Santos R; Ursu O; Gaulton A; Bento AP; Donadi RS; Bologa CG; Karlsson A; Al-Lazikani B; Hersey A; Oprea TI; Overington JP A comprehensive map of molecular drug targets. Nat. Rev. Drug Discovery 2017, 16, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gaitonde SA; González-Maeso J. Contribution of heteromerization to G protein-coupled receptor function. Curr. Opin. Pharmacol 2017, 32, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Guo H; An S; Ward R; Yang Y; Liu Y; Guo XX; Hao Q; Xu TR Methods used to study the oligomeric structure of G-protein-coupled receptors. Biosci. Rep 2017, 37, BSR20160547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Hiller C; Kühhorn J; Gmeiner P. Class A G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J. Med. Chem 2013, 56, 6542–6559. [DOI] [PubMed] [Google Scholar]; (b) Soriano A; Ventura R; Molero A; Hoen R; Casadó V; Cortés A; Fanelli F; Albericio F; Lluís C; Franco R; Royo M. Adenosine A2A receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A-D2 receptor heteromers. J. Med. Chem 2009, 52, 5590–5602. [DOI] [PubMed] [Google Scholar]; (c) Daniels DJ; Lenard NR; Etienne CL; Law P-Y; Roerig SC; Portoghese PS Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 19208–19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Busnelli M; Kleinau G; Muttenthaler M; Stoev S; Manning M; Bibic L; Howell LA; McCormick PJ; Di Lascio S; Braida D; Sala M; Rovati GE; Bellini T; Chini B. Design and characterization of superpotent bivalent ligands targeting oxytocin receptor dimers via a channel-like structure. J. Med. Chem 2016, 59, 7152–7166. [DOI] [PubMed] [Google Scholar]
- (8).Hübner H; Schellhorn T; Gienger M; Schaab C; Kaindl J; Leeb L; Clark T; Möller D; Gmeiner P. Structure-guided development of heterodimer-selective GPCR ligands. Nat. Commun 2016, 7, 12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Shonberg J; Scammells PJ; Capuano B. Design strategies for bivalent ligands targeting GPCRs. ChemMedChem 2011, 6, 963–974. [DOI] [PubMed] [Google Scholar]; (b) Pérez-Benito L; Henry A; Matsoukas MT; Lopez L; Pulido D; Royo M; Cordomí A; Tresadern G; Pardo L. The size matters? A computational tool to design bivalent ligands. Bioinformatics, 2018, doi: 10.1093/bioinformatics/bty422. [DOI] [PMC free article] [PubMed]
- (10).Jörg M; May LT; Mak FS; Lee KCK; Miller ND; Scammells PJ; Capuano B. Synthesis and pharmacological evaluation of dual acting ligands targeting the adenosine A2A and dopamine D2 receptors for the potential treatment of parkinson’s disease. J. Med. Chem 2015, 58, 718–738. [DOI] [PubMed] [Google Scholar]
- (11).(a) Kumar V; Moritz AE; Keck TM; Bonifazi A; Ellenberger MP; Sibley CD; Free RB; Shi L; Lane JR; Sibley DR; Newman AH Synthesis and pharmacological characterization of novel trans-cyclopropylmethyl-linked bivalent ligands that exhibit selectivity and allosteric pharmacology at the dopamine D3 receptor (D3R). J. Med. Chem 2017, 60, 1478–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mohr K; Schmitz J; Schrage R; Tränkle C; Holzgrabe U. Molecular alliance-from orthosteric and allosteric ligands to dualsteric/bitopic agonists at G protein coupled receptors. Angew. Chem. Int. Ed 2013, 52, 508–516. [DOI] [PubMed] [Google Scholar]
- (12).(a) Guo W; Shi L; Javitch JA The fourth transmembrane segment forms the interface of the dopamine D2 receptor homodimer. J. Biol. Chem 2003, 278, 4385–4388. [DOI] [PubMed] [Google Scholar]; (b) Kern A; Albarran-Zeckler R; Walsh HE; Smith RG Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron 2012, 73, 317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bonaventura J; Navarro G; Casadó-Anguera V; Azdad K; Rea W; Moreno E; Brugarolas M; Mallol J; Canela EI; Lluís C; Cortés A; Volkow ND; Schiffmann SN; Ferré S; Casadó V. Allosteric interactions between agonists and antagonists within the adenosine A2A receptor-dopamine D2 receptor heterotetramer. Proc. Natl. Acad. Sci. USA 2015, 112, E3609–E3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ferré S; Ciruela F; Canals M; Marcellino D; Burgueño J; Casadó V; Hillion J; Torvinen M; Fanelli F; de Benedetti P; Goldberg SR; Bouvier M; Fuxe K; Agnati LF; Lluís C; Franco R; Woods A. Adenosine A2A-dopamine D2 receptor-receptor heteromers. Targets for neuro-psychiatric disorders. Parkinsonism Relat. Disord 2004, 10, 265–271. [DOI] [PubMed] [Google Scholar]
- (15).(a) Kaczor A; Jörg M; Capuano B. The dopamine D2 receptor dimer and its interaction with homobivalent antagonists: homology modeling, docking and molecular dynamics. J. Mol. Model 2016, 22, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Carli M; Kolachalam S; Aringhieri S; Rossi M; Giovannini L; Maggio R; Scarselli M. Dopamine D2 receptors dimers: How can we pharmacologically target them? Curr. Neuropharmacol 2018, 16, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Berque-Bestel I; Lezoualc’h F; Jockers R. Bivalent ligands as specific pharmacological tools for G protein-coupled receptor dimers. Curr. Drug Discov. Technol 2008, 5, 312–318. [DOI] [PubMed] [Google Scholar]
- (17).Tabor A; Weisenburger S; Banerjee A; Purkayastha N; Kaindl JM; Hübner H; Wei L; Grömer TW; Kornhuber J; Tschammer N; Birdsall NJ; Mashanov GI; Sandoghdar V; Gmeiner P. Visualization and ligand-induced modulation of dopamine receptor dimerization at the single molecule level. Sci. Rep 2016, 6, 33233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pulido D; Albericio F; Royo M. Controlling multivalency and multimodality: up to pentamodal dendritic platforms based on diethylenetriaminepentaacetic acid cores. Org. Lett 2014, 16, 1318–1321. [DOI] [PubMed] [Google Scholar]
- (19).Carriba P; Navarro G; Ciruela F; Ferré S; Casadó V; Agnati L; Cortés A; Mallol J; Fuxe K; Canela EI; Lluís C; Franco R. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat. Methods 2008, 5, 727–733. [DOI] [PubMed] [Google Scholar]
- (20).(a) Jin C; Mayer LD; Lewin AH; Rehder KS; Brine GA Practical synthesis of p-aminophenethylspiperone (NAPS), a high-affinity, selective D2-dopamine receptor antagonist. Synth. Commun 2008, 38, 816–823. [Google Scholar]; (b) Albizu L; Cottet M; Kralikova M; Stoev S; Seyer R; Brabet I; Roux T; Bazin H; Bourrier E; Lamarque L; Breton C; Rives ML; Newman A; Javitch J; Trinquet E; Manning M; Pin JP; Mouillac B; Durroux T. Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol 2010, 6, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cordomí A; Navarro G; Aymerich MS; Franco R. Structures for G-protein-coupled receptor tetramers in complex with G proteins. Trends Biochem. Sci 2015, 40, 548–551. [DOI] [PubMed] [Google Scholar]
- (22).Kang YS; Son JH; Hwang IC; Ahn KH Synthesis of a bis(oxazoline)-N-carboxylate ligand and its Zn(II) complex that shows C–H···Cl hydrogen bonding. Polyhedron 2006, 25, 3025–3031. [Google Scholar]
- (23).Casadó V; Cortés A; Ciruela F; Mallol J; Ferré S; Lluís C; Canela EI; Franco R. Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: The receptor–dimer cooperativity index. Pharmacol. Ther 2007, 116, 343–354. [DOI] [PubMed] [Google Scholar]
- (24).(a) Casadó V; Cortés A; Mallol J; Pérez-Capote K; Ferré S; Lluís C; Franco R; Canela EI GPCR homomers and heteromers: A better choice as targets for drug development than GPCR monomers? Pharmacol. Ther 2009, 124, 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Casadó V; Ferrada C; Bonaventura J; Gracia E; Mallol J; Canela EI; Lluís C; Cortés A; Franco R. Useful pharmacological parameters for G-protein-coupled receptor homodimers obtained from competition experiments. Agonist–antagonist binding modulation. Biochem. Pharmacol 2009, 78, 1456–1463. [DOI] [PubMed] [Google Scholar]
- (25).Schröder R; Schmidt J; Blättermann S; Peters L; Janssen N; Grundmann M; Seemann W; Kaufel D; Merten N; Drewke C; Gomeza J; Milligan G; Mohr K; Kostenis E. Applying label-free dynamic mass redistribution technology to frame signaling of G protein–coupled receptors noninvasively in living cells. Nat. Protoc 2011, 6, 1748–1760. [DOI] [PubMed] [Google Scholar]
- (26).Navarro G; Cordomí A; Casadó-Anguera V; Moreno E; Cai N-S; Cortés A; Canela EI; Dessauer CW; Casadó V; Pardo L; Lluís C; Ferré S. Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase. Nat. Commun 2018, 9, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Guitart X; Navarro G; Moreno E; Yano H; Cai NS; Sánchez-Soto M; Kumar-Barodia S; Naidu YT; Mallol J; Cortés A; Lluís C; Canela EI; Casadó V; McCormick PJ; Ferré S. Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: the dopamine D1-D3 receptor heterotetramer. Mol. Pharmacol 2014, 86, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Moreno E; Quiroz C; Rea W; Cai NS; Mallol J; Cortés A; Lluís C; Canela EI; Casadó V; Ferré S. Functional μ-opioid-galanin receptor heteromers in the ventral tegmental area. J. Neurosci 2017, 37, 1176–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Orru M; Bakešová J; Brugarolas M; Quiroz C; Beaumont V; Goldberg SR; Lluís C; Cortés A; Franco R; Casadó V; Canela EI; Ferré S. Striatal pre- and postsynaptic profile of adenosine A2A receptor antagonists. PLoS One 2011, 6, e16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chien EY; Liu W; Zhao Q; Katritch V; Han GW; Hanson MA; Shi L; Newman AH; Javitch JA; Cherezov V; Stevens RC Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Marti-Renom MA; Stuart AC; Fiser A; Sanchez R; Melo F; Sali A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct 2000, 29, 291–325. [DOI] [PubMed] [Google Scholar]
- (32).Wang S; Che T; Levit A; Shoichet BK; Wacker D; Roth BL Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Manglik A; Kruse AC; Kobilka TS; Thian FS; Mathiesen JM; Sunahara RK; Pardo L; Weis WI; Kobilka B. Granier, K.; S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Huang J; Chen S; Zhang JJ; Huang XY Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat. Struct. Mol. Biol 2013, 20, 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).de Vries SJ; van Dijk M; Bonvin AM The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc 2010, 5, 883–897. [DOI] [PubMed] [Google Scholar]
- (36).Cordomi A; Caltabiano G; Pardo L. Membrane protein simulations using AMBER force field and Berger lipid parameters. J. Chem. Theory Comput 2012, 8, 948–958. [DOI] [PubMed] [Google Scholar]
- (37).Pronk S; Pall S; Schulz R; Larsson P; Bjelkmar P; Apostolov R; Shirts MR; Smith JC; Kasson PM; van der Spoel D; Hess B; Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.