

Abstract
X-linked adrenoleukodystrophy (X-ALD) is the most common peroxisomal disorder. It is caused by defects in the ATP-binding cassette subfamily D member 1 (ABCD1) gene, resulting in impaired peroxisomal β-oxidation of very-long-chain fatty acids (VLCFAs). As an X-linked recessive disease, female X-ALD carriers are typically asymptomatic. In the present study, a 7-year-old girl was diagnosed with cerebral ALD. Brain magnetic resonance imaging revealed asymmetric demyelination of bilateral white matter. Plasma VLCFAs level showed a substantial increase. Whole exome and Sanger sequencing revealed an ABCD1 c.919C>T (p.Q307X) heterozygous pathogenic mutation, which was inherited from the asymptomatic mother. X chromosome inactivation (XCI) analysis revealed that the normal paternal X chromosome was almost completely inactivated. Thus, the maternal ABCD1 mutation and paternal XCI were responsible for causing the disease in the patient. XCI may be one reason female X-ALD carriers can be symptomatic.
Keywords: X-linked adrenoleukodystrophy, ATP-binding cassette subfamily D member 1, X chromosome inactivation, whole exome sequencing, very-long-chain fatty acid
Introduction
X-linked adrenoleukodystrophy (X-ALD), caused by mutations in the ATP-binding cassette subfamily D member 1 (ABCD1) gene, is the most common peroxisomal disorder. ABCD1 is located on Xq28 and encodes the peroxisomal transporter of very-long-chain fatty acids (VLCFAs) (1). Defects in ABCD1 protein prevent β-oxidation of VLCFAs, leading to its accumulation in tissue and plasma (1). According to age of onset, affected site and progression rate, multiple phenotypes of X-ALD are recognized, including cerebral ALD, adrenomyeloneuropathy, Addison's and asymptomatic (2).
As in the case of numerous types of X-linked recessive disease, female carriers are assumed to remain asymptomatic; however, ~20% of X-ALD female carriers develop symptoms, usually in their thirties, and the incidence rate is up to 88% in those aged >60 years (3,4). In general, female X-ALD carriers display adrenomyeloneuropathy-like phenotype. The primary clinical manifestations of this phenotype are progressive spastic gait, sensory deficit and bladder dysfunction, commonly without adrenal insufficiency (4,5).
The present report describes the case of a 7-year-old girl with cerebral ALD. We aimed to discover the possible pathogenesis of this patient in the present study.
Materials and methods
Sample collection
Peripheral blood (3 ml) was collected from a female patient with X-ALD (age, 8 years in 2021), as well as from members of their family (Fig. 1A). The ages of family members I1, I2, II1 and II2 were 62, 60, 36 and 35 years, respectively. The present study was approved by the Ethics Committee of the Shengjing Hospital of China Medical University (approval no. 2021PS209K). Written informed consent was obtained from the patient's parents and the other adults in the family.
Figure 1.
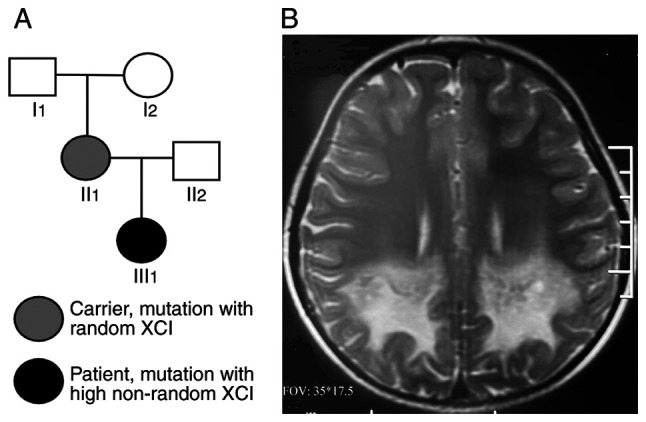
Pedigree and brain MRI of the patient. (A) Pedigree of the X-linked adrenoleukodystrophy family. III1 indicates the patient; square, male; circle, female. (B) Brain MRI of the patient revealed asymmetric demyelination of bilateral white matter. MRI, magnetic resonance imaging; XCI, X chromosome inactivation.
Biochemical measurements
Serum or plasma (0.5 ml) was collected after centrifugation of blood samples at 1,200 x g for 5 min at room temperature. Plasma adrenocorticotrophic hormone (ACTH) and serum cortisol levels were measured according to a chemiluminescence method (6), and serum VLCFAwas measured by gas chromatography and mass spectrometry (7) at Jinyu Medical Laboratory Center.
Whole-exome and Sanger sequencing
Genomic DNA was extracted using QIAamp DNA Mini kit (Qiagen China Co., Ltd.). The concentration of extracted DNA was quantified with Nanodrop 2000 (NanoDrop; Thermo Fisher Scientific, Inc.). Genomic DNA was fragmented to an average size of 150 bp using a S220 Focusedultrasonicator (Covaris, LLC). The DNA library was prepared using the GenCap NGS Fast DNA Library Prep Set for Illumina (cat. no. MG16001M; MyGenostics Technologies Inc.) according to the manufacturer's instructions. The DNA library was captured using a whole-exome capture kit (GenCap Human Gene Enrichment Kit; cat. no. MG16101M, MyGenostics Technologies Inc.). The captured DNA was eluted with buffer, and amplified for 13 cycles using the following program: 95˚C for 4 min (1 cycle); 98˚C for 30 sec, 65˚C for 30 sec, 72˚C for 30 sec (13 cycles); 72˚C for 5 min (1 cycle). The PCR product was purified using SPRI beads (Beckman Coulter, Inc.) according to the manufacturer's protocol. The enriched libraries were sequenced using 150-bp paired-end reads on the Illumina HiSeq X Ten platform (Illumina, Inc.). Raw data were stored in the FASTQ format. Illumina sequencing adapters and low-quality reads (<80 bp) were filtered. Clean reads were aligned to the UCSC hg19 human reference genome using the Burrows-Wheeler Alignment tool 0.7.17 (http://maq.sourceforge.net). Duplicated reads were removed using Picard (broadinstitute.github.io/picard). Insertion, deletion and single nucleotide polymorphism (SNP) variants were detected and filtered using the Genome Analysis Toolkit 4.0.8.1 (https://www.broadinstitute.org/gatk). The identified variants were annotated using ANNOVAR (http://annovar.openbioinformatics.org/en/latest) and assessed via the following databases: 1000 Genomes(https://www.internationalgenome.org), Exome Aggregation Consortium (http://gnomad.broadinstitute.org), Human Gene Mutation Database(http://www.hgmd.cf.ac.uk/ac/index.php), Mutation Taster (https://www.mutationtaster.org), Sorting Intolerant From Tolerant (http://provean.jcvi.org/index.php), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) and Genomic Evolutionary Rate Profiling (http://mendel.stanford.edu/SidowLab/downloads/ger). Variation sites were identified by comparing DNA sequences with the corresponding GenBank reference sequences using Mutation Surveyor software (SoftGenetics, Inc.). The pathogenicity of mutations was assessed in accordance with the American College of Medical Genetics and Genomics Guidelines (ACMG) (8). The mutation was also confirmed via Sanger sequencing on ABI Prism 3730 Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.) using the following primers: Forward, 5'-TGTGTGAGTGGCACTGGGAGA-3' and reverse, 5'-GCTCCAGCATAACATACCACA-3'. SNP haplotype analysis was performed using Infinium Asian Screening Array-24 v1.0 kit (Illumina, Inc.). In brief, DNA samples were incubated with corresponding reagents in the kit at 37˚C overnight for PCR amplification. The amplified DNA was then fragmented in the heat block, precipitated with isopropyl alcohol, resuspended after centrifugation and hybridized with the chip at 48˚C overnight, followed bywashing, extension and chip dying. The prepared chipwasscanned in Iscan chip scanner (Illumina, Inc.). Data were uploaded to ChromGo (Yikon Genomics) for analysis.
X chromosome inactivation (XCI) analysis in peripheral blood
Peripheral blood (1 ml) was used for genomic DNA extraction (Qiagen China Co., Ltd.). The XCI pattern was determined by quantifying the CAG repeat in exon 1 of the androgen receptor gene using a commercial kit according to the manufacturer's instructions (XCI analysis kit, MyGenosticsGenCap Enrichment Technologies). In brief, when the X chromosome is inactivated, the binding site of the methylation-sensitive restriction endonuclease HpaII, which is located near the CAG repeat, is methylated and cannot be cleaved, whereas the non-methylated site of the active X chromosome can be cleaved by HpaII. Therefore, if the genomic DNA is digested with HpaII and then amplified via PCR, only the inactivated X chromosome produces the expected bands (9).
Results
Patient clinical information description
At the age of 7 years, from April 2020 the patient (III1; Fig. 1A) began to experience occasional confusion and needed to be called several times before answering. She was found to have a poor sense of direction and failed to find her way home when she went out. Her eyesight declined and she had 30˚ strabismus. She was afraid of moving to higher positions and her ability to balance worsened. Her electroencephalogram revealed slow waves. Brain magnetic resonance imaging (MRI) revealed asymmetric demyelination of bilateral white matter, suggesting the acute phase of ALD (Fig. 1B). The patient did not receive any treatment. Eight months later (December 2020), she was unable to hear, see or speak and became paralyzed. In May 2021, the patient's mother (II1) visited the obstetric clinic (Shengjing Hospital of China Medical University, Shenyang, China) and wished to have a healthy child via assisted reproductive technology. The clinical information, particularly the MRI, demonstrated that she was affected with ALD.
Patient exhibits high plasma VLCFA levels
Biochemical measurement analysis demonstrated that the C26:0 and C26:0/C22:0 levels in III1 were notably increased (Table I). Moreover, C24:0/C22:0 and ACTH and cortisol levels in III1 were slightly elevated. By contrast, VLCFAs, ACTH and cortisol levels were close to normal in II1 and I2, with C26:0 level and C26:0/C22:0 slightly increased in II1. The results showed the patient exhibited high plasma VLCFA levels.
Table I.
Results of biochemical measurement.
| Type | Sample ID | III1 | II1 | I2 | Reference value |
|---|---|---|---|---|---|
| ACTH | 73.560↑ | 18.240 | 10.760 | 7.200-63.400 pg/ml | |
| Cortisol | 26.180↑ | 8.390 | 9.790 | 4.260-24.850 µg/dl | |
| VLCFA | C22:0 | 50.800 | 53.000 | 62.000 | ≤96.300 nmol/ml |
| C24:0 | 91.000 | 63.400 | 57.100 | ≤91.400 nmol/ml | |
| C26:0 | 3.850 ↑↑ | 1.320 ↑ | 0.780 | ≤1.300 nmol/ml | |
| C24:0/C22:0 | 1.790 ↑ | 1.200 | 0.920 | ≤1.390 | |
| C26:0/C22:0 | 0.076 ↑↑ | 0.025 ↑ | 0.013 | ≤0.023 |
ACTH, adrenocorticotrophic hormone; VLCFA, very-long-chain fatty acid.
Patient harbors a heterozygous ABCD1 mutation
Whole-exome sequencing performed on II1, II2 and III1 (Fig. 2A). II1 and III1 harbored the ABCD1 c.919C>T (p.Q307X) heterozygous mutation, which was confirmed using Sanger sequencing (Fig. 2B). Based on ACMG guidelines, this mutation was identified as pathogenic. The pathogenic mutation in the patient was inherited from her mother who did not demonstrate any symptoms. In addition, SNP haplotype analysis was constructed for the linkage analysis (Fig. 2C); a total of15 available SNP sites upstream of the gene and 15 sites downstream were selected for the haplotype construction. These findings indicated that the patient harbors a heterozygous ABCD1 mutation from her asymptomatic mother.
Figure 2.

Sequencing analysis. (A) Mutation was identified by whole exome sequencing, suggested a heterozygous c.919C>T (p.Q307X) variation of ATP-binding cassette subfamily D member 1 (red box). (B) SNP haplotype linkage analysis of the family. The numbers represent the relative distance in kb up-(blue) and downstream (orange) of gene. (C) Mutation was positive in the patient III1 and mother II1, but not in I2 or II2, as shown by Sanger sequencing.
Paternal X chromosome is inactivated in the patient
Although both II1 and III1 harbored the same ABCD1 heterozygous mutation, III1 showed obvious clinical manifestations, whereas II1 was an asymptomatic carrier. As an X-linked recessive disease, female ABCD1 heterozygous carriers are typically asymptomatic, especially in childhood (4). To identify the pathogenic cause in III1, other pathogenic ABCD1 mutations were excluded. As XCI is involved in ALD, the polymorphism of the CAG repeat in exon 1 of the androgen receptor gene was investigated. X chromosome in III1 displayed highly non-random inactivation, implying that her paternal X chromosome was almost completely inactivated (Fig. 3). This indicated that the severe clinical symptoms in III1 were caused by maternal ABCD1 mutation and paternal XCI.
Figure 3.

XCI analysis. From the undigested chart, the 277 bp fragment in patient III1 was from her father II2 and the 288 bp fragment was from her mother II1. From the HpaII-digested chart, patient demonstrated skewed XCI and paternal X chromosome was almost completely inactivated. X-axis represents the fragment size (bp) and Y-axis represents the peak value. XCI, X chromosome inactivation.
Discussion
The present study describes the case of a patient with X-ALD caused by maternal ABCD1 mutation and paternal XCI. The incidence of X-ALD is 1 in 20,000-50,000 individuals (10). According to the age of onset, affected site and progression rate, X-ALD can be classified into four major groups (2): i) Cerebral ALD, which is characterized by demyelination of white matter of the brain; ii) adrenomyeloneuropathy, with spinal cord demyelination and axonal degeneration; iii) Addison-like phenotype due to adrenocortical insufficiency and iv) asymptomatic type. Childhood cerebral ALD and adrenomyeloneuropathy are the most common types observed during childhood and adulthood, with incidences of 31-35 and 20-60%, respectively (11,12).
Onset of childhood cerebral ALD usually occurs at the age of 3-10 years, with a peak age onset of 7 years, and is characterized by progressive deterioration of cognitive, behavioral, and motor function. The patient initially presents with learning and behavioral problems, as well as attention deficit, followed by cognitive decline, ataxia, cortical blindness, deafness and seizures within 6 months to 2 years. This can progress to a vegetative state and death within years of diagnosis (1,8). The lower the onset age, the faster the disease progression (13). Brain MRI shows demyelination in the white matter of the brain and this finding can precede symptoms (13). In the present study, the patient's clinical manifestations was typical, including hearing, vision and behavioral abnormality, which progressed to the vegetative state rapidly. The subsequent genetic analysis identified the ABCD1 heterozygous pathogenic mutation.
To date, >900 variants of ABCD1 have been reported (14). Although Campopiano et al (15) addressed the genotype-phenotype correlation in a large family, most studies have showed weak correlation between genotype and phenotype (1,2). Thus, ongoing clinical symptoms and prognosis cannot be determined by the patient genotype. It is accepted that other factors, including genetic, epigenetic or environmental, may affect rate of progression (2).
As in the case of many X-linked diseases, X-ALD female carriers typically remain asymptomatic; however, 18-88% of female carriers develop symptoms (4). The incidence rate of X-ALD in ABCD1 heterozygous female carriers is associated with age; 18% of female carriers aged <40 years develop symptoms and this increases to 88% of those aged >60 years (4). Horn et al (16) also found that nearly all female carriers aged >50 years develop neurological symptoms. One of the earliest descriptions of an adrenomyeloneuropathy-like X-ALD phenotype was a female patient (17).
In X-ALD, XCI is an important factor. XCI refers to silencing of one of the two X chromosomes in mammalian female cells that ensures equal expression of X-linked genes between female and male. This process occurs during early development; one X chromosome is transcriptionally silenced while the other remains active (18,19). Which X chromosome is inactivated is usually random; therefore, ~50% of all cells express genes from the paternal-derived X chromosome and the remaining 50% from the maternal chromosome (5). When 80% of cells demonstrate a preferential inactivation of the same X chromosome, it is defined as skewed XCI (5). Extremely skewed XCI implies inactivation of paternal or maternal-derived X chromosome in >95% of all cells (20). To date, whether skewed XCI is associated with symptomatic X-ALD female carriers is unclear. Maier et al (21) reported that the manifestation of symptoms in 22 X-ALD carriers was associated with skewed XCI and that there was a significant correlation between the extent of skewing and the severity of neurological abnormality. By contrast, Watkiss et al (22) and Salsano et al (5) did not find any association between neurological manifestation and XCI pattern. The present study suggested that extremely skewed XCI served an important role in the patient's severe clinical presentation. In general, XCI skewing appears to favor the inactive mutant allele, which may explain why female carriers may be asymptomatic in severe X-linked disorders with a highly skewed XCI (18,19). However, ABCD1 mutations in X-ALD demonstrate a proliferative advantage rather than a disadvantage, caused by XCI skewing in favor of the normal X chromosome (5). The present findings were consistent with this hypothesis as the normal paternal X chromosome was almost completely inactivated. However, in the present study, the mechanism by which the paternal X chromosome was inactivated was not clarified. Further experiments and samples (from this and other families) will be needed to study the mechanism in-depth.
In addition to X-ALD, XCI serves an important role in other common X-linked recessive diseases. Yang et al (23) found skewed XCI in a female patient who had a heterozygous missense mutation in the FIX gene and a complete clinical manifestation of moderate hemophilia B was noted. A total of 28% of heterozygous females with FVIII or FIX variants have clinical manifestations and exhibit skewed XCI (24). In Becker muscular dystrophy, Viggiano et al (25) reported that skewed XCI serves a crucial role in the onset of symptoms in female carriers. Skewed XCI also appears to have a correlation with a severe phenotype in Duchenne muscular dystrophy (26). He et al (9) found that XCI was implicated in symptomatic female carriers of dystrophinopathies and XCI analysis of amniocytes may be useful in predicting the risk of dystrophinopathy in fetal carriers. Thus, the incidence of skewed XCI may be common and contribute to the manifestation of symptoms in carriers of recessive X-linked disorders (18,19).
In conclusion, X-AD in the present patient was caused by both maternal ABCD1 mutation and paternal XCI. This suggests that XCI may be a key factor in X-ALD.
Acknowledgements
Not applicable.
Funding Statement
Funding: The present study was supported by the 345 Talent Project of Shengjing Hospital (grant no. M0298).
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the Sequence Read Archive database (submission number: SUB11494821, Bio Sample accession number: SAMN28555541; https://www.ncbi.nlm.nih.gov/sra/PRJNA841819).
Authors' contributions
ZL provided obstetrical service, collected the samples and clinical information, performed the biochemical measurements, and wrote the paper. GL performed the sequencing and analyzed the data, provided genetic counseling, and revised the manuscript. ZL and GL confirm the authenticity of all the raw data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by the Ethics Committee of the Shengjing Hospital of China Medical University (approval no. 2021PS209K). Written informed consent was obtained from the patient's parents.
Patient consent for publication
Consent for publication was obtained from the patient's parents. Patient personal information was removed from the picture.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Zhu J, Eichler F, Biffi A, Duncan CN, Williams DA, Majzoub JA. The changing face of adrenoleukodystrophy. Endocr Rev. 2020;41:577–593. doi: 10.1210/endrev/bnaa013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palakuzhiyil SV, Christopher R, Chandra SR. Deciphering the modifiers for phenotypic variability of X-linked adrenoleukodystrophy. World J Biol Chem. 2020;11:99–111. doi: 10.4331/wjbc.v11.i3.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt S, Träber F, Block W, Keller E, Pohl C, von Oertzen J, Schild H, Schlegel U, Klockgether T. Phenotype assignment in symptomatic female carriers of X-linked adrenoleukodystrophy. J Neurol. 2001;248:36–44. doi: 10.1007/s004150170267. [DOI] [PubMed] [Google Scholar]
- 4.Engelen M, Barbier M, Dijkstra IM, Schür R, de Bie RM, Verhamme C, Dijkgraaf MG, Aubourg PA, Wanders RJ, van Geel BM, et al. X-linked adrenoleukodystrophy in women: A cross-sectional cohort study. Brain. 2014;137:693–706. doi: 10.1093/brain/awt361. [DOI] [PubMed] [Google Scholar]
- 5.Salsano E, Tabano S, Sirchia SM, Colapietro P, Castellotti B, Gellera C, Rimoldi M, Pensato V, Mariotti C, Pareyson D, et al. Preferential expression of mutant ABCD1 allele is common in adrenoleukodystrophy female carriers but unrelated to clinical symptoms. Orphanet J Rare Dis. 2012;7(10) doi: 10.1186/1750-1172-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ye YJ, Liu B, Qin BZ. Clinical analysis of patients of cirrhosis complicated with adrenal insufficiency. Eur Rev Med Pharmacol Sci. 2016;20:2667–2672. [PubMed] [Google Scholar]
- 7.Hadj Ahmed S, Koubaa N, Kharroubi W, Zarrouk A, Mnari A, Batbout F, Gamra H, Hammami S, Lizard G, Hammami M. Identification of long and very long chain fatty acids, plasmalogen-C16:0 and phytanic acid as new lipid biomarkers in Tunisian coronary artery disease patients. Prostaglandins Other Lipid Mediat. 2017;131:49–58. doi: 10.1016/j.prostaglandins.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for molecular pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He WB, Du J, Xie PY, Zhou S, Zhang YX, Lu GX, Lin G, Li W, Tan YQ. X-chromosome inactivation pattern of amniocytes predicts the risk of dystrophinopathy in fetal carriers of DMD mutations. PrenatDiagn. 2019;39:603–608. doi: 10.1002/pd.5473. [DOI] [PubMed] [Google Scholar]
- 10.Olgac A, Kasapkara ÇS, Derinkuyu B, Yüksel D, Çetinkaya S, Aksoy A, Ceylaner S, Güleray N, Yeşilipek A, Aydın Hİ, et al. Retrospective evaluation of patients with X-linked adrenoleukodystrophy with a wide range of clinical presentations: A single center experience. J Pediatr Endocrinol Metab. 2021;34:1169–1179. doi: 10.1515/jpem-2021-0032. [DOI] [PubMed] [Google Scholar]
- 11.Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJ, Aubourg P, Poll-The BT. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis. 2012;7(51) doi: 10.1186/1750-1172-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohn A, Polidori N, Aiello C, Rizzo C, Giannini C, Chiarelli F, Cappa M. ABCD1 gene mutation in an Italian family with X-linkedadrenoleukodystrophy: Case series. Endocrinol Diabetes Metab Case Rep. 2021;2021:20–0125. doi: 10.1530/EDM-20-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moser HW, Loes DJ, Melhem ER, Raymond GV, Bezman L, Cox CS, Lu SE. X-Linked adrenoleukodystrophy: Overview and prognosis as a function of age and brain magnetic resonance imaging abnormality. A study involving 372 patients. Neuropediatrics. 2000;31:227–239. doi: 10.1055/s-2000-9236. [DOI] [PubMed] [Google Scholar]
- 14. ALD info: The information platform to all aspects of adrenoleukodystrophy and the worldwide registry for ABCD1 variants. https://adrenoleukodystrophy.info/. Accessed March 1, 2022. [Google Scholar]
- 15.Campopiano R, Femiano C, Chiaravalloti MA, Ferese R, Centonze D, Buttari F, Zampatti S, Fanelli M, Amatori S, D'Alessio C, et al. A large family with p.Arg554His mutation in ABCD1: Clinical features and genotype/phenotype correlation in female carriers. Genes (Basel) 2021;12(775) doi: 10.3390/genes12050775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horn MA, Retterstøl L, Abdelnoor M, Skjeldal OH, Tallaksen CM. Adrenoleukodystrophy in Norway: High rate of de novo mutations and age-dependent penetrance. Pediatr Neurol. 2013;48:212–219. doi: 10.1016/j.pediatrneurol.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Penman RW. Addison's disease in association with spastic paraplegia. Br Med J. 1960;1(402) doi: 10.1136/bmj.1.5170.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shvetsova E, Sofronova A, Monajemi R, Gagalova K, Draisma HHM, White SJ, Santen GWE, Chuva de Sousa Lopes SM, Heijmans BT, van Meurs J, et al. Skewed X-inactivation is common in the general female population. Eur J Hum Genet. 2019;27:455–465. doi: 10.1038/s41431-018-0291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira G, Dória S. X-chromosome inactivation: Implications in human disease. J Genet. 2021;100(63) [PubMed] [Google Scholar]
- 20.Wang Z, Yan A, Lin Y, Xie H, Zhou C, Lan F. Familial skewed x chromosome inactivation in adrenoleukodystrophy manifesting heterozygotes from a Chinese pedigree. PLoS One. 2013;8(e57977) doi: 10.1371/journal.pone.0057977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maier EM, Kammerer S, Muntau AC, Wichers M, Braun A, Roscher AA. Symptoms in carriers of adrenoleukodystrophy relate to skewed X inactivation. Ann Neurol. 2002;52:683–688. doi: 10.1002/ana.10376. [DOI] [PubMed] [Google Scholar]
- 22.Watkiss E, Webb T, Bundey S. Is skewed X inactivation responsible for symptoms in female carriers for adrenoleucodystrophy? J Med Genet. 1993;30:651–654. doi: 10.1136/jmg.30.8.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang C, Yu Z, Zhang W, Cao L, Ouyang W, Hu F, Zhang P, Bai X, Ruan C. A novel missense mutation, p. Phe360Cys, in FIX gene results in haemophilia B in a female patient with skewed X-inactivation. Haemophilia. 2018;24:e68–e70. doi: 10.1111/hae.13423. [DOI] [PubMed] [Google Scholar]
- 24.Miller CH, Bean CJ. Genetic causes of haemophilia in women and girls. Haemophilia. 2021;27:e164–e179. doi: 10.1111/hae.14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viggiano E, Picillo E, Ergoli M, Cirillo A, Del Gaudio S, Politano L. Skewed X-chromosome inactivation plays a crucial role in the onset of symptoms in carriers of Becker muscular dystrophy. J Gene Med. 2017;19 doi: 10.1002/jgm.2952. [DOI] [PubMed] [Google Scholar]
- 26.Viggiano E, Ergoli M, Picillo E, Politano L. Determining the role of skewed X-chromosome inactivation in developing muscle symptoms in carriers of Duchenne muscular dystrophy. Hum Genet. 2016;135:685–698. doi: 10.1007/s00439-016-1666-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the Sequence Read Archive database (submission number: SUB11494821, Bio Sample accession number: SAMN28555541; https://www.ncbi.nlm.nih.gov/sra/PRJNA841819).