

Abstract
Multidrug resistance (MDR) is the main cause of clinical treatment failure and poor prognosis in cancer. Targeting P-glycoprotein (P-gp) has been regarded as an effective strategy to overcome MDR. In this work, we reported our preclinical studies of the triazolo[1,5-a]pyrimidine-based compound WS-716 as a highly potent, specific, and orally active P-gp inhibitor. Through direct binding to P-gp, WS-716 inhibited efflux function of P-gp and specifically reversed P-gp-mediated MDR to paclitaxel (PTX) in multiple resistant cell lines, without changing its expression or subcellular localization. WS-716 and PTX synergistically inhibited formation of colony and 3D spheroid, induced apoptosis and cell cycle arrest at G2/M phase in resistant SW620/Ad300 cells. In addition, WS-716 displayed minimal effect on the drug-metabolizing enzyme cytochrome P4503A4 (CYP3A4). Importantly, WS-716 increased sensitivity of both pre-clinically and clinically derived MDR tumors to PTX in vivo with the T/C value of 29.7% in patient-derived xenograft (PDX) models. Relative to PTX treatment alone, combination of WS-716 and PTX caused no obvious adverse reactions. Taken together, our preclinical studies revealed therapeutic promise of WS-716 against MDR cancer, the promising data warrant its further development for cancer therapy.
KEY WORDS: Multidrug resistance (MDR); ATP-Binding cassette; P-gp inhibitors; Triazolo[1,5-a]pyrimidine; Drug combination; Preclinical studies; Cancer therapy
Graphical abstract
Preclinical studies reveal that WS-716 is a highly potent, specific and orally active P-glycoprotein inhibitor and increases sensitivity of both pre-clinically and clinically derived MDR tumors to PTX in vivo.
1. Introduction
Acquired multidrug resistance (MDR) developed during therapy leads to relapse of cancer1. P-glycoprotein (P-gp or ABCB1) encoded by ATP-binding cassette subfamily B member 1 (ABCB1) gene, is an ATP-dependent drug efflux pump for xenobiotic compounds with broad substrate specificity2. P-gp is responsible for decreased drug accumulation in MDR cells and often mediates the development of resistance to various chemotherapeutics in cancer3, 4, 5, 6. One of the major reasons for resistance to chemotherapy is overexpression of P-gp, which increases the efflux of xenobiotics and decreases intracellular concentrations of anticancer drugs simultaneously using the energy derived from ATP hydrolysis7. The overexpression of P-gp confers significant resistance to a wide variety of chemotherapeutic substrates including PTX, etoposide vinblastine, anthracyclines, cisplatin, and fluorouracil8, 9, 10, 11. In structure, P-gp is composed of 1280 amino acids, including two transmembrane domains (TMDs) and two nucleotide-binding domains12,13. The TMDs are mainly responsible for substrate recognition and transport, while the nucleotide-binding domains are associated with the energy-generating step of ATP hydrolysis, which could induce the conformational changes of TMDs to pump substrates out of the cells12,13.
The drug-binding pocket, formed by hydrophobic and aromatic amino acid residues, is positioned at the interface of the two TMDs, which can accommodate at least two substrates simultaneously due to its large cavity14,15. Considering the property of the flexible drug pocket with low specificity, P-gp substrates differ greatly in size, structure, and function. Most of the P-gp substrates include two planar aromatic rings and a positively charged nitrogen atom, which provides important structural information for the development of novel P-gp inhibitors16, 17, 18. P-gp is overexpressed in many types of MDR cancer cells19, 20, 21, targeting P-gp has been regarded as an effective strategy to overcome MDR in cancer22,23 Particularly, inhibition of the efflux function of P-gp has shown promise in reversing MDR. To date, numerous P-gp inhibitors have been developed24, 25, 26, 27, such as verapamil (VPM)28, 29, 30, cyclosporine A31, dexverapamil32, valspodar33, tariquidar34, laniquidar35, and some of them have advanced into clinical trials7,36. Despite some P-gp inhibitors could significantly increase the efficacy of anticancer drugs in MDR cancer, none of the P-gp inhibitors have been approved for clinical use due to the drawbacks such as toxicity, interrupting drug–drug interactions, and so forth37. Therefore, the development of highly potent and specific P-gp inhibitors with good safety and favorable drug-like properties is still urgently needed. Our research group has long been committed to the development of P-gp inhibitors38, 39, 40. In this work, we report our preclinical results of the triazolo[1,5-a]pyrimidine-based compound WS-716, a highly potent, specific, and orally active P-gp inhibitor.
2. Materials and methods
2.1. Reagents and antibodies
WS-716 (HPLC purity: 99.37%), primary antibodies used were anti-P-gp (Cat# 13342 for Western blot analysis, Cell Signaling Technology, Danvers, MA, USA; Cat# HA500474 for immunohistochemistry (IHC) analysis, HUABIO, Hangzhou, China), anti-β-actin (Cat# TA-09, Zhongshan Golden Bridge Biotechnology, Beijing, China). PTX injection and VPM hydrochloride (Cat# MB1346-S) were purchased from Tongli Pharmaceutical Co., Ltd. (Hainan, China) and Meilun Biotechnology (Dalian, China), respectively. For in vivo studies, WS-716 was suspended in an oily solvent containing 90% corn oil, 5% castor oil, and 5% dimethylsulfoxide (DMSO) for oral administration.
2.2. Cell lines and cell culture
The human colon cancer cell line SW620 and its MDR cell line SW620/Ad300 were kindly supplied by Dr. Susan Bates's laboratory (National Institutes of Health, MD, USA). The ABCB1-overexpressing KB-C2 cell line was established by a stepwise exposure of KB-3-1 cells, a parental human epidermoid carcinoma cell line, to increasing concentrations (up to 2 μg/mL) of colchicine41. The human epidermal carcinoma cell line KB-3-1 and its drug-resistant ATP binding cassette subfamily C member 1 (ABCC1)-overexpressing cell line KB/CV60 were maintained in medium with 1 μg/mL of cepharanthine and 60 ng/mL of vincristine42. The colon cancer cell line S1 and its ATP-binding cassette subfamily G member 2 (ABCG2)-overexpressing subline S1-M1-80 were maintained in the presence of 80 μmol/L of mitoxantrone43. HEK293/pcDNA3.1 and HEK293/ABCB1 cells were generated by transfecting the HEK293 cells with empty vector and full length ABCB1 gene-containing vector, respectively, and then selected in a medium containing 2 mg/mL G418. HEK293/pcDNA3.1, HEK293/ABCC1, and HEK293/ABCG2 cells lines were established by transfecting HEK293 cells with either the empty pcDNA3.1 vector or the vector containing full length ABCC1 (HEK293/ABCC1) and ABCG2 (HEK293/ABCG2) DNA, respectively, and were cultured in medium containing 2 mg/mL of G418 (Enzo Life Sciences, Farmingdale, NY, USA)44. All cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (Biological Industries, Kibbutz Beit Haemek, Israel), 100 U/mL penicillin, and 100 μg/mL streptomycin, and cultured in an incubator at 37 °C with 5% CO2. All drug-resistant cell lines were grown in a drug-free culture medium for more than 2 weeks prior to their use.
2.3. Specimen collection
One patient diagnosed with inoperable hepatic metastasis of human epidermal growth factor receptor 2-positive advanced gastric adenocarcinoma was enrolled in this study. The study was conducted under the approval of Ethics Committee of Henan Cancer Hospital (Ethics Approval License: 2020012) and the patient provided informed consent. This patient was treated with sintilimab, trastuzumab, and capecitabine plus oxaliplatin as first line therapy. After seven courses, the primary lesion at the fundus of the stomach showed progressive disease. Before second line therapy, the patient underwent further diagnostic endoscopy, and the tumor tissue was obtained from the primary lesion.
2.4. Methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay
The MTT assay was used to explore the MDR-reversal effects of WS-716 and VPM in vitro. Cells were seeded into 96-well plates and preincubated with or without the reversal agents (100 μL/well) for 4 h. Subsequently, different concentrations of PTX were added into the designated wells for another 72-h incubation. 20 μL MTT solution (5 mg/mL) was added into each well and then further incubated for 4 h. After removal of the medium with MTT solution, 150 μL DMSO was added into each well to dissolve the formazan crystals, and the absorbance was measured at 570 nm by ELx 800 Universal microplate reader (Bio-Tek, Inc., Winooski, VT, USA). IC50 was calculated by SPSS software. The data were shown as the mean ± SD obtained from three independent assays. The reversal fold (RF) was calculated by dividing the IC50 of chemotherapeutic drug in the absence of reversal agent with the IC50 of chemotherapeutic drug in the presence of reversal agent.
2.5. Western blot analysis
Total protein was extracted from cells using radioimmunoprecipitation assay lysis buffer (Beyotime Biotechnology, Shanghai, China) containing protease and phosphatase inhibitor cocktail (Biotool, Houston, TX, USA) on the ice. Proteins in samples were separated using the sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrophoretically transferred to polyvinylidene fluoride membrane. The membrane was blocked by 5% nonfat milk in Tris buffered saline with Tween-20 and then incubated with the primary antibody at 4°C overnight. Then, the membrane was probed with horseradish peroxide-conjugated secondary antibody (Cell Signaling Technology) for 2 h at room temperature (RT). The membrane was developed with enhanced chemiluminescence.
2.6. Ultra-high performance liquid chromatography (UPLC)
The intracellular concertation of PTX was detected by UPLC39. About 5 × 106 SW620 or SW620/Ad300 cells were seeded in 100-mm dish and incubated in the presence or absence of WS-716 (10 and 20 μmol/L) or VPM (4 μmol/L) for 4 h. Then, the medium was replaced with a fresh medium containing PTX (20 μmol/L) or PTX plus WS-716 (20 μmol/L) for another 3-h incubation. Then, cells were collected, washed, and resuspended in 1 mL phosphate buffered saline (PBS) followed by being lysed by sonication. Intracellular PTX released in PBS was extracted by ethyl acetate. The retention time was 1.56 min. The standard curve was established using standard PTX samples from 31 to 8000 ng/mL. The concentration of intracellular PTX was calculated according to the standard curve.
2.7. Drug accumulation and efflux assay
The accumulation assay was performed as previously described45. SW620 or SW620/Ad300 cells were seeded into 24-well plate with 1 × 104 cells/well and incubated overnight. After attached to the plates, the cells were first incubated with or without WS-716 or VPM for 2 h. The medium was then replaced by a medium containing 5 nmol/L tritium-labeled paclitaxel ([3H]-PTX) with WS-716 and VPM for further 2-h incubation. After that, the medium was discarded, and the cells were washed with ice-cold PBS three times. At last, the cells were collected and then transferred to the scintillation fluid. Radioactivity was measured using the Packard TRI-CARB1 190A liquid scintillation analyzer (Packard Instrument, Downers Grove, IL, USA). As for [3H]-PTX efflux assay, after the addition of 5 nmol/L [3H]-PTX for 2 h, cells were incubated in [3H]-free medium for another 2 h, during which each radioactivity data was collected at 0, 30, 60, and 120 min, respectively.
2.8. Immunofluorescence assay
Cells were seeded on the coverslips in the 24-well plate and treated with WS-716 (20 μmol/L). After 76 h, cells were fixed by 4% formaldehyde diluted in PBS and then permeabilized with 0.1% Triton X-100. After the rinse, cells were blocked with 5% bull serum albumin in PBS and then incubated with anti-P-gp primary antibody at 4°C overnight. After the rinse, cells were incubated with Alexa Fluor 488 rabbit anti-goat IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 2 h at RT. Cell nuclei were stained by 10 μg/mL Hoechst 33258 (Beyotime Biotechnology, Shanghai, China). Images were obtained by laser scanning confocal microscopy (Olympus, FV10i, Olympus Corporation, Tokyo, Japan)46.
2.9. ATPase assay
The effect of WS-716 on the hydrolytic activity of ATPase was examined by ATPase assay as described before45. P-gp crude membrane vesicles prepared from High Five insect cells were purchased from BD Biosciences (San Jose, CA, USA) and 10 μg membrane was incubated in the presence or absence of sodium orthovanadate (0.3 mmol/L) in assay buffer for 3 min. After that, 0–40 μmol/L varying concentrations of WS-716 or VPM were added to the assay buffer respectively. The reaction was initiated once adding 5 mmol/L of Mg-ATP followed by the incubation at 37 °C for 20 min, and then was terminated by adding 100 μL 5% SDS solution. The ATPase activity due to P-gp was determined from the amount of inorganic phosphate released detected at 880 nm using a spectrophotometer.
2.10. Cellular thermal shift assay
Cellular thermal shift assay was applied to identify the binding affinity of WS-716 with P-gp. The mechanism is that, heating the cell lysate to different temperatures will leads to the denaturation and precipitation of proteins that are not bound to a ligand. Adding drugs to these cellular mixtures alters the thermal melting curve of the proteins as more become bound to the drug. SW620/Ad300 cells (approximately 3 × 107) were washed and resuspended with PBS containing complete protease inhibitor cocktail. The cell suspension was freeze-thawed five times using liquid nitrogen. Cell lysate was centrifugated at 15,000 × g for 20 min at 4 °C. The supernatant was divided into two aliquots, with one aliquot being treated with WS-716 (200 μmol/L) and the other aliquot with the solvent control containing an equal amount of DMSO (1%, v/v). After 30 min incubation at RT, the respective supernatants were divided into equal aliquots and heated individually at different temperatures for 3 min. The heated supernatants were centrifuged at 15,000 × g for 20 min at 4 °C to removed denatured proteins. The supernatants were collected and analyzed by Western blot.
2.11. Docking analysis
Maestro v11.1 (Schrödinger LLC, MA, USA) was used for performing molecular docking simulations. The structure of WS-716 was constructed using the entry editor, and the energy of the structure was minimized in Macromodel v11.5 module using OPLS3 force field. A ligand preparation process was then carried out in LigPrep v4.1 module according to the following parameters to generate a low-energy 3D structure: different protonation states at pH 7.0 ± 2.0, and all possible tautomers and ring conformations. The prepared ligand was subjected to dock into the P-gp model. The human-mouse chimeric P-gp cryo-EM structure (Protein data bank ID: 6FN1) in complex with zosuquidar is available47, and the protein structure was prepared using the Protein Preparation Wizard implemented in Maestro v11.5. The docking grid (length: 30 Å) was generated at the inhibitor binding site by selecting the bound zosuquidar molecules as the centroid. Flexible docking of WS-716 and P-gp was performed by Glide v7.4 extra precision method. To further get an optimal simulation of binding, the best-docked pose from Glide extra precision docking was subjected to induced-fit docking by Glide v 7.4 in Maestro v11.1. The docked conformations of the tested ligand were ranked based on docking score, glide energy, as well as by analyzing the hydrogen bonding and hydrophobic interactions at binding site, and the best-docked pose of the ligand–protein complex was selected for graphical analysis.
2.12. CYP3A4 assay
The effect of WS-716 on human CYP3A4 was investigated using CYP3A4 activity assay kit (#K701, BioVison, Milpitas, CA, USA). According to the manufacturer, tested compounds, P450 Reaction Mix and CYP3A4 Assay Buffer were mixed and incubated for 10 min at 37 °C to allow the test ligands to interact with CYP3A4 in the absence of P450 catalytic turnover. The reaction was started by adding the CYP3A4 substrate/NADP+, and the fluorescence was immediately measured every 2 min at Ex/Em = 531/595 nm in kinetic mode for 38 min at 37 °C. Ketoconazole provided in the kit was used as a positive CYP3A4 inhibitor control. The IC50 value was calculated using GraphPad Prism 8 software.
2.13. Colony formation assay
SW620/Ad300 cells were seeded in 6-well plates (1000 cells/well) and treat with WS-716 (20 μmol/L, 4 h), PTX (1 μmol/L, 2 h), or the combination (preincubated with WS-716 for 2 h before being treated PTX plus WS-716 for 2 h). After removing the drug, cells were cultured for additional 7 days. Colonies were then fixed with methanol for 20 min and stained with 0.1% (w/v) crystal violet for 30 min. The excess dye was removed and washed with ultrapure water. The plates were air-dried, and the images were taken with the ChemiDoc system (Bio-Rad, Hercules, CA, USA), the counts of colonies were analyzed using Image J software.
2.14. 3D spheroid assay
SW620/Ad300 cells were treated with WS-716 (20 μmol/L, 4 h), PTX (1 μmol/L, 2 h), or the combination (preincubated with WS-716 for 2 h before being treated with PTX plus WS-716 for 2 h). Then, cells were suspended in DMEM/F12 medium supplemented with B27 (2%, v/v), EGF (20 ng/mL), bFGF (20 ng/mL), and heparin (40 μg/mL) and seeded into 96-well plate with low-attachment surface. Cells were cultured for another 10 days, and the diameter of 3D spheroids was monitored on Day 5 and Day 10.
2.15. Apoptosis analysis
FITC Annexin V apoptosis detection kit (BD Biosciences) was used to detect apoptosis. SW620/Ad300 cells were treated with WS-716 (20 μmol/L, 4 h), PTX (1 μmol/L, 2 h), or the combination (preincubated with WS-716 for 2 h before being treated PTX plus WS-716 for 2 h). Twenty-four hours later, cells were collected and washed with PBS. Binding buffer containing Annexin V-FITC and propidium iodide was added, and cells were dyed for 15 min in dark before analyzed by a flow cytometer (NovoCyte, ACEA Biosciences, Agilent, Santa Clara, CA, USA).
2.16. Mitochondrial membrane potential analysis
The JC-1 (Meilun Biotechnology), a fluorescent dye, was used to measure the change of mitochondrial membrane potential (Δψm)48. SW620/Ad300 cells were treated with WS-716 (20 μmol/L, 4 h), PTX (1 μmol/L, 2 h), or the combination (preincubated with WS-716 for 2 h before being treated PTX plus WS-716 for 2 h). Twenty-four hours later, cells were collected and washed before staining by JC-1 according to the manufacturer's instruction. JC-1 monomer with green fluorescence was detected by a flow cytometer.
2.17. Cytosolic protein extract preparation
Cytosolic protein extracts were prepared using Mitochondria Isolation Kit for Cell and Tissue (Yeasen Biotechnology, Shanghai, China). Briefly, cells were washed twice with PBS and cell pellets were resuspended in 800 μL ice-cold reagent A on ice for 15 min. The lysates were centrifuged at 1000 × g for 10 min. Centrifuge was repeated for another time. The supernatant (cytosolic protein extracts) was subjected to Western blot to detected cytochrome c released in cytoplasm.
2.18. Cell cycle analysis
SW620/Ad300 cells were treated with WS-716 (20 μmol/L, 4 h), PTX (1 μmol/L, 2 h), or the combination (preincubated with WS-716 for 2 h before being treated PTX plus WS-716 for 2 h). Twenty-four hours later, cells were collected and washed carefully with PBS and fixed with 70% ethanol at −20 °C. After a week, the cells were washed with PBS and stained with propidium iodide solution containing RNase A using the Cell Cycle Analysis Kit (KeyGEN, Nanjing, China) for 20 min. Cell cycle progression was analyzed using a flow cytometer.
2.19. Cell-derived xenograft (CDX) studies
The sensitization effect of WS-716 in vivo was measured using nude mice MDR xenograft models. The animal experiment was performed in accordance with guidelines of Committee of Zhengzhou University (Ethics approval license: WSQ20190628). Briefly, 8 × 106 SW620/Ad300 cells were inoculated subcutaneously into the right forelimb of each male BALB/c nude mice (13–15 g, aged 4–6 weeks, Silikejingda, Hunan, China). The mice were randomized into five groups (n = 5) and administrated with: (i) vehicle (containing 90% corn oil, 5% castor oil and 5% DMSO, 10 mL/kg/day, intragastric injection), (ii) WS-716 (100 mg/kg/day, intragastric injection), (iii) PTX (5 mg/kg/3 days, intravenous injection) PTX plus VPM (4 mg/kg/day, intragastric injection), (iv) PTX plus WS-716. After the therapy for 15 days, when the maximum tumor diameter reached 15 mm, animals were euthanized by carbon dioxide, tumor and liver tissues were excised and fixed in 4% paraformaldehyde for further analysis.
2.20. Hematoxylin and eosin (HE) staining
The deparaffinized sections were stained by hematoxylin for 5 min, rinsed, and differentiated by 1% (v/v) hydrochloric acid in alcohol. Then sections were incubated in Eosin solution. After rinse and paraffined, slides were mounted in neutral balsam.
2.21. TdT-mediated dUTP nick end labeling (TUNEL) assay
Apoptosis was detected by TUNEL staining according to the manufacturer's instructions (Meilun Biotechnology). Briefly, 4-μm tumor sections were incubated with proteinase K (20 μg/mL) for 20 min at RT. After the rinse, the sections were incubated with TdT enzyme and FITC-12-dUTP for 1 h at 37 °C in a humidified chamber. Subsequently, tumor sections were sealed with antifluorescence attenuation sealant and the fluorescence of positive staining was examined by fluorescence microscopy.
2.22. PDX studies
The animal experiment was performed in accordance with guidelines of Committee of Laboratory Animal Center of Zhengzhou University (Ethics approval license: ZZU-LAC20210115[18]). Gastric cancer material from The Affiliated Cancer Hospital of Zhengzhou University (Henan cancer Hospital) was collected in medium 1640 supplemented with 100 U/mL penicillin, and 100 μg/mL streptomycin, stored at 4 °C and received within 4 h from diagnostic endoscopy. The biopsy specimen was then cut into pieces of approximately 3 mm3 with surgical scissors under sterile conditions. Female NOD/SCID mice were anesthetized with 5% inhaled isoflurane in 95% oxygen. Tumor fragments were transplanted into the right forelimb of female NOD/SCID mice. PDXs were passaged and expanded for 3 generations (F0, F1, and F2) until production of a cohort of 20 mice. When the average tumor volume reached 150 mm3, mice were assigned to four groups (n = 5) and treated with following regimens: (i) vehicle (containing 90% corn oil, 5% castor oil and 5% DMSO, 10 mL/kg/day, intragastric injection), (ii) WS-716 (100 mg/kg/day, intragastric injection), (iii) PTX (5 mg/kg/3 days, intravenous injection), (iv) PTX plus WS-716. After the therapy for 13 days, when the maximum tumor diameter reached 15 mm, animals were euthanized by carbon dioxide. Tumor growth in vivo was monitored every two days by caliper measurements, with volume calculated using Eq. (1):
| (1) |
Relative tumor volume (RTV) was calculated as Eq. (2):
| (2) |
where V0 is the tumor volume on Day 1 and Vt is the tumor volume on Day 13. Relative tumor proliferation rate (T/C, %) was calculated according to Eq. (3) by evaluating the antitumor activity of the treatment.
| (3) |
where CRTV and TRTV is RTV value of vehicle group and treatment group, respectively.
2.23. IHC analysis
Primary anti-P-gp monoclonal antibody, SP Kit (Cat# SP-9000, Zhongshan Golden Bridge Biotechnology) and 3,3′-diaminobenzidine Horseradish Peroxidase Color Development Kit (Cat# ZLI-9017, Zhongshan Golden Bridge Biotechnology) were used in IHC analysis. Paraffin tissue blocks were cut into 4-μm slices and mounted on a glass slide. The slices were dewaxed in xylene and hydrated through a graded series of ethanol and PBS. Then, the slices were immersed in a 10 mmol/L citric acid antigen retrieval solution and incubated in a microwave oven for 10 min for antigen retrieval. The samples were incubated with a 3% (v/v) H2O2 for 20 min to inactivate endogenous peroxidase, after which they were blocked using 10% goat serum and incubated at RT for 40 min. Then, the slices were incubated with a rabbit anti-P-gp monoclonal antibody (1:500) overnight at 4 °C. The negative control slice incubated with equal volume of PBS under the same condition. Then, the slices were incubated with the secondary antibody for 1 h prior to color development and counterstaining with hematoxylin for 2 min.
2.24. Blood biochemical assay and routine analysis
At the end of therapy, blood was gained from the eyeballs of mice. Routine blood analysis was performed using animal blood analyzer (HEMAVET 950FS, Drew Scientific, TX, USA). The serum was obtained by centrifugation (452 × g, 10 min) at RT. The total protein, albumin, direct bilirubin, aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, creatinine, and urea in serum measured using assay kit from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
2.25. Statistical analysis
All experiments were repeated at least three times. One-way analysis of variance was used to evaluate differences. The statistical significance is determined at P < 0.05.
3. Results and discussion
3.1. WS-716 reversed P-gp-mediated MDR in vitro
To examine the cellular toxicity of WS-716 (Fig. 1A), we first evaluated its cytotoxicity against PTX-resistant SW620/Ad300 cells with over-expressed P-gp (Fig. 1B) using the MTT assay. As shown in Fig. 1C and Supporting Information Fig. S1, WS-716 had a minimal inhibition toward SW620/Ad300 and SW620 cells with the cell survival rate over 90% even at 20 μmol/L. Therefore, the nontoxic concentration (20 μmol/L) was used in the following assays to characterize its MDR reversal ability represented by the RF. The well-characterized competitive P-gp inhibitor VPM was used as a positive control. Initially, we explored the reversal ability of WS-716 against SW620/Ad300 cells. As shown in Fig. 1D, WS-716 (20 μmol/L) significantly increased the sensitivity of SW620/Ad300 cells to PTX. We further investigated whether WS-716 decreased the resistance of SW620/Ad300 to PTX in a dose-dependent manner. As shown in Table 1, the IC50 values of PTX in parental SW620 cells and resistant SW620/Ad300 cells were 7.69 and 4233.18 nmol/L, respectively, indicating that SW620/Ad300 cells exhibited a 550-fold resistance to PTX. Satisfactorily, when combined with WS-716 at 10 and 20 μmol/L, respectively, the corresponding IC50 values against SW620/Ad300 cells were 89.58 and 46.65 nmol/L, respectively, significantly lower than that when PTX was used alone. By contrast, co-treatment of SW620/Ad300 cells with PTX and VPM (4 μmol/L), the corresponding IC50 value was 303.53 nmol/L. The results showed that WS-716 had acceptable potency in reversing MDR of SW620/Ad300 cells to PTX. We also found that co-administration with WS-716 or VPM had little effect on the sensitivity of SW620 cells to PTX due to the absence of P-gp on cytomembrane (Table 1). The data further confirmed that WS-716 increased sensitization of SW620/Ad300 cells to PTX by modulating P-gp.
Figure 1.

WS-716 reversed multidrug resistance mediated by P-glycoprotein (P-gp). (A) Chemical structure of WS-716. (B) P-gp was up-regulated in SW620/Ad300 cells. (C) WS-716 and verapamil (VPM) below 30 μmol/L had little inhibitory effect on the viability of SW620/Ad300 cells. (D) WS-716 significantly increased the sensitivity of SW620/Ad300 cells to paclitaxel (PTX) at 20 μmol/L. (E) P-gp was up-regulated in KB-C2 cells. (F) P-gp was over-expressed in HEK293/ABCB1 cells. β-Actin was used as the loading control. Data are presented as the mean ± SD, n = 3.
Table 1.
Effect of WS-716 on reversing paclitaxel (PTX) resistance against SW620/Ad300 cells.
| Treatment | IC50 of PTX (nmol/L)a [RF]b |
|
|---|---|---|
| SW620 | SW620/Ad300 | |
| PTX | 7.69 ± 2.78 [1.00] | 4233.18 ± 499.10 [1.00] |
| PTX + WS-716 (10 μmol/L) | 6.19 ± 1.52 [1.24] | 89.58 ± 5.31 [47.26] |
| PTX + WS-716 (20 μmol/L) | 4.49 ± 0.35 [1.71] | 46.65 ± 6.98 [90.74] |
| PTX + VPM (4 μmol/L) | 7.11 ± 2.88 [1.08] | 303.53 ± 47.93 [13.95] |
Data are presented as the mean ± SD (n = 3).
The RF was calculated by dividing the IC50 of PTX in the absence of reversal agent with the IC50 of PTX in the presence of reversal agent.
Inspired by above interesting results, we next detected the reversal activity of WS-716 against another MDR cell line KB-C2 with overexpressed P-gp (Fig. 1E). As shown in Table 2, WS-716 at 10 and 20 μmol/L remarkably increased the sensitivity of KB-C2 cells to PTX with the IC50 values of 8.77 and 5.25 nmol/L, respectively, significantly lower than that of PTX used alone. The reversal ability of WS-716 in KB-C2 cells was comparable to that of VPM (IC50 = 7.05 nmol/L). To further confirm the P-gp-mediated MDR of WS-716, we established the ABCB1 transfected HEK293/ABCB1 cells and then examined the reversal effect of WS-716. After transfection, P-gp was up-regulated in HEK293/ABCB1 cells compared to the empty vector-transfected HEK293/pcDNA3.1 (Fig. 1F). As shown in Table 3, the HEK293/ABCB1 cells had a 47.36-fold resistance to PTX. WS-716 significantly sensitized HEK293/ABCB1 cells to PTX in a concentration-dependent manner, but had little effect on HEK293/pcDNA3.1 cells. Particularly, WS-716 at 20 μmol/L almost completely reversed the resistance of HEK293/ABCB1 to PTX (IC50 = 3.21 nmol/L, RF = 28.47). Thus, WS-716 significantly reversed P-gp-mediated MDR in multiple PTX-resistant cell lines.
Table 2.
WS-716 significantly sensitizes KB-C2 cells to paclitaxel (PTX).
| Treatment | IC50 of PTX (nmol/L)a [RF]b |
|
|---|---|---|
| KB-3-1 | KB-C2 | |
| PTX | 4.54 ± 1.59 [1.00] | 1886.37 ± 243.05 [1.00] |
| PTX + WS-716 (10 μmol/L) | 3.51 ± 0.73 [1.29] | 8.77 ± 1.42 [215.09] |
| PTX + WS-716 (20 μmol/L) | 1.82 ± 0.62 [2.49] | 5.25 ± 1.27 [359.31] |
| PTX + VPM (4 μmol/L) | 3.84 ± 1.25 [1.18] | 7.05 ± 2.81 [267.57] |
Data are presented as the mean ± SD, n = 3.
The RF was calculated by dividing the IC50 of PTX in the absence of reversal agent with the IC50 of PTX in the presence of reversal agent.
Table 3.
Effect of WS-716 on reversing paclitaxel (PTX) resistance in HEK293/ABCB1 cells.
| Treatment | IC50 of PTX (nmol/L)a [RF]b |
|
|---|---|---|
| HEK293/pcDNA3.1 | HEK293/ABCB1 | |
| PTX | 1.93 ± 0.06 [1.00] | 91.40 ± 23.32 [1.00] |
| PTX + WS-716 (10 μmol/L) | 1.77 ± 0.10 [1.09] | 13.09 ± 5.59 [6.98] |
| PTX + WS-716 (20 μmol/L) | 1.41 ± 0.20 [1.37] | 3.21 ± 2.03 [28.47] |
| PTX + VPM (4 μmol/L) | 1.62 ± 0.16 [1.19] | 10.37 ± 5.47 [8.81] |
Data are presented as the mean ± SD, n = 3.
The RF was calculated by dividing the IC50 of PTX in the absence of reversal agent with the IC50 of PTX in the presence of reversal agent.
To explore the specificity of WS-716 in reversing MDR, we determined the reversal effect of WS-716 in drug resistant cancer cells overexpressing ABCC1 or ABCG2 transporters and in HEK293 cells transfected with ABCC1 or ABCG2 gene. In this experiment, we also determined the effect of ONO-1078 and Ko 143, which are inhibitors of ABCC149 and ABCG250 transporters, respectively, in the same cell lines, as positive controls. As shown in Table 4, the KB/CV60 cells showed 158.89-fold resistance to vincristine compared with the parental KB-3-1 cells. WS-716 (10 and 20 μmol/L) did not alter the efficacy of vincristine in both of the parental KB-3-1 cells and the ABCC1-overexpressing KB/CV60 cells, while the efficacy of vincristine was significantly increased by 20 μmol/L of ONO-1078 in the KB/CV60 cells (Table 4). The ABCG2-overexpressed S1-M1-80 cells were 605.92-fold resistant to mitoxantrone compared to the parental S1 cells that do not express the ABCG2 transporter (Table 5). WS-716 (10 and 20 μmol/L) had little effect on the reversal activity of mitoxantrone in both of the parental S1 cells and the ABCG2-overexpressed S1-M1-80 cells. In contrast, Ko 143 (10 μmol/L) did not significantly alter the efficacy of mitoxantrone in the parental S1 cells, whereas it significantly enhanced the efficacy of mitoxantrone in the S1-M1-80 cells (Table 5).
Table 4.
Effect of WS-716 on reversing ABCC1-mediated multidrug resistance in KB/CV60 cells.
| Treatment | IC50 (nmol/L)a [RF]b |
|
|---|---|---|
| KB-3-1 | KB/CV60 | |
| Vincristine | 2.15 ± 0.19 [1.00] | 341.62 ± 33.05 [1.00] |
| Vincristine + WS-716 (10 μmol/L) | 2.25 ± 0.23 [0.96] | 361.03 ± 31.42 [0.95] |
| Vincristine + WS-716 (20 μmol/L) | 2.17 ± 0.22 [0.99] | 355.82 ± 31.27 [0.96] |
| Vincristine + ONO-1078 (20 μmol/L) | 2.58 ± 0.25 [0.83] | 12.82 ± 0.21 [26.65] |
Data are presented as the mean ± SD, n = 3.
The RF was calculated by dividing the IC50 of vincristine in the absence of reversal agent with the IC50 of vincristine in the presence of reversal agent.
Table 5.
Effect of WS-716 on reversing ABCG2-mediated multidrug resistance in S1-M1-80 cells.
| Treatment | IC50 (μmol/L)a [RF]b |
|
|---|---|---|
| S1 | S1-M1-80 | |
| Mitoxantrone | 0.25 ± 0.02 [1.00] | 151.48 ± 15.38 [1.00] |
| Mitoxantrone + WS-716 (10 μmol/L) | 0.29 ± 0.03 [0.86] | 159.58 ± 15.31 [0.95] |
| Mitoxantrone + WS-716 (20 μmol/L) | 0.24 ± 0.05 [1.04] | 146.65 ± 16.98 [1.03] |
| Mitoxantrone + Ko 143 (10 μmol/L) | 0.21 ± 0.02 [1.19] | 0.53 ± 0.04 [285.81] |
Data are presented as the mean ± SD, n = 3.
The RF was calculated by dividing the IC50 of mitoxantrone in the absence of reversal agent with the IC50 of mitoxantrone in the presence of reversal agent.
We further evaluated the reversal efficacy of WS-716 in transfected cells overexpressing ABCC1 or ABCG2 transporters. The transfection of HEK293 cells with the genes coding for ABCC1 or ABCG2 significantly decreased the efficacy of vincristine or mitoxantrone compared to HEK293 cells transfected with an empty pcDNA3.1 vector, respectively (Table 6). WS-716 (10 and 20 μmol/L) produced no increase in the efficacy of vincristine or mitoxantrone compared to that of the ABCC1 inhibitor, ONO-1078, or Ko 143 (Table 6), which were consistent with the results from the drug-resistant ABCC1- or ABCG2-overexpressed cancer cell lines. These data indicated that WS-716 failed to reverse ABCC1- or ABCG2-mediated MDR. Taken together, our results indicated that WS-716 specifically reversed P-gp-mediated MDR in vitro.
Table 6.
Effect of WS-716 on reversing ABCC1- or ABCG2-mediated multidrug resistance in transfected HEK293 cells.
| Treatment | IC50 (nmol/L)a [RF]b |
||
|---|---|---|---|
| HEK293/pcDNA3.1 | HEK293/ABCC1 | HEK293/ABCG2 | |
| Vincristine | 2.83 ± 0.26 [1.00] | 88.09 ± 9.32 [1.00] | – |
| Vincristine + WS-716 (10 μmol/L) | 2.72 ± 0.21 [1.04] | 85.92 ± 8.59 [1.02] | – |
| Vincristine + WS-716 (20 μmol/L) | 2.96 ± 0.20 [0.96] | 82.23 ± 9.03 [1.07] | – |
| Vincristine + ONO-1078 (20 μmol/L) | 2.65 ± 0.16 [1.07] | 4.59 ± 0.27 [19.19] | – |
| Mitoxantrone | 14.35 ± 1.65 [1.00] | – | 314.04 ± 23.32 [1.00] |
| Mitoxantrone + WS-716 (10 μmol/L) | 15.80 ± 1.28 [0.91] | – | 323.42 ± 25.59 [0.97] |
| Mitoxantrone + WS-716 (20 μmol/L) | 16.58 ± 1.82 [0.87] | – | 305.62 ± 32.03 [1.03] |
| Mitoxantrone + Ko 143 (10 μmol/L) | 14.73 ± 1.62 [0.97] | – | 16.91 ± 1.47 [18.57] |
–Not applicable.
Data are presented as the mean ± SD, n = 3.
The RF was calculated by dividing the IC50 of chemotherapeutic agent in the absence of reversal agent with the IC50 of chemotherapeutic agent in the presence of reversal agent.
3.2. WS-716 increased intracellular PTX accumulation by intercepting its efflux
As an efflux pump, P-gp decreases the intracellular accumulation of chemotherapeutic drugs to develop MDR. We herein assessed the effect of WS-716 on the intracellular accumulation of PTX in SW620/Ad300 cells using UPLC and the efflux of [3H]-PTX assay. Following pre-treatment with WS-716 or VPM for 72 h, SW620 and SW620/Ad300 cells were exposed to PTX for 2 h for detection. As illustrated in Fig. 2A, the intracellular concentration of PTX in SW620 was 4949.68 nmol/L, while the intracellular concentration of PTX in SW620/Ad300 cells was 188.59 nmol/L. However, compared to the control in absence of the reversal agents, WS-716 increased the intracellular PTX in a concentration-dependent manner. Particularly, WS-716 at 20 μmol/L significantly increased intracellular accumulation of PTX in SW620/Ad300 cells. Moreover, we also evaluated the effect of WS-716 on the intracellular accumulation of [3H]-PTX. As shown in Fig. 2B, WS-716 (20 μmol/L) significantly increased the accumulation of [3H]-PTX in SW620/Ad300 cells comparable to that treated with VPM (4 μmol/L), the result was consistent with that of UPLC. Next, the effect of WS-716 on the efflux function of P-gp was performed by comparing the concentration of [3H]-PTX between different groups after four different time points. As shown in Fig. 2C, like VPM, WS-716 slightly decreased the [3H]-PTX efflux in SW620 cells. As shown in Fig. 2D, WS-716 and VPM led to 60.12% and 69.39% loss of [3H]-PTX, respectively, in SW620/Ad300 cells after treatment for 120 min, indicating that WS-716 and VPM could inhibit efflux function of P-gp. Collectively, WS-716 increased the accumulation of PTX by inhibiting the efflux function of P-gp in SW620/Ad300 cells.
Figure 2.

WS-716 increased the intracellular accumulation of paclitaxel (PTX) by intercepting its efflux. (A) WS-716 increased intracellular PTX in SW620/Ad300 cells. (B) WS-716 increased the accumulation of [3H]-PTX. (C) WS-716 slightly reduced the efflux of PTX in SW620 cells. (D) WS-716 significantly reduced the efflux of PTX in SW620/Ad300 cells. Data are presented as the mean ± SD, n = 3. #P < 0.05, ##P < 0.01.
3.3. WS-716 stimulated ATP hydrolysis without affecting expression or subcellular localization of P-gp
As a hydrolase on the surface of the cell membrane, decreased drug efflux may be due to the downregulation or translocation of protein or the inactivation of the ATPase of P-gp, thus inhibiting the efflux function of P-gp is a promising strategy to circumvent MDR. To elucidate the mechanism of WS-716, we examined the effect of WS-716 on the expression of P-gp in SW620/Ad300 cells by Western blot. As illustrated in Fig. 3A, incubation with WS-716 (10 and 20 μmol/L) for 76 h did not change the protein levels of P-gp in SW620/Ad300 cells. Next, after treatment of SW620/Ad300 cells with WS-716 for 76 h, the effect of WS-716 on subcellular localization of P-gp was measured by immunofluorescence. As shown in Fig. 3B, P-gp (green) was uniformly distributed on the cell membrane, indicating that there was no obvious alteration of P-gp subcellular localization in SW620/Ad300 cells. It has been well recognized that the efflux function of P-gp is linked to ATP hydrolysis stimulated in the presence of P-gp substrates such as VPM51. We speculated that WS-716 may block the efflux function of P-gp by regulating the activity of its ATPase. We therefore assessed the effect of WS-716 on the basal activity of P-gp ATPase. The crude membrane of P-gp was incubated with different concentrations of WS-716 (0–40 μmol/L). As shown in Fig. 3C, WS-716 stimulated ATP hydrolysis of P-gp. Particularly, after treatment with WS-716 at 20 μmol/L, the P-gp-stimulation effect reached the maximal value, being around 2.5-fold of the basal activity.
Figure 3.

WS-716 stimulated ATP hydrolysis without changing the expression or subcellular localization of P-glycoprotein (P-gp). (A) WS-716 did not change the protein level of P-gp in SW620/Ad300 cells. (B) Effect of WS-716 on the subcellular localization of P-gp (green) after treatment for 76 h. The cell nuclei (blue) were stained by Hoechst 33258. The scale bar is 10 μm. (C) WS-716 stimulated ATP hydrolysis and reached the maximal value at 20 μmol/L. Data are presented as the mean ± SD, n = 3.
3.4. WS-716 directly bound to P-gp and binding model analysis
To determine the drug target engagement between WS-716 and P-gp, the cellular thermal shift assay was performed52. SW620/Ad300 cells were lysed, incubated with 1% DMSO or WS-716 (200 μmol/L), and then heated to different temperatures. It is widely recognized that direct binding of the compound to the protein of interest could increase thermal stability. As shown in Fig. 4A, compared to the solvent control, WS-716 could significantly enhance the thermal stability of P-gp, showing the target engagement of WS-716 to P-gp in SW620/Ad300 cells.
Figure 4.

WS-716 binds directly to P-glycoprotein (P-gp). (A) The interaction of WS-716 and P-gp was assessed through cellular thermal shift assay. (B) The ribbon diagram of P-gp with the binding location of WS-716 at the internal cavity. (C) The 2D bonding interaction of WS-716 within P-gp (PDB code: 6FN1). (D) The 3D ligand–receptor interaction diagram of WS-716 and P-gp.
To reveal the possible binding model of WS-716 with P-gp, the molecular modeling was performed using the zosuquidar and UIC2 Fab complex of human-mouse chimeric P-gp as the docking template (Protein Data Bank code: 6FN1). The induced-fit docking-simulated best docking pose of WS-716 exhibited a docking score of −11.067 kcal/mol, indicating a relatively low binding free energy53 in complex with P-gp. WS-716 was mainly stabilized within the binding cavity in the transmembrane domain of P-gp (Fig. 4B), which is lined by residues Trp231, Leu235, Met298, Phe302, Tyr306, Asn720, Leu723, Gln724, Phe727, Ser765, Phe769, Gln837, Met875, Ala986, Gln989, Val990, and Phe993 (Fig. 4C). Two π–π stacking interactions were predicted between the imidazole ring of WS-716 and the phenyl ring of Phe302 (4.09 Å), and between the triazole ring of WS-716 and the benzyl side chain of Phe769 (4.84 Å). The triazole ring of WS-716 was also involved in hydrogen bonding interaction at the N4 position with the side chain of Asn720 (–N···H2N–Asn720, 2.10 Å) (Fig. 4C and D).
3.5. WS-716 did not affect the activity of CYP3A4
The substrate and tissue distribution of P-gp and CYP3A4 are partially overlapped, and therefore P-gp inhibitors always inhibit the activity of the drug-metabolizing enzyme CYP3A4, thus causing unexpected drug–drug interactions and side effects28, 29, 30, 31, 32. To explore whether WS-716 could affect human CYP3A4, the CYP3A4 activity assay was performed using the well-known CYP3A4 inhibitor ketoconazole and P-gp inhibitor VPM as the positive controls. As shown in Fig. 5A, at 30 μmol/L, ketoconazole almost completely inhibited the activity of CYP3A4, VPM also showed moderate inhibition. In contrast, WS-716 at 30 μmol/L showed weak inhibitory activity toward CYP3A4. As shown in Fig. 5B, ketoconazole and VPM dose-dependently inhibited the activity of CYP3A4 with the IC50 values of 0.067 and 3.25 μmol/L, respectively, while WS-716 displayed minimal effect on the activity of CYP3A4. These data strongly suggested that WS-716 may be a less toxic P-gp inhibitor compared to VPM.
Figure 5.

WS-716 had little effect on the activity of cytochrome P4503A4 (CYP3A4). (A) The effect of WS-716, VPM and ketoconazole on the reaction kinetics of fluorogenic substrate metabolism in human liver microsomes containing CYP3A4 during 40 min at 37 °C. (B) Dose–response curve of WS-716, VPM, and ketoconazole on CYP3A4 at 37 °C. Data are presented as the mean ± SD, n = 3.
3.6. WS-716 enhanced the inhibitory activity of PTX on the formation of colony and stemness of SW620/Ad300 cells
To further interrogate the underlying mechanisms on how WS-716 increased the sensitivity of SW620/Ad300 cells to PTX, we examined the effect of WS-716 alone or its combination with PTX on the cell proliferative potential of SW620/Ad300 cells using the colony formation assay. As shown in Fig. 6A, WS-716 significantly enhanced the inhibitory activity of PTX on the colony formation of SW620/Ad300 cells, stronger than that of WS-716 and PTX alone. It has been reported that drug resistant cells are always endowed with high stemness, rendering cancer stem cells less sensitive to chemotherapeutic drugs54. Given the prominent role of stemness in the initiation of MDR, we also tested the effect of WS-716 on the stemness using culture of 3D spheroids. Notably, compared with PTX, combination of WS-716 and PTX significantly reduced the average diameter of SW620/Ad300 spheroids, especially on Day 10 (Fig. 6B). These data suggested that co-treatment with WS-716 and PTX significantly reduced the proliferation and stemness of SW620/Ad300 cells.
Figure 6.

WS-716 enhanced the inhibitory effect of paclitaxel (PTX) on the formation of colonies and 3D spheroids. (A) The colony-formation of SW620/Ad300 cells after treatment with WS-716 (20 μmol/L), PTX (1 μmol/L), or their combination on Day 7. (B) The 3D spheroid-formation of SW620/Ad300 cells after treatment of WS-716 (20 μmol/L), PTX (1 μmol/L), or their combination on Day 5 and Day 10. The scale bar is 200 μm. Data are presented as the mean ± SD, n = 3. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. NC, #P < 0.05.
3.7. WS-716 enhanced PTX-induced apoptosis of SW620/Ad300 cells
PTX kills cancer cells through the induction of apoptosis55. To examine the effect of WS-716 on the apoptosis induced by PTX, the Hoechst 33258 staining was used to visualize the cell nuclei of SW620/Ad300 after treatment with WS-716, PTX, or their combination. As shown in Fig. 7A, PTX and the combination of WS-716 and PTX induced highly concentrated chromosomes (strong blue fluorescence) in SW620/Ad300 cells, suggesting the occurrence of cell death. Flow cytometry (FCM) analysis further confirmed that WS-716 significantly enhanced the apoptotic effect of SW620/Ad300 cells induced by PTX with an apoptotic rate of 44.94%, higher than that treated with WS-716 and PTX alone (Fig. 7B). Co-treatment of SW620/Ad300 cells with WS-716 and PTX significantly downregulated PARP, and upregulated cleaved caspase-9, -3, -7, cleaved PARP, NLRP3, cleaved caspase-1, Gasdermin D, as well as increased the ratio of LC3BII to LC3BI (Fig. 7C). The data suggested that combination of WS-716 and PTX may synergistically inhibit the survival of SW620/Ad300 cells by inducing apoptosis, pyroptosis, and autophagy. Apoptosis is initiated through the intrinsic and extrinsic pathways. To elucidate the apoptotic mechanism induced by WS-716 and PTX, we detected the mitochondrial membrane potential using the JC-1 staining. As shown in Fig. 7D, combination of WS-716 and PTX led to 27.62% of cells with a high JC-1 monomer level (P < 0.001), indicating a low mitochondrial membrane potential and damaged mitochondria. Mechanistically, we found that pro-apoptotic Bax and cytochrome c in cytoplasm were up-regulated, while antiapoptotic Bcl-2 and Bcl-xL were down-regulated following treatment with WS-716 and PTX, compared to NC and mono-treatment (Fig. 7E). These data suggested that WS-716 could enhance the capability of PTX to regulate Bcl-2 family, increase mitochondrial membrane permeability, promote the release of cytochrome c to cytoplasm, activating caspase-9, -3, -7 and PARP to provoke apoptosis.
Figure 7.

WS-716 enhanced the apoptosis induced by paclitaxel (PTX) through the mitochondrial pathway in SW620/Ad300 cells. SW620/Ad300 cells were treated with WS-716 (20 μmol/L, 4 h), PTX (1 μmol/L, 2 h)or combination of WS-716 and PTX. (A) Damaged cell nucleus of SW620/Ad300 was detected by Hoechst 33258 staining. The scale bar is 100 μm. (B) Apoptosis was measured by FCM. (C) Expression of cleaved caspase-9, -3, -7, PARP, NLRP3, cleaved caspase-1, Gasdermin D, and LC3B was measured by Western blot. (D) The mitochondrial membrane potential was evaluated by FCM. (E) Bax, Bcl-2, Bcl-xL, and cytochrome c in cytoplasm expression were analyzed by Western blot at 24 h. β-Actin was used as a loading control. Data are represented as mean ± SD, n = 3. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. NC, #P < 0.05.
3.8. WS-716 potentiated the ability of PTX to induce cell cycle arrest at G2/M phase through activating CDK1
It is well established that PTX inhibits depolymerization and promotes microtubular assembly by binding to β-tubulin, thus inducing cell cycle arrest at G2/M phase and hampering mitosis1,56. Accordingly, we speculated that combination of WS-716 and PTX may change the cell cycle distribution of SW620/Ad300 cells. To verify our hypothesis, the cell-cycle progression was monitored by FCM. As depicted in Fig. 8A, PTX decreased the frequency of cells during G0/G1 phase but increased the frequency during S or G2/M phase (P < 0.001). In contrast, when combined with WS-716, PTX significantly reduced the proportion of SW620/Ad300 cells during G0/G1 phase (P < 0.05), and arrested cell cycle progression at G2/M phase (Fig. 8B). To elucidate the underlying mechanisms, we detected the expression of CDK2 and CDK1 which are essential for G1/S and G2/M phase transition, respectively. The results showed that the combination of WS-716 and PTX blocked phosphorylation of CDK2 at threonine 160 (Thr160). Then, we found CDK2 inhibitors p21 and p27 that prevent CDK2 activation by CDK-activating kinase (CAK, the complex of CDK7 and cyclin H) phosphorylation at Thr160 were up-regulated when PTX was combined with WS-716. WS-716, when in combined with PTX, led to phosphorylation of threonine 161 (Thr161) and dephosphorylation of tyrosine 15 (Tyr15) to activate CDK1 (Fig. 8C). Further, phosphorylation of CDK1 at Thr161 was found to be associated with upregulation of cyclin H but not CDK7 (Fig. 8C). These results suggested that WS-716 intensified PTX-induced G2/M arrest of the cell cycle through CDK2 inactivation and CDK1 activation.
Figure 8.

WS-716 potentiates the ability of paclitaxel (PTX) to induce cycle arrest at G2/M phase. SW620/Ad300 cells were treated with WS-716 (20 μmol/L, 4 h),PTX (1 μmol/L, 2 h) or combination of WS-716 and PTX. (A, B) The distribution of cell cycle was examined by FCM. (C) Expression of p-CDK2, total CDK2, p-CDK1, total CDK1, p21, p27, cyclin H, and CDK7 were analyzed by Western blot at 24 h. β-Actin was used as a loading control. Data are represented as mean ± SD, n = 3. ∗P < 0.05, ∗∗∗P < 0.001 vs. NC, #P < 0.05.
3.9. WS-716 synergistically increased the efficacy of PTX in CDX models
To examine the synergistic activity of WS-716 and PTX in vivo, SW620/Ad300 cells were injected subcutaneously to establish xenografts in nude mice. When the average volume of the subcutaneous tumors reached about 100 mm3, the mice were sorted into five different treatment groups (n = 5), and administrated with vehicle, WS-716, PTX, PTX plus VPM, or PTX plus WS-716 for fifteen days. Mice were monitored for tumor volume and body weight every two days. As shown in Fig. 9A, combination therapy of WS-716 and PTX produced improved antitumor effects on xenograft models bearing SW620/Ad300, compared to vehicle and the groups receiving PTX and WS-716 alone, respectively. Of note, the PTX/WS-716 combination showed better efficacy than PTX/VPM combination. The combination of PTX/WS-716 also significantly reduced the average tumor weight compared to vehicle (Fig. 9B and C). During the therapy, no mortality or significant loss of body weight was observed (Fig. 9D). Next, tumor tissues from animals were analyzed by HE or TUNEL staining. As shown in Fig. 9E, in the combination group, severe necrosis characterized by nuclear pyknosis (shrunken and dark), karyorrhexis (nuclear fragmentation), and karyolysis (dark purple, HE staining) were observed, and the cytoplasm and cell borders were beyond recognition (HE staining, the upper panel). Particularly, cavities of about 50 μm formed following removal of the necrotic tissue (HE analysis). Consistently, the results of TUNEL revealed that there was remarkable apoptosis (green) in tumor tissue of the combination group than that in vehicle or mono-therapy groups (the middle panel). To further evaluate the safety of WS-716, the liver tissues were analyzed by HE analysis. There was no obvious lesion in the animal liver (Fig. 9E, the bottom panel), suggesting that the combination regimen did not result in obvious toxicity. These data confirmed that WS-716 synergistically increased the efficacy of PTX in xenograft models bearing SW620/Ad300 cells without extra toxicity.
Figure 9.

WS-716 significantly improves the inhibitory efficacy of paclitaxel (PTX) in vivo. Mice bearing SW620/Ad300 xenograft were treated with vehicle (10 mL/kg/day), WS-716 (100 mg/kg/day), PTX (5 mg/kg/3 days), PTX plus VPM (4 mg/kg/day) or PTX plus WS-716 for 15 days. (A) Tumor volume during the therapy. (B) Tumor weight on Day 15. (C) Photographs of tumors on Day 15. (D) Body weight of mice during the therapy. (E) HE and TUNEL analysis of tumor and liver tissues. Data are presented as the mean ± SD (n = 5). ∗P < 0.05 vs. vehicle. Scale bar is 100 μm.
3.10. WS-716 synergistically increased the efficacy of PTX in PDX models
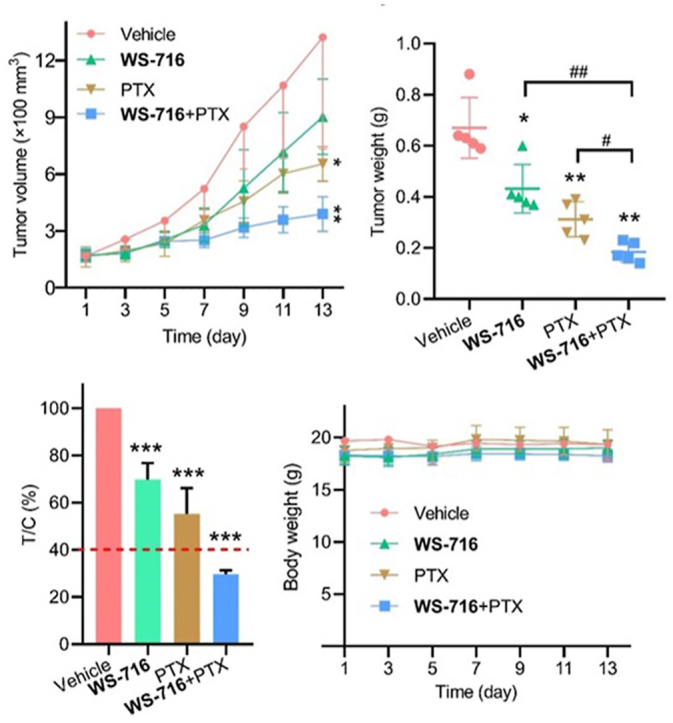
Because of the capability of WS-716 in increasing the sensitivity of cultured MDR tumor cells to PTX in vitro and in vivo, we further investigated that whether WS-716 could sensitize the MDR tumors derived from patients to PTX. At present, PDX models are considered to be effective models that better recapitulate the biological characteristics of human tumors in vivo57. To this end, a 69-year-old female patient with advanced human epidermal growth factor receptor 2-positive gastric cancer was included in the study, who had received trastuzumab, capecitabine plus oxaliplatin and sintilimab as first-line therapy for seven cycles before progressive disease. The tumor tissue obtained through diagnostic endoscopy before second-line therapy was implanted subcutaneously in female NOD/SCID mice (Fig. 10A). The results of pathological analysis validated that the primary tumor is a gastric adenocarcinoma of the fundic gland, and glandular architecture can be seen in tumors from both F0 and F2 xenografts (Fig. 10B, HE staining). Since this gastric cancer patient was treated with capecitabine plus oxaliplatin prior to disease progression, the tumors may have developed MDR. Further IHC analysis in Fig. 10B and Supporting Information Fig. S2, and Western blot analysis in Fig. 10C confirmed that P-gp were expressed in the primary, F0 and F2 tumor tissue. So far, we have successfully established PDX models of MDR cancer mediated by P-gp, and F2 xenografts were used to evaluate the pharmacodynamics of WS-716 combined with PTX. The F2 tumor-bearing mice were divided into four groups (n = 5) and administrated with vehicle, WS-716, PTX or PTX/WS-716 combination for thirteen days. As shown in Fig. 10D and E, the combined therapy of PTX and WS-716 significantly suppressed progression of MDR cancer and showed antitumor superiority over PTX or WS-716 treatment alone. After treatment, the tumor volume and weight were significantly reduced, showing significant difference compared to other groups receiving single therapy. To further determine the sensitization effect of WS-716 to PTX, we calculated the relative tumor proliferation rate (T/C), and the treatment is effective when T/C is below 40%. As depicted in Fig. 10F and G, the T/C value of the combination group was 29.725 ± 1.55%, significantly lower than those receiving WS-716 (69.67 ± 7.13%), PTX (55.23 ± 10.94%) and the vehicle group. Similar to the results from CDX models (Fig. 9D), all these therapies did not cause weight loss in the PDX models (Fig. 10H).
Figure 10.

WS-716 enhances the antitumor activity of paclitaxel (PTX) in the multidrug resistance (MDR) patient-derived xenograft (PDX) models. Mice bearing PDX were treated with vehicle (10 mL/kg/day), PTX (5 mg/kg/3 days), WS-716 (100 mg/kg/day) or PTX plus WS-716. Tumor volume and animal body weight were measured periodically. (A) Workflow of establishment of MDR PDX models. (B) HE staining was used for pathological validation and IHC analysis was used for detection of P-gp expression in tumor tissues from the patient (primary tumor), F0 (F0 tumor), and F2 xenografts (F2 tumor). (C) Expression of P-gp in F0 and F2 tumor was detected using Western blot. (D) Tumor volume during treatment. (E) Tumor weight on Day 13. (F) The relative tumor proliferation rate (T/C) of treatment groups (The red dotted line: 40%). (G) Photographs of tumors on Day 13. (H) Body weight during treatment. Data are presented as the mean ± SD, n = 5. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. vehicle, #P < 0.05, #P < 0.01. Scale bar is 100 μm.
Blood biochemical analysis was conducted to explore the safety of WS-716 combined with PTX. The total protein, albumin, direct bilirubin, aspartate transaminase, alkaline phosphatase, alanine transaminase in serum as shown in Fig. 11A were measured to evaluate liver function. The levels of creatinine and urea in serum as shown in Fig. 11B were used to evaluate renal function at the end of therapy. The results revealed that no significant change was found between each group, suggesting WS-716, PTX or their combination did not interfere with liver and renal functions. As myelosuppression is the major adverse reaction of PTX, blood routine analysis was conducted. As seen in Table 7, compared to vehicle, PTX and WS-716 combined with PTX caused significant reduction of neutrophils, but there was no significant difference in neutrophil count between the PTX group and the combination group. In addition, histological analysis in Supporting Information Fig. S3 revealed that no organic lesions were observed in heart, liver, spleen, lung and kidney at the end of therapies. Our results clarified that, WS-716 synergistically increased the therapeutic efficacy of PTX without obvious adverse reactions in PDX models.
Figure 11.

Hepatic and renal functions were evaluated at the end of treatment. (A) Concentrations of total protein, albumin, direct bilirubin, aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase in serum of the mice were used to evaluate hepatic function of mice. (B) The concentrations of creatinine and urea were used to evaluate renal function of mice. Data are presented as mean ± SD, n = 3.
Table 7.
Blood routine analysis.
| Item | Valuea |
|||
|---|---|---|---|---|
| Vehicle | WS-716 | PTX | WS-716 + PTX | |
| White blood cell count, 109/L | 3.49 ± 1.04 | 2.74 ± 1.26 | 2.37 ± 1.68 | 1.83 ± 0.67 |
| Neutrophil count, 109/L | 2.49 ± 1.04 | 1.41 ± 0.68 | 0.83 ± 0.65∗ | 0.64 ± 0.40∗ |
| Lymphocyte count, 109/L | 0.74 ± 0.55 | 0.82 ± 0.61 | 1.22 ± 0.80 | 0.86 ± 0.40 |
| Monocyte count, 109/L | 0.28 ± 0.13 | 0.47 ± 0.30 | 0.23 ± 0.19 | 0.21 ± 0.10 |
| Eosimophil count, 109/L | 0.02 ± 0.01 | 0.05 ± 0.04 | 0.08 ± 0.08 | 0.08 ± 0.10 |
| Basophil count, 109/L | 0 | 0 | 0.02 ± 0.03 | 0.04 ± 0.06 |
| Percentage of granulocyte, % | 73.83 ± 12.00 | 52.91 ± 16.79 | 30.19 ± 12.75 | 34.92 ± 23.36 |
| Percentage of lymphocyte, % | 19.11 ± 10.52 | 29.00 ± 11.06 | 56.29 ± 11.35∗ | 48.20 ± 19.47 |
| Percentage of monocyte, % | 6.40 ± 3.05 | 12.60 ± 5.40 | 10.25 ± 3.81 | 11.33 ± 2.87 |
| Percentage of eosimophil, % | 0.60 ± 0.25 | 0.90 ± 1.12 | 2.36 ± 1.95 | 3.66 ± 3.66 |
| Percentage of basophil, % | 0.07 ± 0.05 | 0.27 ± 0.37 | 0.91 ± 0.35 | 1.90 ± 2.00 |
| Red blood cell, 1012/L | 9.16 ± 0.29 | 8.273 ± 0.422 | 7.75 ± 2.12 | 7.547 ± 2.06 |
| Hemoglobin concentration, g/L | 14.63 ± 0.61 | 12.97 ± 0.67 | 11.77 ± 3.40 | 11.27 ± 2.94 |
| Red blood cell specific volume, % | 60.00 ± 2.32 | 53.87 ± 1.53 | 46.80 ± 12.77 | 43.27 ± 12.18 |
| Mean corpusular hemoglobin, pg | 15.85 ± 0.07 | 15.67 ± 0.68 | 15.13 ± 0.95 | 15.00 ± 0.61 |
| Mean corpusular hemoglobin concentration, g/L | 24.37 ± 0.32 | 24.07 ± 0.64 | 28.37 ± 6.18 | 26.20 ± 1.08 |
| Red blood cell volume distribution, % | 16.30 ± 0.44 | 16.03 ± 0.35 | 17.13 ± 1.21 | 15.87 ± 0.67 |
| Platelet count, 1012/L | 1.48 ± 2.45 | 1.28 ± 0.26 | 1.14 ± 0.21 | 0.85 ± 0.11 |
| Mean platelet volume, fl | 5.00 ± 0.30 | 5.07 ± 0.31 | 5.43 ± 0.25 | 5.43 ± 0.21 |
Data are presented as the mean ± SD (n = 3), ∗P < 0.05 vs. vehicle.
Collectively, these data suggest that WS-716 safely increased the sensitivity of clinically derived MDR tumor with overexpressed P-gp to PTX, the combined treatment of WS-716 and PTX may have therapeutic potential in patients with disease progression following chemotherapy.
4. Conclusions
In this work, we reported the preclinical results of the triazolo[1,5-a]pyrimidine-based highly potent, specific, safe and orally active P-gp inhibitor WS-716 through our drug discovery program. WS-716, when in combination with PTX, could almost completely and specifically reversed P-gp-mediated MDR in multiple drug-resistant cancer cell lines including SW620/Ad300, KB-C2, and HEK293/ABCB1, while had little impact on the reversal ability of ABCC1- or ABCG2-mediated MDR. WS-716 could reverse the MDR of SW620/Ad300 cells to PTX by inhibiting the efflux function of P-gp. Mechanistically, WS-716 directly bound to P-gp and stimulated the ATP hydrolysis of P-gp without changing its protein levels and subcellular localization. WS-716 enhanced the inhibitory activity of PTX on formation of colony and stemness as well as the PTX-induced apoptosis of SW620/Ad300 cells. WS-716 potentiated the ability of PTX to trigger apoptosis by regulating members of Bcl-2 family and to block cell cycle at G2/M phase through inactivating CDK2 and activating CDK1. WS-716 displayed minimal effect on CYP3A4, suggesting low risk of drug–drug interactions when combined with anticancer drugs. WS-716 enhanced the therapeutic efficacy of PTX in CDX models. Importantly, WS-716, when in combined with PTX, effectively inhibited tumor growth of the clinically derived MDR tumors that progressed after first-line therapies without obvious side effects. Taken together, the triazolo[1,5-a]pyrimidine represents a novel scaffold for developing P-gp inhibitors, and WS-716 specifically reversed P-gp-mediated MDR in vitro and in vivo. For patients who have received first-line chemotherapy, combining WS-716 with chemotherapeutic agents may increase treatment response in the second-line chemotherapy. Our study provides a new strategy for posterior line therapy after chemotherapy failure.
Acknowledgements
We are thankful to Dr. Tanaji T. Talele (St. John's University, New York, NY, USA) for providing the computing resources for docking analysis and Suneet Shukla, Suresh V. Ambudkar (Laboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, 20892, USA) for providing ABCB1 crude membrane. We would like to thank the financial support from the National Natural Science Foundation of China (Nos. 81973177, 82103560 and 82103996), Medical Science and Technique Foundation of Henan Province (Nos. 2018020486 and SB201901101, China), Science and Technique Foundation of Henan Province (Nos. 202102310413, China), Natural Science Foundation of Henan Province (Nos. 222300420069, 212300410270 and 212300410253, China), 1000 Talents Program of Central plains (No. 204200510023, China), Young and Middle-aged Health and Technology Innovation Leading Talent Project of Henan Province (No. YXKC2020008, China), Program for Science & Technology Innovation Talents in Universities of Henan Province (No. 21HASTIT045, China), and State Key Laboratory of Esophageal Cancer Prevention & Treatment (No. Z2020000X, China).
Author contributions
Saiqi Wang: methodology, validation, investigation, writing-original draft, writing-review & editing, funding acquisition. Qiuxu Teng: methodology, validation, investigation. Shuai Wang: synthesis of the compound. Zining Lei: investigation, visualization. Huihui Hu: validation. Huifang Lv: resources. Beibei Chen: resources. Jianzheng Wang: resources. Xiaojing Shi: resources. Weifeng Xu: resources. Hongmin Liu: conceptualization, project administration. Xiaobing Chen: supervision, funding acquisition, project administration. Zhe-Sheng Chen: supervision, writing-review and editing. Bin Yu: supervision, writing-review and editing.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.03.023.
Contributor Information
Xiao-Bing Chen, Email: zlyychenxb0807@zzu.edu.cn.
Zhe-Sheng Chen, Email: chenz@stjohns.edu.
Bin Yu, Email: yubin@zzu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Fuchs D.A., Johnson R.K. Cytologic evidence that taxol, an antineoplastic agent from Taxus brevifolia, acts as a mitotic spindle poison. Cancer Treat Rep. 1978;62:1219–1222. [PubMed] [Google Scholar]
- 2.Robey R.W., Pluchino K.M., Hall M.D., Fojo A.T., Bates S.E., Gottesman M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 2018;18:452–464. doi: 10.1038/s41568-018-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gottesman M.M. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg M.F., Callaghan R., Ford R.C., Higgins C.F. Structure of the multidrug resistance P-glycoprotein to 2.5 nm resolution determined by electron microscopy and image analysis. J Biol Chem. 1997;272:10685–10694. doi: 10.1074/jbc.272.16.10685. [DOI] [PubMed] [Google Scholar]
- 5.Gottesman M.M., Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 6.Aller S.G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong J., Qin Z., Zhang W.D., Cheng G., Yehuda A.G., Ashby C.R., Jr., et al. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: an update. Drug Resist Updat. 2020;49:100681. doi: 10.1016/j.drup.2020.100681. [DOI] [PubMed] [Google Scholar]
- 8.Yuan J., Yin Z., Tan L., Zhu W., Tao K., Wang G., et al. Interferon regulatory factor-1 reverses chemoresistance by downregulating the expression of P-glycoprotein in gastric cancer. Cancer Lett. 2019;457:28–39. doi: 10.1016/j.canlet.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Xia L.L., Tang Y.B., Song F.F., Xu L., Ji P., Wang S.J., et al. DCTPP1 attenuates the sensitivity of human gastric cancer cells to 5-fluorouracil by up-regulating MDR1 expression epigenetically. Oncotarget. 2016;7:68623–68637. doi: 10.18632/oncotarget.11864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Z., Peng X.X., Kim I.W., Shukla S., Si Q.S., Robey R.W., et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007;67:11012–11020. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- 11.Mi Y.J., Liang Y.J., Huang H.B., Zhao H.Y., Wu C.P., Wang F., et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010;70:7981–7991. doi: 10.1158/0008-5472.CAN-10-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binkhathlan Z., Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets. 2013;13:326–346. doi: 10.2174/15680096113139990076. [DOI] [PubMed] [Google Scholar]
- 13.Chen C.J., Chin J.E., Ueda K., Clark D.P., Pastan I., Gottesman M.M., et al. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell. 1986;47:381–389. doi: 10.1016/0092-8674(86)90595-7. [DOI] [PubMed] [Google Scholar]
- 14.Jones P.M., George A.M. A new structural model for P-glycoprotein. J Membr Biol. 1998;166:133–147. doi: 10.1007/s002329900455. [DOI] [PubMed] [Google Scholar]
- 15.Loo T.W., Bartlett M.C., Clarke D.M. Suppressor mutations in the transmembrane segments of P-glycoprotein promote maturation of processing mutants and disrupt a subset of drug-binding sites. J Biol Chem. 2007;282:32043–32052. doi: 10.1074/jbc.M706175200. [DOI] [PubMed] [Google Scholar]
- 16.Silva R., Vilas-Boas V., Carmo H., Dinis-Oliveira R.J., Carvalho F., de Lourdes Bastos M., et al. Modulation of P-glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol Ther. 2015;149:1–123. doi: 10.1016/j.pharmthera.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z., Shi T., Zhang L., Zhu P., Deng M., Huang C., et al. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: a review of the past decade. Cancer Lett. 2016;370:153–164. doi: 10.1016/j.canlet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 18.Feng S., Zhou H., Wu D., Zheng D., Qu B., Liu R., et al. Nobiletin and its derivatives overcome multidrug resistance (MDR) in cancer: total synthesis and discovery of potent MDR reversal agents. Acta Pharm Sin B. 2020;10:327–343. doi: 10.1016/j.apsb.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gottesman M.M., Fojo T., Bates S.E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 20.Baumert C., Hilgeroth A. Recent advances in the development of P-gp inhibitors. Anti Cancer Agents Med Chem. 2009;9:415–436. doi: 10.2174/1871520610909040415. [DOI] [PubMed] [Google Scholar]
- 21.Chen T., Wang C., Liu Q., Meng Q., Sun H., Huo X., et al. Dasatinib reverses the multidrug resistance of breast cancer MCF-7 cells to doxorubicin by downregulating P-gp expression via inhibiting the activation of ERK signaling pathway. Cancer Biol Ther. 2015;16:106–114. doi: 10.4161/15384047.2014.987062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai C.L., Tiwari A.K., Wu C.P., Su X.D., Wang S.R., Liu D.G., et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Z., Tiwari A.K., Shukla S., Robey R.W., Singh S., Kim I.W., et al. Sildenafil reverses ABCB1- and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. 2011;71:3029–3041. doi: 10.1158/0008-5472.CAN-10-3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuruo T., Iida H., Tsukagoshi S., Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981;41:1967–1972. [PubMed] [Google Scholar]
- 25.Minderman H., O'Loughlin K.L., Pendyala L., Baer M.R. VX-710 (biricodar) increases drug retention and enhances chemosensitivity in resistant cells overexpressing P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Clin Cancer Res. 2004;10:1826–1834. doi: 10.1158/1078-0432.ccr-0914-3. [DOI] [PubMed] [Google Scholar]
- 26.Kathawala R.J., Gupta P., Ashby C.R., Jr., Chen Z.S. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat. 2015;18:1–17. doi: 10.1016/j.drup.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Wu X., Yin C., Ma J., Chai S., Zhang C., Yao S., et al. Polyoxypregnanes as safe, potent, and specific ABCB1-inhibitory pro-drugs to overcome multidrug resistance in cancer chemotherapy in vitro and in vivo. Acta Pharm Sin B. 2021;11:1885–1902. doi: 10.1016/j.apsb.2020.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pennock G.D., Dalton W.S., Roeske W.R., Appleton C.P., Mosley K., Plezia P., et al. Systemic toxic effects associated with high-dose verapamil infusion and chemotherapy administration. J Natl Cancer Inst. 1991;83:105–110. doi: 10.1093/jnci/83.2.105. [DOI] [PubMed] [Google Scholar]
- 29.Bechtel L.K., Haverstick D.M., Holstege C.P. Verapamil toxicity dysregulates the phosphatidylinositol 3-kinase pathway. Acad Emerg Med. 2008;15:368–374. doi: 10.1111/j.1553-2712.2008.00088.x. [DOI] [PubMed] [Google Scholar]
- 30.Koski A., Raki M., Nokisalmi P., Liikanen I., Kangasniemi L., Joensuu T., et al. Verapamil results in increased blood levels of oncolytic adenovirus in treatment of patients with advanced cancer. Mol Ther. 2012;20:221–229. doi: 10.1038/mt.2011.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beksac M., Akan H., Koc H., Ilhan O., Erturk S., Guneyli A., et al. P-glycoprotein expression in refractory hematological neoplasms and circumvention of resistance with verapamil or cyclosporine A containing protocols. Med Oncol Tumor Pharmacother. 1992;9:101–105. [PubMed] [Google Scholar]
- 32.Mickisch G.H., Noordzij M.A., vd Gaast A., Gebreamlack P., Kohrmann K.U., Mogler-Drautz E., et al. Dexverapamil to modulate vinblastine resistance in metastatic renal cell carcinoma. J Cancer Res Clin Oncol. 1995;121 Suppl 3:R11–R16. doi: 10.1007/BF02351065. [DOI] [PubMed] [Google Scholar]
- 33.Tidefelt U., Liliemark J., Gruber A., Liliemark E., Sundman-Engberg B., Juliusson G., et al. P-Glycoprotein inhibitor valspodar (PSC 833) increases the intracellular concentrations of daunorubicin in vivo in patients with P-glycoprotein-positive acute myeloid leukemia. J Clin Oncol. 2000;18:1837–1844. doi: 10.1200/JCO.2000.18.9.1837. [DOI] [PubMed] [Google Scholar]
- 34.Kelly R.J., Draper D., Chen C.C., Robey R.W., Figg W.D., Piekarz R.L., et al. A pharmacodynamic study of docetaxel in combination with the P-glycoprotein antagonist tariquidar (XR9576) in patients with lung, ovarian, and cervical cancer. Clin Cancer Res. 2011;17:569–580. doi: 10.1158/1078-0432.CCR-10-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bardelmeijer H.A., Ouwehand M., Beijnen J.H., Schellens J.H., van Tellingen O. Efficacy of novel P-glycoprotein inhibitors to increase the oral uptake of paclitaxel in mice. Invest New Drugs. 2004;22:219–229. doi: 10.1023/B:DRUG.0000026248.45084.21. [DOI] [PubMed] [Google Scholar]
- 36.Gottesman M.M., Ludwig J., Xia D., Szakacs G. Defeating drug resistance in cancer. Discov Med. 2006;6:18–23. [PubMed] [Google Scholar]
- 37.Jackson J., Leung D., Burt H. The use of ultrasound to increase the uptake and cytotoxicity of dual taxane and P-glycoprotein inhibitor loaded, solid core nanoparticles in drug resistant cells. Ultrasonics. 2020;101:106033. doi: 10.1016/j.ultras.2019.106033. [DOI] [PubMed] [Google Scholar]
- 38.Wang S., Wang S.Q., Teng Q.X., Yang L., Lei Z.N., Yuan X.H., et al. Structure-based design, synthesis, and biological evaluation of new triazolo[1,5-a]pyrimidine derivatives as highly potent and orally active ABCB1 modulators. J Med Chem. 2020;63:15979–15996. doi: 10.1021/acs.jmedchem.0c01741. [DOI] [PubMed] [Google Scholar]
- 39.Yuan S., Wang B., Dai Q.Q., Zhang X.N., Zhang J.Y., Zuo J.H., et al. Discovery of new 4-indolyl quinazoline derivatives as highly potent and orally bioavailable P-glycoprotein inhibitors. J Med Chem. 2021;64:14895–14911. doi: 10.1021/acs.jmedchem.1c01452. [DOI] [PubMed] [Google Scholar]
- 40.Wang S., Wang S.Q., Teng Q.X., Lei Z.N., Chen Z.S., Chen X.B., et al. Discovery of the triazolo[1,5-a]pyrimidine-based derivative WS-898 as a highly efficacious and orally bioavailable ABCB1 inhibitor capable of overcoming multidrug resistance. J Med Chem. 2021;64:16187–16204. doi: 10.1021/acs.jmedchem.1c01498. [DOI] [PubMed] [Google Scholar]
- 41.Akiyama S., Fojo A., Hanover J.A., Pastan I., Gottesman M.M. Isolation and genetic characterization of human KB cell lines resistant to multiple drugs. Somat Cell Mol Genet. 1985;11:117–126. doi: 10.1007/BF01534700. [DOI] [PubMed] [Google Scholar]
- 42.Aoki S., Chen Z.S., Higasiyama K., Setiawan A., Akiyama S., Kobayashi M. Reversing effect of agosterol A, a spongean sterol acetate, on multidrug resistance in human carcinoma cells. Jpn J Cancer Res. 2001;92:886–895. doi: 10.1111/j.1349-7006.2001.tb01177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyake K., Mickley L., Litman T., Zhan Z., Robey R., Cristensen B., et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999;59:8–13. [PubMed] [Google Scholar]
- 44.Zhang Y.K., Zhang H., Zhang G.N., Wang Y.J., Kathawala R.J., Si R., et al. Semi-synthetic ocotillol analogues as selective ABCB1-mediated drug resistance reversal agents. Oncotarget. 2015;6:24277–24290. doi: 10.18632/oncotarget.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang W., Fan Y.F., Cai C.Y., Wang J.Q., Teng Q.X., Lei Z.N., et al. Olmutinib (BI1482694/HM61713), a novel epidermal growth factor receptor tyrosine kinase inhibitor, reverses ABCG2-mediated multidrug resistance in cancer cells. Front Pharmacol. 2018;9:1097. doi: 10.3389/fphar.2018.01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang S.Q., Wang C., Chang L.M., Zhou K.R., Wang J.W., Ke Y., et al. Geridonin and paclitaxel act synergistically to inhibit the proliferation of gastric cancer cells through ROS-mediated regulation of the PTEN/PI3K/Akt pathway. Oncotarget. 2016;7:72990–73002. doi: 10.18632/oncotarget.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alam A., Kung R., Kowal J., McLeod R.A., Tremp N., Broude E.V., et al. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc Natl Acad Sci U S A. 2018;115:E1973–E1982. doi: 10.1073/pnas.1717044115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S.Q., Zhou K.R., Shi X.L., Lv H.F., Bie L.Y., Zhao W.J., et al. Steroidal dimer by 001 inhibits proliferation and migration of esophageal cancer cells via multiple mechanisms. Cancer Chemother Pharmacol. 2019;83:179–189. doi: 10.1007/s00280-018-3715-4. [DOI] [PubMed] [Google Scholar]
- 49.Nagayama S., Chen Z.S., Kitazono M., Takebayashi Y., Niwa K., Yamada K., et al. Increased sensitivity to vincristine of MDR cells by the leukotriene D4 receptor antagonist, ONO-1078. Cancer Lett. 1998;130:175–182. doi: 10.1016/s0304-3835(98)00132-3. [DOI] [PubMed] [Google Scholar]
- 50.Stacy A.E., Jansson P.J., Richardson D.R. Molecular pharmacology of ABCG2 and its role in chemoresistance. Mol Pharmacol. 2013;84:655–669. doi: 10.1124/mol.113.088609. [DOI] [PubMed] [Google Scholar]
- 51.Sharom F.J., Yu X., Chu J.W., Doige C.A. Characterization of the ATPase activity of P-glycoprotein from multidrug-resistant Chinese hamster ovary cells. Biochem J. 1995;308 Pt 2:381–390. doi: 10.1042/bj3080381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinez Molina D., Jafari R., Ignatushchenko M., Seki T., Larsson E.A., Dan C., et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 53.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 54.Shibue T., Weinberg R.A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu A., Wang H., Hou X., Ma Y., Yang G., Hou Y., et al. Combinatory antitumor therapy by cascade targeting of a single drug. Acta Pharm Sin B. 2020;10:667–679. doi: 10.1016/j.apsb.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schiff P.B., Fant J., Horwitz S.B. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 57.Corso S., Isella C., Bellomo S.E., Apicella M., Durando S., Migliore C., et al. A comprehensive PDX gastric cancer collection captures cancer cell-intrinsic transcriptional MSI traits. Cancer Res. 2019;79:5884–5896. doi: 10.1158/0008-5472.CAN-19-1166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.